

Abstract
Fascioliasis, a neglected foodborne disease caused by liver flukes (genus Fasciola), affects more than 200 million people worldwide. Despite technological advances, little is known about the molecular biology and biochemistry of these flukes. We present the draft genome of Fasciola gigantica for the first time. The assembled draft genome has a size of ∼1.04 Gb with an N50 and N90 of 129 and 149 kb, respectively. A total of 20 858 genes were predicted. The de novo repeats identified in the draft genome were 46.85%. The pathway included all of the genes of glycolysis, Krebs cycle, and fatty acid metabolism but lacked the key genes of the fatty acid biosynthesis pathway. This indicates that the fatty acid required for survival of the fluke may be acquired from the host bile. It may be hypothesized that the relatively larger F. gigantica genome did not evolve through genome duplications but rather is interspersed with many repetitive elements. The genomic information will provide a comprehensive resource to facilitate the development of novel interventions for fascioliasis control.
Introduction
Fascioliasis, caused by trematodes of the genus Fasciola, is an important foodborne parasitic disease belonging to the group of neglected tropical diseases (NTDs) defined by the WHO.1Fasciola hepatica and/or Fasciola gigantica infection is prevalent in over 600 million domestic ruminants worldwide (cattle, sheep, pig, donkey, buffalo, and goats), causing major economic losses of about US$3 billion p.a.2 Fascioliasis has remarkable latitudinal, longitudinal, and altitudinal distribution due to its ability to adapt to different environments and habitats, including extreme climatic conditions. F. gigantica is found in the tropical regions of Africa, Asia, and the Middle East, where it affects 25–100% of total cattle populations. It is also prevalent in the livestock populations of India, Pakistan, Indonesia, Indochina, and the Philippines. In addition, fascioliasis has been reported in the human population in 51 different countries from five continents; this indicates the geographical expansion of the problem.3−6 It has affected 2.4–17 million people and has put approximately 180 million people at risk globally.7−10 The major human fascioliasis endemic areas include Africa, Europe, the Middle East (including Egypt), Southeast Asia, and Latin America; the highest prevalence at 72–100% is observed in Bolivian Altiplano.11,12 Interestingly, the parasite is better adapted to human hosts in hyperendemic areas.3 Most cases of human fascioliasis are reported on F. hepatica,3,6,11,12 although a few reports on F. gigantica causing human infection are available.13−15
The adult F. gigantica is hermaphroditic and is capable of self-fertilization. The life cycle of Fasciola involves an intermediate host snail of the family Lymnaeidae and a mammalian definitive host. The infection starts on ingesting food contaminated with the larval stage of F. gigantica, i.e., metacercariae, which are found floating freely in fresh water or attached to water plants. The metacercariae exist in the duodenum of the mammalian host and then migrate to the liver through the intestinal wall; the adults mature in the biliary ducts. The eggs are passed into the intestine and then excreted out through feces.3 When the young flukes migrate through the liver, they cause clinical symptoms, such as abdominal pain, weight loss, fever, nausea, vomiting, hepatomegaly, hepatic tenderness, and eosinophilia. The infection causes extensive damage to the liver and may lead to portal cirrhosis. Long-term infection by Fasciola results in chronic stimulation of the bile duct epithelium due to the excretory-secretory (ES) products released from parasites into the host bile environment.16 These ES products have key roles in feeding behavior, detoxification of bile components, and immune evasion by liver flukes.16 Transcriptome data sets for F. gigantica include substantial representation of ES products, suggesting a role in the infection mechanism of this parasite.17 The WHO has recommended triclabendazole, a benzimidazole compound, as the drug of choice for the treatment of fascioliasis as it is active against key parasite stages, i.e., early juvenile, juvenile, and adult stages. However, recent studies have suggested that F. hepatica has gained resistance to triclabendazole in several countries.18−21 In principle, foodborne trematodes can be effectively controlled using multiple interventions implemented simultaneously across sectors.
Recently, genomes from helminth flukes, including Schistosoma japonicum,22Schistosoma mansoni,23Schistosoma haematobium,24,25Opisthorchis viverrini,26Opisthorchis felineus,27Clonorchis sinensis,28,29 and F. hepatica(30,31) have been sequenced. While the present manuscript was under communication, a genome of F. gigantica was also published;32 however, our genome was first submitted and published as a preprint article (https://www.biorxiv.org/content/10.1101/451476v1.full). These genome sequences shed light on how these organisms survive in the host environment and show their metabolic pathways for adapting to host conditions. The F. hepatica genome is one of the largest pathogen genomes sequenced to date.33 The noncoding region of the F. hepatica genome was presumed to be involved in gene regulation, while the genome size was correlated to its complex life cycle and various developmental stages. The foodborne trematodes, including F. hepatica, are generally metabolically less constrained than schistosomes and cestodes.34 The presence of endobacteria, Neorickettsia, that causes chronic illness in a variety of species, including humans, in the reproductive tissues and eggs of F. hepatica suggests a possible mechanism for vertical transmission to the mammalian host. However, its presence in the oral sucker, which helps the flukes to anchor to the biliary tract lining, further suggests a probable mechanism for horizontal transmission.34
Here, we report the draft sequence, assembly, and analysis of the F. gigantica genome. It is one of the largest parasitic genomes to be sequenced. The genomic information provides a resource to facilitate the development of novel interventions for fascioliasis control.
