

Abstract
The metabolism of macro and micronutrients is a complex and highly regulated biological process. An imbalance in the metabolites and their signaling networks can lead to non-resolving inflammation and consequently to the development of chronic inflammatory-associated diseases. Therefore, identifying the accumulated metabolites and altered pathways during inflammatory disorders would not only serve as ‘real time’ markers, but also help in the development of nutritional therapeutics. In this review, we explore recent research that has delved into elucidating the effects of carbohydrate/calorie restriction, protein malnutrition, lipid emulsions and micronutrient deficiencies on metabolic health and inflammation. Moreover, we describe the integrated stress response in terms of amino acid starvation and lipemia, and how this modulates new age diseases such as inflammatory bowel disease and atherosclerosis. Lastly, we explain the latest research on metaflammation and inflammaging. This review focuses on multiple signaling pathways, including, but not limited to, the FGF21-β-hydroxybutryate-NLRP3 axis, the GCN2-eIF2α-ATF4 pathway, the von Hippel-Lindau/Hypoxia-inducible transcription factor pathway and the TMAO-PERK-FoxO1 axis. Additionally, throughout the review, we explain how the gut microbiota responds to altered nutrient status and also how antimicrobial peptides generated from nutrient-based signaling pathways can modulate the gut microbiota. Collectively, it must be emphasized that metabolic starvation and inflammation are strongly regulated by both environmental (i.e. nutrition, gut microbiome) and non-environmental (i.e. genetics) factors, which can influence the susceptibility to inflammatory disorders.
Keywords: Ketogenic Diet, Lipid Emulsions, Integrated Stress Response, Micronutrient Deficiency, Metaflammation, Inflammaging
1. Introduction
The metabolism of calorie-rich macronutrients (carbohydrates, fats, and proteins) in coordination with zero calorie micronutrients (vitamins and minerals) is a complex and highly regulated biological process. In addition to the major pathways of macronutrient metabolism, the existence of alternate pathways, such as the salvage and anaplerotic/cataplerotic pathways, adds another layer of complexity. Any imbalance of these delicate signaling networks can lead to non-resolving, sterile inflammation and consequently to the development of chronic inflammatory-associated metabolic diseases. While acute inflammation is characterized by elevated pro-inflammatory cytokines and acute phase proteins, which can induce classic symptoms of inflammation such as calor (i.e. heat) and rubor (i.e. redness), low-grade chronic inflammation is largely asymptomatic with no clear signs of inflammation at either systemic or histological levels. Consequently, the lack of clear symptoms can cause long-lasting, irreversible inflammatory damage to tissues, building up to the new age disorders such as insulin resistance, hyperlipidemia, obesity, hypertension, and non-alcoholic steatohepatitis, which are hallmark symptoms of metabolic syndrome. Therefore, identifying the metabolites accumulated and the derailed pathways during inflammatory disorders would not only serve as ‘real time’ markers, but would also provide avenues for the incorporation of nutritional therapeutics to curtail inflammation. This review delves into the various signaling mechanisms and metabolic adaptations that arise due to nutrient excess and deficiencies. Additionally, we explore how those responses affect multiple disease states, including inflammatory bowel disease (IBD) and metabolic syndrome, and also how an altered nutrient status can perturb the gut microbiota. Lastly, this review describes several nutritional therapeutics that are being implemented to dampen inflammation and to treat chronic diseases.
2. Engineering Personalized Macronutrient Diets
2.1. Carbohydrate Starvation: The FGF21-β-hydroxybutryate-NLRP3 Axis
Fasting is frequently advertised as one of the ‘best medicines’ and therapeutic options for promoting weight loss and longevity. This voluntary, behavioral practice can include dry/intermittent fasting, which involves either the complete abstention of food and water for a given period of time or the partial restriction of only particular substances. Metabolically, the rate of gluconeogenesis significantly increases during early fasting, whereas prolonged fasting progresses toward a state of ketosis. Hepatic β-oxidation of fatty acids into acetyl CoA, the precursor for ketone bodies (i.e. β-hydroxybutryate), is the adaptive mechanism to replace glucose as the primary energy source for the central nervous system and to spare protein catabolism during carbohydrate restriction [1]. More specifically, ketone bodies are transported to extrahepatic tissues, like skeletal and heart muscles, and are then oxidized by mitochondrial enzymes in the tricarboxylic acid (TCA) cycle to generate ATP [2]. Intriguingly, the liver itself is unable to utilize ketone bodies due to the absence of the enzyme, succinyl-CoA:3-ketacid CoA transferease, which emphasizes how the liver acts as an altruistic organ [3]. In addition to prolonged fasting, enhanced exercise performance can promote ketonemia (i.e. elevated systemic ketone bodies) as a fuel source for the muscles [4]. Comparatively, ketoacidosis is a common characteristic that is exhibited in severe and uncontrolled diabetes, which subsequently results in hypoglycemia [5, 6]. Likewise, prolonged exposure of ketone bodies in the brain results in insulinemia and a decrease in glucose production, thus dysregulating overall energy homeostasis [7]. This emphasizes that acute ketogenesis can have beneficial effects, whereas chronic exposure to ketone bodies can negatively alter host physiology.
As an alternative method to transiently raise systemic ketone body levels, most individuals practice the ketogenic diet (KD), which involves stringently limiting carbohydrate intake but increasing dietary fat and protein consumption. This carbohydrate-restricted diet is widely recommended as an intervention against non-alcoholic fatty liver disease (NAFLD), as this dietary approach can (i) decrease hepatic de novo lipogenesis, (ii) alleviate low-grade chronic inflammation, (iii) abate hyperglycemia, (iv) elevate systemic β-hydroxybutryate levels, which reflects an increase in β-oxidation, and (v) promote the abundance of folate-producing Streptococcus in the gut, which further upregulates folate-dependent one-carbon (-CH3) metabolism (Figure 1A) [8, 9]. Additionally, KD is highly prescribed to treat individuals with refractory epilepsy (as reviewed in [10]), where it has been recently shown that the KD-induced neuroprotective effects are mediated by the gut microbiota diminishing γ-glutamyltranspeptidase activity and γ-glutamyl acid levels, which subsequently increases γ-Aminobutyric acid inhibitory neurotransmission signaling [11]. Likewise, KD has been shown to shift the gut microbiota profile toward ‘beneficial bacteria’ (i.e. Akkermansia muciniphila, Lactobacillus), which was associated with increased cerebral blood flow and β-amyloid peptide clearance (Figure 1A), both of which can reduce the risk for Alzheimer’s disease [12]. Alongside, a modified Mediterranean-KD formula shifts the gut microbiota composition to an increased abundance of Enterobacteriaceae and Akkermansia, but reduced Bifidobacterium, which was associated with improved cerebrospinal fluid biomarkers in Alzheimer’s disease patients [13]. Recently, KD has been recognized as a possible co-therapeutic with the glutamine antagonist, 6-diazo-5-oxo-L-norleucine, [14] and also as an adjuvant during standard chemoradiation [15] to restrict glucose availability to glioblastoma cancer cells. Considering that glioblastoma is associated with immunosuppression [16] and that certain gut bacteria (i.e. Bacteroides fragilis) also exhibit immunosuppressive abilities [17], it would be intriguing to explore whether the KD-mediated neuroprotective effects could also be due to modifications in immune responses directed from the gut microbiota.
Figure 1: Metabolic, renal and neural health are impacted by a ketogenic diet.
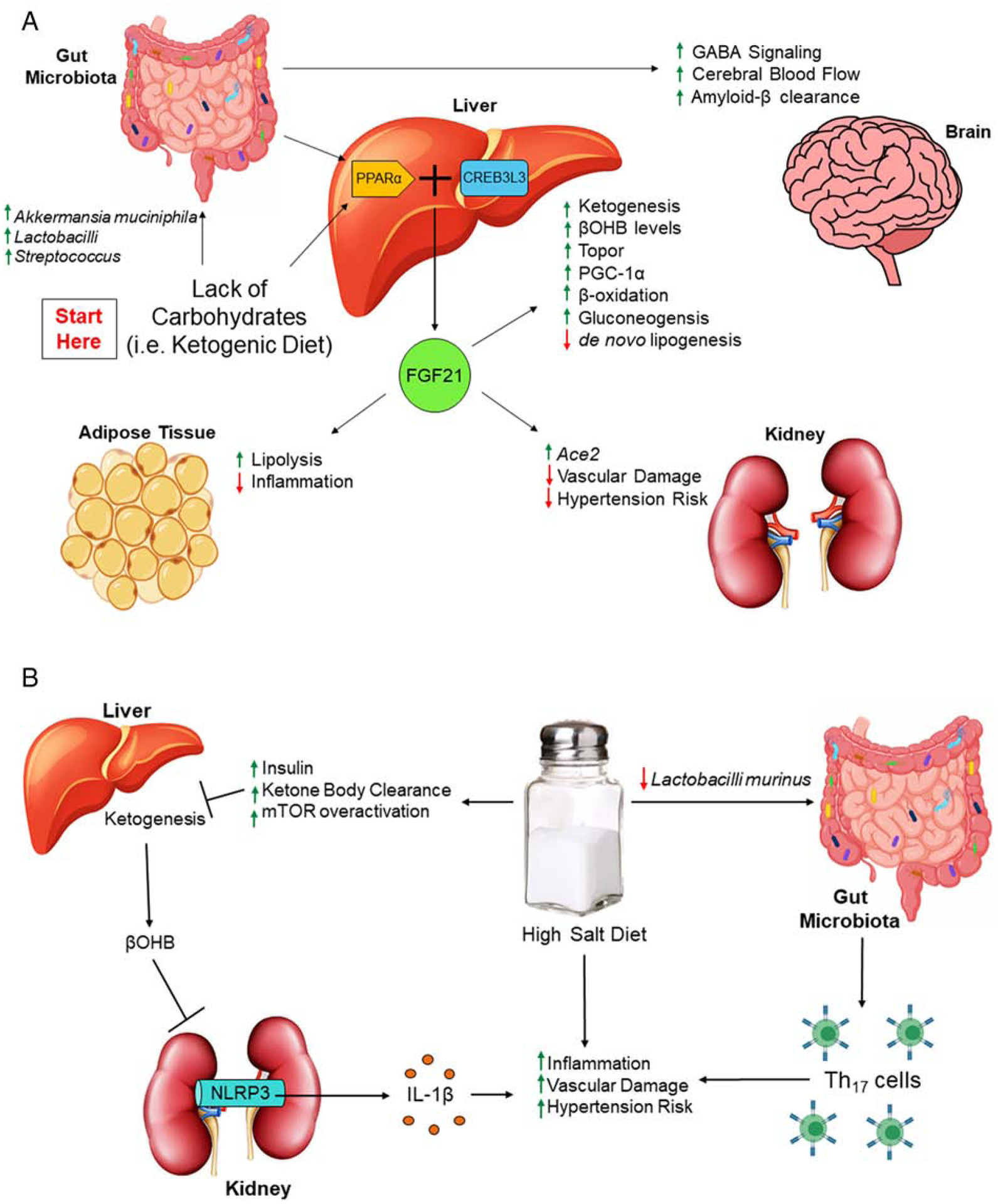
(A) Lack of carbohydrates can perturb the gut microbiota, shifting the composition to ‘beneficial’ bacteria. Through the gut-brain axis, a ketogenic diet (KD) improves neuronal function, including increasing GABA signaling, cerebral blood flow and clearance of amyloid-β. In the gut-liver axis, KD promotes PPARα activation of FGF21, where this hepatokine induces benefits to the liver, the kidney and adipose tissue. (B) One of the hepatic benefits is ketogenesis and the production of βOHB, which can inhibit NLRP3-mediated IL-1β secretion and reduce overall inflammation. However, a high salt diet can impede ketogenesis through the promotion of insulin-mediated ketone body clearance and mTOR activation, resulting in diminished βOHB and increased vascular damage, which can increase the overall risk for hypertension. In addition to this pathway, high salt intake can modulate the gut microbiota to promote Th17 responses, which is also associated with increased hypertension risk. Key words: γ-Aminobutyric acid (GABA), peroxisome proliferator activated receptor α (PPARα), cAMP responsive element-binding protein 3-like 3 (CREB3L3), fibroblast growth factor 21 (FGF21), angiotensin converting enzyme 2 (ACE2), PPARγ coactivator 1α (PGC-1α), β-hydroxybutryate (βOHB), mammalian target of rapamycin (mTOR), Nod-like receptor family pyrin domain containing 3 (NLRP3), interleukin-1β (IL-1β).
In line, it has been suggested that the gut microbiota is required for hepatic ketogenesis, as fasted germ-free mice that lack the gut microbiota exhibit impaired ketogenesis [18]. In particular, Crawford et al. [18] show that gut microbiota-dependent ketogenesis is associated with peroxisome proliferator activated receptor α (PPARα), a nuclear transcription factor that regulates central metabolic processes, including fatty acid metabolism and phospholipid homeostasis [19]. PPARα networks with nutrient sensors, such as AMP-activated protein kinase (AMPK), mammalian target of rapamycin (mTOR), and PPARγ coactivator 1α (PGC-1α); coordinates with cAMP responsive element-binding protein 3-like 3 (CREB3L3); and induces the hepatokine, fibroblast growth factor 21 (FGF21). Firstly, inhibition of mTORC1 is required for the fasting-induced activation of PPARα [20], where then, in an auto-loop manner, CREB3L3 and PPARα synergistically activate FGF21, which stimulates lipolysis in white adipose tissue (WAT) and ketogenesis in the liver, while also promoting topor, a short-term hibernation-like state that conserves energy (Figure 1A) [21–23]. Moreover, FGF21 augments hepatic PGC-1α expression and enhances hepatic fatty acid oxidation, TCA flux and gluconeogenesis (Figure 1A) [24]. Despite these short-term benefits, long-term KD results in impaired FGF21 signaling, followed with the development of dyslipidemia, glucose intolerance, insulin resistance, hepatic macrophage infiltration, and steatosis [25, 26]. Interestingly, senescence-accelerated prone mice fed a carbohydrate-restricted diet for 50 weeks exhibited an elevated abundance of inflammatory-inducing gut bacteria (i.e. Enterobacteria) and an increase rate in aging [27]. Additionally, feeding KD to mice with liver-specific knockout of carnitine palmitoyltransferase 2 (Cpt2L−/−, obligate enzyme in mitochondrial long chain fatty acid β-oxidation), results in hepatomegaly, liver damage, complete absence of adipose triglyceride stores, and a shortened lifespan [28]. Moreover, activation of ketogenesis in Cpt2L−/− mice through fasting results in hepatic oxidative stress and steatosis mediated by PPARα target genes, including FGF21 [28]. These recent reports emphasize that, during times of nutrient restriction, lack of hepatic fatty acid oxidation results in escalated adipocyte lipolysis through procatabolic hepatokines like FGF21, which redefines the role of β-oxidation and thus, ketone bodies, on metabolic and hepatic health. Unlike the liver, though, KD reduces inflammation and does not impact insulin responsiveness in WAT [25, 29], suggesting that KD benefits could be tissue specific.
