

Abstract
Oral cancer, predominantly oral squamous cell carcinoma (OSCC), is the eighth-most common cancer worldwide, with a 5-y survival rate <50%. There are numerous risk factors for oral cancer, among which periodontal disease is gaining increasing recognition. The creation of a sustained dysbiotic proinflammatory environment by periodontal bacteria may serve to functionally link periodontal disease and oral cancer. Moreover, traditional periodontal pathogens, such as Porphyromonas gingivalis, Fusobacterium nucleatum, and Treponema denticola, are among the species most frequently identified as being enriched in OSCC, and they possess a number of oncogenic properties. These organisms share the ability to attach and invade oral epithelial cells, and from there each undergoes its own unique molecular dialogue with the host epithelium, which ultimately converges on acquired phenotypes associated with cancer, including inhibition of apoptosis, increased proliferation, and activation of epithelial-to-mesenchymal transition leading to increased migration of epithelial cells. Additionally, emerging properties of structured bacterial communities may increase oncogenic potential, and consortia of P. gingivalis and F. nucleatum are synergistically pathogenic within in vivo oral cancer models. Interestingly, however, some species of oral streptococci can antagonize the phenotypes induced by P. gingivalis, indicating functionally specialized roles for bacteria in oncogenic communities. Transcriptomic data support the concept that functional, rather than compositional, properties of oral bacterial communities have more relevance to cancer development. Collectively, the evidence is consistent with a modified polymicrobial synergy and dysbiosis model for bacterial involvement in OSCC, with driver mutations generating a conducive microenvironment on the epithelial boundary, which becomes further dysbiotic by the synergistic action of bacterial communities.
Keywords: OSCC, P. gingivalis, S. gordonii, T. denticola, F. nucleatum, polymicrobial synergy and dysbiosis
Introduction
Worldwide, over 400,000 patients are diagnosed with oral cancer each year, and remarkably the 5-y survival rate remains at only 50% (Marsh et al. 2011). The poor prognosis can be partially explained by the characteristic asymptomatic presentation in the early stages; by the time that the patient has developed painful symptoms, the carcinoma is in its late stages (Markopoulos 2012). Oral cancers can originate as lymphomas in the lymphatic tissue of the tonsils and base of tongue, as carcinomas within salivary gland tissue, but most commonly as squamous cell carcinomas in areas of the mouth containing stratified squamous epithelium. Carcinomas of the oropharynx (including the base of the tongue) are generally referred to as oropharyngeal squamous cell carcinomas, in which human papillomavirus (HPV) infection is a major risk factor (Ganly et al. 2019). Cancers of the oral squamous cells (oral squamous cell carcinoma [OSCC]), which account for about 90% of oral carcinomas, present most frequently on the tongue, lips, floor of the mouth, and gingiva (Bagan et al. 2010). Around 75% of OSCC can be attributed to tobacco smoking, which increases the risk for developing oral cancer by 6-fold (Markopoulos 2012). Smoking also provides an encouraging environment for periodontal pathogens and is an independent risk factor for periodontal disease. Alcohol is another significant risk factor for oral cancer, and the combined risk for those who smoke and drink is increased 15-fold (Markopoulos 2012). Gingival squamous cell carcinoma is particularly interesting because the traditional risk factors of smoking and alcohol consumption are not associated with this malignancy. The lesions mimic the appearance of periodontal disease and thus tend to go untreated (Seoane et al. 2006). Tooth loss as a result of bone loss in periodontal disease is an independent risk factor for head and neck, gastric, and colorectal cancer (CRC; Meyer et al. 2008; Shi et al. 2018). Additionally, colonization by periodontal pathogens has recently been identified as a risk factor for OSCC independent of alcohol, smoking, and HPV (Ganly et al. 2019), and increased colonization by the periodontal pathogen Porphyromonas gingivalis has been correlated with gingival squamous cell carcinoma (Katz et al. 2011). The association between the oral microbiota (the collection of microorganisms found in the oral cavity) and cancers of the head and neck region has been extensively studied in recent years via both culture-dependent and culture-independent methodology, as summarized in the Table. What has emerged is a picture of enrichment of particular organisms, such as Fusobacterium nucleatum, Treponema denticola, and P. gingivalis, with a decrease in the oral streptococci (with the exception of Streptococcus anginosus; Nieminen et al. 2018; Chang, Geng, et al. 2019).
The notion that bacteria may be involved in the development of oral tumors is not new; for example, Treponema pallidum was considered an etiologic agent of OSCC in the early part of the 20th century. However, with the subsequent recognition of the importance of viruses in carcinogenesis, the idea fell into abeyance, and it was not until Helicobacter pylori was established as a cause of gastric cancer in the 1990s that the potential for a carcinogenic role of bacteria became rehabilitated (Polk and Peek 2010; Whitmore and Lamont 2014). Mechanistically, there are a number of broadly defined categories by which bacteria could contribute to tumor growth and development. These include modulation of the balance of host cell proliferation and death; disruption of immune surveillance; and alteration of the metabolism of host-produced compounds, nutritional substrates, or pharmaceuticals (Garrett 2015). Oral bacteria such as P. gingivalis, F. nucleatum, andT. denticola exhibit properties consistent with these criteria and can increase epithelial cell proliferation while inhibiting apoptosis, alter the inflammatory microenvironment, and produce carcinogenic metabolites. These organisms are discussed further due to their frequent positive association in OSCC as well the availability of mechanistic studies that have shown cancer-associated phenotypes. Potentially tumorigenic host-bacteria interactions are the focus of this review.
