

Abstract
A general method is described for the coupling of (hetero)aryl bromides with O-alkyl sulfamate esters. The protocol relies on catalytic amounts of nickel and photoexcitable iridium complexes and proceeds under visible light at ambient temperature. This technology engages a broad range of simple and complex O-alkyl sulfamate ester substrates under mild conditions. Furthermore, it is possible to avoid undesirable N-alkylation, which was found to plague palladium-based protocols for N-arylation of O-alkyl sulfamate esters. These investigations represent the first use of sulfamate esters as nucleophiles in transition metal-catalyzed C–N coupling processes.
Graphical abstract

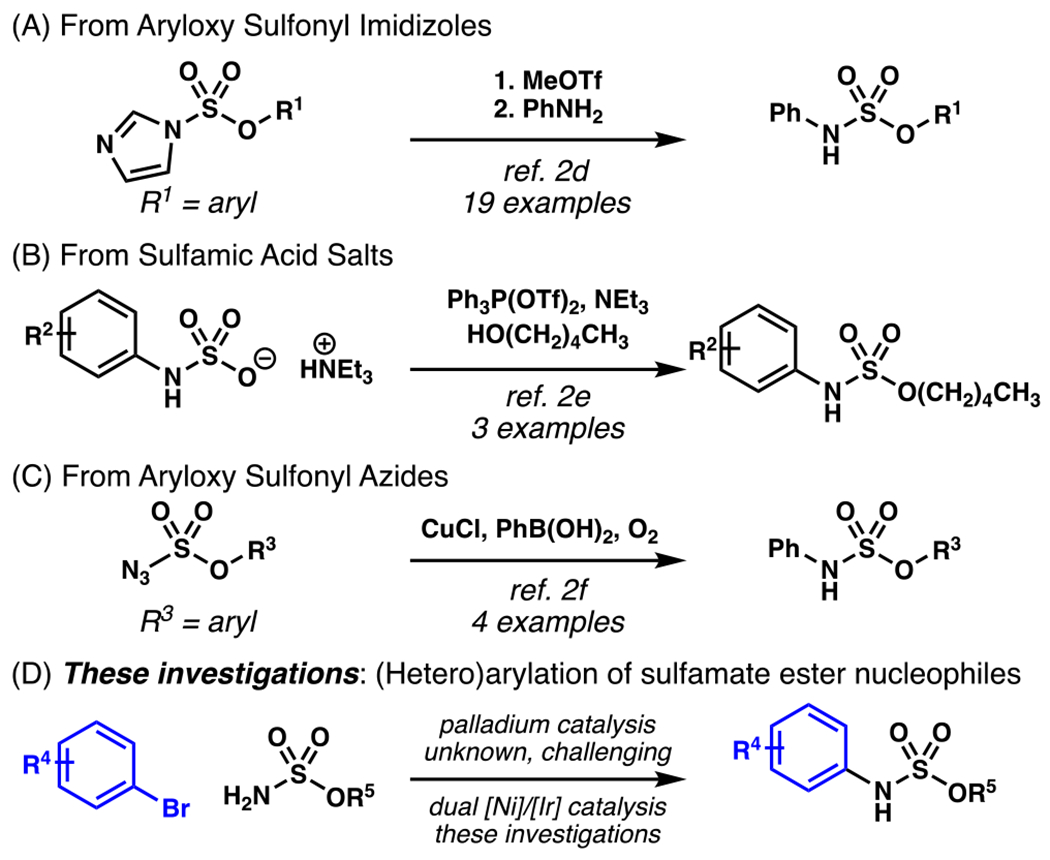
Nitrogen-containing small molecules, including arylamines, are valuable given their importance as bioactive agents.1 While a variety of methods exist to access many N-alkyl, N-aryl, N-acyl, and N-sulfonyl aniline derivatives, examples of N-aryl sulfamate esters remain limited, as user-friendly and efficient preparations of N-aryl sulfamate esters have only recently emerged (Scheme 1).2 Unfortunately, these strategies engage multistep reaction sequences and use highly reactive reagents, which may detract from their utility. Furthermore, using these approaches, preparations of sulfamate esters featuring Lewis basic N-heteroaromatic substituents are exceedingly rare, despite the importance of heteroaryl motifs in medicinally-relevant small molecules.3 Given these limitations, a transition metal-catalyzed protocol capable of coupling easily prepared primary sulfamate esters with widely available (hetero)aryl halide electrophiles would expand the accessibility of elusive N-(hetero)aryl sulfamate esters.4
Scheme 1.
