

Abstract
Background
In 20-30% of patients suspected of a familial colorectal cancer (CRC) syndrome, no underlying genetic cause is detected. Recent advances in whole exome sequencing (WES) have generated evidence for new CRC-susceptibility genes including POLE, POLD1 and NTHL1 but many patients remain unexplained.
Methods
Whole exome sequencing was performed on DNA from nine patients from five different families with familial clusters of CRC in which traditional genetic testing failed to yield a diagnosis. Variants were filtered by minor allele frequencies, followed by prioritization based on in silico prediction tools, and the presence in cancer susceptibility genes or genes in cancer-associated pathways. Effects of frameshift variants on protein structure were modeled using I-Tasser.
Results
One known pathogenic variant in POLD1 was detected (p.S478N), together with variants in 17 candidate genes not previously associated with CRC. Additional in silico analysis using SIFT, PROVEAN and PolyPhen in the 14 missense variants indicated a possible damaging effect in nine of 14 variants. Modeling of the insertions/deletions showed a damaging effect of two variants in NOTCH2 and CYP1B1.
Discussion
One family was explained by a mutation in a known familial CRC gene. In the remaining four families, the most promising candidates found are a frameshift NOTCH2 and a missense RAB25 variant. This study provides potential novel candidate variants in unexplained familial CRC patients, however, functional validation is imperative to confirm the role of these variants in CRC tumorigenesis. Additionally, while whole exome sequencing enables detection of variants throughout the exome, other causes explaining the familial phenotype such as multiple single nucleotide polymorphisms accumulation to a polygenic risk or epigenetic events, might be missed with this approach.
Keywords: Familial colorectal cancer, POLD1, candidate variants
INTRODUCTION
Approximately 25-35% of all CRCs are estimated to have a heritable component, either a pathogenic variant in a high-risk CRC susceptibility gene (3-5%) or a positive family history (20-30%) with an unknown genetic cause [1, 2]. These unexplained patients might carry rare, dominantly inherited variants, or multiple moderately penetrant variants that act synergistically [2].
Determining whether a patient may carry a genetic variant causing familial CRC was historically based on family history, through the revised Amsterdam Criteria for Lynch Syndrome (LS), which were developed to determine whether families were likely to have pathogenic variants in the DNA mismatch repair (MMR) genes [3]. LS tumors characteristically exhibit microsatellite instability (MSI), due to the loss of expression of one or more MMR proteins [4]. However, approximately half of the families fulfilling Amsterdam Criteria do not have a LS phenotype in their tumors [5, 6]. These CRC-enriched families, often referred to as “familial colorectal cancer-type X”, have specific features compared to LS, such as lower incidence of CRC, more tumors in the left colon, fewer extracolonic cancers, and higher mean age of onset (57.3 years as opposed to the average 49.7 years for LS patients) [5, 6]. This seemingly familial clustering of CRC is generally not linked to any one specific gene, though studies in the past have indicated CENPE, KIF24, GALNT12, HNRNPA0, WIF1, ZNF367, GABBR2 and BMP4 as candidate genes, possibly explaining a percentage of these patients [7, 8]. More importantly, previous studies showed that unexplained familial clustering of CRC is likely not a single syndrome, and it is unlikely that we will find the same genetic defects across affected families [9].
Recently, germline and somatic variants in the exonuclease domains of the POLE and POLD1 genes are described to associate with early-onset CRC, endometrial cancer and adenomatous polyps [10, 11]. This variable phenotype has recently been named polymerase proofreading associated polyposis (PPAP), a syndrome with high penetrance and dominant inheritance [11]. PPAP tumors have a specific phenotype with very high mutational burden, exceeding 100 variants/Mb (ultramutated), with a specifically elevated TCT>TAT and TCG>TTG mutational signature [11, 12]. Furthermore, whole exome sequencing efforts in early-onset and/or familial CRCs recently described the recessive NTHL1-associated polyposis syndrome, in which patients presented with adenomas and a spectrum of cancers including colorectal, breast, endometrial, duodenal, skin, prostate and pancreatic cancer [13, 14].
Although family history is an important indicator of a germline genetic defect, recent studies have shown that no more than 20% of CRCs occurring before age 50, regardless of family history, can be explained by a variant in a high-risk CRC susceptibility gene [15, 16]. Identification of the underlying genetic cause in early-onset and/or familial CRC patients is of utmost importance for proper clinical management of the patient and their families. Given the wide spectrum of possible causal variants in these patients, multigene panels or whole-exome sequencing is advised [15, 16].
In this study we aimed to identify the underlying genetic cause in five families who were part of a large familial gastrointestinal cancer registry, who had undergone routine genetic testing in the past (between 2004 and 2014), and had unexplained familial CRCs, using whole exome sequencing. We also analyzed the candidate variants using in silico protein structure modeling in order to predict whether specific variants will affect protein structure and function.
MATERIAL AND METHODS
Patient cohort
All patients provided informed consent for Institutional Review Board-approved research studies at a single institution (Baylor Scott & White, Dallas, TX). Leukocyte DNA was collected from nine patients belonging to five different families (Figure 1A–E, Table 1). Three families showed a dominant pattern of inheritance, with multiple affected family members in multiple generations. Two families (A and C), were compatible with a recessive pattern of inheritance. Family A consisted of two affected sisters (CRC45 and CRC46). Family C consisted of an affected index patient (CRC14) and both parents presented with non-colorectal carcinomas (squamous cell carcinoma of the tonsil and basal cell carcinoma, Figure 1). Patients were previously tested for germline variants in PTEN (family A), MUTYH (Family B, E) and/or the MMR genes (Family C and D) using commercially-available testing.
Figure 1: Pedigrees of tested patients.
