

Are the tau pathologies of CTE unique? Arena et al. show subtypes of tau proteins that accumulate in neurons are different from those in astrocytes in CTE, overlapping with patterns found in ageing and Alzheimer’s disease. This highlights the challenge of distinguishing tau pathologies in CTE from other neurodegenerative diseases.
Keywords: chronic traumatic encephalopathy, tau, ageing-related tau astrogliopathy, traumatic brain injury, TBI
Abstract
Traumatic brain injury (TBI) is a risk factor for neurodegenerative disease, including chronic traumatic encephalopathy (CTE). Preliminary consensus criteria define the pathognomonic lesion of CTE as patchy tau pathology within neurons and astrocytes at the depths of cortical sulci. However, the specific tau isoform composition and post-translational modifications in CTE remain largely unexplored. Using immunohistochemistry, we performed tau phenotyping of CTE neuropathologies and compared this to a range of tau pathologies, including Alzheimer’s disease, primary age-related tauopathy, ageing-related tau astrogliopathy and multiple subtypes of frontotemporal lobar degeneration with tau inclusions. Cases satisfying preliminary consensus diagnostic criteria for CTE neuropathological change (CTE-NC) were identified (athletes, n = 10; long-term survivors of moderate or severe TBI, n = 4) from the Glasgow TBI Archive and Penn Neurodegenerative Disease Brain Bank. In addition, material from a range of autopsy-proven ageing-associated and primary tauopathies in which there was no known history of exposure to TBI was selected as non-injured controls (n = 32). Each case was then stained with a panel of tau antibodies specific for phospho-epitopes (PHF1, CP13, AT100, pS262), microtubule-binding repeat domains (3R, 4R), truncation (Tau-C3) or conformation (GT-7, GT-38) and the extent and distribution of staining assessed. Cell types were confirmed with double immunofluorescent labelling. Results demonstrate that astroglial tau pathology in CTE is composed of 4R-immunoreactive thorn-shaped astrocytes, echoing the morphology and immunophenotype of astrocytes encountered in ageing-related tau astrogliopathy. In contrast, neurofibrillary tangles of CTE contain both 3R and 4R tau, with post-translational modifications and conformations consistent with Alzheimer’s disease and primary age-related tauopathy. Our observations establish that the astroglial and neurofibrillary tau pathologies of CTE are phenotypically distinct from each other and recapitulate the tau immunophenotypes encountered in ageing and Alzheimer’s disease. As such, the immunohistochemical distinction of CTE neuropathology from other mixed 3R/4R tauopathies of Alzheimer’s disease and ageing may rest solely on the pattern and distribution of pathology.
Introduction
There is increasing recognition of the association between exposure to traumatic brain injury (TBI) and risk of subsequent neurodegenerative disease, in particular chronic traumatic encephalopathy (CTE). First described as ‘punch drunk syndrome’ in 1928 (Martland, 1928) and later ‘dementia pugilistica’ (Millspaugh, 1937), progressive neurodegeneration in the context of TBI was historically considered a consequence of participation in boxing. However, more recent descriptions of the neurodegenerative pathology of dementia pugilistica, now recognized as CTE, in non-boxer athletes exposed to repetitive mild TBI (Corsellis et al., 1973; Geddes et al., 1999; Omalu et al., 2011; Saing et al., 2012; McKee et al., 2013, 2016; Smith et al., 2013, 2019; Stewart et al., 2016; Johnson et al., 2017; Mez et al., 2017; Wilson et al., 2017; Lee et al., 2019) and late survivors of moderate or severe TBI (Johnson et al., 2012; Zanier et al., 2018; Kenney et al., 2018 ) have brought the potential lifelong consequences of TBI exposure to wider attention. Nevertheless, the current consensus criteria for CTE neuropathological assessment remain preliminary and there are few detailed accounts of its similarities and differences with other tauopathies.
While intraneuronal tau aggregates in the form of neurofibrillary tangles (NFTs) have long been described in dementia pugilistica/CTE, more recent studies also note the presence of pathological astroglial tau accumulation in the form of thorn-shaped astrocytes (TSA) (Ikeda et al., 1995, 1998; Schmidt et al., 2001; McKee et al., 2009, 2013; Saing et al., 2012; Kanaan et al., 2016; Mez et al., 2017). Indeed, the importance of this mixed neuronal and astroglial pathology in CTE is reflected in preliminary consensus diagnostic criteria for the disease, which propose the pathognomonic lesion as ‘phosphorylated-tau aggregates in neurons, astrocytes, and cell processes around small vessels in an irregular pattern at the depths of the cortical sulci’ (McKee et al., 2016). This sulcal lesion is suggested as sufficiently unique to distinguish CTE from other tau-associated neurodegenerative diseases including Alzheimer’s disease, frontotemporal lobar degeneration characterized by tau inclusions (FTLD-tau)—including progressive supranuclear palsy (PSP), corticobasal degeneration (CBD) and Pick’s disease (Cairns et al., 2007; Montine et al., 2012; Kovacs, 2015), as well as the pathologies of primary age-related tauopathy (PART) and ageing-related tau astrogliopathy (ARTAG) (Crary et al., 2014; Kovacs et al., 2016). However, there remains debate regarding whether tau found at sulcal depths is solely found in individuals with a history of head impacts, or if it can occur in individuals with no contact sport or TBI history (Iverson et al., 2019).
Beyond the potentially unique distribution of tau pathologies in CTE, little is known regarding tau composition and post-translational modifications. Of the six tau isoforms containing either 3 or 4 microtubule-binding repeat domains (3R versus 4R tau) (Goode et al., 2000), NFTs in Alzheimer’s disease and PART contain both 3R and 4R tau (Espinoza et al., 2008; Santa-Maria et al., 2012; Crary et al., 2014). In contrast, while the Pick bodies and ramified astrocytes of Pick’s disease contain primarily 3R tau (Irwin et al., 2016), the astrocytic tau pathologies of PSP, CBD and ARTAG are composed of 4R tau (Cairns et al., 2007; Ferrer et al., 2014). Regarding CTE pathology, biochemical analysis of material from two former boxers with dementia pugilistica/CTE reported NFTs comprising both 3R and 4R tau, similar to Alzheimer’s disease (Schmidt et al., 2001; McKee et al., 2013). A further case series indicates that tau in CTE displays conformations and phospho-epitopes comparable to those observed early in NFT maturation in Alzheimer’s disease (Kanaan et al., 2016). Nonetheless, comprehensive assessment of the phosphorylation, truncation and conformation of both glial and neuronal tau pathologies in CTE has not previously been assessed, particularly within the context of other established tauopathies (Ferrer et al., 2014; Kovacs, 2015; Irwin et al., 2016).
Here, we report immunohistochemical observations on extensive tau phenotypic analysis of autopsy-derived material from individuals with known CTE neuropathologies and histories of exposure to either repetitive sport-related mild TBI, or moderate/severe TBI, when compared to ageing-associated and primary neurodegenerative tauopathies. Specifically, applying antibodies specific for tau isoform, multiple phospho-epitopes, caspase-mediated truncation, and novel monoclonal antibodies capable of differentiating 3R/4R tau in Alzheimer’s disease from the conformationally distinct FTLD-tau (Gibbons et al., 2018, 2019), we demonstrate that while the NFTs and astroglial tau pathologies of CTE differ immunohistochemically from each other, they recapitulate features of Alzheimer’s disease/PART and ARTAG, respectively.
Materials and methods
Cohort demographics
All cases were obtained from the Glasgow TBI Archive, Department of Neuropathology, Queen Elizabeth University Hospital, Glasgow, UK or the University of Pennsylvania Center for Neurodegenerative Disease Research (CNDR) Brain Bank, Philadelphia, PA, USA. Brain tissue was acquired by means of planned donation or at routine diagnostic autopsy. Ethical approval for use of tissue in this study was provided by the West of Scotland Research Ethics Committee (Project ID 225271); and the Greater Glasgow and Clyde Biorepository (Application Number 340), as well as the institutional review board of the University of Pennsylvania.
Cases were selected with a history of participation in contact sports: American football (n = 4), rugby (n = 3), soccer (n = 2), or boxing (n = 1), or a remote history of single moderate or severe TBI caused by assault (n = 1), motor vehicle collision (n = 1) or fall (n = 1). One additional case sustained one mild and one moderate TBI caused by falls (n = 1). Cases were selected with previously confirmed CTE neuropathological change (CTE-NC) (Lee et al., 2019) based on the preliminary consensus criteria for the neuropathological evaluation of CTE (McKee et al., 2016). Notably, in addition to CTE neuropathology, multiple cases also displayed co-morbid neuropathologies (n = 8) as has previously been reported (Lee et al., 2019). Clinical, demographic and neuropathological information, including integrated clinicopathological diagnoses (Lee, 2018) for all cases is presented in Table 1.
Table 1.