Compared to acetoacetate and acetone, β-hydroxybutryate (β-OHB) is the central ketone body that is elevated from KD, fasting and exercise. Numerically, implementing KD can raise serum β-OHB levels above 2 mM, prolonged fasting can have β-OHB levels reach 6–8 mM, and 90 min of intense exercise can elevate β-OHB to 1–2 mM levels [30]. Similar to KD being utilized as a therapeutic for NAFLD and neuronal disorders, exogenous supplementation of sodium β-OHB is the only therapeutic available for those with multiple acyl-CoA dehydrogenase deficiency, which is a severe form of inborn errors of metabolism with impaired mitochondrial fatty acid oxidation [31, 32]. Administration of exogenous β-OHB has also been shown to boost global histone acetylation and thus, expression, of oxidative stress reliving genes due to its histone deacetylase (HDAC) inhibitory property [33, 34]. Additionally, β-OHB has been demonstrated to antagonize the short chain fatty acid receptor, GPR41, further suppressing Gβγ-PLCβ-MAPK signaling, sympathetic activity and overall metabolic rate [35]. Notwithstanding, β-OHB is highly known for its unique ability to suppress activation of the Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome [36]. Specifically, Youm et al. [36] show that β-OHB, but not acetoacetate nor structurally-related short chain fatty acids (SFCAs; i.e. butyrate), prevents potassium efflux and reduces oligomerization of the apoptosis-associated speck-like protein with a caspase-recruitment domain, thus attenuating caspase-1 activation and interleukin (IL)-1β secretion. Recently, Feng et al. demonstrate that gut microbiota ablation via antibiotics treatment lowers intestinal barrier function, which is associated with enhanced NLRP3 activiation and autophagy [37]. Comparatively, Yao et al. report that mutation of NLRP3 results in reshaping of the gut microbiota through the elevation of antimicrobial peptides, which further induces regulatory T cells and protects against gut inflammation [38]. Considering that the structurally-related, gut metabolite, butyrate, has been demonstrated to promote the differentiation of regulatory T cells [39], it would be intriguing for future studies to explore whether β-OHB is capable of alleviating intestinal inflammation by modulating the gut-lymphatic system.
Since NLRP3 is one of the hallmark targets against many inflammatory disorders, this has opened the door for supplementing β-OHB into the diet, instead of inducing ketonemia through KD (as reviewed in [40]). Additionally, β-OHB is indicated as a novel therapeutic candidate for the treatment of stress-related mood disorders and to act as an antidepressant through its inhibition of NLRP3 [41]. This coincides with exogenous β-OHB possessing multiple neuroprotective effects, such as minimizing oxidative stress, maintaining mitochondrial function, and promoting brain-derived neurotrophic factor-mediated neuronal regeneration (as further reviewed in [42]). Furthermore, it has also been shown that β-OHB exerts anti-hypertensive effects by blocking renal NLRP3 activity, where intake of a high-salt diet actually lowers circulating β-OHB levels (Figure 1B) [43]. This may be due, at least in part, to high-salt intake elevating insulin levels and promoting insulin resistance [44], where increased levels of insulin can suppress the rate of ketogenesis by inducing ketone body clearance (Figure 1B) [45]. Likewise, increased insulin can lead to mTOR overactivation, and thus inhibition of ketogenesis (Figure 1B) [46]. Comparatively, Wilck et al. demonstrate that high-salt intake depletes Lactobacillus murinus in the gut, which is associated with elevated Th17 cells and blood pressure (Figure 1B) [47], thus suggesting that this could be another plausible mechanism for how overconsumption of salt increases hypertension risk. Since metabolic syndrome constitutes of hyperinsulinemia, insulin resistance, and gut dysbiosis, this may also explain the inability for ketogenesis in obese and NAFLD patients. Despite the benefits reported regarding β-OHB, Chriett et al. recently suggest for a reassessment on β-OHB as they observe that this ketone body does not exhibit the property of HDAC inhibition and might actually be a pro-inflammatory molecule, at least, in comparison to the structurally-related SCFA, butyrate [48].
2.2. Protein Malnutrition, Colitis and Hypertension
Similar to individuals with NAFLD that are recommended to implement KD, those with chronic kidney disease that are prone to protein intolerance are strategically suggested to practice protein-restricted diets [49]. Likewise, a low-protein diet is a newly suggested concept to alleviate obesity and type 2 diabetes, as restraining the consumption of amino acids improves glucose sensitivity and normalizes body weight [50], which was reported to be independent of alterations in the gut microbiota induced by a low-protein diet [51]. Recently, Li et al. demonstrate that a periodized low-protein, high carbohydrate diet regimen promotes the abundance of Bacteroidetes and Akkermansia, which is associated with improved metabolic health [52]. Similarly, Fan et al. observe that, in an adult pig model, moderate protein restriction improves intestinal health through the increased abundance of ‘beneficial’ bacteria and preserved gut permeability; yet, they also show that when protein intake decreases below the ‘moderate’ protein consumption that bacterial richness diminishes in the ileum [53]. This overall suggests that the gut microbiota does play some part in the metabolic benefits when consuming a low-protein diet, but there is a certain range where restricting protein intake becomes beneficial.
Comparatively, those with ulcerative colitis have diminished L-arginine levels, where administration of this semi-essential amino acid helps alleviate ulcerative colitis (Figure 2) [54, 55], which suggests for a protein-enriched diet. Similarly, tryptophan degradation is more prone in those with IBD, where its product, quinolinic acid, and overall tryptophan deficiency could be aggravating disease activity [56], further supporting for amino acid supplementation as a possible therapeutic. Other tryptophan metabolites, including kynurenine, indole-3-aldehyde, and indole-3-acetic acid, can act as ligands for the aryl hydrocarbon receptor (AhR), a critical regulator of immunity, inflammation, and intestinal barrier function (as reviewed in [57]). Lanis et al. [58] identify that tryptophan metabolism mediates interferon-γ-induction of indoleamine 2,3-dioxygenase 1, an enzyme that catalyzes the conversion of tryptophan to kynurenine, where kynurenine subsequently activates an AhR response element on the promotor region of the IL-10 receptor 1. The upregulation of IL-10 signaling, specifically in intestinal epithelia, exerts anti-inflammatory effects, which further alleviates IBD (Figure 2) [58]. Alongside, Lactobacilli in the gut microbiota has been shown to preserve gut mucosa health by crosstalking with tryptophan metabolism. Specifically, Qi et al. show that the production of L-Ornithine from arginine-consuming Lactobacilli promotes kynurenine levels and subsequently AhR signaling (Figure 2) [59].
Figure 2: Dietary therapeutics and their mechanism(s) for alleviating IBD.
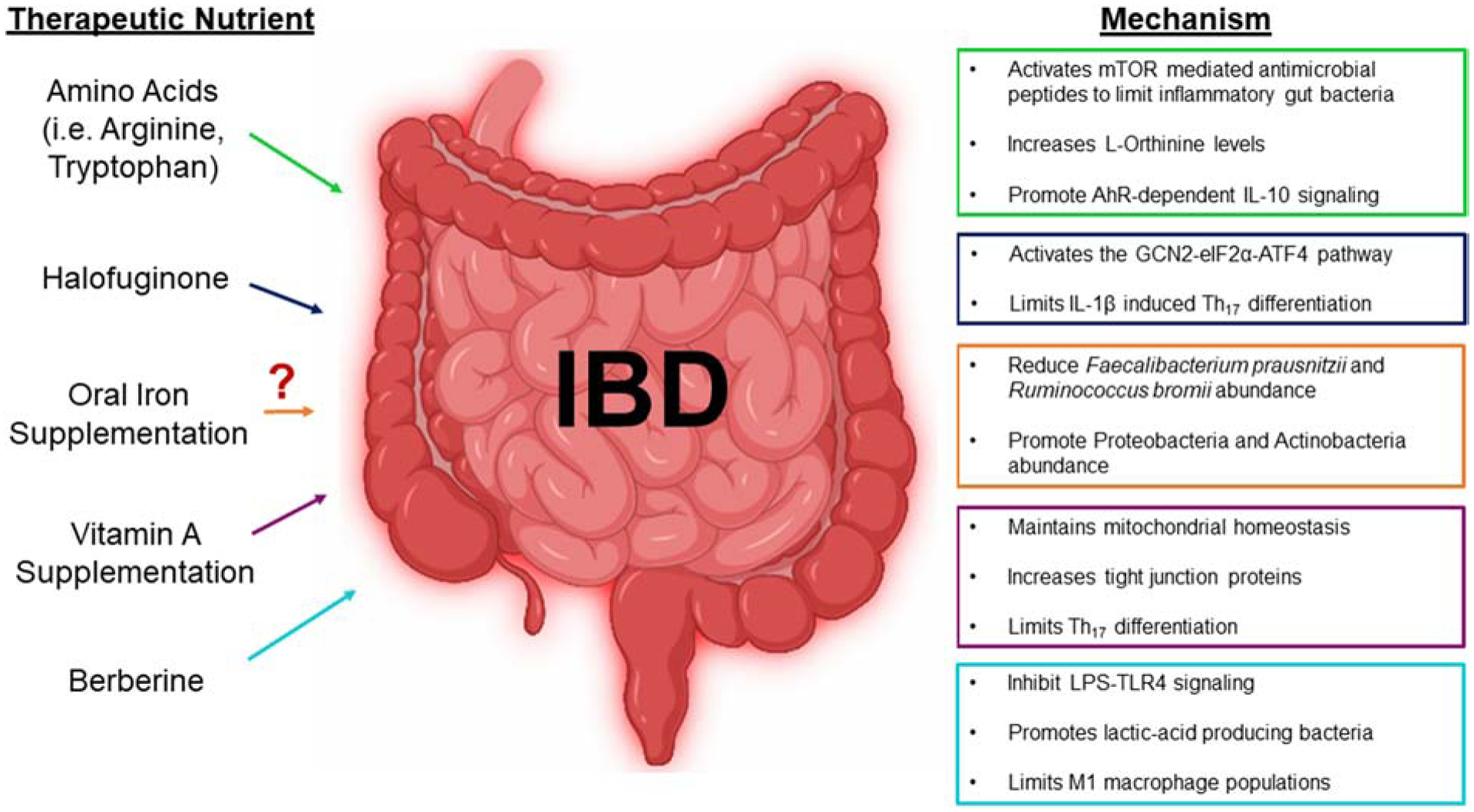
Intestinal inflammation is associated with severe malabsorption of various nutrients such as amino acids, iron and vitamin A. This has introduced supplementing these lost nutrients as therapeutics. While amino acids and vitamin A have shown positive results, recent research questions the intake of oral iron. As an alternative to resupplying nutrients, introducing the plant alkaloid, halofuginone, has been indicated to activate the GCN2 receptor that responds to nutrient starvation and limit inflammation. Additionally, supplementing herbs, like berberine, has been highlighted for anti-inflammatory properties. Key words: mammalian target of rapamycin (mTOR), aryl hydrocarbon receptor (AhR), interleukin 10 (IL-10), General Controlled Nonderepressible 2 (GCN2), eukaryotic translation initiator factor 2α (eIF2α), activating transcription factor 4 (ATF4), interleukin-1β (IL-1β), lipopolysaccharide (LPS), Toll-like receptor 4 (TLR4), pro-inflammatory macrophages (M1).
In addition to modulating IL-10 signaling, dietary tryptophan uptake into intestinal epithelia can result in the activation of mTOR, either through nutrient sensing and/or through the tryptophan nicotinamide pathway, where the production of antimicrobial peptides affects the composition of the gut microbial community and reduces inflammation (Figure 2) [60]. Intriguingly, Hashimoto et al. [60] demonstrate that deficiency of angiotensin converting enzyme 2 (Ace2) impairs intestinal tryptophan homeostasis and aggravates colitis, which is most likely due to the susceptibility of low-grade chronic inflammation mediated by the gut microbiota. As a side note, Pan et al. [61] recently show that FGF21 induces Ace2, which promotes the conversion of angiotensin II to angiotensin-(1–7), and thus, inhibits hypertension and reverses vascular damage (Figure 1A). Coming full circle, FGF21 signaling initiates hepatic ketogenesis and prompts renal Ace2 expression, both of which alleviate hypertension through NLRP3 inhibition and diminished angiotensin II, respectively (Figure 1). Moreover, Ace2 promoting tryptophan-activation of mTOR could possibily be a regulative feedback system to properly modulate ketogenesis so that hyperketonemia is impeded, while also preventing gut microbiota-induced intestinal inflammation. Future studies are necessary to elucidate the interplay among FGF21, Ace2 and ketogenesis in response to carbohydrate and protein-restricted diets.
2.3. Lipid Emulsions: Parenteral Nutrition and PNALD
So far, we have reviewed the acute vs. chronic effects of a carbohydrate-restricted diet, while also emphasizing the dramatic outcomes that can occur from single amino acid deficiency. Comparatively, the lifestyle intervention of restricting dietary fat intake, such as saturated and trans fats, is known to improve overall metabolic health and to treat NAFLD [62, 63]. It must be reminded, however, that fat is still an important macronutrient as they serve as a dense source of cellular energy and are the building blocks of cell membranes, amongst also influencing cellular communication, supplying essential fatty acids and much more. In fact, patients with intestine failure abnormalities that are unable to assimilate nutrients are succumbed to total parenteral nutrition (TPN), where intravenous lipid emulsions serve as the alternative caloric and nutrient source [64, 65]. Despite these emulsions playing a critical role to improve metabolic efficiency and to prevent essential fatty acid and fat soluble vitamin deficiencies for TPN-treated patients, fats that are given intravenously bypass the metabolism and packaging systems (i.e. chylomicrons) that naturally occur within the small intestinal lumen; therefore, hypertriglyceridemia and hypercholesterolemia, along with lipid and glucose intolerance, are common side-effects with long-term TPN [64, 66]. Collectively, the hyperlipidemia generated from prolonged use of TPN can lead to parenteral nutrition-associated liver disease (PNALD), which can progress to hepatic failure [67]. Likewise, PN patients are more prone to parenteral nutrition-associated cholestasis, as manifested by elevated bilirubin levels, which is further associated with transaminitis [67].
