

Abstract
Purpose of review
Diffuse intrinsic pontine glioma (DIPG) is a fatal childhood brainstem malignancy. Despite advances in understanding of the molecular underpinnings of the tumor in the past decade, the dismal prognosis of DIPG has thus far remained unchanged. This review seeks to highlight promising therapeutic targets within three arenas: DIPG cell-intrinsic vulnerabilities, immunotherapeutic approaches to tumor clearance, and microenvironmental dependencies that promote tumor growth.
Recent findings
Promising therapeutic strategies from recent studies include epigenetic modifying agents such as histone deacetylase inhibitors, bromodomain and extra-terminal motif (BET) protein inhibitors, and CDK7 inhibitors. Tumor-specific immunotherapies are emerging. Key interactions between DIPG and normal brain cells are coming to light, and targeting critical microenvironmental mechanisms driving DIPG growth in the developing childhood brain represents a new direction for therapy.
Summary
Several DIPG treatment strategies are being evaluated in early clinical trials. Ultimately, we suspect that a multifaceted therapeutic approach utilizing cell-intrinsic, microenvironmental, and immunotherapeutic targets will be necessary for eradicating DIPG.
Keywords: diffuse intrinsic pontine glioma, epigenetics, H3K27M, immunotherapy, microenvironment
INTRODUCTION
Diffuse intrinsic pontine glioma (DIPG) is a devastating childhood brainstem tumor and a leading cause of pediatric brain tumor-related death. The median survival of DIPG is 9–11 months with a 99% 5-year mortality [1,2,3■■]. DIPG is classified as a subtype of diffuse midline glioma, differentiated by its origin in the pons. The tumor’s dismal prognosis arises in part due to this origination in the brainstem where many critical nervous system functions such as respiration are regulated, thus rendering surgical tumor resection impossible. In addition, DIPG diffusely infiltrates pontine tissue and often invades into more distant brain regions [4,5■]. DIPG resistance to conventional chemotherapeutic agents used for brain tumors, such as temozolomide, has added to the challenge of even moderately effective treatment [6,7]. Radiotherapy is presently the sole treatment that has demonstrated clinical efficacy in prolonging life, conferring an average of 3 months survival benefit; the median survival is only 6 months without standard radio-therapy. DIPG still remains universally fatal.
In the past decade, studies of tumor biopsy and autopsy samples have vastly increased our understanding of DIPG’s underlying molecular pathogenesis [8]. Eighty-percent of DIPG tumors and a high percentage of gliomas in other midline structures such as thalamus and spinal cord were found to harbor lysine-to-methionine substitutions (K27M) in genes encoding histone H3 [9–11] - these findings mark the first known associations between histone mutations and cancer. The findings that histone mutations delineated a tumor subgroup with unique pathophysiology and prognosis led to the reclassification of H3K27M-mutated DIPG in the 2016 WHO classification of tumors of the central nervous system as diffuse midline glioma with K27M mutation, a shift from the previous anatomical and histological classifications [12]. The H3K27M histone mutation results in dysfunction of the polycomb repressive complex-2 (PRC2) methyltransferase complex and global hypomethylation of the lysine at position 27 of the H3 protein (H3K27), with consequent dysregulation of gene expression [11,13–15]. In DIPG, this broad disruption of epigenetic regulation fosters oncogenesis, in some cases with secondary associated mutations in classical oncogenic pathways [13,16–18]. Recent advances from patient-derived DIPG cell cultures and orthotopic xenograft models have identified promising drug targets that are being investigated further in ongoing preclinical and clinical trials [19,20■]. However, the complexity of DIPG molecular pathogenesis and resistance to conventional therapeutics highlights the necessity of multipronged approaches to DIPG treatment to improve outcomes. Here, we discuss a triad of emerging therapeutic strategies - targeting cell intrinsic vulnerabilities, immunotherapy targeting tumor-specific antigens, and targeting glioma-promoting interactions in the brain microenvironment - and hypothesize that exploiting DIPG vulnerabilities in all three arenas will be necessary to eradicate this devastating tumor.
DEVELOPMENTAL ORIGINS
DIPG arises in a specific spatio-temporal pattern, typically during middle childhood, suggesting tumor cells arise from dysregulation of a normal neurodevelopmental process. Thus, the formation of DIPG is likely dependent on both a susceptible cell of origin and microenvironmental signaling that promotes or enables tumor formation. The histological findings of enrichment of an early oligodendroglial precursor cell at the age and location of DIPG origination (ventral pons) first suggested that the tumor may arise from precursor cells in the oligodendroglial lineage [21], cells that are engaged in myelin development throughout childhood and adolescence. Recent transcriptional and chromatin landscape studies demonstrate that oligodendroglial lineage genes are activated at both the epigenetic and transcriptional levels in DIPG [20■■,22,23]. In addition, DIPG single cell sequencing studies reveal that the proliferative stem-like cells of primary DIPG tumors closely resemble oligodendroglial precursors; this oligodendrocyte precursor cell (OPC)-like, self-renewing population of malignant cells represents the tumor-initiating cell (or ‘cancer stem cell’) population in DIPG [24■■]. Several excellent studies have examined the effect of H3K27M mutations in potentiating malignant growth in precursor cells [17,25■,26,27], and clarified differences in DNA methylation and clinical phenotype between H3.1K27M and H3.3K27M DIPG [28–30]. H3.1 K27M tumors originate exclusively in pons, whereas H3.3 K27M mutations give rise to pontine tumors in the case of DIPG, as well as other diffuse midline tumors such as thalamic or spinal cord high-grade gliomas (HGGs) [31,32] - future studies will elaborate on the significance of these anatomical and epigenetic profiles with regards to the origin or treatment of DIPG.
