

Abstract
Genetic screening has identified numerous variants of the endosomal solute carrier family 9 member A6 (SLC9A6)/(Na+,K+)/H+ exchanger 6 (NHE6) gene that cause Christianson syndrome, a debilitating X-linked developmental disorder associated with a range of neurological, somatic, and behavioral symptoms. Many of these variants cause complete loss of NHE6 expression, but how subtler missense substitutions or nonsense mutations that partially truncate its C-terminal cytoplasmic regulatory domain impair NHE6 activity and endosomal function are poorly understood. Here, we describe the molecular and cellular consequences of six unique mutations located in the N-terminal cytoplasmic segment (A9S), the membrane ion translocation domain (L188P and G383D), and the C-terminal regulatory domain (E547*, R568Q, and W570*) of human NHE6 that purportedly cause disease. Using a heterologous NHE6-deficient cell expression system, we show that the biochemical, catalytic, and cellular properties of the A9S and R568Q variants were largely indistinguishable from those of the WT transporter, which obscured their disease significance. By contrast, the L188P, G383D, E547*, and W570* mutants exhibited variable deficiencies in biosynthetic post-translational maturation, membrane sorting, pH homeostasis in recycling endosomes, and cargo trafficking, and they also triggered apoptosis. These findings broaden our understanding of the molecular dysfunctions of distinct NHE6 variants associated with Christianson syndrome.
Keywords: apoptosis, endosome, endocytosis, membrane transport, neurological disease, pH regulation, protein sorting, sodium-proton exchange, structure–function
Introduction
Genetic variants or aberrant expressions of SLC9A6 (solute carrier family 9, member A6), an X chromosome gene encoding the alkali cation (Na+,K+)/proton (H+) exchanger NHE6 isoform, have been linked to multiple neurological conditions, including Christianson syndrome (CS)2 (1–24), autism spectrum disorder (25, 26), schizophrenia (25), chromesthesia (i.e. auditory-visual synesthesia) (27), idiopathic Parkinson's disease (28), and Alzheimer's disease (29, 30). Evidence establishing a direct causal relationship between altered gene function and phenotype is strongest for CS, a debilitating neurodevelopmental disorder manifested in males by moderate to severe intellectual disability, autistic-like behavior, epilepsy, microcephaly, mutism, hypotonia, sensory and motor dysfunction, neurodegeneration, and a shortened life span (1, 15). Female carriers exhibit variable penetrance and milder symptoms (1, 21, 22, 31). A CS-like phenotype is also reiterated in Slc9a6/Nhe6 knockout (KO) male mice (Nhe6−/Y), including atrophy and degeneration of cortical, hippocampal, and especially cerebellar Purkinje neurons that parallel deficits in visuospatial memory and motor dexterity, and increased mortality prior to weaning (32–34). Likewise, heterozygous Nhe6 KO female mice (Nhe6−/+) display a comparable, albeit milder, range of symptoms (35).
The precise mechanisms by which altered NHE6 activity cause CS are not fully understood. The NHE6 gene is widely transcribed, with highest expression in the central nervous system (CNS), which likely explains the marked neurological morbidity of CS (36, 37). Within cells, NHE6 resides in vesicles along the recycling endosomal pathway (37–39) where it operates as an electroneutral H+ efflux pathway to moderate intraluminal acidification driven by electrogenic vacuolar H+-ATPase (V-ATPase) pumps and 2Cl−/H+ exchangers, the latter providing a counterion shunt to accumulate endosomal chloride that reduces the luminal positive charge and facilitates acidification. Collectively, these ion carriers attain an intraluminal pH that ranges between ∼6.0 and 6.6 along this pathway (39–42). This tempered acidification enables efficient dissociation and dephosphorylation of many activated ligand–receptor complexes internalized from the cell surface (43). The unbound dephosphorylated receptors, in turn, can be salvaged and returned to the plasma membrane for reuse while the freed ligands are transferred to more acidic late endosomes/multivesicular bodies (pH ∼5.0–5.5) and lysosomes (pH ∼4.6–5.0) for degradation by acid hydrolases (43–47). Proper acidification of endosomes is also important for their efficient movement within cells (41, 48), although the mechanisms linking intraluminal acidification to the vesicular trafficking machinery are not fully understood (49, 50). In the case of early/recycling endosomes, suppressing NHE6 expression in cervical cancer HeLa cells by small interfering RNA (siRNA) caused excessive endosomal acidification that correlated with impaired trafficking of a subset of clathrin-dependent recycling endosomal cargo (51). In polarized hepatoma HepG2 cells, siRNA knockdown of NHE6 selectively decreased the intraluminal pH of subapical recycling endosomes accompanied by gradual loss of apical surface bile canalicular proteins and lipids (52). Thus in CS, it has been postulated that defects in NHE6 will compromise endosomal pH homeostasis and cargo trafficking in many tissues, but especially the CNS, leading to pleiotropic pathophysiological disturbances. This is supported by studies of mouse hippocampal neurons where disruption of NHE6 expression resulted in overacidification of early and recycling endosomes and diminished signaling from the tropomyosin or tyrosine receptor kinase B (TrkB) (33) and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (53, 54) that correlated with reduced neurite outgrowth and branching, synapse density and maturation, and circuit strength.
More than 80 genetic variants of NHE6 have been identified thus far (1–23) (also see ClinVar (RRID:SCR_006169) and DECIPHER (RRID:SCR_006552)), many of which are frameshift and nonsense mutations or mRNA-splicing defects in the ion translocation domain (amino acids 25–533) that are predicted to result in total loss of NHE6 protein. However, the consequences on endosomal and cellular function of other CS-linked NHE6 mutations that may not lead to a complete loss of protein expression or function have been less well-documented (53, 55–57). These include missense substitutions, small in-frame deletions, or nonsense mutations in the C-terminal regulatory domain that may generate protein products with altered functional properties (i.e. dominant-negative or constitutively-active) which, alongside other genetic factors, could contribute to reported variations in disease severity of CS patients (15).
In this report, we describe the consequences of previously uncharacterized CS-associated variants of NHE6 on various molecular and cellular parameters in transfected cells. Our results show that whereas some variants appear benign, others impair NHE6 protein biosynthetic maturation and membrane sorting to different extents, but ultimately they adversely affect recycling endosomal function and cell viability. Collectively, our findings provide greater insight into the dynamics of NHE6 dysfunction in CS and should prove valuable in the future design and development of potential therapeutic strategies for treating this neurodevelopmental disorder.
Results
To broaden our understanding of the molecular and cellular mechanisms underlying this disorder, we examined six reported inherited and de novo mutations located in the short N-terminal cytoplasmically-located segment (amino acids 1–24) (c.25G→T:pAla9 → Ser, A9S) (3), the transmembrane ion translocation domain (amino acids 25–533) (c.563T→C:Leu188 → Pro, L188P; c.1148G→A:Gly383 → Asp, G383D) (5, 15), and the longer C-terminal cytoplasmic regulatory domain (amino acids 534–701) (c.1639G→T: Glu547 → Ter, E547*; c.1703G→A:Arg568 → Gln, R568Q; c.1710G→A:Trp570 → Ter, W570*) (4, 12, 15, 19, 25, 58) of the longest splice-variant of human NHE6 (NM_001042537.1, also called NHE6v1 but hereafter simply referred to as NHE6). The positions of these amino acids are indicated in the predicted planar transmembrane organization of NHE6 (Fig. 1A) based on the proposed topology of NHE1 (59, 60). These mutations occur at highly-conserved sites in NHE6 orthologs from mammals to fruit flies, suggestive of functional importance (Fig. 1B). Transcripts containing the C-terminal premature stop codon mutations (E547* and W570*) remove the bulk of the cytoplasmic regulatory domain (i.e. the last 155 and 132 amino acids, respectively) and may be subject to nonsense-mediated mRNA decay (61). However, if translated, such truncations might remove putative signal sequences important for the membrane trafficking and/or regulation of the transporter and therefore merit further investigation as little is known about this region of the transporter.
Figure 1.

Location of missense and nonsense mutations in SLC9A6/NHE6 variants of patients with Christianson syndrome. A, schematic planar drawing of the predicted membrane topology of the longest splice-variant of mammalian NHE6 and location of the mutations (yellow circles). Two consensus N-linked glycosylation sites (128NVT and 145NVS) within extracellular loop 2 have been verified experimentally (data not shown) and are illustrated in the drawing. B, phylogenetic comparison of the primary sequence of segments containing the various mutations in NHE6. The affected residues are shaded in black.
Structural modeling of the L188P and G383D variants
Of the aforementioned six mutations, two (L188P and G383D) were localized within the transmembrane ion translocation domain, which afforded an opportunity to assess the theoretical structural and functional implications of these substitutions by structure homology modeling. In this regard, we previously described a putative structural model of the human NHE6 N-terminal transmembrane region (encompassing residues 74–540) in its monomeric state using the spatial atomic coordinates of the outward-facing conformation of the Thermus thermophilus Na+/H+ antiporter NapA (57, 62). However, mammalian NHEs, like their bacterial homologs, normally form homodimers (63). A homodimer ribbon model of NHE6 is now displayed in its frontal and top configurations in Fig. 2A. In the wildtype (WT) exchanger, Leu188 is adjacent to two consecutive proline residues (Pro189 and Pro190) in the middle of membrane-spanning helix 4 (M4). The latter di-proline motif would typically be expected to interrupt and break a helix (64). However, subjecting the helix to molecular dynamics (MD) simulations (for description, see “Experimental procedures”) stretched out the segment containing the proline residues due to the absence of H-bonds in this location but did not break the helical formation completely (illustrated in monomer, Fig. 2B, left panel). Although very rare, it is possible to have two consecutive prolines without disrupting a helix (65, 66). However, replacement of Leu188 with proline results in three consecutive prolines, which is predicted by MD simulations to unravel the helix, resulting in a much shorter helical segment (Fig. 2B, right panel). Because the homologous M4 segment in the plasmalemmal NHE1 isoform is thought to form a critical part of the ion translocation pathway (60, 67, 68), it is very likely that the L188P substitution affects the functionality of the transporter.
Figure 2.

Structure homology modeling of transmembrane NHE6 variants associated with Christianson syndrome. A, front (membrane aspect, left panel) and top (extracellular aspect, right panel) views of a 3D structure homology model of dimeric human NHE6 based on the crystal structure of the bacterial T. thermophilus Na+/H+ antiporter NapA (TtNapA) (Protein Data Bank accession code 5bz3; 2.30 Å, 15% identity, 27% similarity), which provided the broadest coverage, highest resolution, and best spatial fit compared with other crystallized bacterial Na+/H+ antiporters. The proposed structure includes only the membrane-spanning helices (M2–M12; amino acids 74–540) that aligned with homologous segments of TtNapA. The top view includes the locations of the transmembrane-localized residues Leu-188 and Gly-383 mutated in CS. B, molecular dynamics simulation of structural changes predicted to occur in TM4 (highlighted in cyan) upon substitution of Leu-188 with Pro (L188P). The monomeric forms of NHE6 WT and L188P are illustrated. C and D, structural perturbations predicted to occur in the intramembranous re-entrant (R) loop between helices M8 and M9 upon substitution of Gly-383 with Asp (G383D). C, upper and lower panels show front and top views, respectively, of Gly-383, which is packed tightly against amino acids Phe-373, Ala-376, and Glu-377 (top left). Mutation of Gly-383 to Asp would result in steric clashes between these residues and interfere with the packing between these segments of the R-loop. D, G383D substitution would also disrupt interactions of R-loop residues Trp-379, Phe-381, and especially Thr-382 with residues Asp-92 and Ile-296 in helix M7 and residues Ile-330, Phe-331, and Ser-334 in helix M8.
The consequences of the G383D mutation are also predicted to significantly perturb protein structure and function. In the modeled WT structure, Gly383 is located close to the bend of an intramembranous re-entrant loop (R-loop) between helices M8 and M9 and packed tightly against amino acids Phe373, Ala376, and Glu377 (Fig. 2C), a segment strongly implicated in ion permeation (69, 70). Replacing Gly383 with an aspartic acid residue would result in steric clashes between these residues and interfere with the packing between these segments of the R-loop (Fig. 2C). Furthermore, this substitution would also disrupt interactions of R-loop residues Trp379, Phe381, and especially Thr382 with residues Asp292 and Ile296 in helix M7 and residues Ile330, Phe331, and Ser334 in helix M8 (Fig. 2D). Notably, amino acid Asp292 is invariant in all mammalian NHEs, and its homologous residue in human NHE1 (i.e. Asp267) is critical for catalytic activity (71). Thus, it is probable that the G383D mutation would also disrupt the conformation and function of NHE6.
Biosynthetic maturation and stability of the NHE6 variants
To empirically visualize and evaluate the functional significance of the individual variants, we engineered them into the WT human NHE6 cDNA that was fused at its C terminus to either enhanced green fluorescent protein (GFP), monomeric Cherry fluorescent protein (ChFP), or influenza hemagglutinin (HA)-epitope (NHE6GFP, NHE6ChFP, or NHE6HA, respectively). We previously showed that the molecular and cellular properties of these chimeric constructs were indistinguishable from unmodified NHE6 when examined in AP-1 cells, a Chinese hamster ovary-derived cell line lacking detectable levels of endogenous NHE6, and therefore serve as a useful cell model system to study this transporter (53).
To assess their biosynthetic maturation, mammalian expression plasmids containing the NHE6GFP constructs were transiently transfected in AP-1 cells, and their total cellular expression was monitored over a 48-h period by Western blotting. Consistent with previous results (56), the homodimeric WT transporter fractionated as multiple bands that correspond to its dissociated, newly-synthesized core-glycosylated (∼85–90 kDa), and fully-glycosylated (∼110–130 kDa) monomeric species, and its fully-glycosylated dimeric form (>250 kDa) that does not fully dissociate under standard SDS-PAGE conditions (Fig. 3A) (53). Dimeric assembly was observed for all constructs, suggesting that formation of their quaternary structures was not grossly perturbed. However, whereas post-translational oligosaccharide maturation proceeded normally for A9S and R568Q, the ratio of fully-glycosylated (monomer and dimer) to total NHE6 protein expression was noticeably impaired for the other four mutants compared with WT (Fig. 3A), showing significant reductions (∼70–90%; two-way ANOVA, p < 0.05) at peak expression (between 24 and 32 h post-transfection) as quantified by densitometry (see “Experimental procedures” for details) (Fig. 3B). Total protein expression of these latter mutants was also noticeably diminished at 48 h post-transfection compared with WT, indicative of increased protein clearance.