While there is less known about how parenteral nutrition-associated cholestasis affects the gut microbiota, the relationship among TPN, PNALD and the microbiota has been emerging. Generally, TPN induces gut dysbiosis and alters bacterial colonization in the intestinal tract, resulting in reduced microbial diversity [68]. Alongside, TPN promotes bacterial translocation due to an impaired mucosal barrier (as evident by reduced intestinal alkaline phosphate levels) and also alters the bacterial composition, as described with an increase in the abundances of Bacteroidetes and Tenericutes [69]. To study PNALD, El Kasmi et al. [70] developed a murine model by using dextran sulfate sodium (DSS) followed by continuous infusion of soy lipid-based TPN solution, which results in the upregulation of lipopolysaccharide-toll-like receptor 4 (LPS-TLR4) signaling, and thereafter, the activation of Kupffer cell-induced liver injury. The same group [71] performed microbiome analysis in their PNALD murine model and observed that these mice have an increased abundance of Erysipelotrichaceae, a Gram-positive bacteria generally associated with inflammatory-related disorders of the gastrointestinal tract [72]. Noteworthy, removal of the soy lipid-based emulsion reduces Erysipelotrichaceae and attenuates PNALD, whereas supplementation of the soy-derived plant sterol to a fish-oil emulsion restores Erysipelotrichaceae abundance and PNALD [71]. These opposite outcomes between soy- and fish-oil emulsions are generally explained by their distinct fatty acid and phytosterol compositions; fish-oil emulsions contain anti-inflammatory, omega-3 fatty acids and no levels of phytosterols, whereas pro-inflammatory, omega-6 fatty acids and phytosterols are highly abundant in soybean-based lipid emulsions [73–77]. Therefore, fish-oil supplementation has become a popular nutritional strategy to promote health, including in terms of alleviation/preventation of liver diseases such as PNALD and NAFLD [78], along with cardiovascular diseases [79]. Intriugingly, Miller et al. [80] recently report the first evidence of the harmful effects of feeding an omega-3-rich diet. Specifically, they observed that overconsumption of omega-3 fatty acids in mice elevates peroxidated lipid metabolites and potentiates lipotoxicity in WAT as evident by increased fibrosis, lipofuscin and reduced anti-inflammatory markers; yet, this diet was very benficial in terms other metabolic functions, including improved glucose sensitivity and reduced body weight [80]. Using lipidomics, Miller et al. [80] unravel that the accumulation of lipid metabolites differ between the brain and adipose tissue, which would explain why other studies have observed beneficial effects when enriching omega-3 fatty acids in the diet to protect against neurodisorders and to promote neurodevelopment [81–83].
3. Integrated Stress Response by Amino Acid Deprivation and Lipemia
3.1. Background
Earlier we mentioned about how amino acid deficiencies can increase susceptibility to inflammation and gut dysbiosis due to lowered mTOR activity. mTOR is one of the factors associated with the global integrated stress response (ISR), a cytoprotective operation that identifies and responds to stress indicators, including the unfolded protein response and nutrient deprivation [84]. There are four known ISR kinase sensors: protein kinase R, heme-regulated inhibitor, PKR-like endoplasmic reticulum kinase (PERK), and general controlled nonderepressible 2 (GCN2) kinase, which orchestrate responses toward viral double-stranded RNA, heme deficiency, ER stress, and amino acid depletion, respectively [85]. Activation of any kinase triggers the phosphorylation of the eukaryotic translation initiator factor 2α (eIF2α), which results in the inhibition of overall protein synthesis and promotion of autophagy [86]; yet, eIF2α can selectively stimulate certain genes (i.e. ATF4, CHOP) for mRNA translation [87, 88], where amino acid deprivation, in particular, activates the GCN2-eIF2α-ATF4 pathway to increase amino acid biosynthesis [87]. Additionally, upregulation of eIF2α and ATF4 has been associated with selective translation of inflammatory cytokines [89], suggesting a coupling effect of metabolic stress and inflammation. This section of the review will focus on GCN2 and PERK in different disease conditions associated with altered nutrient status.
3.2. GCN2 and IBD
Gut inflammation is associated with altered digestion, absorption and barrier function, as evident by reduced villi lenghth and crypt depth, and increased gut permeability [90]. Hence, it is highly probable to exhibit nutrient deprivation in cases of IBD, where the uncharged tRNA resulting from amino acid deficiency would be sensed by colonic GCN2 [91]. Comparatively, DSS-induced colitis in GCN2- or eIF2α-deficient mice worsens disease severity compared to wild-type controls, where this is most aggravated during GCN2 deficiency [85]. Ravindran et al. [85] demonstrate that the exacerbated colitis is associated with an increase in Th17 response in CD11c+ antigen presenting cells and intestinal epithelia. Alongside, GCN2 deficient colitic mice have a defect in autophagy, resulting in elevated reactive oxygen species (ROS) activation of inflammasome-mediated IL-1β production [85]. Therapeutically, administrating the plant alkaloid derivative, halofuginone, can activate the GCN2-eIF2α-ATF4 pathway and inhibit Th17 differentiation (Figure 2) [92], supporting the evidence that GCN2 is capable of alleviating gut-lymphatic associated inflammation. Comparatively, administration of the amino acid, serine, was demonstrated to alleviate DSS-induced colitis, which was associated with reduced GCN2 expression, increased Clostridia abundance, and diminished apoptosis [93]. This indicates that the association of amino acid starvation, GCN2 and gut inflammation is not necessarily reciprocal.
3.3. GCN2 and Hepatic Steatosis
Along with influencing the gastrointestinal tract, amino acid deprivation has been linked to programming the liver toward a steatosis state, which emphasizes the gut-liver axis. For example, dietary arginine or threonine deficiency was found to induce hepatic triglyceride accumulation [94]. Likewise, eliminating asparagine and glutamine through asparaginase and glutaminase, respectively, promotes transient hepatic lipid accumulation [95]. Mechanistically, asparaginase treatment activates GCN2, which subsequently triggers the amino acid stress response as a protective mechanism against asparaginase-mediated hepatotoxicity, where GCN2 deficient mice are prone to lipid accumulation and DNA damage in the liver after asparaginase administration [96, 97]. More recently, Wilson et al. demonstrate that asparaginase-treated GCN2 deficient mice have significantly diminished hepatic and WAT expression of FGF21, which is associated with impaired mitochondrial function in the liver and adipose tissue [98]. Since FGF21 promotes fatty acid oxidation and alleviates obesity, insulin resistance and hyperlipidemia, this suggests that GCN2-induced FGF21 is a safeguard against hepatic steatosis during asparaginase therapy. In line, leucine deprivation induces the GCN2-FGF21 axis, which represses lipogenic enzymes and lipid deposition in the liver [99, 100]. This further corresponds with previous reports that amino acid-deficient diets and low-protein diets increases FGF21 expression through GCN2-eIF2α-ATF4 and PPARα signaling [101, 102]. Intriguingly, while amino acid starvation (i.e. leucine) and asparaginase activation of GCN2 is protective against hepatic lipid accumulation, GCN2 seems to have opposing effects when a high-fat diet (HFD) is introduced. Specifically, HFD-fed GCN2-deficient mice have alleviated steatosis and insulin resistance compared to their control counterparts, which was further correlated with reduction of de novo lipogenesis and lessened oxidative stress [103]. This emphasizes that the therapeutic potential of GCN2 is context-dependent.
3.4. PERK, Metabolic Dysfunction and Atherosclerosis
It has been more recognized that the ER functions as a nutrient sensor for glucose, amino acids and free fatty acids [104]. Under conditions that challenge ER function, the unfolded protein response is activated, which consists of three ER-associated proteins: (1) inositol-requiring enzyme-1, (2) activating transcription factor (ATF)-6, and (3) PERK [105]. If the ER disruption is not resolved, this can result in liver dysfunction and metabolic consequences, such as microvesicular steatosis and atherosclerosis [88, 106, 107]. In terms of atherosclerosis, fatty acid binding protein-4 has been proven to be necessary for lipid-induced macrophage ER stress [108]. Recently, Onat et al. [88] explicitly demonstrate that PERK is also activated in response to lipotoxic signals and aggravates atherosclerotic plaques. Specifically, administrating the saturated fatty acid, palmitate, activates PERK-eIF2α-ATF4 signaling, which subsequently augments Lon protease 1 (LONP1) expression [88]. LONP1 is a mitochondrial-associated protease that degrades unfolded proteins and also serves as a chaperone that regulates mitophagy and ROS generation [109–111]. Onat et al. [88] show that LONP1 degrades phosphatase and tensin-induced putative kinase 1, while also blocking Parkin-mediated mitophagy. This further results in activation of the NLRP3 inflammasome, overall prompting hyperlipidemia-induced inflammation and aggravating atherosclerosis [88]. By blocking or inhibiting PERK, this suppresses mitochondrial ROS-induced inflammasome activation and reduces atherosclerosis [88], demonstrating that PERK is a possible therapeutic target against atherosclerotic plaques. This also emphasizes that components of nutrients, like lipids, can act as danger associated molecular patterns (DAMPs), where its elevation during chronic nutrient excess, as seen during obesity, leads to sterile inflammation and cardiovascular occlusions. More recently, it has been understood that gut metabolites can also act as DAMPs. Choline and carnitine from the diet, for example, is converted to trimethylamine (TMA) by the gut microbiota, which is eventually converted to trimethylamine-oxide (TMAO) by host hepatic flavin-containing monooxygenase 3 (FMO3) [112]. TMAO, in particular, has been identified as another DAMP that activates PERK; specifically, TMAO promotes PERK-induced upregulation of the transcription factor, FoxO1, which is a high value target of metabolic diseases [113]. Inhibiting FMO3 or manipulating the gut microbiota can impede metabolic dysfunction mediated by the PERK-FoxO1 axis [113]. Considering that TMAO is strongly associated with insulin resistance and atherosclerosis [114–116], this newly released mechanism further solidifies PERK as a novel target against metabolic disease and atherosclerosis.
4. Micronutrient Scarcity
4.1. Iron Deficiency and Infection
Iron is an indispensable nutrient for the survival of almost all aerobic organisms and bacteria except Lactobacilli and Borrelia species; yet, iron consumption can fluctuate depending on the type of diet an individual follows. Compared to the high quantities of heme-iron found in meat, fish and poultry, vegetarians that generally consume low levels of non-heme-iron content from vegetables, fruits and nuts are at greater risk of developing iron deficiency [117]. This dangerous likelihood has an increased probability due to plant-derived phytate and polyphenols inhibiting iron absorption, which is why vegetarians have a near 2-fold increased requirement for iron [118–120]. On the contrary, individuals that suffer from iron overload disorders (i.e. hemochromatosis) require iron chelation therapy in order to minimize its toxicity and its capacity to generate oxidative stress (as reviewed in [121]). Similarly, low iron conditions can institute a natural defense against local and systemic infections prompt by pathogens that require iron for growth and survival [122]. This involves several actions, including transferring iron from circulating iron-binding proteins (i.e. transferrin) to cytoplasmic storages (i.e. ferritin) and degrading the sole iron exporter, ferroportin (Fpn), through the master iron regulatory hormone, hepcidin [123]. While these actions of nutritional immunity and thereafter, hypoferremia, can limit iron from extracellular bacteria, this has been consequently correlated with the promotion of the intracellular pathogen, Salmonella, which specifically replicates in macrophages. Intriguingly, Lim et al. recently demonstrate that the iron requirement for Salmonella is for ROS generation and not to maintain nutrient status [124]. Moreover, they reveal that Salmonella proliferate in macrophages via a Salmonella-containing vacuole (SCV), where the host can blunt this infection by hepcidin promoting the internalization and degradation of Fpn located on the SCV, which subsequently increases the cytosolic iron content of macrophages [124]. Additionally, it has been shown that, in response to Salmonella infection, iron-regulatory proteins (IRPs) promote the induction of lipocalin 2 [125], an innate immune protein that sequesters iron-laden siderophores to prevent bacterial iron acquisition (as reviewed in [126]). However, despite this defensive mechanism, Salmonella enterica serovar Typhimurium (S. Typhimurium) is still capable of colonizing the gastrointestinal tract and outcompeting the microbiota with ease. Behnsen et al. [127] uncover that the virulence of S. Typhimurium is due to IL-22 suppressing the growth of commensal Enterobacteriaceae in the inflamed gut. Intriguingly, Forbester et al. [128] recently demonstrate through intestinal organoids that IL-22 can protect against S. Typhimurium infection through its antimicrobial activity. This suggests that IL-22 can act as a double edge sword in S. Typhimurium infection.
4.2. Iron Malabsorption and Hypoxia Stress Response
While both IRP-1 and IRP-2 are needed to coordinate post-transcriptional regulation of iron metabolism, is has been more recognized that IRP-1 is rapidly activated by H2O2, distinguishing its responsibility toward iron metabolism and oxidative stress [129, 130]. Notwithstanding, ferrous iron can react with H2O2 through Fenton chemistry to generate toxic hydroxyl radicals [122], emphasizing that there is a strong interplay between ferrous iron and O2, which further correlates to stress pathways like hypoferremia and hypoxia, respectively. This is further supported by the iron-dependent modulation of the von Hippel-Lindau/Hypoxia-inducible transcription factor pathway. Specifically, iron is required to activate prolyl hydroxylases that promote HIF-1α degradation, whereas iron deprivation or chelation allows for HIF-1α to translocate into the nucleus, resulting in expression of genes regulated by the hypoxia-response elements [131]. Intruigingly, mice that lack a microbiota have diminished intestinal HIF expression, where Kelly et al. show that butyrate, produced by gut bacteria, increases O2 consumption, which preserves HIF and gut barrier function [132]. On the contrary, Das et al. demonstrate that the gut microbiota blocks HIF-2α expression but increases ferritin levels, which causes iron malabsorption [133]. Ironically, iron deficiency is known to promote HIF-2α mediated induction of divalent metal transporter 1 to enhance intestinal iron absorption [134], while further upregulating Fpn [135]. Detrimentally, genetic ablation of Fpn in intestinal epithelia induces iron deficiency anemia (IDA) through the loss of intestinal iron absorption and limited erythropoiesis [136]. This is further associated with cardiomegaly and perturbed cardiac function that is independent of cardiac hypoxia; yet, it must be emphasized that IDA in this model still activates hypoxia transcriptional stress responses (i.e. HIF-2α) in the intestine in attempt to rebalance iron homeostasis [136].
4.3. Hypoferremia and IBD
Patients with IBD are commonly found to exhibit IDA, where the bloody diarrhea and impaired iron absorption can be caused by tissue inflammation. This inflammatory tone results in the upregulation of acute phase proteins and pro-inflammatory cytokines (i.e. IL-6), where IL-6, in particular, promotes the expression of hepcidin, later leading to the degradation of Fpn and thus, induction of hypoferremia [137]. While oral iron supplementation would seem like the logical therapeutic to counteract hypoferremia, recent research suggests that this ‘solution’ could be problematic and may actually worsen intestinal inflammation. Mahalhal et al. [138] uncover that increasing the amount of dietary iron exacerbates both clinical and histological severity of DSS-induced colitis in mice; in particular, doubling the amount of dietary iron significantly aggravates intestinal inflammation, while also altering the gut microbiota via diminishing the Firmicutes and Bacteroidetes, and promoting opportunistic pathogens, like Proteobacteria and Actinobacteria (Figure 2) [138]. Likewise, it has been clinically reported that oral iron administration perturbs the gut microbiota, including dimishing Faecalibacterium prausnitzii and Ruminococcus bromii abundance (Figure 2) [139]. This is detrimental since Faecalibacterium prausnitzii is one of the most abundant bacteria in the healthy human microbiota and a candidate for next generation probiotics due to its anti-inflammatory and butyrate-producing capabilities [140, 141]. Similarly, Ruminococcus bromii can degrade resistant starch particles [142], where fermentation of starches generates SCFAs, including the ‘beneficial’ butyrate [143]; therefore, reduction of Faecalibacterium prausnitzii and Ruminococcus bromii explains, in part, why IBD patients have significantly attenuated butyrate levels. This has introduced supplementing butyrate-producing bacteria as a therapeutic for IBD cases, and there have been promising results of improved intestinal mucosal integrity [144]; yet, Zhang et al. document that introducing butyrate-producing bacterium (i.e. Anaerostipes hadrus) actually accelerates gut dysbiosis and aggravates colitis in DSS-treated mice [145]. In particular, it was demonstrated that administrating Anaerostipes hadrus does not result in butyrate elevation in colitic mice [145]; therefore, one could argue that reducing butyrate levels could be an adaptive mechanism during colitis pathogenesis. Contradictory results on the benefits vs. detrimental butyrate effects have generated the ‘Butyrate Paradox’ (as reviewed in [146]), which still needs to be fully elucidated. As an easier method to avoid the detrimental effects of oral iron supplementation, it has been suggested to feed iron intravenously to IBD patients, which has been associated with less frequent hospitalizations [147].