CELL-INTRINSIC VULNERABILITIES
The discovery of heterozygous, clonal K27M histone mutations found in the majority of DIPG tumors has led to a greater understanding of the underlying transcriptional dysfunction that potentiates tumor growth [33,34]. The vast majority of these mutations arise in the histone genes H3F3A or HIST1H3B encoding the H3.3 or H3.1 variants, respectively [9–11,13,14,16,18,35]. Both H3K27M mutation variants result in global reduction of repressive lysine 27 trimethylation (H3K27me3), resulting in aberrant transcription central to DIPG oncogenesis [13–17]. Initial studies revealed that interaction between the H3K27M oncohistone and the enhancer of zeste homologue-2 (EZH2) component of the H3K27 methyltransferase complex, polycomb repressive complex 2 (PRC2), leads to global hypomethylation of K27 in wild type H3 histones in the cell [11,13,14]. Dysfunction in PRC2 activity is seen even after the methyltransferase complex has dissociated from H3K27M oncohistones [36■,37■■,38], with the loss of methylation leading to subsequent oncogenic gene activation in the glioma cells [11,13,14,36■] (Fig. 1). Therefore, epigenetic modifying therapies which restore normal patterns of trimethylation may be a promising class of agents for DIPG.
FIGURE 1.
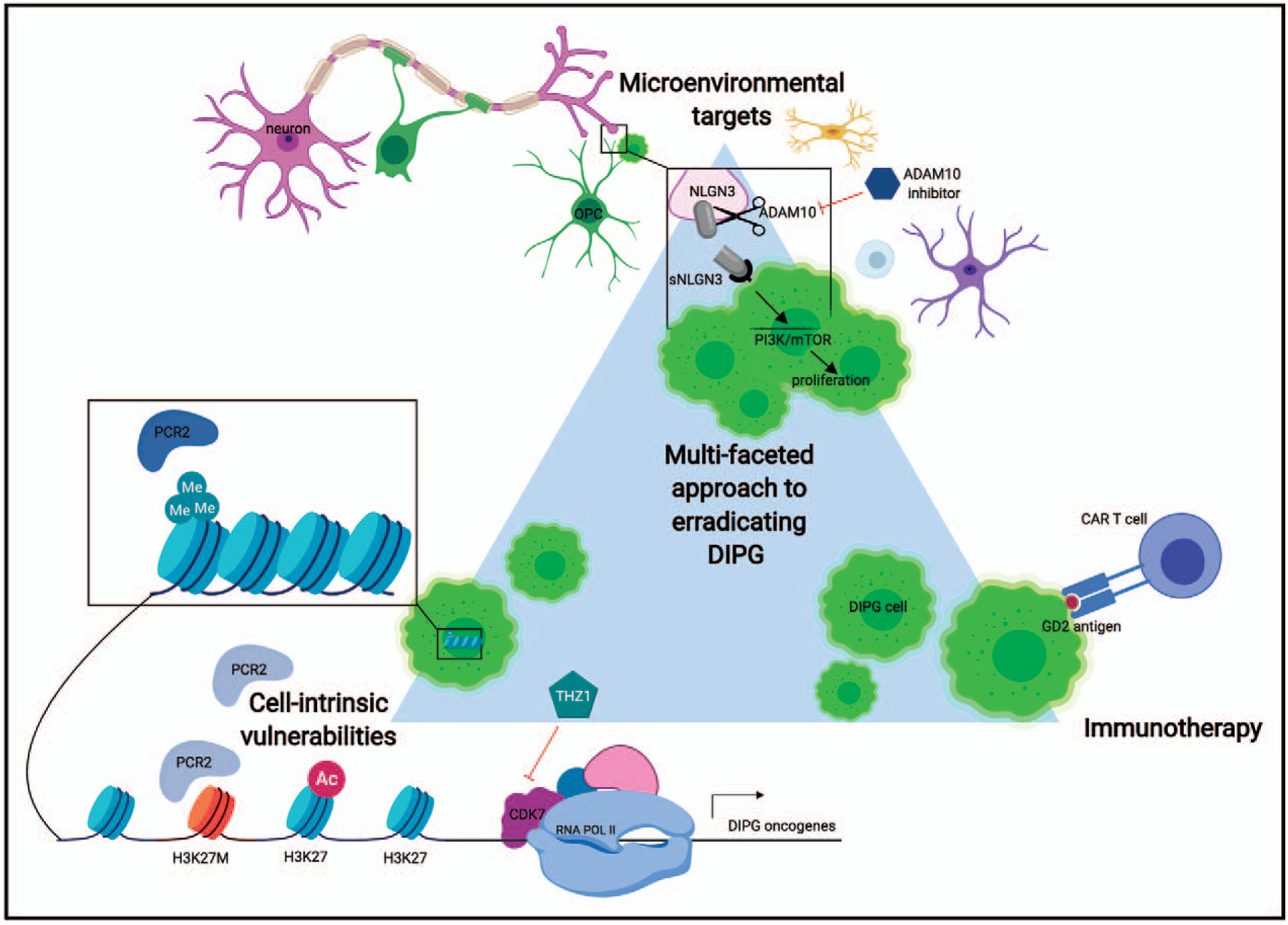
Diffuse intrinsic pontine glioma is a heterogeneous tumor that lacks effective therapies. Eradicating this tumor may require a multifaceted approach with combinatorial therapies that target distinct tumor vulnerabilities. Here, we illustrate one therapeutic example in three distinct domains where studies have shown promising targeting of diffuse intrinsic pontine glioma. One cell-intrinsic therapy that targets the aberrant transcription resulting from H3K27M-mediated polycomb repressive complex-2 dysfunction is THZ1, an inhibitor of CDK7. CDK7 is responsible for phosphorylation of RNA pol II required for transcription initiation, and thus THZ1 may inhibit the increased transcription of diffuse intrinsic pontine glioma oncogenes that potentiates tumorigenesis. In immunotherapy, one of the most promising targets is the widely expressed diffuse intrinsic pontine glioma antigen GD2. Anti-GD2 chimeric antigen receptor T cells have demonstrated dramatic tumor clearance in xenograft models, and this strategy will be pursued in clinical trials. Diffuse intrinsic pontine glioma cells initiate and proliferate due to signals in the developing brain microenvironment, and strategies to inhibit protumorgenic signals will be important in containing tumor growth. Here, we illustrate the proliferation effect of neuronal-activity regulated neuroligin-3 secretion through the mammalian target of rapamycin (mTOR)/PI3K pathway. Inhibition of ADAM10 cleavage of neuroligin-3 prevents its activity-regulated secretion from neurons and oligodendrocyte precursor cells (OPCs) and causes a stark reduction in diffuse intrinsic pontine glioma proliferation. ADAM10 inhibitors will be used in future clinical trials for diffuse intrinsic pontine glioma. Diffuse intrinsic pontine glioma cells = neon green, neuron = pink, OPC = dark green, chimeric antigen receptor-T cell = purple, WT H3K27 histone = light blue, H3K27M histone = orange, active polycomb repressive complex-2 = dark blue, inactive polycomb repressive complex-2 = gray, ADAM10 inhibitor = dark blue hexagon, THZ1 (CDK7 inhibitor) = turquoise pentagon. Illustration created using Biorender.