Porphyromonas gingivalis
P. gingivalis is a keystone pathogen in periodontitis (Hajishengallis et al. 2011; Lamont et al. 2018), and many of the pathogenic mechanisms that impinge on tissue integrity and disrupt protective immune responses are potentially relevant to tumorigenesis. Moreover, immunohistochemistry and other detection methods have identified increased colonization of P. gingivalis in OSCC, esophageal squamous cell carcinoma, and gingival squamous cell carcinoma (Katz et al. 2011; Gao et al. 2016; Sztukowska et al. 2016). In vitro, P. gingivalis engages gingival epithelial cells (GECs) in a complex molecular dialogue, a major thread of which involves subversion of host signaling pathways by bacterial effectors such as the FimA-component fimbriae and the SerB serine phosphatase to promote bacterial entry, intracellular trafficking, and survival (Tribble et al. 2006; Takeuchi et al. 2016; Lee, Roberts, Choi, et al. 2018). Comprehensive analyses of host transcriptional responses to P. gingivalis invoke a pattern of enhanced cell survival and proliferation (Handfield et al. 2005; Geng et al. 2017; Geng et al. 2019): phenotypes that have been verified by a number of laboratories. Indeed, P. gingivalis utilizes multiple mechanisms to suppress host cell death and stimulate proliferation. In primary cultures of GECs, P. gingivalis activates the Jak1/Akt/Stat3 signaling hub that controls intrinsic mitochondrial apoptosis pathways (Yilmaz et al. 2004; Mao et al. 2007). At the mitochondrial membrane, the activity of proapoptotic effectors such as Bad is reduced, while the ratio of antiapoptotic factors such as Bcl2 to proapoptotic factors such as Bax is increased (Yao et al. 2010). Downstream caspases, including caspase 9 and the executioner caspase 3, are consequently suppressed. Additionally, P. gingivalis can modulate expression of microRNAs (miRs) in epithelial cells, and upregulation of miR-203 leads to inhibition of the proapoptotic signaling molecule SOCS3 (Moffatt and Lamont 2011). A major antiapoptotic effector molecule of P. gingivalis, as established by the Yilmaz group, is the secreted enzyme nucleoside diphosphate kinase, which can function as an ATPase and prevent ATP-dependent apoptosis mediated through the purinergic receptor P2X7 (Yilmaz et al. 2008). Another antiapoptotic function of nucleoside diphosphate kinase involves phosphorylation of heat shock protein 27 (HSP27), which curtails cytochrome C release and caspase 9 activation (Lee, Roberts, Atanasova, et al. 2018). Recently it has become apparent that P. gingivalis possesses a variety of kinase and phosphatase enzymes, some of which can function within host cells. Of particular relevance to cell survival, activation of the multipurpose transcriptional regulator FOXO1 by dephosphorylation of serine residues induces antiapoptotic programs in epithelial cells, and knockdown of FOXO1 abrogates P. gingivalis–induced resistance to cell death (Wang et al. 2015).
Along with prolonged cell survival, increased proliferation is a feature of P. gingivalis–infected epithelial cells. Signaling induced by the FimA fimbrial protein accelerates progression of primary GECs through the S-phase of the cell cycle by manipulation of cyclin/CDK activity (cyclin-dependent kinase) and by reducing the level of the p53 tumor suppressor (Kuboniwa et al. 2008). The gingipains of P. gingivalis may also contribute to cell proliferation through proteolytic activation of β-catenin and disassociation of the β-catenin destruction complex. The accumulation of active β-catenin fragments in the nucleus drives the activity of the β-catenin-dependent, pro-proliferative TCF/LEF promoter (Zhou et al. 2015). In oral squamous carcinoma cells, P. gingivalis can increase cell proliferation by regulating cyclin D1 expression through the miR-21/PDCD4/AP-1 negative feedback signaling pathway (Chang, Wang, et al. 2019). Additionally, in oral tumor cells, P. gingivalis can increase expression of α-defensins, which have been found to elevate proliferation through intersecting with epidermal growth factor receptor signaling (Hoppe et al. 2016).
Recently, P. gingivalis has been shown to induce at least a partial epithelial-to-mesenchymal transition (EMT) in GECs (Fig. 1). EMT is a cellular program through which epithelial cells shed their tight junctions in favor of an individual mesenchymal phenotype. EMT is important for embryogenesis and wound healing but, if uncontrolled, ultimately leads to increased migration/invasion and cancer cell stemness. As befits its importance to the cell, EMT is controlled by a complex regulatory network that funnels through a series of transcription factors, such as ZEB1, ZEB2, SNAI1, and Twist. These factors induce EMT by downregulating epithelial cell tight junction proteins (e.g., E-cadherin, ZO-1) while upregulating mesenchymal characteristics (e.g., N-cadherin, MMP-9, vimentin) and are a target of P. gingivalis (Ha et al. 2015; Sztukowska et al. 2016; Lee et al. 2017; Abdulkareem et al. 2018; Ohshima et al. 2019). ZEB1 is upregulated in a FimA-dependent manner through a pathway that involves GSK-3β in primary GECs (Sztukowska et al. 2016; Lee et al. 2017). ZEB2, however, is regulated in a FimA-independent manner involving gingipain processing and activation of β-catenin with dephosphorylation and activation of FOXO1 (Ohshima et al. 2019). Although the extent and duration of EMT induced by P. gingivalis remain to be determined, epithelial cell infection does lead to an increase in stemness, as evidenced by upregulation of the stem cell markers CD44 and CD133 and enhanced migration (Ha et al. 2015; Sztukowska et al. 2016; Lee et al. 2017; Abdulkareem et al. 2018; Ohshima et al. 2019). Invasion and potentially metastasis of epithelial cells can be facilitated by host matrix metalloproteinase (MMP) enzymes, which degrade extracellular matrix and basement components. P. gingivalis has been shown to upregulate production of several MMPs, including MMP-1, MMP-2, MMP-7, MMP-9, and MMP-10, from primary and transformed oral epithelial cells (Inaba et al. 2014; Ha et al. 2015; Ha et al. 2016; Sztukowska et al. 2016; Lee et al. 2017). In invasive OSCC lines, P. gingivalis gingipains can stimulate proteinase-activated receptor 2 (PAR2) and PAR4 to increase MMP-9 proenzyme expression through ERK1/2-Ets1, p38/HSP27, and NF-kB pathways (Inaba et al. 2014). Subsequently, in a 2-hit mechanism, gingipains process the proenzyme to active MMP9, ensuring an increase in cellular invasion (Inaba et al. 2014; Inaba et al. 2015).
Figure 1.

Potential mechanisms by which Porphyromonas gingivalis could affect cancer-associated processes in gingival epithelial cells. Peach: healthy epithelial cells. Blue: epithelial cells that have acquired an antiapoptotic phenotype. Purple: epithelial cells that have acquired an accelerated proliferation phenotype. Green: epithelial cells that have undergone epithelial-to-mesenchymal transition and have acquired an invasive phenotype. Note that for simplicity other contributing host and environmental factors are not depicted.