Demographic and clinical summary of cases
| Case | Group | Age | Sex | Sport / TBI exposure | Integrated clinicopathologic diagnosis | PMI, h | Source | Anatomical region examined |
|---|---|---|---|---|---|---|---|---|
| 1 | CTE-NC | 40s | M | Football | CBD | 7 | Penn-CNDR | Frontal |
| 2 | CTE-NC | 60s | M | Football | CBD | 3 | Penn-CNDR | Frontal |
| 3 | CTE-NC | 70s | M | Football | DLB | 18 | Penn-CNDR | Frontal |
| 4 | CTE-NC | 80s | M | Football | FTLD-TDP | 7 | Penn-CNDR | Frontal |
| 5 | CTE-NC | 60s | M | Boxing | CTE | 24 | GTBIA | Temporal |
| 6 | CTE-NC | 70s | M | Rugby | CTE | 12 | GTBIA | Insular |
| 7 | CTE-NC | 70s | M | Rugby | AD | 48 | GTBIA | Frontal |
| 8 | CTE-NC | 70s | M | Rugby | Mixed AD/VaD | 48 | GTBIA | Frontal |
| 9 | CTE-NC | 50s | M | Soccer | CTE | Unknown | GTBIA | Insular |
| 10 | CTE-NC | 80s | M | Soccer | AD | 24 | GTBIA | Frontal |
| 11 | CTE-NC | 50s | M | sTBI | Remote TBI - no NDD | 108 | GTBIA | Frontal |
| 12 | CTE-NC | 60s | M | sTBI | Remote TBI - no NDD | 24 | GTBIA | Temporal |
| 13 | CTE-NC | 70s | M | sTBI | CTE | 24 | GTBIA | Temporal |
| 14 | CTE-NC | 70s | M | Mild and moderate TBI | PDD | 7.5 | Penn-CNDR | Angular |
| 15 | ARTAG, PART | 50s | M | No | No NDD | 80.5 | GTBIA | Frontal |
| 16 | ARTAG, PART | 80s | M | No | PART | 19 | Penn-CNDR | Temporal |
| 17 | ARTAG, PART | 70s | M | No | No NDD | 17 | Penn-CNDR | Temporal |
| 18 | PART | 60s | M | No | No NDD | 11 | Penn-CNDR | Temporal |
| 19 | ARTAG, low ADNC | 70s | F | No | No NDD | 18 | Penn-CNDR | Amygdala |
| 20 | ARTAG, low ADNC | 70s | F | No | No NDD | 19 | Penn-CNDR | Amygdala |
| 21 | ARTAG, low ADNC | 70s | F | No | No NDD | 18 | Penn-CNDR | Temporal |
| 22 | ARTAG, low ADNC | 80s | M | No | No NDD; CVD | 7 | Penn-CNDR | Amygdala |
| 23 | AD | 60s | M | No | AD | 13.5 | Penn-CNDR | Angular |
| 24 | AD | 60s | M | No | AD | 5 | Penn-CNDR | Frontal |
| 25 | AD | 70s | M | No | AD | 4 | Penn-CNDR | Angular |
| 26 | AD | 70s | M | No | AD | 8.5 | Penn-CNDR | Angular |
| 27 | AD | 70s | F | No | AD | 11 | Penn-CNDR | Temporal |
| 28 | AD | 80s | F | No | AD | 6 | GTBIA | Cingulate |
| 29 | FTLD-Tau | 50s | M | No | PiD | 11 | Penn-CNDR | Temporal |
| 30 | FTLD-Tau | 60s | M | No | PiD | Unknown | Penn-CNDR | Temporal |
| 31 | FTLD-Tau | 70s | M | No | PiD | 22 | Penn-CNDR | Frontal |
| 32 | FTLD-Tau | 70s | M | No | PiD | 4 | Penn-CNDR | Angular |
| 33 | FTLD-Tau | 80s | F | No | PiD | 24 | Penn-CNDR | Frontal |
| 34 | FTLD-Tau | 50s | M | No | PiD | 10 | Penn-CNDR | Frontal |
| 35 | FTLD-Tau | 70s | M | No | PSP | 14 | Penn-CNDR | Frontal |
| 36 | FTLD-Tau | 80s | M | No | PSP | 23 | Penn-CNDR | Angular |
| 37 | FTLD-Tau | 70s | M | No | PSP | 23 | Penn-CNDR | Frontal |
| 38 | FTLD-Tau | 60s | M | No | PSP | 13 | Penn-CNDR | Angular |
| 39 | FTLD-Tau | 70s | M | No | PSP | 17 | Penn-CNDR | Temporal |
| 40 | FTLD-Tau | 70s | M | No | PSP | 19 | Penn-CNDR | Frontal |
| 41 | FTLD-Tau | 60s | F | No | CBD | 16 | Penn-CNDR | Temporal |
| 42 | FTLD-Tau | 70s | M | No | CBD | 11 | Penn-CNDR | Angular |
| 43 | FTLD-Tau | 50s | M | No | CBD | 41 | Penn-CNDR | Frontal |
| 44 | FTLD-Tau | 70s | M | No | CBD | 7.5 | Penn-CNDR | Temporal |
| 45 | FTLD-Tau | 80s | M | No | CBD | 11 | Penn-CNDR | Frontal |
| 46 | FTLD-Tau | 70s | M | No | CBD | 5 | Penn-CNDR | Frontal |
AD = Alzheimer’s disease; ADNC = Alzheimer’s disease neuropathological change; CTE-NC = CTE-neuropathological change; CVD = cerebrovascular disease; DLB = dementia with Lewy bodies; F = female; FTLD-TDP = frontotemporal lobar degeneration with TDP-43 inclusions; GTBIA = Glasgow TBI archive; M = male; NDD = neurodegenerative disease; PDD = Parkinson’s disease dementia; Penn-CNDR = University of Pennsylvania Center for Neurodegenerative Disease Research; PiD = Pick’s disease; PMI = post-mortem interval; sTBI = single moderate or severe TBI; VaD = vascular dementia.
To permit comparisons of CTE tau pathologies with those of established neurodegenerative disease, material from patients without documented history of TBI or participation in contact sport was selected that met neuropathological criteria for the diagnosis of Alzheimer’s disease (Braak stage V or VI; n = 6) or FTLD-tau as Pick’s disease (n = 6), PSP (n = 6), or CBD (n = 6). In addition, non-demented control cases without history of TBI and with known ageing-related tau pathologies were selected including: PART (n = 1), ARTAG and PART (n = 3) and ARTAG with low Alzheimer’s disease neuropathological change (n = 4) (Cairns et al., 2007; Montine et al., 2012; Crary et al., 2014; Kovacs et al., 2016; McKee et al., 2016) (Table 1).
Brain tissue handling
Whole brains from the Glasgow TBI archive were fixed in 10% formol saline at autopsy for a minimum of 2 weeks prior to dissection. Standardized anatomical sampling, tissue processing and paraffin embedding were performed as previously described (Graham et al., 1995). From the University of Pennsylvania CNDR Brain Bank, tissue blocks cut from fresh brains were fixed overnight in 70% ethanol and 150 mM sodium chloride or 10% neutral buffered formalin and processed to paraffin as previously described (Toledo et al., 2014). From each case, 8-µm tissue sections were prepared from regions with stereotypical CTE neuropathology at the depths of cortical sulci. For comparison, regions displaying hallmark, disease-specific pathologies were selected from non-trauma control cases of Alzheimer’s disease (NFTs), PART (NFTs), Pick’s disease (Pick bodies and ramified astrocytes), PSP (tufted astrocytes), CBD (astrocytic plaques) and ARTAG (TSA) (Cairns et al., 2007; Montine et al., 2012; Crary et al., 2014; Kovacs et al., 2016).
Single immunohistochemical labelling
Serial tissue sections for all cases were subjected to deparaffinization and rehydration to H2O before being immersed in 3% aqueous H2O2 (15 min) to quench endogenous peroxidase activity. Antigen retrieval was performed via microwave pressure cooker in either Tris/EDTA or citrate buffer, with or without formic acid pretreatment, as optimized for each antibody (Supplementary Table 1). Sections were blocked using normal horse serum (Vector Labs) in OptimaxTM buffer (BioGenex) for 30 min followed by incubation in the primary antibody overnight at 4°C. Specifically, a panel of tau antibodies (Supplementary Table 1) was applied targeting multiple phospho-epitopes including S202 (CP13) (Jicha et al., 1999), S396/S404 (PHF1) (Greenberg et al., 1992; Otvos et al., 1994), S212/T214 (AT100) (Hoffmann et al., 1997; Zheng-Fischhofer et al., 1998) and S262; 3 or 4 microtubule-binding domain repeats (RD3 and RD4) (de Silva et al., 2003); and caspase-cleaved tau at Asp421 (Tau-C3) (Gamblin et al., 2003). In addition we applied the recently characterized antibodies GT-7 and GT-38 (Gibbons et al., 2018, 2019). Evidence from co-immunofluorescence studies in human tissue with FTLD-tau and Alzheimer’s disease-tau suggest that GT-38 requires the presence of both 3R and 4R tau. Moreover, it was demonstrated that GT-38 binding requires a pathological conformation of Alzheimer’s disease-tau since chemical denaturation leads to a reduction of GT-38 binding (Gibbons et al., 2018).
