

Abstract
Diazeniumdiolates of structure RHN—N(O)═NOR’ are of interest as prodrug (caged) forms of the bioeffectors nitric oxide (NO) and nitroxyl (HNO). Previous work has focused on examples possessing α-branched R groups, with IPA/NO (R = isopropyl) being the smallest examined to date. To probe the effect of minimizing alkyl group size on the chemistry of IPA/NO, we prepared the corresponding methyl derivative, MA/NO, as a sodium salt that was highly unstable but that could be trapped in very low overall yield as the stable O2 -benzyl derivative. To prepare enough for efficient characterization, we devised an alternate synthesis involving a novel N- dealkylation route. CH3HN—N(O)═NOBn, synthesized in high yield and crystallized as the Z-isomer as determined by X-ray crystallography, was observed to exist as a mixture of two isomeric forms in 11:1 ratio in dynamic equilibrium in solution phase. Similar results were seen for the O2-ethyl derivative, whose two equilibrium constituents were partially separated by HPLC to reveal essentially identical UV and mass spectra, indicating them to be Z- and E- isomers of CH3HN—N(O)═NOEt. The results could lead the way to a fuller understanding of the chemistry of the acyclic E-diazeniumdiolates.
Graphical Abstract
INTRODUCTION
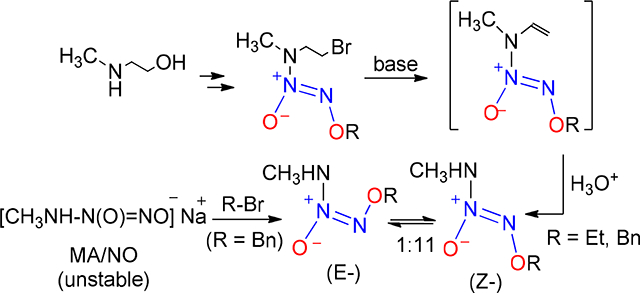
We are pursuing an increasingly refined understanding of the fundamental physicochemical properties of the N-bound diazeniumdiolates1, compounds of structure R1R2N-N(O)=NOR3, as a basis for rationally designing therapeutically useful prodrugs of nitric oxide2 (NO) and nitroxyl3 (HNO) as well as novel tools for probing the chemical biology of these important bioeffectors.4 One aspect that has so far been difficult to elucidate fully concerns the factors that control the cis/trans stereochemistry of the oxygens in the -N(O)=NOR3 group. Arulsamy et al have very recently addressed this issue as it relates to compounds in which the diazeniumdiolate nitrogen is attached to carbon,5,6 but so far we know of only one report in which an E configuration has been confirmed in a nitrogen-bound diazeniumdiolate; this was an O2-protected IPA/NO7 analog, a special case (R1 isopropyl, R2 = H, R3 = 2-bromoethyl) in which base-induced removal of the N- H proton leads to an anion whose E isomer is trapped by an intramolecular alkylation process to form cyclic product 28 (see Scheme 1). In an effort to obtain a macroscopically observable pair of acyclic Z/E isomers in sufficient equilibrium concentrations that the E form’s properties can be directly characterized and compared with those of the Z isomer, we hypothesized that minimizing steric interactions among the R groups might achieve that objective. Here we provide evidence that replacing the isopropyl group of IPA/NO anion (1) in Scheme 1 with methyl (R3 = benzyl or ethyl) provides just such an equilibrium mixture of isomers.
Scheme 1.
Synthesis of a crystallographically confirmed E diazeniumdiolate, 2
RESULTS AND DISCUSSION
Direct Synthesis of MA/NO.
Preparation of primary amine diazeniumdiolates has been an enduring challenge on account of their low thermodynamic stabilities.9 Initial attempts towards the synthesis of MA/NO anion involved the reaction of NO with a solution of methylamine in ether-acetonitrile, precooled to −80 °C. Filtration of the resulting methylammonium salt in very low yield and subsequent treatment with sodium methoxide afforded the corresponding sodium salt (3) as a white solid in about 3% overall yield (Scheme 2). All the manipulations were performed at −80 °C and the solids were found to be thermally unstable, often decomposing explosively at ambient temperature. Nevertheless, the powder survived long enough in cold conditions to allow anion 3 to be trapped by reaction with benzyl bromide at −80 °C to produce O2-benzyl derivative 4 in trace quantities. Evidently, however, a more convenient alternative synthetic route for the preparation of caged MA/NO prodrugs was desired for systematic studies of MA/NO’s chemistry.
Scheme 2.