Results and Discussion
De Novo Genome Assembly and Annotation
To avoid technical difficulties in assembly, genomic DNA was isolated from a single adult fluke and one each of the shotgun sequencing library and mate-pair DNA library were constructed with a library size of approximately 350 bp. The Paired-end and Mate-pair libraries were sequenced using HiSeq 2500 to generate 32.7 and 1.7 Gb of data, respectively. The raw reads were then quality-filtered and adapter-trimmed. The filtered high-quality reads were assembled using SOAPdenovo-v1.5.2 program. This primary assembly was further used for gap filling by Paired-end and Mate-pair reads using GapCloser. Further, SSPACE-v2.0 was used for scaffolding. The resultant assembly was used in Chromosomer-v0.1.4a for further improvement of the assembly. The assembled draft genome obtained was 40 381 scaffolds with a genome size of 1.04 Gb (Table 1), which was similar to that of the F. hepatica genome and much larger than the genomes of other parasitic flukes (Table 2). The N50 and N90 values were 129 and 149 kb, respectively. A total of 16 465 scaffolds were larger than 10 kb size, resulting in 978.97 Mb of genome length comprising 94.11% of the genome assembly. The completeness of the genome was estimated to be 51.3%, which consisted of 48.1% complete and single copy and 3.2% complete and duplicate copy. Fragments were estimated to be 12.3% using BUSCO2.0. In comparison, the BUSCO completeness of the published genomes of Platyhelminthes ranges from 20 to 73%, as reported in WormBase database (http://parasite.wormbase.org/species.html#Platyhelminthes). The chromosome set of F. gigantica comprises 10 pairs of chromosomes, and the karyotype consists of the chromosomes with 2M, 4Sm, 3St, and 1T.35
Table 1. Assembly Features.
| description | F. gigantica |
|---|---|
| genome assembly size | 1040 230 724 bp [1.04 Gb] |
| number of scaffolds | 40 381 |
| longest scaffold length | 1 127 280 bp |
| average size of scaffolds | 25 760 bp |
| number of genes | 20 858 |
| mean protein length | 264 aa |
| number of coding exons | 54 948 |
| mean number of coding exons per gene | 3 |
| coding exons combined length | 16 599 815 |
| number of introns | 35 695 |
| mean intron length | 2612 |
Table 2. Comparison of the Nuclear Genome Assemblies of F. gigantica and Related Parasitic Flukes.
| F. gigantica (present work) | F. gigantica(32) | F. hepatica(31) | F. hepatica(30) | O. viverrini(26) | C. sinensis(28,29) | S. japonicum(22) | S. haematobium(24,25) | S. mansoni(23) | |
|---|---|---|---|---|---|---|---|---|---|
| genome size | 1.04 Gb | 1.13 Gb | 1.13 Gb | 1.27 Gb | 634.5 Mb | 320.5 Mb | 397 Mb | 385 Mb | 364 Mb |
| number of genes | 20 858 | 13 940 | 14 851 | 22 676 | 16 379 | 28 407 | 13 469 | 13 073 | 13 184 |
| mean number of exons per gene | 3 | 5.9 | 3.18 | 5.3 | 5.8 | 7.7 | 5.3 | 5.4 | 6 |
| mean exon length (bp) | 302.5 | 1376 | 257 | 303 | 254 | 312 | 222 | 246 | 222 |
| mean intron length (bp) | 2612 | 3982 | NA | 3700 | 3531 | 359 | 2059 | 2442 | 2407 |
| total GC content (%) | 43.76 | 41.80 | 44 | 47.80 | 34.06 | 33.50 | 34.30 | 34.70 |
Repeat Annotation
The de novo method-predicted F. gigantica specific repeats to be 487 374 279 bp, accounting for 46.85% of the entire genome. The total number of repeat sequences identified was represented in 40 381 scaffolds. The repeat unit length ranged from 12 to 2 253 045 bp. We have identified 21.26% LINEs, 6.76% LTR elements, 45.93% total interspersed repeats, and 15.09% of unclassified repeats, as summarized in Table 3. The details of the repetitive elements are provided in Table S1.
Table 3. Summary of the De Novo Repeats Identified.
| description | number of elements | length occupied in bp |
|---|---|---|
| SINEs | 67 024 | 11 130 748 |
| MIRs | 1689 | 186 234 |
| LINEs | 469 965 | 221 172 212 |
| LINE2 | 12 439 | 4 408 849 |
| L3/CR1 | 134 130 | 66 193 633 |
| LTR elements | 130 496 | 70 357 405 |
| ERV_class I | 344 | 32 937 |
| ERV_class II | 2413 | 579 624 |
| DNA elements | 63 140 | 18 072 908 |
| TcMar-Tigger | 176 | 53 598 |
| unclassified | 697 673 | 157 005 240 |
| total interspersed repeats | 477 738 513 | |
| small RNA | 35 107 | 6 310 113 |
| satellites | 45 457 | 7 519 158 |
| simple repeats | 68 849 | 3 254 749 |
| low complexity | 2024 | 92 148 |
Gene Prediction and Annotation
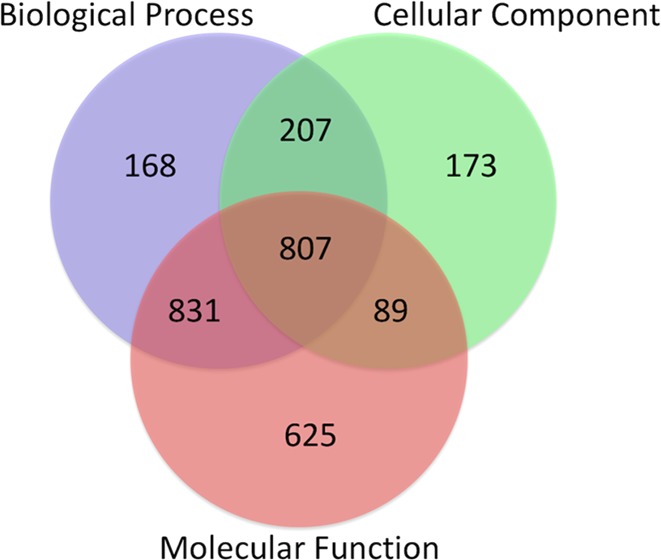
The draft genome was further used for gene prediction to identify protein coding genes using S. mansoni as the model species. A total of 20 858 genes were predicted with an average gene length of 795 bp and 264 aa. Of them, 59% (12 285 genes) were found to have homology with NCBI NR database, and 13.9% (2900 genes) were classified with gene ontology (GO) terms (details provided in Table S2). The annotation of genes showed the highest hits against F. hepatica (5248), followed by O. viverrini (1389). A total of 2900 genes were annotated with 5641 GO terms distributed in three GO subvocabularies [i.e., cellular component (CC), biological process (BP), and molecular function (MF)]. A total of 2013 genes were classified as BP, 2352 genes as MF, and 1276 genes as CC. Out of the total of 20 858 genes, 807 genes have been found to have all three categories of GO terms (Figures 1 and 2). Genes associated with similar functions were assigned to the same GO functional group. Further, the proteins for F. gigantica and F. hepatica were compared using Blast with 90% identity and were found to have a 65.3% similarity. Out of the total genes similar in both genomes, only 3688 genes were found to have GO terms, which included 1403 CC, 2474 BP, and 3143 MF; the details are mentioned in Figure S1.