Access to N-(Hetero)aryl Sulfamate Esters Remains Limited Despite Recent Strategic Innovations
Transition metal-catalyzed C(sp2)–N bond-forming reactions have revolutionized access to arylamines, and are among the most practiced synthetic manipulations in both academic and industrial settings.5 The majority of transformations developed to convert (hetero)aryl halide substrates to valuable aniline derivatives feature palladium-based catalysts.6 In addition to traditional aliphatic amines, advances in the Buchwald-Hartwig reaction have enabled the efficient coupling of a variety of less nucleophilic substrates, including amides, carbamates, sulfonamides, and sulfamides.7 While sulfamate esters are employed as electrophilic components in a variety of transition metal-mediated cross-coupling manifolds, 8 sulfamate esters have not been described as viable nucleophiles within these transformations. Owing to their low nucleophilicity, even examples of their use as substrates in SNAr reactions is rare.2g
Given that similarly nucleophilic sulfonamide and sulfamide substrates have been previously disclosed as effective components in palladium-catalyzed C(sp2)–N coupling reactions,7 we questioned whether sulfamate esters would serve as effective nucleophiles under palladium-mediated Buchwald-Hartwig amination conditions. Unfortunately, using these established protocols, the N-arylation of pentyl sulfamate (1a) was found to proceed in poor yield (Scheme 2). Furthermore, N-alkylated byproducts 3a and 3b were observed to form via a competitive decomposition pathway, indicating a potential barrier to the elucidation of an efficient palladium-catalyzed protocol.
Scheme 2.

When Applied to Sulfamate Esters, Palladium-Catalyzed N-Arylation Protocols Generate Undesired Byproducts
As compared to palladium-mediated C(sp2)–N bond forming methods, fewer technologies rely on nickel catalysis.9 Such transformations often requires a challenging reductive elimination step from Ni(II)–amido complexes, which can demonstrate increased thermal stability.10 Recently, photochemically-driven, nickel-catalyzed strategies have emerged as an approach to facilitate C(sp2)–N bond formation under mild conditions at ambient temperatures.11 We hypothesized that the mild conditions offered by photochemically-driven nickel catalysis might bypass the deleterious sulfamate ester N-alkylation processes observed when palladium catalysts were employed (Scheme 2). Despite the remarkable potential offered by this dual catalytic approach, the range of nitrogen-based nucleophiles has been limited to those previously known to efficiently engage in palladium-catalyzed processes. To date, only amines, sulfonamides, amides and carbamates, which are substrates tolerated in analogous palladium-mediated processes, have been employed in these light-enabled, nickel-catalyzed reactions.11
Herein disclosed is the first general N-(hetero)arylation reaction featuring sulfamate esters as nucleophilic components, and provides broad access to O-alkyl sulfamate esters bearing both N-aryl and N-heteroaryl substituents. This protocol employs readily available substrates and reagents, and proceeds under mild conditions. This process highlights the chemically distinct reactivity afforded by the dual catalyzed reaction manifold, and provides access to N-(hetero)aryl sulfamate esters, which have traditionally been underexplored, presumably owing to their challenging preparation.
Initial investigations to develop this N-(hetero)arylation process employed pentyl sulfamate (1a) as a model substrate, and 4-(trifluoromethyl)bromobenzene as an electrophile (Table 1). Utilizing previously reported conditions,11d,e 1.5 equiv of sulfamate 1a was treated with 1.0 equiv of bromoarene, photoactive [Ir(ppy)2(dtbbpy)]PF6 (1 mol %), NiBr2•glyme (5 mol %) and tetramethylguanidine (TMG) as a base, and furnished desired N-arylated 2a in 18% yield (entry 1). Importantly, formation of undesired N-alkylated byproducts 3a and 3b was suppressed, as unreacted 1a primarily accounted for the remaining mass balance using this dual catalytic manifold. When three equivalents of DBU were employed, full conversion to desired N-aryl sulfamate 2a was observed (entry 4).12 Either sulfamate ester or aryl bromide could be used as the limiting substrate (entries 4, 6), which may be an important consideration depending on the relative accessibilities of the requisite starting materials. Equimolar amounts of each substrate could be employed, albeit with a slight decrease in the reaction yield (entry 7).