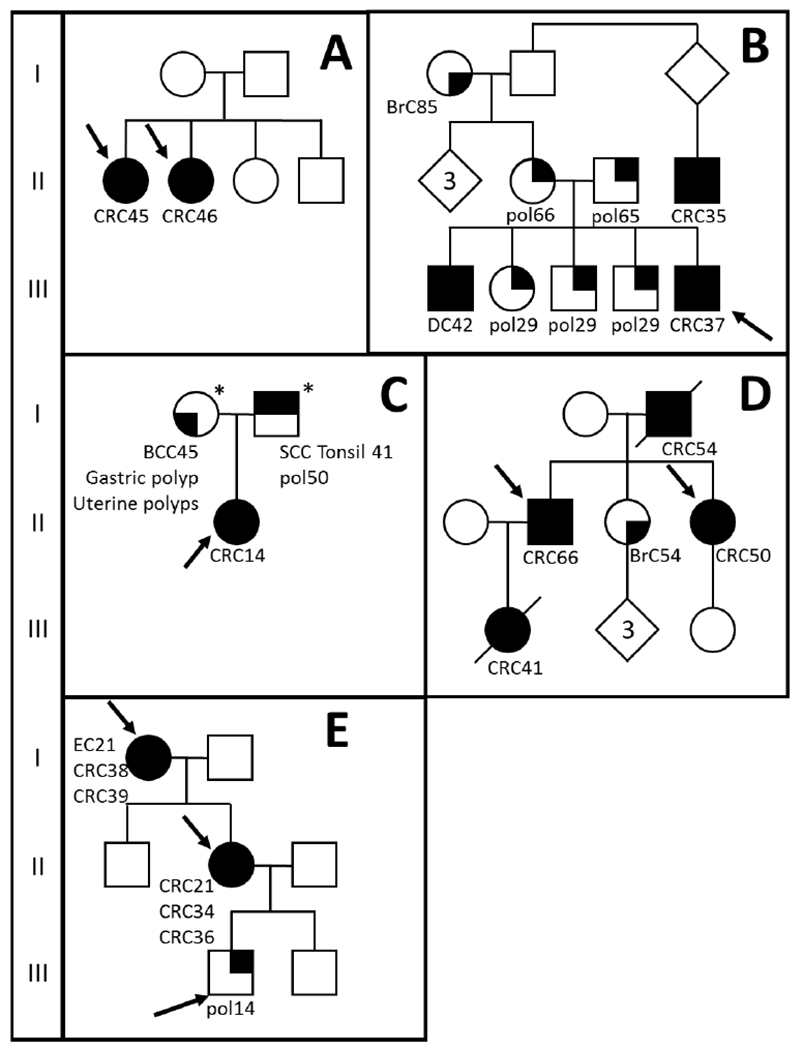
Pedigrees of six tested families (A-E). Squares represent males, circles represent females and diamonds is undisclosed gender. Phenotype is shown as tumor type followed by age of onset. Patients presented with colorectal cancer (CRC; fully filled symbol), polyps (pol, right top corner), breast cancer (BrC, bottom right corner), basal cell carcinoma (BCC bottom left corner) or squamous cell carcinoma (SCC, top left corner). EC = endometrial cancer, DC = duodenal cancer. Arrows indicate patients sequenced with whole exome sequencing.*DNA available for co-segregation study
Table 1:
Patient characteristics
| Characteristics | Value |
|---|---|
| Families | 5 (9 patients) |
| Patient characteristics | |
| Male | 3 patients |
| Female | 6 patients |
| Age of onset (years) | 34.8 (range 14-66) |
| Phenotype per patient | |
| CRC | 7 |
| EC and CRC | 1 |
| Adenomatous polyps | 1 |
| Family history | |
| First-degree relatives with CRC | 4 |
| No CRC family history | 1 |
| Pattern of inheritance | |
| Dominant | 3 |
| Recessive/de novo | 2 |
CRC= colorectal cancer, EC = endometrial cancer
Whole exome sequencing
Whole exome sequencing, alignment and annotation were conducted by the Novogene Corporation. Sequencing libraries were generated using Agilent SureSelect Human All exon kit (Agilent Technologies, CA, USA) with an input of 1μg of genomic DNA, according to the manufacturer’s protocol. DNA was sheared (Covaris, Massachussetts, USA), followed by blunt-end conversion, adenylation of the 3’-end and adapter ligation. The libraries were hybridized with biotin labeled probes and captured with magnetic beads. Captured libraries were enriched and purified using an AMPure XP system (Beckman Coulter, Beverly, USA). Sequencing was performed on an Illumina platform.
Reads were mapped to the human reference genome (hg37) using the Burrows-Wheeler Aligner (BWA, version 0.7.8-r455). The resulting BAM files were sorted using SAMtools. Duplicate reads were marked using Picard (version 1.111), followed by variant calling using GATK (v3.8).
Data analysis
Variant annotation was done using ANNOVAR [17]. Variant allele frequencies in the population were given using large available consortia including the 1000 Genome Project, Exome Aggregation Consortium (ExAC) and the Exome Sequencing Project (ESP). Through ANNOVAR the databases dbSNP, COSMIC, OMIM, GWAS catalog and HGMD were used to find reported information on the variant. In silico prediction SIFT, Polyphen, MutationAssessor, LRT and CADD scores (v1.0) were used to predict the effect of the variant on protein function. GERP++ scores were used to assess conservation of the affected base.
Prioritization of variants
All exonic non-synonymous variants (including nonsense, frameshift or in-frame insertions and deletions) were filtered by minor allele frequency (MAF) as reported by ExAC score (ExAC_ALL, MAF >0.01 excluded). Additionally, intronic variants predicted to affect splicing with a MAF <0.01 were included. Variants present in segmental duplicated regions were excluded from the analysis, as were known benign variants. If multiple family members were screened, all variants that were not present in all affected family members were excluded.
All remaining variants were prioritized according to the following characteristics: 1) scaled CADD (CADD_Phred) score of 18 or higher; 2) at least one of the following key words found after annotation of the gene in the DAVID bioinformatic resource: cancer, repair, apoptosis, wnt, suppressor, TGF, cell cycle, polymerase, colorectal; and 3) variants present in genes that are expressed in normal colonic tissue according to the protein atlas. All variants were visually inspected in the integrative genomics viewer (IGV).
All variants present in possible tumor suppressor genes (TSGs) according to the TSG database (https://bioinfo.uth.edu/TSGene/), as well as all variants present in known CRC-susceptibility genes, recently published candidate familial CRC genes, or in previously described multigene cancer sequencing panels were validated with Sanger sequencing. A list of these suspected CRC-genes is provided in Supplemental Table 1.
Variant validation
In total, 18 variants resulting from prioritization were validated with Sanger sequencing. One variant in the FAT4 gene could not be confirmed and was excluded from further analysis. For family C, leukocyte DNA of both unaffected parents was tested for the variants detected in the index patient. Primers sequences for all validated variants are presented in Supplemental Table 2. The BigDye® Direct Cycle Sequencing kit (Applied Biosystems®, Foster City, CA, USA) was used for PCR, post-PCR cleanup and cycle sequencing. The resulting product was run on the 3130XL Genetic Analyzer (Applied Biosystems®, Foster City, CA, USA). Sequences were analyzed using Chromas.