Figure 3.

Assessment of the biosynthetic maturation of NHE6 variants. A, AP-1 cells transiently expressing NHE6GFP WT or CS-linked variants were lysed at the indicated time points (6–48 h) post-transfection. Equal amounts of proteins (20 μg) were subjected to SDS-PAGE and immunoblotting with a polyclonal anti-GFP antibody. NHE6 migrates as multiple bands: higher molecular weight bands represent the fully-glycosylated (fg) and core-glycosylated (cg) dimeric (d) forms of the exchanger that do not fully dissociate under SDS-PAGE conditions, whereas lower molecular weight bands represent fully-glycosylated and core-glycosylated forms of the dissociated monomeric (m) protein. The blots were stripped and reprobed with a mouse monoclonal anti-GAPDH antibody to control for protein loading. B, ratios of fully-glycosylated protein (monomer and dimer)/total NHE6 protein (fg/total) were quantified by densitometry of X-ray films exposed in the linear range and analyzed using ImageJ software. Values represent the mean ± S.D. of three different experiments. Significance was determined by two-way ANOVA with a post hoc Tukey test. The NHE6 variants clustered into two groups: 1) WT, A9S, and R568Q and 2) L188P, G383D, E547*, and W570*, with variants within each cluster yielding similar statistical values. Population means of the NHE6 variants are significantly different (F value = 26.4, p value = 1.5 × 10−16). Population means as a function of time are significantly different (F value = 48.6, p value = 1.9 × 10−19). § indicates significance (p < 0.01) of the means of NHE6 variants within a cluster relative to the 12-h time point. Asterisks indicate significance (★, p < 0.05, and ★★, p < 0.01) of the means between clusters of NHE6 variants at the indicated time points.
The stability of the NHE6 constructs was further examined by pulse–chase experiments. After 24 h of transient transfection, cells were treated with cycloheximide for an additional 4–24 h to inhibit de novo protein synthesis while monitoring levels of the previously synthesized transporters as a function of time. As shown in Fig. 4, levels of the core-glycosylated forms of WT, A9S, and R568Q at 24 h post-transfection matured to their fully-glycosylated states within 4 h after cycloheximide treatment and then remained relatively stable over the ensuing 20-h period. By contrast, the L188P, G383D, E547*, and W570* mutants showed reduced oligosaccharide maturation and abundance over the course of the pulse–chase period. As a control for protein loading, we measured the expression of the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and found it to be constant over the treatment period. These results are consistent with reduced stability of these variants.
Figure 4.
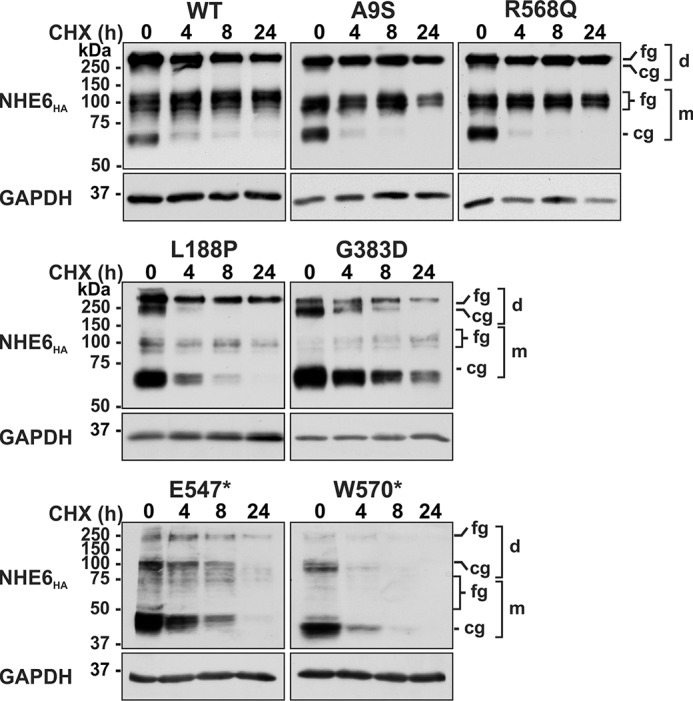
Assessment of the protein stability of NHE6 variants. AP-1 cells were transiently transfected with NHE6HA WT of CS-linked variants for 24 h and then treated with 150 μg/ml cycloheximide (CHX) for the indicated time points and lysed, and equal amounts of protein (20 μg) were analyzed by Western blotting using a mouse monoclonal anti-HA antibody. Blots were reprobed with a mouse monoclonal anti-GAPDH antibody to control for loading. Blots are representative images from four separate experiments. fg, fully-glycosylated; cg, core-glycosylated; d, dimeric; m, monomeric.
The delayed processing and enhanced turnover of the L188P, G383D, E547*, and W570* mutants could arise by redirecting the defective transporters to proteasomes and/or lysosomes. To assess this possibility, another pulse–chase experiment was performed with transiently-transfected cells (24 h) expressing the various NHE6 constructs incubated with cycloheximide and inhibitors of proteasomal (MG132) (72) or lysosomal (leupeptin plus pepstatin, LeuP) (73, 74) proteolysis over an 8-h period. As expected, both types of inhibitors had no appreciable effect on WT, A9S, and R568Q levels compared with vehicle-treated controls (Fig. 5). By comparison, suppression of proteasomal, but not lysosomal, proteolysis partially reduced the cellular loss of L188P, G383D, E547*, and W570* and resulted in the appearance of a unique high-molecular-weight NHE6 band above the fully-glycosylated dimeric band (indicated by an asterisks) for each of these variants. Although the nature of this band was not examined empirically, it likely represents polyubiquitinated or possibly aggregated forms of these variants that accumulated upon proteasomal inhibition. The absence of an effect of the lysosomal protease inhibitors leupeptin and pepstatin, which are only moderately cell-permeant, prompted us to test the effect of the lysosomotropic agent chloroquine, a weak base that readily permeates and neutralizes acidic organelles such as lysosomes and, as such, nonselectively represses the actions of resident acid hydrolases (73, 75). Under these conditions, chloroquine mildly impeded their clearance at 4 h, but this was not sustained at the 8-h time point relative to diluent-treated controls (Fig. 6). Collectively, these data implicate a preferential role for the endoplasmic reticulum–associated degradation (ERAD) pathway (76, 77), and to a lesser extent lysosomes, in clearance of these variants.
Figure 5.

Effect of proteasomal and lysosomal inhibitors on cellular clearance of NHE6 variants. AP-1 cells were transiently transfected with NHE6HA WT or CS-linked variants for 24 h and then treated with 150 μg/ml cycloheximide (CHX) for the indicated time points in the presence of diluent (DMSO) and the proteasomal inhibitor MG-132 (40 μm) or the lysosomal inhibitor leupeptin/pepstatin (LeuP, 100 μg/ml). Total-cell lysates were analyzed by Western blotting with a mouse monoclonal HA antibody. Membranes were also probed for β-tubulin expression as a loading control. The immunoblots are representative of three separate experiments. fg, fully-glycosylated; cg, core-glycosylated; d, dimeric; m, monomeric.
Figure 6.

Effect of the lysosomotropic agent chloroquine on cellular clearance of NHE6 variants. AP-1 cells were transiently transfected with NHE6HA WT or CS-linked variants for 24 h and then treated with 150 μg/ml cycloheximide for the indicated time points in the presence of diluent (H2O) or chloroquine (CQ, 500 μm). Total-cell lysates were analyzed by Western blotting with a mouse monoclonal HA antibody. Membranes were also probed for β-tubulin expression as a loading control. The immunoblots are representative of four separate experiments. fg, fully-glycosylated; cg, core-glycosylated; d, dimeric; m, monomeric.
Subcellular distribution of NHE6 variants
To visualize the subcellular distribution of the various NHE6 constructs, dual-labeling fluorescence confocal microscopy was performed with NHE6ChFP and organellar markers in transiently-transfected AP-1 cells at 48 h. This time point was chosen based on the extent of the biosynthetic maturation of the WT transporter that showed nominal core-glycosylation at 48 h post-transfection (presented in Fig. 3), indicating that the bulk of newly-synthesized NHE6 had exited the ER and reached a steady-state subcellular distribution. Representative images are shown in Fig. 7A. As expected, the WT transporter was present in tubulovesicular carriers distributed throughout the cell, with fluorescence signals concentrated in the para-Golgi/pericentriolar region but also dispersed peripherally in a spatial pattern that overlapped extensively with Alexa FluorTM 488–conjugated transferrin (Tf-AF488), a marker of both slow Rab11-dependent (pericentriolar) and fast Rab4-dependent (peripheral) recycling endosomes (78, 79). The fraction of WT NHE6ChFP that overlapped with Tf-AF488 in several cells was quantified by calculating the thresholded Manders' co-localization coefficient (MCC) (80–82), which revealed a high degree of co-occurrence (mean ± S.D., WT-Tf, 0.709 ± 0.062, n = 6) (Fig. 7B). An identical pattern was also observed for the A9S variant (A9S-Tf: 0.622 ± 0.038, n = 7, one-way repeated measures ANOVA with a post hoc Dunnett's test, p > 0.05). The R568Q mutant was similarly distributed but also tended to accrue more visibly at the cell surface, which resulted in a small, but statistically significant, decrease in its co-localization with internalized Tf-AF488 (R568Q-Tf: 0.519 ± 0.052, n = 6, p < 0.001) compared with WT. The L188P and G383D mutants showed a more restricted and reduced overlap with Tf-AF488–labeled endosomes, with the signals overlapping only partially in the pericentriolar region and markedly less in the cell periphery. This was reflected by significant decreases in their MCC values for co-localization with Tf-AF488 compared with WT (L188P-Tf: 0.522 ± 0.039, n = 8; G383D-Tf: 0.467 ± 0.105, n = 7; p < 0.001). Instead, sizable fractions of these mutants were dispersed in a diffuse latticed pattern suggestive of partial accumulation in the endoplasmic reticulum (ER). This was confirmed by enhanced overlap with the ER-resident protein calnexin (CANX) (L188P–CANX: 0.410 ± 0.060, n = 11; G383D–CANX: 0.551 ± 0.096, n = 10; p < 0.001), whereas WT, A9S, and R568Q showed minimal co-localization with CANX (WT–CANX: 0.269 ± 0.036, n = 8; A9S–CANX: 0.311 ± 0.057, n = 8; R568Q–CANX: 0.257 ± 0.028, n = 8) (Fig. 8, A and B). The fluorescence signals for E547* and W570* showed even lower co-localization with Tf-AF488 (E547*-Tf, 0.312 ± 0.042, n = 7 and W570*-Tf: 0.348 ± 0.059, n = 6; p < 0.001) (Fig. 7, A and B) and a more prominent overlap with CANX (E547*–CANX: 0.648 ± 0.042, n = 8; W570*–CANX: 0.645 ± 0.066, n = 9, p < 0.001) (Fig. 8, A and B) compared with WT, consistent with significant accumulation in the ER.
Figure 7.

Subcellular detection of NHE6 variants in recycling endosomes in transfected AP-1 cells. A, AP-1 cells were transiently transfected with mCherry fluorescent protein-tagged NHE6 (NHE6ChFP) WT or CS-linked variants. Forty eight hours post-transfection, cells were incubated with the recycling endosomal marker Alexa Fluor 488–conjugated transferrin (Tf-AF488, 10 μg/ml) for 45 min, fixed in 4% paraformaldehyde, mounted onto glass slides, and examined by confocal microscopy. Images show each channel individually, with merged images of the NHE6ChFP and Tf-AF488 channels. Scale bars represent 10 μm. B, quantitation of the degree of NHE6 overlapping with Tf-AF488 as determined by calculating the thresholded Mander's coefficient (M1) using ImageJ software and the JACoP plugin. Data are plotted as a box chart, with the central white square indicating the mean, the box representing the S.E., and the error bars showing the S.D. (n = 6–8 cells). Significance from WT was determined by one-way repeated measures ANOVA (F value = 6479.8, p value = 5.6 × 10−9), with a post hoc Dunnett's test, ★★★, p < 0.001.
Figure 8.

Subcellular localization of certain CS variants in the endoplasmic reticulum in transfected AP-1 cells. A, AP-1 cells were transiently transfected with mCherry fluorescent protein-tagged NHE6 (NHE6ChFP) WT or CS-linked variants. Forty eight hours post-transfection, cells were immunostained for endogenous CANX, fixed in 4% paraformaldehyde, mounted onto glass slides, and examined by confocal microscopy. Images show each channel individually, with merged images of the NHE6ChFP and CANX channels. Scale bars represent 10 μm. B, quantitation of the degree of NHE6 overlapping with CANX as determined by calculating the thresholded Mander's coefficient (M1) using ImageJ software and the JACoP plugin. Data are plotted as a box chart, with the central white square indicating the mean, the box representing the S.E., and the error bars showing the S.D. (n = 6–8 cells). Significance from WT was determined by one-way repeated measures ANOVA (F value = 3012.9, p value = 1.7 × 10−10), with a post hoc Dunnett's test, ★★★, p < 0.001.
