

Abstract
Gestational Bisphenol A (BPA) exposure is associated with low birth weight. We hypothesized that the low birth weight is the consequence of reduced placental efficiency and a function of BPA-induced inflammatory, oxidative, lipotoxic, angiogenic, steroidal and fibrotic changes involving epigenetic alterations. Placentomes were collected during early (day 65) and mid (day 90) gestation (term ~147 days) from control and BPA (gestational day 30–90)-treated pregnant sheep. BPA treatment: reduced placental efficiency and fetal weight; increased interleukin 8, lipid peroxidation marker, antioxidants, aromatase, 17 alpha-hydroxylase, estrogen receptor 2, insulin like growth factor (IGF) 2 receptor and IGF binding proteins (IGFBP), and histone deacetylase 1 and 2; reduced tumor necrosis factor alpha and IGF1 receptor at early gestation (Day 65). Gestational BPA-induced mid-gestational changes include: reduced angiogenic factor hypoxia inducible factor 1 alpha; increased IL1beta, oxidative stress markers, triglyceride, 17alpha hydroxylase, IGFBP 1, DNA methyltransferase 3A and histone deacetylase 1. These findings indicate that gestational BPA, either acting directly or by altering steroidal input, produces early/mid-gestational-specific epigenetic changes culminating in placental disruptions at several levels, in keeping with time-specific/time-lagged pregnancy-associated changes in placental efficiency and fetal weight. The reduced early-gestational placental efficiency may be a function of increased inflammation/oxidative stress and reduced IGF bioavailability with the mid-gestational restoration of placental efficiency likely driven by improved IGF bioavailability and the time-lagged response to antioxidant increase. This compensation, the result of time-lagged response to increases in negative mediators of placental function must have failed with pregnancy advancement to explain the low birthweight outcome.
Keywords: female reproduction, Endocrine disruptors, oxidative stress, inflammation, angiogenesis, lipotoxicity, epigenetics
Introduction
Developmental insults resulting from disease state, stress, nutritional excess/deficiency or environmental factors, can lead to poor birth outcomes such as intrauterine growth restriction (IUGR), preterm birth and low birth weight, risk factors for adult onset diseases as posit by developmental origin of health and disease (DoHaD) hypothesis (Barker, 2004). The increase in the incidences of the non-communicable diseases coinciding with the rapid industrialization over the 20th century (Neel and Sargis, 2011) has resulted in environmental chemicals especially those with endocrine disrupting potential being viewed as major developmental insults. National Health and Nutrition Examination Survey (NHANES) conducted by Centers for Disease Control and Prevention shows that most Americans have detectable levels of many such environmental endocrine disrupting chemicals (EDCs) (CDC, 2014). Bisphenol A (BPA), one such EDC, is found in >90% of the US population including pregnant women (Calafat et al., 2005; Calafat et al., 2008). Commonly found in many household plastics, BPA in addition to having estrogenic (Vom Saal, 2016) and antiandrogenic (Lee et al., 2003a) properties has been shown to disrupt thyroid hormone action (Ahmed, 2016) and enhance insulin secretion (Alonso-Magdalena et al., 2006). Several human cohort and animal studies have linked gestational exposure of BPA to adverse birth outcomes (Savabieasfahani et al., 2006; Lee et al., 2014; Peretz et al., 2014; Veiga-Lopez et al., 2015a).
Considering that placenta serves as the conduit of nutrients to the fetus (Wooding and Burton, 2008), any disruption in placental homeostasis could affect fetal growth trajectory leading to IUGR and small for gestational age babies such as that seen under conditions of maternal malnutrition, maternal stress and exposure to environmental insults (Nugent and Bale, 2015). Disruptions in placental dysfunctions may be facilitated via reduced nutrient availability, diminished vascular supply, and/or compromised hormonal, inflammatory, oxidant and metabolic states. While evidence exists that gestational exposure to EDCs including BPA is associated with disrupted maternal/fetal cord blood inflammatory/oxidative stress states (Veiga-Lopez et al., 2015b; Watkins et al., 2015; Huang et al., 2017; Kelley et al., 2019) and altered lipid homeostasis (Geetharathan and Josthna, 2016; Shoaito et al., 2019), the extent to which this also involves the placenta is unknown.
Understanding the mechanisms by which gestational BPA disrupts placental function is critical for developing preventive strategies. Considering that placenta is a steroidogenic organ (Strauss et al., 1996), a possible mechanism of BPA action is via disruption of placental steroidal milieu. Recent demonstration that gestational exposure to phthalate, another EDC with antiandrogenic and estrogenic properties, is associated with disruption of maternal and fetal steroidal milieu in humans (Sathyanarayana et al., 2017) supports such assertions. Likewise, BPA exposure during gestation has also been shown to induce steroidal changes during pregnancy (Saadeldin et al., 2018; Kolatorova et al., 2018) and these steroidal changes were found to be associated with BPA induced alterations in expression of placental steroidogenic enzymes (Saadeldin et al., 2018). Steroids by their virtue of being powerful programming agents (Padmanabhan et al., 2010) can induce placental dysfunctions via epigenetic mechanisms (Maccani and Marsit, 2009; Robins et al., 2011; Nugent and Bale, 2015; Marsit, 2016; Yang et al., 2019). In fact, animal studies suggest placental tissues are more susceptible to epigenetic changes than embryonic tissues (Nelissen et al., 2011).
Disruptions in any of the several mediators of placental function can compromise placental growth and processes that are important to meet the metabolic demands of the growing fetus leading to IUGR and low birth weight offspring (Sharma et al., 2016; Woods et al., 2018). Considering that BPA reduces placental cell proliferation (Benachour and Aris, 2009; Elmetwally et al., 2018), genes that regulate placental growth and development such as the members of the insulin-like growth factor (IGF) axis consisting of IGF1 and 2 and IGF binding proteins (IGFBPs) (Haywood et al., 2019) are also potential targets of BPA action. For instance, gestational exposure to polyaromatic hydrocarbons and polybrominated diphenyl ethers was found to be associated with reduced cord blood levels of IGFs (Xu et al., 2013) and BPA with reduced methylation of IGF2 promoter (Montrose et al., 2018) lending support to such possibility.
Due to ethical considerations and the risks associated with early pregnancy sampling of placental tissue, placental research in humans are generally limited to those collected at the time of delivery (Grigsby, 2016). Studies utilizing animal models are therefore of translational value and help fill this knowledge gap for understanding early placental changes associated with maternal exposures (Grigsby, 2016), critical for identifying targets for early interventions. Utilizing the sheep model of gestational BPA treatment that exhibits low birth weight (Savabieasfahani et al., 2006), we tested the hypothesis that gestational treatment with environmentally relevant levels of BPA, increases negative mediators of placental function, disrupts angiogenesis and placental steroidogenesis and contributes to reduced placental efficiency. Furthermore, we hypothesized that such placental compromises are associated with epigenetic modifications and reduced expression of genes involved in placental growth. Documentation of placental compromises resulting from gestational BPA treatment may explain the low birth weight associated with pregnancies from BPA exposures (Savabieasfahani et al., 2006; Veiga-Lopez et al., 2015a).
Methods
Approach:
Utilizing sheep placentae collected during early and mid-gestation coinciding with major organizational windows of fetal organ differentiation (Padmanabhan and Veiga-Lopez, 2014) this study addressed the impact of prenatal BPA treatment on mediators of placental function such as angiogenesis, inflammation, oxidative stress, lipid accumulation, fibrosis, and steroidal milieu. In addition, as many of these changes are regulated transcriptionally and EDCs can bring about epigenetic alterations (Alonso-Magdalena et al., 2016), impact of gestational BPA exposure on gene expression of enzymes that mediate epigenetic alterations were also examined.
Animals and prenatal treatments
All procedures were performed under the approved protocol from the Institutional Animal Care and Use Committee at the University of Michigan. Suffolk ewes aged 2–3 years purchased locally were maintained, bred and prenatally treated as described before (Manikkam et al., 2004; Veiga-Lopez et al., 2013). Treatment groups involved mothers that received only the vehicle (corn oil; Control group) and BPA-treated animals that received 0.5 mg/kg /·day of BPA (purity ≥99%, cat. no. 239658; Aldrich Chemical, Milwaukee, WI) dissolved in corn oil and administered daily through subcutaneous (sc) injections. BPA administration was carried out from days 30 through 90 of gestation (term: ∼147 days). This mode of administration was found to result in umbilical arterial levels of ~2.6 ng/ml of free/uncongugated BPA on gestational day 90 (Veiga-Lopez et al., 2013), well within the range observed in umbilical cord blood of human pregnancy cohorts (Gerona et al., 2013; Veiga-Lopez et al., 2015a, Lee et al., 2018) and maternal urine (Lee et al., 2018).
Tissue collection
Sheep are characterized by cotyledonary placenta. Unlike human, where a single large area of contact exists between maternal and fetal vascular systems, sheep have numerous smaller areas of contacts called placentome. Depending on progression of placental differentiation they are classed as type A, B, C and D (Vatnick et al., 1991). All dams were anesthetized with 20–30 mL of pentobarbital iv (Nembutol sodium solution, 50 mg/mL; Abbott Laboratories, Chicago, IL). Placentomes were collected while dams were maintained under general anesthesia with isofluorane (1%–2%; Halocarbon Laboratories, River Edge, NJ). Through an abdominal midline incision, uterus was exposed and the uterine wall incised to access the fetus. Dams were administered a barbiturate overdose (Fatal Plus; Vortech Pharmaceuticals, Dearborn, Michigan) and fetuses with the placenta removed for harvest of the placentomes. Placentomes were collected, washed, weighed and the type of placentomes identified as described before (Vatnick et al., 1991). To address early perturbations, Type A placentomes, representing the earliest stage of placental differentiation, were used for the analysis in this study. Placentomes were collected from control (C) and BPA-treated fetuses on gestational day 65 (GD65; n = 8, each for C and BPA) and gestational day 90 (GD90; n = 8 and 5, respectively for C and BPA), quick frozen, and stored at −80°C until processing. One type A placentome from each animal was used in the present study. The fetal sex distribution in the control groups were male = 3 and females = 5 at both gestational time points. In the BPA group the distribution was males = 3 and female = 5 at GD65 and male = 1 and female = 4 at GD90.
Real time PCR analysis
The markers of inflammation (proinflammatory cytokines interleukin [IL] 1 beta [IL1B], IL6, IL8, tumor necrosis factor alpha [TNF], chemokine ligand 2 [CCL2] and macrophage marker CD68), antioxidants (glutathione reductase [GSR], superoxide dismutase (SOD) 1 and 2), angiogenic markers (hypoxia inducible factor 1 alpha [HIF1A] and vascular endothelial growth factor [VEGF]), steroidogenic enzymes (aromatase [CYP19] and 17 alpha hydroxylase [CYP17]) and steroid receptors (androgen [AR], estrogen [ESR1 and ESR2] and progesterone [PGR] receptors), epigenetic enzymes (DNA methyltrasnferases [DNMT 1, 2 and 3]; histone methylases [SUV39H1 and EZH2], histone demethylase [KDM1A], histone deacetylases [HDAC1, 2 and 3] and histone acetyltransferases [EP300]) and IGF family members (IGF1 and 2, IGF receptors [IGF1R and IGF2R] and IGF binding proteins [IGFB1, 2, 3 and 4] were measured through gene expression analysis. Total RNA was isolated using Trizol reagent (Life Technologies, Carlsbad, CA), DNAse treated and purified using RNAeasy kit (Qiagen, Germantown, MD) as per manufacturer’s instructions. RNA was quantified using Nanodrop and 260/280 ratios were generally ≥ 1.8. About 1000ng of total RNA was subjected to cDNA synthesis using the SuperScript VILO kit as per manufacturer’s instructions (Life Technologies). Gene expression analysis was carried using SYBRgreen based real time RT-PCR on a BioRad myiQ iCycler instrument as described before (Puttabyatappa et al., 2017). Oligonucleotide primers for the genes under study were either designed using Primer Express software (Life Technologies, Carlsbad, CA) or from previously published reports (Supplemental Table 1). The relative amount of each transcript was calculated using the ΔΔCT method and normalized to the endogenous reference gene ribosomal protein L19 (RPL19).