For the validated variants, the following Genbank reference sequences were used: NM_006207 (PDGFRL), NM_001099691 (TGFA), NM_002185(IL7R), NM_001383 (DPH1), NM_024503 (HIVEP3), NM_080685 (PTPN13), NM_001200001 (NOTCH2), NM_133477 (SYNPO2), NM_000234 (LIG1), NM_020387 (RAB25), NM_001164 (APBB1), NM_001256849 (POLD1), NM_015466 (PTPN23), NM_015466 (PHB), NM_000104 (CYP1B1), NM_015541 (LRIG1) and NM_001127208 (TET2).
Sequence homology-based SNP prediction using SIFT and PROVEAN
Of the 17 variants remaining after validation, pathogenicity of the 14 missense non-synonymous single nucleotide polymorphisms (nsSNPs) was ascertained using sequence homology-based prediction tools such as SIFT (http://sift.jcvi.org/) and PROVEAN (Protein Variation Effect Analyzer) (http://provean.jcvi.org) [18]. The methodology used was based on the procedure previously published with some modifications [19]. For SIFT, substitutions at a position with normalized probabilities of ≤0.05 in a tolerance index were predicted to be damaging, whereas those with normalized probabilities ≥0.05 were predicted to be tolerated [20]. Likewise, in PROVEAN, a nsSNP present in the coding region of a gene was predicted to be “deleterious” if the prediction score was below threshold value (cutoff is −2.5), and “neutral” if the predicted score was above the cutoff value.
Structural homology-based SNP prediction using PolyPhen-2
Seven missense nsSNPs that returned with incongruent scores (i.e., deleterious/damaging in one and tolerant/neutral in other or vice-versa in sequence-homology based SNP prediction methods such as SIFT/Provean) were subjected to PolyPhen-2 (Polymorphism Phenotyping-2) analysis (http://genetics.bwh.harvard.edu/pph2/). PolyPhen-2 is an online tool that predicts the putative functional consequences of a missense nsSNP on the structure and function of a human protein, using physical and comparative considerations [21]. The output of the PolyPhen-2 is a position-specific independent count (PSIC) score for every amino acid variant with respect to wild type variant.
Identification of conserved residues and sequence motifs using Consurf
The Uniprot amino acid sequence of the sixteen proteins in FASTA format was used as input for computational analysis of conserved sequences and motifs using Consurf web server [22], which perform evolutionary conservation analysis based on a Bayesian algorithm. The output of Consurf analysis shows degree of conservation of an amino acid residue in the test protein by means of color coding (conservation scores: 1–4 variable, 5-6 intermediate, and 7–9 conserved). Exposed and buried residues with high conservation levels were respectively scored as functional and structural residues in the amino acid sequence. The structural and functional motif (pfam motifs) present in the protein were predicted using the MOTIF Search tool (http://www.genome.jp/tools/motif/).
Computational 3D structural modelling
For the variants in Notch2, Synpo2 and Cyp1b1 proteins, 3D structure models were built using I-Tasser (http://zhanglab.ccmb.med.umich.edu/I-TASSER/), which employs an integrated combinatorial approach comprising comparative modeling, threading, and ab-initio modelling [23] using the procedure adopted by Dakal et al [24]. For the large protein (>1500 aa length) where the modelling cannot be done by I-Tasser, SWISS-MODEL (https://swissmodel.expasy.org/) was used.
Comparison sporadic CRC
To investigate which gene ontology (GO) pathways are affected in our familial cohort (nine patients described above together with one patient from family F, see supplementary Figure 1) as compared to a sporadic CRC cohort, an unbiased comparison was made as described in the supplemental data.
RESULTS
Variant prioritization
To identify the underlying genetic cause in patients with suspected familial CRC, whole exome sequencing was performed on nine patients from five different families with clustering of colon cancer (average age of onset 34.8 years, Table 1). All sequenced samples had an average sequencing depth on target of 133x (range: 107-167), with on average 22,763 coding and splice site variants (range: 22,316-23,358) per exome (Supplemental Table 3). Variants were filtered according to Figure 2, first on minor allele frequency (MAF) and then on their presence in all sequenced affected family members (Supplemental Table 4). Variants with an in silico prediction tool CADD score higher than 18 were prioritized. All variants in genes associated with cancer, DNA repair, apoptosis, Wnt (signaling), suppressor, TGF (TGF-beta signaling), cell cycle, polymerase and colorectal (cancer) according to the DAVID annotation tool were prioritized. Variants in genomic duplicated regions, in genes not expressed in normal colonic tissue, and variants previously reported to be benign, were excluded. All variants were visually inspected with the integrative genomics viewer (IGV). After prioritization, an average of 29 variants remained per family (family A: 26, B: 48, C: 40, D19, E: 14, see Supplemental Table 4 and 5). Of these variants, all variants known to be present in a predicted TSG, as well as variants in CRC-related pathways were validated using Sanger sequencing (Table 2). Additionally, the predicted pathogenic variants in TGFA, LIG1, APBB1 and CYP1B1 were also confirmed, because these genes were present in the KEGG pathway ‘Pathways in Cancer’ (TGFA), linked to DNA repair pathways (LIG1, APBB1) or associated before with CRC or CRC syndromes (CYP1B1).
Figure 2: Filtering and prioritization of variants.