To biochemically assess the ability of the NHE6 variants to traffic to the plasma membrane, plasmalemmal proteins were extracted using a cell-surface biotinylation procedure and analyzed by Western blotting. We previously showed that ∼5% of the total cellular pool of NHE6 is located at the cell surface of transfected AP-1 cells (57). Because of the low levels of expression of some of the NHE6 variants, we loaded varying amounts of protein in each lane (as indicated) so that their signal intensities would be comparable and easily visualized within a single X-ray film exposure of the immunoblots. As presented in Fig. 9A, all mutants were detected at the plasma membrane, although the fractional surface abundances of L188P, G383D, E547*, and W570* were noticeably reduced compared with WT, A9S, and R568Q after taking into account differences in gel protein loading. Notably, despite their lower levels, the L188P, G383D, E547*, and W570* mutants at the cell surface were largely fully-glycosylated (monomer and dimer). This indicates that these variants have the potential to fold properly and undergo oligosaccharide maturation along the exocytic pathway, although at greatly reduced efficiency. The absence of the core-glycosylated forms (monomer and dimer) in the biotinylated fractions is consistent with their intracellular location (i.e. mainly ER). To compare their plasmalemmal abundances more quantitatively, we used a cell-based enzyme-linked immunosorbent assay (ELISA). For these experiments, AP-1 cells were transiently transfected with NHE6 constructs containing a triple-Flag epitope-tag inserted in the first exofacial loop as described previously (53). Consistent with cell-surface biotinylation, the WT and A9S variants showed equivalent levels of surface expression. The plasmalemmal abundance of R568Q was detectably higher, consistent with image analyses, but this increase did not reach statistical significance (Fig. 9B). By contrast, surface abundances of the other mutants were markedly reduced by ∼55–70%, especially for G383D, E547*, and W570* (*, p < 0.05, one-way repeated measures ANOVA followed by a post hoc Dunnett's test).
Figure 9.

Assessment of the functional properties of NHE6 variants. A, biochemical determination of plasma membrane trafficking of NHE6GFP WT or CS-linked variants using a cell-surface biotinylation assay. Cell-surface proteins were labeled with N-hydroxysulfosuccinimidyl–SS–biotin in AP-1 cells expressing the NHE6GFP constructs after 48 h. Total-cell lysates (left panel; protein loading ranged from 10 to 50 μg of protein per sample as indicated below the blot) and biotinylated fractions (right panel; representing 20–100% of plasma membrane proteins extracted per sample) were examined by Western blotting with polyclonal anti-GFP and monoclonal anti-GAPDH antibodies. Representative blots from three experiments are shown. B and C, surface expression and endocytosis of external triple flag tag–labeled NHE6 (3FNHE6HA) constructs in transiently transfected (48 h) AP-1 cells using a cell-based ELISA. Mean intensity fluorescence (M.I.F.) units were determined as a function of the cellular protein concentration and then normalized as percentage (M.I.F. units for WT (100%): 25,100 ± 6,348, n = 4). The surface expression of each construct at time 0 min (before the start of internalization) is charted in B (n = 3–4 experiments). Significance from WT-expressing cells was determined using a one-way repeated measures ANOVA (F value = 463.3, p value = 0.0022), with a post hoc Dunnett's test; *, p < 0.05. Percentage internalization of NHE6 constructs normalized to the zero time point are presented in C and represent the mean ± S.D. (n = 3–4 experiments). The NHE6 variants clustered into two groups: 1) WT, A9S, and R568Q, and 2) L188P, G383D, E547*, and W570*, with variants within each cluster yielding similar statistical values. Significance from WT cells at the 5- and 15-min time points was determined using a one-way ANOVA (F value = 9.43, p value = 4.48 × 10−5), with a post hoc Tukey test; ★, p < 0.05. D, transferrin uptake in HeLa cells transiently transfected (48 h) with GFP or NHE6GFP constructs. The initial uptake (5 min) of Alexa 633–conjugated transferrin (Tf-AF633) was measured in 1 × 104 GFP-positive HeLa cells per experiment by flow cytometry (M.I.F. units for GFP control: 10,204 ± 1554, n = 4). Data were normalized as a percentage and displayed as percent change from GFP control cells. Significance from control cells was determined using a one-way repeated measures ANOVA (F value = 320.7, p value = 3.8 × 10−4), with a post hoc Dunnett's test; ★★, p < 0.001. E, recycling endosomal pH (pHe) was measured in AP-1 cells in the absence or presence of transiently transfected (48 h) NHE6ChFP constructs by fluorescence ratio image analysis of the internalized pH-sensitive probe FITC-conjugated human transferrin (Tf–FITC). Data represent the average endosomal pHe per cell pooled from three separate experiments (8–12 cells per construct/experiment; n = 24–36). Significance was determined by one-way ANOVA (F value = 40.02, p value = 0), with a post-hoc Tukey test; ★★, p < 0.001. Data in B, D, and E are plotted as box charts, with the central white square indicating the mean; the box representing the S.E.; and the error bars showing the S.D. fg, fully-glycosylated; cg, core-glycosylated; d, dimeric; m, monomeric.
Based on the sensitivity of the ELISA, we proceeded to measure endocytosis of the different NHE6 variants. For these experiments, cells transiently expressing the various constructs were placed on ice and incubated with a primary mouse monoclonal anti-Flag antibody and secondary goat anti-mouse antibody to label cell surface 3FNHE6HA, and then endocytosis of the labeled pool was initiated by incubating the cells at 37 °C over a 15-min period. As shown in Fig. 9C, the levels of internalization of surface-labeled A9S and R568Q were equivalent to WT, whereas L188P, G383D, E547*, and W570* were translocated more slowly (p < 0.05), indicative of their differential handling by the endocytic trafficking machinery.
Effect of NHE6 variants on recycling endosomal cargo trafficking and pH
Previous studies have shown that overexpression and knockdown of NHE6 promoted up- and down-regulation, respectively, of clathrin-mediated endocytosis of certain integral plasma membrane proteins, such as the ligand-activated transferrin receptor (Tf–TfR) but not the epidermal growth factor receptor, in a manner that depended on its ability to modulate intravesicular pH (51, 53).
To determine the effect of the NHE6 variants on internalization of Tf, we measured the initial rate of uptake (5-min period) of Alexa FluorTM 633-conjugated Tf (Tf-AF633) by flow cytometry in live HeLa cells transiently expressing with GFP alone (control) or GFP-tagged constructs of NHE6 as described previously (53). For these experiments, HeLa cells were used because their TfR levels are severalfold higher than in AP-1 cells and therefore provided a stronger signal-to-noise ratio for detecting the initial uptake of Tf-AF633 (53). The patterns of protein biosynthesis and oligosaccharide maturation for the various NHE6 constructs in transfected HeLa cells were equivalent to those observed in AP-1 cells (data not shown). Equivalent numbers of live GFP-positive HeLa cells (104 GFP+ cells) were analyzed for each construct; live cells were distinguished from nonviable cells by their ability to exclude the membrane-impermeant fluorescent dye 7-amino-actinomycin D. HeLa cells overexpressing WT, A9S, and R568Q exhibited significant increases (∼25–35%) in Tf-AF633 uptake compared with control GFP-only expressing cells (p < 0.001, one-way repeated measures ANOVA followed by a post hoc Dunnett's test) (Fig. 9D). However, this up-regulation was greatly diminished or absent in cells expressing the L188P, G383D, E547*, and W570* mutants (p ≥ 0.05).
The regulation of endosomal trafficking by NHE6 is dependent on its ability to regulate endosomal pHe, although the underlying mechanism linking transporter function to vesicle trafficking is unknown. NHE6, like other NHE family members, is a secondary active transporter that moves monovalent cations down their respective concentration gradients, and thus at steady-state it should operate as a “H+ leak” pathway (i.e. alkalinizing mechanism) to counter acidification driven by the vacuolar H+-ATPase. To examine whether this property is affected, we measured the pHe of recycling endosomes in situ by fluorescence ratiometric image analysis (FRIA) (83) using the pH-sensitive fluorophore fluorescein isothiocyanate (FITC) conjugated to transferrin (Tf–FITC) as a probe. Briefly, AP-1 cells were transiently transfected (48 h) with ChFP alone or the different NHE6ChFP constructs, followed by a 60-min incubation with Tf–FITC at 37 °C to allow for sufficient accumulation of the probe throughout the recycling endosomal pathway. The steady-state pHe spectrum of vesicles or puncta under each condition was determined by analyzing fluorescence signals pooled from a total of 25–36 cells (∼2700–5300 vesicles collected from 8 to 12 cells per transfectant per experiment, repeated three times). In each case, the pHe spectrum of the vesicles exhibited a unimodal distribution (see Fig. S1). When the data were expressed as the mean recycling endosomal pHe per cell, the level for AP-1 cells lacking NHE6 was very acidic at pHe 5.79 ± 0.11 (mean ± S.D., n = 27 cells) (Fig. 9E). By contrast, endosomal pHe values for WT-, A9S-, and R568Q-expressing cells were significantly more alkaline and equivalent to each other relative to parental AP-1 cells (WT: 6.23 ± 0.16, n = 28; A9S: 6.22 ± 0.19, n = 34; R568Q: 6.20 ± 0.15, n = 28; p < 0.001, one-way ANOVA followed by a post hoc Tukey test). In contrast, endosomal pHe levels of L188P- and G383D-expressing cells were intermediate and significantly different from both control and WT-transfectants (L188P: 6.05 ± 0.15, n = 36; G383D: 5.96 ± 0.13, n = 34; p < 0.001). These intermediate pHe values could reflect incomplete distribution of these NHE6 variants throughout the recycling endosomal compartments containing Tf–FITC (i.e. the averaged pHe of NHE6-containing and noncontaining recycling endosomes), but they may also result from impairment of their catalytic activity (i.e. diminished H+ efflux) in recycling endosomes or a combination of both possibilities. Finally, the E547* and W570* variants had no significant effect on recycling endosomal pHe compared with parental AP-1 cells (AP-1: 5.79 ± 0.11, n = 27; E547*: 5.90 ± 0.13, n = 35; W570*: 5.88 ± 0.12, n = 25; p > 0.05), an expected outcome given their pronounced retention in the ER.
Because the L188P, G383D, E547*, and W570* variants showed significant accumulation in the ER, we next evaluated whether they influenced the luminal pHe of this compartment. To this end, AP-1 cells were co-transfected with the various mCherry-tagged NHE6 (NHE6ChFP) constructs and a ratiometric pH-sensitive GFP (i.e. pHluorin2 or pH2) (84) containing an ER signal sequence from calreticulin at its N terminus and an ER retention sequence (KDEL) at its C terminus (construct called ERpH2). ER pHe in AP-1 cells was 7.28 ± 0.11 (mean ± S.D., n = 8 cells) (Fig. S2), a value comparable with resting ER pHe (pH 7.1–7.4) reported for other cell types (85–87). As expected, this level was unchanged in cells co-expressing recycling endosome-sorted WT (7.23 ± 0.12, n = 14), A9S (7.22 ± 0.10, n = 13), or R568Q (7.29 ± 0.13, n = 14). ER pH was also unaffected by the L188P (7.26 ± 0.15, n = 16), G383D (7.16 ± 0.18, n = 13), E547* (7.29 ± 0.11, n = 15), and W570* (7.24 ± 0.13, n = 14) variants, indicating that they are not demonstrably active when retained in the ER. Additional control experiments using cytoplasmic-localized phluorin2 (CytopH2) showed that cytoplasmic pH (pHc 7.59 ± 0.17, n = 21) was also unaltered in the presence of NHE6ChFP WT or its variants (see Fig. S3).
Effect of NHE6 variants on cell viability
Previous studies have revealed that genetic disruption of NHE6 expression causes degeneration of a subset of neurons within the cortex, hippocampus, and especially the cerebellum, which exhibits extensive Purkinje cell loss with age (32, 34, 35). Neurons in other brain regions, despite the loss of NHE6 expression, do not appear as severely affected. The reason for these differences in cell viability is unclear. AP-1 cells, which also lack NHE6, also show little evidence of cell death under normal culture conditions (53). However, ectopic expression of a CS-linked in-frame deletion variant of NHE6 (p.E287–S288del, Δ287ES288) in AP-1 cells and in primary mouse hippocampal neurons could induce programmed cell death (53). Thus, AP-1 cells serve as a useful proxy for assessing the potential in vitro consequences of NHE6 variants on cell survival. To this end, AP-1 cells were transiently transfected with GFP alone or GFP-tagged NHE6 variants for 48 h. These cells then were incubated in the presence of the fluorescent annexin V–allophycocyanin conjugate (annexin V–APC) and propidium iodide (PI), and GFP-positive cells (104 cells per experiment) were analyzed by flow cytometry to determine the fraction of apoptotic cells as described previously (53). Annexin V–APC-positive and PI-negative cells represent the early stages of apoptosis, whereas annexin V–APC/PI–double-positive cells are late apoptotic or necrotic, and PI-only–positive cells are considered necrotic. As presented in Fig. 10, a significantly higher proportion of early apoptotic cells was detected in cells expressing L188P, G383D, E547*, and W570* (∼22–27%) compared with WT, A9S, or R568Q transfectants (∼12–14%) (p < 0.001). At this time point, there were no significant increases in late apoptotic or necrotic cells among the CS-linked variants relative to WT. Curiously, expression of WT, A9S, and R568Q also significantly increased the percentage of apoptotic cells relative to AP-1 cells (p < 0.001), but not as greatly as the other four defective variants. The reason for this is unclear and will require further study. Nevertheless, these findings suggest that L188P, G383D, E547*, and W570* have the potential to reduce cell viability relative to WT, although the underlying mechanisms remain obscure.
Figure 10.

Apoptosis is enhanced in AP-1 cells expressing L188P, G383D, E547*, and W570* mutants. Measurement of apoptosis by flow cytometry analysis of AP-1 cells transfected with NHE6GFP WT and CS-linked variants is shown. Forty eight hours post-transfection, cells were labeled with annexin V–APC and PI, and 1 × 104 GFP-positive cells were examined by flow cytometry for each transfectant. A, annexin V and PI double-negative cells represent viable cells. B, PI-positive cells represent necrotic cells. C, annexin V–positive cells represent early apoptotic cells. D, annexin V and PI double-positive cells represent late apoptotic cells. Results are shown as mean ± S.D. of three independent experiments. Significance was determined by one-way ANOVA (A, F value = 27.7, p value = 8.5 × 10−8; B, F value = 2.1, p value = 0.11; C, F value = 40.9, p value = 4.9 × 10−9; D, F value = 5.4, p value = 0.0025), with a post hoc Tukey test; ★, p < 0.05; ★★, p < 0.01; and ★★★, p < 0.001.