Chronic inflammation has emerged as a major contributor to tumor growth and spread, mainly through modulation of the tumor microenvironment by cytokines and chemokines and through differential receptor expression (Sahingur and Yeudall 2015). The ability of P. gingivalis to incite prolonged, dysregulated inflammation could account for the epidemiologic associations between periodontitis and OSCC (Sahingur and Yeudall 2015; Michaud et al. 2017). In OSCC cell lines and primary GECs, P. gingivalis can upregulate programmed death-ligand 1 (PD-L1, B7-H1) and B7-DC, receptors that lead to anergy and apoptosis of activated T cells and contribute to tumor cells’ resistance to host immune responses (Groeger et al. 2011). In OSCC cells, P. gingivalis stimulates the release of a variety of chemokines/cytokines, including IL-8, IL-6, TGF-β1, and TNF-α (Yee et al. 2014; Ha et al. 2016; Abdulkareem et al. 2018). IL-8 can increase MMP production and cell invasiveness, as well as stimulate proliferation through transactivation of the EGF receptor (Joh et al. 2005). In addition, the IL-23/IL-17 axis, which is strongly protumorigenic, at least in CRC (Grivennikov et al. 2012), can be induced by P. gingivalis (Cheng et al. 2016). Interestingly, in primary GECs, P. gingivalis adopts a more stealth-like behavior through a process known as localized immune paralysis (Darveau et al. 1998). Release of a serine phosphatase (SerB) intracellularly results in antagonism of IL-8 production through dephosphorylation of the serine 538 residue of the p65 subunit of NF-κB (Takeuchi et al. 2013). While this may restrain tumor progression, the effect may be offset by inhibition of the angiostatic cytokines CXCL9, CXCL10, and CXCL11, which could promote neovascularization of tumors and increased tumor growth or metastasis (Jauregui et al. 2013; Sahingur and Yeudall 2015).
In vivo evidence supports a role for P. gingivalis in the development of oral carcinomas. In the 4NQO tongue squamous cell carcinoma model, P. gingivalis–treated mice developed more and larger tumors on the tongue as compared with the carcinogen-alone group (Wu et al. 2018). The development of squamous cell carcinoma was associated with enhanced free fatty acid production in the tongue and the serum of 4NQO-treated mice, which is a shift also seen in oral cancer.
Fusobacterium nucleatum
While F. nucleatum is prevalent in a healthy microbiota, several studies have found that F. nucleatum is significantly enriched in patients with disease, whether that be periodontal disease, preterm delivery of low birth weight infants, head and neck cancer, or CRC (Han et al. 2014). The potential role of F. nucleatum in cancers has been investigated in both in vitro and in vivo studies. F. nucleatum produces an adhesin, FadA, which is crucial for attachment and subsequent invasion of epithelial cells (Xu et al. 2007). FadA is thought to play a major role in CRC by binding to E-cadherin on CRC cells, thus activating β-catenin signaling and differentially regulating inflammatory and oncogenic responses (Rubinstein et al. 2013). The FadA-E-cadherin axis also upregulates annexin A1, a modulator of Wnt/β-catenin-based proliferative signaling in CRC cells (Rubinstein et al. 2019). Localization of what is primarily an oral organism with developing tumors in the gastrointestinal tract may be accomplished by another fusobacterial adhesin, Fap2, which binds to Gal-GalNac, abundant on CRC cell surfaces (Abed et al. 2016). Fap2 can also immunosuppress tumor-infiltrating lymphocytes, which are essential for immune responses to tumors. Specifically, Fap2 binds and activates the inhibitory immunoreceptor TIGIT, which is expressed by T and natural killer cells (Gur et al. 2015). Further compromising antitumor immunity, F. nucleatum activates the human inhibitory receptor CEACAM1, which also suppresses the activities of T and natural killer cells (Gur et al. 2019). Clearly then, F. nucleatum can significantly affect cell signaling and tumor immunity with relevance to CRC. The extent to which these properties may pertain to OSCC is a vein of information ready to be mined. Studies that have been performed establish the ability of F. nucleatum to induce nuclear localization of NF-κB in GECs while increasing secretion of IL-1β via activation of the NLRP3 inflammasome and caspase 1 (Bui et al. 2016). Release of endogenous secondary danger-associated molecular patterns, such as apoptosis-associated speck-like protein (ASC) and high-mobility group box 1 protein (HMGB1), further amplifies inflammation. P38 is also activated by F. nucleatum, which leads to increased secretion of MMP9 and MMP13 (Uitto et al. 2005). Moreover, F. nucleatum can induce an EMT program in OSCC cells through upregulation of TGF-β, TNFα, and EGF signaling (Abdulkareem et al. 2018).
In vivo, coinfection with F. nucleatum and P. gingivalis exacerbated tumor development in the murine 4NQO tongue squamous cell carcinoma model (Binder Gallimidi et al. 2015). The infected group had larger, more invasive tumors, with increased expression of cell cycle progression marker cyclin D1 (Binder Gallimidi et al. 2015). There was also an increase in phosphorylation of STAT3 in the infected group, which led to increased expression of IL-6 (Binder Gallimidi et al. 2015). Furthermore, in a CRC model, F. nucleatum increases the size and number of tumors that develop in C57Bl/6 Apcmin/+ mice (Kostic et al. 2013).
Treponema denticola
In health, T. denticola is found in low abundance; however, in periodontal disease, T. denticola is one of the most abundant organisms (Dashper et al. 2011). Similarly, an increased abundance of T. denticola has been associated with esophageal squamous cell carcinoma and OSCC, and similarly correlated with an increased risk of CRC (Narikiyo et al. 2004). T. denticola is highly proteolytic, with dentilisin (chymotrypsin-like proteinase) being the primary secreted protease (Fenno 2012). The presence of dentilisin is strongly correlated with early-stage mobile tongue squamous cell carcinoma, and high expression of dentilisin is associated with increased tumor invasion, tumor size, and recurrence in patients <60 y of age (Listyarifah et al. 2018). Dentilisin can degrade IL-8 and TNFα (Deng et al. 2001; Jo et al. 2014) and cleaves pro-MMP8 and pro-MMP9 to their active forms (Nieminen et al. 2018). In a 2-hit mechanism, dentilisin degrades tissue inhibitors of MMPs, TIMP1 and TIMP2, contributing to an overall more proteolytic environment favoring invasion of epithelial cells.
Oral Microbial Communities
The preceding text describes how individual species could affect carcinogenesis; however, in the oral cavity, bacteria assemble into multispecies, spatially constrained communities known as biofilms. Within these communities, functional specialization of bacterial species emerges, and in periodontitis it is pathogenicity at the community level, or nososymbiocity, which is thought to determine the potential for disease (Lamont et al. 2018). One current theory of periodontal disease etiology, the PSD model (polymicrobial synergy and dysbiosis), holds that synergistic interactions within the polymicrobial community shape and stabilize a dysbiotic microbiota, which perturbs host homeostasis. Disease is caused by reciprocally reinforced interactions between such physically and metabolically integrated polymicrobial communities and a dysregulated host inflammatory response (Lamont et al. 2018). While periodontitis and cancer are clearly distinct diseases, they do share an underlying similarity in that they are in essence wounds that fail to heal (Cugini et al. 2013). Community perturbations consistent with a PSD model have been proposed for tumor development (Flynn et al. 2016; Bornigen et al. 2017). Combinations of oral bacterial species are consistently identified in OSCC lesions (see Table), resonating with the idea that community-wide properties may promote tumorigenesis. The implications of the source materials found in the Table should also be noted. Oral rinse, saliva, and serum levels indicate only the presence of particular organisms; they do not indicate the spatial relationship to the tumor. Tumor scraping, homogenate, and oral swabs indicate surface colonization and/or penetration of the tumor site, whereas biopsy sections stained for immunohistochemistry show intracellular localization of the bacteria.