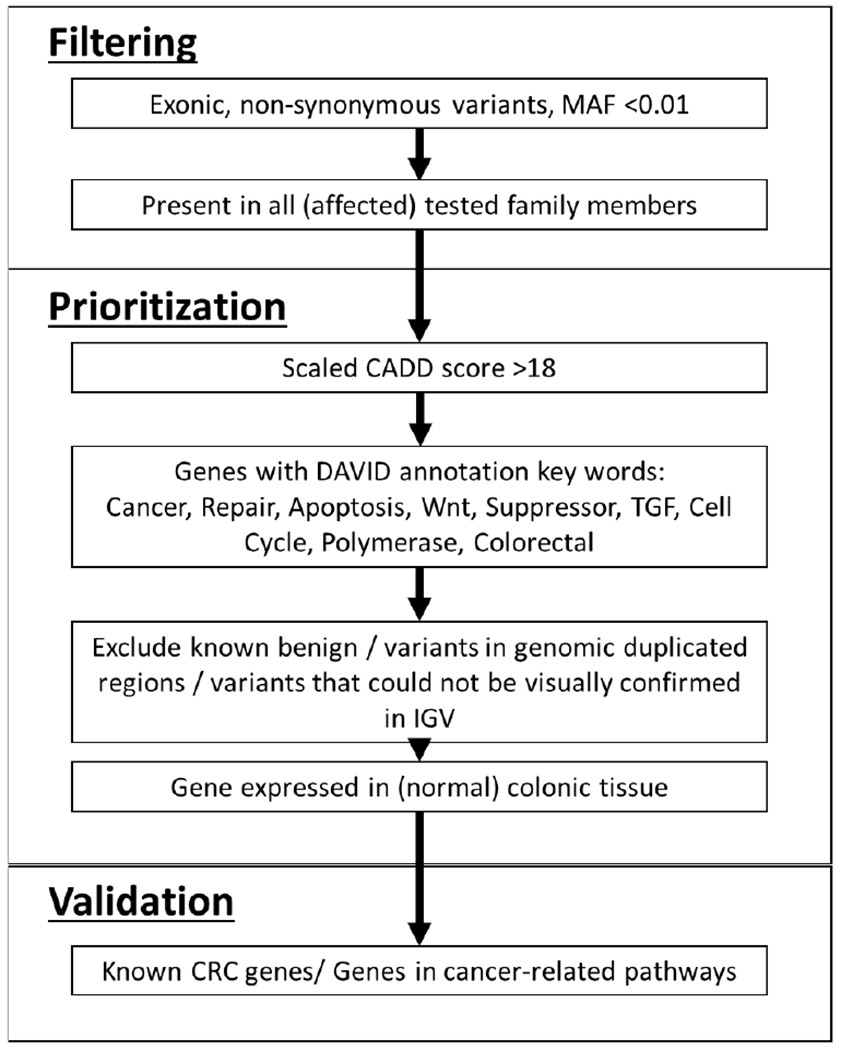
MAF = minor allele frequency, IGV; integrative genomics viewer, CRC = colorectal cancer
Table 2:
Candidate variants validated with Sanger Sequencing
| Fam | Pres | Gene | Variant | MAF | CADD_PHRED | Additional Information | |
|---|---|---|---|---|---|---|---|
| A | 2/2 | PDGFRL | c.371G>A | p.(R124H) | 0,0021 | 19,1 | TSG |
| A | 2/2 | TGFA | c.377G>A | p.(R127Q) | 0,0002 | 28,8 | KEGG pathways in cancer |
| A | 2/2 | IL7R | c.644G>T | p.(G215V) | . | 19,0 | Gene list |
| B | 1/1 | PTPN23 | c.483C>A | p.(F161L) | 0,0027 | 29,6 | TSG |
| B | 1/1 | PHB | c.128G>T | p.(R43L) | 0,0051 | 19,2 | TSG |
| B | 1/1 | CYP1B1 | c.1064_1076del | p.(R355Hfs) | 0,0002 | 37,0 | Associated with CRC |
| B | 1/1 | LRIG1 | c.455G>C | p.(T152R) | 0,0017 | 19,8 | TSG |
| B | 1/1 | TET2 | c.2599T>C | p.(Y867H) | 0,0069 | 20,4 | TSG*/gene list |
| C | 1/1ǂ | DPH1 | c.746C>G | p.(P249R) | 0,0051 | 24,7 | TSG |
| C | 1/1ǂ | HIVEP3 | c.6722G>A | p.(R2241Q) | 0,0022 | 21,7 | TSG |
| C | 1/1ǂ | PTPN13 | c.7157C>T | p.(T2381I) | 0,0091 | 23,2 | TSG |
| C | 1/1ǂ | NOTCH2 | c.2786delG | p.(G929Efs) | . | . | TSG*/gene list |
| C | 1/1± | SYNPO2 | c.1583_1585dup | p.(D528dup) | 1,65E-05 | . | TSG |
| C | 1/1± | LIG1 | c.1003C>T | p.(L335F) | 0,0012 | 21,2 | Associated with DNA repair pathways BER, NER and MMR |
| D | 2/2 | RAB25 | c.59A>G | p.(E20G) | 0,0048 | 28,9 | TSG |
| D | 2/2 | APBB1 | c.760G>A | p.(D254N) | 0,0002 | 31,0 | Associated with DSB-R and apoptosis |
| E | 3/3 | POLD1 | c.1433G>A | p.(S479N) | . | 25,2 | Gene list |
Pres - number of patients within a family that carries the variant / total number of patients tested in the family; Fs = frameshift, dup = duplication, MAF = minor allele frequency, TSG = tumor suppressor gene, with TSG* indicating genes with TUSON p-value <0.001 predicted to be a tumor suppressor gene, CRC = colorectal cancer, BER = base excision repair, NER = nucleotide excision repair, MMR = mismatch repair, DSB-R = double strand break repair.
Only one family carried a known pathogenic variant previously described in CRC, POLD1 c.1433G>A, p.S478N. This variant was found in family E, in all three tested family members. Family E had a severe phenotype, with three affected generations with the first cancers at age 31 (I-1) and 21 (II-1). In the third generation, patients were screened by colonoscopies due to positive family history.
Besides the POLD1 variant, no other variant in a known high-risk or moderate-risk CRC susceptibility gene was found. However, since IL7R, NOTCH2 and TET2 genes are present in pathways involved in tumorigenesis (immune response, Notch signaling and cell cycle respectively), these were therefore included in the gene list (Supplemental Table 1).
Co-segregation analysis in the family
In family C, only the affected proband (CRC14) was tested, but leukocyte DNA was available from both unaffected parents. All variants detected in the patient were found in either the father (DPH1, HIVEP3, PTPN13, NOTCH2) or the mother (LIG1).
In silico analysis with SIFT, PROVEAN and PolyPhen
To ascertain pathogenicity of the 14 missense variants, prediction tools SIFT and PROVEAN were used (Table 3, Supplemental Table 6). In 7/14 variants, SIFT and PROVEAN scores were concordant. The seven discordant variants were also analyzed with PolyPhen-2. In total, five variants were predicted to be neutral (TGFA, HIVEP3, PTPN23, PHB and LRIG1), and nine variants were predicted to affect protein function.
Table 3:
In silico prediction of missense variants
| Family | Gene | Variant | SIFT/PROVEAN | POLYPHEN |
|---|---|---|---|---|
| A | PDGFRL | p.(R124H) | Damaging/Deleterious | |
| A | TGFA | p.(R127Q) | Tolerated/Neutral | |
| A | IL7R | p.(G215V) | Damaging/Deleterious | |
| B | PTPN23 | p.(F161L) | Tolerated/Deleterious | Neutral |
| B | PHB | p.(R43L) | Tolerated/Deleterious | Neutral |
| B | LRIG1 | p.(T152R) | Tolerated/Deleterious | Neutral |
| B | TET2 | p.(Y867H) | Damaging/Neutral | Deleterious |
| C | DPH1 | p.(P249R) | Damaging/Deleterious | |
| C | HIVEP3 | p.(R2241Q) | Damaging/Neutral | Neutral |
| C | PTPN13 | p.(T2381I) | Damaging/Neutral | Deleterious |
| C | LIG1 | p.(L335F) | Damaging/Deleterious | |
| D | RAB25 | p.(E20G) | Damaging/Deleterious | |
| D | APBB1 | p.(D254N) | Tolerated/Deleterious | Deleterious |
| E | POLD1 | p.(S479N) | Damaging/Deleterious |
Prediction of variant effect of the prediction tools used
Structural modeling of indel/frameshift variants of NOTCH2, SYNPO2 and CYP1B1
For the structural modeling of the indel/frameshift variants of NOTCH2, SYNPO2 and CYP1B1, the amino acid sequence of each protein was subjected to I-Tasser (or SWISS-MODEL) based integrated structural modeling that uses template search, alignment, threading and ab-initio modeling [19, 24].