4.4. Vitamin A Deficiency and IBD
The acute phase response is the early activation of the innate immune system, where a type of stress (i.e. infection) triggers the production of local pro-inflammatory cytokines (i.e. IL-6, TNF-α) that further stimulates acute phase proteins and immune cells to resolve the initial stress (as reviewed in [148]). If this transit inflammation is unresolved, then this results in chronic, sterile inflammation that can severely cause tissue damage, impair nutrient status, and potentiate various diseases. In particular, long-term gut inflammation can impede the absorption of essential micronutrients, including minerals and fat-soluble vitamins. Vitamin A deficiency, for example, is associated with anemia of inflammation during IBD [149], where its restoration has proven to be a dietary therapeutic on multiple levels. For one, pretreating rats with a high vitamin A diet protects against colonic mitochondrial damage during colitis [150]. Specifically, vitamin A promotes the upregulation of the mitochondrial transcription factors, NFR-1 and TFAM, which inhibits inflammatory and necrotic inflictions on the colon (Figure 2) [150]. Moreover, DSS-induced colitis in vitamin A-deficient mice worsens the colitis phenotype and delays the intestinal restitution [151].
The aggravated colitis in vitamin A-deficient mice was associated with altered colonic crypt function and elevated dendritic cells (DCs) [151]. Generally, DCs in the small intestine produce retinoic acid (RA) from vitamin A (retinol), which plays an important role in immune tolerance through managing the differentiation of anti-inflammatory cells [152]. Recent studies have shown that RA reprograms the gut-lymphoid system to restrain anti-inflammatory regulatory T cells from converting into pro-inflammatory Th17 cells (Figure 2) [153, 154]. In fact, RA cooperates with IL-2 to maintain forkhead box P3 (transcription factor and master regulator in regulatory T cell development) expression even after stimulation of Th17 polarization [154]. Along with minimizing Th17 levels, RA signaling promotes IL-22-dependent antimicrobial responses; however, this protection is blocked by commensal bacteria in the Clostridia class [155]. Specifically, some Clostridia can limit RA levels by suppressing retinol dehydrogenase 7 expression, where this impairment in immune response allows for easy colonization of S. Typhimurium [155]. This highlights the importance of RA in innate immunity and protection against inflammation/infection.
In line, RA restoration via pharmacologic blockage of the RA-catabolizing enzyme, CYP26A1, in APCMin/+ mice confers protection against intestinal inflammation and intestinal tumor burden [153]. In a similar manner, RA supplementation diminishes inflammation markers and elevates anemia indicator levels during LPS-induced anemia of inflammation [156]. This is further associated with RA-mediated downregulation of hepcidin and TLR4 signaling, with a reciprocal upregulation of Fpn and serum iron levels. [156]. Moreover, vitamin A supplementation was associated with enhanced tight junction protein expressions (i.e. ZO-1, Occludin) in LPS-treated mice that would be prone to increased intestinal epithelial permeability (Figure 2) [157]. These results suggest for vitamin A and RA to be nutrition-based therapeutics against IBD. In addition to protecting the intestine, RA has been further shown to have anti-inflammatory effects in adipose tissue, suggesting that RA can also be a nutritional strategy against obesity and its complications [158].
5. Metabolic-Immune-Microbiota Networks
Metaflammation and Immunologic Responses
Obesity has become one of the unintended consequences from industrialization, economic growth and mechanized transportation. In particular, the combination of a sedentary lifestyle with overconsumption of high-fat foods has become the fuel for metabolic disease, which is progressively becoming more common in children. It is recently understood that obesity itself and thus, overnutrition, induces an inflammatory state termed ‘metaflammation’ [159]. In the similar fashion to where metabolic cells respond to nutrient starvation, low-grade, chronic metaflammation is the result from cells reacting toward excess nutrients and energy [159]. One of the responses associated with obesity-related metaflammation is the activation of toll-like receptors (TLRs) in adipose tissue, which upregulates cytokines in both MyD88 independent and dependent fashions [160]. Intruigingly, Kim et al. demonstrate that TLR activation is greater in diet-induced obese mice compared to ob/ob mice that are profoundly obese due to a genetic mutation in the leptin receptor [160]. Along with LPS-TLR4 signaling, it has been recently uncovered that the high-mobility group B1 (HMGB1; a damage-associated molecular pattern released from necrotic cells) activates the TLR4/MD-2 complex, which is necessary for HMGB1-mediated production of pro-inflammatory cytokines [161]. Likewise, Ghosh et al. show that adipose tissue-derived HMGB1 activates TLR9 located on recruiting circulating plasmacytoid DCs, which results in type I inteferons helping in polarization of pro-inflammatory (M1) macrophages [162]. In line, lack of CD1d expression on anti-inflammatory (M2) macrophages switches these immunes cells to M1 macrophages, where they exacerbate metaflammation through natural killer T cell activation of Th1 responses and inhibition of M2 polarization [163]. Moreover, obesity is associated to promote a subset of IL-6 receptor expressing natural killer cells that promote inflammation and insulin resistance in adipose tissue [164].
Along with an imbalance in macrophage and other immune cell subpopulations, cyclic guanosine monophosphate signaling is disrupted in inflamed WAT due to increased cytokine production (i.e. TNFα) from NF-κB signaling, further resulting in diminished adipogenesis [165]. Interestingly, deletion of the transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI; a receptor for the TNF superfamily) protects against HFD-induced metaflammation independent of adipogenesis, where Sanyal et al. demonstrate that TACI deficiency promotes the recruitment of M2 macrophages and other protective innate immune cells [166]. Interestingly, there is an emerging speculation that dying adipocytes may play a role in metaflammation through its release of pro-inflammatory contents such as cell-free DNA, which induces TLR9-mediated macrophage accumulation [167]. Overall, targeting immune cells, more particularly M1 macrophages, is a strong therapeutic possibility for preventing metaflammation. One option is to blunt heme oxygenase-1 expression on macrophages, as its absence is associated with resistance against obesity-mediated inflammation [168].
Metabolic Endotoxemia and the Gut Microbiota
While the subject of inflammation in metabolic disease has been examined in detail, especially in terms of immunological responses (as further reviewed in [169–171]), the role of the microbiome has only recently been delineated. In 2007, Cani et al. [172] observe that mice fed a HFD display microbiotal dysbiosis, which is associated with increased quantities of LPS-producing gut bacteria, a 2–3 fold increase in plasma endotoxin levels, and concomitant insulin resistance and obesity. The authors appropriately described this condition as “metabolic endotoxemia”, or the chronic, low-grade inflammatory response attributed to diet-induced endotoxin (i.e. LPS) production and circulation. LPS is a major outer surface membrane component predominantly present in Gram-negative bacteria, and an endotoxin that is recognized by an assortment of immunoreceptors and accessory proteins, including TLR4, LPS-binding protein, and CD14 [173]. The activation and colocalization of intestinal TLR4 and CD14, respectively, by LPS increases gut barrier permeability resulting in a ‘leaky gut’ [174], which allows this endotoxin to translocate into systemic circulation and to trigger metaflammation through downstream activation of c-Jun N-terminal kinases in macrophages [175]. In line, a diet rich in saturated dietary lipids has been suggested to modulate the gut microbiota to promote endotoxin-mediated TLR4 signaling, and subsequent macrophage infiltration and inflammation in WAT [176]. Overnutrition or ‘Western Diet’ promotion of LPS-driven metaflammation is also associated with worsened sepsis severity; yet, in terms of this disease, this aggravation may be independent of changes in the microbiome, but more due to direct regulation of the innate immune system [177].
Additional studies have examined the extent to which TLR4 and CD14-mediated signaling contributes to metaflammation pathogenicity. Deletion of MyD88, a downstream effector for TLR4-signaling, has been found to be protective against metaflammation [178]. Luche et al. also observe in vitro and in vivo that CD14 deficiency ameliorates the effects of HFD-induced, LPS-driven metabolic endotoxemia and subsequently, abrogates preadipocyte proliferation [179], thus overall suggesting to target TLR4 as a potential therapeutic; however, other reports make this option less ambiguous. For example, the ablation of TLR4 has been found to promote the expression of lipogenic genes (i.e. Fasn, Scd1, Dgat1, Pparγ) in fasting conditions, which demonstrates that this immune receptor is important in regulating lipid metabolism through inhibition of lipogenesis [180]. More recently, Lu et al. [181] finds that mice with intestinal epithelial cell specific deletion of TLR4 ultimately develop metabolic disease and maintain a pro-inflammatory phenotype. Intriguingly, TLR4 defeciency did not impact PPAR-regulated metabolic gene expressions, where its activation through a PPAR agonist profoundly reversed the effects of metabolic syndrome in TLR4-deficient mice [181]. These findings implicate PPAR as a potential therapeutic target in remedying metaflammation.
Compared to targeting the later effects of LPS, the simple administration of broad-spectrum antibiotics to HFD-fed mice to alter their gut microbiota profile can decrease intestinal lumen LPS content, blunt adipose tissue inflammation, and improve metabolic parameters, demonstrating that targeting the gut microbiota may be the front line therapeutic to alleviate metabolic endotoxemia and metaflammation [182]. One dietary approach to modulate the gut microbiota would be to introduce probiotics, where Xue et al. demonstrate that this can reverse the effects of gut dysbiosis, improve gut barrier integrity and alleviate endotoxemia-induced metaflammation, which is further associated with a delay or protection against NAFLD [183]. Likewise, Ghadimi et al. [184] recently show that Bifidobacteria, a common probiotic bacterium, dampens iron overload- and LPS-associated metaflammation. Specifically, Bifidobacteria was demonstrated to downregulate hepcidin levels and inhibit TLR4 signaling in response to excessive LPS [184], which suggests that promoting the bloom of this commensal could be a potent therapeutic.
6. Inflammaging and the Gut Microbiota
Background
Inflammaging refers to the ‘chronic, low-grade inflammation that characterizes aging’ [169, 170, 185]. In general, it is commonly associated that aging increases the risk for developing certain diseases, including hyperinsulinemia and obesity. There are many theories on the process of inflammaging. Franceschi et al. [169, 170] states that inflammaging is due to the lack of disposing endogenous, misplaced or altered molecules that generally increase during aging, resulting in an ‘autoreactive/autoimmune’ process that fuels age-associated chronic diseases that can accelerate the aging process. In line, inflammaging has been associated to be macrophage centered, where hyperinsulinemia, for example, augments pro-inflammatory M1 macrophages and suppresses anti-inflammatory M2 macrophages, where this inflammatory state promotes the pathogenesis of insulin resistance and extracellular matrix deposition in adipose tissue [186]. Moreover, the inflammatory-based macrophages are not rendered their normal bactericidal status, which can result in the inhibition of macrophages from limiting pro-inflammatory bacteria; this is further proven as macrophages from aged germ-free mice maintain antimicrobial activity [187]. This section of the review will delve further into the relationship between inflammaging and the gut microbiota.
Gut Bacteria as an Inflammatory Age Marker
There is an emerging theory that age-associated gut dysbiosis increases susceptibility toward inflammatory disorders. When comparing the initial bacteria that colonizes the gut in adults, elderly, centenarians (age 99–104) and semi-supercentenarians (age 105–109), the cumulative abundance of the three dominated families, Bacteroidaceae, Lachnospiraceae, and Ruminococcaceae, decreases with age [188]. Likewise, Firmicutes are found to be more dominant in young individuals, whereas the Bacteroidetes are more profound in the gut microbiota of the elderly [189]. Moreover, an aged microbiota in mice has been associated with lower levels of Akkermansia (anti-inflammatory bacteria) and higher levels of Proteobacteria (pro-inflammatory bacteria) [190]. Comparatively, rendering germ-free mice that lack the gut microbiota do not exhibit an age-related increase of inflammatory cytokines, which might contribute to their increased longetivity compared to their conventional counterparts; yet, when co-housing germ-free with aged conventional mice, this reverts the protective effects observed in the germ-free mice, thus emphasizing the role of the gut microbiota in inflammaging [187]. Similarily, when transferring the gut microbiota from old conventional to young germ-free mice, these mice become more prone to inflammation due to their introduced age-associated dysbiotic bacteria and the shift toward pro-inflammatory macrophages [190]. Intriguingly, Kundu et al. [191] recently show that performing fecal microbiota transplantation from old conventional mice to young germ-free mice actually promotes neurogenesis, intestinal growth, and hepatic FGF21-signaling, where it is suggested that the enrichment of butyrate-producing bacteria in the elder mice confers benefits to the young receipents. This emphasizes that an ‘older’ gut microbiota does not necessarily equal deleterious effects on the host and that, in fact, an aged gut microbiota may possess benefits that are lacked in a young host.
Multiple therapeutic strategies have arisen to dampen age-associated inflammation. For one, anti-TNFα therapy can be used to reverse the age-associated microbiota changes and blunt the age-related inflammatory cytokine levels [187]. Additionally, the probiotic strain, Lactobacillus acidophilus DDS-1, has been shown to diminish the production of inflammatory cytokines, which was associated with an increase in Firmicutes and a decrease in Bacteroidetes abundance in aged mice [192]. Lastly, while not explicitly described for treating inflammaging, the Akkermansia muciniphila protein, Amuc_100, has been demonstrated to improve gut barrier function, promote beneficial bacteria that alleviates obesity and type 2 diabetes, and has also been proven safe for human consumption [193]. Future studies should examine whether Amuc_100 could be a potential therapeutic against age-related metabolic disorders, including inflammaging.
7. Nutritional Interventions: Suppling Essentials for the Gut Microbiota
So far, we have delved into understanding how the host responds to metabolic starvation and inflammation, and intertwining the role of the gut microbiota in those responses. In this last section, we will delve into potential nutritional therapeutics such as cruciferous vegetables and their role in modulating the gut microbiota and regulating inflammatory responses. This also includes exploring common prebiotics and the rise of herbel medicines that are being suggested as novel prebiotics.