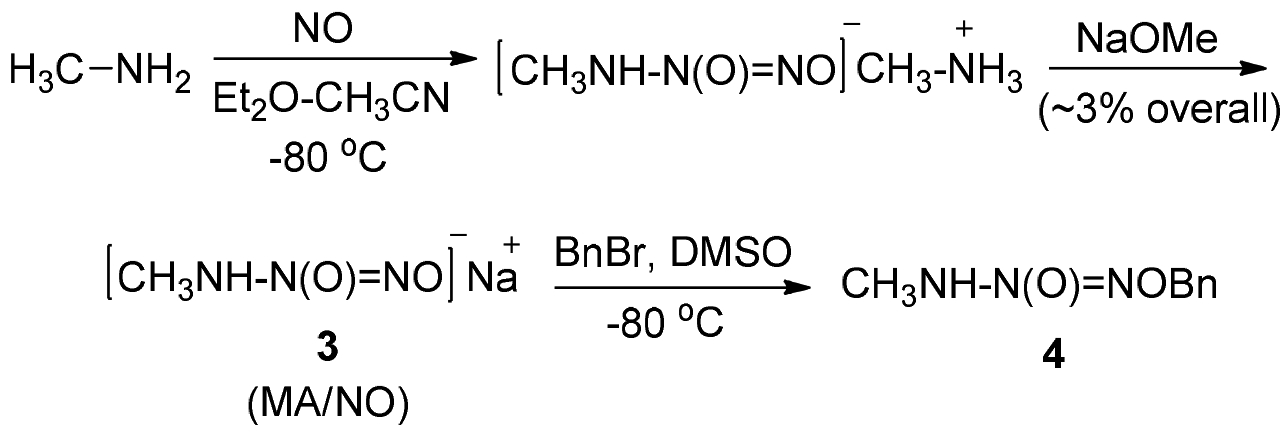
Reaction of methylamine with NO
Dehydrohalogenation-Mediated Preparation of O2-Protected MA/NO.
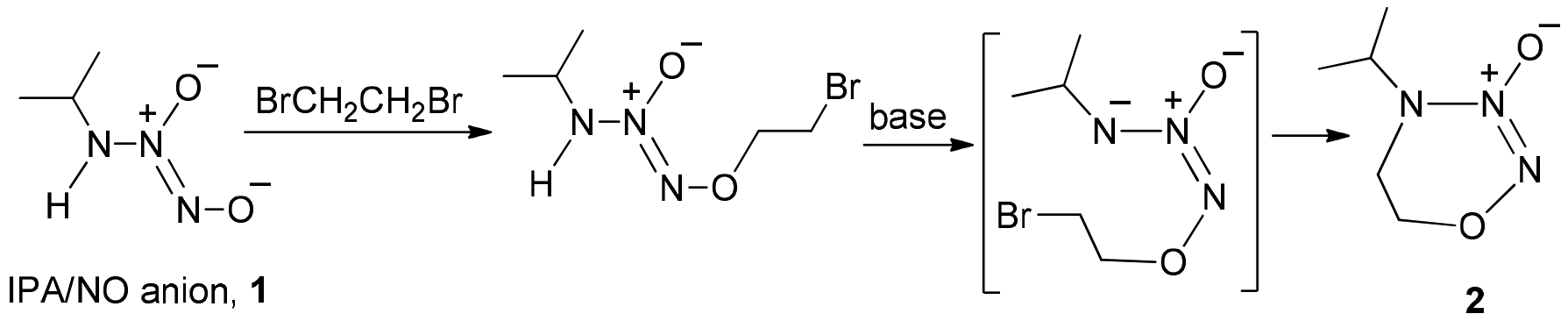
Accordingly, we devised the dealkylation route as outlined in Scheme 3 for the preparation of derivatized MA/NO analogs starting with the sodium salt of N-diazeniumdiolated 2-(methylamino)ethanol,10 6, used as a synthon. Preparation of the O2-benzyl-protected diazeniumdiolate 7 and subsequent bromination with NBS/Ph3P afforded the corresponding N-bromoethyl analog 8. Recently, we reported the use of 2,8,9-trimethyl-2,5,8,9-tetraaza-1-phosphabicyclo-[3.3.3]-undecane (Verkade’s super base) for dehydrohalogenation of diazeniumdiolate-containing compounds under mild conditions.11 Reaction of 8 with this base in acetonitrile was facile, but the desired N- vinyl derivative 9 was found to be unstable and could not be isolated. Acidic hydrolysis of the in-situ generated 9 with p-toluenesulfonic acid, followed by purification of the crude reaction mixture, afforded the O2-benzyl protected MA/NO (4, 69% isolated yield) whose physicochemical properties were identical to those of the material obtained in the low- temperature benzylation of MA/NO 3.
Scheme 3.