Discussion
CS patients display a wide range of neurological and somatic symptoms (1, 15), with their acuteness ostensibly governed by the nature of the mutation as well as other intrinsic compensatory or aggravating influences. The findings reported herein reveal differences in the biochemical properties and cellular handling of several SLC9A6/NHE6 variants associated with CS. Of the six variants examined, four (L188P, G383D, E547*, and W570*) showed varying degrees of impairment in post-translational oligosaccharide maturation, protein stability, and membrane targeting. L188P and G383D partially accumulated in the ER, but measurable fractions of L188P and to a lesser extent G383D were fully-glycosylated and sorted to a pericentriolar endosomal compartment. By contrast, both E547* and W570* were largely retained in the ER, although minor fractions did exit the ER and became fully-glycosylated. However, in each case their trafficking to the cell surface and internalization into recycling endosomes was compromised, albeit to different extents (WT > L188P > G383D ≥ E547* ≈ W570*). Each variant exhibited correspondingly diminished abilities to stimulate the internalization of endosomal cargo (i.e. ligand-activated transferrin receptor), to regulate acidification of recycling endosomes, and to maintain cell viability. Remarkably, two of the purported CS variants, A9S (3, 13, 25) and R568Q (4, 25), exhibited properties that were essentially indistinguishable from the WT transporter that obscures their disease significance, at least when examined in AP-1 cells. Although this experimental cell system has proven informative in deciphering NHE6 function, we cannot exclude the possibility that any deleterious consequences of the A9S and R568Q variants may be more subtle and manifested only in certain neuronal cell types. However, during the preparation of this manuscript, Ouyang and co-workers (88) reported that mice genetically manipulated by CRISPR/Cas9 technology to harbor the homologous human NHE6 A9S variant (i.e. mouse A11S) appeared normal in terms of brain morphology, NHE6 subcellular localization, and intra-endosomal pH of hippocampal neurons. Although a behavioral assessment of these mice was not described, we envision that human A9S and possibly the R568Q variants are likely benign and possibly misattributed as the cause of the patients' neurological symptoms. This suggestion is further supported by the common occurrence of both variants in the general human population (genome aggregation database, gnomAD; RRID:SCR_014964).
Preferential degradation of defective NHE6 variants by ERAD
A biochemical feature of mature WT NHE6 is the covalent attachment of N-linked oligosaccharides (53). In unpublished studies,3 we have identified two neighboring asparagine residues located in canonical N-glycosylation motifs (Asn–Xaa–Ser/Thr) in the second extracellular loop, Asn128–Val–Thr and Asn145–Val–Ser, that serve as the sole targets for N-glycan addition. Post-translational addition of N-glycans (as well as O-glycans) in the ER and Golgi is known to facilitate intracellular processes such as the folding, multisubunit assembly, membrane trafficking, and function of many glycoproteins (89–91). As described herein, the L188P, G383D, E547*, and W570* variants showed marked reductions in their N-oligosaccharide maturation and membrane targeting, especially the latter three, and correlated with increased retention in the ER and cellular clearance. These hypo-glycosylated variants assembled correctly as homodimers, indicating that their quaternary structures were not grossly perturbed. Instead, their glycosylation impairments are more consistent with localized misfolding, as was predicted for the L188P and G383D variants by structural modeling analyses. Aberrant conformations of N-glycoproteins, which can include exposed hydrophobic segments, improper disulfide bond formation, and immature glycans, are recognized by the ER protein quality control machinery that encompasses multiple systems capable of detecting discrete structural malformations situated in the ER lumen, within the membrane (i.e. L188P and G383D), or facing the cytoplasm (i.e. E547* and W570*) (92–94). Misshapen proteins are then subjected to repeated cycles of deglucosylation–reglucosylation of the initial core N-glycan structure (Glc3–Man9–GlcNAc2) and binding to the lectin chaperones calnexin and calreticulin, which assist in protein refolding until the proper conformation is attained (95, 96). However, deformed proteins refractory to the refolding process after a prolonged ER residency are targeted for ERAD by proteasomes (93). This enhanced proteasomal proteolysis was evident for the L188P, G383D, E547*, and W570* variants. In the case of the E547* and W570* truncation variants, aside from possible misfolding, their pronounced accumulation in the ER might also result from the loss of one or more specific ER-export signal motifs (97, 98). Absence of such signals could delay their exit from the ER and favor retrotranslocation to the proteasome (99). The fate of the minor fraction of these variants that did escape ERAD, possibly in part due to overexpression, could be degradation in lysosomes. However, blocking lysosomal proteolysis with leupeptin/pepstatin or chloroquine did not substantially prevent their clearance, suggesting that other pathways such as cellular expulsion in exosomes might be an alternate route (100). Such a mechanism has been proposed for another CS-linked variant p.Gly218 → Arg (57) and remains to be tested for these variants.
Defective NHE6 variants disrupt recycling endosomal pH homeostasis and trafficking
The severity of sorting defects for L188P, G383D, E547*, and W570* correlated with corresponding deficits in recycling endosomal function, as revealed by excessive luminal acidification and reduced uptake of recycling endosomal cargo (e.g. ligand-activated transferrin receptor) relative to WT transfectants. Optimal acidification of organelles is required not only for efficient biochemical processing of their contents, but also for effective sorting and trafficking of proteins and lipids along the biosynthetic and endocytic pathways (48, 101–103). Yet how intravesicular pH influences vesicle movement is not fully understood. Recent studies have indicated that the vacuolar H+-ATPase pump functions not only in organellar acidification but also as a luminal pH-sensor and binding platform for members of the ADP-ribosylation factor family of small GTPases involved in vesicular trafficking (49, 50, 104). Curiously, despite the broad organellar distribution of the vacuolar H+-ATPase, inhibiting its activity with pharmacological agents disrupted only vesicular movement between the early and late endosome/lysosomal compartments, while trafficking along the recycling endosomal pathway was relatively unaffected (49). This raises the possibility that other endosomal pH-regulatory transporters may compensate and play more prominent multifunctional roles in vesicular trafficking along other endomembrane pathways. In keeping with this notion, the yeast Saccharomyces cerevisiae endomembrane (Na+,K+)/H+ exchanger Nhx1/Vps44 was found to form a complex with the GTPase-activating protein Gyp6 and one of its effectors Ypt6, the homolog of the mammalian Rab6 GTPase, and served to coordinate endosomal shuttling between the Golgi and prevacuolar (late endosomes) compartments (105). Loss of Nhx1 caused hyperacidification and disruption of endosomal protein trafficking and fusion to the vacuole (106–109). In an analogous manner, mammalian NHE6 might fulfill a similar role in pH-sensing and modulation of vesicular trafficking along the recycling endosomal pathway, a process that is diminished by loss of NHE6 function (33, 51–53). Indeed, we found that excessive acidification and impaired trafficking of recycling endosomes were most pronounced for E547*- and W570*-expressing cells at levels comparable with those of NHE6-deficient AP-1 cells. This would be expected as these variants are not properly sorted to recycling endosomes. By contrast, values for luminal pHe and trafficking of L188P-containing endosomes were intermediate between WT transfectants and AP-1 cells, indicating that this variant retained some activity to counter H+ influx driven by the vacuolar H+-ATPase. Leu188 is a highly-conserved amino acid in transmembrane helix M4, a segment previously identified as a critical part of the ion permeation pore of the plasmalemmal NHE1 isoform (60, 67, 68). Thus, insertion of an additional proline residue in this region is likely to cause steric or rotamer constraints on the geometry of M4 that compromises catalytic activity. G383D also exhibited some nominal ability to elevate recycling endosomal pHe above that measured in AP-1 cells, but this level was insufficient to significantly stimulate uptake of transferrin. The crippling effect of the G383D mutation was predicted from the structural homology modeling analyses and consistent with earlier findings (69) showing that nonpolar and polar amino acid substitutions of the homologous Gly residue in NHE1 (i.e. human Gly352 and rat Gly356) greatly reduced its catalytic activity (∼10% of WT) as well as the binding of amiloride- and benzoylguanidine-based antagonists. Thus, all four variants elicit deleterious effects on recycling endosomal function.
Expression of defective NHE6 variants reduces cell viability
Progressive cerebellar atrophy, loss of Purkinje neurons, and motor regression are common features of a majority (∼60%) of CS patients examined thus far (1, 2, 6, 7, 10, 14). Less severe neuronal loss and gliosis have been documented in other brain regions, such as the cerebral cortex and hippocampus (6). Similar degeneration of the cerebellum and mild undergrowth and atrophy of the hippocampus, striatum, and cortex were also observed in Slc9a6 knockout mice (32, 34, 35). The variable penetrance and seemingly selective deterioration of certain neuronal populations upon loss of NHE6 function, despite its broad expression throughout the CNS, is not well-understood but may reflect the nature of the mutation as well as other ill-defined genetic modifier effects or compensatory mechanisms. The latter could include expression of its close paralog NHE9/SLC9A9, which is also important for neurological function, as defects in NHE9 expression have been linked to attention-deficit hyperactivity and autism spectrum disorders in humans (26, 110–114) and autism-like behavior in mice (115, 116). Using the AP-1 cell line as a surrogate model for assessing cell viability, we found that AP-1 cells lacking NHE6 showed only nominal cell death (<10%). Surprisingly, expressing WT, A9S, and R568Q raised this percentage (∼23%), but it was further elevated in the presence of the L188P, G383D, E547*, and W570* variants (33–40%). The mechanisms by which NHE6 modulates cell viability have yet to be elucidated. However, insights can be gleaned from recent studies showing that NHE6 and the neurotrophin TrkB receptor co-localize in recycling endosomes of cultured mouse hippocampal neurons (33). The Trk family of neurotrophin receptors are known positive regulators of nervous system development, survival, and plasticity (117–119). Notably, loss of NHE6 results in overacidification of recycling endosomes and diminished TrkB receptor signaling, neurite branching, synapse number, and circuit connectivity (33). Remarkably, these morphological and functional deficits could be rescued by activation of residual TrkB receptors with high levels of exogenous brain-derived neurotrophic factor (33). Thus, we posit that NHE6 impairment of neurotrophin Trk receptor-mediated prosurvival signals may shift the equilibrium toward neurotrophin p75NTR receptor-mediated proapoptotic signals (120–122) leading to degeneration and death of certain neuronal populations. Likewise, imbalances of endosomal trafficking and signaling of prosurvival growth factor receptor and proapoptotic death- or dependence-receptor signaling pathways in non-neuronal cells (123–126) may also explain the observed changes in survival of AP-1 cells expressing the various NHE6 constructs.
Aside from imbalances in prosurvival versus proapoptotic signaling pathways, ER stress (127) may also be another factor that decreases the function and viability of certain cells possessing the CS-linked variants. Accumulation of malformed proteins in the ER initiates the unfolded protein response and ERAD in order to restore cellular homeostasis (128, 129). However, prolonged ER stress can activate several protein kinases, including glycogen synthase kinase-3β (GSK-3β) (130) and inositol-requiring kinase 1 (IRE1)/apoptosis signal-regulating kinase 1 (ASK1)/c-Jun N-terminal kinase signaling cascade (127, 131), ultimately stimulating caspase-mediated cell death. Post-mitotic neurons are particularly vulnerable to ER stress, as they are unable to dissipate any accumulated misfolded proteins through cell division. This inability to undergo mitosis may be an additional contributing factor in the progression of CS as well as many other neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and Huntington's disease (132).
In summary, our results provide additional insight into the molecular mechanisms by which different NHE6 variants linked to CS impair recycling endosomal pH homeostasis and cargo trafficking, and ultimately cell viability. These analyses will guide future investigations to delineate the molecular network of proteins that interact with NHE6 and influence receptor trafficking, synaptic plasticity, and learning deficits. Such knowledge has the potential to identify novel diagnostic and treatment options for CS that may also be applicable to other disorders such as autism and attention deficit hyperactivity disorders.
Experimental procedures
Antibodies and reagents
Mouse monoclonal anti-hemagglutinin (HA) antibody was purchased from Covance Inc. (Berkeley, CA); rabbit polyclonal anti-HA and mouse monoclonal anti-GAPDH were obtained from Abcam Inc. (Cambridge, MA); mouse monoclonal anti-Flag M2 and anti-β-tubulin antibodies were from Sigma; rabbit polyclonal anti-GFP antibody was from Life Technologies, Inc.; and rabbit polyclonal anti-calnexin was from Enzo Life Sciences, Inc. Horseradish peroxidase-conjugated secondary IgG antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). All Alexa Fluor®-conjugated secondary antibodies, Alexa Fluor® 488- and 633-conjugated transferrin (Tf-AF488 and Tf-AF633, respectively), and Tf-FITC were purchased from Molecular Probes (Eugene, OR). α-Minimum essential medium (α-MEM), fetal bovine serum, penicillin/streptomycin, and trypsin-EDTA were purchased from Wisent (Saint-Bruno, Quebec, Canada). All other chemical and reagents were obtained from BioShop Canada (Burlington, Ontario, Canada), Sigma, or Thermo Fisher Scientific and were of the highest grade available.
Recombinant DNA constructs and mutagenesis
The long transcript splice-variant of human NHE6 (NHE6v1; NCBI NM_001042537) was engineered to contain the influenza virus HA (YPYDVPDYAS) epitope at its extreme C terminus (NHE6HA), as described previously (56). NHE6HA was then used as a template to introduce a triple-Flag epitope (AAADYKDDDDKGDYKDDDDKGDYKDDDDKAAA) in the first extracellular loop immediately after residue Met53 (3FNHE6HA) (56). Enhanced GFP and monomeric ChFP were also fused to the C terminus of WT NHE6 (NHE6GFP and NHE6ChFP). The various tagged NHE6 constructs were then used as templates to engineer the various CS-linked mutations by PCR mutagenesis. Insertion of the different epitope tags in the various positions did not alter the biochemical properties or cellular distribution of exogenous NHE6 compared with the endogenous protein (56). All constructs were sequenced to ensure that no additional mutations were introduced during PCR.