Table.
Studies Showing Positive Associations of Oral Bacteria in Cancer of the Head and Neck Region.
| Cancer | Oral Bacteria | Source Material | Association | Reference |
|---|---|---|---|---|
| OSCC | P. gingivalis | Saliva | Increased presence in patients with OSCC vs. health | Galvão-Moreira and da Cruz (2016) |
| OSCC | P. gingivalis | Biopsy | Increased presence of P. gingivalis in OSCC biopsies | Sztukowska et al. (2016) |
| GSCC | P. gingivalis | Biopsy | Increased presence of P. gingivalis in GSCC biopsy samples vs. health | Katz et al. (2011) |
| Orodigestive cancer | P. gingivalis | Serum | Increased P. gingivalis serum antibody levels were associated with increased likelihood of mortality | Ahn et al. (2012) |
| ESCC | P. gingivalis | Oral rinse | Increased P. gingivalis was associated with increased risk of ESCC | Peters et al. (2017) |
| ESCC | P. gingivalis | Biopsy | Increased association in patients with ESCC and correlated with increased mortality | Gao et al. (2016) |
| OSCC | Capnocytophaga gingivalis, Prevotella melaninogenica, Streptococcus mitis | Saliva | Increased presence in saliva of patients with OSCC | Mager et al. (2005) |
| OSCC | Veillonella, Fusobacterium, Prevotella, Porphyromonas, Actinomyces, Streptococcus | Tumor scraping | Positive correlation between bacteria and OSCC | Nagy et al. 1998) |
| OSCC | Streptococcus | Homogenized tumor | Increased abundance in patients with OSCC | Mukherjee et al. (2017) |
| OSCC | Peptostreptococcus stomatis, Streptococcus salivarius, Streptococcus gordonii, Gemella haemolysans, Gemella morbillorum, Johnsonella ignava and Streptococcus parasanguinis | Homogenized tumor | Increased abundance in patients with OSCC | Pushalkar et al. (2012) |
| OSCC | P. gingivalis, F. nucleatum | Subgingival plaque and homogenized tumor | Increased colonization of cancerous and paracancerous tissue vs. healthy sites | Chang, Geng, et al. (2019) |
| OSCC | Fusobacteria, Peptostreptococcus, Filifactor | Tongue scraping | Increased colonization in patients with OSCC | Zhao et al. (2017) |
| OSCC | Fusobacteria, Bacteroidetes, Filifactor, Streptococci | Oral rinse | Increased colonization by Fusobacteria, Bacteroidetes, and Filifactor was positively associated with OSCC and stage, whereas Streptococci were negatively associated | Yang et al. (2018) |
| OSCC | Fusobacteria, Prevotella, Streptococcus | Oral rinse | Positive association between Fusobacteria and Prevotella with OSCC and negative association with Streptococcus. This association was independent of HPV status | Ganly et al. (2019) |
| OSCC | Streptococcus anginosus, Prevotella, Veillonella, Fusobacterium, | Direct culture | Positively associated with OSCC | Hooper et al. (2006) |
| OSCC | F. nucleatum, Streptococci | Oral swab | Positive correlation with F. nucleatum, negative correlation with Streptococci | Schmidt et al. (2014) |
| OSCC | T. denticola | Biopsy | Positive association of dentilisin with OSCC and stage | Listyarifah et al. (2018) |
| ESCC and OSCC | T. denticola | Biopsy | Positive association of dentilisin and OSCC | Nieminen et al. (2018) |
| ESCC | T. denticola | Saliva | Increased colonization in patients with ESCC | Narikiyo et al. (2004) |
ESCC, esophageal squamous cell carcinoma; F. nucleatum, Fusobacterium nucleatum; GSCC, gingival squamous cell carcinoma; HPV, human papillomavirus; OSCC, oral squamous cell carcinoma; P. gingivalis, Porphyromonas gingivalis; T. denticola, Treponema denticola.
In the 4NQO model, F. nucleatum and P. gingivalis synergistically promote cancer progression (Binder Gallimidi et al. 2015). Another feature of the PSD model is that the microbial roster is of less relevance to nososymbiocity than the presence of combinations of functional genes, as communities of different compositions can exhibit similar functions (Lamont et al. 2018). Support for this concept in OSCC comes from a study of the microbiota and transcriptome in the 4NQO mouse model. Whereas variability in community dynamics was observed, the metatranscriptome revealed patterns of metabolic signatures consistently present in OSCC. These include nitrogen transport, response to stress, interspecies interactions, Wnt pathway modulation, and amino acid and lipid biosynthesis (Stashenko et al. 2019). Similarly, a pilot study of human OSCC tumors found metabolic activities better correlated with disease than did community microbial composition (Yost et al. 2018), and a comparison of microbiotas associated with OSCC in different countries revealed functional rather than compositional similarities (Perera et al. 2018).
Interactions among bacterial constituents of communities can be antagonistic as well as synergistic, and numerous cases of antagonisms have been documented among oral bacteria (Duran-Pinedo et al. 2014; Lamont et al. 2018; Ohshima et al. 2019). Although there are conflicting reports in the literature (see Table; Mukherjee et al. 2017), in general, most oral streptococcal species tend to be underrepresented in the microbiotas associated with OSCC (Al-Hebshi et al. 2019; Stashenko et al. 2019). This can be interpreted to indicate reduced fitness of these organisms in a tumorigenic environment. However, a further interpretation is that these organisms are eubiotic and help maintain homeostasis at mucosal membranes, and in their absence the microbiota becomes increasingly tumorigenic. In that regard, S. gordonii can reprogram epithelial cell global transcriptional patterns such that the subsequent response to P. gingivalis is diminished; furthermore, S. gordonii can prevent P. gingivalis–induced GEC proliferation (Lamont et al. 2018). Moreover, S. gordonii can antagonize P. gingivalis–induced ZEB2 production and associated cell migration by inhibiting the activation of the FOXO1 transcription factor through the TAK1-NLK negative regulatory pathway, as shown in Figure 2 (Ohshima et al. 2019). Hence, while S. gordonii is an accessory pathogen in periodontal disease (Lamont et al. 2018), this species may be a homeostatic commensal in oral cancer, an illustration of the importance of environmental context for bacterial functionality.
Figure 2.