Table 1.
Optimization of Sulfamate Ester Arylation
 | |||||
|---|---|---|---|---|---|
| entrya | photocatalyst [mol %] | 1a [equiv] | aryl halide [equiv] | base [equiv] | yield [%]b |
| 1 | [Ir(ppy)2(dtbbpy)]PF6 | 1.5 | 1.0 | TMG [1.5] | 18 |
| 2 | [Ir(ppy)2(dtbbpy)]PF6 | 1.5 | 1.0 | DBU [1.5] | 67 |
| 3 | [Ir(ppy)2(dtbbpy)]PF6 | 1.5 | 1.0 | DBU [2.0] | 80 |
| 4 | [Ir(ppy)2(dtbbpy)]PF6 | 1.5 | 1.0 | DBU [3.0] | 96 |
| 5 | [Ir(ppy)2(dtbbpy)]PF6 | 1.0 | 1.5 | DBU [2.0] | 72 |
| 6 | [Ir(ppy)2(dtbbpy)]PF6 | 1.0 | 1.5 | DBU [3.0] | > 98 |
| 7 | [Ir(ppy)2(dtbbpy)]PF6 | 1.0 | 1.0 | DBU [3.0] | 84 |
| 8 | fac-Ir(ppy)3 | 1.0 | 1.5 | DBU [3.0] | 20 |
| 9 | [Ir(ppy)2(bpy)]PF6 | 1.0 | 1.5 | DBU [3.0] | > 98 |
| 10 | [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 | 1.0 | 1.5 | DBU [3.0] | 9 |
| 11 | [Ru(bpy)3](PF6)2 | 1.0 | 1.5 | DBU [3.0] | 48 |
| 12 | 4-CzIPN | 1.0 | 1.5 | DBU [3.0] | 70 |
| 13 | none | 1.0 | 1.5 | DBU [3.0] | < 5 |
General reaction conditions: 1.0–1.5 equiv 1a, 1.0–1.5 equiv 4-bromobenzotrifluoride, photocatalyst (1 mol %), NiBr2•glyme (5 mol %), base (1.0–3.0 equiv), MeCN (0.25 M with respect to limiting reagent), with stirring in front of two 34W blue Kessil lamps for 24 h.
Isolated yield.
A variety of photochemical mediators can be used to drive this transformation, yet trends in their efficacy are limited. The yield of this transformation does not improve with increases in the oxidizing power of the excited state iridium complex (entries 6, 8–10; see supporting information for a compilation of experimentally-derived E1/2III*/II estimates). To date, the most effective photoexcitable metal complexes have triplet state energies (ET) in the range of 46.2–49.2 kcal/mol, a trend that could be anticipated for a reaction proceeding through triplet sensitization (entries 6, 9, 11; see supporting information for a compilation of ET values). Importantly, this N-arylation reaction relies on the presence of a photochemical mediator (entry 13), confirming that the process does not proceed via direct excitation of an intermediate nickel species.13
This optimized dual catalytic protocol effectively transforms a variety of ortho-, meta-, and para-substituted electron-neutral and electron-deficient bromobenzene derivatives, including those featuring nitriles, esters, and fluorides, are transformed in good yields (Scheme 3). Furthermore, chloride and boron substituents remain intact over the course of the reaction. These functional groups are important in cross-coupling technologies, and offer the opportunity for further manipulation.
Scheme 3. A Wide Range of (Hetero)aryl Bromides are Installed onto Sulfamate Estersa.

aGeneral reaction conditions: 1.0 equiv pentyl sulfamate (1a), 1.5 equiv (hetero)aryl bromide, photocatalyst (1 mol %), NiBr2•glyme (5 mol %), DBU (3.0 equiv), MeCN (0.25 M), with stirring in front of two 34W blue Kessil lamps for 12–72 h. bReaction performed with 10 mol % NiBr2•glyme. cReactions performed with NiBr2•glyme (10 mol %) and dtbbpy (4 mol %). dReaction performed in iPrOAc (0.25 M).