7.1. Cruciferous Vegetables
Broccili, cauliflower and cabbage are a few examples of vegetables that belong to the Brassicaceae family. Brassicaceae plants contain myrosinases, glucosinolate hydrolyzing enzymes that generate bioactive isothiocyanates (ITCs) [194]. Myrosinase activity is heat-labile and thus, inactivated at temperatures above 60 degrees Celsius [195], while also being very sensitive to pH changes [196]; therefore, when these plants are cooked for human consumption and then enter the digestive system, myrosinase activity is inhibited. To maximize and retain the production of ITCs, it is recommended to stir-fry or steam the Brassicaceae plants instead of boiling [197]. Intriguingly, dietary intake of Brassicaceae plants and the production of ITCs has been demonstrated to provide chemoprotective effects (as reviewed in [198]), suggesting that myrosinase activity is somehow maintained in the host after consumption. Recent research has attributed the gut microbiota for supplying myrosinase, where ingestion of broccoli, for example, was associated with improved hydrolysis of glucosinolates to ITCs due to increased myrosinase activity [199, 200]; however, a study by Budnowski et al. suggests that the biotransformation from glucosinolates to ITCs is minimal in a human microbiota associated mouse model [201]. Alongside, there have been various reports on the alterations in the gut microbiota after ingestion of cruciferous vegetables in both animal and human studies, including an increase in Akkermansia, Ruminococcaceae and Bacteroidetes [199, 202], along with lowered Firmicutes [202] and sulphate-reducing bacteria (i.e. Clostridia) [203]. Moreover, it has also been reported that ingestion of a Brassica-rich diet promoted the butyrate-producing bacteria, Eubacterium rectale and Faecalibacterium prausnitzii, which was correlated to diminished inflammation, as evident by increased IL-10 levels and regulatory T cells [204]. This matches a similar report that observed sulforaphane, a type of ITC, had rebalanced the gut microbiota (i.e. increase Bacteroides fragilis), which assisted in providing better gut barrier function and decreased inflammatory responses [205]. Furthermore, fermented Brassica foods (i.e. sauerkraut and kimchi) were demonstrated to promote the abundance of Lactobacilli in the gut [203], which has presented Brassica plants as a potential prebiotic option.
Two of the most extensively studied ITCs is indole-3-carbinol (I3C) and 3,3’-diindolylmethane (DIM). I3C has been studied for its anti-cancer properties (as reviewed in [206]), and more recently for its influence on the gut-lymphatic system, especially in terms of IBD pathogenesis. For example, I3C administration in colitic mice was observed to abate gut dysbiosis and promote butyrate-producing bacteria, along with suppressing inflammation through increased IL-22 production [207]. Similarily, DIM was demonstrated to ameloriate non-alcoholic steatohepatitis in mice by shifiting toward regulatory T cell predominance and minimizing Th17 populations [208]. The protective effects of I3C and DIM have been attributed to them being agonists for AhR signaling [208, 209]. In line, DSS-induced colitis in AhR-deficient mice aggravates pathogenesis due to over-immune activation and elevated bacterial burden; interestingly, administrating a broad-spectrum antibiotic to colitic AhR-deficient mice prevents the aggravated inflammation [210]. Despite these benefits of ITCs, some glucosinolates have been suggested to be genotoxins [211]. Recently, Gronke et al. solidified glucosinolates to be a source of genotoxic stress in intestinal epithelial cells [212]. This study utilized a colitis-associated colon cancer model, which involves inducing DNA damage via the pro-carcinogen, azoxymethane, and triggering colitis through DSS. The goal was to determine the effects of ITCs such as I3C and 1-methoxy-3-indolylmethyl alcohol (1-MIM-OH) on the DNA damage response (DDR), where IL-22, in particular, is an essential requirement in activating DDR after DNA damage. While I3C did not trigger the DDR, 1-MIM-OH activated AhR-signaling and thereafter IL-22 production, which presented this glucosinolate as a genotoxin and modulator of the DDR; yet, it must be emphasized that IL-22 provided protection against the genotoxic stress [212]. Overall, the differences in gut microbiota alterations and immunological responses of Brassica vegetables could be, in part, contributed to the distinct plant microbiomes and genotypes from the various species in the Brassicaceae family [213]. Therefore, future studies should explore the specificity of each distinct Brassica plant and their effects on the host, which will help pave for directing appropriate nutritional therapeutics.
7.2. Prebiotics and Herbal Medicines
Prebiotics, such as fructans, fructo-oligosaccharides, and galacto-oligosaccharides are non-viable substrates that promote the blooming of ‘beneficial’ gut bacteria (i.e. Bifidobacterium and Lactobacilli) [214]; yet, there is also the possibility of cross-feeding, where fermented product(s) generated from the ‘good’ bacteria could promote the growth of ‘bad’ bacteria [215]. Similarily, the distinct molecular structure of various prebiotics makes their susceptibility to fermentation different from each other, including the output of SCFA production. For example, inulin is the prebiotic polysaccharide from chicory root, where its main fermentated metabolite is butyrate compared to acetate and propionate [216]. Inulin has been extensively proven to promote the blooming of Bifidobacterium and Lactobacilli [216–219], which is associated with protecting against HFD-induced metabolic syndrome [220], strengthening gut barrier function [221] and alleviating endotoxemia [217]. Mechanistically, inulin is demonstrated to induce IL-22 expression in intestinal epithelial cells, which diminished bacterial burden [220], and is also shown to promote anti-inflammatory responses in a gut microbiota-dependent manner [217, 222]. Intruigingly, the degree of inulin benefits seems to be chain length-dependent, where Li et al. report that long-chain inulin polysaccharides have a more effective inhibition of metabolic endotoxemia compared to short-chain inulin [217]. They suggest that the superiority of long-chain inulin may be due its capability of being fermented by certain bacteria, such as Bacteroides [217].
While prebiotics have been shown to alleviate metabolic and inflammatory diseases with the help of the gut microbiota (as reviewed in [223]), current research questions whether prebiotics are truly beneficial (as reviewed in [224]). To highlight two of our findings, we have observed that inulin aggravates intestinal inflammation through NLRP3-activation [225] and that feeding inulin to a subset of gut dysbiotic mice induces cholestatic hepatocellular carcinoma [226]. Hence, this suggests that under certain contexts, fermentable prebiotic fibers can be detrimental. Therefore, there is an urgency to look for other prebiotic candidates. One rising star is herbal medicines, as they have been presented to promote butyrate- and propionate-producing gut bacteria like Bifidobacterium spp., Lactobacillus spp., and Bacteroides spp while also blunting the growth of opportunitistic pathogens such as Citrobactera freundii and Klebsiella pneumonia [227, 228]. In particular, traditional herbs like Rhizoma Atractylodis Macrocephalae and Flos Lonicera, display anti-obesity effects and inhibition against metabolic endotoxemia, which is associated with alterations in gut flora, including increased abundance of Bacteriodetes, Lactobacillus, and Akkermansia [229, 230]. Along with promoting beneficial gut bacteria, the mulberry leaf herb has been demonstrated to ameliorate insulin resistance and diabetes by minimizing non-esterified fatty acid signaling [231]. Additionally, fermented green tea extract increases glucose tolerance and attenuates hepatic steatosis, which is associated to changes in the Firmicutes/Bacteroidetes and Bacteroides/Prevotella ratios [232]. Moreover, Ganoderma lucidum, a tranditional Chinese medicinal mushroom, demonstrates anti-diabetic effects through modulation of the gut microbiota, as proven through fecal microbiota transfer from herb-fed to HFD-fed mice, which has also suggested for this herb to be a prebiotic agent against metabolic disorders associated with the gut microbiota [233]. Furthermore, the Chinese herb, berberine, has been extensively studied for its potent effects in lowering metabolic stressors, diminishing inflammation and alleviating colitis; specifically, berberine reduces M1 macrophages and inhibits hepatic LPS-TLR4 signaling, while also promoting lactic-acid producing bacteria and probiotic-associated bacteria and decreasing pathogenic bacteria (Figure 2) [234–237]. Interestingly, the gut metabolite of berberine, oxyberberine, was indicated to achieve a superior effect on alleviating DSS-induced colitis compared to berberine [235]. Considering that berberine has a poor bioavailability after oral ingestion [238], oxyberberine may be a new front-line candidate as a prebiotic against gut-associated inflammation and metabolic diseases.
8. Conclusion
Decades ago, nutrition, microbiology, immunology and biochemistry were studied as their own distinct disciplinary. Now, the rapid development in technology has merged all these fields into a combined network. This collaboration has advanced the understanding on how nutrient excess and starvation orchestrate inflammatory signaling pathways, which can further lead to altered susceptibility to several chronic diseases. Moreover, this knowledge has prompted for nutritional therapeutics to counteract the negative outcomes that occur due to altered macro and micronutrient metabolisms, including exploring the gut microbiota. Considering the recent literature that has been collected, it must emphasized that environmental (i.e. nutrition, gut microbiome) and non-environmental (i.e. genetics) factors can alter the hosts response to metabolic starvation and inflammation.
Acknowledgements
M Vijay-Kumar is supported by R01 grant from the National Institutes of Health (NIH) [grant number CA219144]. B Joe is supported by NIH R01 grant [grant number HL1430820].
Abbreviation Key:
- 1-MIM-OH
1-methoxy-3-indolylmethyl alcohol
- ACE2
angiotensin converting enzyme 2
- AhR
aryl hydrocarbon receptor
- AMPK
AMP-activiated protein kinase
- ATF
activating transcription factor
- β-OHB
β-hydroxybutryate
- CREB3L3
cAMP responsive element-binding protein 3-like 3
- DAMP
danger associated molecular pattern
- DDR
DNA damage response
- DIM
3,3’-diindolylmethane
- DSS
dextran sulfate sodium
- eIF2α
eukaryotic translation initiator factor 2α
- FGF21
fibroblast growth factor 21
- FMO3
flavin-containing monooxygenase 3
- Fpn
ferroportin
- GCN2
general controlled nonderepressible 2
- HDAC
histone deacetylase
- HFD
high-fat diet
- HIF
hypoxia-inducible transcription factor
- I3C
indole-3-carbinol
- IBD
inflammatory bowel disease
- IDA
iron deficiency anemia
- IL
interleukin
- IRP
iron-regulatory protein
- ISR
integrated stress response
- ITC
isothiocyanate
- KD
ketogenic diet
- LONP1
Lon protease 1
- M1
pro-inflammatory macrophage
- M2
anti-inflammatory macrophage
- mTOR
mammalian target of rapamycin
- NAFLD
non-alcoholic fatty liver disease
- NLRP3
Nod-like receptor family pyrin domain containing 3
- PERK
PKR-like endoplasmic reticulum kinase
- PGC-1α
PPARγ coactivator 1α
- PNALD
parenteral nutrition-associated liver disease
- PPARα
peroxisome proliferator activated receptor α
- ROS
reactive oxygen species
- SCFA
short-chain fatty acid
- SCV
Salmonella-containing vacuole
- S. Typhimurium
Salmonella enterica serovar Typhimurium
- TCA
tricarboxylic acid
- TMA
trimethylamine
- TMAO
trimethylamine-oxide
- TPN
total parenteral nutrition
- WAT
white adipose tissue
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Kerndt PR, Naughton JL, Driscoll CE, Loxterkamp DA. Fasting: the history, pathophysiology and complications. West J Med. 1982;137:379–99. [PMC free article] [PubMed] [Google Scholar]
- [2].Dedkova EN, Blatter LA. Role of beta-hydroxybutyrate, its polymer poly-beta-hydroxybutyrate and inorganic polyphosphate in mammalian health and disease. Front Physiol. 2014;5:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fukao T, Song X-Q, Mitchell GA, Yamaguchi S, Sukegawa K, Or T, et al. Enzymes of Ketone Body Utilization in Human Tissues: Protein and Messenger RNA Levels of Succinyl-Coenzyme A (CoA):3-Ketoacid CoA Transferase and Mitochondrial and Cytosolic Acetoacetyl-CoA Thiolases. Pediatric Research. 1997;42:498–502. [DOI] [PubMed] [Google Scholar]
- [4].Evans M, Cogan KE, Egan B. Metabolism of ketone bodies during exercise and training: physiological basis for exogenous supplementation. J Physiol. 2017;595:2857–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Thewjitcharoen Y, Plianpan P, Chotjirat A, Nakasatien S, Chotwanvirat P, Wanothayaroj E, et al. Clinical characteristics and outcomes of care in adult patients with diabetic ketoacidosis: A retrospective study from a tertiary diabetes center in Thailand. J Clin Transl Endocrinol. 2019;16:100188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Watkins PJ, Hill DM, Fitzgerald MG, Malins JM. Ketonaemia in uncontrolled diabetes mellitus. Br Med J. 1970;4:522–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Carneiro L, Geller S, Hébert A, Repond C, Fioramonti X, Leloup C, et al. Hypothalamic sensing of ketone bodies after prolonged cerebral exposure leads to metabolic control dysregulation. Scientific Reports. 2016;6:34909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mardinoglu A, Wu H, Bjornson E, Zhang C, Hakkarainen A, Rasanen SM, et al. An Integrated Understanding of the Rapid Metabolic Benefits of a Carbohydrate-Restricted Diet on Hepatic Steatosis in Humans. Cell Metab. 2018;27:559–71 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cotter DG, Ercal B, Huang X, Leid JM, d’Avignon DA, Graham MJ, et al. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J Clin Invest. 2014;124:5175–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].D’Andrea Meira I, Romao TT, Pires do Prado HJ, Kruger LT, Pires MEP, da Conceicao PO. Ketogenic Diet and Epilepsy: What We Know So Far. Front Neurosci. 2019;13:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Olson CA, Vuong HE, Yano JM, Liang QY, Nusbaum DJ, Hsiao EY. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell. 2018;173:1728–41 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ma D, Wang AC, Parikh I, Green SJ, Hoffman JD, Chlipala G, et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Scientific Reports. 2018;8:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nagpal R, Neth BJ, Wang S, Craft S, Yadav H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine. 2019;47:529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mukherjee P, Augur ZM, Li M, Hill C, Greenwood B, Domin MA, et al. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Communications Biology. 2019;2:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].van der Louw E, Olieman JF, van den Bemt P, Bromberg JEC, Oomen-de Hoop E, Neuteboom RF, et al. Ketogenic diet treatment as adjuvant to standard treatment of glioblastoma multiforme: a feasibility and safety study. Ther Adv Med Oncol. 2019;11:1758835919853958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mangani D, Weller M, Roth P. The network of immunosuppressive pathways in glioblastoma. Biochem Pharmacol. 2017;130:1–9. [DOI] [PubMed] [Google Scholar]