After rinsing, sections were incubated in a biotinylated universal secondary antibody (Vector Labs) for 30 min, followed by the avidin-biotin complex for 30 min (Vector Labs). Visualization was achieved using the DAB peroxidase substrate kit (Vector Labs). Sections were counterstained with haematoxylin, followed by rinsing, dehydration, and coverslipping using Cytoseal™ 60. Tissue sections from a case with neuropathologically confirmed Alzheimer’s disease were included as a positive control in all staining procedures. Omission of the primary antibody using the same Alzheimer’s disease case was performed in parallel to control for non-specific binding. Notably, three cases [Cases 9 (CTE neuropathology), 11 (CTE neuropathology) and 15 (ARTAG/PART)] (Table 2) failed to demonstrate immunoreactivity to antibodies specific for 3R and 4R tau due to fixation sensitivity, as has been reported previously with these antibodies (Espinoza et al., 2008; Ferrer et al., 2014).
Table 2.
Tau phenotype semiquantitative scores: 3R, 4R and conformation
| Case | 3R |
4R |
GT − 7 |
GT − 38 |
||||
|---|---|---|---|---|---|---|---|---|
| TSA | NFT | TSA | NFT | TSA | NFT | TSA | NFT | |
| 1 CTE-NC | − | ++ | +++ | +++ | – | ++ | – | ++ |
| 2 CTE-NC | − | ++ | +++ | +++ | – | ++ | – | ++ |
| 3 CTE-NC | − | +++ | +++ | +++ | – | +++ | – | +++ |
| 4 CTE-NC | + | ++ | +++ | +++ | – | ++ | + | ++ |
| 5 CTE-NC | − | +++ | +++ | +++ | – | +++ | – | +++ |
| 6 CTE-NC | − | +++ | +++ | +++ | – | +++ | – | +++ |
| 7 CTE-NC | + | +++ | +++ | +++ | – | +++ | – | +++ |
| 8 CTE-NC | − | +++ | +++ | +++ | + | +++ | + | +++ |
| 9 CTE-NC | x | x | x | x | x | x | x | x |
| 10 CTE-NC | − | +++ | +++ | +++ | – | +++ | – | +++ |
| 11 CTE-NC | x | x | x | x | – | +++ | – | +++ |
| 12 CTE-NC | − | ++ | ++ | ++ | – | ++ | – | ++ |
| 13 CTE-NC | − | +++ | ++ | ++ | + | +++ | + | +++ |
| 14 CTE-NC | − | +++ | +++ | +++ | + | ++ | – | ++ |
| 15 ARTAG, PART | x | x | x | x | – | +++ | – | +++ |
| 16 ARTAG, PART | − | + | +++ | +++ | – | +++ | – | +++ |
| 17 ARTAG, PART | − | +++ | +++ | +++ | – | +++ | – | +++ |
| 18 PART | N/A | ++ | N/A | ++ | N/A | ++ | N/A | ++ |
| 19 ARTAG, low ADNC | − | +++ | +++ | +++ | – | ++ | – | +++ |
| 20 ARTAG, low ADNC | + | ++ | +++ | +++ | + | +++ | + | +++ |
| 21 ARTAG, low ADNC | − | +++ | +++ | +++ | – | +++ | – | +++ |
| 22 ARTAG, low ADNC | − | +++ | +++ | +++ | – | +++ | – | +++ |
| 23 AD | +++ | +++ | +++ | +++ | ||||
| 24 AD | +++ | +++ | +++ | +++ | ||||
| 25 AD | +++ | +++ | +++ | +++ | ||||
| 26 AD | +++ | +++ | +++ | +++ | ||||
| 27 AD | +++ | +++ | +++ | +++ | ||||
| 28 AD | ++ | ++ | +++ | +++ | ||||
|
| ||||||||
| Pick bodies | Ramified astro | Pick bodies | Ramified astro | Pick bodies | Ramified astro | Pick bodies | Ramified astro | |
|
| ||||||||
| 29 PiD | +++ | +++ | − | ++ | – | – | – | – |
| 30 PiD | +++ | N/A | − | N/A | – | N/A | – | N/A |
| 31 PiD | +++ | +++ | − | − | – | – | – | – |
| 32 PiD | +++ | N/A | − | N/A | – | N/A | – | N/A |
| 33 PiD | +++ | N/A | − | N/A | – | N/A | – | N/A |
| 34 PiD | +++ | +++ | − | − | – | – | – | – |
|
| ||||||||
| Tufted astrocytes | Tufted astrocytes | Tufted astrocytes | Tufted astrocytes | |||||
|
| ||||||||
| 35 PSP | − | +++ | − | − | ||||
| 36 PSP | − | +++ | − | − | ||||
| 37 PSP | − | ++ | − | − | ||||
| 38 PSP | − | +++ | − | − | ||||
| 39 PSP | − | ++ | − | − | ||||
| 40 PSP | − | +++ | − | − | ||||
|
| ||||||||
| Astrocytic plaques | Astrocytic plaques | Astrocytic plaques | Astrocytic plaques | |||||
|
| ||||||||
| 41 CBD | − | +++ | − | − | ||||
| 42 CBD | − | +++ | − | − | ||||
| 43 CBD | − | +++ | − | − | ||||
| 44 CBD | − | +++ | − | − | ||||
| 45 CBD | − | +++ | − | − | ||||
| 46 CBD | − | +++ | − | − | ||||
Semiquantitative scoring: − = absent, or nearly absent (≤5% concordance with extent of PHF1); + = minimal (5–30% concordance); ++ = moderate (30–70% concordance); +++ = extensive (>70% concordance); × = no immunoreactivity.
AD = Alzheimer’s disease; ADNC = Alzheimer’s disease neuropathological change; CTE-NC = CTE neuropathological change; N/A = not applicable.
Double immunofluorescent labelling
Serial tissue sections from a subset of cases (CTE n = 5, ARTAG n = 2, Alzheimer’s disease n = 2) were selected for double labelling immunofluorescence to confirm and validate morphological identification of cell types (astrocytes versus neurons) as identified by both PHF1 and GT-38. Specifically, sections were labelled with combinations of tau antibodies (PHF1 or GT-38) and cell-type specific markers, namely MAP2 for neurons and GFAP for astrocytes using established protocols (Johnson et al., 2016). Briefly, following deparaffinization and rehydration, antigen retrieval was performed as described above and tissue blocked in the relevant species-specific serum (1%) (Vector Labs). Primary antibodies were applied serially for 20 h (4°C) specific for PHF1 (1:100) or GT-38 (1:100), followed by glial fibrillary acidic protein (GFAP) (Abcam; 1:200) or the microtubule-associated protein 2 (MAP2) (Abcam; 1:200). After rising, the corresponding Alexa Fluor® (Invitrogen) secondary antibody was applied at 1:500 in a 2% species-specific blocking solution for 2 h at room temperature. Serial sections of positive control tissue (Alzheimer’s disease) were subjected to the entire procedure with omission of subsets of primary antibodies to control for non-specific immunofluorescence. Following rinsing, all double fluorescent-immunolabelled sections were incubated in TrueView autofluorescence quenching reagent (Vector Labs) for 5 min at room temperature before being rinsed and coverslipped using VECTASHIELD® mounting medium (Vector Labs).
Analysis of immunohistochemical findings
Using a standardized approach, the extent of immunoreactivity for each antibody was scored relative to that observed using an index antibody in each individual case, as has been described previously (Ferrer et al., 2014). Specifically, a semiquantitative score was used to denote the extent of immunoreactivity relative to that of PHF1 as: absent or nearly absent (<5% concordance with the extent of PHF1 immunoreactivity); minimal (5–30% concordance); moderate (30–70% concordance); or extensive (>70% concordance). PHF1 was selected as the index antibody given its widely reported use for the identification of tau pathologies across neurodegenerative diseases and recommended use for the identification of CTE pathology in preliminary consensus criteria (McKee et al., 2016). Under this protocol, scoring does not reflect the number of positive cells or permit comparisons of the extent of pathology between cases, but rather reflects the relative extent to which a particular antibody recognizes the burden of tau pathology as identified via PHF1 in each field of interest. Glial versus neuronal pathologies were distinguished based on characteristic cellular morphologies. A subset of sections was reviewed and scored independently by two observers (J.D.A. and V.E.J.), with good interrater reliability (Cohen’s κ = 0.71). Where there was a discrepancy in scoring, cases were jointly reviewed and a consensus score reached.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material.
Results
Consistent with prior descriptions of CTE neuropathology (McKee et al., 2016), each TBI case displayed PHF1-positive neurons and astrocytes in a patchy and perivascular distribution concentrated at the depths of cortical sulci (Fig. 1). Astrocytes typically displayed thorn-shaped morphologies, with short, thickened processes (Ikeda et al., 1995; Kovacs et al., 2016, 2017a), frequently in the immediate subpial region at the sulcal depth, in addition to being observed in a patchy and perivascular distribution within deeper layers of cortex. Neuronal tau pathology at the depths of cortical sulci in cases with CTE neuropathology displayed the morphology of NFTs, consistent with historical and contemporary descriptions (Corsellis et al., 1973; Geddes et al., 1996, 1999; Omalu et al., 2005; McKee et al., 2009, 2013, 2016). The distinctive cellular morphologies of CTE astrocytes and neurons were confirmed via double immunofluorescence labelling on a subset of cases as described. Specifically, NFTs identified via PHF1 co-localized with MAP2, but not GFAP. Conversely, TSAs were observed to co-localize with GFAP but not MAP2, consistent with findings in Alzheimer’s disease and ARTAG (Supplementary Figs 1–3).