Due to the prevalence of heteroaromatic motifs in biologically-relevant small molecules, heteroaryl bromides were also investigated (Scheme 3). Given our interest in functionalized pyridines,14 we were pleased to find that 2-, 3-, and 4-bromopyridines were all effective arylating agents using the disclosed protocol. Moreover, this procedure transforms a range of other 6-membered heteroaryl bromides, including pyrimidine, quinoline, and azaindole substrates.
Predictably, electron-rich (hetero)arylbromides were more challenging cross-coupling partners. Fortunately, once 4,4’-di(tert-butyl)-2,2’-bipyridine is included in the reaction conditions as a ligand for nickel, the transformation of these substrates becomes more efficient. This observation mirrors that noted during the development of photocatalytically-driven nickel-promoted N-arylation of sulfonamides,11d and may prove to be a general tactic to enable the use of more challenging aryl electrophiles in photochemically-promoted nickel-catalyzed C–heteroatom coupling reactions. Using this modification, the protocol can effectively engage 4-substituted aryl bromides featuring electron-donating groups (2p–2s), as well as some 5-membered heterocyclic bromides as electrophiles. Of note, a benzothiophene (2t), a thiophene (2u), and a benzothioazole (2v) are effectively installed on sulfamate ester 1a under prolonged reaction times. These substrates overcome the common tendency of sulfur to react with nickel catalysts to deactivate them or to engage in C–S bond activation.15
This technology efficiently N-arylates complex O-alkyl sulfamate esters derived from primary and secondary alcohols, even those involving oxidatively sensitive functional groups (Scheme 4). Secondary alcohol-derived sulfamate esters based on a steroid (trans-andosterone, 2y), a vasodilator (pentoxifylline, 2z), a triterpene (enoxolone, 2aa) and an analgesic (menthol, 2ab) are efficiently transformed under the standard reaction conditions, as is a primary alcohol-derived sulfamate ester based on a diterpene (dehydroabietic acid, 2ac). The evaluated substrates highlight the compatibility of ketones, α,β-unsaturated ketones, methyl xanthines, esters, and acetals afforded by these mild reaction conditions. This protocol is not an efficient strategy to N-arylate O-aryl sulfamate esters (see supporting information for details).
Scheme 4. Evaluation of Complex Sulfamate Ester Substrates.

aGeneral reaction conditions: 1.0 equiv sulfamate, 1.5 equiv (hetero)aryl bromide, photocatalyst (1 mol %), NiBr2•glyme (5 mol %), DBU (3.0 equiv), MeCN (0.25 M), with stirring in front of two 34W blue Kessil lamps for 24–48 h. See Supplementary Information for specific experimental details.
This protocol may prove useful for the evaluation of sulfamate esters during lead optimization. N-aryl sulfamate esters have been assessed during the investigation of analogues of topiramate, an FDA approved anticonvulsant that incorporates a sulfamate ester.16 Indeed, topiramate undergoes arylation to furnish N-phenyl topiramate (2ad). This approach represents a marked improvement in efficiency over the previously reported route,14 and makes use of commercially available topiramate as a substrate.
Herein, the N-(hetero)arylation of sulfamate esters has been shown to proceed using a dual photochemically-driven, nickel-catalyzed reaction manifold. This approach marks the first use of sulfamate esters as nucleophiles in a C–N coupling reaction, and avoids potentially competitive and undesired N-alkylation processes. This general technology enables the installation of a diverse range of aryl and heteroaryl substituents onto both simple and complex O-alkyl sulfamate ester substrates, which we expect will facilitate future evaluation of N-(hetero)aryl sulfamate esters. As such, this method compliments synthetic strategies to prepare N-substituted sulfamate esters, and expands the breadth of accessible nitrogen-containing small molecules.