- [17].Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Crawford PA, Crowley JR, Sambandam N, Muegge BD, Costello EK, Hamady M, et al. Regulation of myocardial ketone body metabolism by the gut microbiota during nutrient deprivation. Proc Natl Acad Sci U S A. 2009;106:11276–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee SS, Chan WY, Lo CK, Wan DC, Tsang DS, Cheung WT. Requirement of PPARalpha in maintaining phospholipid and triacylglycerol homeostasis during energy deprivation. J Lipid Res. 2004;45:2025–37. [DOI] [PubMed] [Google Scholar]
- [20].Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010;468:1100–4. [DOI] [PubMed] [Google Scholar]
- [21].Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–37. [DOI] [PubMed] [Google Scholar]
- [22].Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–25. [DOI] [PubMed] [Google Scholar]
- [23].Nakagawa Y, Satoh A, Tezuka H, Han SI, Takei K, Iwasaki H, et al. CREB3L3 controls fatty acid oxidation and ketogenesis in synergy with PPARalpha. Sci Rep. 2016;6:39182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, et al. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci U S A. 2009;106:10853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Asrih M, Altirriba J, Rohner-Jeanrenaud F, Jornayvaz FR. Ketogenic Diet Impairs FGF21 Signaling and Promotes Differential Inflammatory Responses in the Liver and White Adipose Tissue. PLoS One. 2015;10:e0126364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Garbow JR, Doherty JM, Schugar RC, Travers S, Weber ML, Wentz AE, et al. Hepatic steatosis, inflammation, and ER stress in mice maintained long term on a very low-carbohydrate ketogenic diet. Am J Physiol Gastrointest Liver Physiol. 2011;300:G956–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].He C, Wu Q, Hayashi N, Nakano F, Nakatsukasa E, Tsuduki T. Carbohydrate-restricted diet alters the gut microbiota, promotes senescence and shortens the life span in senescence-accelerated prone mice. J Nutr Biochem. 2019;78:108326. [DOI] [PubMed] [Google Scholar]
- [28].Lee J, Choi J, Scafidi S, Wolfgang MJ. Hepatic Fatty Acid Oxidation Restrains Systemic Catabolism during Starvation. Cell Rep. 2016;16:201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jornayvaz FR, Jurczak MJ, Lee HY, Birkenfeld AL, Frederick DW, Zhang D, et al. A high-fat, ketogenic diet causes hepatic insulin resistance in mice, despite increasing energy expenditure and preventing weight gain. Am J Physiol Endocrinol Metab. 2010;299:E808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fischer T, Och U, Klawon I, Och T, Gruneberg M, Fobker M, et al. Effect of a Sodium and Calcium DL-beta-Hydroxybutyrate Salt in Healthy Adults. J Nutr Metab. 2018;2018:9812806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fischer T, Elpers C, Och U, Fobker M, Marquardt T. Ketone body therapy with D/L-beta-hydroxybutyric acid solution in severe MADD. Mol Genet Metab Rep. 2019;20:100491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978;14:105–13. [DOI] [PubMed] [Google Scholar]
- [35].Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, et al. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl Acad Sci U S A. 2011;108:8030–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Feng Y, Huang Y, Wang Y, Wang P, Song H, Wang F. Antibiotics induced intestinal tight junction barrier dysfunction is associated with microbiota dysbiosis, activated NLRP3 inflammasome and autophagy. PLoS One. 2019;14:e0218384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yao X, Zhang C, Xing Y, Xue G, Zhang Q, Pan F, et al. Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nature Communications. 2017;8:1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–50. [DOI] [PubMed] [Google Scholar]
- [40].Harvey C, Schofield GM, Williden M. The use of nutritional supplements to induce ketosis and reduce symptoms associated with keto-induction: a narrative review. PeerJ. 2018;6:e4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yamanashi T, Iwata M, Kamiya N, Tsunetomi K, Kajitani N, Wada N, et al. Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Scientific Reports. 2017;7:7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wood TR, Stubbs BJ, Juul SE. Exogenous Ketone Bodies as Promising Neuroprotective Agents for Developmental Brain Injury. Dev Neurosci. 2018;40:451–62. [DOI] [PubMed] [Google Scholar]
- [43].Chakraborty S, Galla S, Cheng X, Yeo JY, Mell B, Singh V, et al. Salt-Responsive Metabolite, beta-Hydroxybutyrate, Attenuates Hypertension. Cell Rep. 2018;25:677–89 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wan Z, Wen W, Ren K, Zhou D, Liu J, Wu Y, et al. Involvement of NLRP3 inflammasome in the impacts of sodium and potassium on insulin resistance in normotensive Asians. Br J Nutr. 2018;119:228–37. [DOI] [PubMed] [Google Scholar]
- [45].Keller U, Lustenberger M, Stauffacher W. Effect of insulin on ketone body clearance studied by a ketone body “clamp” technique in normal man. Diabetologia. 1988;31:24–9. [DOI] [PubMed] [Google Scholar]
- [46].Blagosklonny MV. Fasting and rapamycin: diabetes versus benevolent glucose intolerance. Cell Death & Disease. 2019;10:607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017;551:585–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chriett S, Dąbek A, Wojtala M, Vidal H, Balcerczyk A, Pirola L. Prominent action of butyrate over β-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Scientific Reports. 2019;9:742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mandayam S, Mitch WE. Dietary protein restriction benefits patients with chronic kidney disease. Nephrology (Carlton). 2006;11:53–7. [DOI] [PubMed] [Google Scholar]
- [50].Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J Physiol. 2018;596:623–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pak HH, Cummings NE, Green CL, Brinkman JA, Yu D, Tomasiewicz JL, et al. The Metabolic Response to a Low Amino Acid Diet is Independent of Diet-Induced Shifts in the Composition of the Gut Microbiome. Scientific Reports. 2019;9:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li Z, Rasmussen TS, Rasmussen ML, Li J, Henriquez Olguin C, Kot W, et al. The Gut Microbiome on a Periodized Low-Protein Diet Is Associated With Improved Metabolic Health. Front Microbiol. 2019;10:709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fan P, Liu P, Song P, Chen X, Ma X. Moderate dietary protein restriction alters the composition of gut microbiota and improves ileal barrier function in adult pig model. Scientific Reports. 2017;7:43412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Coburn LA, Horst SN, Allaman MM, Brown CT, Williams CS, Hodges ME, et al. L-Arginine Availability and Metabolism Is Altered in Ulcerative Colitis. Inflamm Bowel Dis. 2016;22:1847–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Coburn LA, Gong X, Singh K, Asim M, Scull BP, Allaman MM, et al. L-arginine supplementation improves responses to injury and inflammation in dextran sulfate sodium colitis. PLoS One. 2012;7:e33546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nikolaus S, Schulte B, Al-Massad N, Thieme F, Schulte DM, Bethge J, et al. Increased Tryptophan Metabolism Is Associated With Activity of Inflammatory Bowel Diseases. Gastroenterology. 2017;153:1504–16 e2. [DOI] [PubMed] [Google Scholar]
- [57].Sugihara K, Morhardt TL, Kamada N. The Role of Dietary Nutrients in Inflammatory Bowel Disease. Front Immunol. 2018;9:3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lanis JM, Alexeev EE, Curtis VF, Kitzenberg DA, Kao DJ, Battista KD, et al. Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol. 2017;10:1133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Qi H, Li Y, Yun H, Zhang T, Huang Y, Zhou J, et al. Lactobacillus maintains healthy gut mucosa by producing L-Ornithine. Communications Biology. 2019;2:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012;487:477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pan X, Shao Y, Wu F, Wang Y, Xiong R, Zheng J, et al. FGF21 Prevents Angiotensin II-Induced Hypertension and Vascular Dysfunction by Activation of ACE2/Angiotensin-(1–7) Axis in Mice. Cell Metab. 2018;27:1323–37 e5. [DOI] [PubMed] [Google Scholar]
- [62].Perdomo CM, Fruhbeck G, Escalada J. Impact of Nutritional Changes on Nonalcoholic Fatty Liver Disease. Nutrients. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sarin HV, Lee JH, Jauhiainen M, Joensuu A, Borodulin K, Männistö S, et al. Substantial fat mass loss reduces low-grade inflammation and induces positive alteration in cardiometabolic factors in normal-weight individuals. Scientific Reports. 2019;9:3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Raman M, Almutairdi A, Mulesa L, Alberda C, Beattie C, Gramlich L. Parenteral Nutrition and Lipids. Nutrients. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Fell GL, Nandivada P, Gura KM, Puder M. Intravenous Lipid Emulsions in Parenteral Nutrition. Adv Nutr. 2015;6:600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Calkins KL, Venick RS, Devaskar SU. Complications associated with parenteral nutrition in the neonate. Clin Perinatol. 2014;41:331–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Christensen RD, Henry E, Wiedmeier SE, Burnett J, Lambert DK. Identifying patients, on the first day of life, at high-risk of developing parenteral nutrition-associated liver disease. J Perinatol. 2007;27:284–90. [DOI] [PubMed] [Google Scholar]
- [68].Dahlgren AF, Pan A, Lam V, Gouthro KC, Simpson PM, Salzman NH, et al. Longitudinal changes in the gut microbiome of infants on total parenteral nutrition. Pediatr Res. 2019;86:107–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wan X, Bi J, Gao X, Tian F, Wang X, Li N, et al. Partial Enteral Nutrition Preserves Elements of Gut Barrier Function, Including Innate Immunity, Intestinal Alkaline Phosphatase (IAP) Level, and Intestinal Microbiota in Mice. Nutrients. 2015;7:6294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].El Kasmi KC, Anderson AL, Devereaux MW, Fillon SA, Harris JK, Lovell MA, et al. Toll-like receptor 4-dependent Kupffer cell activation and liver injury in a novel mouse model of parenteral nutrition and intestinal injury. Hepatology. 2012;55:1518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Harris JK, El Kasmi KC, Anderson AL, Devereaux MW, Fillon SA, Robertson CE, et al. Specific microbiome changes in a mouse model of parenteral nutrition associated liver injury and intestinal inflammation. PLoS One. 2014;9:e110396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kaakoush NO. Insights into the Role of Erysipelotrichaceae in the Human Host. Front Cell Infect Microbiol. 2015;5:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jones CJ, Calder PC. Influence of different intravenous lipid emulsions on fatty acid status and laboratory and clinical outcomes in adult patients receiving home parenteral nutrition: A systematic review. Clin Nutr. 2018;37:285–91. [DOI] [PubMed] [Google Scholar]
- [74].Pironi L, Agostini F, Guidetti M. Intravenous lipids in home parenteral nutrition. World Rev Nutr Diet. 2015;112:141–9. [DOI] [PubMed] [Google Scholar]
- [75].Puder M, Valim C, Meisel JA, Le HD, de Meijer VE, Robinson EM, et al. Parenteral fish oil improves outcomes in patients with parenteral nutrition-associated liver injury. Ann Surg. 2009;250:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Calkins KL, DeBarber A, Steiner RD, Flores MJ, Grogan TR, Henning SM, et al. Intravenous Fish Oil and Pediatric Intestinal Failure-Associated Liver Disease: Changes in Plasma Phytosterols, Cytokines, and Bile Acids and Erythrocyte Fatty Acids. JPEN J Parenter Enteral Nutr. 2018;42:633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Calkins KL, Dunn JC, Shew SB, Reyen L, Farmer DG, Devaskar SU, et al. Pediatric intestinal failure-associated liver disease is reversed with 6 months of intravenous fish oil. JPEN J Parenter Enteral Nutr. 2014;38:682–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Yuan F, Wang H, Tian Y, Li Q, He L, Li N, et al. Fish oil alleviated high-fat diet-induced non-alcoholic fatty liver disease via regulating hepatic lipids metabolism and metaflammation: a transcriptomic study. Lipids Health Dis. 2016;15:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Siscovick DS, Barringer TA, Fretts AM, Wu JH, Lichtenstein AH, Costello RB, et al. Omega-3 Polyunsaturated Fatty Acid (Fish Oil) Supplementation and the Prevention of Clinical Cardiovascular Disease: A Science Advisory From the American Heart Association. Circulation. 2017;135:e867–e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Miller JL, Blaszkiewicz M, Beaton C, Johnson CP, Waible S 2nd, Dubois AL, et al. A peroxidized omega-3-enriched polyunsaturated diet leads to adipose and metabolic dysfunction. J Nutr Biochem. 2019;64:50–60. [DOI] [PubMed] [Google Scholar]
- [81].Geng X, Yang B, Li R, Teng T, Ladu MJ, Sun GY, et al. Effects of Docosahexaenoic Acid and Its Peroxidation Product on Amyloid-beta Peptide-Stimulated Microglia. Mol Neurobiol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bi J, Chen C, Sun P, Tan H, Feng F, Shen J. Neuroprotective effect of omega-3 fatty acids on spinal cord injury induced rats. Brain Behav. 2019;9:e01339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Yang B, Li R, Woo T, Browning JD Jr., Song H, Gu Z, et al. Maternal Dietary Docosahexaenoic Acid Alters Lipid Peroxidation Products and (n-3)/(n-6) Fatty Acid Balance in Offspring Mice. Metabolites. 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Xia X, Lei L, Qin W, Wang L, Zhang G, Hu J. GCN2 controls the cellular checkpoint: potential target for regulating inflammation. Cell Death Discovery. 2018;4:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ravindran R, Loebbermann J, Nakaya HI, Khan N, Ma H, Gama L, et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature. 2016;531:523–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wengrod JC, Gardner LB. Cellular adaptation to nutrient deprivation: crosstalk between the mTORC1 and eIF2alpha signaling pathways and implications for autophagy. Cell Cycle. 2015;14:2571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, et al. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010;29:2082–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Onat UI, Yildirim AD, Tufanli O, Cimen I, Kocaturk B, Veli Z, et al. Intercepting the Lipid-Induced Integrated Stress Response Reduces Atherosclerosis. J Am Coll Cardiol. 2019;73:1149–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Gameiro PA, Struhl K. Nutrient Deprivation Elicits a Transcriptional and Translational Inflammatory Response Coupled to Decreased Protein Synthesis. Cell Rep. 2018;24:1415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Peuhkuri K, Vapaatalo H, Korpela R. Even low-grade inflammation impacts on small intestinal function. World J Gastroenterol. 2010;16:1057–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wek RC, Ramirez M, Jackson BM, Hinnebusch AG. Identification of positive-acting domains in GCN2 protein kinase required for translational activation of GCN4 expression. Mol Cell Biol. 1990;10:2820–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule-Smith A, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324:1334–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zhang H, Hua R, Zhang B, Zhang X, Yang H, Zhou X. Serine Alleviates Dextran Sulfate Sodium-Induced Colitis and Regulates the Gut Microbiota in Mice. Front Microbiol. 2018;9:3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Nishi H, Yamanaka D, Kamei H, Goda Y, Kumano M, Toyoshima Y, et al. Importance of Serum Amino Acid Profile for Induction of Hepatic Steatosis under Protein Malnutrition. Scientific Reports. 2018;8:5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Durden DL, Salazar AM, Distasio JA. Kinetic analysis of hepatotoxicity associated with antineoplastic asparaginases. Cancer Res. 1983;43:1602–5. [PubMed] [Google Scholar]