Figure 1.

Sulcal depth astrocytic and neuronal tau pathologies in CTE. (A and B) PHF1 immunohistochemistry reveals NFT and TSA pathology concentrated at the depths of cortical sulci of a former American football player (Case 3) and (C and D) chronic survivor of a single severe TBI (Case 11). (B and D) High magnification images from the same sulci showing perivascular pathology composed of mixed neuronal and astrocytic populations consistent with the preliminary diagnostic criteria for CTE. Scale bars = 100 µm.
As expected, neurodegenerative disease control subjects displayed the hallmark and cell-specific tau pathologies characteristic for each diagnosis as identified by PHF1. Specifically, FTLD-tau controls demonstrated Pick bodies and, in some cases, ramified astrocytes in Pick’s disease, tufted astrocytes in PSP and astrocytic plaques in CBD. Alzheimer’s disease cases had NFTs in a bilaminar cortical distribution in addition to diffuse neuritic threads. Controls with ageing-related pathologies displayed limited cortical NFT pathology consistent with PART or low Alzheimer’s disease neuropathological change, as well as TSAs consistent with ARTAG in white matter, perivascular, grey matter, subpial, or subependymal distributions.
Astroglial pathology of CTE contains 4R tau only, whereas neurofibrillary tangles are composed of both 3R and 4R tau
Tau positive astrocytes within regions of CTE neuropathology were composed of 4R tau only in virtually all cases (Figs 2, 3 and Table 2). Specifically, in cases demonstrating adequate immunoreactivity, robust and consistent immunoreactivity to RD4 was observed in astrocytes within the subpial region, as well as those extending to deeper cortical layers. In contrast, astrocytic immunoreactivity to the RD3 antibody was absent in all but two cases where just minimal staining was observed. Notably, TSAs of CTE were morphologically indistinguishable from those within non-injured control cases with ARTAG, which were also composed almost entirely of 4R tau, consistent with previous reports (Lopez-Gonzalez et al., 2013; Ferrer et al., 2018) (Fig. 3). Several cases with CTE neuropathology displayed TSA elsewhere in the tissue sections examined, including within subcortical white matter, subependymal, and subpial regions, in keeping with descriptions of ARTAG. These astrocytes were morphologically indistinguishable from those in the sulcal depths associated with stereotypical CTE neuropathology and displayed the same pattern of selective 4R tau immunoreactivity.
Figure 2.

3R versus 4R tau immunoreactivity in CTE, Alzheimer’s disease, PART and Pick’s disease. (A) Sulcal depth CTE neuropathology with prominent subpial TSAs in addition to patchy and perivascular TSAs and NFTs within the deeper layers of cortex (Case 10; PHF1 staining). (B) Higher magnification of box in A displaying perivascular astrocytic and neuronal tau pathologies. (C) Immunoreactivity-specific for 3R tau in the same region as in B showing perivascular NFTs, but an absence of immunoreactivity within astrocytes. (D) In contrast, immunohistochemistry specific for 4R tau identified cells with both neuronal and astrocytic morphologies in the same region. (E) Consistent with previous descriptions, cases meeting diagnostic criteria for Alzheimer’s disease (AD; Case 25) and PART (Case 15) displayed neurofibrillary tangles in the cortex that were immunoreactive for 3R and 4R tau. In contrast, Pick bodies of Pick’s disease (PiD) within the dentate granule cells of Case 29 were composed of only 3R tau. Scale bars = 100 µm in A–D, 50 µm in E.
Figure 3.

3R versus 4R tau immunoreactivity within the astrocytic pathologies of CTE, ARTAG, PSP, CBD and Pick’s disease. Representative examples of serial sections showing immunoreactivity specific for 4R tau, but not 3R tau, in the TSAs in cases of both CTE (Case 6) and ARTAG (Case 21), tufted astrocytes in a case with PSP (Case 35) and astrocytic plaques within a case of CBD (Case 41). The ramified astrocytes observed in just one case of Pick’s disease (PiD; Case 29) were immunoreactive for 3R and 4R tau. Scale bars = 50 µm.
In contrast, ramified astrocytes of Pick’s disease were composed of 3R tau, with a subset of cells in just one of the six cases also displaying 4R immunoreactivity, as has been reported previously in a subset of cases (Arai et al., 2001; Ferrer et al., 2014; Irwin et al., 2016). The tufted astrocytes in PSP and astrocytic plaques in CBD controls displayed immunoreactivity for 4R tau only (Fig. 3).
While astrocytes in CTE were typically 4R tau-immunoreactive/3R tau-negative, NFTs in these regions were immunoreactive for both 3R and 4R tau isoforms, similar to those of Alzheimer’s disease and PART (Fig. 2 and Table 2) and distinct from Pick bodies (3R positive only) (Fig. 2).
Post-translational modification of tau in CTE neuropathology is consistent with ARTAG and Alzheimer’s disease
The post-translational modifications of tau within astrocytes and NFTs of CTE were observed to recapitulate those of ARTAG and Alzheimer’s disease with respect to all antibodies assessed. Specifically, tau immunoreactive astrocytes in CTE exhibited phosphorylation at residues S202 (CP13), S212/T214 (AT100), S262, and S396/S404 (PHF1) (Fig. 4 and Table 3). Typically, CP13, AT100, and PHF1 displayed dense cytoplasmic staining throughout the cell body. In contrast, pS262 immunoreactivity displayed both robust and confluent immunoreactivity (Fig. 4), as well as a more dot-like pattern of immunoreactivity in a subset of cases/cells that was often concentrated in the peripheral processes of the astrocyte (Supplementary Fig. 4). Caspase-mediated truncation at D421 (Tau-C3), however, was virtually never seen in astrocytes in CTE, with just minimal cells observed in a single case. Again, all findings were consistent across TSA in subpial and deeper cortical astrocytes within the sulcal depth. This profile of staining was also indistinguishable from that observed in non-injured, ARTAG control cases (Fig. 4). In contrast with the astroglial pathologies of CTE, ARTAG, Pick’s disease and CBD, which did not typically demonstrate immunoreactivity to Tau-C3, subsets of tufted astrocytes in two PSP cases demonstrated Tau-C3 immunoreactivity, consistent with previous reports (Fig. 4) (Ferrer et al., 2014).
Figure 4.
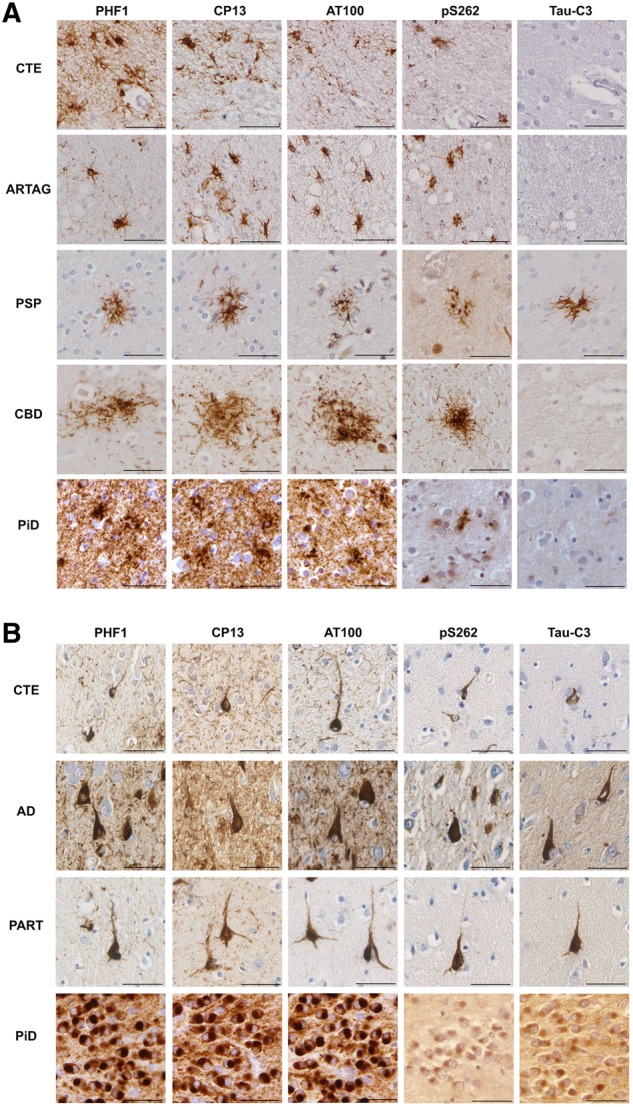
Post-translational modifications of tau within astrocytes and neurons in CTE, Alzheimer's disease, PART, ARTAG, PSP, CBD and Pick’s disease. (A) Representative examples of serial sections showing the TSAs of CTE (Case 3) and ARTAG (Case 22), astrocytic plaques of CBD (Case 41) and ramified astrocytes in a case of Pick’s disease (PiD; Case 29) all displayed robust immunoreactivity to phospho-epitope antibodies PHF1, CP13, AT100 and pS262, but not Tau-C3 (truncation at D421). In contrast with all other astrocytic tau pathologies examined, the tufted astrocytes of PSP in a subset of cases (Case 35 pictured here) were labelled with Tau-C3, indicative of truncation as described. (B) Representative examples of serial sections showing the neurofibrillary tangles of CTE (Case 5), Alzheimer’s disease (AD; Case 28) and PART (Case 15) with immunoreactivity to the phospho-epitope antibodies PHF1, CP13, AT100 and pS262, as well as Tau-C3 indicating truncation at D421. In addition, while Pick bodies (Case 29) also demonstrated the same post-translational modifications, immunoreactivity to p262 and Tau-C3 was noted in just one case as shown here within the dentate granule cells, where immunoreactivity was notably less robust. Scale bars = 50 µm.