Figure 1.
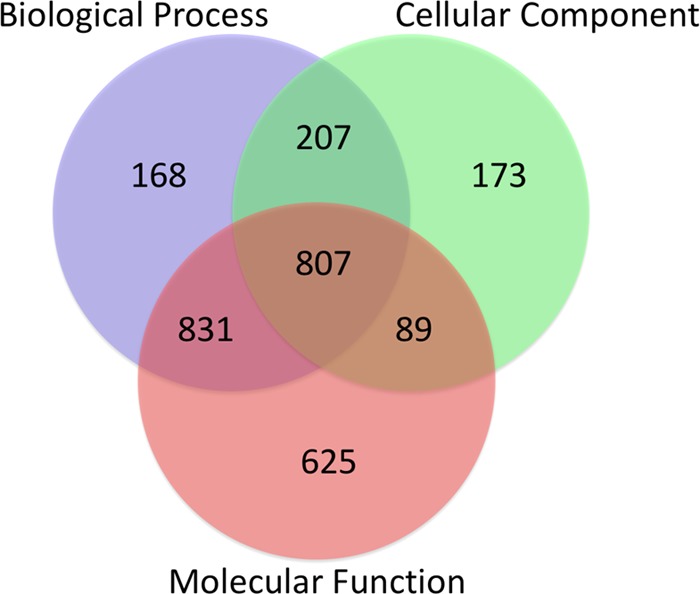
Graphical representation of the distribution of genes assigned to GO terms. The proportion of 5371 F. gigantica proteins with functional information in different GO categories is shown as the biological process, molecular function, and cellular component.
Figure 2.

GO classification of genes in cellular components, molecular function, and biological process.
The ES proteins found were cathepsin proteases (which include cathepsin L-like proteases, cathepsin B-like proteases, and cathepsin D-like proteases), glutathione transferase, fatty acid-binding protein, and glyceraldehyde-3-phosphate dehydrogenase.16,36,37 A total of 23 blast hits against ES proteins were identified from the Blast results, in which cathepsin protein was found predominantly (Table S2). Cathepsin B and L cysteine proteases are important antigens produced in trematodes, mainly in genus Fasciola, and play an important role in parasite nutrition, immune evasion, and host invasion.38,39 A total of 46 GO terms was assigned, and 4 genes had missing GO terms (Table S2). The significantly enriched proteins are classified in the following GO terms: proteolysis, cysteine-type endopeptidase activity, and regulation of catalytic activity.31 The GO terms of ES proteins were classified into 0 CC, 19 MP, and 13 BP. Earlier studies have suggested that cathepsins help the parasite to survive inside the host gall bladder and bile duct. Trematodes encode various subfamilies of cathepsins, which, in turn, provide insight into host–parasite relationships and developmentally regulated expression with the passage of the parasites through the host in the life cycle.40 Proteases may help in the activation of cathepsins, which, in turn, facilitate the digestion of host tissues, releasing essential amino acids.22 Of the 20 858 predicted proteins, about 28% (5783) did not have sufficient similarity to proteins in other organisms to justify the provision of functional assignments or known functions. They were classified as hypothetical proteins.
Annotation of Conserved Domains
The search made against InterPro database provided 14 487 InterPro hits, 4810 InterPro hits with GO terms, and 6371 nonhits. The GO terms in InterPro were merged, which resulted in 9039 GO before merging, 12 285 GO after merging, 20 351 confirmed IPS GO, and 1608 too general IPS GO.
The analysis revealed that 5205 protein sequences were categorized into 1591 domains and 2448 families. InterPro domains/families were sorted according to the assigned gene sequences; the distribution of the top 20 InterPro domains is represented in Figure 3. The most abundant domain (IPR000477) reverse transcriptase domain was obtained with 1155 annotated gene sequences, followed by (IPR001584) integrase catalytic core with 235 annotated gene sequences and (IPR000719) protein kinase domain with 481 annotated gene sequences. The InterPro families’ distribution is represented in Figure 4, and the top 5 families identified are (IPR036691) Endonuclease/exonuclease/phosphatase superfamily, (IPR027124) SWR1-complex protein, (IPR027417) P-loop containing nucleoside triphosphate hydrolase, (IPR036397) ribonuclease H superfamily, and (IPR012337) ribonuclease H-like superfamily.
Figure 3.

Representation of the 20 most abundant InterPro domains revealed by InterProScan (IPS) annotation.
Figure 4.

Representation of the 20 most abundant InterPro families revealed by InterProScan annotation.
The search found 2084 Pfam domains in 6693 genes, in which the reverse transcriptase domain [PF00078] and integrase, catalytic core [PF00665] domains were highly represented by 989 and 175 genes, respectively. The details of the conserved domains/families are provided in Table S3.
Pathway Analysis
KAAS was used to carry out ortholog and mapping of the genes to the biological pathways. The annotated genes were compared against those available in the kyoto encyclopedia of genes and genomes (KEGG) database using BLASTx with a default threshold bit score value and an expected threshold. The total assigned KO IDs were 1343 of 4016 genes that were mapped to respective pathways (details provided in Table S2). The mapped genes represented a metabolic pathway of major biomolecules, such as carbohydrates, amino acids, and other pathways.