- [96].Bunpo P, Dudley A, Cundiff JK, Cavener DR, Wek RC, Anthony TG. GCN2 protein kinase is required to activate amino acid deprivation responses in mice treated with the anti-cancer agent L-asparaginase. J Biol Chem. 2009;284:32742–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wilson GJ, Bunpo P, Cundiff JK, Wek RC, Anthony TG. The eukaryotic initiation factor 2 kinase GCN2 protects against hepatotoxicity during asparaginase treatment. Am J Physiol Endocrinol Metab. 2013;305:E1124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Wilson GJ, Lennox BA, She P, Mirek ET, Al Baghdadi RJ, Fusakio ME, et al. GCN2 is required to increase fibroblast growth factor 21 and maintain hepatic triglyceride homeostasis during asparaginase treatment. Am J Physiol Endocrinol Metab. 2015;308:E283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].De Sousa-Coelho AL, Relat J, Hondares E, Perez-Marti A, Ribas F, Villarroya F, et al. FGF21 mediates the lipid metabolism response to amino acid starvation. J Lipid Res. 2013;54:1786–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Guo F, Cavener DR. The GCN2 eIF2alpha kinase regulates fatty-acid homeostasis in the liver during deprivation of an essential amino acid. Cell Metab. 2007;5:103–14. [DOI] [PubMed] [Google Scholar]
- [101].Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC, et al. FGF21 is an endocrine signal of protein restriction. J Clin Invest. 2014;124:3913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].De Sousa-Coelho AL, Marrero PF, Haro D. Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem J. 2012;443:165–71. [DOI] [PubMed] [Google Scholar]
- [103].Liu S, Yuan J, Yue W, Bi Y, Shen X, Gao J, et al. GCN2 deficiency protects against high fat diet induced hepatic steatosis and insulin resistance in mice. Biochim Biophys Acta Mol Basis Dis. 2018;1864:3257–67. [DOI] [PubMed] [Google Scholar]
- [104].Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16:396–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell. 2008;15:829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med. 2009;15:1383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Pryde KR, Taanman JW, Schapira AH. A LON-ClpP Proteolytic Axis Degrades Complex I to Extinguish ROS Production in Depolarized Mitochondria. Cell Rep. 2016;17:2522–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Goto-Yamada S, Mano S, Nakamori C, Kondo M, Yamawaki R, Kato A, et al. Chaperone and protease functions of LON protease 2 modulate the peroxisomal transition and degradation with autophagy. Plant Cell Physiol. 2014;55:482–96. [DOI] [PubMed] [Google Scholar]
- [111].Bezawork-Geleta A, Brodie EJ, Dougan DA, Truscott KN. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Scientific Reports. 2015;5:17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Janeiro MH, Ramirez MJ, Milagro FI, Martinez JA, Solas M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Chen S, Henderson A, Petriello MC, Romano KA, Gearing M, Miao J, et al. Trimethylamine N-Oxide Binds and Activates PERK to Promote Metabolic Dysfunction. Cell Metab. 2019;30:1141–51 e5. [DOI] [PubMed] [Google Scholar]
- [114].Miao J, Ling AV, Manthena PV, Gearing ME, Graham MJ, Crooke RM, et al. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat Commun. 2015;6:6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell. 2015;163:1585–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Randrianarisoa E, Lehn-Stefan A, Wang X, Hoene M, Peter A, Heinzmann SS, et al. Relationship of Serum Trimethylamine N-Oxide (TMAO) Levels with early Atherosclerosis in Humans. Scientific Reports. 2016;6:26745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Chieppa M, Giannelli G. Immune Cells and Microbiota Response to Iron Starvation. Front Med (Lausanne). 2018;5:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Saunders AV, Craig WJ, Baines SK, Posen JS. Iron and vegetarian diets. Med J Aust. 2013;199:S11–6. [DOI] [PubMed] [Google Scholar]
- [119].Ma Q, Kim EY, Lindsay EA, Han O. Bioactive dietary polyphenols inhibit heme iron absorption in a dose-dependent manner in human intestinal Caco-2 cells. J Food Sci. 2011;76:H143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Trumbo P, Yates AA, Schlicker S, Poos M. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101:294–301. [DOI] [PubMed] [Google Scholar]
- [121].Mobarra N, Shanaki M, Ehteram H, Nasiri H, Sahmani M, Saeidi M, et al. A Review on Iron Chelators in Treatment of Iron Overload Syndromes. Int J Hematol Oncol Stem Cell Res. 2016;10:239–47. [PMC free article] [PubMed] [Google Scholar]
- [122].Gozzelino R, Arosio P. Iron Homeostasis in Health and Disease. Int J Mol Sci. 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Hennigar SR, McClung JP. Nutritional Immunity: Starving Pathogens of Trace Minerals. Am J Lifestyle Med. 2016;10:170–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Lim D, Kim KS, Jeong JH, Marques O, Kim HJ, Song M, et al. The hepcidin-ferroportin axis controls the iron content of Salmonella-containing vacuoles in macrophages. Nat Commun. 2018;9:2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Nairz M, Ferring-Appel D, Casarrubea D, Sonnweber T, Viatte L, Schroll A, et al. Iron Regulatory Proteins Mediate Host Resistance to Salmonella Infection. Cell Host Microbe. 2015;18:254–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Golonka R, Yeoh BS, Vijay-Kumar M. The Iron Tug-of-War between Bacterial Siderophores and Innate Immunity. J Innate Immun. 2019;11:249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Behnsen J, Jellbauer S, Wong CP, Edwards RA, George MD, Ouyang W, et al. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity. 2014;40:262–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Forbester JL, Lees EA, Goulding D, Forrest S, Yeung A, Speak A, et al. Interleukin-22 promotes phagolysosomal fusion to induce protection against Salmonella enterica Typhimurium in human epithelial cells. Proc Natl Acad Sci U S A. 2018;115:10118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Pantopoulos K, Hentze MW. Rapid responses to oxidative stress mediated by iron regulatory protein. EMBO J. 1995;14:2917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Pantopoulos K, Hentze MW. Activation of iron regulatory protein-1 by oxidative stress in vitro. Proceedings of the National Academy of Sciences. 1998;95:10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest. 2007;117:1926–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe. 2015;17:662–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Das NK, Schwartz AJ, Barthel G, Inohara N, Liu Q, Sankar A, et al. Microbial Metabolite Signaling Is Required for Systemic Iron Homeostasis. Cell Metab. 2020;31:115–30 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Mastrogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, Peyssonnaux C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest. 2009;119:1159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Taylor M, Qu A, Anderson ER, Matsubara T, Martin A, Gonzalez FJ, et al. Hypoxia-inducible factor-2alpha mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology. 2011;140:2044–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Schwartz AJ, Converso-Baran K, Michele DE, Shah YM. A genetic mouse model of severe iron deficiency anemia reveals tissue-specific transcriptional stress responses and cardiac remodeling. J Biol Chem. 2019;294:14991–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Mahalhal A, Williams JM, Johnson S, Ellaby N, Duckworth CA, Burkitt MD, et al. Oral iron exacerbates colitis and influences the intestinal microbiome. PLoS One. 2018;13:e0202460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Lee T, Clavel T, Smirnov K, Schmidt A, Lagkouvardos I, Walker A, et al. Oral versus intravenous iron replacement therapy distinctly alters the gut microbiota and metabolome in patients with IBD. Gut. 2017;66:863–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Martin R, Miquel S, Benevides L, Bridonneau C, Robert V, Hudault S, et al. Functional Characterization of Novel Faecalibacterium prausnitzii Strains Isolated from Healthy Volunteers: A Step Forward in the Use of F. prausnitzii as a Next-Generation Probiotic. Front Microbiol. 2017;8:1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Zhou L, Zhang M, Wang Y, Dorfman RG, Liu H, Yu T, et al. Faecalibacterium prausnitzii Produces Butyrate to Maintain Th17/Treg Balance and to Ameliorate Colorectal Colitis by Inhibiting Histone Deacetylase 1. Inflamm Bowel Dis. 2018. [DOI] [PubMed] [Google Scholar]
- [142].Ze X, Duncan SH, Louis P, Flint HJ. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012;6:1535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Cerqueira FM, Photenhauer AL, Pollet RM, Brown HA, Koropatkin NM. Starch Digestion by Gut Bacteria: Crowdsourcing for Carbs. Trends Microbiol. 2019. [DOI] [PubMed] [Google Scholar]
- [144].Geirnaert A, Calatayud M, Grootaert C, Laukens D, Devriese S, Smagghe G, et al. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Scientific Reports. 2017;7:11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Zhang Q, Wu Y, Wang J, Wu G, Long W, Xue Z, et al. Accelerated dysbiosis of gut microbiota during aggravation of DSS-induced colitis by a butyrate-producing bacterium. Sci Rep. 2016;6:27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Singh V, Yeoh BS, Vijay-Kumar M. Feed your gut with caution! Transl Cancer Res. 2016;5:S507–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Stein J, Haas JS, Ong SH, Borchert K, Hardt T, Lechat E, et al. Oral versus intravenous iron therapy in patients with inflammatory bowel disease and iron deficiency with and without anemia in Germany - a real-world evidence analysis. Clinicoecon Outcomes Res. 2018;10:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Cray C, Zaias J, Altman NH. Acute phase response in animals: a review. Comp Med. 2009;59:517–26. [PMC free article] [PubMed] [Google Scholar]
- [149].Soares-Mota M, Silva TA, Gomes LM, Pinto MA, Mendonca LM, Farias ML, et al. High prevalence of vitamin A deficiency in Crohn’s disease patients according to serum retinol levels and the relative dose-response test. World J Gastroenterol. 2015;21:1614–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Reifen R, Levy E, Berkovich Z, Tirosh O. Vitamin A exerts its antiinflammatory activities in colitis through preservation of mitochondrial activity. Nutrition. 2015;31:1402–7. [DOI] [PubMed] [Google Scholar]
- [151].Okayasu I, Hana K, Nemoto N, Yoshida T, Saegusa M, Yokota-Nakatsuma A, et al. Vitamin A Inhibits Development of Dextran Sulfate Sodium-Induced Colitis and Colon Cancer in a Mouse Model. Biomed Res Int. 2016;2016:4874809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Iwata M, Yokota A. Retinoic acid production by intestinal dendritic cells. Vitam Horm. 2011;86:127–52. [DOI] [PubMed] [Google Scholar]
- [153].Penny HL, Prestwood TR, Bhattacharya N, Sun F, Kenkel JA, Davidson MG, et al. Restoring Retinoic Acid Attenuates Intestinal Inflammation and Tumorigenesis in APCMin/+ Mice. Cancer Immunol Res. 2016;4:917–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Tejon G, Manriquez V, De Calisto J, Flores-Santibanez F, Hidalgo Y, Crisostomo N, et al. Vitamin A Impairs the Reprogramming of Tregs into IL-17-Producing Cells during Intestinal Inflammation. Biomed Res Int. 2015;2015:137893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Grizotte-Lake M, Zhong G, Duncan K, Kirkwood J, Iyer N, Smolenski I, et al. Commensals Suppress Intestinal Epithelial Cell Retinoic Acid Synthesis to Regulate Interleukin-22 Activity and Prevent Microbial Dysbiosis. Immunity. 2018;49:1103–15 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Han L, Liu Y, Lu M, Wang H, Tang F. Retinoic acid modulates iron metabolism imbalance in anemia of inflammation induced by LPS via reversely regulating hepcidin and ferroportin expression. Biochem Biophys Res Commun. 2018;507:280–5. [DOI] [PubMed] [Google Scholar]
- [157].He C, Deng J, Hu X, Zhou S, Wu J, Xiao D, et al. Vitamin A inhibits the action of LPS on the intestinal epithelial barrier function and tight junction proteins. Food Funct. 2019;10:1235–42. [DOI] [PubMed] [Google Scholar]
- [158].Karkeni E, Bonnet L, Astier J, Couturier C, Dalifard J, Tourniaire F, et al. All-trans-retinoic acid represses chemokine expression in adipocytes and adipose tissue by inhibiting NF-kappaB signaling. J Nutr Biochem. 2017;42:101–7. [DOI] [PubMed] [Google Scholar]
- [159].Gregor MF, Hotamisligil GS. Inflammatory Mechanisms in Obesity. Annual Review of Immunology. 2011;29:415–45. [DOI] [PubMed] [Google Scholar]
- [160].Kim SJ, Choi Y, Choi YH, Park T. Obesity activates toll-like receptor-mediated proinflammatory signaling cascades in the adipose tissue of mice. J Nutr Biochem. 2012;23:113–22. [DOI] [PubMed] [Google Scholar]
- [161].He M, Bianchi ME, Coleman TR, Tracey KJ, Al-Abed Y. Exploring the biological functional mechanism of the HMGB1/TLR4/MD-2 complex by surface plasmon resonance. Molecular Medicine. 2018;24:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Ghosh AR, Bhattacharya R, Bhattacharya S, Nargis T, Rahaman O, Duttagupta P, et al. Adipose Recruitment and Activation of Plasmacytoid Dendritic Cells Fuel Metaflammation. Diabetes. 2016;65:3440–52. [DOI] [PubMed] [Google Scholar]
- [163].Zhang H, Xue R, Zhu S, Fu S, Chen Z, Zhou R, et al. M2-specific reduction of CD1d switches NKT cell-mediated immune responses and triggers metaflammation in adipose tissue. Cell Mol Immunol. 2018;15:506–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Theurich S, Tsaousidou E, Hanssen R, Lempradl AM, Mauer J, Timper K, et al. IL-6/Stat3-Dependent Induction of a Distinct, Obesity-Associated NK Cell Subpopulation Deteriorates Energy and Glucose Homeostasis. Cell Metab. 2017;26:171–84 e6. [DOI] [PubMed] [Google Scholar]
- [165].Sanyal A, Naumann J, Hoffmann LS, Chabowska-Kita A, Ehrlund A, Schlitzer A, et al. Interplay between Obesity-Induced Inflammation and cGMP Signaling in White Adipose Tissue. Cell Rep. 2017;18:225–36. [DOI] [PubMed] [Google Scholar]
- [166].Liu L, Inouye KE, Allman WR, Coleman AS, Siddiqui S, Hotamisligil GS, et al. TACI-Deficient Macrophages Protect Mice Against Metaflammation and Obesity-Induced Dysregulation of Glucose Homeostasis. Diabetes. 2018;67:1589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Nishimoto S, Fukuda D, Higashikuni Y, Tanaka K, Hirata Y, Murata C, et al. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci Adv. 2016;2:e1501332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Jais A, Einwallner E, Sharif O, Gossens K, Lu TT, Soyal SM, et al. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell. 2014;158:25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nature Reviews Endocrinology. 2018;14:576–90. [DOI] [PubMed] [Google Scholar]