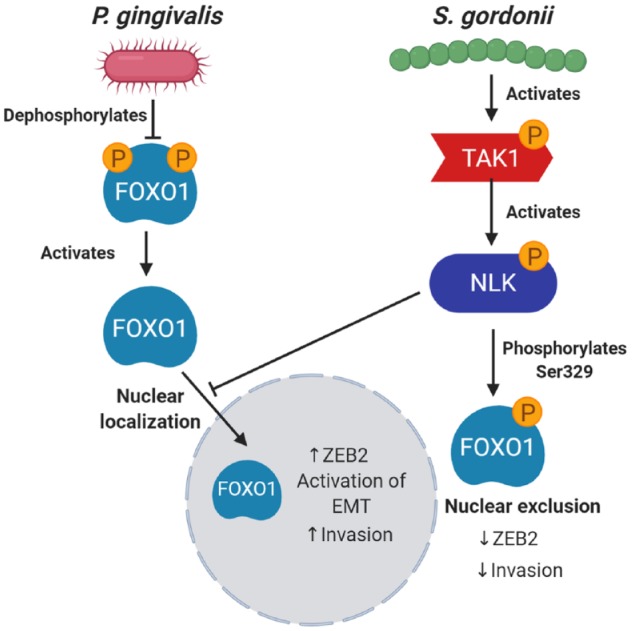
Antagonistic interactions of Streptococcus gordonii on Porphyromonas gingivalis through activation of the TAK1-NLK host kinase cascade. P. gingivalis can dephosphorylate FOXO1 on serine residues, which prevents translocation from the nucleus to the cytoplasm, thus enhancing activity. When S. gordonii is present, the TAK1-NLK1 pathway is activated, which supersedes the effect of P. gingivalis and increases phosphorylation of FOXO1 on Ser329, thus allowing translocation of FOXO1 to the cytoplasm, where it is inactive. EMT, epithelial-to-mesenchymal transition.
Perspectives
Cancer is a multistep, generally slowly progressing disease involving multiple genetic and environmental predisposing factors, which may operate in a temporally defined manner. In such a situation, demarcating the role of an individual component, such as the microbiota, is challenging, and the role is likely to be nuanced and context dependent. As with other oral diseases that have a significant microbial component, in particular periodontitis, it is likely that the role for bacteria in OSCC will involve the community as the pathogenic unit. Expansion of the PSD model, similar to that proposed for CRC (Flynn et al. 2016), is one conceptual framework that could accommodate the involvement of oral bacteria in OSCC (Fig. 3). Importantly, periodontal bacterial communities are pathogenic only in a susceptible host (Hajishengallis 2015). In this scenario, driver mutations begin to establish a tumor microenvironment that selects for a microbial community in which gram-negative anaerobes are enriched. Collectively these organisms suppress programmed cell death and induce uncontrolled epithelial cell proliferation with a more mesenchymal, migratory phenotype. Dysbiotic inflammation further contributes to oncogenesis while sustaining colonization by inflammophilic organisms such as P. gingivalis through a reciprocating feed-forward loop. However, the role of particular bacteria as drivers themselves or as passengers turned drivers (Al-Hebshi et al. 2019) is equally plausible, and much work is required to discriminate among these possibilities, including conducting large longitudinal and intervention studies. While the focus of this review has been on dysbiosis of the oral microbiota as it relates to OSCC, recent evidence suggests that such changes can also be associated with nonoral tumors. There is an increased risk for gastric cancer in patients with periodontal disease and tooth loss, and patients with esophageal, gastric, pancreatic, and colorectal cancers can show oral microbial dysbiosis (Mascitti et al. 2019). Our current appreciation of the extent of the involvement of oral bacteria in cancer may represent only the tip of the iceberg.
Figure 3.

Schematic representation of polymicrobial synergy and dysbiosis model for oral squamous cell carcinoma. In health, the host responses to homeostatic communities are a eubiotic balance of proliferation with programmed cell death. Driver mutations lead to dysregulation of host processes, which can also be manipulated by organisms associated with a dysbiotic community. As the tumor microenvironment is established, anaerobic, gram-negative organisms are enriched as a result of hypoxic proinflammatory conditions. As organisms such as Porphyromonas gingivalis, Fusobacterium nucleatum, and Treponema denticola accumulate, the tumor cells can acquire an invasive phenotype through epithelial-to-mesenchymal transition, as well as increased resistance to chemotherapeutic drugs.
Author Contributions
Z.R. Fitzsimonds, C.J. Rodriguez-Hernandez, J. Bagaitkar, R.J. Lamont, contributed to conception and design, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Acknowledgments
We thank the National Institutes of Health for support through DE012505, DE023193, DE011111, DE017921 (R.J.L.), DE0280 31, DE028296, GM125504 (J.B and R.J.L.), and DE028166 (Z.R.F.).