Oxidative stress measures
Oxidative stress markers Thiobarbituric Acid Reactive Substances (TBARS, a lipid peroxidation marker) and oxidized protein tyrosine moieties, 3-nitrotyrosine (NY) and o, o’-dityrosine (DY) were quantified in placentome tissue extracts. TBARS was quantified utilizing commercially available colorimetric assay kit (Cayman Chemicals, Ann Arbor, MI). Oxidized tyrosine moieties were measured through isotope dilution liquid chromatography electrospray ionization tandem mass spectrometry (HPLC-ESI-MS/MS) as described previously (Vivekanandan-Giri et al., 2011). Briefly, the placental proteins were precipitated with ice-cold trichloroacetic acid (10% vol/vol) and then delipidated with the mixture containing water, methanol, water-washed diethyl ether (1:3:7; vol/vol/vol). Known amounts of isotopically labeled internal standards13C6-Y and 13C6-NY and 13C12-o, o'-DY were added and precipitated proteins were hydrolyzed at 110°C for 24 hours in 4M methanesulfonic acid solution saturated with 1% benzoic acid. Samples were then subjected to solid-phase extraction and the amount of oxidized amino acids was quantified by HPLC-ESI-MS/MS with multiple reactions monitoring by integrating peak areas of the labeled standards and the analytes. The levels of the oxidized amino acids in each sample were then normalized to the amino acid tyrosine content in the respective samples and expressed as the ratio of the oxidized product over the total tyrosine. The Intra- and inter-assay coefficients for TBARS assay were 5.6% and 10.2%, respectively and the measurable range is 0.0625–50 μM. Intra-assay coefficients of variation for NY and DY were 8.47% and 8.16%, respectively.
Ectopic lipid accumulation
Lipid accumulation in liver were assessed by Oil Red O staining of placentome cryosections and triglyceride content using a commercially available assay kit as described before (Puttabyatappa et al., 2017). The measurable range for the triglyceride assay is 1.1 to 2000 mg/dL. All samples were assessed in a single assay and the intra-assay coefficient of variance for triglyceride assay was 3.53 ± 0.08.
Sircol collagen assay
The acid-soluble collagen content in the placentome was measured using a sirius red dye binding (Sircol) assay following the protocol described before (Coentro et al., 2017). Briefly, the acid-soluble collagen was extracted from 100 mg placentome homogenates incubated with 0.5 M acetic acid for 4 h at 4 °C and centrifuged to remove tissue debris. The soluble collagen was incubated with 1 mL 0.1% Direct Red 80 in saturated picric acid dye reagent solution for 1 h at room temperature. The collagen-dye complex was precipitated by centrifugation at 10,000 g for 10 min, and the supernatant was removed. The resulting pellets were dissolved in 1 mL 0.5 M sodium hydroxide, and the relative absorbance was measured in a 96-well plate at 540 nm using a microplate reader (Molecular Devices SpectraMax, San Jose, CA). Collagen content was normalized to total protein content determined as described before (Puttabyatappa et al., 2017) in placentome protein lysates prepared in parallel from 100 mg of tissue. All samples were assayed at the same time and the intra-assay coefficients of variance for Sircol assay was 2.81 ± 1.01. The measurable range for this assay is 31.25 to 500 μg/mL.
Statistical analysis
Heterogeneity of variance was tested with Fisher’s test and when required data were log-transformed. The means from C and BPA-treated groups at their respective time points (GD65 and GD90) were compared using two-tailed Student’s t- test (Prism 6 software, GraphPad, La Jolla, CA) and the threshold for significance was set as p ≤ 0.05. Data were also subjected to effect size analysis (Cohen, 1992) and computed Cohen’s d value ≥ 0.8 was considered large magnitude changes induced by BPA treatment.
Results
Effect of gestational BPA exposure on placental differentiation, weight and efficiency, and fetal weight:
There were no significant changes in number of Type A or B+C+D placentomes at GD65 or GD 90 (Figure 1A and 1B), although a trend for a large magnitude (d= 0.9; p = 0.09) increase in number of type A placentomes was evident in the BPA-treated sheep by Cohen’s effect size analysis at GD65. There was no effect of gestational BPA treatment on the weight of Type A or other (B+C+D) placentomes at either gestational time points (Figure 1C and D). In contrast, gestational BPA treatment induced a significant decrease in total fetal weight at GD65 (p = 0.04) but not at GD90 (Figure 1E). Consistent with this reduction, the placental efficiency calculated as ratio of fetal/placental weights was also significantly reduced at GD65 (p = 0.04) and not at D90 in animals exposed to BPA during gestation relative to controls (Figure 1F).
Figure 1:
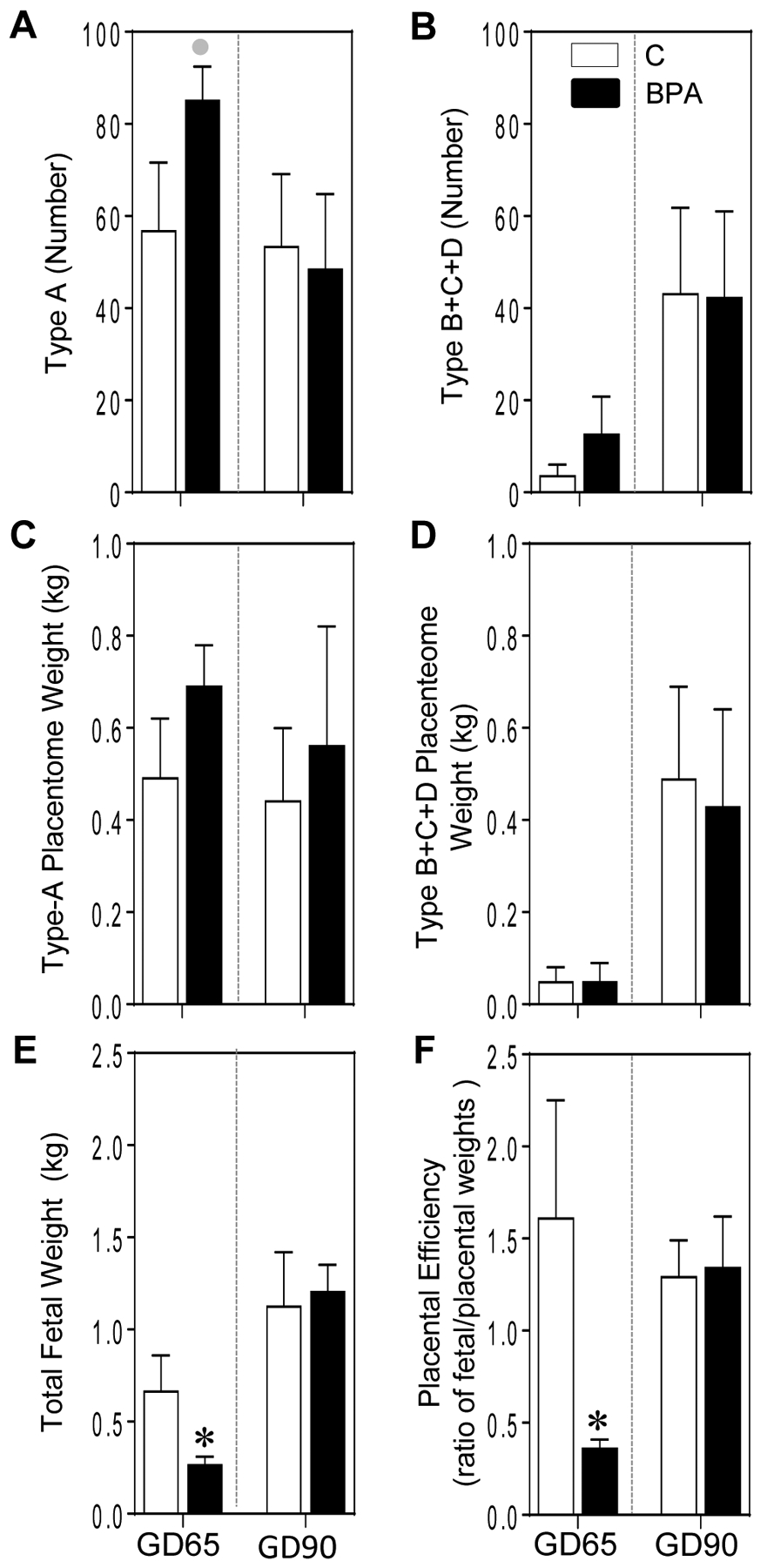
Mean ± SEM of number (A and B) and weight (C and D) of type A and B+C+D placentomes, fetal weight (E) and placental efficiency (F) at GD65 and GD90 from control (C) and BPA-treated pregnant sheep are shown. * = p < 0.05 by Student’s t-test and grey circles indicate large effect size by Cohen’s effect size analysis.
Effect of gestational BPA treatment on early placental inflammatory status:
Gestational BPA treatment significantly increased interleukin (IL) 8 (IL8; p = 0.005) but decreased tumor necrosis factor alpha (TNF) mRNA expression at GD65 (p = 0.04) (Figure 2A). In contrast at GD90, gestational BPA significantly increased IL1B mRNA (p = 0.04) and showed a trend for a large magnitude decrease (d= 2.0; p = 0.06) in TNF mRNA expression (Figure 2A). No changes at either time points were evident for the expression of cytokines chemokine ligand 2 (CCL2) and IL6 and macrophage marker CD68 between C and BPA-treated groups.
Figure 2:
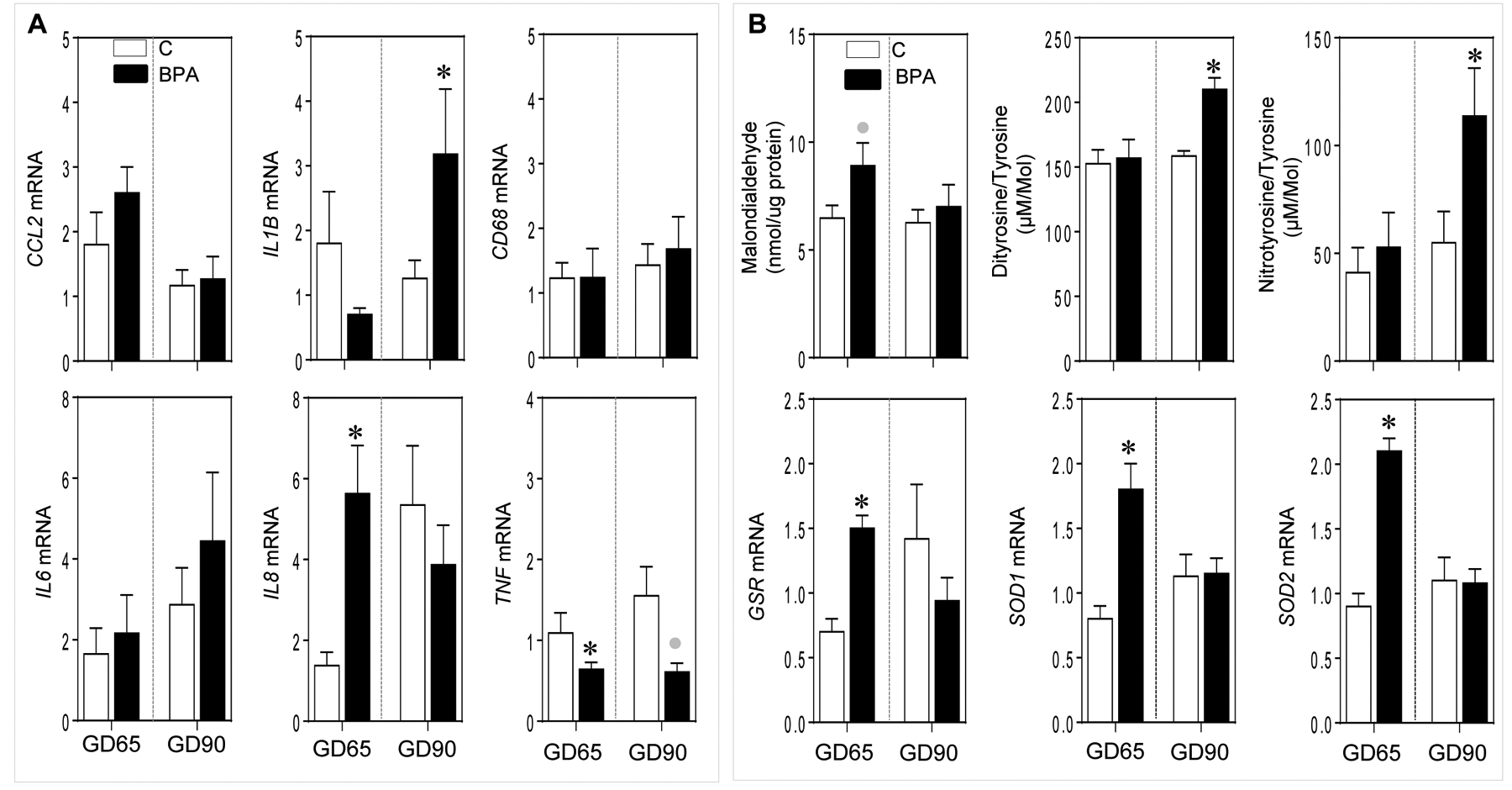
Mean ± SEM of fold change in mRNA expression for inflammatory cytokine and macrophage marker genes (Panel A) and oxidative stress markers malondialdehyde, dityrosine and nitrotyrosine (Panel B, top) and fold change in mRNA expression of antioxidants genes (Panel B, bottom) at GD65 and GD90 from control (C) and BPA exposed pregnant sheep are shown. * = p < 0.05 by Student’s t-test and grey circles indicate large effect size by Cohen’s effect size analysis.