The Notch2 protein is a single-pass transmembrane protein. Based on structural analysis, and on pfam motif analysis, the basic structure of the human Notch2 protein appears to be comprised of 35 epidermal growth factor (EGF) repeats, 3 copies of a Lin-12/Notch/Glp motif (1423..1456 aa, 1463..1497 aa, and 1501..1534 aa; possibly in the extracellular region), 3–4 Ankyrin repeats, and single motifs for DUF3454 (2382..2444 aa), NOD (1539..1594 aa) and NODP (1618..1672 aa). Notch2 protein also contains a PEST sequence for proteolytic processing, a nuclear localization signal (NLS), and several putative phosphorylation sites [25]. The wild type translated protein encoded by NOTCH2 is 2471 aa in length, while the detected frameshift variant in patient C (NOTCH2 c.2786delG) would result in a stop gain at aa 930, resulting in C-terminal loss (Figure 3).
Figure 3: Modeling of Notch2 protein structure.
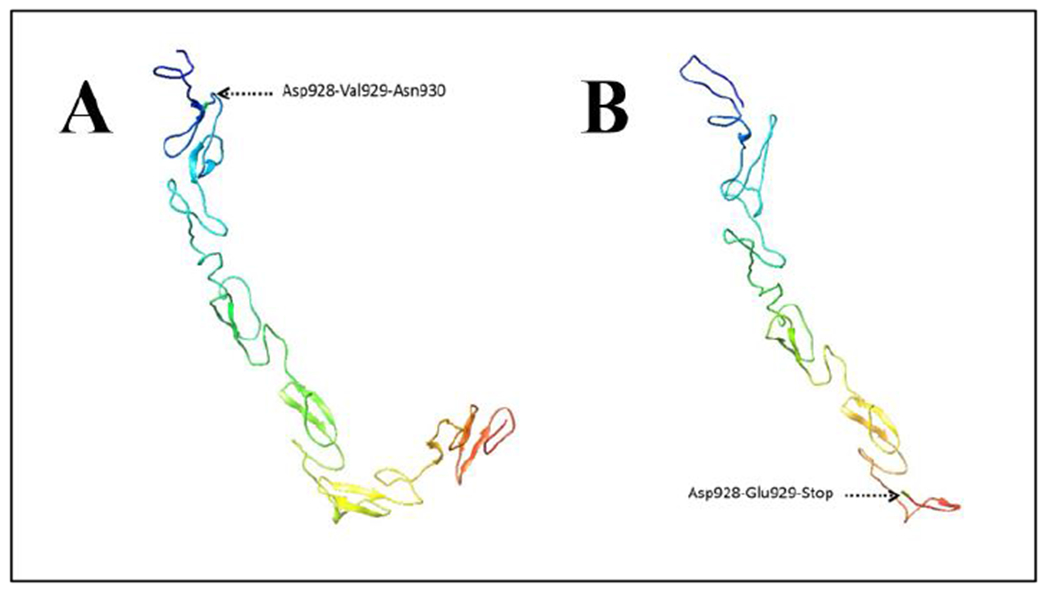
A: Partial structure of wildtype Notch2 protein. Asp928-Val929-Asn930 shows location where the variant was present in the mutant protein. B: Partial structure mutant Notch2 protein. Asp-Glu929-Stop is the result of the frameshift mutation.
Another small frameshift deletion in current study was found in the CYP1B1 gene. While the WT protein is 543 aa (Uniprot ID Q16678), the frameshift variant causes a stop gain producing a protein of 422 aa (p.Arg355Hisfs) (Figure 4A and B). The Cyp1b1 protein has a single pfam motif, namely p450 spanning from aa’s 51 to 519 in the wild type protein. The truncated mutated protein (p.Arg355Hisfs) would result in partial p450 motif loss.
Figure 4: Modeling of Cyp1b1 and Synpo2 protein structure.
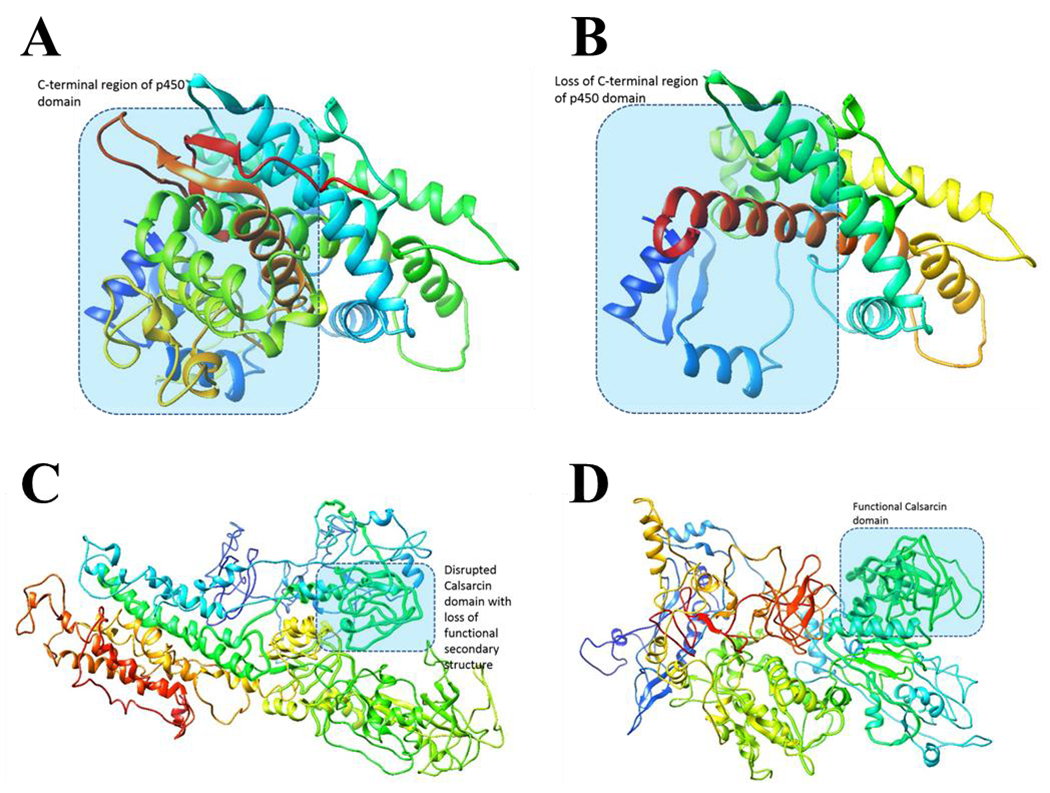
A; Structure wildtype Cyp1b1, B; Structure mutant Cyp1b1, C; Structure wildtype Synpo2, D; Structure mutant Synpo2
Finally, the in-frame duplication in the SYNPO2 gene (c.1583_1585dup) is not expected to entail any drastic structural and functional defect in the mutant protein (Figure 4C and D). The crystal structure of Synpo2 protein is not resolved, and hence, there is no structure model of the protein available in PDB database. We modeled both wild type and mutant variants of the Synpo 2 protein using I-Tasser. Considering that the residues flanking the insertion position in protein sequence are not evolutionarily conserved, we assume that the mutant variant would retain normal functions. However, since the insertion is predicted to lie in the Calsarcin domain (390..594 aa) we cannot exclude the possibility of structure-based functional defects. Structural modeling showed loss of secondary structural features of the Calsarcin domain in the mutant protein. While this could indicate that the variant affects protein function, further structural biology and experimental evidence is needed.