Table 3.
Tau phenotype semiquantitative scores: post-translational modifications
| Case | PHF − 1 |
CP13 |
AT100 |
pS262 |
Tau−C3 |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| TSA | NFT | TSA | NFT | TSA | NFT | TSA | NFT | TSA | NFT | |
| 1 CTE-NC | +++ | +++ | +++ | +++ | + | +++ | +++ | +++ | – | + |
| 2 CTE-NC | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | – | + |
| 3 CTE-NC | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | – | – |
| 4 CTE-NC | +++ | +++ | +++ | +++ | ++ | ++ | +++ | +++ | – | ++ |
| 5 CTE-NC | +++ | +++ | +++ | +++ | +++ | +++ | + | +++ | – | + |
| 6 CTE-NC | +++ | +++ | +++ | +++ | +++ | +++ | ++ | +++ | – | + |
| 7 CTE-NC | +++ | +++ | +++ | +++ | +++ | +++ | + | +++ | – | + |
| 8 CTE-NC | +++ | +++ | +++ | +++ | +++ | +++ | ++ | +++ | – | + |
| 9 CTE-NC | +++ | +++ | +++ | +++ | +++ | +++ | – | + | x | x |
| 10 CTE-NC | +++ | +++ | +++ | +++ | +++ | +++ | ++ | +++ | + | ++ |
| 11 CTE-NC | +++ | +++ | +++ | +++ | +++ | +++ | − | +++ | – | + |
| 12 CTE-NC | +++ | +++ | +++ | +++ | ++ | +++ | – | +++ | – | + |
| 13 CTE-NC | +++ | +++ | +++ | +++ | +++ | +++ | ++ | +++ | – | ++ |
| 14 CTE-NC | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | – | + |
| 15 ARTAG, PART | +++ | +++ | +++ | +++ | +++ | +++ | + | ++ | + | ++ |
| 16 ARTAG, PART | +++ | +++ | +++ | +++ | +++ | +++ | ++ | +++ | – | +++ |
| 17 ARTAG, PART | +++ | +++ | +++ | +++ | +++ | +++ | + | +++ | – | + |
| 18 PART | N/A | +++ | N/A | +++ | N/A | ++ | N/A | ++ | N/A | – |
| 19 ARTAG, low ADNC | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | – | + |
| 20 ARTAG, low ADNC | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | – | ++ |
| 21 ARTAG, low ADNC | +++ | +++ | +++ | +++ | +++ | +++ | ++ | +++ | – | – |
| 22 ARTAG, low ADNC | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | – | – |
| 23 AD | +++ | +++ | ++ | +++ | ++ | |||||
| 24 AD | +++ | +++ | +++ | +++ | ++ | |||||
| 25 AD | +++ | +++ | +++ | +++ | + | |||||
| 26 AD | +++ | +++ | +++ | +++ | + | |||||
| 27 AD | +++ | +++ | +++ | +++ | ++ | |||||
| 28 AD | +++ | +++ | +++ | +++ | +++ | |||||
|
| ||||||||||
| Pick bodies | Ramified astro | Pick bodies | Ramified astro | Pick bodies | Ramified astro | Pick bodies | Ramified astro | Pick bodies | Ramified astro | |
|
| ||||||||||
| 29 PiD | +++ | +++ | +++ | +++ | +++ | +++ | ++ | + | ++ | − |
| 30 PiD | +++ | N/A | +++ | N/A | +++ | N/A | − | N/A | ++ | N/A |
| 31 PiD | +++ | +++ | +++ | +++ | +++ | +++ | − | − | − | − |
| 32 PiD | +++ | N/A | +++ | N/A | +++ | N/A | − | N/A | − | N/A |
| 33 PiD | +++ | N/A | +++ | N/A | +++ | N/A | − | N/A | − | N/A |
| 34 PiD | +++ | +++ | +++ | +++ | +++ | +++ | − | − | − | − |
|
| ||||||||||
| Tufted astrocytes | Tufted astrocytes | Tufted astrocytes | Tufted astrocytes | Tufted astrocytes | ||||||
|
| ||||||||||
| 35 PSP | +++ | +++ | ++ | ++ | + | |||||
| 36 PSP | +++ | +++ | +++ | + | +++ | |||||
| 37 PSP | +++ | +++ | +++ | + | − | |||||
| 38 PSP | +++ | +++ | +++ | + | − | |||||
| 39 PSP | +++ | +++ | ++ | − | − | |||||
| 40 PSP | +++ | +++ | +++ | ++ | − | |||||
|
| ||||||||||
| Astrocytic plaques | Astrocytic plaques | Astrocytic plaques | Astrocytic plaques | Astrocytic plaques | ||||||
|
| ||||||||||
| 41 CBD | +++ | +++ | + | + | − | |||||
| 42 CBD | +++ | +++ | +++ | − | − | |||||
| 43 CBD | +++ | +++ | ++ | − | − | |||||
| 44 CBD | +++ | +++ | ++ | ++ | − | |||||
| 45 CBD | +++ | +++ | +++ | + | − | |||||
| 46 CBD | +++ | +++ | +++ | +++ | − | |||||
Semi-quantitative scoring: − = absent, or nearly absent (≤5% concordance with extent of PHF1); + = minimal (5–30% concordance); ++ = moderate (30–70% concordance); +++ = extensive (>70% concordance); × = no immunoreactivity.
AD = Alzheimer’s disease; ADNC = Alzheimer’s disease neuropathological change; CTE-NC = chronic traumatic encephalopathy neuropathological change; N/A = not applicable; PiD = Pick’s disease.
Notably, NFTs in CTE also displayed robust cytoplasmic immunoreactivity for all tau phospho-epitopes including S202 (CP13), S212/T214 (AT100), S262 and S396/S404 (PHF1) (Fig. 4). Moreover, a subpopulation of NFTs in CTE demonstrated evidence of caspase-mediated truncation of tau at D421 (Tau-C3) (Fig. 4), consistent with that observed previously (Kanaan et al., 2016), and in Alzheimer’s disease here and in prior reports (Gamblin et al., 2003).
FTLD-tau controls were consistent with prior characterizations performed using these antibodies (Buee and Delacourte, 1999; Guillozet-Bongaarts et al., 2007; Ferrer et al., 2014), and displayed immunoreactivity for antibodies recognizing phosphorylation at residues S202 (CP13), S212/T214 (AT100), S262 (p262), and S396/S404 (PHF1). Notably, in one case of Pick’s disease, Pick bodies appeared weakly immunoreactive for p262 (Fig. 4). In contrast, neurons were otherwise negative for tau p262 in all other five Pick’s disease cases. Notably, previous work has demonstrated conflicting results with regard to pS262 immunoreactivity in Pick’s disease (Probst et al., 1996; Delacourte et al., 1998; Ferrer et al., 2002; Zhukareva et al., 2002; Irwin et al., 2016; Falcon et al., 2018). However, greater immuno-reactivity has been reported in ethanol versus formalin fixed tissue (Irwin et al., 2016), consistent with our observations. Moreover, the intensity of pS262 immunoreactivity was reported as greater in cases with 4R tau inclusions (Zhukareva et al., 2002).
Neurofibrillary tangles, but not astrocytes, in CTE show a similar conformational profile to those in Alzheimer’s disease
Recently developed conformation-selective antibodies, GT-7 and GT-38, have been shown to detect a conformation-dependent epitope present in tau within the inclusions of Alzheimer’s disease requiring both 3R and 4R tau, but not 3R or 4R tau only inclusions of other primary tauopathies, with both antibodies labelling Alzheimer’s disease-tau in a phosphorylation-independent manner (Gibbons et al., 2018, 2019). In CTE neuropathology, both GT-7 and GT-38 showed moderate to strong labelling of NFTs at the depths of sulci, consistent with Alzheimer’s disease (Fig. 5 and Table 2). However, in contrast with NFTs, astrocytes within CTE were negative for GT-7 or GT-38 in all but four cases in which there was occasional and minimal positivity to one or the other antibody (Table 2). These cells did not differ in their morphology, and no notable differences in fixation, clinical history or anatomic distribution distinguished them from the rest of the cohort. Double immunofluorescence labelling in a subset of cases confirmed neuron-specific co-localization of GT-38 with MAP-2, and an absence of co-localization with GFAP (Supplementary Figs 1 and 3).