Effect of gestational BPA treatment on early placental oxidative stress markers:
Gestational BPA treatment significantly increased oxidative stress markers dityrosine (p = <0.001) and nitrotyrosine (p = 0.04) at GD90 but not at GD65 (Figure 2B). No change in lipid peroxidation marker TBARS product malondialdehyde was evident at although a trend for a large magnitude increase (d = 1.0; p = 0.06) in TBARS in the BPA-treated group was evident at GD65 by effect size analysis.
Gestational BPA treatment significantly increased the expression of antioxidant genes glutathione reductase (GSR; p = 0.001), superoxide dismutase (SOD) 1 (p = 0.007), and SOD2 (p = <0.01) at GD65 but not at GD90 (Figure 2B).
Effect of gestational BPA treatment on early placental lipid and collagen accumulation:
Gestational BPA treatment significantly increased triglyceride accumulation in the placentome by GD90 (p = 0.003) but not at GD65 (Figure 3A). Consistent with this, there was a trend for a large magnitude increase (d = 1.1; p= 0.07) in collagen accumulation in the placentomes of BPA-treated animals at GD90 and not at GD65 (Figure 3B).
Figure 3:
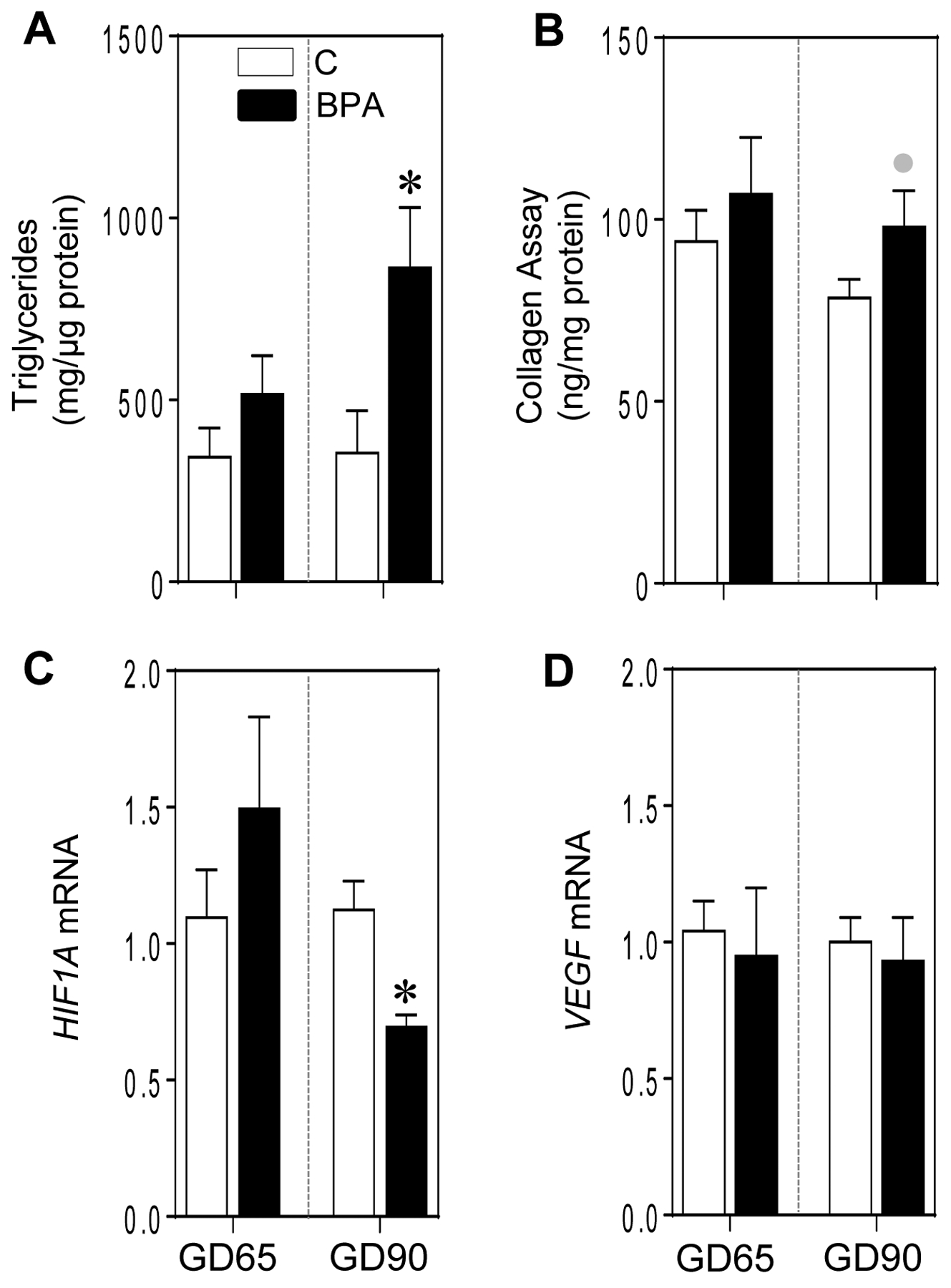
Mean ± SEM of placentome triglyceride (A) and collagen (B) content and fold change in mRNA expression of angiogenic genes (C and D) at GD65 and GD90 from control (C) and BPA exposed pregnant sheep are shown. * = p < 0.05 by Student’s t-test and grey circles indicate large effect size by Cohen’s effect size analysis.
Effect of gestational BPA treatment on early placental angiogenesis:
The effect of gestational BPA treatment on placental angiogenesis was examined by examination of genes that promote vascularization. There was a significant reduction in hypoxia inducible factor 1 alpha (HIF1A) at GD90 (p = 0.01) but not at GD65 (Figure 3C). There was no effect of gestational BPA treatment on vascular endothelial growth factor (VEGF) expression at either time point (Figure 3D).
Effect of gestational BPA treatment on early placental steroidogenic enzyme and steroid receptor expression:
The effect of gestational BPA treatment on steroidogenic components was examined by assessing the gene expression of steroidogenic enzymes and steroid receptors. Gestational BPA treatment significantly increased expression of steroidogenic enzymes 17α-hydroxylase, 17,20-lyase (CYP17; p = < 0.001) and aromatase (CYP19; p = 0.02) at GD65 (Figure 4A and B). In contrast at GD90, gestational BPA treatment significantly increased CYP17 expression only (p = 0.02) (Figure 4A). Relative to steroid receptors, BPA treatment had no impact on androgen receptor (AR) expression at both time points (Figure 4C). In contrast, gestational BPA treatment resulted in a significant increase in estrogen receptor 2 (ESR2) (p = <0.001) and a non-significant large magnitude increase (d= 0.8; p = 0.14) in estrogen receptor 1 (ESRI) as well as progesterone receptor (PGR) (d = 0.8; p = 0.15) at GD65 but not GD90 in the BPA-treated animals (Figure 4D–F).
Figure 4:
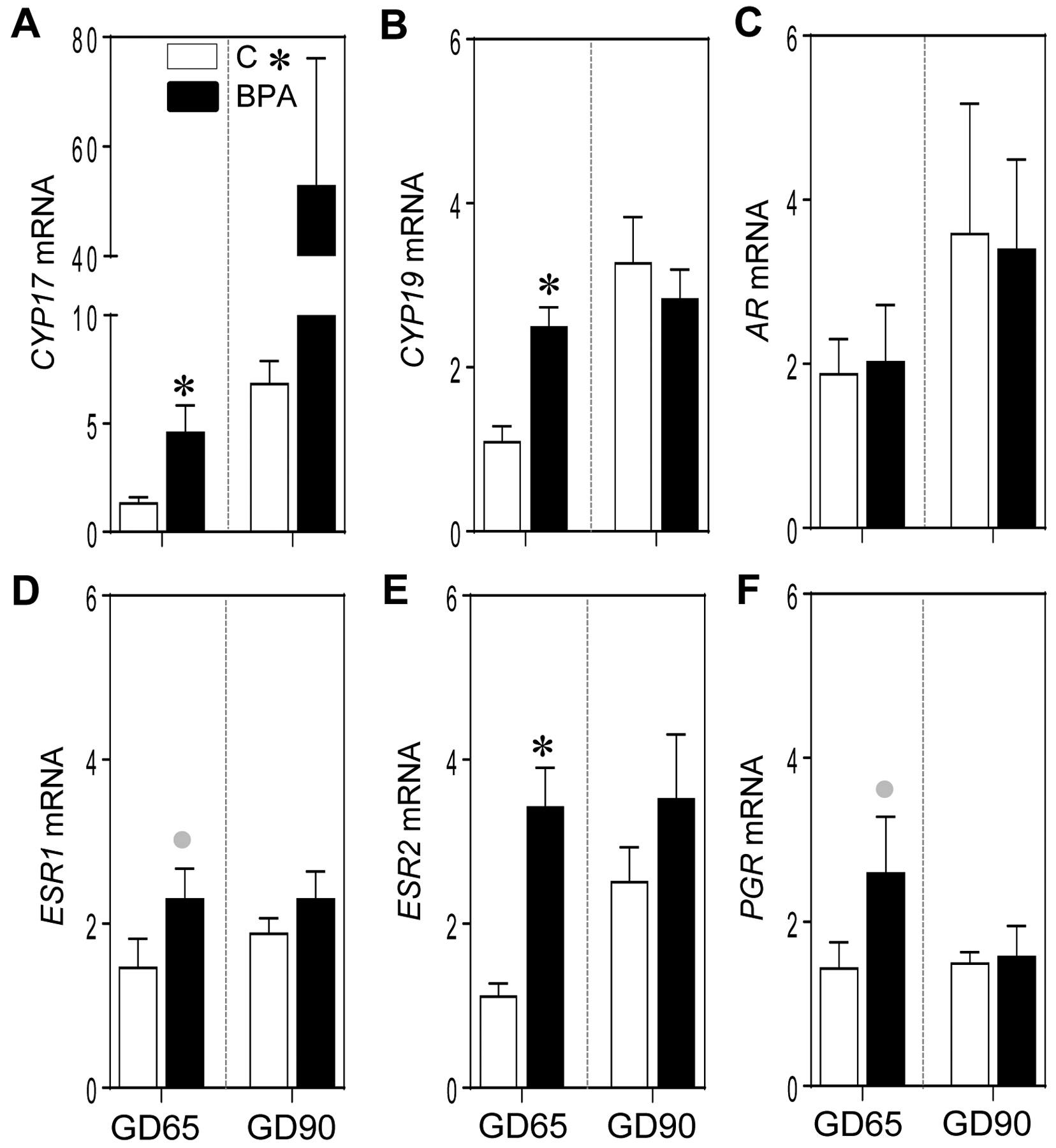
Mean ± SEM of fold change in mRNA expression of steroidogenic genes (CYP17 and CYP19; A and B, respectively) and steroid receptors (AR, ESR1, ESR2 and PGR; C, D, E and F, respectively) at GD65 and GD90 from control (C) and BPA exposed pregnant sheep are shown. * = p < 0.05 by Student’s t-test and grey circles indicate large effect size by Cohen’s effect size analysis.
Effect of gestational BPA treatment on placental expression of IGF family members:
Gestational BPA treatment induced non-significant large magnitude increases in IGF2 expression at both GD65 (d = 0.81; p = 0.11) and 90 (d = 0.92; p = 0.13) and IGF1 (d = 1.1; p = 0.08) only at GD90 (Figure 5A and B). As for the expression of receptors for IGFs, BPA treatment resulted in a significant reduction in the expression of IGF1 receptor (IGF1R) at GD65 (p = 0.02) with only a non-significant large magnitude increase (d = 0.9; p = 0.12) at GD90 (Figure 5C). In contrast, the IGF2 receptor (IGF2R) was significantly increased at GD65 (p = 0.01) with a trend for a large magnitude increase (d = 1.2; p = 0.07) at GD90 (Figure 5D). Relative to the expression of proteins that regulate IGF bioavailability, gestational BPA treatment increased significantly the expression of IGFBP1 (p = 0.004) and IGFBP3 (p = 0.03) at GD65 and expression of IGFBP2 (p = 0.02), IGFBP3 (p = 0.04) and IGFBP4 (p = 0.04) at GD90 (Figure 5E–H).