- [170].Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and ‘Garb-aging’. Trends Endocrinol Metab. 2017;28:199–212. [DOI] [PubMed] [Google Scholar]
- [171].Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542:177–85. [DOI] [PubMed] [Google Scholar]
- [172].Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–72. [DOI] [PubMed] [Google Scholar]
- [173].Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. 2013;45:e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. 2013;182:375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Caesar R, Tremaroli V, Kovatcheva-Datchary P, Cani PD, Backhed F. Crosstalk between Gut Microbiota and Dietary Lipids Aggravates WAT Inflammation through TLR Signaling. Cell Metab. 2015;22:658–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Napier BA, Andres-Terre M, Massis LM, Hryckowian AJ, Higginbottom SK, Cumnock K, et al. Western diet regulates immune status and the response to LPS-driven sepsis independent of diet-associated microbiome. Proc Natl Acad Sci U S A. 2019;116:3688–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Everard A, Geurts L, Caesar R, Van Hul M, Matamoros S, Duparc T, et al. Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat Commun. 2014;5:5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Luche E, Cousin B, Garidou L, Serino M, Waget A, Barreau C, et al. Metabolic endotoxemia directly increases the proliferation of adipocyte precursors at the onset of metabolic diseases through a CD14-dependent mechanism. Mol Metab. 2013;2:281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Pang S, Tang H, Zhuo S, Zang YQ, Le Y. Regulation of fasting fuel metabolism by toll-like receptor 4. Diabetes. 2010;59:3041–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Lu P, Sodhi CP, Yamaguchi Y, Jia H, Prindle T Jr., Fulton WB, et al. Intestinal epithelial Toll-like receptor 4 prevents metabolic syndrome by regulating interactions between microbes and intestinal epithelial cells in mice. Mucosal Immunol. 2018;11:727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–81. [DOI] [PubMed] [Google Scholar]
- [183].Xue L, He J, Gao N, Lu X, Li M, Wu X, et al. Probiotics may delay the progression of nonalcoholic fatty liver disease by restoring the gut microbiota structure and improving intestinal endotoxemia. Sci Rep. 2017;7:45176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Ghadimi D, Yoness Hassan MF, Folster-Holst R, Rocken C, Ebsen M, de Vrese M, et al. Regulation of hepcidin/iron-signalling pathway interactions by commensal bifidobateria plays an important role for the inhibition of metaflammation-related biomarkers. Immunobiology. 2019. [DOI] [PubMed] [Google Scholar]
- [185].Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Kumar D, Shankar K, Patel S, Gupta A, Varshney S, Gupta S, et al. Chronic hyperinsulinemia promotes meta-inflammation and extracellular matrix deposition in adipose tissue: Implications of nitric oxide. Mol Cell Endocrinol. 2018;477:15–28. [DOI] [PubMed] [Google Scholar]
- [187].Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe. 2017;21:455–66 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Biagi E, Franceschi C, Rampelli S, Severgnini M, Ostan R, Turroni S, et al. Gut Microbiota and Extreme Longevity. Curr Biol. 2016;26:1480–5. [DOI] [PubMed] [Google Scholar]
- [189].Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4586–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Fransen F, van Beek AA, Borghuis T, Aidy SE, Hugenholtz F, van der Gaast-de Jongh C, et al. Aged Gut Microbiota Contributes to Systemical Inflammaging after Transfer to Germ-Free Mice. Front Immunol. 2017;8:1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Kundu P, Lee HU, Garcia-Perez I, Tay EXY, Kim H, Faylon LE, et al. Neurogenesis and prolongevity signaling in young germ-free mice transplanted with the gut microbiota of old mice. Sci Transl Med. 2019;11. [DOI] [PubMed] [Google Scholar]
- [192].Vemuri R, Gundamaraju R, Shinde T, Perera AP, Basheer W, Southam B, et al. Lactobacillus acidophilus DDS-1 Modulates Intestinal-Specific Microbiota, Short-Chain Fatty Acid and Immunological Profiles in Aging Mice. Nutrients. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med. 2017;23:107–13. [DOI] [PubMed] [Google Scholar]
- [194].Narbad A, Rossiter JT. Gut Glucosinolate Metabolism and Isothiocyanate Production. Mol Nutr Food Res. 2018;62:e1700991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Tian S, Liu X, Lei P, Zhang X, Shan Y. Microbiota: a mediator to transform glucosinolate precursors in cruciferous vegetables to the active isothiocyanates. J Sci Food Agric. 2018;98:1255–60. [DOI] [PubMed] [Google Scholar]
- [196].Hanschen FS, Klopsch R, Oliviero T, Schreiner M, Verkerk R, Dekker M. Optimizing isothiocyanate formation during enzymatic glucosinolate breakdown by adjusting pH value, temperature and dilution in Brassica vegetables and Arabidopsis thaliana. Scientific Reports. 2017;7:40807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Baenas N, Marhuenda J, Garcia-Viguera C, Zafrilla P, Moreno DA. Influence of Cooking Methods on Glucosinolates and Isothiocyanates Content in Novel Cruciferous Foods. Foods. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Fofaria NM, Ranjan A, Kim SH, Srivastava SK. Mechanisms of the Anticancer Effects of Isothiocyanates. Enzymes. 2015;37:111–37. [DOI] [PubMed] [Google Scholar]
- [199].Liu X, Wang Y, Hoeflinger JL, Neme BP, Jeffery EH, Miller MJ. Dietary Broccoli Alters Rat Cecal Microbiota to Improve Glucoraphanin Hydrolysis to Bioactive Isothiocyanates. Nutrients. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Wu Y, Shen Y, Zhu Y, Mupunga J, Zou L, Liu C, et al. Broccoli ingestion increases the glucosinolate hydrolysis activity of microbiota in the mouse gut. Int J Food Sci Nutr. 2019;70:585–94. [DOI] [PubMed] [Google Scholar]
- [201].Budnowski J, Hanske L, Schumacher F, Glatt H, Platz S, Rohn S, et al. Glucosinolates Are Mainly Absorbed Intact in Germfree and Human Microbiota-Associated Mice. J Agric Food Chem. 2015;63:8418–28. [DOI] [PubMed] [Google Scholar]
- [202].Kaczmarek JL, Liu X, Charron CS, Novotny JA, Jeffery EH, Seifried HE, et al. Broccoli consumption affects the human gastrointestinal microbiota. J Nutr Biochem. 2019;63:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Kellingray L, Tapp HS, Saha S, Doleman JF, Narbad A, Mithen RF. Consumption of a diet rich in Brassica vegetables is associated with a reduced abundance of sulphate-reducing bacteria: A randomised crossover study. Mol Nutr Food Res. 2017;61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Tanaka S, Yamamoto K, Yamada K, Furuya K, Uyeno Y. Relationship of Enhanced Butyrate Production by Colonic Butyrate-Producing Bacteria to Immunomodulatory Effects in Normal Mice Fed an Insoluble Fraction of Brassica rapa L. Appl Environ Microbiol. 2016;82:2693–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].He C, Huang L, Lei P, Liu X, Li B, Shan Y. Sulforaphane Normalizes Intestinal Flora and Enhances Gut Barrier in Mice with BBN-Induced Bladder Cancer. Mol Nutr Food Res. 2018;62:e1800427. [DOI] [PubMed] [Google Scholar]
- [206].Popolo A, Pinto A, Daglia M, Nabavi SF, Farooqi AA, Rastrelli L. Two likely targets for the anti-cancer effect of indole derivatives from cruciferous vegetables: PI3K/Akt/mTOR signalling pathway and the aryl hydrocarbon receptor. Semin Cancer Biol. 2017;46:132–7. [DOI] [PubMed] [Google Scholar]
- [207].Busbee PB, Menzel L, Alrafas HR, Dopkins N, Becker W, Miranda K, et al. Indole-3-carbinol prevents colitis and associated microbial dysbiosis in an IL-22-dependent manner. JCI Insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Liu Y, She W, Wang F, Li J, Wang J, Jiang W. 3, 3’-Diindolylmethane alleviates steatosis and the progression of NASH partly through shifting the imbalance of Treg/Th17 cells to Treg dominance. Int Immunopharmacol. 2014;23:489–98. [DOI] [PubMed] [Google Scholar]
- [209].Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci U S A. 1991;88:9543–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147:629–40. [DOI] [PubMed] [Google Scholar]
- [211].Schumacher F, Florian S, Schnapper A, Monien BH, Mewis I, Schreiner M, et al. A secondary metabolite of Brassicales, 1-methoxy-3-indolylmethyl glucosinolate, as well as its degradation product, 1-methoxy-3-indolylmethyl alcohol, forms DNA adducts in the mouse, but in varying tissues and cells. Arch Toxicol. 2014;88:823–36. [DOI] [PubMed] [Google Scholar]
- [212].Gronke K, Hernandez PP, Zimmermann J, Klose CSN, Kofoed-Branzk M, Guendel F, et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature. 2019;566:249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Wassermann B, Rybakova D, Müller C, Berg G. Harnessing the microbiomes of Brassica vegetables for health issues. Scientific Reports. 2017;7:17649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Gibson GR, Hutkins R, Sanders ME, Prescott SL, Reimer RA, Salminen SJ, et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat Rev Gastroenterol Hepatol. 2017;14:491–502. [DOI] [PubMed] [Google Scholar]
- [215].Davani-Davari D, Negahdaripour M, Karimzadeh I, Seifan M, Mohkam M, Masoumi SJ, et al. Prebiotics: Definition, Types, Sources, Mechanisms, and Clinical Applications. Foods. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Baxter NT, Schmidt AW, Venkataraman A, Kim KS, Waldron C, Schmidt TM. Dynamics of Human Gut Microbiota and Short-Chain Fatty Acids in Response to Dietary Interventions with Three Fermentable Fibers. mBio. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Li LL, Wang YT, Zhu LM, Liu ZY, Ye CQ, Qin S. Inulin with different degrees of polymerization protects against diet-induced endotoxemia and inflammation in association with gut microbiota regulation in mice. Sci Rep. 2020;10:978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Vandeputte D, Falony G, Vieira-Silva S, Wang J, Sailer M, Theis S, et al. Prebiotic inulin-type fructans induce specific changes in the human gut microbiota. Gut. 2017;66:1968–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Costabile A, Kolida S, Klinder A, Gietl E, Bauerlein M, Frohberg C, et al. A double-blind, placebo-controlled, cross-over study to establish the bifidogenic effect of a very-long-chain inulin extracted from globe artichoke (Cynara scolymus) in healthy human subjects. Br J Nutr. 2010;104:1007–17. [DOI] [PubMed] [Google Scholar]
- [220].Zou J, Chassaing B, Singh V, Pellizzon M, Ricci M, Fythe MD, et al. Fiber-Mediated Nourishment of Gut Microbiota Protects against Diet-Induced Obesity by Restoring IL-22-Mediated Colonic Health. Cell Host Microbe. 2018;23:41–53 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Schroeder BO, Birchenough GMH, Stahlman M, Arike L, Johansson MEV, Hansson GC, et al. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe. 2018;23:27–40 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Chassaing B, Miles-Brown J, Pellizzon M, Ulman E, Ricci M, Zhang L, et al. Lack of soluble fiber drives diet-induced adiposity in mice. Am J Physiol Gastrointest Liver Physiol. 2015;309:G528–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Choque Delgado GT, Tamashiro W. Role of prebiotics in regulation of microbiota and prevention of obesity. Food Res Int. 2018;113:183–8. [DOI] [PubMed] [Google Scholar]
- [224].Golonka RM, Yeoh BS, Vijay-Kumar M. Author Correction: Dietary Additives and Supplements Revisited: the Fewer, the Safer for Gut and Liver Health. Current Pharmacology Reports. 2019;5:317-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Singh V, Yeoh BS, Walker RE, Xiao X, Saha P, Golonka RM, et al. Microbiota fermentation-NLRP3 axis shapes the impact of dietary fibres on intestinal inflammation. Gut. 2019;68:1801–12. [DOI] [PubMed] [Google Scholar]
- [226].Singh V, Yeoh BS, Chassaing B, Xiao X, Saha P, Aguilera Olvera R, et al. Dysregulated Microbial Fermentation of Soluble Fiber Induces Cholestatic Liver Cancer. Cell. 2018;175:679–94 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Peterson CT, Sharma V, Uchitel S, Denniston K, Chopra D, Mills PJ, et al. Prebiotic Potential of Herbal Medicines Used in Digestive Health and Disease. J Altern Complement Med. 2018;24:656–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Li D, Wang P, Wang P, Hu X, Chen F. Gut microbiota promotes production of aromatic metabolites through degradation of barley leaf fiber. J Nutr Biochem. 2018;58:49–58. [DOI] [PubMed] [Google Scholar]
- [229].Wang JH, Bose S, Kim HG, Han KS, Kim H. Fermented Rhizoma Atractylodis Macrocephalae alleviates high fat diet-induced obesity in association with regulation of intestinal permeability and microbiota in rats. Sci Rep. 2015;5:8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Wang JH, Bose S, Kim GC, Hong SU, Kim JH, Kim JE, et al. Flos Lonicera ameliorates obesity and associated endotoxemia in rats through modulation of gut permeability and intestinal microbiota. PLoS One. 2014;9:e86117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Sheng Y, Zheng S, Ma T, Zhang C, Ou X, He X, et al. Mulberry leaf alleviates streptozotocin-induced diabetic rats by attenuating NEFA signaling and modulating intestinal microflora. Scientific Reports. 2017;7:12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Seo DB, Jeong HW, Cho D, Lee BJ, Lee JH, Choi JY, et al. Fermented green tea extract alleviates obesity and related complications and alters gut microbiota composition in diet-induced obese mice. J Med Food. 2015;18:549–56. [DOI] [PubMed] [Google Scholar]
- [233].Chang C-J, Lin C-S, Lu C-C, Martel J, Ko Y-F, Ojcius DM, et al. Ganoderma lucidum reduces obesity in mice by modulating the composition of the gut microbiota. Nature Communications. 2015;6:7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Liao Z, Xie Y, Zhou B, Zou B, Xiao D, Liu W, et al. Berberine ameliorates colonic damage accompanied with the modulation of dysfunctional bacteria and functions in ulcerative colitis rats. Appl Microbiol Biotechnol. 2020;104:1737–49. [DOI] [PubMed] [Google Scholar]
- [235].Li C, Ai G, Wang Y, Lu Q, Luo C, Tan L, et al. Oxyberberine, a novel gut microbiota-mediated metabolite of berberine, possesses superior anti-colitis effect: Impact on intestinal epithelial barrier, gut microbiota profile and TLR4-MyD88-NF-kappaB pathway. Pharmacol Res. 2019;152:104603. [DOI] [PubMed] [Google Scholar]
- [236].Liu Y, Liu X, Hua W, Wei Q, Fang X, Zhao Z, et al. Berberine inhibits macrophage M1 polarization via AKT1/SOCS1/NF-kappaB signaling pathway to protect against DSS-induced colitis. Int Immunopharmacol. 2018;57:121–31. [DOI] [PubMed] [Google Scholar]
- [237].Liu D, Zhang Y, Liu Y, Hou L, Li S, Tian H, et al. Berberine Modulates Gut Microbiota and Reduces Insulin Resistance via the TLR4 Signaling Pathway. Exp Clin Endocrinol Diabetes. 2018;126:513–20. [DOI] [PubMed] [Google Scholar]
- [238].Liu CS, Zheng YR, Zhang YF, Long XY. Research progress on berberine with a special focus on its oral bioavailability. Fitoterapia. 2016;109:274–82. [DOI] [PubMed] [Google Scholar]