Significantly, some genomic areas in H3K27M-mutant DIPG cells exhibit paradoxically increased methylation. Although the H3K27M oncohistone results in dose-dependent inhibition of PRC2 function, strong PRC2 targets (with high concentrations of H3K27me3, CpG islands, or low levels of H3.3K27M as seen in one study) are able to maintain recruitment of PRC2 [37■■,39,40■]. This repressive gene silencing also plays a role in DIPG development, and should be accounted for in future therapeutics.
Histone deacetylase and demethylase inhibitors (Panobinostat)
Inhibitors of histone deacetylases (HDAC) are effective against HGGs and other pediatric central nervous tumors [41,42], and a DIPG drug screen of small molecules found robust anti-DIPG activity of the HDAC inhibitor panobinostat in vitro [19]. Histone acetyltransferases lead to lysine acetylation, most frequently on N-terminal tails - in the case of DIPG, it has been speculated that acetylation may disrupt interactions between H3K27M and PRC2 and thus normalize the chromatin environment [43]. Panobinostat was found to increase global H3 acetylation in a dose-dependent manner and partially rescue the H3K27 hypo-trimethylation phenotype. DIPG cells exposed to panobinostat also had lower expression of proliferation associated genes (e.g., MYC oncogene), decreased cell proliferation and increased cell death, and DIPG xenografted mice treated with panobinostat exhibited prolonged survival [19]. Another study showed a modest benefit of panobinostat in a murine DIPG model measured by reduced tumor cell proliferation [44]. In a more recent study, panobinostat treatment was found to lead to increased expression of endogenous retroviral elements, possibly providing an alternative mechanism for DIPG sensitivity to HDAC inhibition [45■].
With demonstration of preclinical benefit, panobinostat was moved into clinical trials. A current Phase I trial is testing side effects and optimal dosage of panobinostat in children with DIPG (NCT02717455). Preliminary results reported in abstract form have been encouraging, but it is early to draw conclusions. Although a promising treatment, the current challenges of panobinostat treatment for DIPG is the demonstrated evolution of resistance shown in preclinical studies [19] and limited but present brain penetration of drug. As is a challenge for many DIPG treatments, the intact blood brain barrier surrounding the tumor can make drug delivery challenging, and alternative delivery strategies such as direct intratumoral delivery by convection enhanced delivery are in development (NCT03566199) [46–48]. Combination therapies with panobinostat are promising, including reports of clinical benefit when panobinostat was used in conjunction with reirradiation in DIPG [49].
H3K27 demethylase inhibitor (GSKJ4)
In addition to inhibiting the loss of acetylation to normalize transcription, another strategy to address impaired H3K27 trimethylation in the context of H3K27M-induced PRC2 dysfunction is to stabilize the trimethyl mark on wild type H3K27. GSKJ4, which inhibits the K27 demethylase JMJD3, stabilizes H3K27 methylation in tumor cells and demonstrates antitumor activity [50]. However, inhibition of DIPG growth using this tool compound was seen at concentrations not attainable in clinical settings [19].
Enhancer of zeste homologue-2 inhibitors
In specific cases where tumor suppressor genes such as p16 are intact, the paradoxical increase in trimethylation and subsequent gene silencing described above promotes tumor growth [26,51]. H3K27M-expressing DIPG cell lines were found to require PRC2 methylation for proliferation and small-molecule EZH2 inhibitors arrest cell growth by increasing transcription of the tumor-suppressor protein p16INK4A normally silenced by PRC2 [26,40■]. However, while no significant cytotoxic impact of EZH2 inhibition by Tazemetostat (EPZ-6438) was found in pediatric GBM/DIPG cells with H3.3 mutations - this therapy may yet prove effective in combination with other cytotoxic epigenetic modulating drugs [52].
Transcriptional regulation
Given the central role of transcriptional dysregulation in DIPG, an additional strategy has been to directly target efficient transcription by RNA polymerase II complex. Targeting of activating bromodomain proteins with JQ1 has been effective in preclinical models, with BRD4 inhibition shown to disrupt aberrant transcription in DIPG [20■■,36■]. Furthermore, phosphorylation of the C-terminal tail of RNA pol II is required for transcription initiation and CDK7 blockade with THZ1 has importantly shown therapeutic efficacy in DIPG cells resistant to HDAC inhibitor therapy [20■■] (Fig. 1). Combining treatments of epigenetic and transcriptional disruption demonstrates a synergistic effect in disrupting DIPG cell viability [20■■,53]. Although the epigenetic targets highlighted above have shown promising preclinical efficacy, the therapies are by nature nonspecific, and may have effects on other cells types. Targeting pathways that are specific to DIPG may be another important avenue to contain tumor growth and spread.