Figure 5:
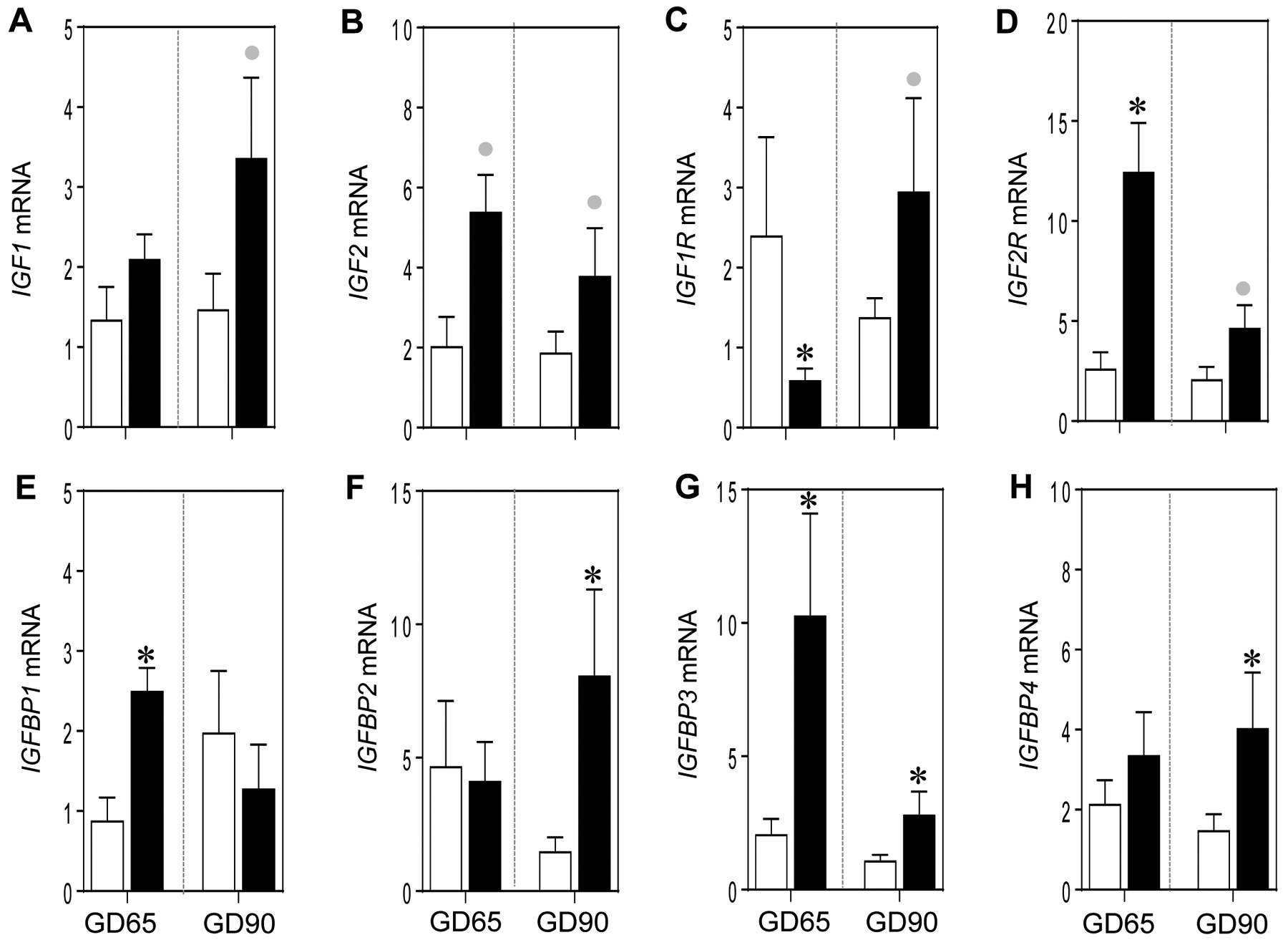
Mean ± SEM of fold change in mRNA expression of IGF family members: IGF1 (A) and IGF2 (B), IGF receptors (IGF1R and IGF2R; C and D, respectively) and binding proteins (IGFBP1–4; E-H, respectively) at GD65 and GD90 from control (C) and BPA exposed pregnant sheep are shown. * = p < 0.05 by Student’s t-test and grey circles indicate large effect size by Cohen’s effect size analysis.
Effect of gestational BPA treatment on early placental epigenetic machinery:
Epigenetic alterations affect expression of key genes required for placental function and usually involves covalent modulation of DNA and histones through methylation and acetylation (Kouzarides, 2007; Smith and Meissner, 2013). These changes are driven at the level of DNA by the enzymes belonging to the DNA methyltransferase (DNMT) family of which DNMT1, DNMT3A and DNMT3B are important (Smith and Meissner, 2013). Histone modification through methylation involves methyltransferases SMYD3, SUV39H1 and enhancer of zeste 2 (EZH2) and demethylase KDM1A. Another major histone modification involves acetylation that enhances gene expression and is driven by histone acetyltransfease EP300 (Roth et al., 2001). On the contrary reduction of gene expression is driven by histone deacetylases (HDAC) (Marks et al., 2003). The effects of EDC exposure including BPA in enhancing DNA methylation and histone modification in various bodily systems including placenta have been studied (Kundakovic and Champagne, 2011; Robins et al., 2011; Hogg et al., 2012; Nahar et al., 2015; LaRocca et al., 2016; Barouki et al., 2018). In the current study, the impact of gestational BPA treatment on placental epigenetic machinery was determined by examining the expression of enzymes that bring about modification of DNA and histones. Among the DNA methyl transferases (DNMTs), gestational BPA treatment significantly increased DNMT3A at GD90 (p = 0.02) but not GD65, while having no effect on DNMT1 and DNMT3B at both time points (Figure 6A–C). Gestational BPA treatment did not have any effect on histone methyl transferases SUV38H1 and EZH2 and histone demethylase KDM1A (Figure 6D–F) at both time points. Among the histone deacetylase (HDAC) enzymes, the expression of HDAC1 was significantly elevated by BPA treatment at both GD65 and GD90 (p = 0.004 and 0.005, respectively), while HDAC2 was only significantly increased at GD65 (p = 0.003) (Figure 6G and H). Cohen’s effect size analysis revealed large magnitude non-significant increase in HDAC2 (d = 1.1; p = 0.14) at GD90, a trend for an increase in HDAC3 at GD65 (d = 1.2; p = 0.055) and a non-significant large magnitude increase in HDAC3 at GD90 (d = 1.2; p = 0.18) (Figure 6H and I). In line with the changes in HDAC but only at GD65, BPA treatment, by effect size analysis, also induced a large magnitude non-significant increase (d = 1.0; p = 0.09;) in expression of histone acetyl transferase enzyme EP300 (Figure 6J).
Figure 6:
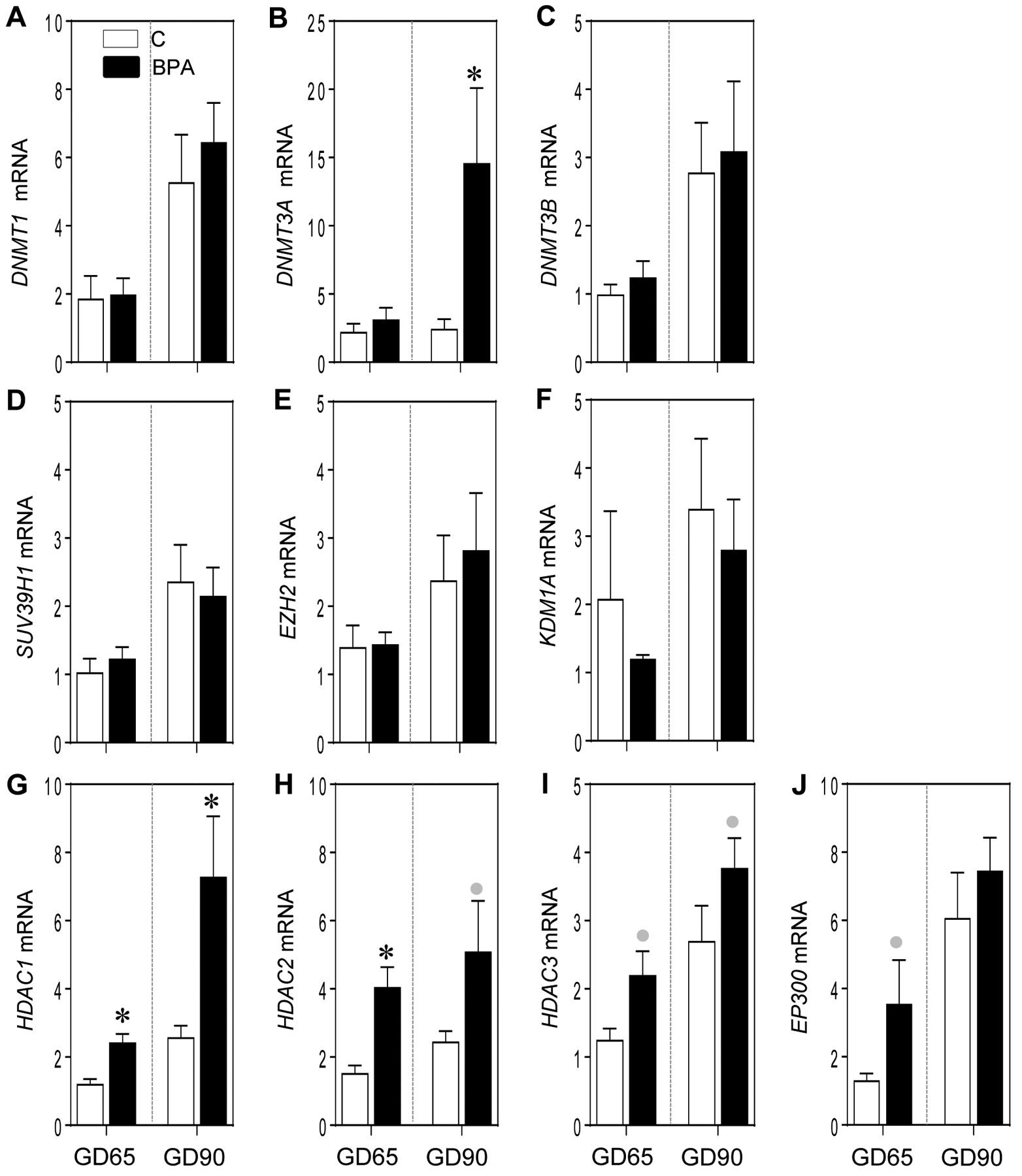
Mean ± SEM of fold change in mRNA expression of epigenetic modifying enzymes: DNA methyl transferases (DNMT1, DNMT3A and DNMT3B; A, B and C, respectively), histone methyl transferases (SUV39H1 and EZH2, D and E, respectively) and histone demethylase (KDM1A; F), histone deacetylases (HDAC1, HDAC2 and HDAC3; G, H and I, respectively) and histone acetyl transferase (EP300; J) at GD65 and GD90 from control (C) and BPA exposed pregnant sheep are shown. * = p < 0.05 by Student’s t-test and grey circles indicate large effect size by Cohen’s effect size analysis.
Discussion
The findings from this study indicate that gestational BPA treatment increases several negative mediators of placental function and that the impact on these negative mediators differ between mid – (day 90) and early (day 65) gestation. Changes in steroidogenic enzymes and receptors, altered IGF bioavailability, inflammatory and oxidative stress markers, lipotoxicity and epigenetic enzymes induced by gestational BPA-treatment are all possible mechanisms through which placental function and fetal growth is compromised. The finding that placental efficiency and fetal weight are reduced at day 65 but not at day 90 suggests some compensatory mechanism such as latency in response to day 65 increase in antioxidants may be in place to overcome the disruptive effects of BPA. The implication of these findings relative to BPA-associated placental dysfunction is discussed below.