Cell culture
Chinese hamster ovary AP-1 and HeLa cells were cultured in α-MEM supplemented with 10% fetal bovine serum, penicillin (100 units/ml), streptomycin (100 μg/ml), and 25 mm NaHCO3 (pH 7.4), as described previously (56).
Western blotting
For Western blotting analyses, AP-1 and HeLa cells were grown in 10-cm dishes and transiently transfected with 5 μg of plasmid DNA encoding NHE6GFP or NHE6HA WT or mutant constructs using Lipofectamine2000TM (Invitrogen) according to the manufacturer's recommended procedure. Cell lysates were prepared following 6–48 h post-transfection (as indicated in the figure legends) by washing cells twice on ice with ice-cold PBS, followed by scraping in 0.5 ml of lysis buffer (1% NP-40, 0.25% sodium deoxycholate, PBS supplemented with protease inhibitor mixture (Roche Diagnostics)). Lysates were incubated for 30 min on a rocker at 4 °C and then centrifuged at 16,000 × g for 20 min at 4 °C to pellet the nuclei and cellular debris. Ten to 50 μg of protein (as indicated in the figure legends) from the resulting supernatants were diluted in SDS-sample buffer (50 mm Tris-HCl, pH 6.8, 1% SDS, 50 mm DTT, 10% glycerol, 1% bromphenol blue), subjected to 9% SDS-PAGE, and then transferred to polyvinylidene fluoride membranes (Millipore, Nepean, Ontario, Canada) for immunoblotting. The membranes were blocked with 5% nonfat skim milk for 1 h and then incubated with the specified primary antibodies (rabbit polyclonal GFP 1:8000, mouse monoclonal HA 1:5000, β-tubulin 1:10,000, or GAPDH 1:50,000) in PBS containing 0.1% Tween 20, followed by extensive washes and incubation with goat anti-mouse (1:5000) or goat anti-rabbit (1:8000) horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h. Immunoreactive bands were detected using Western LightningTM Plus–ECL blotting detection reagents (PerkinElmer Life Sciences). Quantification of immunoreactive signals was determined by densitometry of X-ray films exposed in the linear range and analyzed using ImageJ software.
Cell-surface biotinylation
AP-1 cells expressing NHE6GFP WT or mutant constructs were cultured in 10-cm dishes to subconfluence, placed on ice, and washed three times with ice-cold PBS containing 1 mm MgCl2 and 0.1 mm CaCl2, pH 8.0 (PBS-CM). Next, cells were incubated at 4 °C for 30 min with the membrane-impermeable reagent N-hydroxysulfosuccinimidyl–SS-biotin (0.5 mg/ml) (Thermo Fisher Scientific, Rockford, IL). Cells were washed and incubated twice in quenching buffer (50 mm glycine in PBS-CM) for 7 min each on ice to remove unreacted biotin. After two more washes in PBS-CM, the cells were lysed for 30 min on ice and then centrifuged at 16,000 × g for 20 min at 4 °C to remove insoluble cellular debris. A fraction of the resulting supernatant was removed, and this represents the total fraction. The remaining supernatant was incubated with 100 μl of 50% NeutrAvidin®–agarose resin slurry (Thermo Fisher Scientific, Whitby, Ontario, Canada) in lysis buffer overnight at 4 °C to extract biotinylated membrane proteins. The proteins were then resolved by SDS-PAGE and analyzed by Western blotting.
Immunofluorescence confocal microscopy
AP-1 cells were cultured on fibronectin-coated 18-mm glass coverslips, transfected with the NHE6 constructs, and fixed post-transfection (at indicated times) with 4% paraformaldehyde for 20 min at room temperature. To examine the subcellular distribution of the NHE6 constructs, AP-1 cells were co-transfected with the various NHE6ChFP constructs and different organellar markers. To label transferrin-containing recycling endosomes, AP-1 cells were transfected with NHE6ChFP constructs for 48 h and then incubated with Alexa Fluor® 488-conjugated transferrin (Tf–AF488) for 45 min in serum-free α-MEM, washed, fixed, and mounted. To visualize the ER, AP-1 cells expressing NHE6ChFP constructs were fixed 48 h after transfection and labeled with rabbit polyclonal anti-CANX followed by goat anti-rabbit Alexa Fluor® 488-conjugated secondary antibodies, washed, and mounted. Cells were examined by laser-scanning confocal microscopy using the ZEN software of a Zeiss LSM 780 microscope equipped with a photomultiplier tube (PMT) detector, with images acquired using a ×63/1.4 NA oil immersion objective lens.
Fluorescence-based endocytosis assay
Cell-surface expression and internalization of the 3FNHE6HA constructs was determined using an ELISA, as described previously (53). Briefly, AP-1 cells were grown in 10-cm dishes and transiently transfected with 6 μg of empty plasmid vector (pcDNA3) or vectors containing 3FNHE6HA WT or mutant DNA constructs. Twenty four h post-transfection, cells were transferred to 12-well plates and grown for an additional 12–24 h. Cells were chilled on ice, washed with ice-cold PBS-CM, pH 7.4, blocked in 10% goat serum/PBS-CM, and then incubated with a mouse monoclonal anti-Flag antibody (1:3000) (Sigma) on ice. Internalization of the bound antibody was initiated by incubating the cells with warm (37 °C) α-MEM for the indicated time points and terminated by placing the plates on ice. Cell were washed and labeled with goat anti-mouse HRP-conjugated secondary antibody (1:1000) (GE Healthcare). After extensive washes with PBS-CM, cells were treated on ice with Amplex® Red reagent (Invitrogen). Aliquots were transferred to 96-well plates, and fluorescence readings were taken with a POLARstar OPTIMA (BMG Labtech. Inc, Offenburg, Germany) plate reader using 544-nm excitation and 585-nm emission wavelengths. All experiments were performed in triplicate and repeated at least three times. Results were expressed as a percentage of the fluorescence recorded prior to internalization, after subtraction of the value measured with empty vector-transfected cells.
Measurements of organellar pH
Recycling endosomes
AP-1 cells were grown in 6-well plates and transfected with 1 μg of ChFP alone (control) or the NHE6ChFP constructs using Lipofectamine2000 according to the manufacturer's instructions. Twenty four hours after transfection, cells were transferred to fibronectin-coated (2 μg/ml in PBS, pH 7.4, overnight at 4 °C) FluoroDishesTM (World Precision Instruments, Inc.) and grown overnight. Before loading with transferrin, cells were serum-starved in serum-free α-MEM for 1 h at 37 °C and 5% CO2. Next, the cells were incubated with FITC-conjugated human transferrin (Tf–FITC) (Molecular Probes) at 6.6 μg/ml in serum-free α-MEM for 1 h at 37 °C and 5% CO2. Cells were subsequently washed twice with warm α-MEM supplemented with 10% fetal bovine serum, keeping the second wash for 1 min to allow for internalization of remaining plasma-membrane bound Tf–FITC. Next, cells were washed three times in ice-cold PBS-CM (PBS containing 1 mm MgCl2 and 0.1 mm CaCl2, pH 7.4) and imaged in the same solution at room temperature. Endosomal pH was measured by single-cell FRIA using a Carl Zeiss MicroImaging AxioObserver Z1 inverted microscope with a Plan-Apochromat ×63/1.4 numerical aperture oil immersion objective and equipped with a X-Cite 120Q fluorescence illumination system (Lumen Dynamics Group Inc.) and an Evolve 512 electron-multiplying charged-coupled device camera (Photometrics Technology). MetaFluor software (Molecular Devices) was used for image acquisition and vesicle selection. Images were acquired at 490- and 440-nm excitation wavelengths. Average intensities of fluorescent puncta (0.3–3 μm in diameter) (570-nm emission using a long-pass 525-nm emission filter) were obtained for both the 440 and 490 images, and the 490/440 ratios were calculated.
Calibration curves of fluorescence as a function of vesicular pH values were performed in situ in AP-1 cells containing Tf–FITC by clamping the vesicular pH between 5 and 7.8 in K+-rich medium (135 mm KCl, 10 mm NaCl, 20 mm Hepes or 20 mm MES, 1 mm MgCl2, and 0.1 mm CaCl2) with 10 μm nigericin, 10 μm monensin, 0.4 μm bafilomycin, and 20 μm CCCP and recording the 490/440 fluorescence ratios, as described above. The calibration curves and Gaussian distributions of vesicular pH values were plotted with OriginPro 8 software (OriginLab, Northampton, MA).
Endoplasmic reticulum
To measure pH in the ER, an ER-targeted pH-sensitive probe was engineered by replacing Dendra 2 in Dendra2_ER-5 (a gift from Michael Davidson, Addgene plasmid no. 57716) by the pH-sensitive GFP pHluorin2 using the AgeI and BspEI restriction sites. This modified construct contains an ER signal sequence from calreticulin at its N terminus and an ER retention sequence (KDEL) at its C terminus (named ERpH2). AP-1 cells were grown in 6-well plates and transfected with 1 μg of ERpH2 alone or together with 1 μg of ChFP NHE6ChFP WT or mutants using Lipofectamine2000 according to the manufacturer's instructions. Twenty four hours after transfections, cells were transferred to fibronectin-coated FluoroDishesTM (World Precision Instruments, Inc.) and grown overnight. ER pH was measured by single-cell FRIA at 37 °C, using a Zeiss LSM 780 confocal microscope, equipped with a PMT detector. Images were acquired with a ×63/1.4 NA oil immersion objective lens by sequential line scanning at 405 and 488 excitation wavelengths, with emission set at 500–550 for both channels. All cells were imaged in a heated chamber (37 °C, 5% CO2) at ×3 zoom, and the laser power, resolution, speed of scanning, digital gain and offset, pinhole opening, and line averaging were identical for both channels. Average intensities of fluorescent puncta or regions of interest (0.3 to 3 μm in diameter) were obtained for both the 405 and 488 channels using the MetaXpress software (Molecular Devices, Downingtown, PA), and 488/405 ratios were calculated. Calibration curves of fluorescence as a function of pHe were performed in situ in AP-1 cells expressing ERpH2 by clamping the intracellular pH between 5 and 7.8 in K+-rich medium (135 mm KCl, 10 mm NaCl, 20 mm Hepes or 20 mm MES, 1 mm MgCl2, and 0.1 mm CaCl2) with 10 μm nigericin, 10 μm monensin, 0.4 μm bafilomycin, and 20 μm CCCP and recording the 488/405 fluorescence ratios, as described above. The calibration curves and Gaussian distributions of pH values were plotted with OriginPro 8 software (OriginLab, Northampton, MA).
Flow cytometry
To measure transferrin uptake by flow cytometry, HeLa cells were transfected with GFP alone or the NHE6GFP constructs using FuGENE6 (Promega). Twenty four hours after transfection, the cells were serum-depleted for 2 h, then incubated with Alexa Fluor® 633-conjugated transferrin (Tf-AF633, 10 μg/ml) for 5 min at 37 °C, and followed by washes to remove unbound transferrin. Cells were detached from the plates by trypsinization, and 5 μl of the cell viability dye 7-amino-actinomycin D (7-AAD, eBioscience) was added to each cell suspension. Cells were analyzed by flow cytometry using a FACS Aria Sorter (BD Biosciences). A gate was set around the GFP-positive cells, and the amount of Tf-AF633 taken up by 104 GFP-expressing live cells (i.e. 7-AAD negative) was measured using the FACS Diva software (BD Biosciences).
Apoptosis assays
Apoptosis in AP-1 cells was measured using a flow cytometry assay that measures changes in plasma membrane asymmetry binding of annexin V–allophycocyanin conjugate (annexin V–APC) to phosphatidylserine and permeability to propidium iodide (PI) as described previously (53). Briefly, AP-1 cells were grown in 6-cm dishes and transfected with 1 μg of GFP or 4 μg of NHE6v1GFP WT or mutant constructs using Lipofectamine2000 (Invitrogen). Forty eight hours post-transfection, the cells were washed twice with warm (37 °C) PBS, detached using Cell Dissociation Buffer (Gibco), and then collected by centrifugation. The cell pellets were resuspended in PBS and labeled using the annexin V–APC apoptosis detection kit (eBioscience) according to the manufacturer's instructions. After labeling, cells were placed on ice, and 1 × 104 GFP-positive cells were examined on a BDTMLSR II flow cytometer, and the percentage of annexin V-positive cells (i.e. early apoptotic), PI-positive (i.e. necrotic), and annexin V/PI-double positive cells (i.e. late apoptotic) was determined.
MD simulation
The construction of a homology structure model of monomeric human NHE6 (transcript splice-variant 1) was described in our previous publication (57). The objective of the simulation was to characterize the potential effects of the L188P mutation located in membrane-spanning helix M4 and the G383D mutation in the re-entrant loop between helices M8 and M9 of NHE6. The mutations were introduced into the homology model by using the program Coot (133). Energy minimization and MD simulation were carried out with the GROMACS package (134). First, the topology for the NHE6 model was generated with the GROMACS pdb2gmx module and the OPLS all-atom force field parameters (135). The protein was then placed in a periodic cubic simulation box, with the minimum distance of 1.0 nm to the box edge. For system solvation, the SPC/E model was used (136). After charge neutralization with counterions, the system was energy-minimized to the target Fmax of no greater than 1000 kJ mol−1 nm−1 by using the steepest descent algorithm implemented in GROMACS. To constrain covalent bond lengths, the LINear Constraint Solver (LINCS) algorithm was applied. To model long-range electrostatic interactions, the Particle Mesh Ewald method was used. Short-range van der Waals and Coulomb cutoff distances were both 1.0 nm. For the production run, the V-rescale thermostat and the Parrinello-Rahman barostat were used to couple the system to a constant temperature of 300 K and a pressure of 1 bar. The total simulation period was 10 ns, and the equations of motion were integrated with the leap-frog algorithm in time steps of 2 fs.
Statistical analyses
Statistical analyses were performed using OriginPro software. The data are presented as the mean ± S.D. unless stated otherwise. Means of multiple samples were compared using either a one-way, one-way repeated measures, or two-way ANOVA followed by post hoc Tukey or Dunnett's test, as indicated. Data (where n ≥ 6) were analyzed for statistical outliers using a Grubbs test. A few outliers were detected for some experiments that did not significantly affect the mean, so all the data were retained in the final calculations. A minimum p value of <0.05 was considered significant.