Footnotes
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
ORCID iDs: Z.R. Fitzsimonds  https://orcid.org/0000-0002-6523-4911
https://orcid.org/0000-0002-6523-4911
R.J. Lamont
https://orcid.org/0000-0002-3147-5039
References
- Abdulkareem AA, Shelton RM, Landini G, Cooper PR, Milward MR. 2018. Periodontal pathogens promote epithelial-mesenchymal transition in oral squamous carcinoma cells in vitro. Cell Adh Migr. 12(2):127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abed J, Emgard JE, Zamir G, Faroja M, Almogy G, Grenov A, Sol A, Naor R, Pikarsky E, Atlan KA, et al. 2016. Fap2 mediates Fusobacterium nucleatum colorectal adenocarcinoma enrichment by binding to tumor-expressed Gal-GalNac. Cell Host Microbe. 20(2):215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Segers S, Hayes RB. 2012. Periodontal disease, Porphyromonas gingivalis serum antibody levels and orodigestive cancer mortality. Carcinogenesis. 33(5):1055–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hebshi NN, Borgnakke WS, Johnson NWJ. 2019. The microbiome of oral squamous cell carcinomas: a functional perspective. Curr Oral Health Rep. 6(2):145–160. [Google Scholar]
- Bagan J, Sarrion G, Jimenez Y. 2010. Oral cancer: clinical features. Oral Oncol. 46(6):414–417. [DOI] [PubMed] [Google Scholar]
- Binder Gallimidi A, Fischman S, Revach B, Bulvik R, Maliutina A, Rubinstein AM, Nussbaum G, Elkin M. 2015. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget. 6(26):22613–22623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornigen D, Ren B, Pickard R, Li J, Ozer E, Hartmann EM, Xiao W, Tickle T, Rider J, Gevers D, et al. 2017. Alterations in oral bacterial communities are associated with risk factors for oral and oropharyngeal cancer. Sci Rep. 7(1):17686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui FQ, Johnson L, Roberts J, Hung SC, Lee J, Atanasova KR, Huang PR, Yilmaz O, Ojcius DM. 2016. Fusobacterium nucleatum infection of gingival epithelial cells leads to NLRP3 inflammasome-dependent secretion of IL-1beta and the danger signals ASC and HMGB1. Cell Microbiol. 18(7):970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Geng F, Shi X, Li Y, Zhang X, Zhao X, Pan Y. 2019. The prevalence rate of periodontal pathogens and its association with oral squamous cell carcinoma. Appl Microbiol Biotechnol. 103(3):1393–1404. [DOI] [PubMed] [Google Scholar]
- Chang C, Wang H, Liu J, Pan C, Zhang D, Li X, Pan Y. 2019. Porphyromonas gingivalis infection promoted the proliferation of oral squamous cell carcinoma cells through the mir-21/PDCD4/AP-1 negative signaling pathway. ACS Infect Dis. 5(8):1336–1347. [DOI] [PubMed] [Google Scholar]
- Cheng WC, van Asten SD, Burns LA, Evans HG, Walter GJ, Hashim A, Hughes FJ, Taams LS. 2016. Periodontitis-associated pathogens P. gingivalis and A. actinomycetemcomitans activate human CD14(+) monocytes leading to enhanced TH17/IL-17 responses. Eur J Immunol. 46(9):2211–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cugini C, Klepac-Ceraj V, Rackaityte E, Riggs JE, Davey ME. 2013. Porphyromonas gingivalis: keeping the pathos out of the biont. J Oral Microbiol. 5:19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveau RP, Belton CM, Reife RA, Lamont RJ. 1998. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. 66(4):1660–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashper SG, Seers CA, Tan KH, Reynolds EC. 2011. Virulence factors of the oral spirochete Treponema denticola. J Dent Res. 90(6):691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng QD, Han Y, Xia X, Kuramitsu HK. 2001. Effects of the oral spirochete Treponema denticola on interleukin-8 expression from epithelial cells. Oral Microbiol Immunol. 16(3):185–187. [DOI] [PubMed] [Google Scholar]
- Duran-Pinedo AE, Baker VD, Frias-Lopez J. 2014. The periodontal pathogen Porphyromonas gingivalis induces expression of transposases and cell death of streptococcus mitis in a biofilm model. Infect Immun. 82(8):3374–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenno JC. 2012. Treponema denticola interactions with host proteins. J Oral Microbiol [epub ahead of print 21 Feb 2012]. doi: 10.3402/jom.v4i0.9929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn KJ, Baxter NT, Schloss PD. 2016. Metabolic and community synergy of oral bacteria in colorectal cancer. mSphere. 1(3):e00102-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvão-Moreira LV, da Cruz MC. 2016. Oral microbiome, periodontitis and risk of head and neck cancer. Oral Oncol. 53:17–19. [DOI] [PubMed] [Google Scholar]
- Ganly I, Yang L, Giese RA, Hao Y, Nossa CW, Morris LGT, Rosenthal M, Migliacci J, Kelly D, Tseng W, et al. 2019. Periodontal pathogens are a risk factor of oral cavity squamous cell carcinoma, independent of tobacco and alcohol and human papillomavirus. Int J Cancer. 145(3):775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Li S, Ma Z, Liang S, Shan T, Zhang M, Zhu X, Zhang P, Liu G, Zhou F, et al. 2016. Presence of Porphyromonas gingivalis in esophagus and its association with the clinicopathological characteristics and survival in patients with esophageal cancer. Infect Agent Cancer. 11:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS. 2015. Cancer and the microbiota. Science. 348(6230):80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng F, Liu J, Guo Y, Li C, Wang H, Wang H, Zhao H, Pan Y. 2017. Persistent exposure to Porphyromonas gingivalis promotes proliferative and invasion capabilities, and tumorigenic properties of human immortalized oral epithelial cells. Front Cell Infect Microbiol. 7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng F, Wang Q, Li C, Liu J, Zhang D, Zhang S, Pan Y. 2019. Identification of potential candidate genes of oral cancer in response to chronic infection with Porphyromonas gingivalis using bioinformatical analyses. Front Oncol. 9:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, et al. 2012. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 491(7423):254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeger S, Domann E, Gonzales JR, Chakraborty T, Meyle J. 2011. B7-H1 and B7-DC receptors of oral squamous carcinoma cells are upregulated by Porphyromonas gingivalis. Immunobiology. 216(12):1302–1310. [DOI] [PubMed] [Google Scholar]
- Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, Enk J, Bar-On Y, Stanietsky-Kaynan N, Coppenhagen-Glazer S, et al. 2015. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor tigit protects tumors from immune cell attack. Immunity. 42(2):344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur C, Maalouf N, Shhadeh A, Berhani O, Singer BB, Bachrach G, Mandelboim O. 2019. Fusobacterium nucleatum supresses anti-tumor immunity by activating CEACAM1. Oncoimmunology. 8(6):e1581531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha NH, Park DG, Woo BH, Kim DJ, Choi JI, Park BS, Kim YD, Lee JH, Park HR. 2016. Porphyromonas gingivalis increases the invasiveness of oral cancer cells by upregulating IL-8 and MMPs. Cytokine. 86:64–72. [DOI] [PubMed] [Google Scholar]