Gestational BPA exposure on placental inflammation
As placenta forms the maternal-fetal interface where maternal cells encounter fetal cells which are akin to an allograft, the establishment and maintenance of pregnancy involves preserving a balance between placental immune reactivity and suppression (Redline, 2007). Any disruption of this balance would compromise the normal inflammatory cascade contributing to the poor birth outcomes (Trevisanuto et al., 2013) such as that seen in pathological conditions like preeclampsia and gestational diabetes (Kumar et al., 2013; Syngelaki et al., 2016; Rambaldi et al., 2019). EDCs including BPA have been shown to alter maternal and neonatal cord blood inflammatory cytokines (Ferguson et al., 2015b; Huang et al., 2017; Kelley et al., 2019). The finding of increased secretion of IL6 by placental explants following treatment with tetrabromobisphenol A (Arita et al., 2018), a brominated BPA derivative with similar endocrine disrupting action as BPA (Borghoff et al., 2016), is supportive of BPA-induced disruptions in placental inflammatory processes as well. The significant increase in inflammatory cytokines IL8 at early and IL1B at mid-gestation in BPA-treated animals (this study) is consistent with this premise. The increase in the expression of IL8 may have contributed to the reduced placental efficiency and fetal weight evidenced during early pregnancy. On the contrary, the significant large magnitude decrease at GD65 and non-significant large magnitude decrease in TNF expression in the BPA-treated group is not consistent with a pro-inflammatory role of TNF (Idriss and Naismith, 2000).
Gestational BPA exposure on placental oxidative stress
Normal placental function is associated with balanced production of reactive species and antioxidants (Wang et al., 1991) that helps prevent induction of oxidative stress. In pregnancies associated with poor placental function such as pre-eclampsia, fetal growth restriction and preterm labor this balance is disrupted leading to development of oxidative stress (Al-Gubory et al., 2010). In general, conditions associated with proinflammatory state are usually accompanied by oxidative stress (Chatterjee, 2016). In human, high BPA levels are associated with Increase in maternal and fetal oxidative stress markers, di and nitrotyrosine (Veiga-Lopez et al., 2015b). Gestational BPA treatment also increases oxidative stress markers in sheep and rats (Veiga-Lopez et al., 2015b). Furthermore, BPA treatment has been found to increase oxidative stress in studies with placental explants (Basak et al., 2018) as well as JEG3 and BeWo cells (Ponniah et al., 2015; Perez-Albaladejo et al., 2017). Our findings at the placental level of large magnitude albeit non-significant increase in TBARS at GD65 and significant large magnitude increases in di- and nitro-tyrosine at GD90 in gestational BPA-treated sheep are consistent with these other findings. However, the reason behind these time-dependent changes with increase in lipid peroxidation marker at early gestation, and nitrative and oxidative stress markers at mid gestation is not clear. The significant large magnitude increase in antioxidants only at GD65 by BPA treatment may be reflective of compensatory response to overcome the increase in oxidative stress evident at that time and may have contributed to the normalization of fetal weight by GD90. Lagged response to increase in oxidative stress markers coupled with unchanged antioxidant levels at GD90 may result in failure of this compensatory process as pregnancy progresses and contribute to the low birth offspring of gestational BPA treated animals (Savabieasfahani et al., 2006).
Gestational BPA exposure on placental lipotoxicity and fibrosis
While lipids are essential for maintenance of placental structure and function (Delhaes et al., 2018), increase in placental lipid accumulation in conditions such as obesity (Calabuig-Navarro et al., 2017; Delhaes et al., 2018) can lead to adverse programming. Abnormal placental lipid metabolism and lipid accumulation have also been observed following: 1) in vitro treatment of rat trophoblast cells (Xu et al., 2006) and in vivo treatment of rats (Xu et al., 2008) with phthalates metabolites and 2) treatment of JEG3, a placental cell line with bisphenol A diglycidyl ether, a synthesis product of BPA (Marqueno et al., 2019). Therefore, it is not surprising that gestational BPA treatment produced a significant large magnitude increase in placental lipid accumulation at mid-gestation.
Chronic activation of cellular repair mechanisms due to various insults can lead to increased production of extracellular matrix such as collagen resulting in placental fibrosis such as that seen with placental vaculopathies and IUGR (Chen et al., 2005; Ohmaru-Nakanishi et al., 2018). Insults that can lead to fibrosis include inflammation, oxidative stress and lipid accumulation (Zeisberg and Kalluri, 2013), all of which have been observed in gestational BPA-treated sheep (this study). Consistent with this premise, a large magnitude albeit non-significant increase in collagen accumulation was observed at mid- gestation in BPA-treated sheep, a finding requiring further validation with larger sample sizes. These emerging fibrotic changes at mid gestation may amplify with progression of pregnancy and contribute to the low birth weight offspring of BPA-treated sheep (Savabieasfahani et al., 2006).
Effect of BPA treatment on placental angiogenesis
Placental angiogenesis and vasculature changes markedly with the progression of gestation and pathogenesis associated with placental insufficiencies such as preeclampsia are associated with compromised angiogenesis and vascular functions (Charnock-Jones et al., 2004; Kaufmann et al., 2004; Mayhew et al., 2004). That endocrine disruptors such as phthalates and BPA are also associated with reduced angiogenic markers in maternal circulation (Ferguson et al., 2015a) suggest that they have the potential to affect placental angiogenesis. In support, gestational BPA-treatment alters placental vasculature in mouse placenta (Tait et al., 2015). The significant large magnitude decrease in angiogenesis promoter HIF1A found at mid-gestation in the BPA-treated sheep is in agreement with compromised angiogenesis. Considering estrogens promote placental angiogenesis (Albrecht and Pepe, 2010), the adverse impact of BPA - an estrogen mimic - is surprising and may relate to other properties of BPA. The lack of impact of gestational BPA treatment on VEGF expression was unexpected considering pregnancies associated with IUGR have reduced levels of VEGF transcripts (Ravikumar et al., 2019). A possibility to consider is that the VEGF disruption may be manifest during later stage of gestation, since HIF1A is known to exert its proangiogenic role by increasing VEGF expression (Zimna and Kurpisz, 2015).
Effect of BPA treatment on placental steroidal milieu
Apart from its function as a conduit between the mother and fetus, placenta is also a steroidogenic organ as well as a target of steroid action (Strauss et al., 1996; Miller and Auchus, 2011). Conditions associated with placental dysfunction are associated with altered steroidogenic enzyme expression in humans (Hogg et al., 2013). Maternal exposure to plasticizers such as phthalates are associated with alterations in maternal and fetal steroidal milieu (Sathyanarayana et al., 2017). While BPA treatment reduces aromatase activity in placental cell line JEG3 (Perez-Albaladejo et al., 2017), there is no consensus on its effect on CYP19 expression with one study showing no effect (Nativelle-Serpentini et al., 2003) and another showing reduced expression (Chu et al., 2018). BPA treatment has been shown to reduce placental CYP19 expression in term trophoblast cultures (Chu et al., 2018). The finding of a significant large magnitude increase in placental CYP19 expression in gestational BPA treated sheep is inconsistent with these in vitro findings from human placenta. These discrepancies may be a function of species differences (human vs. sheep), time of placental collection (early in sheep vs term in human), treatment duration (chronic in vivo in sheep vs acute in vitro in human) or their organizational (this study) vs. activational (in vitro studies in human) effects.
In contrast to the report that human placenta does not express CYP17 (Miller and Auchus, 2011), sheep placenta not only expresses CYP17 but BPA treatment induces a significant large magnitude increase in its expression at both GD65 and GD90 time points. While effects of BPA in increasing CYP17 have been shown in other cell types (Zhou et al., 2008), to our knowledge this is the first report of such induction in placental level. Effects of BPA on placental expression of steroidal receptors have not been previously examined. The significant large magnitude increase in ESR2 and non-significant large magnitude increases in ESR1 and PGR at early gestation by BPA treatment is supportive of paracrine actions of steroids at the placental level. Similar changes in placental steroid receptor mRNAs were seen in rats following treatment with the estrogen precursor, testosterone (Sun et al., 2012). Since placenta can aromatize testosterone to estradiol, the similarity in outcomes in terms of steroid receptor expression in both situations suggest that the regulation may be via estrogenic actions in both instances. The findings from this study point to BPA-induced disruption of placental steroidal biosynthesis and action. Whether the compromised steroid milieu is a contributor to the placental dysfunctions discussed above or vice versa remains to be determined.
Effect of BPA treatment on placental expression of IGF family members
Placenta also expresses insulin-like growth factors 1 and 2, its receptors and binding proteins (Han et al., 1996; Forbes and Westwood, 2008). While fetal IGF1 stimulates fetal growth by promoting anabolic events and DNA synthesis, placental IGFs influence fetal growth by promoting normal placental development/function and allowing proper transfer of nutrients to the fetus (Klauwer et al., 1997; Forbes and Westwood, 2008). Consistent with its key role, decreased placental expression of IGF1 accompanies pregnancies with fetal growth restriction (Koutsaki et al., 2011; Martin-Estal et al., 2016). Studies in mice indicate over expression of IGF2 leads to placental and fetal overgrowth, while its deletion, the opposite (DeChiara et al., 1990; Gicquel and Le Bouc, 2006). Exposure to EDCs such as polyaromatic hydrocarbons and polybrominated diphenyl ethers during pregnancy is associated with low levels of cord blood IGFs (Xu et al., 2013), possibly reflecting impaired placental expression. However, in the current study, such a decrease in IGF ligand expression was not observed at the early time points studied. An obvious possibility is that the large magnitude albeit non-significant increase in placental IGF2 expression observed at gestational day 65 in the present study is countered by the significant decrease in IGF1R expression and significant increase in expression of IGFBP1 and 3 leading to reduced IGF bioavailability, which would be consistent with the reduced placental efficiency and fetal weight evident in BPA-treated sheep at this time point. In contrast, the large magnitude non-significant increases in IGF1, IGF2, IGF1R and IGF2R at fetal day 90 may in unison have countered the significant increase in IGFBP 2–4 found at this time leading to increased IGF bioavailability, supportive of a compensatory response to overcome the fetal growth retardation seen at fetal day 65.
Effect of BPA treatment on placental expression of epigenetic modifying enzymes
The disruption in the mediators of placental function such as steroidogenic enzymes, steroid receptors and IGF family members evident in this study involves disruption of normal gene expression. Regulation of gene transcription involves epigenetic modifications and facilitated via changes in DNA methylation, histone modification and/or non-coding RNAs. Exposure to BPA has been shown to induce epigenetic modifications in several systems (Kundakovic and Champagne, 2011; Robins et al., 2011; Hogg et al., 2012; Nahar et al., 2015; LaRocca et al., 2016; Barouki et al., 2018; Strakovsky and Schantz, 2018). Findings from the current study with the gestational BPA-treated animals that manifest increase in DNMT3A – a de novo methyltransferase - and HDACs that removes the acetyl group from histone proteins and reduces accessibility of DNA to transcription factors, is consistent with BPA’s ability to induce epigenetic modifications. Studies with animal models have provided evidence that placental DNA methylation plays an important role in placental and fetal growth (Koukoura et al., 2012; Vrooman et al., 2016). In pregnancies affected by IUGR, placental IGF1 mRNA is decreased and the IGFBPs expression increased and these changes are associated with hypermethylation of IGF1 and hypomethylation of IGFBPs and IGF1R (Desgagne et al., 2014; Nawathe et al., 2016; Jia et al., 2017). Preeclampsia is also associated with hypermethylation of IGF1 promoter which is mediated by DNMT1 (Ma et al., 2018). In stark contrast, the large magnitude increase in IGF mRNA seen in the current study is not consistent with the observed changes in epigenetic enzyme. One possibility is that in BPA-treated sheep this regulation is exerted at the receptor level with the increase in HDAC1 contributing to BPA induced reduction in placental IGF1R during early gestation. Such an association between HDAC1 and IGF1R has been demonstrated in vascular smooth-muscle cells (Kavurma et al., 2007). These findings raise the possibility that the epigenetic regulation of IGF1R expression may be a function of changes in histone acetylation rather than DNA methylation.
Contrary to the dissociation in epigenetic enzyme changes and IGF, the reduced TNF and HIF1A expression observed at mid gestation in this study is in line with the observed increases in epigenetic enzymes and their predicted regulation. The findings from this study are also consistent with the decreased expression of TNF (Hermsdorff et al., 2013) and HIF1A (Walczak-Drzewiecka et al., 2010) found to accompany the increased DNA methylation of their promoters shown in other cell types.
While available evidence indicates that BPA exposure is associated with changes to placental DNA methylation, micro-RNA expression, and genomic imprinting (Strakovsky and Schantz, 2018), no reports exist regarding histone modifications at the placental level. Findings from the current study showing significant increase in histone deacetylase HDAC1 and HDAC2 and non-significant large magnitude increase in HDAC3 and histone acetylase EP300 indicate that effects of BPA on placental epigenome involve histone modifications. Since histone acetylation is viewed to be a prerequisite for TNF expression in some cells (Lee et al., 2003b) the decrease in TNF observed in this study maybe the result of increased HDACs that reduce acetylation of histones.