Figure 5.

Neurofibrillary tangles, but not astrocytes, are immunoreactive for antibodies that detect a conformation-dependent epitope of tau in Alzheimer’s disease. PHF1 immunohistochemistry revealed sulcal depth astrocytic and neuronal tau pathology in CTE (Case 8), including prominent clusters of perivascular and subpial TSAs (top left; black arrows). However, GT-7 and GT-38 antibodies labelled NFTs in CTE, but not TSAs, on serial tissue sections (top middle and top right). GT-7 and GT-38 reliably detected NFTs in Alzheimer’s disease (Case 28), but failed to label the characteristic pathologies of ARTAG (Case 19), Pick’s disease (PiD; Case 29), PSP (Case 35) or CBD (Case 41). Scale bars = 100 µm in CTE (low power); = 50 µm in CTE (high power), Alzheimer’s disease (AD), Pick’s disease, ARTAG, PSP and CBD.
As anticipated, NFTs in non-injured Alzheimer’s disease and PART control cases demonstrated robust immunoreactivity for both antibodies (Fig. 5 and Table 2), while Pick bodies and the glial profiles of ARTAG, CBD and PSP were negative for both GT-7 and GT-38 (Fig. 5). Notably, the ramified astrocytes within the single Pick’s disease case that were immunoreactive for both 3R and 4R tau, were not immunoreactive for GT-7 or GT-38. Among TBI cases with co-morbid diagnoses of FTLD-tau, both GT-7 and GT-38 antibodies stained the characteristic NFTs of CTE but not the adjacent FTLD-tau disease-associated pathologies within the same tissue section.
Discussion
Here, we performed immunohistochemical characterization of tau phenotypes within the cellular constituents of CTE in patients with known exposure to repetitive mild TBI or moderate/severe TBI. Intriguingly, it was found that the tau species within NFTs and immediately adjacent astrocytes in CTE are phenotypically distinct. Specifically, the NFTs of CTE displayed an immunophenotype that mirrored that seen in Alzheimer’s disease or PART, including being both 3R and 4R immunoreactive and positive for antibodies previously shown to bind to a conformation-dependent epitope present within the tau inclusions of Alzheimer’s disease. In contrast, the astroglial component of CTE was solely 4R immunoreactive and without evidence of Alzheimer’s disease conformation, which was indistinguishable from ARTAG. Notably, while the neuronal and glial pathologies of CTE showed similarity with those of Alzheimer’s disease, PART and ARTAG, they were distinct from those of the primary FTLD tauopathies of Pick’s disease, CBD and PSP. As such, our data suggest that while pattern and distribution of involvement might be distinct, the tau pathologies of CTE show considerable overlap with the immunoreactivity profiles of both ageing-related tau pathologies and Alzheimer’s disease.
Typically, we observed tau immunoreactive astrocytes within CTE to have the characteristic morphology of TSA, in keeping with those encountered in ARTAG, in both the non-injured control material studied here and in multiple other reports (Lopez-Gonzalez et al., 2013; Kovacs et al., 2016, 2017a, 2018a). Notably, TSAs have long been described in material from patients with and without concomitant neurodegenerative disease. In particular, their appearance in association with increased age led to the recognition of the specific entity recently defined as ARTAG, wherein TSAs are described in subpial, subependymal and perivascular distributions (Ikeda et al., 1995, 1998; Ikeda, 1996; Schultz et al., 2004; Lace et al., 2012; Kovacs et al., 2013, 2016). While the clinical significance of ARTAG has yet to be fully explored (Kovacs et al., 2017b), the morphological resemblance between astrocytes immunoreactive for phosphorylated tau in both CTE and ARTAG raises the possibility that these entities share common pathogenic mechanisms (Kovacs et al., 2016, 2017b, 2018a; Liu et al., 2016; Goldfinger et al., 2018; Forrest et al., 2019). Our data demonstrate that the astrocytes of CTE and ARTAG not only share similar morphologies, but display indistinguishable tau immunophenotypes with respect to the panel of antibodies applied. Specifically, consistent with previous reports characterizing ARTAG (Kovacs et al., 2016; Ferrer et al., 2018) and one limited description in CTE (McKee et al., 2013), TSAs in CTE were typically 4R tau-immunoreactive/3R tau-negative, with only very occasional cells displaying immunoreactivity for 3R tau in a subset of cases. Further, astrocytes in CTE demonstrated a profile of immunoreactivity for multiple phospho-epitopes of tau consistent with descriptions of ARTAG here and previously (Lopez-Gonzalez et al., 2013; Ferrer et al., 2018).
Beyond this astroglial pathology, the regionally co-existing NFTs in CTE appeared morphologically and phenotypically consistent with those of Alzheimer’s disease and PART, composed of both 3R and 4R tau, and immunoreactive for tau hyperphosphorylated at multiple phospho-epitopes, including S202, S212/T214, S262, and S396/S404. Notably, characterization by others has highlighted additional shared features of tau including phosphatase-activating domain exposed conformation (TNT1 antibody), tau oligomers (TOC1), and truncation at D421 (Tau-C3) (Kanaan et al., 2016). Here we demonstrate that a subset of NFTs in Alzheimer’s disease, PART and CTE demonstrate caspase-mediated truncation at D421. Interestingly, Tau-C3 positivity has previously been reported as relatively sparse in CTE neuropathology when compared with Alzheimer’s disease (Kanaan et al., 2016). However, the diminished total burden of Tau-C3 immunoreactivity may reflect a virtual absence of immunoreactivity in TSA, as observed here. Truncation of tau at D421 has been reported as an early event in the evolution of NFTs in Alzheimer’s disease (Gamblin et al., 2003; Rissman et al., 2004; Cotman et al., 2005), although it may not be essential for filament formation (Delobel et al., 2008). Moreover, experimental data indicate caspase-mediated truncation may promote polymerization and seeding of full-length tau (Abraha et al., 2000; Berry et al., 2003; Gamblin et al., 2003; Rissman et al., 2004), as well as contribute to neurotoxicity via apoptosis (Fasulo et al., 2000, 2005; Chung et al., 2001). As such, the relative absence of Tau-C3 immunoreactivity in TSAs of CTE may have implications as to the potential pathological nature of tau immunoreactive TSAs in CTE.
The astrocytic tau pathologies of both CTE and ARTAG also failed to display immunoreactivity for the recently developed tau antibodies, GT-7 and GT-38, previously demonstrated to detect a conformation-dependent epitope of tau in Alzheimer’s disease in a phosphorylation-independent manner (Gibbons et al., 2018, 2019). Furthermore, these antibodies failed to label pathologies composed of tau with either 3R or 4R isoforms only (Gibbons et al., 2018, 2019). Thus, the observation that both GT-7 and GT-38 bind to NFTs in CTE, but not sulcal TSAs, further supports the observation that astrocytic tau is 4R only and differs from that of the adjacent NFTs. Indeed, while the characteristic pathologies of FTLD-tau were not immunoreactive for either GT-7 or GT-38, as previously characterized in detail (Gibbons et al., 2018, 2019), the NFTs of CTE, Alzheimer’s disease and PART were consistently immunoreactive for both.
The panel of antibodies examined herein were selected for their previous extensive characterization and reported differences across a range of neurodegenerative diseases. As no immunolabel examined thus far can morphologically or phenotypically differentiate individual cells in CTE from those of other established tauopathies, the distinguishing features of this pathology remain dependent on the overall pattern and distribution of pathology when using immunohistochemistry (McKee et al., 2016). Notably, this panel is not exhaustive, and the use of immunohistochemistry to explore additional, and potentially distinguishing tau phenotypes, including ubiquitination and acetylation, will be important. Notably, a recent study based on cryo-electron microscopy (cryo-EM) analysis of temporal lobe tissue from three cases of known CTE suggests that the tau filament structure of CTE is distinct from that of Alzheimer’s disease (Falcon et al., 2019). However, it is noteworthy that all three cases demonstrated clinical or neuropathological evidence of other neurodegenerative disease, including Parkinson’s disease, FTLD and/or motor neuron disease, in one instance associated with C9orf72 mutation (Falcon et al., 2019). Given the diversity and heterogeneity of co-morbid pathologies reported with CTE here and elsewhere (Mez et al., 2017; Lee et al., 2019), it will be important to extend cryo-EM studies to a wider range of trauma-associated cases. Nonetheless, these data suggest that while tau in CTE might differ from that of other established neurodegenerative disease, in particular Alzheimer’s disease, this may only be detectable by means beyond established immuno-histochemical or biochemical approaches.