F. gigantica can obtain energy from both aerobic and anaerobic metabolism.41 The adult metabolism is anaerobic, and juvenile metabolism is almost aerobic. It is also evident that all liver flukes inhabit the bile duct, which is anaerobic, but for the survival in the intermediate host, biochemical pathways of aerobic metabolism play crucial roles. The glycolytic pathway shows the presence of all of the key enzymes, such as hexokinase [EC: 2.7.1.1], enolase [EC: 4.2.1.11], pyruvate kinase [EC: 2.7.1.40], and lactate dehydrogenase [EC: 1.1.1.27] (Figure S2). Some of the genes involved in energy metabolism were absent, indicating that the adult worms utilize the glucose exogenously from the glycolytic pathway or may absorb nutrients from the host under anaerobic conditions.28 All of the genes of the Krebs cycle were present (Figure S3). In the fatty acid metabolism pathway, all of the genes encoding enzymes were present (Figure S4). In contrast, only three enzymes, acetyl-CoA carboxylase/biotin carboxylase 1 [EC: 6.4.1.2 6.3.4.14], 3-oxoacyl-[acyl-carrier-protein] synthase II [EC: 2.3.1.179], and long-chain acyl-CoA synthetase [EC: 6.2.1.3], were present for the fatty acid biosynthesis pathway (Figure S5). It is known that the fatty acid-binding proteins in liver flukes play a crucial role in utilizing the fatty acid produced by the host bile. Therefore, liver flukes do not need to synthesize their fatty acids endogenously.28 The genes of fatty acid metabolism were present, but certain genes of the fatty acid biosynthesis pathway were missing. This indicates that the fatty acid required for the survival of the fluke may be acquired from the host bile.
Analysis of Orthologous Groups
F. gigantica and F. hepatica genomes were predicted to have 20 858 and 33 454 proteins, which resulted in 9365 clusters. A total of 6241 core genes (i.e., in the cluster, multiple copies of genes are present) and 5654 single copies of gene clusters were identified between the two genomes using OrthoVenn (Figure 5A). In addition, 905 and 2219 unique ortholog clusters were deciphered in F. gigantica and F. hepatica genome, respectively.
Figure 5.
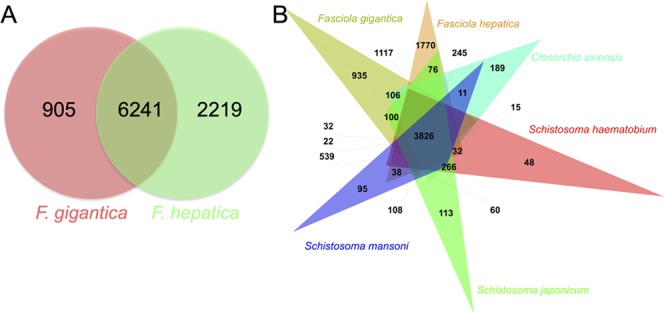
Venn diagram showing the phylogenetic distribution of orthologous protein families. (A) Between F. gigantica and F. hepatica. (B) Between F. hepatica, F. gigantica, S. mansoni, and O. viverrini.
Similarly, we compared six genomes, i.e., F. gigantica, F. hepatica, S. mansoni, S. japonicum, S. haematobium, and C. sinensis. The total predicted proteins for S. mansoni, S. japonicum, S. haematobium, and C. sinensis were 11 774, 12 738, 11 140, and 13 634, respectively. Total clusters generated were 14 288, out of which 11 138 orthologous clusters were common in at least two species, and 1863 were single copy gene clusters. The total number of clusters identified in each genome is 7664, 10 289, 8298, 8010, 8455, and 7664, respectively. The core genes identified were 3826 from all of the six species, as shown in Figure 5B. The unique orthologous clusters identified in F. gigantica, F. hepatica, S. mansoni, S. japonicum, S. haematobium, and C. sinensis were 935, 1770, 95, 113, 48, and 189, respectively. Details are provided in Table S4.
Conclusions
F. gigantica is a major parasite of livestock worldwide, causing huge economic losses to agriculture and 2.4–17 million human infections annually. We studied the draft genome of the organism, which is among the largest known parasitic genomes at 1.04 Gb. The relatively larger genome size suggests that F. gigantica genome did not evolve through whole-genome duplications but rather interspersed with many repetitive elements, such as DNA transposons, SINEs, and LINEs. Detailed comparative genome sequencing will provide answers to the large genome size of this parasite. The genomic information will provide new insights into its adaptation to the host environment, and external selection pressures and will help in the development of novel therapies for fascioliasis control.
Methods
DNA Isolation
F. gigantica flukes were collected from the liver of naturally infected cattle from the Bara Bazar slaughterhouse, Shillong, India (latitude- 25.5 724 472; longitude- 91.87 45 219). The whole worm was washed with 70% ethanol, followed by rinsing several times with 1× phosphate buffer saline. Individual flukes were immediately frozen in liquid nitrogen and stored at −80 °C until processing for genomic DNA extraction. A single individual worm was crushed in liquid nitrogen to isolate its genomic DNA using the standard phenol–chloroform extraction method. The quality and integrity of the isolated DNA were checked on 0.8% Agarose gel and a Nanodrop spectrofluorimeter.
DNA Library Construction and Sequencing
One shotgun sequencing library and one Mate-pair DNA library were constructed according to the Illumina Sample Preparation Guide (Illumina, San Diego, CA). The shotgun Paired-end sequencing library with an insert size of approximately 350 bp was prepared using the TruSeq Nano DNA Library Prep Kit for Illumina. Briefly, 200 ng of DNA was fragmented by Covaris M220 to generate a mean fragment distribution of 300–400 bp. Covaris shearing generates dsDNA fragments with 3′ or 5′ overhangs that were then subjected to End Repair Mix to convert the overhangs into blunt ends. The 3′ to 5′ exonuclease activity of this mix removes the 3′ overhangs, and the 5′ to 3′ polymerase activity fills in the 5′ overhangs. A single “A” base was then added to the ends of the polished DNA fragments followed by adapter ligation to ensure a low formation rate of chimera (concatenated template). Indexing adapters were ligated to the ends of the DNA fragments to prepare them for hybridization onto a flow cell. The ligated products were size-selected using Agencourt AMPure XP beads (Beckman Coulter Life Sciences) and polymerase chain reaction (PCR)-enriched with the Illumina adapter index PCR primer for six cycles.