Linking BPA induced placental changes with placental efficiency and fetal growth
The effects of BPA, a xenoestrogen, may be exerted by direct action at the placental level or mediated via changes in placental steroidal milieu and involve changes in the epigenetic machinery. Steroids are classic programming agents (Padmanabhan et al., 2010). Available evidence suggests that nuclear hormone receptors through which steroid hormones signal and epigenetic regulatory enzymes can regulate each another and cooperatively modulate expression of downstream genes (Kato et al., 2011; Vilahur et al., 2014; Kappil et al., 2016; Vrooman et al., 2016). A possibility to consider is that the plethora of placental disruptions seen in gestational BPA-treated animals - summarized in Figure 7 - may all be mediated by altered steroidal input, the result of changes at the steroidogenic biosynthetic level as well as steroid receptor level or alternatively all the other BPA-induced placental compromise may underlie the disruption in steroid homeostasis with both scenario involving changes in the epigenome. Importantly the effects of BPA on the placenta appear to vary depending on the stage of the pregnancy and consistent with a latency in final outcomes. During early gestation, BPA induced increase in inflammation and oxidative stress, reduced levels of TNF, a cytokine with role in promoting placental function (Jaattela et al., 1988; Basu et al., 2016; Pavlov et al., 2016), and reduced bioavailability of IGFs may be the main drivers for reduced placental efficiency and fetal weight. During the mid-gestation, lag in action of increased expression of antioxidants observed during early-gestation and large magnitude albeit non-significant mid-gestational increase in both IGFs and its receptors may have aided in circumventing the increase in IGFBPs leading to restoration of placental efficiency and fetal weight. However, the mid-gestational increases in inflammatory, oxidative and lipotoxic state, reduced angiogenesis and increased fibrosis is suggestive of failure to sustain such compensation with pregnancy progression and could be the basis for poor birth outcomes in prenatal BPA-treated sheep (Savabieasfahani et al., 2006), a premise that needs to be tested in future investigations.
Figure 7:
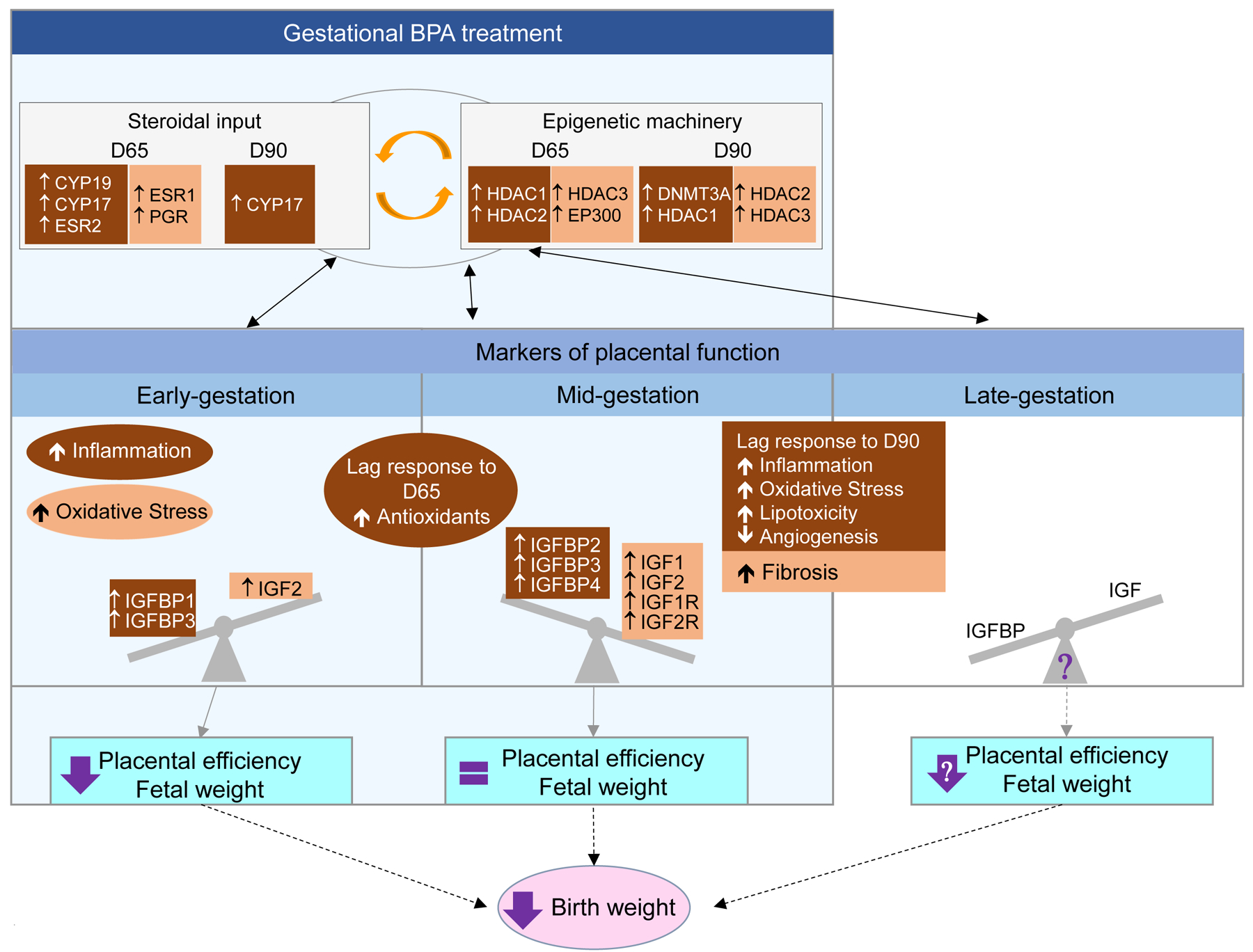
Schematic summarizing the effects of gestational BPA treatment during early, mid and late gestation. The light blue shaded area represents the period of BPA treatment spanning early to mid-gestation (gestational day 30–90; term ~147 days). Outcomes evident during this period may be due to direct effects of BPA or indirectly mediated by the changes in steroid input – function of changes in steroidogenic enzymes and steroid receptors, epigenetic machinery and mediators of placental function. Outcomes extending into late gestation likely represent the latent effects induced by BPA during early- / mid-gestation and programmed through changes in steroidal and epigenetic changes. Significant changes are shown in brown shaded areas with white letters and non-significant large magnitude effects as determined by Cohens effect size analysis are shown in orange shaded areas with black letters.
Strengths and limitations
The major strengths of this study include: 1) the use of an precocial large animal model with similar in utero developmental time line as humans, 2) treatment with environmentally relevant levels of BPA leading to achievement of fetal levels of BPA comparable to that observed in human pregnancy cohorts (Gerona et al., 2013; Lee et al., 2018), 3) focus on early and mid-gestation placenta, which is difficult to accomplish in humans, and 4) integrative assessment of plethora of mediators of placental function in unison. Notwithstanding these, the data presented here should be interpreted considering some of the limitations. These include small sample size that did not allow achievement of statistical significance with some key variables (eg. IGF), consideration of fetal sex as a variable in view of the sexual dimorphism existing at the placental level (Kalisch-Smith et al., 2017) and only gene expression analysis of inflammatory mediators, antioxidants, IGF family members, and angiogenic, steroidogenic and epigenetic markers. While further studies with larger sample size and protein and/or activity assessment of inflammation, angiogenesis, steroidal and epigenetic changes are required to fully delineate the final outcome, the coordinated directionality of changes amongst mediators of the placental phenome emphasize compromised placental dysfunction.
In conclusion, the findings from this study demonstrate that gestational BPA exposure impairs mediators of placental function at several levels and involves changes in epigenetic mechanisms, a finding of potential relevance to humans during pregnancy.
Supplementary Material
Acknowledgments
We are grateful to Mr Douglas Doop for his expert animal care, facility management, and help with generation of the experimental animals; Dr. Almudena Veiga-Lopez, Mr. Jim Lee, Ms Carol Herkimer, Ms Alexandra Spencer, and Ms Genevieve Ray for assistance with prenatal BPA treatment and/or the collection and processing of placentomes.
This work was supported by National Institutes of Health grant NIEHS R01 ES016541 to VP. MP’s efforts during this study was supported by Ruth L. Kirschstein Institutional Training Grant T32 ES007062.
References:
- Ahmed RG, 2016. Maternal bisphenol A alters fetal endocrine system: Thyroid adipokine dysfunction. Food Chem Toxicol 95, 168–174. [DOI] [PubMed] [Google Scholar]
- Al-Gubory KH, Fowler PA, Garrel C, 2010. The roles of cellular reactive oxygen species, oxidative stress and antioxidants in pregnancy outcomes. Int J Biochem Cell Biol 42, 1634–1650. [DOI] [PubMed] [Google Scholar]
- Albrecht ED, Pepe GJ, 2010. Estrogen regulation of placental angiogenesis and fetal ovarian development during primate pregnancy. Int J Dev Biol 54, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A, 2006. The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance. Environ Health Perspect 114, 106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Magdalena P, Rivera FJ, Guerrero-Bosagna C, 2016. Bisphenol-A and metabolic diseases: epigenetic, developmental and transgenerational basis. Environ Epigenet 2, dvw022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita Y, Pressman M, Getahun D, Menon R, Peltier MR, 2018. Effect of Tetrabromobisphenol A on expression of biomarkers for inflammation and neurodevelopment by the placenta. Placenta 68, 33–39. [DOI] [PubMed] [Google Scholar]
- Barker DJ, 2004. The developmental origins of adult disease. Journal of the American College of Nutrition 23, 588S–595S. [DOI] [PubMed] [Google Scholar]
- Barouki R, Melen E, Herceg Z, Beckers J, Chen J, Karagas M, Puga A, Xia Y, Chadwick L, Yan W, Audouze K, Slama R, Heindel J, Grandjean P, Kawamoto T, Nohara K, 2018. Epigenetics as a mechanism linking developmental exposures to long-term toxicity. Environ Int 114, 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak S, Srinivas V, Duttaroy AK, 2018. Bisphenol-A impairs cellular function and alters DNA methylation of stress pathway genes in first trimester trophoblast cells. Reprod Toxicol 82, 72–79. [DOI] [PubMed] [Google Scholar]
- Basu J, Agamasu E, Bendek B, Salafia CM, Mishra A, Benfield N, Prasad P, Mikhail M, 2016. Placental tumor necrosis factor-alpha protein expression during normal human gestation. J Matern Fetal Neonatal Med 29, 3934–3938. [DOI] [PubMed] [Google Scholar]
- Benachour N, Aris A, 2009. Toxic effects of low doses of Bisphenol-A on human placental cells. Toxicol Appl Pharmacol 241, 322–328. [DOI] [PubMed] [Google Scholar]
- Borghoff SJ, Wikoff D, Harvey S, Haws L, 2016. Dose- and time-dependent changes in tissue levels of tetrabromobisphenol A (TBBPA) and its sulfate and glucuronide conjugates following repeated administration to female Wistar Han Rats. Toxicol Rep 3, 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabuig-Navarro V, Haghiac M, Minium J, Glazebrook P, Ranasinghe GC, Hoppel C, Hauguel de-Mouzon S, Catalano P, O’Tierney-Ginn P, 2017. Effect of Maternal Obesity on Placental Lipid Metabolism. Endocrinology 158, 2543–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham LL, 2005. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ Health Perspect 113, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL, 2008. Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ Health Perspect 116, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC., 2014. Fourth National Report on Human Exposure to Environmental Chemicals. US Department of Health and Human Services Centres for Disease Control and Prevention, Atlanta, GA, https://www.cdc.gov/exposurereport/pdf/fourthreport.pdf (accessed 09/17/2019). [Google Scholar]
- Charnock-Jones DS, Kaufmann P, Mayhew TM, 2004. Aspects of human fetoplacental vasculogenesis and angiogenesis. I. Molecular regulation. Placenta 25, 103–113. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, 2016. Oxidative stress, inflammation, and disease. Oxidative Stress and Biomaterials; Elsevier, pp. 35–58. [Google Scholar]
- Chen CP, Yang YC, Su TH, Chen CY, Aplin JD, 2005. Hypoxia and transforming growth factor-beta 1 act independently to increase extracellular matrix production by placental fibroblasts. J Clin Endocrinol Metab 90, 1083–1090. [DOI] [PubMed] [Google Scholar]
- Chu PW, Yang ZJ, Huang HH, Chang AA, Cheng YC, Wu GJ, Lan HC, 2018. Low-dose bisphenol A activates the ERK signaling pathway and attenuates steroidogenic gene expression in human placental cells. Biol Reprod 98, 250–258. [DOI] [PubMed] [Google Scholar]
- Coentro JQ, Capella-Monsonis H, Graceffa V, Wu Z, Mullen AM, Raghunath M, Zeugolis DI, 2017. Collagen Quantification in Tissue Specimens. Methods Mol Biol 1627, 341–350. [DOI] [PubMed] [Google Scholar]
- Cohen J, 1992. A power primer. Psychol Bull 112, 155–159. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Efstratiadis A, Robertson EJ, 1990. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345, 78–80. [DOI] [PubMed] [Google Scholar]
- Delhaes F, Giza SA, Koreman T, Eastabrook G, McKenzie CA, Bedell S, Regnault TRH, de Vrijer B, 2018. Altered maternal and placental lipid metabolism and fetal fat development in obesity: Current knowledge and advances in non-invasive assessment. Placenta 69, 118–124. [DOI] [PubMed] [Google Scholar]
- Desgagne V, Hivert MF, St-Pierre J, Guay SP, Baillargeon JP, Perron P, Gaudet D, Brisson D, Bouchard L, 2014. Epigenetic dysregulation of the IGF system in placenta of newborns exposed to maternal impaired glucose tolerance. Epigenomics 6, 193–207. [DOI] [PubMed] [Google Scholar]
- Elmetwally MA, Halawa AA, Lenis YY, Tang W, Wu G, Bazer FW, 2018. Effects of Bisphenol-A on proliferation and expression of genes related to synthesis of polyamines, interferon tau and insulin-like growth factor 2 by ovine trophectoderm cells. Reprod Toxicol 78, 90–96. [DOI] [PubMed] [Google Scholar]