Supplementary Material
ACKNOWLEDGMENT
Funding was provided by the National Institutes of Health (R35GM128741-01). Characterization data were obtained on instrumentation secured with funding from the NSF (CHE-0923097, ESI-MS, George Dubay, the Duke Dept. of Chemistry Instrument Center), or the NSF, the NIH, HHMI, the North Carolina Biotechnology Center and Duke University (Duke Magnetic Resonance Spectroscopy Center). We thank Dr. Peter Silinski (Duke University) for performing high-resolution mass spectrometry.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Full experimental details, copies of NMR spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Roughley SD; Jordan AM The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]; (b) Brown DG; Boström J Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reaction Gone? J. Med. Chem 2016, 59, 4442–4458. [DOI] [PubMed] [Google Scholar]
- (2).For a general review of the physical properties of, preparation of, and uses of sulfamate esters, see:; (a) Spillane W; Malaubier J-B Sulfamic Acid and its N- and O-Substituted Derivatives. Chem. Rev 2014, 114, 2507–2586. [DOI] [PubMed] [Google Scholar]; For reports detailing the preparation of N-substituted sulfamate esters, including select examples of N-(hetero)aryl sulfamates, see:; (b) Ingram LJ; Desoky A; Ali AM; Taylor SD O- and N-Sulfations of Carbohydrates Using Sulfuryl Imidazolium Salts. J. Org. Chem 2009, 74, 6479–6485. [DOI] [PubMed] [Google Scholar]; (c) Reuillon T; Bertoli A; Griffin RJ; Miller DC; Golding BT Efficacious N-Protection or O-Aryl Sulfamates with 2,4-Dimethoxybenzyl Groups. Org. Biomol. Chem 2012, 10, 7610–7617. [DOI] [PubMed] [Google Scholar]; (d) Yang B; Sun Z; Liu C; Cui y.; Guo Z; Ren Y; Lu Z; Knapp S O-(Aminosulfonylation) of Phenols and an Example of Slow Hydrolytic Release. Tetrahedron Lett. 2014, 55, 6658–6661. [Google Scholar]; (e) Blackburn JM; Short MA; Castanheiro T; Ayer SK; Muellers TD; Roizen JL Synthesis of N-Substituted Sulfamate Esters from Sulfamic Acid Salts by Activation with Triphenylphosphine Ditriflate. Org. Lett 2017, 19, 6012–6015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Won S-Y; Kim S-E; Kwon Y-J; Shin I; Ham J; Kim W-S Chan-Lam Coupling Reaction of Sulfamoyl Azides with Arylboronic Acids for Synthesis of Unsymmetrical N-arylsulfamides. RSC Adv. 2019, 9, 2493–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an example of an SNAr reaction using a sulfamate ester, see:; (g) Kim SJ; Jung M-H; Yoo KH; Cho J-H; Oh C-H Synthesis and Antibacterial Activities of Novel Oxazolidinones Having Cyclic Sulfonamide Moieties. Biorg. Med. Chem. Lett 2008, 18, 5815–5818. [DOI] [PubMed] [Google Scholar]
- (3).Taylor RD; MacCoss M; Lawson ADG Rings in Drugs. J. Med. Chem 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]
- (4).For recent examples of the utility of N-substituted sulfamate esters, see:; (a) Short MA; Blackburn JM, Roizen JL Sulfamate Esters Guide Selective Radical-Mediated Chlorination of Aliphatic C–H Bonds. Angew. Chem. Int. Ed 2018, 57, 296–299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sathyamoorthi S; Banerjee S; Du Bois J; Burns NZ; Zare RN Site-Selective Bromination of sp3C–H Bonds. Chem. Sci 2018, 9, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ayer SK; Roizen JL Sulfamate Esters Guide C(3)-Selective Xanthylation of Alkanes. J. Org. Chem 2019, 84, 3508–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Del Castillo E; Martìnez MD; Bosnidou AE; Duhamel T; O’Broin CQ; Zhang H; Escudero-Adán