The Mate-pair sequencing library was prepared using the Illumina Nextera Mate-pair Sample Preparation Kit. Briefly, 4 μg of the high-quality gDNA was tagmented using Mate-pair transposomes. Using Zymo Genomic DNA Clean & Concentrator kit (Zymo Research), the tagmented DNA was purified and then fragmented for circularization by repairing the ends by strand displacement reactions. Short fragments less than 1500 bp were removed using Ampure XP bead clean up steps. Precise size selection was carried out using Pippin prep system to select 8–11 kb fragments, followed by clean-up using Zymo clean Genomic DNA Clean & Concentrator Kit. The DNA fragments were then self-circularized by an intramolecular ligation, and noncircularized DNA was removed by DNA exonuclease treatment. The large circularized DNA fragments were physically sheared to smaller sized fragments (approximately 300–1000 bp) in Covaris using a defined shearing parameter. The sheared DNA fragments (Mate-pair fragments) containing the biotinylated junction adapter were purified by binding to streptavidin magnetic beads, and the unwanted, un-biotinylated molecules were removed through a series of washes. The streptavidin bead bound fragments were then subjected to end repair, A-tailing, Illumina adapter ligation, and final PCR enrichment for the Mate-pair fragments that have TruSeq DNA adapters on both of the ends.
The library validation was carried out using Tape Station 4200 (Agilent Technologies) using the D1000 Screen Tape assay kit. The Paired-end sequencing run was performed on HiSeq 2500 (Illumina) using 2 × 125 bp read chemistry.
Genome Assembly
The whole-genome sequencing was carried out for Paired-end and Mate-pair library using HiSeq. 2500 with 2 × 125 bp chemistry. The raw Mate-pair reads were extracted using an in-house script based on their orientation and presence of the junction adapter between read1 and read2. The reads having the junction adapter in between the reads were used as Mate-pair reads.42 The raw reads were adapter-trimmed and quality-filtered using Trimmomatic (v 0.35)43 with a minimum read length cut-off of 100 bp. The assembly of Paired-end and Mate-pair reads was carried out using SOAPDenovo (v1.5.2) with an optimized 57 kmer length. After the primary assembly, GapCloser was used for gap filling and scaffolding with both Paired-end and Mate-pair libraries. Further, scaffolding was carried out using SSPACE-v2.0.5 The resultant assembly was used with the available genome of F. hepatica (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/002/763/495/GCA_002763495.1_F_hepatica_1.0.allpaths.pg) using Chromosomer-v0.1.4a.5 The assembled draft genome was used in downstream analysis. The completeness of the genome was estimated using BUSCO2.0. De novo repeat identification was performed using RepeatModeler v1.0.10. The de novo repeat libraries were constructed using the draft genome with RepeatModeler, which contains two repeat finding programs (RECON and RepatScout). This resulted in a repeat library with classified repeat families that was used in RepeatMasker v4.0.6 as the repeat library, on the draft genome to identify the de novo repeats.
Gene Annotation
The draft genome of F. gigantica was used for gene prediction using Augustus v3.2.144 with the gene model parameters tuned for Schistosoma; the rest of the parameters were kept as default. Functional annotations of the predicted genes were performed using BLASTx program, keeping an e value 1 × 10–6 against the NCBI NR database. BLASTx determines the homologous sequences for the genes against NR database. Homologs of F. gigantica-predicted protein sequences were identified using BLAST, and the functional domains were identified using InterPro. The results of BLAST searches were used as an input to Blast2GO PRO.45 On the basis of the BLAST hits obtained, GO annotation was performed to obtain the GO terms and classify them into BP, MF, and CC. The GO terms associated with each of the BLAST results (mapping step) and the GO annotation assigned to the query (annotation step) were obtained. Further, the conserved domain/motifs were identified using InterProScan (IPS), an online plugin of BLAST2GO that combines various protein signature recognition methods with the Interpro database. The resulting GO terms were merged with the GO term results obtained from the above annotation step. The protein coding gene sequences of F. gigantica and F. hepatica (PRJEB6687) (downloaded from WormBase WBPS10: http://parasite.wormbase.org) were aligned using Blastn to identify the similarity in the protein coding genes. The F. hepatica genes were used as a database for the Blast against F. gigantica protein with an e value of ×10–5.
Pathway Analysis
To identify the potential involvement of the predicted genes of F. gigantica in biological pathways, the predicted genes were aligned to the KEGG pathway database using the kyoto encyclopedia of genes and genomes (KEGG) automatic annotation server.46−48 KEGG analysis includes KEGG Orthology (KO) assignments and Corresponding Enzyme Commission (EC) numbers and metabolic pathways of predicted genes using KEGG automated annotation server KAAS (http://www.genome.jp/kaas-bin/kaas_main). The genes’ distribution under the respective EC number was used to map them to the KEGG biochemical pathways. This process provides an overview of the different metabolic processes active within an organism and enables further understanding of the biological functions of the genes.
Identification of Orthologous Groups
The protein sequences of F. hepatica, S. mansoni, S. japonicum, S. haematobium, and, C. sinensis were obtained from the WormBase Parasite database (http://parasite.wormbase.org). Protein sequences of F. gigantica and F. hepatica were used to perform an all-against-all comparison using BLASTP with orthoVenn at default parameters.49 The core genes and unique genes were identified between F. gigantica and F. hepatica genomes. The ortholog analysis was also performed with F. hepatica, S. mansoni, S. japonicum, S. haematobium, and C. sinensis. This enabled us to elucidate the function and evolution of protein across the six species.