Author contributions
A. I., A. B., and J. P. data curation; A. I., A. B., J. P., A. M. B., and J. O. formal analysis; A. I. and J. O. supervision; A. I., R. A. M., and J. O. validation; A. I., A. B., J. P., and J. O. investigation; A. I., J. P., A. M. B., and J. O. visualization; A. I., A. B., J. P., A. M. B., and J. O. methodology; A. I., A. B., J. P., R. A. M., and J. O. writing-review and editing; A. M. B. software; R. A. M. and J. O. conceptualization; R. A. M. and J. O. funding acquisition; R. A. M. and J. O. project administration; J. O. resources.
Supplementary Material
Acknowledgments
We are grateful for the services provided by the McGill Life Sciences Advanced BioImaging Facility and the Flow Cytometry Facility and by Genome Québec for DNA sequencing. The platforms were supported by funding from the Canadian Foundation for Innovation.
This work was supported by Canadian Institutes of Health Research Grants PJT-155976 (to J. O.), PJT-162365 (to A. M. B.), and MOP-133611 and PJT-155976 (to R. A. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3.
A. Ilie and J. Orlowski, unpublished data.
- CS
- Christianson syndrome
- AP-1
- Chinese hamster ovary (CHO) cells deficient in the Na+/H+ exchanger 1 and 6 isoforms
- CANX
- calnexin
- ChFP
- monomeric Cherry fluorescent protein
- CNS
- central nervous system
- ER
- endoplasmic reticulum
- ERAD
- endoplasmic reticulum–associated degradation
- FRIA
- fluorescence ratiometric image analysis
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GFP
- green fluorescent protein
- HA
- influenza virus hemagglutinin epitope
- LeuP
- leupeptin/pepstatin
- M.I.F.
- mean intensity fluorescence
- TCL
- total-cell lysate
- Tf
- transferrin
- TfR
- transferrin receptor
- TrkB
- tropomyosin or tyrosine receptor kinase B
- ANOVA
- analysis of variance
- ELISA
- enzyme-linked immunosorbent assay
- Tf–FITC
- FITC-conjugated human transferrin
- PI
- propidium iodide
- HRP
- horseradish peroxidase
- α-MEM
- α-minimum essential medium
- MD
- molecular dynamics
- 7-AAD
- 7-amino-actinomycin D
- CCCP
- carbonyl cyanide m-chlorophenylhydrazone
- Tf-AF488
- Alexa FluorTM 488–conjugated transferrin
- MCC
- Manders' co-localization coefficient
- PMT
- photomultiplier tube.
References
- 1. Christianson A. L., Stevenson R. E., van der Meyden C. H., Pelser J., Theron F. W., van Rensburg P. L., Chandler M., and Schwartz C. E. (1999) X-linked severe mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellar atrophy in a large South African kindred is localised to Xq24-q27. J. Med. Genet 36, 759–766 10.1136/jmg.36.10.759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gilfillan G. D., Selmer K. K., Roxrud I., Smith R., Kyllerman M., Eiklid K., Kroken M., Mattingsdal M., Egeland T., Stenmark H., Sjøholm H., Server A., Samuelsson L., Christianson A., Tarpey P., et al. (2008) SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am. J. Hum. Genet. 82, 1003–1010 10.1016/j.ajhg.2008.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fichou Y., Bahi-Buisson N., Nectoux J., Chelly J., Héron D., Cuisset L., and Bienvenu T. (2009) Mutation in the SLC9A6 gene is not a frequent cause of sporadic Angelman-like syndrome. Eur. J. Hum. Genet 17, 1378–1380 10.1038/ejhg.2009.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tarpey P. S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O'Meara S., Latimer C., Dicks E., Menzies A., Stephens P., Blow M., Greenman C., Xue Y., Tyler-Smith C., et al. (2009) A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 41, 535–543 10.1038/ng.367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hu H., Wrogemann K., Kalscheuer V., Tzschach A., Richard H., Haas S. A., Menzel C., Bienek M., Froyen G., Raynaud M., Van Bokhoven H., Chelly J., Ropers H., and Chen W. (2009) Mutation screening in 86 known X-linked mental retardation genes by droplet-based multiplex PCR and massive parallel sequencing. Hugo J. 3, 41–49 10.1007/s11568-010-9137-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garbern J. Y., Neumann M., Trojanowski J. Q., Lee V. M., Feldman G., Norris J. W., Friez M. J., Schwartz C. E., Stevenson R., and Sima A. A. (2010) A mutation affecting the sodium/proton exchanger, SLC9A6, causes mental retardation with tau deposition. Brain 133, 1391–1402 10.1093/brain/awq071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schroer R. J., Holden K. R., Tarpey P. S., Matheus M. G., Griesemer D. A., Friez M. J., Fan J. Z., Simensen R. J., Stromme P., Stevenson R. E., Stratton M. R., and Schwartz C. E. (2010) Natural history of Christianson syndrome. Am. J. Med. Genet. A 152A, 2775–2783 10.1002/ajmg.a.33093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tzschach A., Ullmann R., Ahmed A., Martin T., Weber G., Decker-Schwering O., Pauly F., Shamdeen M. G., Reith W., and Oehl-Jaschkowitz B. (2011) Christianson syndrome in a patient with an interstitial Xq26.3 deletion. Am. J. Med. Genet. A 155A, 2771–2774 10.1002/ajmg.a.34230 [DOI] [PubMed] [Google Scholar]
- 9. Takahashi Y., Hosoki K., Matsushita M., Funatsuka M., Saito K., Kanazawa H., Goto Y., and Saitoh S. (2011) A loss-of-function mutation in the SLC9A6 gene causes X-linked mental retardation resembling Angelman syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156, 799–807 10.1002/ajmg.b.31221 [DOI] [PubMed] [Google Scholar]
- 10. Mignot C., Héron D., Bursztyn J., Momtchilova M., Mayer M., Whalen S., Legall A., Billette de Villemeur T., and Burglen L. (2013) Novel mutation in SLC9A6 gene in a patient with Christianson syndrome and retinitis pigmentosum. Brain Dev. 35, 172–176 10.1016/j.braindev.2012.03.010 [DOI] [PubMed] [Google Scholar]
- 11. Riess A., Rossier E., Krüger R., Dufke A., Beck-Woedl S., Horber V., Alber M., Gläser D., Riess O., and Tzschach A. (2013) Novel SLC9A6 mutations in two families with Christianson syndrome. Clin. Genet. 83, 596–597 10.1111/j.1399-0004.2012.01948.x [DOI] [PubMed] [Google Scholar]
- 12. Schuurs-Hoeijmakers J. H., Vulto-van Silfhout A. T., Vissers L. E., van de Vondervoort I. I., van Bon B. W., de Ligt J., Gilissen C., Hehir-Kwa J. Y., Neveling K., del Rosario M., Hira G., Reitano S., Vitello A., Failla P., Greco D., et al. (2013) Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J. Med. Genet. 50, 802–811 10.1136/jmedgenet-2013-101644 [DOI] [PubMed] [Google Scholar]
- 13. Redin C., Gérard B., Lauer J., Herenger Y., Muller J., Quartier A., Masurel-Paulet A., Willems M., Lesca G., El-Chehadeh S., Le Gras S., Vicaire S., Philipps M., Dumas M., Geoffroy V., et al. (2014) Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 51, 724–736 10.1136/jmedgenet-2014-102554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bosemani T., Zanni G., Hartman A. L., Cohen R., Huisman T. A., Bertini E., and Poretti A. (2014) Christianson syndrome: spectrum of neuroimaging findings. Neuropediatrics 45, 247–251 10.1055/s-0033-1363091 [DOI] [PubMed] [Google Scholar]
- 15. Pescosolido M. F., Stein D. M., Schmidt M., El Achkar C. M., Sabbagh M., Rogg J. M., Tantravahi U., McLean R. L., Liu J. S., Poduri A., and Morrow E. M. (2014) Genetic and phenotypic diversity of NHE6 mutations in Christianson syndrome. Ann. Neurol. 76, 581–593 10.1002/ana.24225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zanni G., Barresi S., Cohen R., Specchio N., Basel-Vanagaite L., Valente E. M., Shuper A., Vigevano F., and Bertini E. (2014) A novel mutation in the endosomal Na+/H+ exchanger NHE6 (SLC9A6) causes Christianson syndrome with electrical status epilepticus during slow-wave sleep (ESES). Epilepsy Res. 108, 811–815 10.1016/j.eplepsyres.2014.02.009 [DOI] [PubMed] [Google Scholar]
- 17. Tzschach A., Grasshoff U., Beck-Woedl S., Dufke C., Bauer C., Kehrer M., Evers C., Moog U., Oehl-Jaschkowitz B., Di Donato N., Maiwald R., Jung C., Kuechler A., Schulz S., Meinecke P., et al. (2015) Next-generation sequencing in X-linked intellectual disability. Eur. J. Hum. Genet. 23, 1513–1518. 10.1038/ejhg.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mercimek-Mahmutoglu S., Patel J., Cordeiro D., Hewson S., Callen D., Donner E. J., Hahn C. D., Kannu P., Kobayashi J., Minassian B. A., Moharir M., Siriwardena K., Weiss S. K., Weksberg R., and Snead O. C. 3rd. (2015) Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia 56, 707–716 10.1111/epi.12954 [DOI] [PubMed] [Google Scholar]
- 19. Coorg R., and Weisenberg J. L. (2015) Successful treatment of electrographic status epilepticus of sleep with felbamate in a patient with SLC9A6 mutation. Pediatr. Neurol. 53, 527–531 10.1016/j.pediatrneurol.2015.07.007 [DOI] [PubMed] [Google Scholar]
- 20. Trump N., McTague A., Brittain H., Papandreou A., Meyer E., Ngoh A., Palmer R., Morrogh D., Boustred C., Hurst J. A., Jenkins L., Kurian M. A., and Scott R. H. (2016) Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J. Med. Genet. 53, 310–317 10.1136/jmedgenet-2015-103263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sinajon P., Verbaan D., and So J. (2016) The expanding phenotypic spectrum of female SLC9A6 mutation carriers: a case series and review of the literature. Hum. Genet. 135, 841–850 10.1007/s00439-016-1675-5 [DOI] [PubMed] [Google Scholar]
- 22. Masurel-Paulet A., Piton A., Chancenotte S., Redin C., Thauvin-Robinet C., Henrenger Y., Minot D., Creppy A., Ruffier-Bourdet M., Thevenon J., Kuentz P., Lehalle D., Curie A., Blanchard G., Ghosn E., et al. (2016) A new family with an SLC9A6 mutation expanding the phenotypic spectrum of Christianson syndrome. Am. J. Med. Genet. A 170, 2103–2110 10.1002/ajmg.a.37765 [DOI] [PubMed] [Google Scholar]
- 23. Liu J., Tong L., Song S., Niu Y., Li J., Wu X., Zhang J., Zai C. C., Luo F., Wu J., Li H., Wong A. H. C., Sun R., Liu F., and Li B. (2018) Novel and de novo mutations in pediatric refractory epilepsy. Mol. Brain 11, 48 10.1186/s13041-018-0392-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Padmanabha H., Saini A. G., Sahu J. K., and Singhi P. (2017) Syndrome of X-linked intellectual disability, epilepsy, progressive brain atrophy and large head associated with SLC9A6 mutation. BMJ Case Rep. 2017, bcr-2017-222050 10.1136/bcr-2017-222050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Piton A., Gauthier J., Hamdan F. F., Lafrenière R. G., Yang Y., Henrion E., Laurent S., Noreau A., Thibodeau P., Karemera L., Spiegelman D., Kuku F., Duguay J., Destroismaisons L., Jolivet P., et al. (2011) Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol. Psychiatry 16, 867–880 10.1038/mp.2010.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwede M., Garbett K., Mirnics K., Geschwind D. H., and Morrow E. M. (2014) Genes for endosomal NHE6 and NHE9 are misregulated in autism brains. Mol. Psychiatry 19, 277–279 10.1038/mp.2013.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tilot A. K., Kucera K. S., Vino A., Asher J. E., Baron-Cohen S., and Fisher S. E. (2018) Rare variants in axonogenesis genes connect three families with sound-color synesthesia. Proc. Natl. Acad. Sci. U.S.A. 115, 3168–3173 10.1073/pnas.1715492115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hauser M. A., Li Y. J., Xu H., Noureddine M. A., Shao Y. S., Gullans S. R., Scherzer C. R., Jensen R. V., McLaurin A. C., Gibson J. R., Scott B. L., Jewett R. M., Stenger J. E., Schmechel D. E., Hulette C. M., and Vance J. M. (2005) Expression profiling of substantia nigra in Parkinson disease, progressive supranuclear palsy, and frontotemporal dementia with parkinsonism. Arch. Neurol. 62, 917–921 10.1001/archneur.62.6.917 [DOI] [PubMed] [Google Scholar]