Secondary genetic alterations
Although several human DIPG tumors are found only to exhibit the histone mutation, H3K27M is not sufficient to induce tumor formation in DIPG models and promotes tumor growth only in combination with mutant p53 and activated platelet-derived growth factor receptor α in experimental settings explored thus far [17,25■,54–56]. In addition, the H3.1K27M and H3.3K27M DIPG subgroups are characteristically associated with differing cooperating genetic alterations [3■■,11,18,35,57]. H3.3K27M tumors associate with mutations in TP53 while H3.1K27M tumors often harbor mutations in ACVR1 or have PI3K pathway dysfunction [18,25■,28–30,58]. A meta-analysis of over 1000 pediatric high -grade glioma and DIPG cases demonstrated multiple altered pathways previously unrecognized in subsets of tumors such as miRNA regulation, wingless-related integration site (Wnt) pathway and splicing machinery [3■■]. Other subclonal alterations occasionally found in H3.1 K27M DIPGs include PIK3CA mutation, phosphatase and tensin homolog (PTEN) loss, PPM1D mutation, and amplification of cell cycle genes including CCND1, CDK4, and CDK6 [59,60].
Several studies have investigated targeting of these biologically relevant pathways in DIPG. For example, the majority of DIPG tumors exhibit dysfunction in PI3K/Ak strain transforming (AKT)/mammalian target of rapamycin (mTOR) signaling pathway [61,62], and dual mTOR inhibitors have shown preclinical efficacy in vitro [20■■,63] as well as in a xenograft model [64]. mTOR inhibitors have also shown synergistic effects with CDK4/6 inhibitors which prevent cell cycle progression as well as with mitrochondrial inhibitors although efficacy has been limited in orthotopic xenograft models and in clinical settings, likely due to tumor heterogeneity and poor blood brain barrier penetrance [65,66]. With regard to targeting P53, one study has shown that the inhibition of mutant PPM1D enhances DNA damage response and growth suppressive effects of ionizing radiation in DIPG [67]. Recent studies have additionally demonstrated the ability of ALK2 inhibitors to cross the blood-brain barrier and produced modest preclinical efficacy in ACVR1 mutant DIPGs as a single treatment [58,68]. Due to the subclonal nature of secondary mutations, targeted monotherapies will not likely be curative on their own; however, a combinatorial strategy targeting several pathways alongside other treatments may be a necessity to eliminate these heterogeneous tumors.
IMMUNOTHERAPY
Harnessing the host immune response has been critical in improving the outcomes of several malignancies in the past decade. However, the immunosuppressive microenvironment of DIPG may present a challenge for therapeutics that rely on endogenous immune responses. Below we outline recent studies that utilize immunotherapeutic targets in DIPG.
Peptide vaccines
A recent peptide vaccine induces mutation-specific, cytotoxic T-cell-mediated and Th1-cell-mediated immune responses in a mouse model (H3K27M presented on major histocompatibility complex class II) [69]. Another study using autologous dendritic cell vaccines demonstrated that this therapy was safe and generated a DIPG-specific immune response [70]. In contrast to other brain tumors in which peptide vaccines are under investigation, there is very minimal immune infiltration in DIPG. DIPG cells and DIPG-associated macrophages express fewer cytokines and chemokines than in adult glioblastoma [71,72]. This may represent a substantial obstacle to the efficacy of vaccine-mediated strategies, as the lack of lymphocytes and non-inflammatory phenotype of DIPG-associated microglia/macrophages may indicate minimal ability for peptide vaccination to stimulate sufficient endogenous lymphocyte expansion and migration to the tumor site.
Adoptive T-cell therapies
Adoptive T-cell therapies have recently emerged as an extremely promising approach particularly for hematological malignancies [73,74], and a case report in glioblastoma suggests that engineered T cells have the potential to achieve a profound, albeit temporary, response even in advanced stages of disease [75]. Due to their overwhelming prevalence in DIPG, tumor-specific H3K27M peptides represent a target of great interest for engineered T cell receptor (TCR) development. A recent study has demonstrated that the creation of H3.3K27M-specific TCRs was able to kill human leukocyte antigen-A2+H3.3K27M+glioma cells and suppress the progression of glioma xenografts in mice [76]. These studies show promise for developing safe and effective T-cell-based immunotherapeutic strategies.
Chimeric antigen receptors (CARs) represent another approach to adoptive T-cell therapies, and recent work identified extremely high expression of the disialoganglioside GD2 in patient-derived DIPG cultures [77■■]. Anti-GD2 CAR T cells incorporating the 4–1BBz costimulatory domain caused antigen-specific cytotoxicity to DIPG cells [78] (Fig. 1). Patient-derived H3-K27M+ diffuse midline glioma orthotopic xenograft models showed near-complete tumor clearance and substantially improved survival after peripheral administration of GD2-targeted CAR T cells [77■■]. Although the majority of mice tolerated the treatment well, the on-target, on-tumor inflammation causes pontine tissue expansion that can compress the fourth ventricle and result in life-threatening hydrocephalus [77■■]. GD2-targeted CAR T-cell therapy, including the same GD2-directed CAR T-cell construct used in this DIPG preclinical study, has been employed in clinical trials for neuroblastoma (NCT00085930, NCT01822652) and has thus far been well tolerated [79–81]. However, as the brainstem is a precarious neuroanatomical site for swelling, peritumoral edema will be intensively monitored and managed in planned GD2-targeted CAR T-cell therapy clinical trials.
Checkpoint inhibitors
As noted above, the DIPG tumor microenvironment is ‘immune cold’ with minimal programmed death-ligand 1 (PD-L1) expression. A retrospective cohort analysis of children with DIPG who received reirradiation with concomitant programmed cell death protein 1 (PD-1) inhibitor nivolumab demonstrated tolerability and slightly prolonged overall survival (OS) [82]. However, PD-1 inhibitor therapy alone is unlikely to have a robust effect in DIPG and may need to be combined with an additional intervention to enhance the endogenous immune response [71,72].
Immunomodulatory antibodies
Immune modulating monoclonal antibodies are an emerging treatment for a variety of cancers. One such immune modulating antibody, MDV9300 (pidilizumab), has shown efficacy in various pediatric cancers, including hematological malignancies, and was thus tried in DIPG [83]. Pidilizumab is immune modulating humanized IgG1 mAb with secondary inhibitory effects on PD-1, and thus enhances endogenous antitumor immune responses. In a recent clinical trial with 9 patients (NCT 01952769), median event free survival after pidilizumab treatment was 9.3 months (range 6.8–24), median OS was 15.6 months (6.9–28), thus suggesting an improvement in survival. The results from this small study are preliminary, demonstrating that pidilizumab may be well tolerated and possibly active in pediatric patients with DIPG, although a larger sample size will be needed to further confirm these preliminary findings.