Acknowledgments
T.T. Lab is supported by the Department of Biotechnology, Government of India, India [BT/PR24905/ NER/95/901/2017].
Glossary
Abbreviations Used
- F. gigantica
Fasciola gigantica
- F. hepatica
Fasciola hepatica
- NTDs
neglected tropical diseases
- CC
cellular component
- BP
biological process
- MF
molecular function
- IPS
InterProScan
- KO
KEGG orthology
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00980.
Details of GO terms, which included 1403 cellular component (CC), 2474 biological process (BP), and 3143 molecular function (MF) (Figure S1); schematic pathway of glycolysis (Figure S2); schematic pathway of the TCA cycle (Figure S3); schematic pathway of fatty acid degradation (Figure S4); schematic pathway of fatty acid biosynthesis (Figure S5); details of the repeats (Table S1); list of genes classified with GO terms (Table S2); details of the conserved domains/families (Table S3); details of unique orthologous clusters (Table S4) (PDF)
Accession Codes
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession MKHB00000000. The version described in this paper is version MKHB03000000.
Author Contributions
§ T.P. and A.G. contributed equally to the work.
The authors declare no competing financial interest.
Notes
The whole genome from this study has been submitted to/ENA/GenBank/DDBJ Accession number: MKHB00000000 ENA/GenBank/DDBJ Study number: PRJNA339660 ENA/GenBank/DDBJ.
Supplementary Material
References
- FAO/WHO. Multicriteria-Based Ranking for Risk Management of Food-Borne Parasites, 2014; p 287.
- Mas-Coma S.; Bargues M. D.; Valero M. A. Fascioliasis and other plant-borne trematode zoonoses. Int. J. Parasitol. 2005, 35, 1255–1278. 10.1016/j.ijpara.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Mas-Coma S.; Valero M. A.; Bargues M. D. Chapter 2 Fasciola, lymnaeids and human fascioliasis, with a global overview on disease transmission, epidemiology, evolutionary genetics, molecular epidemiology and control. Adv. Parasitol. 2009, 69, 41–146. 10.1016/S0065-308X(09)69002-3. [DOI] [PubMed] [Google Scholar]
- Cwiklinski K.; O’Neill S. M.; Donnelly S.; Dalton J. P. A prospective view of animal and human Fasciolosis. Parasite Immunol. 2016, 38, 558–568. 10.1111/pim.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi K.; Bargues M. D.; O’Neill S.; Mas-Coma S. Fascioliasis: a worldwide parasitic disease of importance in travel medicine. Travel Med. Infect. Dis. 2014, 12, 636–649. 10.1016/j.tmaid.2014.09.006. [DOI] [PubMed] [Google Scholar]
- Fried B.; Abruzzi A. Food-borne trematode infections of humans in the United States of America. Parasitol. Res. 2010, 106, 1263–1280. 10.1007/s00436-010-1807-0. [DOI] [PubMed] [Google Scholar]
- Mas-Coma S.; Valero M. A.; Bargues M. D.. Fascioliasis. In Digenetic Trematodes;Toledo R.; Fried B., Eds.; Springer: New York, 2014; pp 77–114. [Google Scholar]
- Murrell K. D.; Crompton D. W. T.; Motarjemi Y.; Adams M.. Foodborne trematodes and helminths. In Emerging Foodborne Pathogens; Woodhead Publishing: Cambridge, United Kingdom, 2006; pp 222–252. [Google Scholar]
- WHO. Control of Foodborne Trematode Infections; Geneva, Switzerland, 1995.
- Tripathi T.; Suttiprapa S.; Sripa B. Unusual thiol-based redox metabolism of parasitic flukes. Parasitol. Int. 2017, 66, 390–395. 10.1016/j.parint.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Mas-Coma S. Epidemiology of fascioliasis in human endemic areas. J. Helminthol. 2005, 79, 207–216. 10.1079/JOH2005296. [DOI] [PubMed] [Google Scholar]
- Mas C. S.; Angles R.; Strauss W.; Esteban J. G.; Oviedo J. A.; Buchon P. Human fasciolasis in Bolivia: a general analysis and a critical review of existing data. Res. Rev. Parasitol. 1995, 55, 73–79. [Google Scholar]
- Ganaie N., First liver fluke case reported in JK. Rising Kashmir 2016. [Google Scholar]
- Menon P.; Sinha A. K.; Rao K. L. N.; Khurana S.; Lal S.; Thapa B. R. Biliary Fasciola gigantica infestation in a nonendemic area--An intraoperative surprise. J. Pediatr. Surg. 2015, 50, 1983–1986. 10.1016/j.jpedsurg.2015.08.009. [DOI] [PubMed] [Google Scholar]
- Rana S. S.; Bhasin D. K.; Nanda M.; Singh K. Parasitic infestations of the biliary tract. Curr. Gastroenterol. Rep. 2007, 9, 156–164. 10.1007/s11894-007-0011-6. [DOI] [PubMed] [Google Scholar]
- Morphew R. M.; Wright H. A.; LaCourse E. J.; Woods D. J.; Brophy P. M. Comparative proteomics of excretory-secretory proteins released by the liver fluke Fasciola hepatica in sheep host bile and during in vitro culture ex host. Mol. Cell. Proteomics 2007, 6, 963–972. 10.1074/mcp.M600375-MCP200. [DOI] [PubMed] [Google Scholar]
- Young N. D.; Jex A. R.; Cantacessi C.; Hall R. S.; Campbell B. E.; Spithill T. W.; Tangkawattana S.; Tangkawattana P.; Laha T.; Gasser R. B. A portrait of the transcriptome of the neglected trematode, Fasciola gigantica- biological and biotechnological implications. PLoS Neglected Trop. Dis. 2011, 5, e1004 10.1371/journal.pntd.0001004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles G. C. Anthelmintic activity of triclabendazole. J. Helminthol. 1986, 60, 210–212. 10.1017/S0022149X00026110. [DOI] [PubMed] [Google Scholar]