- 29. Prasad H., and Rao R. (2015) The Na+/H+ exchanger NHE6 modulates endosomal pH to control processing of amyloid precursor protein in a cell culture model of Alzheimer disease. J. Biol. Chem. 290, 5311–5327 10.1074/jbc.M114.602219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prasad H., and Rao R. (2018) Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc. Natl. Acad. Sci. U.S.A. 115, E6640–E6649 10.1073/pnas.1801612115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pescosolido M. F., Kavanaugh B. C., Pochet N., Schmidt M., Jerskey B. A., Rogg J. M., De Jager P. L., Young-Pearse T. L., Liu J. S., and Morrow E. M. (2019) Complex neurological phenotype in female carriers of NHE6 mutations. Mol. Neuropsychiatry 5, 98–108 10.1159/000496341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Strømme P., Dobrenis K., Sillitoe R. V., Gulinello M., Ali N. F., Davidson C., Micsenyi M. C., Stephney G., Ellevog L., Klungland A., and Walkley S. U. (2011) X-linked Angelman-like syndrome caused by Slc9a6 knockout in mice exhibits evidence of endosomal–lysosomal dysfunction. Brain 134, 3369–3383 10.1093/brain/awr250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ouyang Q., Lizarraga S. B., Schmidt M., Yang U., Gong J., Ellisor D., Kauer J. A., and Morrow E. M. (2013) Christianson syndrome protein NHE6 modulates TrkB endosomal signaling required for neuronal circuit development. Neuron 80, 97–112 10.1016/j.neuron.2013.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu M., Ouyang Q., Gong J., Pescosolido M. F., Pruett B. S., Mishra S., Schmidt M., Jones R. N., Gamsiz Uzun E. D., Lizarraga S. B., and Morrow E. M. (2017) Mixed neurodevelopmental and neurodegenerative pathology in Nhe6-null mouse model of Christianson syndrome. eNeuro 4, ENEURO.0388-17.2017 10.1523/ENEURO.0388-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sikora J., Leddy J., Gulinello M., and Walkley S. U. (2016) X-linked Christianson syndrome: heterozygous female Slc9a6 knockout mice develop mosaic neuropathological changes and related behavioral abnormalities. Dis. Model. Mech. 9, 13–23 10.1242/dmm.022780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fagerberg L., Hallström B. M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K., Asplund A., Sjöstedt E., Lundberg E., Szigyarto C. A., Skogs M., et al. (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406 10.1074/mcp.M113.035600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deane E. C., Ilie A. E., Sizdahkhani S., Das Gupta M., Orlowski J., and McKinney R. A. (2013) Enhanced recruitment of endosomal Na+/H+ exchanger NHE6 into dendritic spines of hippocampal pyramidal neurons during NMDA receptor–dependent long-term potentiation. J. Neurosci. 33, 595–610 10.1523/JNEUROSCI.2583-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brett C. L., Wei Y., Donowitz M., and Rao R. (2002) Human Na+/H+ exchanger isoform 6 is found in recycling endosomes of cells, not in mitochondria. Am. J. Physiol. Cell Physiol. 282, C1031–C1041 10.1152/ajpcell.00420.2001 [DOI] [PubMed] [Google Scholar]
- 39. Nakamura N., Tanaka S., Teko Y., Mitsui K., and Kanazawa H. (2005) Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J. Biol. Chem. 280, 1561–1572 10.1074/jbc.M410041200 [DOI] [PubMed] [Google Scholar]
- 40. Yamashiro D. J., and Maxfield F. R. (1988) Regulation of endocytic processes by pH. Trends Pharmacol. Sci. 9, 190–193 10.1016/0165-6147(88)90078-8 [DOI] [PubMed] [Google Scholar]
- 41. Casey J. R., Grinstein S., and Orlowski J. (2010) Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 11, 50–61 10.1038/nrm2820 [DOI] [PubMed] [Google Scholar]
- 42. Jentsch T. J., and Pusch M. (2018) CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol. Rev. 98, 1493–1590 10.1152/physrev.00047.2017 [DOI] [PubMed] [Google Scholar]
- 43. Krueger K. M., Daaka Y., Pitcher J. A., and Lefkowitz R. J. (1997) The role of sequestration in G protein–coupled receptor resensitization. Regulation of β2-adrenergic receptor dephosphorylation by vesicular acidification. J. Biol. Chem. 272, 5–8 10.1074/jbc.272.1.5 [DOI] [PubMed] [Google Scholar]
- 44. Harford J., Bridges K., Ashwell G., and Klausner R. D. (1983) Intracellular dissociation of receptor-bound asialoglycoproteins in cultured hepatocytes. A pH-mediated nonlysosomal event. J. Biol. Chem. 258, 3191–3197 [PubMed] [Google Scholar]
- 45. Yamashiro D. J., and Maxfield F. R. (1984) Acidification of endocytic compartments and the intracellular pathways of ligands and receptors. J. Cell. Biochem. 26, 231–246 10.1002/jcb.240260404 [DOI] [PubMed] [Google Scholar]
- 46. Mellman I. (1992) The importance of being acid: the role of acidification in intracellular membrane traffic. J. Exp. Biol. 172, 39–45 [DOI] [PubMed] [Google Scholar]
- 47. Grady E. F., Garland A. M., Gamp P. D., Lovett M., Payan D. G., and Bunnett N. W. (1995) Delineation of the endocytic pathway of substance P and its seven-transmembrane domain NK1 receptor. Mol. Biol. Cell 6, 509–524 10.1091/mbc.6.5.509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weisz O. A. (2003) Acidification and protein traffic. Int. Rev. Cytol. 226, 259–319 10.1016/S0074-7696(03)01005-2 [DOI] [PubMed] [Google Scholar]
- 49. Hurtado-Lorenzo A., Skinner M., El Annan J., Futai M., Sun-Wada G. H., Bourgoin S., Casanova J., Wildeman A., Bechoua S., Ausiello D. A., Brown D., and Marshansky V. (2006) V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat. Cell Biol. 8, 124–136 10.1038/ncb1348 [DOI] [PubMed] [Google Scholar]
- 50. Merkulova M., Păunescu T. G., Azroyan A., Marshansky V., Breton S., and Brown D. (2015) Mapping the H+ (V)-ATPase interactome: identification of proteins involved in trafficking, folding, assembly and phosphorylation. Sci. Rep. 5, 14827 10.1038/srep14827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xinhan L., Matsushita M., Numaza M., Taguchi A., Mitsui K., and Kanazawa H. (2011) Na+/H+ exchanger isoform 6 (NHE6/SLC9A6) is involved in clathrin-dependent endocytosis of transferrin. Am. J. Physiol. Cell Physiol. 301, C1431–C1444 10.1152/ajpcell.00154.2011 [DOI] [PubMed] [Google Scholar]
- 52. Ohgaki R., Matsushita M., Kanazawa H., Ogihara S., Hoekstra D., and van IJzendoorn S. C. (2010) The Na+/H+ exchanger NHE6 in the endosomal recycling system is involved in the development of apical bile canalicular surface domains in HepG2 cells. Mol. Biol. Cell 21, 1293–1304 10.1091/mbc.e09-09-0767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ilie A., Gao A. Y., Reid J., Boucher A., McEwan C., Barrière H., Lukacs G. L., McKinney R. A., and Orlowski J. (2016) A Christianson syndrome-linked deletion mutation (Δ287ES288) in SLC9A6 disrupts recycling endosomal function and elicits neurodegeneration and cell death. Mol. Neurodegener. 11, 63 10.1186/s13024-016-0129-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gao A. Y. L., Ilie A., Chang P. K. Y., Orlowski J., and McKinney R. A. (2019) A Christianson syndrome-linked deletion mutation (Δ287ES288) in SLC9A6 impairs hippocampal neuronal plasticity. Neurobiol. Dis. 130, 104490 10.1016/j.nbd.2019.104490 [DOI] [PubMed] [Google Scholar]
- 55. Roxrud I., Raiborg C., Gilfillan G. D., Strømme P., and Stenmark H. (2009) Dual degradation mechanisms ensure disposal of NHE6 mutant protein associated with neurological disease. Exp. Cell Res. 315, 3014–3027 10.1016/j.yexcr.2009.07.012 [DOI] [PubMed] [Google Scholar]
- 56. Ilie A., Weinstein E., Boucher A., McKinney R. A., and Orlowski J. (2014) Impaired posttranslational processing and trafficking of an endosomal Na+/H+ exchanger NHE6 mutant (Δ370WST372) associated with X-linked intellectual disability and autism. Neurochem. Int. 73, 192–203 10.1016/j.neuint.2013.09.020 [DOI] [PubMed] [Google Scholar]
- 57. Ilie A., Gao A. Y. L., Boucher A., Park J., Berghuis A. M., Hoffer M. J. V., Hilhorst-Hofstee Y., McKinney R. A., and Orlowski J. (2019) A potential gain-of-function variant of SLC9A6 leads to endosomal alkalinization and neuronal atrophy associated with Christianson syndrome. Neurobiol. Dis. 121, 187–204 10.1016/j.nbd.2018.10.002 [DOI] [PubMed] [Google Scholar]
- 58. Santoni F. A., Makrythanasis P., Nikolaev S., Guipponi M., Robyr D., Bottani A., and Antonarakis S. E. (2014) Simultaneous identification and prioritization of variants in familial, de novo, and somatic genetic disorders with VariantMaster. Genome Res. 24, 349–355 10.1101/gr.163832.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Landau M., Herz K., Padan E., and Ben-Tal N. (2007) Model structure of the Na+/H+ exchanger 1 (NHE1): functional and clinical implications. J. Biol. Chem. 282, 37854–37863 10.1074/jbc.M705460200 [DOI] [PubMed] [Google Scholar]
- 60. Nygaard E. B., Lagerstedt J. O., Bjerre G., Shi B., Budamagunta M., Poulsen K. A., Meinild S., Rigor R. R., Voss J. C., Cala P. M., and Pedersen S. F. (2011) Structural modeling and electron paramagnetic resonance spectroscopy of the human Na+/H+ exchanger isoform 1, NHE1. J. Biol. Chem. 286, 634–648 10.1074/jbc.M110.159202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lykke-Andersen S., and Jensen T. H. (2015) Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 16, 665–677 10.1038/nrm4063 [DOI] [PubMed] [Google Scholar]
- 62. Coincon M., Uzdavinys P., Nji E., Dotson D. L., Winkelmann I., Abdul-Hussein S., Cameron A. D., Beckstein O., and Drew D. (2016) Crystal structures reveal the molecular basis of ion translocation in sodium/proton antiporters. Nat. Struct. Mol. Biol. 23, 248–255 10.1038/nsmb.3164 [DOI] [PubMed] [Google Scholar]
- 63. Fafournoux P., Noël J., and Pouysségur J. (1994) Evidence that Na+/H+ exchanger isoforms NHE1 and NHE3 exist as stable dimers in membranes with a high degree of specificity for homodimers. J. Biol. Chem. 269, 2589–2596 [PubMed] [Google Scholar]
- 64. Pace C. N., and Scholtz J. M. (1998) A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75, 422–427 10.1016/S0006-3495(98)77529-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rai R., Aravinda S., Kanagarajadurai K., Raghothama S., Shamala N., and Balaram P. (2006) Diproline templates as folding nuclei in designed peptides. Conformational analysis of synthetic peptide helices containing amino-terminal Pro-Pro segments. J. Am. Chem. Soc. 128, 7916–7928 10.1021/ja060674v [DOI] [PubMed] [Google Scholar]
- 66. Pokkuluri P. R., Dwulit-Smith J., Duke N. E., Wilton R., Mack J. C., Bearden J., Rakowski E., Babnigg G., Szurmant H., Joachimiak A., and Schiffer M. (2013) Analysis of periplasmic sensor domains from Anaeromyxobacter dehalogenans 2CP-C: structure of one sensor domain from a histidine kinase and another from a chemotaxis protein. MicrobiologyOpen 2, 766–777 10.1002/mbo3.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Counillon L., Noël J., Reithmeier R. A., and Pouysségur J. (1997) Random mutagenesis reveals a novel site involved in inhibitor interaction within the fourth transmembrane segment of the Na+/H+ exchanger-1. Biochemistry 36, 2951–2959 10.1021/bi9615405 [DOI] [PubMed] [Google Scholar]
- 68. Slepkov E. R., Rainey J. K., Li X., Liu Y., Cheng F. J., Lindhout D. A., Sykes B. D., and Fliegel L. (2005) Structural and functional characterization of transmembrane segment IV of the NHE1 isoform of the Na+/H+ exchanger. J. Biol. Chem. 280, 17863–17872 10.1074/jbc.M409608200 [DOI] [PubMed] [Google Scholar]
- 69. Khadilkar A., Iannuzzi P., and Orlowski J. (2001) Identification of sites in the second exomembrane loop and ninth transmembrane helix of the mammalian Na+/H+ exchanger important for drug recognition and cation translocation. J. Biol. Chem. 276, 43792–43800 10.1074/jbc.M106659200 [DOI] [PubMed] [Google Scholar]
- 70. Jinadasa T., Josephson C. B., Boucher A., and Orlowski J. (2015) Determinants of cation permeation and drug sensitivity in predicted transmembrane helix 9 and adjoining exofacial re-entrant loop 5 of Na+/H+ exchanger NHE1. J. Biol. Chem. 290, 18173–18186 10.1074/jbc.M115.642199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ding J., Ng R. W., and Fliegel L. (2007) Functional characterization of the transmembrane segment VII of the NHE1 isoform of the Na+/H+ exchanger. Can. J. Physiol. Pharmacol. 85, 319–325 10.1139/y06-081 [DOI] [PubMed] [Google Scholar]
- 72. Lee D. H., and Goldberg A. L. (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8, 397–403 10.1016/S0962-8924(98)01346-4 [DOI] [PubMed] [Google Scholar]
- 73. Seglen P. O., Grinde B., and Solheim A. E. (1979) Inhibition of the lysosomal pathway of protein degradation in isolated rat hepatocytes by ammonia, methylamine, chloroquine and leupeptin. Eur. J. Biochem. 95, 215–225 10.1111/j.1432-1033.1979.tb12956.x [DOI] [PubMed] [Google Scholar]
- 74. Tanaka K., Ikegaki N., and Ichihara A. (1981) Effects of leupeptin and pepstatin on protein turnover in adult rat hepatocytes in primary culture. Arch. Biochem. Biophys. 208, 296–304 10.1016/0003-9861(81)90152-1 [DOI] [PubMed] [Google Scholar]
- 75. Maxfield F. R. (1982) Weak bases and ionophores rapidly and reversibly raise the pH of endocytic vesicles in cultured mouse fibroblasts. J. Cell Biol. 95, 676–681 10.1083/jcb.95.2.676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Meusser B., Hirsch C., Jarosch E., and Sommer T. (2005) ERAD: the long road to destruction. Nat. Cell Biol. 7, 766–772 10.1038/ncb0805-766 [DOI] [PubMed] [Google Scholar]
- 77. Raasi S., and Wolf D. H. (2007) Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin. Cell Dev. Biol. 18, 780–791 10.1016/j.semcdb.2007.09.008 [DOI] [PubMed] [Google Scholar]
- 78. Daro E., van der Sluijs P., Galli T., and Mellman I. (1996) Rab4 and cellubrevin define different early endosome populations on the pathway of transferrin receptor recycling. Proc. Natl. Acad. Sci. U.S.A. 93, 9559–9564 10.1073/pnas.93.18.9559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ullrich O., Reinsch S., Urbé S., Zerial M., and Parton R. G. (1996) Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 135, 913–924 10.1083/jcb.135.4.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Manders E. M. M., Verbeek F. J., and Aten J. A. (1993) Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 169, 375–382 10.1111/j.1365-2818.1993.tb03313.x [DOI] [PubMed] [Google Scholar]