MICROENVIRONMENT
DIPG cells diffusely infiltrate the brainstem and brain, and they receive signals from multiple other cell types in this complex neural environment. The fact that DIPG arises at a particular time point during childhood and in a precise anatomical location indicates that there may be microenvironmental factors in the childhood pons that influence tumor initiation and growth. Oligodendrocyte precursor cells, a putative cell of origin for DIPG, are monopotent stem cells that self-renew and generate myelinating oligodendrocytes. Myelination of the central nervous system is a protracted process that extends over the first 3 decades of life, with predictable topographical and chronological patterns of myelination [84,85]. A discrete wave of ventral pontine myelination in mid-childhood correlates with the peak age of DIPG incidence [21,23].
Neuronal activity can regulate the proliferation of OPCs, resulting in generation of mature myelinating cells that fine-tune neural circuit function and contribute to adaptive changes in neurological function [86–89]. Activity-regulated secretion of brain-derived neurotrophic factor (BDNF) is a key mechanistic component of adaptive myelination [90]. Activity-regulated mechanisms mediating brain plasticity and growth, such as BDNF, can be hijacked by glioma cells that utilize these signals for their own malignant proliferation.
Accordingly, glutamatergic neuronal activity promotes the proliferation and growth of DIPG and other HGGs [91] similar to the effects on the normal cellular counterparts. Two key glioma growth factors are released into the tumor microenvironment in response to neuronal activity - BDNF and, unexpectedly, a postsynaptic adhesion molecule called neuroligin-3 (NLGN3) that has been found to exert a profound effect on tumor proliferation [91]. NLGN3 not only promotes the growth of DIPG and other gliomas, it is required for glioma progression. Patient-derived xenografts of DIPG and other HGGs fail to progress when xenografted into the environment of the NLGN3-deficient brain [92■]. The reason for this surprising dependency on microenvironmental NLGN3 is presently a subject of intense study.
Although the binding partner for NLGN3 is currently unknown, future studies to elucidate and block the receptor or binding partner for NLGN3 may be an effective therapy. NLGN3 binding to this as-of-yet unidentified binding partner stimulates numerous oncogenic signaling pathways, including PI3K-mTOR pathway, Src, and Ras pathways in glioma cells to induce tumor proliferation. NLGN3 is cleaved by the metalloproteinase enzyme ADAM10, and secreted in a strictly activity-regulated manner by both OPCs (a postsynaptic cell type) and neurons. ADAM10 inhibitors prevent the cleavage and release of NLGN3 into the tumor microenvironment, and are thus an effective therapeutic strategy to limit tumor growth in preclinical models of DIPG and other HGGs [92■] (Fig. 1). A clinical trial is in preparation to test ADAM10 inhibition for children with DIPG.
Beyond activity-regulated protein secretion into the tumor microenvironment, several seminal studies have identified neurotransmitters and neuropep-tides that affect adult glioma cell behavior [93–95], although for the purposes of this review we will discuss mechanisms that have shown a role in DIPG. Certain H3K27M DIPGs have been shown to over-express the dopamine receptor D2, and other gliomas such as adult glioblastoma have demonstrated dopamine receptor-dependent growth [96,97]. A recent DIPG case report demonstrates improvement of clinical symptoms with the DRD2/3 antagonist ONC201 [98]. Glutamate also enhances the proliferation and invasion of glioblastoma cells through autocrine/paracrine signaling [99–102], and the effects of this neurotransmitter in DIPG is an area of active investigation. Underscoring the importance of neuron-glioma interactions, the ability of glioma cells to enhance excitability of surrounding neurons may act in a feed-forward mechanism to promote further secretion of protumorigenic factors [99,100].
CONCLUSION
DIPG is a complex tumor due to its molecular pathogenesis and epigenetic dysregulation, as well as critical influences of tumor microenvironmental signals that promote DIPG growth and progression. Additional challenges for DIPG therapy include a largely intact blood-brain barrier that is difficult to penetrate for drug delivery, and limited endogenous immune responses towards DIPG. No therapies for DIPG have proven curative thus far. Promising therapies may involve harnessing distinct angles of DIPG vulnerabilities in combinatorial approaches to eradicate this heterogeneous tumor. Therapies that have been most promising in preclinical work or early clinical trials include epigenetic modifiers such as panobinostat, transcriptional regulators such as THZ1, CAR T immunotherapy, and microenvironmental targets such as ADAM10 inhibition. The enormous progress in understanding DIPG pathobiology, combined with advances in cancer neuroscience and immune-oncology give reason to hope that an effective therapeutic strategy will soon be realized.
KEY POINTS.
DIPG is a relatively common pediatric brain malignancy with a median survival of ~10 months and 99% 5-year mortality.
The H3K27M mutation found in the majority of DIPG cases causes epigenetic dysregulation and aberrant transcription that can be targeted therapeutically to limit expression of DIPG oncogenes.
Targeting protumorigenic microenvironmental signaling may limit DIPG proliferation and represents an important strategy for therapy.
Emerging immunotherapeutic approaches show promise.
A combinatorial approach applying multiple therapeutic strategies may be necessary to eradicate this devastating pediatric tumor.
Acknowledgements
We thank Surya Nagaraja, Shawn Gillespie, Kathryn Taylor, Christopher Mount, and Humsa Venkatesh for critical reading of the article.