EC; Martínez-Belmonte M; Muñiz K Chem. Eur. J 2018, 24, 17225–17229. [DOI] [PubMed] [Google Scholar]; (e) Ma Z-Y; Guo L-N; You Y; Yang F; Hu M; Duan X-H Visible Light Driven Alkylation of C(sp3)–H Bonds Enabled by 1,6-Hydrogen Atom Transfer/Radical Relay Addition. Org. Lett 2019, 21, 5500–5504. [DOI] [PubMed] [Google Scholar]; (f) Kanegusuku ALG; Castanheiro T; Ayer SK; Roizen JL Sulfamyl Radicals Direct Photoredox-Mediated Giese Reactions at Unactivated C(3)–H Bonds. Org. Lett 2019, 21, 6089–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Shu W; Zhang H; Huang Y γ-Alkylation of Alcohols Enabled by Visible-Light Induced 1,6-Hydrogen Atom Transfer. Org. Lett 2019, 21, 6107–6111. [DOI] [PubMed] [Google Scholar]
- (5).For a recent overview, see:; Bariwal J; Van der Eycken E C–N Bond Forming Cross-Coupling Reactions: An Overview. Chem. Soc. Rev 2013, 42, 9283–9303. [DOI] [PubMed] [Google Scholar]
- (6).For a recent review, see:; Ruiz-Castillo P; Buchwald SL Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For early reports on the use of sulfonamides, amides, carbamates, and sulfamides in palladium-mediated Buchwald-Hartwig amination reactions, see:; (a) Hartwig JF; Kawatsura M; Hauck SI; Shaughnessy KH; Alcazar-Roman LM Room-Temperature Palladium-Catalyzed Amination of Aryl Bromides and Chlorides and Extended Scope of Aromatic C–N Bond Formation with a Commercial Ligand. J. Org. Chem 1999, 64, 5575–5580. [DOI] [PubMed] [Google Scholar]; (b) Yin J; Buchwald SL Palladium-Catalyzed Intermolecular Coupling of Aryl Halides and Amides. Org. Lett 2000, 2, 1101–1104. [DOI] [PubMed] [Google Scholar]; (c) Alcaraz L; Bennion C; Morris J; Meghani P; Thom SM Novel N-Aryl and N-Heteroaryl Sulfamide Synthesis via Palladium Cross Coupling. Org. Lett 2004, 6, 2705–2708. [DOI] [PubMed] [Google Scholar]; For select recent improvements, see:; (d) Rosen BR; Ruble JC; Beauchamp TJ; Navarro A Mild Pd-Catalyzed N-Arylation of Methanesulfonamide and Related Nucleophiles: Avoiding Potentially Genotoxic Reagents and Byproducts. Org. Lett 2011, 13, 2564–2567. [DOI] [PubMed] [Google Scholar]; (e) Crawford SM; Lavery CB; Stradiotto M Bippyphos: A Single Ligand with Unprecedented Scope in the Buchwald-Hartwig Amination of (Hetero)aryl Chlorides. Chem. Eur. J 2013, 19, 16760–16771. [DOI] [PubMed] [Google Scholar]; (f) Lavoie CM; MacQueen PM; Stradiatto M Nickel-Catalyzed N-Arylation of Primary Amides and Lactams with Activated (Hetero)aryl Electrophiles. Chem. Eur. J 2016, 22, 18752–18755. [DOI] [PubMed] [Google Scholar]
- (8).For seminal examples detailing the use of sulfamate esters as electrophiles in transition metal-catalyzed coupling processes, see:; (a) Macklin TK; Snieckus V Directed Ortho Metalation Methodology. The N,N-Dialkyl Aryl O-Sulfamate as New Directed Metalation Group and Cross-Coupling Partner for Grignard Reagents. Org. Lett 2005, 7, 2519–2522. [DOI] [PubMed] [Google Scholar]; (b) Wehn PM; Du Bois J Exploring New Uses for C–H Amination: Ni-Catalyzed Cross-Coupling of Cyclic Sulfamates. Org. Lett 2005, 7, 4685–4688. [DOI] [PubMed] [Google Scholar]; For select advances in the use of sulfamate ester electrophiles, see:; (c) Quasdorf KW; Reiner M; Petrova KV; Garg NK Suzuki-Miyaura Coupling of Aryl Carbamates, Carbonates, and Sulfamates. J. Am. Chem. Soc 2009, 131, 17748–17749. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mesganaw T; Silberstein AL; Ramgren SD; Fine Nathel NF; Hong X; Liu P; Garg NK Nickel-Catalyzed Amination of Aryl Carbamates and Sequential Site-Selective Cross-Couplings. Chem. Sci 2011, 2, 1766–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ramgren SD; Silberstein AL; Yang Y; Garg NK Nickel-Catalyzed Amination of Aryl Sulfamates. Angew. Chem. Int. Ed 2011, 50, 2171–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Silberstein A; Ramgren SD; Garg NK Iron-Catalyzed Alkylations of Aryl Sulfamates and Carbamates. Org. Lett 2012, 14, 3796–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Quasdorf KW; Antoft,-Finch A; Liu P; Silberstein AL; Komaromi A; Blackburn T; Ramgren SD; Houk KN; Snieckus V Garg NK J. Am. Chem. Soc 2011, 133, 6352–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Ackermann L; Sandmann R; Song, W. Palladium- and Nickel-Catalyzed Aminations of Aryl Imidazolylsulfonates and Sulfamates. Org. Lett 2011, 13, 1784–1786. [DOI] [PubMed] [Google Scholar]; (i) Molander GA; Shin I Pd-Catalyzed Suzuki–Miyaura Cross-Coupling Reactions between Sulfamates and Potassium Boc-Protected Aminomethyltrifluoroborates. Org. Lett 2013, 15, 2534–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Takise R; Itami K; Yamaguchi J Cyanation of Phenol Derivatives with Aminoacetonitriles by Nickel Catalysis. Org. Lett 2016, 18, 4428–4431. [DOI] [PubMed] [Google Scholar]; (k) Melvin PR; Hazari N; Beromi MM; Shah HP; Williams MJ Pd-Catalyzed Suzuki-Miyaura and Hiyama-Denmark Couplings of Aryl Sulfamates. Org. Lett 2016, 18, 5784–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Kuriyama M; Kujirada S; Tsukuda K; Onomura O Nickel-Catalyzed Deoxygenative Deuteration of Aryl Sulfamates Adv. Synth. Catal 2017, 359, 1043–1048. [Google Scholar]; (m) Gan Y; Wang G; Xie X; Liu Y; Nickel-Catalyzed Cyanation of Phenol Derivatives with Zn(CN)2 Involving C–O Bond Cleavage. J. Org. Chem 2018, 83, 14036–14048. [DOI] [PubMed] [Google Scholar]; (n) Coombs JR; Green RA; Roberts F; Simmons EM; Stevens JM; Wisniewski SR Advances in Base-Metal Catalysis: Development of a Screening Platform for Nickel-Catalyzed Borylations of Aryl (Pseudo)halides with B2(OH)4. Organometallics, 2019, 38, 157–166. [Google Scholar]
- (9).For seminal reports on nickel-catalyzed C–N coupling reactions, see:; (a) Wolfe JP; Buchwald SL Nickel-Catalyzed Amination of Aryl Chlorides. J. Am. Chem. Soc 1997, 119, 6054–6058. [Google Scholar]; (b) Brenner E; Fort Y New Efficient Nickel(0) Catalysed Amination of Aryl Chlorides. Tetrahedron Lett. 1998, 39, 5359–5362. [Google Scholar]; For a recent review, see:; (c) Marín M; Rama RJ; Micasio MC Ni-Catalyzed Amination Reactions: An Overview, Chem. Rec 2016, 16, 1819–1832. [DOI] [PubMed] [Google Scholar]
- (10).(a) Koo K; Hillhouse GL Carbon-Nitrogen Bond Formation by Reductive Elimination From Nickel(II) Amido Alkyl Complexes. Organometallics 1995, 14, 4421–4423. [Google Scholar]; (b) Ilies L; Matsubara T; Nakamura E Nickel-Catalyzed Synthesis of Diarylamines via Oxidatively Induced C−N Bond Formation at Room Temperature. Org. Lett 2012, 14, 5570–5573. [DOI] [PubMed] [Google Scholar]
- (11).(a) Corcoran EB; Pirnot MT; Lin S; Dreher SD; DiRocco DA; Davies IW; Buchwald SL; MacMillan DWC Aryl Amination Using Ligand-Free Ni(II) Salts and Photoredox Catalysis. Science 2016, 353, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Oderinde MS; Jones NH; Juneau A; Frenette M; Aquila B; Tentarelli S; Robbins DW; Johannes JW Highly Chemoselective Iridium Photoredox and Nickel Catalysis for the Cross-Coupling of Primary Aryl Amines with Aryl Halides. Angew. Chem. Int. Ed 2016, 55, 13219–13223. [DOI] [PubMed] [Google Scholar]; (c) Key RJ; Vannucci AK Nickel Dual