- Ferguson KK, McElrath TF, Cantonwine DE, Mukherjee B, Meeker JD, 2015a. Phthalate metabolites and bisphenol-A in association with circulating angiogenic biomarkers across pregnancy. Placenta 36, 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KK, McElrath TF, Mukherjee B, Loch-Caruso R, Meeker JD, 2015b. Associations between Maternal Biomarkers of Phthalate Exposure and Inflammation Using Repeated Measurements across Pregnancy. PLoS One 10, e0135601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes K, Westwood M, 2008. The IGF axis and placental function. a mini review. Horm Res 69, 129–137. [DOI] [PubMed] [Google Scholar]
- Geetharathan T, Josthna P, 2016. Effect of BPA on protein, lipid profile and immuno-histo chemical changes in placenta and uterine tissues of albino rat. International Journal of Pharmaceutical and Clinical Research 8, 260–268. [Google Scholar]
- Gerona RR, Woodruff TJ, Dickenson CA, Pan J, Schwartz JM, Sen S, Friesen MW, Fujimoto VY, Hunt PA, 2013. Bisphenol-A (BPA), BPA glucuronide, and BPA sulfate in midgestation umbilical cord serum in a northern and central California population. Environ Sci Technol 47, 12477–12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gicquel C, Le Bouc Y, 2006. Hormonal regulation of fetal growth. Horm Res 65 Suppl 3, 28–33. [DOI] [PubMed] [Google Scholar]
- Grigsby PL, 2016. Animal Models to Study Placental Development and Function throughout Normal and Dysfunctional Human Pregnancy. Semin Reprod Med 34, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han VK, Bassett N, Walton J, Challis JR, 1996. The expression of insulin-like growth factor (IGF) and IGF-binding protein (IGFBP) genes in the human placenta and membranes: evidence for IGF-IGFBP interactions at the feto-maternal interface. J Clin Endocrinol Metab 81, 2680–2693. [DOI] [PubMed] [Google Scholar]
- Haywood NJ, Slater TA, Matthews CJ, Wheatcroft SB, 2019. The insulin like growth factor and binding protein family: Novel therapeutic targets in obesity & diabetes. Mol Metab 19, 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermsdorff HH, Mansego ML, Campion J, Milagro FI, Zulet MA, Martinez JA, 2013. TNF-alpha promoter methylation in peripheral white blood cells: relationship with circulating TNFalpha, truncal fat and n-6 PUFA intake in young women. Cytokine 64, 265–271. [DOI] [PubMed] [Google Scholar]
- Hogg K, Blair JD, McFadden DE, von Dadelszen P, Robinson WP, 2013. Early onset pre-eclampsia is associated with altered DNA methylation of cortisol-signalling and steroidogenic genes in the placenta. PLoS One 8, e62969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg K, Price EM, Hanna CW, Robinson WP, 2012. Prenatal and perinatal environmental influences on the human fetal and placental epigenome. Clin Pharmacol Ther 92, 716–726. [DOI] [PubMed] [Google Scholar]
- Huang YF, Wang PW, Huang LW, Lai CH, Yang W, Wu KY, Lu CA, Chen HC, Chen ML, 2017. Prenatal Nonylphenol and Bisphenol A Exposures and Inflammation Are Determinants of Oxidative/Nitrative Stress: A Taiwanese Cohort Study. Environ Sci Technol 51, 6422–6429. [DOI] [PubMed] [Google Scholar]
- Idriss HT, Naismith JH, 2000. TNF alpha and the TNF receptor superfamily: structure-function relationship(s). Microsc Res Tech 50, 184–195. [DOI] [PubMed] [Google Scholar]
- Jaattela M, Kuusela P, Saksela E, 1988. Demonstration of tumor necrosis factor in human amniotic fluids and supernatants of placental and decidual tissues. Lab Invest 58, 48–52. [PubMed] [Google Scholar]
- Jia Y, Li T, Huang X, Xu X, Zhou X, Jia L, Zhu J, Xie D, Wang K, Zhou Q, Jin L, Zhang J, Duan T, 2017. Dysregulated DNA Methyltransferase 3A Upregulates IGFBP5 to Suppress Trophoblast Cell Migration and Invasion in Preeclampsia. Hypertension 69, 356–366. [DOI] [PubMed] [Google Scholar]
- Kalisch-Smith JI, Simmons DG, Dickinson H, Moritz KM, 2017. Review: Sexual dimorphism in the formation, function and adaptation of the placenta. Placenta 54, 10–16. [DOI] [PubMed] [Google Scholar]
- Kappil MA, Li Q, Li A, Dassanayake PS, Xia Y, Nanes JA, Landrigan PJ, Stodgell CJ, Aagaard KM, Schadt EE, Dole N, Varner M, Moye J, Kasten C, Miller RK, Ma Y, Chen J, Lambertini L, 2016. In utero exposures to environmental organic pollutants disrupt epigenetic marks linked to fetoplacental development. Environ Epigenet 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Yokoyama A, Fujiki R, 2011. Nuclear receptor coregulators merge transcriptional coregulation with epigenetic regulation. Trends Biochem Sci 36, 272–281. [DOI] [PubMed] [Google Scholar]
- Kaufmann P, Mayhew TM, Charnock-Jones DS, 2004. Aspects of human fetoplacental vasculogenesis and angiogenesis. II. Changes during normal pregnancy. Placenta 25, 114–126. [DOI] [PubMed] [Google Scholar]
- Kavurma MM, Figg N, Bennett MR, Mercer J, Khachigian LM, Littlewood TD, 2007. Oxidative stress regulates IGF1R expression in vascular smooth-muscle cells via p53 and HDAC recruitment. Biochem J 407, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley AS, Banker M, Goodrich JM, Dolinoy DC, Burant C, Domino SE, Smith YR, Song PXK, Padmanabhan V, 2019. Early pregnancy exposure to endocrine disrupting chemical mixtures are associated with inflammatory changes in maternal and neonatal circulation. Sci Rep 9, 5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauwer D, Blum WF, Hanitsch S, Rascher W, Lee PD, Kiess W, 1997. IGF-I, IGF-II, free IGF-I and IGFBP-1, −2 and −3 levels in venous cord blood: relationship to birthweight, length and gestational age in healthy newborns. Acta Paediatr 86, 826–833. [DOI] [PubMed] [Google Scholar]
- Kolatorova L, Vitku J, Hampl R, Adamcova K, Skodova T, Simkova M, Parizek A, Starka L, Duskova M, 2018. Exposure to bisphenols and parabens during pregnancy and relations to steroid changes. Environ Res. 163,115–122. [DOI] [PubMed] [Google Scholar]
- Koukoura O, Sifakis S, Spandidos DA, 2012. DNA methylation in the human placenta and fetal growth (review). Mol Med Rep 5, 883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsaki M, Sifakis S, Zaravinos A, Koutroulakis D, Koukoura O, Spandidos DA, 2011. Decreased placental expression of hPGH, IGF-I and IGFBP-1 in pregnancies complicated by fetal growth restriction. Growth Horm IGF Res 21, 31–36. [DOI] [PubMed] [Google Scholar]
- Kouzarides T, 2007. Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Kumar A, Begum N, Prasad S, Agarwal S, Sharma S, 2013. IL-10, TNF-alpha & IFN-gamma: potential early biomarkers for preeclampsia. Cell Immunol 283, 70–74. [DOI] [PubMed] [Google Scholar]
- Kundakovic M, Champagne FA, 2011. Epigenetic perspective on the developmental effects of bisphenol A. Brain Behav Immun 25, 1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRocca J, Binder AM, McElrath TF, Michels KB, 2016. First-Trimester Urine Concentrations of Phthalate Metabolites and Phenols and Placenta miRNA Expression in a Cohort of U.S. Women. Environ Health Perspect 124, 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BE, Park H, Hong YC, Ha M, Kim Y, Chang N, Kim BN, Kim YJ, Yu SD, Ha EH, 2014. Prenatal bisphenol A and birth outcomes: MOCEH (Mothers and Children’s Environmental Health) study. Int J Hyg Environ Health 217, 328–334. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Chattopadhyay S, Gong EY, Ahn RS, Lee K, 2003a. Antiandrogenic effects of bisphenol A and nonylphenol on the function of androgen receptor. Toxicol Sci 75, 40–46. [DOI] [PubMed] [Google Scholar]
- Lee J, Choi K, Park J, Moon HB, Choi G, Lee JJ, Suh E, Kim HJ, Eun SH, Kim GH, Cho GJ, Kim SK, Kim S, Kim SY, Kim S, Eom S, Choi S, Kim YD, Kim S, 2018. Bisphenol A distribution in serum, urine, placenta, breast milk, and umbilical cord serum in a birth panel of mother-neonate pairs. Sci Total Environ 626, 1494–1501. [DOI] [PubMed] [Google Scholar]
- Lee JY, Kim NA, Sanford A, Sullivan KE, 2003b. Histone acetylation and chromatin conformation are regulated separately at the TNF-alpha promoter in monocytes and macrophages. J Leukoc Biol 73, 862–871. [DOI] [PubMed] [Google Scholar]
- Ma M, Zhou QJ, Xiong Y, Li B, Li XT, 2018. Preeclampsia is associated with hypermethylation of IGF-1 promoter mediated by DNMT1. Am J Transl Res 10, 16–39. [PMC free article] [PubMed] [Google Scholar]
- Maccani MA, Marsit CJ, 2009. Epigenetics in the placenta. Am J Reprod Immunol 62, 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manikkam M, Crespi EJ, Doop DD, Herkimer C, Lee JS, Yu S, Brown MB, Foster DL, Padmanabhan V, 2004. Fetal programming: prenatal testosterone excess leads to fetal growth retardation and postnatal catch-up growth in sheep. Endocrinology 145, 790–798. [DOI] [PubMed] [Google Scholar]
- Marks PA, Miller T, Richon VM, 2003. Histone deacetylases. Curr Opin Pharmacol 3, 344–351. [DOI] [PubMed] [Google Scholar]
- Marqueno A, Perez-Albaladejo E, Flores C, Moyano E, Porte C, 2019. Toxic effects of bisphenol A diglycidyl ether and derivatives in human placental cells. Environ Pollut 244, 513–521. [DOI] [PubMed] [Google Scholar]
- Marsit CJ, 2016. Placental Epigenetics in Children’s Environmental Health. Semin Reprod Med 34, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Estal I, de la Garza RG, Castilla-Cortazar I, 2016. Intrauterine Growth Retardation (IUGR) as a Novel Condition of Insulin-Like Growth Factor-1 (IGF-1) Deficiency. Rev Physiol Biochem Pharmacol 170, 1–35. [DOI] [PubMed] [Google Scholar]
- Mayhew TM, Charnock-Jones DS, Kaufmann P, 2004. Aspects of human fetoplacental vasculogenesis and angiogenesis. III. Changes in complicated pregnancies. Placenta 25, 127–139. [DOI] [PubMed] [Google Scholar]
- Miller WL, Auchus RJ, 2011. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 32, 81–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montrose L, Padmanabhan V, Goodrich JM, Domino SE, Treadwell MC, Meeker JD, Watkins DJ, Dolinoy DC, 2018. Maternal levels of endocrine disrupting chemicals in the first trimester of pregnancy are associated with infant cord blood DNA methylation. Epigenetics 13, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahar MS, Liao C, Kannan K, Harris C, Dolinoy DC, 2015. In utero bisphenol A concentration, metabolism, and global DNA methylation across matched placenta, kidney, and liver in the human fetus. Chemosphere 124, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nativelle-Serpentini C, Richard S, Seralini GE, Sourdaine P, 2003. Aromatase activity modulation by lindane and bisphenol-A in human placental JEG-3 and transfected kidney E293 cells. Toxicol In Vitro 17, 413–422. [DOI] [PubMed] [Google Scholar]