Financial support and sponsorship
The authors gratefully acknowledge support from the National Institute of Neurological Disorders and Stroke (R01NS092597), NIH Director’s Pioneer Award (DP1NS111132), Unravel Pediatric Cancer, McKenna Claire Foundation, Virginia and D.K. Ludwig Fund for Cancer Research, Abbie’s Army Foundation, Defeat DIPG Foundation, ChadTough Foundation, Cure Starts Now Foundation, Alex’s Lemonade Stand Foundation, and the Howard Hughes Medical Institute Medical Fellows Program.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Cooney T, Lane A, Bartels U, et al. Contemporary survival endpoints: an International Diffuse Intrinsic Pontine Glioma Registry study. Neuro Oncol 2017; 19:1279–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffman LM, Veldhuijzen van Zanten SEM, Colditz N, et al. Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): a collaborative report from the International and European Society for Pediatric Oncology DIPG Registries. J Clin Oncol 2018; 36:1963–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.■■.Mackay A, Burford A, Carvalho D, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 2017; 32:520–537.e5. The analysis of more than 1000 cases of pediatric HGGs and diffuse intrinsic pontine glioma (DIPG) identified distinct mutations and pathway dysfunctions which cosegregate with histone-mutant subgroups.
- 4.Caretti V, Bugiani M, Freret M, et al. Subventricular spread of diffuse intrinsic pontine glioma. Acta Neuropathol 2014; 128:605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.■.Qin EY, Cooper DD, Abbott KL, et al. Neural precursor-derived pleiotrophin mediates subventricular zone invasion by glioma. Cell 2017; 170: 845–859.e19. Glioma invasion into the subventricular zone is dependent on pleiotrophin (PTN), a highly enriched growth factor in this region that is secreted by neural precursor cells (NPCs). When NPCs secrete PTN, interaction with HSP90B SPARC/SPARCL1 forms a chemoattractant complex that promotes tumor invasion through the activation of RhoA/ROCK signaling.
- 6.Cohen KJ, Jabado N, Grill J. Diffuse intrinsic pontine gliomas-current management and new biologic insights. Is there a glimmer of hope? Neuro Oncol 2017; 19:1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jalali R, Raut N, Arora B, et al. Prospective evaluation of radiotherapy with concurrent and adjuvant temozolomide in children with newly diagnosed diffuse intrinsic pontine glioma. Int J Radiat Oncol Biol Phys 2010; 77: 113–118. [DOI] [PubMed] [Google Scholar]
- 8.Lin GL, Monje M. A protocol for rapid postmortem cell culture of diffuse intrinsic pontine glioma (DIPG). J Vis Exp 2017; 121:e55360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khuong-Quang D-A, Buczkowicz P, Rakopoulos P, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 2012; 124:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartzentruber J, Korshunov A, Liu X-Y, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482:226–231. [DOI] [PubMed] [Google Scholar]
- 11.Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and nonbrainstem glioblastomas. Nat Genet 2012; 44:251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016; 131:803–820. [DOI] [PubMed] [Google Scholar]
- 13.Bender S, Tang Y, Lindroth AM, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013; 24:660–672. [DOI] [PubMed] [Google Scholar]
- 14.Lewis PW, Müller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013; 340:857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venneti S, Garimella MT, Sullivan LM, et al. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol Zurich Switz 2013; 23:558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan K-M, Fang D, Gan H, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 2013; 27:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Funato K, Major T, Lewis PW, et al. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014; 346:1529–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor KR, Mackay A, Truffaux N, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 2014; 46: 457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grasso CS, Tang Y, Truffaux N, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med 2015; 21:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.■■.Nagaraja S, Vitanza NA, Woo PJ, et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 2017; 31:635–652.e6. DIPG with H3K27M exhibit oncogenic transcriptional aberrancies that can be targeted by bromodomain inhibition or CDK7 blockade, and used synergistically with histone deacetylases (HDAC) inhibitors as a potential therapeutic. Analysis of key regulatory elements (super enhancers) in DIPG suggests its cell of origin may be a precursor cell in the oligodendroglial lineage.
- 21.Monje M, Mitra SS, Freret ME, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A 2011; 108:4453–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ballester LY, Wang Z, Shandilya S, et al. Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am J Surg Pathol 2013; 37:1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tate MC, Lindquist RA, Nguyen T, et al. Postnatal growth of the human pons: a morphometric and immunohistochemical analysis: human postnatal pons development. J Comp Neurol 2015; 523:449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.■■.Filbin MG, Tirosh I, Hovestadt V, et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018; 360:331–335. Single-cell RNA sequencing from six primary H3K27M-glioma demonstrated that H3K27M cells resemble undifferentiated oligodendrocyte precursor cells with distinct stem-cell like programs.
- 25.■.Larson JD, Kasper LH, Paugh BS, et al. Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell 2019; 35:140–155.e7. Induction of H3.3K27M in NPCs along with activating platelet-derived growth factor receptor α (PDGFRα) mutant and Trp53 loss leads to formation of DIPG. H3K27me3 loss in these NPCs drove altered regulation of bivalent promoters that dysregulated key neural development genes.
- 26.Mohammad F, Weissmann S, Leblanc B, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 2017; 23:483–492. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y, Xu C, Pan C, et al. Diffuse intrinsic pontine gliomas exhibit cell biological and molecular signatures of fetal hindbrain-derived neural progenitor cells. Neurosci Bull 2019; 35:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castel D, Philippe C, Calmon R, et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 2015; 130: 815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castel D, Philippe C, Kergrohen T, et al. Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol Commun 2018; 6:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012; 22:425–437. [DOI] [PubMed] [Google Scholar]
- 31.Jones C, Baker SJ. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer 2014; 14:651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones C, Karajannis MA, Jones DTW, et al. Pediatric high-grade glioma: biologically and clinically in need of new thinking. Neuro Oncol 2017; 19:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nikbakht H, Panditharatna E, Mikael LG, et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun 2016; 7:11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vinci M, Burford A, Molinari V, et al. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat Med 2018; 24:1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buczkowicz P, Hoeman C, Rakopoulos P, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 2014; 46:451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.■.Piunti A, Hashizume R, Morgan MA, et al. Therapeutic targeting of polycomb & and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med 2017; 23:493–500. Epigenetic profiling of H3K27M mutant DIPG cells suggests a model where H3K27M excludes polycomb repressive complex 2 (PRC2) binding and induces aberrant H3K27 acetylation at these locations. PRC2 may be then be shunted preferentially to other locations where it represses neuronal differentiation, explaining its requirement in DIPG proliferation.