- Ha NH, Woo BH, Kim DJ, Ha ES, Choi JI, Kim SJ, Park BS, Lee JH, Park HR. 2015. Prolonged and repetitive exposure to Porphyromonas gingivalis increases aggressiveness of oral cancer cells by promoting acquisition of cancer stem cell properties. Tumour Biol. 36(12):9947–9960. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G. 2015. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. 15(1):30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, et al. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 10(5):497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YW, Houcken W, Loos BG, Schenkein HA, Tezal M. 2014. Periodontal disease, atherosclerosis, adverse pregnancy outcomes, and head-and-neck cancer. Adv Dent Res. 26(1):47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handfield M, Mans JJ, Zheng G, Lopez MC, Mao S, Progulske-Fox A, Narasimhan G, Baker HV, Lamont RJ. 2005. Distinct transcriptional profiles characterize oral epithelium-microbiota interactions. Cell Microbiol. 7(6):811–823. [DOI] [PubMed] [Google Scholar]
- Hooper SJ, Crean SJ, Lewis MA, Spratt DA, Wade WG, Wilson MJ. 2006. Viable bacteria present within oral squamous cell carcinoma tissue. J Clin Microbiol. 44(5):1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe T, Kraus D, Novak N, Probstmeier R, Frentzen M, Wenghoefer M, Jepsen S, Winter J. 2016. Oral pathogens change proliferation properties of oral tumor cells by affecting gene expression of human defensins. Tumour Biol. 37(10):13789–13798. [DOI] [PubMed] [Google Scholar]
- Inaba H, Amano A, Lamont RJ, Murakami Y. 2015. Involvement of protease-activated receptor 4 in over-expression of matrix metalloproteinase 9 induced by Porphyromonas gingivalis. Med Microbiol Immunol. 204(5):605–612. [DOI] [PubMed] [Google Scholar]
- Inaba H, Sugita H, Kuboniwa M, Iwai S, Hamada M, Noda T, Morisaki I, Lamont RJ, Amano A. 2014. Porphyromonas gingivalis promotes invasion of oral squamous cell carcinoma through induction of proMMP9 and its activation. Cell Microbiol. 16(1):131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauregui CE, Wang Q, Wright CJ, Takeuchi H, Uriarte SM, Lamont RJ. 2013. Suppression of t-cell chemokines by Porphyromonas gingivalis. Infect Immun. 81(7):2288–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo AR, Baek KJ, Shin JE, Choi Y. 2014. Mechanisms of il-8 suppression by Treponema denticola in gingival epithelial cells. Immunol Cell Biol. 92(2):139–147. [DOI] [PubMed] [Google Scholar]
- Joh T, Kataoka H, Tanida S, Watanabe K, Ohshima T, Sasaki M, Nakao H, Ohhara H, Higashiyama S, Itoh M. 2005. Helicobacter pylori-stimulated interleukin-8 (IL-8) promotes cell proliferation through transactivation of epidermal growth factor receptor (EGFR) by disintegrin and metalloproteinase (ADAM) activation. Dig Dis Sci. 50(11):2081–2089. [DOI] [PubMed] [Google Scholar]
- Katz J, Onate MD, Pauley KM, Bhattacharyya I, Cha S. 2011. Presence of Porphyromonas gingivalis in gingival squamous cell carcinoma. Int J Oral Sci. 3(4):209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al. 2013. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 14(2):207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboniwa M, Hasegawa Y, Mao S, Shizukuishi S, Amano A, Lamont RJ, Yilmaz O. 2008. P. gingivalis accelerates gingival epithelial cell progression through the cell cycle. Microbes Infect. 10(2):122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont RJ, Koo H, Hajishengallis G. 2018. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. 16(12):745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Roberts JS, Atanasova KR, Chowdhury N, Han K, Yilmaz O. 2017. Human primary epithelial cells acquire an epithelial-mesenchymal-transition phenotype during long-term infection by the oral opportunistic pathogen, Porphyromonas gingivalis. Front Cell Infect Microbiol. 7:493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Roberts JS, Atanasova KR, Chowdhury N, Yilmaz O. 2018. A novel kinase function of a nucleoside-diphosphate-kinase homologue in Porphyromonas gingivalis is critical in subversion of host cell apoptosis by targeting heat-shock protein 27. Cell Microbiol. 20(5):e12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Roberts JS, Choi CH, Atanasova KR, Yilmaz O. 2018. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence. 9(1):845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listyarifah D, Nieminen MT, Makinen LK, Haglund C, Grenier D, Hayry V, Nordstrom D, Hernandez M, Yucel-Lindberg T, Tervahartiala T, et al. 2018. Treponema denticola chymotrypsin-like proteinase is present in early-stage mobile tongue squamous cell carcinoma and related to the clinicopathological features. J Oral Pathol Med. 47(8):764–772. [DOI] [PubMed] [Google Scholar]
- Mager DL, Haffajee AD, Devlin PM, Norris CM, Posner MR, Goodson JM. 2005. The salivary microbiota as a diagnostic indicator of oral cancer: a descriptive, non-randomized study of cancer-free and oral squamous cell carcinoma subjects. J Transl Med. 3:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao S, Park Y, Hasegawa Y, Tribble GD, James CE, Handfield M, Stavropoulos MF, Yilmaz O, Lamont RJ. 2007. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol. 9(8):1997–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markopoulos AK. 2012. Current aspects on oral squamous cell carcinoma. Open Dent J. 6:126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D, Suchak K, Moutasim KA, Vallath S, Hopper C, Jerjes W, Upile T, Kalavrezos N, Violette SM, Weinreb PH, et al. 2011. Stromal features are predictive of disease mortality in oral cancer patients. J Pathol. 223(4):470–481. [DOI] [PubMed] [Google Scholar]
- Mascitti M, Togni L, Troiano G, Caponio V, Gissi DB, Montebugnoli L, Procaccini M, Lo Muzio L, Santarelli A. 2019. Beyond head and neck cancer: the relationship between oral microbiota and tumour development in distant organs. Front Cell Infect Microbiol 9:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MS, Joshipura K, Giovannucci E, Michaud DS. 2008. A review of the relationship between tooth loss, periodontal disease, and cancer. Cancer Causes Control. 19(9):895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud DS, Fu Z, Shi J, Chung M. 2017. Periodontal disease, tooth loss, and cancer risk. Epidemiol Rev. 39(1):49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt CE, Lamont RJ. 2011. Porphyromonas gingivalis induction of microRNA-203 expression controls suppressor of cytokine signaling 3 in gingival epithelial cells. Infect Immun. 79(7):2632–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee PK, Wang H, Retuerto M, Zhang H, Burkey B, Ghannoum MA, Eng C. 2017. Bacteriome and mycobiome associations in oral tongue cancer. Oncotarget. 8 (57):97273–97289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy KN, Sonkodi I, Szoke I, Nagy E, Newman HN. 1998. The microflora associated with human oral carcinomas. Oral Oncol. 34(4):304–308. [PubMed] [Google Scholar]