Comparison with sporadic CRC
Finally, to determine whether there was an overrepresentation of mutations in genes in certain pathways within our familial CRC cohort compared to somatic mutations found in a sporadic CRC cohort, an unbiased comparison was performed (See supplemental data). Interestingly, REVIGO analysis showed an overrepresentation of cell-cell adhesion pathways in the familial cohort, compared to the sporadic cohort (Supplemental Figure 2).
DISCUSSION
In this study, we performed whole exome sequencing to search for underlying genetic causes in five families with familial or early-onset CRC (Figure 1). Possible pathogenic variants in known CRC genes or CRC-related pathways were prioritized, resulting in an average of 29 remaining variants per family. Only one family (family E) carried a known pathogenic germline defect, a POLD1 c.1433G>A. This variant has previously been described in two familial CRC families [10]. The Saccharomyces pombe equivalent of this variant was previously described to lead to a 12-fold mutation rate increase compared to wildtype [10]. The previous families showed a less severe phenotype than the family reported in this study, with multiple adenomas detected at ages 26-68 and endometrial and colorectal cancers from age 33-53 [10].
While no other known pathogenic variants or possibly pathogenic variants were detected in known CRC-susceptibility genes, many variants in possible TSGs were found. The two sisters studied from family A carried possible pathogenic variants in the PDGFRL, TGFA and IL7R genes. In silico analyses with SIFT and PROVEAN predicted damaging effects of the variants in PDGFRL and IL7R only. The proposed TSG PDGFRL is commonly somatically deleted in sporadic hepatocellular carcinomas, sporadic CRCs, breast cancer and small cell lung cancers [26, 27].
In family B, only one family member (CRC37) could be analyzed with WES, and four candidate variants were identified, of which one (TET2 c.2599T>C) was predicted to be damaging by secondary in silico analyses. TET2 is an enzyme that converts 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC). TET2 is critical for trophoblast stem cell maintenance by driving expression of epithelial genes [28]. Germline and somatic variants in TET2 have been described in acute myeloid leukemia (AML) [29]. A recent study also reports a germline TET2 variant in a patient with ovarian cancer [30]. Importantly, TET2 has been reported to be crucial for CRC initiation, making it an interesting candidate in a familial CRC context [31]. Additionally, a CYP1B1 frameshift deletion was found and structural modeling predicted that the mutation would render the protein non-functional for p450-related metabolic functions such as drug metabolism, and synthesis of steroids, cholesterol and lipid molecules.
Family C consisted of very early-onset patient (CRC14), with two unaffected parents. It was speculated that this patient could be explained by a recessive syndrome caused by biallelic inactivation of a CRC-susceptibility gene. No homozygous variants, or double hits within a gene (possible compound heterozygous inactivation) were detected. However, 5 heterozygous variants were found in possible TSGs: DPH1, HIVEP3, NOTCH2, PTPN13 and SYNPO2. Additionally, a predicted pathogenic variant was found in the LIG1 gene, which is involved in DNA replication, base-excision repair, nucleotide excision repair and mismatch repair. In silico analyses predicted damaging effects on protein function for the missense variants in DPH1 and PTPN13. Structural modeling showed an unlikely structural effect of the small in-frame duplication in SYNPO2, while modeling of the effect of the NOTCH2 frameshift deletion indicated that this variant results in loss of the C-terminal. Structural analysis showed that the C-terminus is important, since it contains a number of pfam motifs such as EGF, hEGF, EGF_CA, DUF3454, NOD, Notch, Ank, NODP, and cEGF. Additionally, Consurf analysis showed that the C-terminus of the Notch2 protein contains a number of evolutionarily conserved structural (buried) and functional (exposed) motifs. Germline loss-of-function variants in NOTCH2 have previously been described in patients with Alagille Syndrome, where patients present with renal manifestations, cholestatic liver disease and cardiac disease, and in Hadju-Cheney syndrome, a rare disorder with facial anomalies and osteoporosis [32]. Somatic NOTCH1 or NOTCH2 variants are detected in up to 75% of squamous cell carcinomas [33]. Screening of the probands parents determined the presence of this novel variant in the patient’s father. While the father had a history of a squamous cell carcinoma of the tonsil at age 41 as well as a few colonic polyps at age 50, his phenotype did not resemble that of the proband. This could indicate that even though the variant likely affects Notch2 function, it cannot fully explain the proband’s phenotype.
Finally, in family D, exhibiting a dominant pattern of inheritance based on phenotype, we found a possible pathogenic variant in the RAB25 and APBB1 genes. RAB25 is a member of the RAS family and is involved in membrane trafficking and cell survival. Rab25 has been previously described to show a tumor suppressive role in colon carcinogenesis, with loss of Rab25 promoting the development of intestinal neoplasia in mice [34, 35]. In head and neck squamous cell carcinomas Rab25 was shown to play an important role in tumor migration and metastasis [36]. In triple-negative breast cancers it has been described to influence tumor initiation and tumor progression [37]. In the same study, wild type Rab25 was shown to enhance apoptosis and suppress angiogenesis, further confirming a putative tumor suppressive function [37]. While the variant could unfortunately not be analyzed in other relatives to confirm co-segregation with the disease throughout the family, it remains an interesting candidate in familial CRC.