- 37.■■.Stafford JM, Lee C-H, Voigt P, et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 2018; 4:eaau5935. Quantitative proteomics demonstrated a dose-related response between PRC2 inhibition and levels of H3K27M. PRC2 inhibition requires transient interaction with H3K27M but inhibition persists even after PRC2 dissociates from H3K27M-containing chromatin.
- 38.Tatavosian R, Duc HN, Huynh TN, et al. Live-cell single-molecule dynamics of PcG proteins imposed by the DIPG H3.3K27M mutation. Nat Commun 2018; 9:2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang D, Gan H, Cheng L, et al. H3.3K27M mutant proteins reprogram epigenome by sequestering the PRC2 complex to poised enhancers. eLife 2018; 7:e36696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.■.Harutyunyan AS, Krug B, Chen H, et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat Commun 2019; 10:1262. A preclinical report demonstrated that PRC2 is recruited to binding sites normally in K27M mutant cells, but is unable to spread me3 deposition in the presence of the mutant histone, thus resulting in global hypo-me3 and loss of repressive marks.
- 41.Kitange GJ, Mladek AC, Carlson BL, et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin Cancer Res 2012; 18:4070–4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milde T, Kleber S, Korshunov A, et al. A novel human high-risk ependymoma stem cell model reveals the differentiation-inducing potential of the histone deacetylase inhibitor Vorinostat. Acta Neuropathol 2011; 122:637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown ZZ, Müller MM, Jain SU, et al. Strategy for ‘detoxification’ of a cancer-derived histone mutant based on mapping its interaction with the methyltransferase PRC2. J Am Chem Soc 2014; 136:13498–13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hennika T, Hu G, Olaciregui NG, et al. Pre-clinical study of panobinostat in xenograft and genetically engineered murine diffuse intrinsic pontine glioma models. PLoS One 2017; 12:e0169485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.■.Krug B, De Jay N, Harutyunyan AS, et al. Pervasive H3K27 acetylation leads to ERV expression and a therapeutic vulnerability in H3K27M gliomas. Cancer Cell 2019; 35:782–797.e8. Epigenetic analysis of pediatric HGG cells with H3K27M demonstrates lineagespecific gene profiles for the tumor cell of origin. Increased acetylation in H3.3K27M cells induces expression of repetitive elements, such as endogenous retroviral elements (ERVs) which can stimulate innate immune responses - HDAC inhibitors further increase expression of ERVs and may act synergistically to overcome immune evasion.
- 46.Singleton WGB, Bienemann AS, Woolley M, et al. The distribution, clearance, and brainstem toxicity of panobinostat administered by convection-enhanced delivery. J Neurosurg Pediatr 2018; 22:288–296. [DOI] [PubMed] [Google Scholar]
- 47.Souweidane MM, Kramer K, Pandit-Taskar N, et al. Convection-enhanced delivery for diffuse intrinsic pontine glioma: a single-centre, dose-escalation, phase 1 trial. Lancet Oncol 2018; 19:1040–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou Z, Singh R, Souweidane MM. Convection-enhanced delivery for diffuse intrinsic pontine glioma treatment. Curr Neuropharmacol 2017; 15: 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang ZJ, Ge Y, Altinok D, et al. Concomitant use of panobinostat and reirradiation in progressive DIPG: report of 2 cases. J Pediatr Hematol Oncol 2017; 39:e332–e335. [DOI] [PubMed] [Google Scholar]
- 50.Hashizume R, Andor N, Ihara Y, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med 2014; 20:1394–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cordero FJ, Huang Z, Grenier C, et al. Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol Cancer Res 2017; 15:1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wiese M, Schill F, Sturm D, et al. No significant cytotoxic effect of the EZH2 inhibitor tazemetostat (EPZ-6438) on pediatric glioma cells with wildtype histone 3 or mutated histone 3.3. Klin Padiatr 2016; 228:113–117. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Dong W, Zhu J, et al. Combination of EZH2 inhibitor and BET inhibitor for treatment of diffuse intrinsic pontine glioma. Cell Biosci 2017; 7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Misuraca KL, Hu G, Barton KL, et al. A novel mouse model of diffuse intrinsic pontine glioma initiated in Pax3-expressing cells. Neoplasia 2016; 18:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pathania M, De Jay N, Maestro N, et al. H3.3K27M cooperates with Trp53 loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell 2017; 32:684–700.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sin-Chan P, Mumal I, Huang A. Tails of a super histone. Cancer Cell 2019; 35:7–9. [DOI] [PubMed] [Google Scholar]
- 57.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 2014; 46:462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoeman CM, Cordero FJ, Hu G, et al. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat Commun 2019; 10:1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoeman C, Shen C, Becher OJ. CDK4/6 and PDGFRA signaling as therapeutic targets in diffuse intrinsic pontine glioma. Front Oncol 2018; 8:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koschmann C, Farooqui Z, Kasaian K, et al. Multifocal sequencing of a diffuse intrinsic pontine glioma establishes PTEN loss as an early event. NPJ Precis Oncol 2017; 1:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Warren KE, Killian K, Suuriniemi M, et al. Genomic aberrations in pediatric diffuse intrinsic pontine gliomas. Neuro Oncol 2012; 14:326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zarghooni M, Bartels U, Lee E, et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol 2010; 28:1337–1344. [DOI] [PubMed] [Google Scholar]
- 63.Flannery PC, DeSisto JA, Amani V, et al. Preclinical analysis of MTOR complex 1/2 inhibition in diffuse intrinsic pontine glioma. Oncol Rep 2018; 39:455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miyahara H, Yadavilli S, Natsumeda M, et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett 2017; 400:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Asby DJ, Killick-Cole CL, Boulter LJ, et al. Combined use of CDK4/6 and mTOR inhibitors induce synergistic growth arrest of diffuse intrinsic pontine glioma cells via mutual downregulation of mTORC1 activity. Cancer Manag Res 2018; 10:3483–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsoli M, Liu J, Franshaw L, et al. Dual targeting of mitochondrial function and mTOR pathway as a therapeutic strategy for diffuse intrinsic pontine glioma. Oncotarget 2018; 9:7541–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Akamandisa MP, Nie K, Nahta R, et al. Inhibition of mutant PPM1D enhances DNA damage response and growth suppressive effects of ionizing radiation in diffuse intrinsic pontine glioma. Neuro Oncol 2019; 21:786–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carvalho D, Taylor KR, Olaciregui NG, et al. ALK2 inhibitors display beneficial effects in preclinical models of ACVR1 mutant diffuse intrinsic pontine glioma. Commun Biol 2019; 2:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ochs K, Ott M, Bunse T, et al. K27M-mutant histone-3 as a novel target for glioma immunotherapy. Oncoimmunology 2017; 6:e1328340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benitez-Ribas D, Cabezón R, Flórez-Grau G, et al. Immune response generated with the administration of autologous dendritic cells pulsed with an allogenic tumoral cell-lines lysate in patients with newly diagnosed diffuse intrinsic pontine glioma. Front Oncol 2018; 8:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lieberman NAP, DeGolier K, Kovar HM, et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy. Neuro Oncol 2019; 21:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin GL, Nagaraja S, Filbin MG, et al. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol Commun 2018; 6:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6:224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016; 375: 2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chheda ZS, Kohanbash G, Okada K, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med 2018; 215:141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.■■.Mount CW, Majzner RG, Sundaresh S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat Med 2018; 24:572–579. Anti-GD2 chimeric antigen receptor (CAR) T cells incorporating a 4–1BBz costimulatory domain demonstrated tumor killing and cytokine generation in vitro, and dramatic clearance of engrafted diffuse midline glioma in an in-vivo model xenograft model. Anti-GD2 CAR T therapy will soon be in clinical trials for DIPG.