- Narikiyo M, Tanabe C, Yamada Y, Igaki H, Tachimori Y, Kato H, Muto M, Montesano R, Sakamoto H, Nakajima Y, et al. 2004. Frequent and preferential infection of Treponema denticola, Streptococcus mitis, and Streptococcus anginosus in esophageal cancers. Cancer Sci. 95(7):569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminen MT, Listyarifah D, Hagstrom J, Haglund C, Grenier D, Nordstrom D, Uitto VJ, Hernandez M, Yucel-Lindberg T, Tervahartiala T, et al. 2018. Treponema denticola chymotrypsin-like proteinase may contribute to orodigestive carcinogenesis through immunomodulation. Br J Cancer. 118(3):428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima J, Wang Q, Fitzsimonds ZR, Miller DP, Sztukowska MN, Jung YJ, Hayashi M, Whiteley M, Lamont RJ. 2019. Streptococcus gordonii programs epithelial cells to resist ZEB2 induction by Porphyromonas gingivalis. Proc Natl Acad Sci USA. 116(17):8544–8553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera M, Al-Hebshi NN, Perera I, Ipe D, Ulett GC, Speicher DJ, Chen T, Johnson NW. 2018. Inflammatory bacteriome and oral squamous cell carcinoma. J Dent Res. 97(6):725–732. [DOI] [PubMed] [Google Scholar]
- Peters BA, Wu J, Pei Z, Yang L, Purdue MP, Freedman ND, Jacobs EJ, Gapstur SM, Hayes RB, Ahn J. 2017. Oral microbiome composition reflects prospective risk for esophageal cancers. Cancer Res. 77(23):6777–6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polk DB, Peek RM., Jr. 2010. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 10(6):403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushalkar S, Ji X, Li Y, Estilo C, Yegnanarayana R, Singh B, Li X, Saxena D. 2012. Comparison of oral microbiota in tumor and non-tumor tissues of patients with oral squamous cell carcinoma. BMC Microbiol. 12:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein MR, Baik JE, Lagana SM, Han RP, Raab WJ, Sahoo D, Dalerba P, Wang TC, Han YW. 2019. Fusobacterium nucleatum promotes colorectal cancer by inducing wnt/beta-catenin modulator annexin A1. EMBO Rep. 20(4). pii:e47638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. 2013. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its fada adhesin. Cell Host Microbe. 14(2):195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahingur SE, Yeudall WA. 2015. Chemokine function in periodontal disease and oral cavity cancer. Front Immunol. 6:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt BL, Kuczynski J, Bhattacharya A, Huey B, Corby PM, Queiroz EL, Nightingale K, Kerr AR, DeLacure MD, Veeramachaneni R, et al. 2014. Changes in abundance of oral microbiota associated with oral cancer. PLoS One. 9(6):e98741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J, Varela-Centelles PI, Walsh TF, Lopez-Cedrun JL, Vazquez I. 2006. Gingival squamous cell carcinoma: diagnostic delay or rapid invasion? J Periodontol. 77(7):1229–1233. [DOI] [PubMed] [Google Scholar]
- Shi J, Leng W, Zhao L, Deng C, Xu C, Wang J, Wang Y, Peng X. 2018. Tooth loss and cancer risk: a dose-response meta analysis of prospective cohort studies. Oncotarget. 9(19):15090–15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stashenko P, Yost S, Choi Y, Danciu T, Chen T, Yoganathan S, Kressirer C, Ruiz-Tourrella M, Das B, Kokaras A, et al. 2019. The oral mouse microbiome promotes tumorigenesis in oral squamous cell carcinoma. mSystems. 4(4). pii:e00323–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztukowska MN, Ojo A, Ahmed S, Carenbauer AL, Wang Q, Shumway B, Jenkinson HF, Wang H, Darling DS, Lamont RJ. 2016. Porphyromonas gingivalis initiates a mesenchymal-like transition through Zeb1 in gingival epithelial cells. Cell Microbiol. 18(6):844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Hirano T, Whitmore SE, Morisaki I, Amano A, Lamont RJ. 2013. The serine phosphatase SerB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-κB RelA/p65. PLoS Pathog. 9(4):e1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Takada A, Kuboniwa M, Amano A. 2016. Intracellular periodontal pathogen exploits recycling pathway to exit from infected cells. Cell Microbiol. 18(7):928–948. [DOI] [PubMed] [Google Scholar]
- Tribble GD, Mao S, James CE, Lamont RJ. 2006. A Porphyromonas gingivalis haloacid dehalogenase family phosphatase interacts with human phosphoproteins and is important for invasion. Proc Natl Acad Sci USA. 103(29):11027–11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto VJ, Baillie D, Wu Q, Gendron R, Grenier D, Putnins EE, Kanervo A, Firth JD. 2005. Fusobacterium nucleatum increases collagenase 3 production and migration of epithelial cells. Infect Immun. 73(2):1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Sztukowska M, Ojo A, Scott DA, Wang H, Lamont RJ. 2015. Foxo responses to Porphyromonas gingivalis in epithelial cells. Cell Microbiol. 17(11):1605–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore SE, Lamont RJ. 2014. Oral bacteria and cancer. PLoS Pathog. 10(3):e1003933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JS, Zheng M, Zhang M, Pang X, Li L, Wang SS, Yang X, Wu JB, Tang YJ, Tang YL, et al. 2018. Porphyromonas gingivalis promotes 4-nitroquinoline-1-oxide-induced oral carcinogenesis with an alteration of fatty acid metabolism. Front Microbiol. 9:2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Yamada M, Li M, Liu H, Chen SG, Han YW. 2007. FadA from Fusobacterium nucleatum utilizes both secreted and nonsecreted forms for functional oligomerization for attachment and invasion of host cells. J Biol Chem. 282(34):25000–25009. [DOI] [PubMed] [Google Scholar]
- Yang CY, Yeh YM, Yu HY, Chin CY, Hsu CW, Liu H, Huang PJ, Hu SN, Liao CT, Chang KP, et al. 2018. Oral microbiota community dynamics associated with oral squamous cell carcinoma staging. Front Microbiol. 9:862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Jermanus C, Barbetta B, Choi C, Verbeke P, Ojcius DM, Yilmaz O. 2010. Porphyromonas gingivalis infection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Mol Oral Microbiol. 25(2):89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee M, Kim S, Sethi P, Duzgunes N, Konopka K. 2014. Porphyromonas gingivalis stimulates IL-6 and IL-8 secretion in GMSM-K, HSC-3 and H413 oral epithelial cells. Anaerobe. 28:62–67. [DOI] [PubMed] [Google Scholar]
- Yilmaz O, Jungas T, Verbeke P, Ojcius DM. 2004. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun. 72(7):3743–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Yao L, Maeda K, Rose TM, Lewis EL, Duman M, Lamont RJ, Ojcius DM. 2008. ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X7-mediated host-cell apoptosis. Cell Microbiol. 10(4):863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost S, Stashenko P, Choi Y, Kukuruzinska M, Genco CA, Salama A, Weinberg EO, Kramer CD, Frias-Lopez J. 2018. Increased virulence of the oral microbiome in oral squamous cell carcinoma revealed by metatranscriptome analyses. Int J Oral Sci. 10(4):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Chu M, Huang Z, Yang X, Ran S, Hu B, Zhang C, Liang J. 2017. Variations in oral microbiota associated with oral cancer. Sci Rep. 7(1):11773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Sztukowska M, Wang Q, Inaba H, Potempa J, Scott DA, Wang H, Lamont RJ. 2015. Noncanonical activation of beta-catenin by Porphyromonas gingivalis. Infect Immun. 83(8):3195–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]