On average 1.8 patients per family are screened with WES in this study. The high number of variants remaining after excluding variants not present in all sequenced affected family members increased the difficulty of identifying a possible monogenic cause. Screening of 2 or 3 family members, preferably second-degree or more distantly related is advisable for detecting high penetrance genes in these families. However, screening distant related relatives increases the change of sequencing a phenocopy, e.g. a patient displaying the same phenotype but not due to the same genotype. By sequencing multiple patients within a family, it is even possible to determine oligogenic inheritance, although collecting many clinical samples is often not feasible. Additionally, screening of first-degree relatives might increase the change of patients carrying the same genetic CRC-predisposing variant, but will also increase the number of variants which are in both individuals by chance, but that do not increase CRC-risk.
A number of limitations in the present study need to be taken into consideration. Co-segregation of a variant with a CRC phenotype can provide insight into variants detected with WES, but CRC-screening programs for younger (unaffected) generations in CRC enriched families adds complexity in determining which family members are truly affected. Individuals within CRC enriched families are often screened from a younger age, and when adenomas are detected they are removed before they can progress to adenocarcinomas. These patients might have a genetic predisposition, but will not develop CRC. Whether or not to include these individuals as affected family members is arguable, but often a necessity. However, this unreliability needs to be kept in mind when assessing co-segregation within a family. Secondly, while WES could lead to initial candidate variants, functional testing of the variants within this study is lacking and is needed to proof the effect of the variants found. Finally, similar to other studies using targeted or whole exome sequencing, we identified few likely candidate single Mendelian causes of CRC in our enriched families [8, 16, 38]. Therefore, other causes of missing heredity such as single nucleotide polymorphisms possibly contributing risks through polygenic risk, or other epigenetic events, remain plausible sources of familial cancer risks, but can be missed with the current approach.
In conclusion, while previous studies find germline pathogenic variants in known highly penetrant CRC-susceptibility genes in 17-35% of early-onset patients irrespective of family history [15, 39], here we only find one known pathogenic variant, the previously described POLD1 c.1433G>A. In the remaining four families the most promising candidates seem to be a frameshift NOTCH2 variant, although this does not fully co-segregate with the phenotype in the family, and a predicted pathogenic missense variant in RAB25. While these candidate genes are possibly involved in CRC tumorigenesis, functional studies are imperative to confirm their association with CRC, and epistasis between genetic variants is still an open question to be resolved in the future.
Supplementary Material
Supplemental Figure 1: Pedigree of family F
Squares represent males, circles represent females and diamonds is undisclosed gender. Phenotype is shown as tumor type followed by age of onset. Patients presented with colorectal cancer (CRC; fully filled symbol) or polyps (pol, right top corner). Arrows indicate patient sequenced with whole exome sequencing.*DNA available for co-segregation study
Supplemental Figure 2: Enriched terms of familial and sporadic CRC TCGA cohort.
Enriched terms comparing the familial cohort (this study) with the sporadic CRC TCGA data are represented as scatterplots in a two dimensional space to summarize GO terms’semantic similarities (using REVIGO). Sematic space X and Y are used to be able to show distance between dots, but have no intrinsic meaning. The size of the dots indicates the frequency of the GO terms in the underlying GOA database. Distance of the dots from each other indicate the relatedness of the GO terms. Color of the dots indicates the p-value for false discovery rates. P-value is the enrichment p-value computed according to the mHG or HG model. This p-value is not corrected for multiple testing of 15413 GO terms. The most significant changes in the familiar cohort are seen in cell-cell adhesion pathways.
Acknowledgments
Funding: The present work was supported by the CA72851, CA184792, CA187956 and CA202797 grants from the National Cancer Institute, National Institute of Health; grants from the Sammons Cancer Center and Baylor Foundation, as well as funds from the Baylor Scott & White Research Institute, Dallas, TX, USA to AG. This work was also supported by grant CA160911 from the NIH to PG.
Footnotes
Conflicts of interest: The authors have no conflicts to disclose
REFERENCES
- 1.Valle L: Recent Discoveries in the Genetics of Familial Colorectal Cancer and Polyposis. Clin Gastroenterol Hepatol 2017, 15(6):809–819. [DOI] [PubMed] [Google Scholar]
- 2.Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, Bacher J, Bigley C, Nelsen L, Goodfellow PJ et al. : Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol 2017, 3(4):464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vasen HF: Clinical description of the Lynch syndrome [hereditary nonpolyposis colorectal cancer (HNPCC)]. Fam Cancer 2005, 4(3):219–225. [DOI] [PubMed] [Google Scholar]
- 4.Boland CR, Goel A: Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138(6):2073–2087.e2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindor NM, Rabe K, Petersen GM, Haile R, Casey G, Baron J, Gallinger S, Bapat B, Aronson M, Hopper J et al. : Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. Jama 2005, 293(16):1979–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shiovitz S, Copeland WK, Passarelli MN, Burnett-Hartman AN, Grady WM, Potter JD, Gallinger S, Buchanan DD, Rosty C, Win AK et al. : Characterisation of Familial Colorectal Cancer Type X, Lynch syndrome, and non-familial colorectal cancer. British Journal of Cancer 2014, 111(3):598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.FC DAS, Wernhoff P, Dominguez-Barrera C, Dominguez-Valentin M: Update on Hereditary Colorectal Cancer. Anticancer Res 2016, 36(9):4399–4405. [DOI] [PubMed] [Google Scholar]
- 8.Wei C, Peng B, Han Y, Chen WV, Rother J, Tomlinson GE, Boland CR, Chaussabel D, Frazier ML, Amos CI: Mutations of HNRNPA0 and WIF1 predispose members of a large family to multiple cancers. Fam Cancer 2015, 14(2):297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francisco I, Albuquerque C, Lage P, Belo H, Vitoriano I, Filipe B, Claro I, Ferreira S, Rodrigues P, Chaves P et al. : Familial colorectal cancer type X syndrome: two distinct molecular entities? Fam Cancer 2011, 10(4):623–631. [DOI] [PubMed] [Google Scholar]