- 81. Costes S. V., Daelemans D., Cho E. H., Dobbin Z., Pavlakis G., and Lockett S. (2004) Automatic and quantitative measurement of protein–protein colocalization in live cells. Biophys. J. 86, 3993–4003 10.1529/biophysj.103.038422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bolte S., and Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 83. Barriere H., and Lukacs G. L. (2008) Analysis of endocytic trafficking by single-cell fluorescence ratio imaging. Curr. Protoc. Cell Biol. 2008, Chapter 15, Unit 15.13 10.1002/0471143030.cb1513s40 [DOI] [PubMed] [Google Scholar]
- 84. Mahon M. J. (2011) pHluorin2: an enhanced, ratiometric, pH-sensitive green florescent protein. Adv. Biosci. Biotechnol. 2, 132–137 10.4236/abb.2011.23021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kim J. H., Johannes L., Goud B., Antony C., Lingwood C. A., Daneman R., and Grinstein S. (1998) Noninvasive measurement of the pH of the endoplasmic reticulum at rest and during calcium release. Proc. Natl. Acad. Sci. U.S.A. 95, 2997–3002 10.1073/pnas.95.6.2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wu M. M., Llopis J., Adams S., McCaffery J. M., Kulomaa M. S., Machen T. E., Moore H. P., and Tsien R. Y. (2000) Organelle pH studies using targeted avidin and fluorescein-biotin. Chem. Biol. 7, 197–209 10.1016/S1074-5521(00)00088-0 [DOI] [PubMed] [Google Scholar]
- 87. Wu M. M., Grabe M., Adams S., Tsien R. Y., Moore H. P., and Machen T. E. (2001) Mechanisms of pH regulation in the regulated secretory pathway. J. Biol. Chem. 276, 33027–33035 10.1074/jbc.M103917200 [DOI] [PubMed] [Google Scholar]
- 88. Ouyang Q., Joesch-Cohen L., Mishra S., Riaz H. A., Schmidt M., and Morrow E. M. (2019) Functional assessment in vivo of the mouse homolog of the human Ala-9–Ser NHE6 variant. eNeuro 6, ENEURO.0046-19.2019 10.1523/ENEURO.0046-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Helenius A., and Aebi M. (2001) Intracellular functions of N-linked glycans. Science 291, 2364–2369 10.1126/science.291.5512.2364 [DOI] [PubMed] [Google Scholar]
- 90. Ellgaard L., and Helenius A. (2003) Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191 10.1038/nrm1052 [DOI] [PubMed] [Google Scholar]
- 91. Xu C., and Ng D. T. (2015) Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol. 16, 742–752 10.1038/nrm4073 [DOI] [PubMed] [Google Scholar]
- 92. Braakman I., and Hebert D. N. (2013) Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 5, a013201 10.1101/cshperspect.a013201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ruggiano A., Foresti O., and Carvalho P. (2014) ER-associated degradation: protein quality control and beyond. J. Cell Biol. 204, 869–879 10.1083/jcb.201312042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tannous A., Pisoni G. B., Hebert D. N., and Molinari M. (2015) N-Linked sugar-regulated protein folding and quality control in the ER. Semin. Cell Dev. Biol. 41, 79–89 10.1016/j.semcdb.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hammond C., Braakman I., and Helenius A. (1994) Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc. Natl. Acad. Sci. U.S.A. 91, 913–917 10.1073/pnas.91.3.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ruddock L. W., and Molinari M. (2006) N-Glycan processing in ER quality control. J. Cell Sci. 119, 4373–4380 10.1242/jcs.03225 [DOI] [PubMed] [Google Scholar]
- 97. Gomez-Navarro N., and Miller E. (2016) Protein sorting at the ER–Golgi interface. J. Cell Biol. 215, 769–778 10.1083/jcb.201610031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Barlowe C., and Helenius A. (2016) Cargo capture and bulk flow in the early secretory pathway. Annu. Rev. Cell Dev. Biol. 32, 197–222 10.1146/annurev-cellbio-111315-125016 [DOI] [PubMed] [Google Scholar]
- 99. Kincaid M. M., and Cooper A. A. (2007) Misfolded proteins traffic from the endoplasmic reticulum (ER) due to ER export signals. Mol. Biol. Cell 18, 455–463 10.1091/mbc.e06-08-0696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Colombo M., Raposo G., and Théry C. (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 30, 255–289 10.1146/annurev-cellbio-101512-122326 [DOI] [PubMed] [Google Scholar]
- 101. Mellman I., Fuchs R., and Helenius A. (1986) Acidification of the endocytic and exocytic pathways. Annu. Rev. Biochem. 55, 663–700 10.1146/annurev.bi.55.070186.003311 [DOI] [PubMed] [Google Scholar]
- 102. van Weert A. W., Dunn K. W., Geuze H. J., Maxfield F. R., and Stoorvogel W. (1995) Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J. Cell Biol. 130, 821–834 10.1083/jcb.130.4.821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Maxfield F. R., and McGraw T. E. (2004) Endocytic recycling. Nat. Rev. Mol. Cell Biol. 5, 121–132 10.1038/nrm1315 [DOI] [PubMed] [Google Scholar]
- 104. Merkulova M., Bakulina A., Thaker Y. R., Grüber G., and Marshansky V. (2010) Specific motifs of the V-ATPase a2-subunit isoform interact with catalytic and regulatory domains of ARNO. Biochim. Biophys. Acta 1797, 1398–1409 10.1016/j.bbabio.2010.02.009 [DOI] [PubMed] [Google Scholar]
- 105. Ali R., Brett C. L., Mukherjee S., and Rao R. (2004) Inhibition of sodium/proton exchange by a Rab–GTPase-activating protein regulates endosomal traffic in yeast. J. Biol. Chem. 279, 4498–4506 10.1074/jbc.M307446200 [DOI] [PubMed] [Google Scholar]
- 106. Brett C. L., Tukaye D. N., Mukherjee S., and Rao R. (2005) The yeast endosomal Na+(K+)/H+ exchanger Nhx1 regulates cellular pH to control vesicle trafficking. Mol. Biol. Cell 16, 1396–1405 10.1091/mbc.e04-11-0999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bowers K., Levi B. P., Patel F. I., and Stevens T. H. (2000) The sodium/proton exchanger Nhx1p is required for endosomal protein trafficking in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 11, 4277–4294 10.1091/mbc.11.12.4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Qiu Q. S., and Fratti R. A. (2010) The Na+/H+ exchanger Nhx1p regulates the initiation of Saccharomyces cerevisiae vacuole fusion. J. Cell Sci. 123, 3266–3275 10.1242/jcs.067637 [DOI] [PubMed] [Google Scholar]
- 109. Karim M. A., and Brett C. L. (2018) The Na+(K+)/H+ exchanger Nhx1 controls multivesicular body-vacuolar lysosome fusion. Mol. Biol. Cell 29, 317–325 10.1091/mbc.E17-08-0496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. de Silva M. G., Elliott K., Dahl H. H., Fitzpatrick E., Wilcox S., Delatycki M., Williamson R., Efron D., Lynch M., and Forrest S. (2003) Disruption of a novel member of a sodium/hydrogen exchanger family and DOCK3 is associated with an attention deficit hyperactivity disorder-like phenotype. J. Med. Genet. 40, 733–740 10.1136/jmg.40.10.733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Markunas C. A., Quinn K. S., Collins A. L., Garrett M. E., Lachiewicz A. M., Sommer J. L., Morrissey-Kane E., Kollins S. H., Anastopoulos A. D., and Ashley-Koch A. E. (2010) Genetic variants in SLC9A9 are associated with measures of attention-deficit/hyperactivity disorder symptoms in families. Psychiatr. Genet. 20, 73–81 10.1097/YPG.0b013e3283351209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mick E., Todorov A., Smalley S., Hu X., Loo S., Todd R. D., Biederman J., Byrne D., Dechairo B., Guiney A., McCracken J., McGough J., Nelson S. F., Reiersen A. M., Wilens T. E., et al. (2010) Family-based genome-wide association scan of attention-deficit/hyperactivity disorder. J. Am. Acad. Child Adolesc. Psychiatry 49, 898–905.e3 10.1016/j.jaac.2010.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Morrow E. M., Yoo S. Y., Flavell S. W., Kim T. K., Lin Y., Hill R. S., Mukaddes N. M., Balkhy S., Gascon G., Hashmi A., Al-Saad S., Ware J., Joseph R. M., Greenblatt R., Gleason D., et al. (2008) Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223 10.1126/science.1157657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cardon M., Evankovich K. D., and Holder J. L. Jr. (2016) Exonic deletion of SLC9A9 in autism with epilepsy. Neurol. Genet. 2, e62 10.1212/NXG.0000000000000062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Yang L., Faraone S. V., and Zhang-James Y. (2016) Autism spectrum disorder traits in Slc9a9 knock-out mice. Am. J. Med. Genet. B Neuropsychiatr. Genet. 171, 363–376 10.1002/ajmg.b.32415 [DOI] [PubMed] [Google Scholar]
- 116. Ullman J. C., Yang J., Sullivan M., Bendor J., Levy J., Pham E., Silm K., Seifikar H., Sohal V. S., Nicoll R. A., and Edwards R. H. (2018) A mouse model of autism implicates endosome pH in the regulation of presynaptic calcium entry. Nat. Commun. 9, 330 10.1038/s41467-017-02716-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Huang E. J., and Reichardt L. F. (2001) Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736 10.1146/annurev.neuro.24.1.677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Leal G., Afonso P. M., Salazar I. L., and Duarte C. B. (2015) Regulation of hippocampal synaptic plasticity by BDNF. Brain Res. 1621, 82–101 10.1016/j.brainres.2014.10.019 [DOI] [PubMed] [Google Scholar]
- 119. Huang S. H., Wang J., Sui W. H., Chen B., Zhang X. Y., Yan J., Geng Z., and Chen Z. Y. (2013) BDNF-dependent recycling facilitates TrkB translocation to postsynaptic density during LTP via a Rab11-dependent pathway. J. Neurosci. 33, 9214–9230 10.1523/JNEUROSCI.3256-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Friedman W. J. (2000) Neurotrophins induce death of hippocampal neurons via the p75 receptor. J. Neurosci. 20, 6340–6346 10.1523/JNEUROSCI.20-17-06340.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Troy C. M., Friedman J. E., and Friedman W. J. (2002) Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J. Biol. Chem. 277, 34295–34302 10.1074/jbc.M205167200 [DOI] [PubMed] [Google Scholar]
- 122. Zagrebelsky M., Holz A., Dechant G., Barde Y. A., Bonhoeffer T., and Korte M. (2005) The p75 neurotrophin receptor negatively modulates dendrite complexity and spine density in hippocampal neurons. J. Neurosci. 25, 9989–9999 10.1523/JNEUROSCI.2492-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Schenck A., Goto-Silva L., Collinet C., Rhinn M., Giner A., Habermann B., Brand M., and Zerial M. (2008) The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell 133, 486–497 10.1016/j.cell.2008.02.044 [DOI] [PubMed] [Google Scholar]
- 124. Schütze S., Tchikov V., and Schneider-Brachert W. (2008) Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat. Rev. Mol. Cell Biol. 9, 655–662 10.1038/nrm2430 [DOI] [PubMed] [Google Scholar]
- 125. Bredesen D. E., Mehlen P., and Rabizadeh S. (2005) Receptors that mediate cellular dependence. Cell Death Differ. 12, 1031–1043 10.1038/sj.cdd.4401680 [DOI] [PubMed] [Google Scholar]
- 126. Thibert C., and Fombonne J. (2010) Dependence receptors: mechanisms of an announced death. Cell Cycle 9, 2085–2091 10.4161/cc.9.11.11809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tabas I., and Ron D. (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190 10.1038/ncb0311-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Doyle K. M., Kennedy D., Gorman A. M., Gupta S., Healy S. J., and Samali A. (2011) Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med. 15, 2025–2039 10.1111/j.1582-4934.2011.01374.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Viana R. J., Nunes A. F., and Rodrigues C. M. (2012) Endoplasmic reticulum enrollment in Alzheimer's disease. Mol. Neurobiol. 46, 522–534 10.1007/s12035-012-8301-x [DOI] [PubMed] [Google Scholar]
- 130. Brewster J. L., Linseman D. A., Bouchard R. J., Loucks F. A., Precht T. A., Esch E. A., and Heidenreich K. A. (2006) Endoplasmic reticulum stress and trophic factor withdrawal activate distinct signaling cascades that induce glycogen synthase kinase-3β and a caspase-9-dependent apoptosis in cerebellar granule neurons. Mol. Cell. Neurosci. 32, 242–253 10.1016/j.mcn.2006.04.006 [DOI] [PubMed] [Google Scholar]
- 131. Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., and Ron D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 10.1126/science.287.5453.664 [DOI] [PubMed] [Google Scholar]
- 132. Roussel B. D., Kruppa A. J., Miranda E., Crowther D. C., Lomas D. A., and Marciniak S. J. (2013) Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 12, 105–118 10.1016/S1474-4422(12)70238-7 [DOI] [PubMed] [Google Scholar]
- 133. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., and Lindahl E. (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- 135. Jorgensen W. L., Maxwell D. S., and TiradoRives J. (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 10.1021/ja9621760 [DOI] [Google Scholar]
- 136. Berendsen H. J. C., Grigera J. R., and Straatsma T. P. (1987) The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 10.1021/j100308a038 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.