- Gordon D.; Zadoks R.; Skuce P.; Sargison N. Confirmation of triclabendazole resistance in liver fluke in the UK. Vet. Rec. 2012, 171, 159–60. 10.1136/vr.e5381. [DOI] [PubMed] [Google Scholar]
- Kelley J. M.; Elliott T. P.; Beddoe T.; Anderson G.; Skuce P.; Spithill T. W. Current threat of triclabendazole resistance in Fasciola hepatica. Trends Parasitol. 2016, 32, 458–469. 10.1016/j.pt.2016.03.002. [DOI] [PubMed] [Google Scholar]
- Winkelhagen A. J.; Mank T.; de Vries P. J.; Soetekouw R. Apparent triclabendazole-resistant human Fasciola hepatica infection, the Netherlands. Emerging Infect. Dis. 2012, 18, 1028–1029. 10.3201/eid1806.120302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Zheng H.; Chen Y.; Zhang L.; Wang K.; Guo J.; Huang Z.; Zhang B.; Huang W.; Jin K.; Dou T.; Hasegawa M.; Wang L.; Zhang Y.; Zhou J.; Tao L.; Cao Z.; Li Y.; Vinar T.; Brejova B.; Brown D.; Li M.; Miller D. J.; Blair D.; Zhong Y.; Chen Z.; Liu F.; Hu W.; Wang Z. Q.; Zhang Q. H.; Song H. D.; Chen S.; Xu X.; Xu B.; Ju C.; Huang Y.; Brindley P. J.; McManus D. P.; Feng Z.; Han Z. G.; Lu G.; Ren S.; Wang Y.; Gu W.; Kang H.; Chen J.; Chen X.; Chen S.; Wang L.; Yan J.; Wang B.; Lv X.; Jin L.; Wang B.; Pu S.; Zhang X.; Zhang W.; Hu Q.; Zhu G.; Wang J.; Yu J.; Wang J.; Yang H.; Ning Z.; Beriman M.; Wei C. L.; Ruan Y.; Zhao G.; Wang S.; Liu F.; Zhou Y.; Wang Z. Q.; Lu G.; Zheng H.; Brindley P. J.; McManus D. P.; Blair D.; Zhang Q. H.; Zhong Y.; Wang S.; Han Z. G.; Chen Z.; Wang S.; Han Z. G.; Chen Z. The Schistosoma japonicum genome reveals features of host-parasite interplay. Nature 2009, 460, 345–351. 10.1038/nature08140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman M.; Haas B. J.; LoVerde P. T.; Wilson R. A.; Dillon G. P.; Cerqueira G. C.; Mashiyama S. T.; Al-Lazikani B.; Andrade L. F.; Ashton P. D.; Aslett M. A.; Bartholomeu D. C.; Blandin G.; Caffrey C. R.; Coghlan A.; Coulson R.; Day T. A.; Delcher A.; DeMarco R.; Djikeng A.; Eyre T.; Gamble J. A.; Ghedin E.; Gu Y.; Hertz-Fowler C.; Hirai H.; Hirai Y.; Houston R.; Ivens A.; Johnston D. A.; Lacerda D.; Macedo C. D.; McVeigh P.; Ning Z.; Oliveira G.; Overington J. P.; Parkhill J.; Pertea M.; Pierce R. J.; Protasio A. V.; Quail M. A.; Rajandream M. A.; Rogers J.; Sajid M.; Salzberg S. L.; Stanke M.; Tivey A. R.; White O.; Williams D. L.; Wortman J.; Wu W.; Zamanian M.; Zerlotini A.; Fraser-Liggett C. M.; Barrell B. G.; El-Sayed N. M. The genome of the blood fluke Schistosoma mansoni. Nature 2009, 460, 352–358. 10.1038/nature08160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N. D.; Jex A. R.; Li B.; Liu S.; Yang L.; Xiong Z.; Li Y.; Cantacessi C.; Hall R. S.; Xu X.; Chen F.; Wu X.; Zerlotini A.; Oliveira G.; Hofmann A.; Zhang G.; Fang X.; Kang Y.; Campbell B. E.; Loukas A.; Ranganathan S.; Rollinson D.; Rinaldi G.; Brindley P. J.; Yang H.; Wang J.; Wang J.; Gasser R. B. Whole-genome sequence of Schistosoma haematobium. Nat. Genet. 2012, 44, 221–225. 10.1038/ng.1065. [DOI] [PubMed] [Google Scholar]
- Mitreva M. The genome of a blood fluke associated with human cancer. Nat. Genet. 2012, 44, 116–118. 10.1038/ng.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N. D.; Nagarajan N.; Lin S. J.; Korhonen P. K.; Jex A. R.; Hall R. S.; Safavi-Hemami H.; Kaewkong W.; Bertrand D.; Gao S.; Seet Q.; Wongkham S.; Teh B. T.; Wongkham C.; Intapan P. M.; Maleewong W.; Yang X.; Hu M.; Wang Z.; Hofmann A.; Sternberg P. W.; Tan P.; Wang J.; Gasser R. B. The Opisthorchis viverrini genome provides insights into life in the bile duct. Nat. Commun. 2014, 5, 4378 10.1038/ncomms5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ershov N. I.; Mordvinov V. A.; Prokhortchouk E. B.; Pakharukova M. Y.; Gunbin K. V.; Ustyantsev K.; Genaev M. A.; Blinov A. G.; Mazur A.; Boulygina E.; Tsygankova S.; Khrameeva E.; Chekanov N.; Fan G.; Xiao A.; Zhang H.; Xu X.; Yang H.; Solovyev V.; Lee S. M.-Y.; Liu X.; Afonnikov D. A.; Skryabin K. G. New insights from Opisthorchis felineus genome: update on genomics of the epidemiologically important liver flukes. BMC Genomics 2019, 20, 399 10.1186/s12864-019-5752-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Chen W.; Huang Y.; Sun J.; Men J.; Liu H.; Luo F.; Guo L.; Lv X.; Deng C.; Zhou C.; Fan Y.; Li X.; Huang L.; Hu Y.; Liang C.; Hu X.; Xu J.; Yu X. The draft genome of the carcinogenic human liver fluke Clonorchis sinensis. Genome Biol. 2011, 12, R107 10.1186/gb-2011-12-10-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Chen W.; Wang X.; Liu H.; Chen Y.; Guo L.; Luo F.; Sun J.; Mao Q.; Liang P.; Xie Z.; Zhou C.; Tian Y.; Lv X.; Huang L.; Zhou J.; Hu Y.; Li R.; Zhang F.; Lei H.; Li W.; Hu X.; Liang C.; Xu J.; Li X.; Yu X. The carcinogenic liver fluke, Clonorchis sinensis: new assembly, reannotation and analysis of the genome and characterization of tissue transcriptomes. PLoS One 2013, 8, e54732 10.1371/journal.pone.0054732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cwiklinski K.; Dalton J. P.; Dufresne P. J.; La Course J.; Williams D. J.; Hodgkinson J.; Paterson S. The Fasciola hepatica genome: gene duplication and polymorphism reveals adaptation to the host environment and the capacity for rapid evolution. Genome Biol. 2015, 16, 71 10.1186/s13059-015-0632-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty S. N.; Tort J. F.; Rinaldi G.; Fischer K.; Rosa B. A.; Smircich P.; Fontenla S.; Choi Y.