Photoredox Catalysis for the Synthesis of Aryl Amines. Organometallics 2018, 37, 1468–1472. [Google Scholar]; (d) Kim T; McCarver SJ; Lee C; MacMillan DWC Sulfonamidation of Aryl and Heteroaryl Halides through Photosensitized Nickel Catalysis. Angew. Chem. Int. Ed 2018, 57, 3488–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Reddy LR; Kotturi S; Waman Y; Reddy VR; Patel C; Kobarne A; Kuttappan S; N-Arylation of Carbamates through Photosensitized Nickel Catalysis. J. Org. Chem 2018, 83, 13854–13860. [DOI] [PubMed] [Google Scholar]; (f) Koney MO; McTeague TA; Johannes JW Nickel-Catalyzed Photoredox-Mediated Cross-Coupling of Aryl Electrophiles and Aryl Azides. ACS Catal. 2018, 8, 9120–9124. [Google Scholar]
- (12).DBU has previously found use in electrochemically-enabled, nickel-catalyzed C–N couplings:; (a) Li C; Kawamata Y; Nakamura H; Vantourout JC; Liu Z; Hou Q; Bao D; Starr JT; Chen J; Yan M; Baran PS Electrochemically Enabled, Nickel-Catalyzed Amination. Angew. Chem. Int. Ed 2017, 56, 13088–13093. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kawamata Y; Vantourout JC; Hickey DP; Bai P; Chen L; Hou Q; Qiao W; Barman K; Edwards MA; Garrido-Castro AF; deGruyter JN; Nakamura H; Knouse K; Qin C; Clay KJ; Bao D; Li C; Starr JT; Garcia-Irizarry C; Sach N; White HS; Neurock M; Minteer SD; Baran PS Electrochemically Driven, Ni-Catalyzed Aryl Amination: Scope, Mechanism, and Applications. J. Am. Chem. Soc 2019, 141, 6392–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lim C-H; Kudisch M; Liu B; Miyake GM C–N Cross-Coupling via Photoexcitation of Nickel–Amine Complexes. J. Am. Chem. Soc 2018, 140, 7667–7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Laulhé S; Blackburn JM; Roizen JL Selective and Serial Suzuki-Miyaura Reactions of Polychlorinated Aromatics with Alkyl Pinacol Boronic Esters. Org. Lett 2016, 18, 4440–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Laulhé S; Blackburn JM; Roizen JL Exhaustive Suzuki-Miyaura Reactions of Polyhalogenated Heteroarenes with Alkyl Boronic Pinacol Esters. Chem. Commun 2017, 53, 7270–7273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Blackburn JM; Roizen JL Catalytic Strategies to Convert 2-Halopyridines to 2-Alkylpyridines. Asian J. Org. Chem 2019, 8, DOI: 10.1002/ajoc.201900163. [DOI] [Google Scholar]
- (15).For pioneering reports, see; (a) Liebeskind LS; Srogl J Thiol Ester-Boronic Acid Coupling. A Mechanistically Unprecedented and General Ketone Synthesis. J. Am. Chem. Soc 2000, 122, 11260–11261. [Google Scholar]; (b) Liebeskind LS; Srogl J Heteroaromatic Thioether-Boronic Acid Cross-Coupling under Neutral Conditions. Org. Lett 2002, 4, 979–981. [DOI] [PubMed] [Google Scholar]; For reviews, see:; (c) Prokopcova H; Kappe CO The Liebeskind−Srogl C−C Cross-Coupling Reaction. Angew. Chem., Int. Ed 2009, 48, 2276–2286. [DOI] [PubMed] [Google Scholar]; (d) Cheng H-G; Chen H; Liu Y; Zhou Q The Liebeskind−Srogl Cross-Coupling Reaction and Its Synthetic Applications. Asian J. Org. Chem 2018, 7, 490–508. [Google Scholar]
- (16).(a) Maryanoff BE; Nortey SO; Gardocki JF; Shank RP; Dodgson SP Anticonvulsant O-Alkyl Sulfamates. 2,3:4,5-Bis-O-(1-methylethylidene)-β-d-fructopyranose Sulfamate and Related Compounds. J. Med. Chem 1987, 30, 880–887. [DOI] [PubMed] [Google Scholar]; For an example of a biologically active topiramate derivative bearing a N-aryl substituent, see; (b) Yu D; Forman B Bile Acid Receptor Modulators and Methods of Use Thereof. US 2018105533 B2, April 19, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.