- Nawathe AR, Christian M, Kim SH, Johnson M, Savvidou MD, Terzidou V, 2016. Insulin-like growth factor axis in pregnancies affected by fetal growth disorders. Clin Epigenetics 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel BA, Sargis RM, 2011. The paradox of progress: environmental disruption of metabolism and the diabetes epidemic. Diabetes 60, 1838–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelissen EC, van Montfoort AP, Dumoulin JC, Evers JL, 2011. Epigenetics and the placenta. Hum Reprod Update 17, 397–417. [DOI] [PubMed] [Google Scholar]
- Nugent BM, Bale TL, 2015. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol 39, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmaru-Nakanishi T, Asanoma K, Fujikawa M, Fujita Y, Yagi H, Onoyama I, Hidaka N, Sonoda K, Kato K, 2018. Fibrosis in Preeclamptic Placentas Is Associated with Stromal Fibroblasts Activated by the Transforming Growth Factor-beta1 Signaling Pathway. Am J Pathol 188, 683–695. [DOI] [PubMed] [Google Scholar]
- Padmanabhan V, Sarma HN, Savabieasfahani M, Steckler TL, Veiga-Lopez A, 2010. Developmental reprogramming of reproductive and metabolic dysfunction in sheep: native steroids vs. environmental steroid receptor modulators. Int J Androl 33, 394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan V, Veiga-Lopez A, 2014. Reproduction Symposium: developmental programming of reproductive and metabolic health. J Anim Sci 92, 3199–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov OV, Niauri DA, Selutin AV, Selkov SA, 2016. Coordinated expression of TNFalpha- and VEGF-mediated signaling components by placental macrophages in early and late pregnancy. Placenta 42, 28–36. [DOI] [PubMed] [Google Scholar]
- Peretz J, Vrooman L, Ricke WA, Hunt PA, Ehrlich S, Hauser R, Padmanabhan V, Taylor HS, Swan SH, VandeVoort CA, Flaws JA, 2014. Bisphenol a and reproductive health: update of experimental and human evidence, 2007–2013. Environ Health Perspect 122, 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Albaladejo E, Fernandes D, Lacorte S, Porte C, 2017. Comparative toxicity, oxidative stress and endocrine disruption potential of plasticizers in JEG-3 human placental cells. Toxicol In Vitro 38, 41–48. [DOI] [PubMed] [Google Scholar]
- Ponniah M, Billett EE, De Girolamo LA, 2015. Bisphenol A increases BeWo trophoblast survival in stress-induced paradigms through regulation of oxidative stress and apoptosis. Chem Res Toxicol 28, 1693–1703. [DOI] [PubMed] [Google Scholar]
- Puttabyatappa M, Andriessen V, Mesquitta M, Zeng L, Pennathur S, Padmanabhan V, 2017. Developmental Programming: Impact of Gestational Steroid and Metabolic Milieus on Mediators of Insulin Sensitivity in Prenatal Testosterone-Treated Female Sheep. Endocrinology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaldi MP, Weiner E, Mecacci F, Bar J, Petraglia F, 2019. Immunomodulation and preeclampsia. Best Pract Res Clin Obstet Gynaecol. [DOI] [PubMed] [Google Scholar]
- Ravikumar G, Mukhopadhyay A, Mani C, Kocchar P, Crasta J, Thomas T, Dwarkanath P, Thomas A, Kurpad AV, Sridhar TS, 2019. Placental expression of angiogenesis-related genes and their receptors in IUGR pregnancies: correlation with fetoplacental and maternal parameters. J Matern Fetal Neonatal Med, 1–8. [DOI] [PubMed] [Google Scholar]
- Redline RW, 2007. Placental Inflammation. in: Keeling JW, Khong TY (Eds.). Fetal and Neonatal Pathology; Springer London, London, pp. 90–101. [Google Scholar]
- Robins JC, Marsit CJ, Padbury JF, Sharma SS, 2011. Endocrine disruptors, environmental oxygen, epigenetics and pregnancy. Front Biosci (Elite Ed) 3, 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD, 2001. Histone acetyltransferases. Annu Rev Biochem 70, 81–120. [DOI] [PubMed] [Google Scholar]
- Saadeldin IM, Hussein MA, Suleiman AH, Abohassan MG, Ahmed MM, Moustafa AA, Moumen AF, Abdel-Aziz S,A, 2018. Ameliorative effect of ginseng extract on phthalate and bisphenol A reprotoxicity during pregnancy in rats. Environ Sci Pollut Res Int. 25, 21205–21215. [DOI] [PubMed] [Google Scholar]
- Sathyanarayana S, Butts S, Wang C, Barrett E, Nguyen R, Schwartz SM, Haaland W, Swan SH, Team T, 2017. Early Prenatal Phthalate Exposure, Sex Steroid Hormones, and Birth Outcomes. J Clin Endocrinol Metab 102, 1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savabieasfahani M, Kannan K, Astapova O, Evans NP, Padmanabhan V, 2006. Developmental programming: differential effects of prenatal exposure to bisphenol-A or methoxychlor on reproductive function. Endocrinology 147, 5956–5966. [DOI] [PubMed] [Google Scholar]
- Sharma D, Shastri S, Sharma P, 2016. Intrauterine Growth Restriction: Antenatal and Postnatal Aspects. Clin Med Insights Pediatr 10, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoaito H, Petit J, Chissey A, Auzeil N, Guibourdenche J, Gil S, Laprevote O, Fournier T, Degrelle SA, 2019. The Role of Peroxisome Proliferator-Activated Receptor Gamma (PPARgamma) in Mono(2-ethylhexyl) Phthalate (MEHP)-Mediated Cytotrophoblast Differentiation. Environ Health Perspect 127, 27003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Meissner A, 2013. DNA methylation: roles in mammalian development. Nat Rev Genet 14, 204–220. [DOI] [PubMed] [Google Scholar]
- Strakovsky RS, Schantz SL, 2018. Impacts of bisphenol A (BPA) and phthalate exposures on epigenetic outcomes in the human placenta. Environ Epigenet 4, dvy022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JF 3rd, Martinez F, Kiriakidou M, 1996. Placental steroid hormone synthesis: unique features and unanswered questions. Biol Reprod 54, 303–311. [DOI] [PubMed] [Google Scholar]
- Sun M, Maliqueo M, Benrick A, Johansson J, Shao R, Hou L, Jansson T, Wu X, Stener-Victorin E, 2012. Maternal androgen excess reduces placental and fetal weights, increases placental steroidogenesis, and leads to long-term health effects in their female offspring. Am J Physiol Endocrinol Metab 303, E1373–1385. [DOI] [PubMed] [Google Scholar]
- Syngelaki A, Visser GH, Krithinakis K, Wright A, Nicolaides KH, 2016. First trimester screening for gestational diabetes mellitus by maternal factors and markers of inflammation. Metabolism 65, 131–137. [DOI] [PubMed] [Google Scholar]
- Tait S, Tassinari R, Maranghi F, Mantovani A, 2015. Bisphenol A affects placental layers morphology and angiogenesis during early pregnancy phase in mice. J Appl Toxicol 35, 1278–1291. [DOI] [PubMed] [Google Scholar]
- Trevisanuto D, Peruzzetto C, Cavallin F, Vedovato S, Cosmi E, Visentin S, Chiarelli S, Zanardo V, 2013. Fetal placental inflammation is associated with poor neonatal growth of preterm infants: a case-control study. J Matern Fetal Neonatal Med 26, 1484–1490. [DOI] [PubMed] [Google Scholar]
- Vatnick I, Ignotz G, McBride BW, Bell AW, 1991. Effect of heat stress on ovine placental growth in early pregnancy. J Dev Physiol 16, 163–166. [PubMed] [Google Scholar]
- Veiga-Lopez A, Kannan K, Liao C, Ye W, Domino SE, Padmanabhan V, 2015a. Gender-Specific Effects on Gestational Length and Birth Weight by Early Pregnancy BPA Exposure. J Clin Endocrinol Metab 100, E1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Luense LJ, Christenson LK, Padmanabhan V, 2013. Developmental programming: gestational bisphenol-A treatment alters trajectory of fetal ovarian gene expression. Endocrinology 154, 1873–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Pennathur S, Kannan K, Patisaul HB, Dolinoy DC, Zeng L, Padmanabhan V, 2015b. Impact of gestational bisphenol A on oxidative stress and free fatty acids: Human association and interspecies animal testing studies. Endocrinology 156, 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilahur N, Bustamante M, Byun HM, Fernandez MF, Santa Marina L, Basterrechea M, Ballester F, Murcia M, Tardon A, Fernandez-Somoano A, Estivill X, Olea N, Sunyer J, Baccarelli AA, 2014. Prenatal exposure to mixtures of xenoestrogens and repetitive element DNA methylation changes in human placenta. Environ Int 71, 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivekanandan-Giri A, Byun J, Pennathur S, 2011. Quantitative analysis of amino Acid oxidation markers by tandem mass spectrometry. Methods in enzymology 491, 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vom Saal FS, 2016. TRIENNIAL REPRODUCTION SYMPOSIUM: Environmental programming of reproduction during fetal life: Effects of intrauterine position and the endocrine disrupting chemical bisphenol A. J Anim Sci 94, 2722–2736. [DOI] [PubMed] [Google Scholar]
- Vrooman LA, Xin F, Bartolomei MS, 2016. Morphologic and molecular changes in the placenta: what we can learn from environmental exposures. Fertil Steril 106, 930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak-Drzewiecka A, Ratajewski M, Pulaski L, Dastych J, 2010. DNA methylation-dependent suppression of HIF1A in an immature hematopoietic cell line HMC-1. Biochem Biophys Res Commun 391, 1028–1032. [DOI] [PubMed] [Google Scholar]
- Wang YP, Walsh SW, Guo JD, Zhang JY, 1991. Maternal levels of prostacyclin, thromboxane, vitamin E, and lipid peroxides throughout normal pregnancy. Am J Obstet Gynecol 165, 1690–1694. [DOI] [PubMed] [Google Scholar]
- Watkins DJ, Ferguson KK, Anzalota Del Toro LV, Alshawabkeh AN, Cordero JF, Meeker JD, 2015. Associations between urinary phenol and paraben concentrations and markers of oxidative stress and inflammation among pregnant women in Puerto Rico. Int J Hyg Environ Health 218, 212–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooding P, Burton G, 2008. Comparative placentation: structures, functions and evolution. Springer Science & Business Media. [Google Scholar]
- Woods L, Perez-Garcia V, Hemberger M, 2018. Regulation of Placental Development and Its Impact on Fetal Growth-New Insights From Mouse Models. Front Endocrinol (Lausanne) 9, 570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Yekeen TA, Xiao Q, Wang Y, Lu F, Huo X, 2013. Placental IGF-1 and IGFBP-3 expression correlate with umbilical cord blood PAH and PBDE levels from prenatal exposure to electronic waste. Environ Pollut 182, 63–69. [DOI] [PubMed] [Google Scholar]
- Xu Y, Agrawal S, Cook TJ, Knipp GT, 2008. Maternal di-(2-ethylhexyl)-phthalate exposure influences essential fatty acid homeostasis in rat placenta. Placenta 29, 962–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Knipp GT, Cook TJ, 2006. Effects of di-(2-ethylhexyl)-phthalate and its metabolites on the lipid profiling in rat HRP-1 trophoblast cells. Arch Toxicol 80, 293–298. [DOI] [PubMed] [Google Scholar]
- Yang C, Song G, Lim W, 2019. A mechanism for the effect of endocrine disrupting chemicals on placentation. Chemosphere 231, 326–336. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Kalluri R, 2013. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol 304, C216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Liu J, Liao L, Han S, Liu J, 2008. Effect of bisphenol A on steroid hormone production in rat ovarian theca-interstitial and granulosa cells. Mol Cell Endocrinol 283, 12–18. [DOI] [PubMed] [Google Scholar]
- Zimna A, Kurpisz M, 2015. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed Res Int 2015, 549412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.