-J.; Tyagi R.; Hallsworth-Pepin K.; Mann V. H.; Kammili L.; Latham P. S.; Dell’Oca N.; Dominguez F.; Carmona C.; Fischer P. U.; Brindley P. J.; Mitreva M. Genomes of Fasciola hepatica from the Americas Reveal Colonization with Neorickettsia Endobacteria Related to the Agents of Potomac Horse and Human Sennetsu Fevers. PLoS Genet. 2017, 13, e1006537 10.1371/journal.pgen.1006537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y. J.; Fontenla S.; Fischer P. U.; Le T. H.; Costabile A.; Blair D.; Brindley P. J.; Tort J. F.; Cabada M. M.; Mitreva M. Adaptive radiation of the flukes of the family fasciolidae inferred from genome-wide comparisons of key species. Mol. Biol. Evol. 2020, 37, 84–99. 10.1093/molbev/msz204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. W.; Corvo I.; Jones P. M.; George A. M.; Padula M. P.; To J.; Cancela M.; Rinaldi G.; Tort J. F.; Roche L.; Dalton J. P. Collagenolytic activities of the major secreted cathepsin L peptidases involved in the virulence of the helminth pathogen, Fasciola hepatica. PLoS Neglected Trop. Dis. 2011, 5, e1012 10.1371/journal.pntd.0001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvo I.; Cancela M.; Cappetta M.; Pi-Denis N.; Tort J. F.; Roche L. The major cathepsin L secreted by the invasive juvenile Fasciola hepatica prefers proline in the S2 subsite and can cleave collagen. Mol. Biochem. Parasitol. 2009, 167, 41–47. 10.1016/j.molbiopara.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Yin H. Z.; Ye B. Y. [Studies on the karyotypes of Fasciola spp]. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 1990, 8, 124–126. [PubMed] [Google Scholar]
- Kalita J.; Shukla R.; Shukla H.; Gadhave K.; Giri R.; Tripathi T. Comprehensive analysis of the catalytic and structural properties of a mu-class glutathione s-transferase from Fasciola gigantica. Sci. Rep. 2017, 7, 17547 10.1038/s41598-017-17678-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetri P. B.; Shukla R.; Tripathi T. Identification and characterization of glyceraldehyde 3-phosphate dehydrogenase from Fasciola gigantica. Parasitol. Res. 2019, 118, 861–872. 10.1007/s00436-019-06225-w. [DOI] [PubMed] [Google Scholar]
- Tarasuk M.; Vichasri Grams S.; Viyanant V.; Grams R. Type I cystatin (stefin) is a major component of Fasciola gigantica excretion/secretion product. Mol. Biochem. Parasitol. 2009, 167, 60–71. 10.1016/j.molbiopara.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Caffrey C. R.; Goupil L.; Rebello K. M.; Dalton J. P.; Smith D. Cysteine proteases as digestive enzymes in parasitic helminths. PLoS Neglected Trop. Dis. 2018, 12, e0005840 10.1371/journal.pntd.0005840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack C.; Dalton J. P.; Robinson M. W. The phylogeny, structure and function of trematode cysteine proteases, with particular emphasis on the Fasciola hepatica cathepsin L family. Adv. Exp. Med. Biol. 2011, 712, 116–35. 10.1007/978-1-4419-8414-2_8. [DOI] [PubMed] [Google Scholar]
- Tielens A. G. M.; van den Heuvel J. M.; van den Bergh S. G. Changes in energy metabolism of the juvenile Fasciola hepatica during its development in the liver parenchyma. Mol. Biochem. Parasitol. 1982, 6, 277–286. 10.1016/0166-6851(82)90060-3. [DOI] [PubMed] [Google Scholar]
- Leggett R. M.; Clavijo B. J.; Clissold L.; Clark M. D.; Caccamo M. NextClip: an analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 2014, 30, 566–568. 10.1093/bioinformatics/btt702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A. M.; Lohse M.; Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M.; Keller O.; Gunduz I.; Hayes A.; Waack S.; Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A.; Gotz S.; Garcia-Gomez J. M.; Terol J.; Talon M.; Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Kanehisa M.; Sato Y.; Kawashima M.; Furumichi M.; Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M.; Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M.; Furumichi M.; Tanabe M.; Sato Y.; Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Coleman-Derr D.; Chen G.; Gu Y. Q. OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2015, 43, W78–W84. 10.1093/nar/gkv487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.