There is increasing recognition that mixed, often multiple pathologies might co-exist in patients with neurodegenerative disease, including in those with TBI-related neurodegeneration where CTE might serve as the primary pathology driving disease or as a co-morbidity in context of an alternate diagnosis (Lee et al., 2019). Consistent with this, most cases with CTE examined here also met criteria for other tauopathies, including Alzheimer’s disease, PSP, and CBD, as previously reported (Lee et al., 2019). Notably, the morphologies and immunophenotype of CTE were highly consistent across cases, regardless of the presence or extent of co-morbid disease. Moreover, CTE was consistent in phenotype, regardless of the nature of TBI exposure, including in three cases with a remote history of single moderate or severe TBI. These cases add to the limited number of described cases of single TBI associated with CTE neuropathologies, supporting the assertion that it is exposure to TBI rather than severity or number of injuries that serves as the primary risk factor for CTE (Smith et al., 2013, 2019; Maroon et al., 2015).
Collectively, these data indicate the co-existence of distinct tau phenotypes within neurons and astrocytes contributing to CTE neuropathology. Moreover, immunohistochemical observations were notably consistent across all cases examined, composed of diverse TBI exposure histories. While criteria for delineating the extent or stage of disease in CTE have yet to be adequately defined, future explorations of tau immunophenotype in association with disease progression will be of importance to examine.
While the mechanisms driving tau pathology following TBI exposure remain poorly understood, it is possible that the differential neuronal and astrocytic components reflect mechanistically independent pathological processes. Curiously, TSA in both ARTAG and CTE are frequently observed at brain parenchyma–fluid interfaces, including subpial, subependymal and perivascular regions (Ikeda et al., 1995; Geddes et al., 1996, 1999; Kovacs et al., 2016, 2018a; McKee et al., 2016). Moreover, regional correlation of ARTAG with astrocytic expression of connexin-43 and aquaporin 4 has led to speculation that blood–brain barrier dysfunction may be of pathological significance to the development of this pathology (Kovacs et al., 2018b). While blood–brain barrier dysfunction is increasingly recognized as an important acute pathology of TBI, even following con-cussion (Weissberg et al., 2014; Johnson et al., 2018), recent data indicate blood–brain barrier permeability may persist chronically in some individuals after severe TBI (Hay et al., 2015) and has also been described in cases of CTE (Doherty et al., 2016). However, a potential mechanistic relationship between blood–brain barrier dysfunction and pathological astrocytic tau accumulation remains unexplored.
Here we provide new insights into the nature of tau in CTE neuropathological change directly within the context of other neurodegenerative pathologies. Moreover, these data highlight the potential challenges in distinguishing trauma-associated tau pathologies from those of other diseases at the individual cell level using immunohistochemistry alone. Nonetheless, morphological and phenotypic similarities between tau in CTE and those of ARTAG suggests the intriguing possibility of shared pathogenic mechanisms.
Supplementary Material
Acknowledgements
We would like to thank Dr Peter Davies of the Albert Einstein College of Medicine for generously providing the tau antibodies PHF1 and CP13, and Josephine Atkinson of the University of Glasgow for technical assistance with immunohistochemical studies.
Funding
Research reported in this publication was supported by: the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (NIH) under award number, U54NS115322, R01NS092398, R01NS094003 and R01NS038104; NIH National Institute on Aging grants AG010124, AG017586, AG054991, AG09215 and AG17586; NHS Research Scotland; Clinical and Translational Science Awards training grant TL1TR001880 (J.D.A.); and NIH F32AG053036 (G.S.G.).
Competing interests
The authors report no competing interests.
Glossary
- ARTAG =
ageing-related tau astrogliopathy
- CBD =
corticobasal degeneration
- CTE =
chronic traumatic encephalopathy
- FTLD-tau =
frontotemporal lobar degeneration characterized by tau inclusions
- NFT =
neurofibrillary tangle
- PART =
primary age-related tauopathy
- PSP =
progressive supranuclear palsy
- TBI =
traumatic brain injury
- TSA =
thorn-shaped astrocytes
References
- Abraha A, Ghoshal N, Gamblin TC, Cryns V, Berry RW, Kuret J, et al. C-terminal inhibition of tau assembly in vitro and in Alzheimer’s disease. J Cell Sci 2000; 113 (Pt 21): 3737–45. [DOI] [PubMed] [Google Scholar]
- Arai T, Ikeda K, Akiyama H, Shikamoto Y, Tsuchiya K, Yagishita S, et al. Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol 2001; 101: 167–73. [DOI] [PubMed] [Google Scholar]
- Berry RW, Abraha A, Lagalwar S, LaPointe N, Gamblin TC, Cryns VL, et al. Inhibition of tau polymerization by its carboxy-terminal caspase cleavage fragment. Biochemistry 2003; 42: 8325–31. [DOI] [PubMed] [Google Scholar]
- Buee L, Delacourte A.. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol 1999; 9: 681–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007; 114: 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, et al. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis 2001; 8: 162–72. [DOI] [PubMed] [Google Scholar]
- Corsellis JA, Bruton CJ, Freeman-Browne D.. The aftermath of boxing. Psychol Med 1973; 3: 270–303. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Poon WW, Rissman RA, Blurton-Jones M.. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol 2005; 64: 104–12. [DOI] [PubMed] [Google Scholar]
- Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014; 128: 755–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva R, Lashley T, Gibb G, Hanger D, Hope A, Reid A, et al. Pathological inclusion bodies in tauopathies contain distinct complements of tau with three or four microtubule-binding repeat domains as demonstrated by new specific monoclonal antibodies. Neuropathol Appl Neurobiol 2003; 29: 288–302. [DOI] [PubMed] [Google Scholar]
- Delacourte A, Sergeant N, Wattez A, Gauvreau D, Robitaille Y.. Vulnerable neuronal subsets in Alzheimer’s and Pick’s disease are distinguished by their tau isoform distribution and phosphorylation. Ann Neurol 1998; 43: 193–204. [DOI] [PubMed] [Google Scholar]
- Delobel P, Lavenir I, Fraser G, Ingram E, Holzer M, Ghetti B, et al. Analysis of tau phosphorylation and truncation in a mouse model of human tauopathy. Am J Pathol 2008; 172: 123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty CP, O’Keefe E, Wallace E, Loftus T, Keaney J, Kealy J, et al. Blood-brain barrier dysfunction as a hallmark pathology in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 2016; 75: 656–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza M, de Silva R, Dickson DW, Davies P.. Differential incorporation of tau isoforms in Alzheimer’s disease. J Alzheimers Dis 2008; 14: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018; 561: 137–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019; 568: 420–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasulo L, Ugolini G, Cattaneo A.. Apoptotic effect of caspase-3 cleaved tau in hippocampal neurons and its potentiation by tau FTDP-mutation N279K. J Alzheimers Dis 2005; 7: 3–13. [DOI] [PubMed] [Google Scholar]
- Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V, et al. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J Neurochem 2000; 75: 624–33. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Barrachina M, Puig B.. Anti-tau phospho-specific Ser262 antibody recognizes a variety of abnormal hyper-phosphorylated tau deposits in tauopathies including Pick bodies and argyrophilic grains. Acta Neuropathol 2002; 104: 658–64. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Garcia MA, Gonzalez IL, Lucena DD, Villalonga AR, Tech MC, et al. Aging-related tau astrogliopathy (ARTAG): not only tau phosphorylation in astrocytes. Brain Pathol 2018; 28: 965–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Lopez-Gonzalez I, Carmona M, Arregui L, Dalfo E, Torrejon-Escribano B, et al. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol 2014; 73: 81–97. [DOI] [PubMed] [Google Scholar]
- Forrest SL, Kril JJ, Wagner S, Honigschnabl S, Reiner A, Fischer P, et al. Chronic traumatic encephalopathy (CTE) is absent from a european community-based aging cohort while cortical aging-related tau astrogliopathy (ARTAG) is highly prevalent. J Neuropathol Exp Neurol 2019; 78: 398–405. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, et al. Caspase cleavage of tlinking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci USA 2003; 100: 10032–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes JF, Vowles GH, Nicoll JA, Revesz T.. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol 1999; 98: 171–8. [DOI] [PubMed] [Google Scholar]
- Geddes JF, Vowles GH, Robinson SF, Sutcliffe JC.. Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol Appl Neurobiol 1996; 22: 12–6. [PubMed] [Google Scholar]
- Gibbons GS, Banks RA, Kim B, Changolkar L, Riddle DM, Leight SN, et al. Detection of Alzheimer disease (AD)-specific tau pathology in AD and nonAD tauopathies by immunohistochemistry with novel conformation-selective tau antibodies. J Neuropathol Exp Neurol 2018; 77: 216–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons GS, Kim S-J, Robinson JL, Changolkar L, Irwin DJ, Shaw LM, et al. Detection of Alzheimer’s disease (AD) specific tau pathology with conformation-selective anti-tau monoclonal antibody in co-morbid frontotemporal lobar degeneration-tau (FTLD-tau). Acta Neuropathol Commun 2019; 7: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfinger MH, Ling H, Tilley BS, Liu AKL, Davey K, Holton JL, et al. The aftermath of boxing revisited: identifying chronic traumatic encephalopathy pathology in the original Corsellis boxer series. Acta Neuropathol 2018; 136: 973–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, Chau M, Denis PE, Feinstein SC.. Structural and functional differences between 3-repeat and 4-repeat tau isoforms. Implications for normal tau function and the onset of neurodegenetative disease. J Biol Chem 2000; 275: 38182–9. [DOI] [PubMed] [Google Scholar]