- 78.Long AH, Haso WM, Shern JF, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015; 21:581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heczey A, Louis CU, Savoldo B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther J Am Soc Gene Ther 2017; 25:2214–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Louis CU, Savoldo B, Dotti G, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011; 118:6050–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pule MA, Savoldo B, Myers GD, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 2008; 14:1264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kline C, Liu SJ, Duriseti S, et al. Reirradiation and PD-1 inhibition with nivolumab for the treatment of recurrent diffuse intrinsic pontine glioma: a single-institution experience. J Neurooncol 2018; 140:629–638. [DOI] [PubMed] [Google Scholar]
- 83.Fried I, Lossos A, Ben Ami T, et al. Preliminary results of immune modulating antibody MDV9300 (pidilizumab) treatment in children with diffuse intrinsic pontine glioma. J Neurooncol 2018; 136:189–195. [DOI] [PubMed] [Google Scholar]
- 84.Lebel C, Gee M, Camicioli R, et al. Diffusion tensor imaging of white matter tract evolution over the lifespan. NeuroImage 2012; 60:340–352. [DOI] [PubMed] [Google Scholar]
- 85.Yakovlev P, Lecours A. The myelogenetic cycles of regional maturation of the brain In: Minkowski A, editor. Regional development of the brain in early life. Oxford Blackwell; 1967. [Google Scholar]
- 86.Gibson EM, Purger D, Mount CW, et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014; 344:1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hughes EG, Orthmann-Murphy JL, Langseth AJ, Bergles DE. Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat Neurosci 2018; 21:696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McKenzie IA, Ohayon D, Li H, et al. Motor skill learning requires active central myelination. Science 2014; 346:318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mitew S, Gobius I, Fenlon LR, et al. Pharmacogenetic stimulation of neuronal activity increases myelination in an axon-specific manner. Nat Commun 2018; 9:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Geraghty AC, Gibson EM, Ghanem RA, et al. Loss of adaptive myelination contributes to methotrexate chemotherapy-related cognitive impairment. Neuron 2019; 103:250–265.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Venkatesh HS, Johung TB, Caretti V, et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 2015; 161:803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.■.Venkatesh HS, Tam LT, Woo PJ, et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017; 549:533–537. Neuronal activity promotes HGG growth through ADAM10-mediated cleavage and secretion of the synaptic adhesion molecule neuroligin-3 from neurons and OPCs. Inhibition of ADAM10 blocks glioma growth by preventing neuroligin-3 shedding into the tumor microenvironment, and is a potential therapeutic treatment.
- 93.Cuddapah VA, Robel S, Watkins S, Sontheimer H. A neurocentric perspective on glioma invasion. Nat Rev Neurosci 2014; 15:455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Labrakakis C, Patt S, Hartmann J, Kettenmann H. Functional GABA(A) receptors on human glioma cells. Eur J Neurosci 1998; 10:231–238. [DOI] [PubMed] [Google Scholar]
- 95.Synowitz M, Ahmann P, Matyash M, et al. GABA(A)-receptor expression in glioma cells is triggered by contact with neuronal cells. Eur J Neurosci 2001; 14:1294–1302. [DOI] [PubMed] [Google Scholar]
- 96.Dolma S, Selvadurai HJ, Lan X, et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell 2016; 29:859–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu Z, Jiang X, Gao L, et al. Synergistic suppression of glioblastoma cell growth by combined application of temozolomide and dopamine D2 receptor antagonists. World Neurosurg 2019; 128:e468–e477. [DOI] [PubMed] [Google Scholar]
- 98.Hall MD, Odia Y, Allen JE, et al. First clinical experience with DRD2/3 antagonist ONC201 in H3 K27M-mutant pediatric diffuse intrinsic pontine glioma: a case report. J Neurosurg Pediatr 2019; 23:1–7. [DOI] [PubMed] [Google Scholar]
- 99.Buckingham SC, Campbell SL, Haas BR, et al. Glutamate release by primary brain tumors induces epileptic activity. Nat Med 2011; 17:1269–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Campbell SL, Buckingham SC, Sontheimer H. Human glioma cells induce hyperexcitability in cortical networks. Epilepsia 2012; 53:1360–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ishiuchi S, Tsuzuki K, Yoshida Y, et al. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med 2002; 8:971–978. [DOI] [PubMed] [Google Scholar]
- 102.Ishiuchi S, Yoshida Y, Sugawara K, et al. Ca2+–permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J Neurosci 2007; 27:7987–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]