- 10.Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I et al. : Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013, 45(2):136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Briggs S, Tomlinson I: Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol 2013, 230(2):148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shinbrot E, Henninger EE, Weinhold N, Covington KR, Goksenin AY, Schultz N, Chao H, Doddapaneni H, Muzny DM, Gibbs RA et al. : Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res 2014, 24(11):1740–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Spruijt L, van Zelst-Stams WA, Jongmans MC, Gilissen C, Hehir-Kwa JY et al. : A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet 2015, 47(6):668–671. [DOI] [PubMed] [Google Scholar]
- 14.Kuiper RP, Hoogerbrugge N: NTHL1 defines novel cancer syndrome. Oncotarget 2015, 6(33):34069–34070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoffel EM, Koeppe E, Everett J, Ulintz P, Kiel M, Osborne J, Williams L, Hanson K, Gruber SB, Rozek LS: Germline Genetic Features of Young Individuals With Colorectal Cancer. Gastroenterology 2018, 154(4):897–905.e891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dominguez-Valentin M, Nakken S, Tubeuf H, Vodak D, Ekstrøm PO, Nissen AM, Morak M, Holinski-Feder E, Martins A, Møller P et al. : Identification of genetic variants for clinical management of familial colorectal tumors. BMC Medical Genetics 2018, 19:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang K, Li M, Hakonarson H: ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research 2010, 38(16):e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP: Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLOS ONE 2012, 7(10):e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dakal TC, Kala D, Dhiman G, Yadav V, Krokhotin A, Dokholyan NV: Predicting the functional consequences of non-synonymous single nucleotide polymorphisms in IL8 gene. Sci Rep 2017, 7(1):6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ng PC, Henikoff S: SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003, 31(13):3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramensky V, Bork P, Sunyaev S: Human non-synonymous SNPs: server and survey. Nucleic Acids Res 2002, 30(17):3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N: ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 2010, 38(Web Server issue):W529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy A, Kucukural A, Zhang Y: I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols 2010, 5(4):725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dakal TC, Kumar R, Ramotar D: Structural modeling of human organic cation transporters. Comput Biol Chem 2017, 68:153–163. [DOI] [PubMed] [Google Scholar]
- 25.Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie FV, Mansour S, Holder SE, Brain CE, Burton BK et al. : Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat Genet 2011, 43(4):303–305. [DOI] [PubMed] [Google Scholar]
- 26.Guo F- J, Zhang W- J, Li Y- L, Liu Y, Li Y- H, Huang J, Wang J- J, Xie P- L, Li G- C: Expression and functional characterization of platelet-derived growth factor receptor-like gene. World Journal of Gastroenterology : WJG 2010, 16(12):1465–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujiwara Y, Ohata H, Kuroki T, Koyama K, Tsuchiya E, Monden M, Nakamura Y: Isolation of a candidate tumor suppressor gene on chromosome 8p21.3-p22 that is homologous to an extracellular domain of the PDGF receptor beta gene. Oncogene 1995, 10(5):891–895. [PubMed] [Google Scholar]
- 28.Montagner S, Leoni C, Emming S, Chiara GD, Balestrieri C, Barozzi I, Piccolo V, Togher S, Ko M, Rao A et al. : TET2 Regulates Mast Cell Differentiation and Proliferation through Catalytic and Non-catalytic Activities. Cell reports 2016, 15(7):1566–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nazha A, Meggendorfer M, Nadarajah N, Kneen KE, Radivoyevitch T, Przychodzen B, Makishima H, Patel BJ, Sanikommu SR, Hobson S et al. : TET2 Alterations in Myeloid Malignancies, Impact on Clinical Characteristics, Outcome, and Disease Predisposition. Blood 2015, 126(23):1645–1645. [Google Scholar]
- 30.Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MDM, Wendl MC, Zhang Q, Koboldt DC, Xie M, Kandoth C et al. : Integrated analysis of germline and somatic variants in ovarian cancer. Nature Communications 2014, 5:3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Y, Wang G, Liang Z, Yang Y, Cui L, Liu C- Y: Loss of nuclear localization of TET2 in colorectal cancer. Clinical Epigenetics 2016, 8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, Spinner NB: NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet 2006, 79(1):169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, Leigh IM, Collisson EA, Gordon PB, Jakkula L et al. : Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proceedings of the National Academy of Sciences 2011, 108(43):17761–17766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldenring JR, Nam KT: Rab25 as a tumour suppressor in colon carcinogenesis. British Journal of Cancer 2011, 104(1):33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nam KT, Lee HJ, Smith JJ, Lapierre LA, Kamath VP, Chen X, Aronow BJ, Yeatman TJ, Bhartur SG, Calhoun BC et al. : Loss of Rab25 promotes the development of intestinal neoplasia in mice and is associated with human colorectal adenocarcinomas. J Clin Invest 2010, 120(3):840–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amornphimoltham P, Rechache K, Thompson J, Masedunskas A, Leelahavanichkul K, Patel V, Molinolo A, Gutkind JS, Weigert R: Rab25 regulates invasion and metastasis in head and neck cancer. Clin Cancer Res 2013, 19(6):1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng JM, Volk L, Janaki DK, Vyakaranam S, Ran S, Rao KA: Tumor suppressor function of Rab25 in triple-negative breast cancer. Int J Cancer 2010, 126(12):2799–2812. [DOI] [PubMed] [Google Scholar]
- 38.DeRycke MS, Gunawardena SR, Middha S, Asmann YW, Schaid DJ, McDonnell SK, Riska SM, Eckloff BW, Cunningham JM, Fridley BL et al. : Identification of novel variants in colorectal cancer families by high-throughput exome sequencing. Cancer Epidemiol Biomarkers Prev 2013, 22(7):1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pearlman R, Frankel WL, Swanson B, et al. : Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncology 2017, 3(4):464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Pedigree of family F
Squares represent males, circles represent females and diamonds is undisclosed gender. Phenotype is shown as tumor type followed by age of onset. Patients presented with colorectal cancer (CRC; fully filled symbol) or polyps (pol, right top corner). Arrows indicate patient sequenced with whole exome sequencing.*DNA available for co-segregation study
Supplemental Figure 2: Enriched terms of familial and sporadic CRC TCGA cohort.
Enriched terms comparing the familial cohort (this study) with the sporadic CRC TCGA data are represented as scatterplots in a two dimensional space to summarize GO terms’semantic similarities (using REVIGO). Sematic space X and Y are used to be able to show distance between dots, but have no intrinsic meaning. The size of the dots indicates the frequency of the GO terms in the underlying GOA database. Distance of the dots from each other indicate the relatedness of the GO terms. Color of the dots indicates the p-value for false discovery rates. P-value is the enrichment p-value computed according to the mHG or HG model. This p-value is not corrected for multiple testing of 15413 GO terms. The most significant changes in the familiar cohort are seen in cell-cell adhesion pathways.