- Graham DI, Gentleman SM, Lynch A, Roberts GW.. Distribution of beta-amyloid protein in the brain following severe head injury. Neuropathol Appl Neurobiol 1995; 21: 27–34. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P, Schein JD, Binder LI.. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem 1992; 267: 564–9. [PubMed] [Google Scholar]
- Guillozet-Bongaarts AL, Glajch KE, Libson EG, Cahill ME, Bigio E, Berry RW, et al. Phosphorylation and cleavage of tau in non-AD tauopathies. Acta Neuropathol 2007; 113: 513–20. [DOI] [PubMed] [Google Scholar]
- Hay JR, Johnson VE, Young AM, Smith DH, Stewart W.. Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol 2015; 74: 1147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann R, Lee VM, Leight S, Varga I, Otvos L Jr.. Unique Alzheimer’s disease paired helical filament specific epitopes involve double phosphorylation at specific sites. Biochemistry 1997; 36: 8114–24. [DOI] [PubMed] [Google Scholar]
- Ikeda K. Glial fibrillary tangles and argyrophilic threads: classification and disease specificity. Neuropathology 1996; 16: 71–7. [Google Scholar]
- Ikeda K, Akiyama H, Arai T, Nishimura T.. Glial tau pathology in neurodegenerative diseases: their nature and comparison with neuronal tangles. Neurobiol Aging 1998; 19: S85–91. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Akiyama H, Kondo H, Haga C, Tanno E, Tokuda T, et al. Thorn-shaped astrocytes: possibly secondarily induced tau-positive glial fibrillary tangles. Acta Neuropathol 1995; 90: 620–5. [DOI] [PubMed] [Google Scholar]
- Irwin DJ, Brettschneider J, McMillan CT, Cooper F, Olm C, Arnold SE, et al. Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol 2016; 79: 272–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson GL, Luoto TM, Karhunen PJ, Castellani RJ.. Mild chronic traumatic encephalopathy neuropathology in people with no known participation in contact sports or history of repetitive neurotrauma. J Neuropathol Exp Neurol 2019; 78: 615–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jicha GA, Weaver C, Lane E, Vianna C, Kress Y, Rockwood J, et al. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer’s disease. J Neurosci 1999; 19: 7486–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Arena JD, Smith DH.. Traumatic brain injury as a trigger of neurodegeneration. Adv Neurobiol 2017; 15: 383–400. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH.. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 2012; 22: 142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Weber MT, Cullen DK, Siman R, Smith DH.. SNTF immunostaining reveals previously undetected axonal pathology in traumatic brain injury. Acta Neuropathol 2016; 131: 115–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Weber MT, Xiao R, Cullen DK, Meaney DF, Stewart W, et al. Mechanical disruption of the blood-brain barrier following experimental concussion. Acta Neuropathol 2018; 135: 711–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan NM, Cox K, Alvarez VE, Stein TD, Poncil S, McKee AC.. Characterization of early pathological tau conformations and phosphorylation in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 2016; 75: 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney K, Iacono D, Edlow BL, Katz DI, Diaz-Arrastia R, Dams-O’Connor K, et al. Dementia after moderate-severe traumatic brain injury: coexistence of multiple proteinopathies. J Neuropathol Exp Neurol 2018; 77: 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG. Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol 2015; 41: 3–23. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 2016; 131: 87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Lee VM, Trojanowski JQ.. Protein astrogliopathies in human neurodegenerative diseases and aging. Brain Pathol 2017a; 27: 675–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Milenkovic I, Wohrer A, Hoftberger R, Gelpi E, Haberler C, et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 2013; 126: 365–84. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, Robinson JL, Xie SX, Lee EB, Grossman M, Wolk DA, et al. Evaluating the patterns of aging-related tau astrogliopathy unravels novel insights into brain aging and neurodegenerative diseases. J Neuropathol Exp Neurol 2017b; 76: 270–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Xie SX, Robinson JL, Lee EB, Smith DH, Schuck T, et al. Sequential stages and distribution patterns of aging-related tau astrogliopathy (ARTAG) in the human brain. Acta Neuropathol Commun 2018a; 6: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Yousef A, Kaindl S, Lee VM, Trojanowski JQ.. Connexin-43 and aquaporin-4 are markers of ageing-related tau astrogliopathy (ARTAG)-related astroglial response. Neuropathol Appl Neurobiol 2018b; 44: 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lace G, Ince PG, Brayne C, Savva GM, Matthews FE, de Silva R, et al. Mesial temporal astrocyte tau pathology in the MRC-CFAS ageing brain cohort. Dement Geriatr Cogn Disord 2012; 34: 15–24. [DOI] [PubMed] [Google Scholar]
- Lee EB. Integrated neurodegenerative disease autopsy diagnosis. Acta Neuropathol 2018; 135: 643–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Kinch K, Johnson VE, Trojanowski JQ, Smith DH, Stewart W.. Chronic traumatic encephalopathy is a common co-morbidity, but less frequent primary dementia in former soccer and rugby players. Acta Neuropathol 2019; 138: 389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AK, Goldfinger MH, Questari HE, Pearce RK, Gentleman SM.. ARTAG in the basal forebrain: widening the constellation of astrocytic tau pathology. Acta Neuropathol Commun 2016; 4: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gonzalez I, Carmona M, Blanco R, Luna-Munoz J, Martinez-Mandonado A, Mena R, et al. Characterization of thorn-shaped astrocytes in white matter of temporal lobe in Alzheimer’s disease brains. Brain Pathol 2013; 23: 144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroon JC, Winkelman R, Bost J, Amos A, Mathyssek C, Miele V.. Chronic traumatic encephalopathy in contact sports: a systematic review of all reported pathological cases. PLoS One 2015; 10: e0117338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martland H. Punch drunk. JAMA 1928; 91: 1103–7. [Google Scholar]
- McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 2016; 131: 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 2009; 68: 709–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Stein TD, Nowinski CJ, Stern RA, Daneshvar DH, Alvarez VE, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013; 136: 43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA 2017; 318: 360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millspaugh J. Dementia pugilistica. US Nav Med Bull 1937; 297–303. [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012; 123: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omalu B, Hammers JL, Bailes J, Hamilton RL, Kamboh MI, Webster G, et al. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus 2011; 31: E3. [DOI] [PubMed] [Google Scholar]
- Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH.. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 2005; 57: 128–34; discussion 34. [DOI] [PubMed] [Google Scholar]
- Otvos L Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM.. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res 1994; 39: 669–73. [DOI] [PubMed] [Google Scholar]
- Probst A, Tolnay M, Langui D, Goedert M, Spillantini MG.. Pick’s disease: hyperphosphorylated tau protein segregates to the somatoaxonal compartment. Acta Neuropathol 1996; 92: 588–96. [DOI] [PubMed] [Google Scholar]
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 2004; 114: 121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saing T, Dick M, Nelson PT, Kim RC, Cribbs DH, Head E.. Frontal cortex neuropathology in dementia pugilistica. J Neurotrauma 2012; 29: 1054–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santa-Maria I, Haggiagi A, Liu X, Wasserscheid J, Nelson PT, Dewar K, et al. The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Neuropathol 2012; 124: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ.. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol 2001; 101: 518–24. [DOI] [PubMed] [Google Scholar]
- Schultz C, Ghebremedhin E, Del Tredici K, Rub U, Braak H.. High prevalence of thorn-shaped astrocytes in the aged human medial temporal lobe. Neurobiol Aging 2004; 25: 397–405. [DOI] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Stewart W.. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 2013; 9: 211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Trojanowski JQ, Stewart W.. Chronic traumatic encephalopathy - confusion and controversies. Nat Rev Neurol 2019; 15: 179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart W, McNamara PH, Lawlor B, Hutchinson S, Farrell M.. Chronic traumatic encephalopathy: a potential late and under recognized consequence of rugby union? QJM 2016; 109: 11–5. [DOI] [PubMed] [Google Scholar]
- Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL, et al. A platform for discovery: the University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement 2014; 10: 477–84.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissberg I, Veksler R, Kamintsky L, Saar-Ashkenazy R, Milikovsky DZ, Shelef I, et al. Imaging blood-brain barrier dysfunction in football players. JAMA Neurol 2014; 71: 1453–5. [DOI] [PubMed] [Google Scholar]
- Wilson L, Stewart W, Dams-O’Connor K, Diaz-Arrastia R, Horton L, Menon DK, et al. The chronic and evolving neurological consequences of traumatic brain injury. Lancet Neurol 2017; 16: 813–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanier ER, Bertani I, Sammali E, Pischiutta F, Chiaravalloti MA, Vegliante G,. et al. Induction of a transmissible tau pathology by traumatic brain injury. Brain 2018; 141: 2685–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng-Fischhofer Q, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E.. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem 1998; 252: 542–52. [DOI] [PubMed] [Google Scholar]
- Zhukareva V, Mann D, Pickering-Brown S, Uryu K, Shuck T, Shah K, et al. Sporadic Pick’s disease: a tauopathy characterized by a spectrum of pathological tau isoforms in gray and white matter. Ann Neurol 2002; 51: 730–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material.