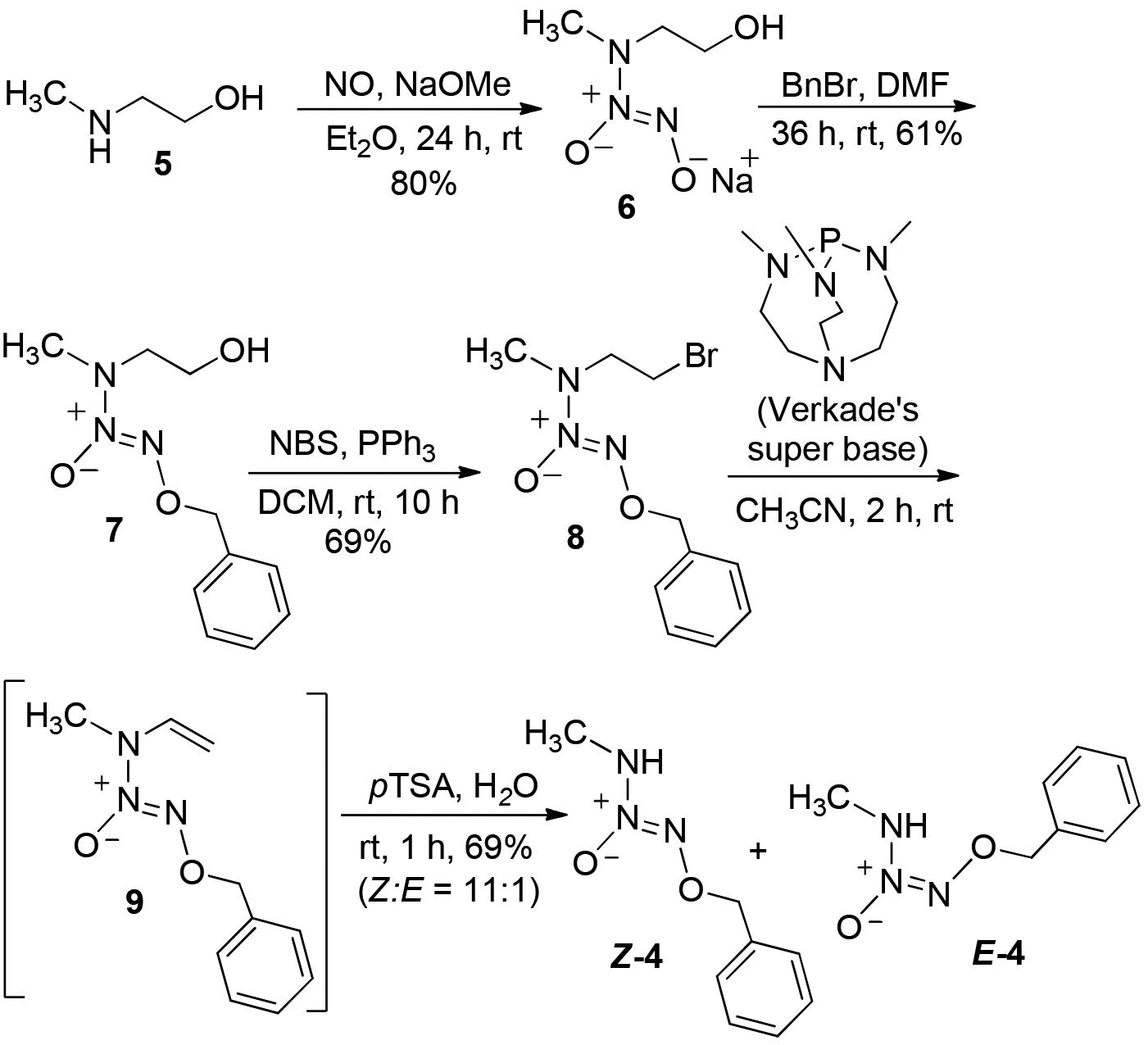
An N-dealkylation route to O2-benzylated MA/NO
Isomer Equilibration in 4.
Careful examination of the 1H NMR spectral pattern of 4 in CDCl3 revealed the presence of a pair of doublets at 2.98 and 3.05 ppm in 11:1 ratio along with the two overlapped singlets at 5.18 ppm indicating the existence of this O2-benzyl MA/NO analog as a mixture of two isomeric forms in solution phase. Similarly, the proton-coupled 13C NMR spectrum of 4 also exhibits two sets of six peaks, further supporting the existence of two isomeric forms of this analog. Recrystallization of the product mixture from diethyl ether at 4 °C followed by X-ray crystallographic analysis revealed that it exists in a Z-conformation (Z-4) (Figure 1A). We believe that the minor isomer exists in E-conformation E-4, although it could not be isolated by recrystallization. Low temperature NMR analysis of a freshly prepared solution of recrystallized Z-4 in CDCl3 at −20 °C revealed the Z:E isomeric ratio of 4 to be about 66:1, indicating the crystalline material to contain no more than 1.5% of E-4 conformer (Figure 1B). At room temperature the sample was found to equilibrate to a mixture of Z-4:E-4 in 11:1 ratio, similar to that of the purified product mixture, indicating the two isomers to exist in a dynamic equilibrium in solution phase at room temperature (See Supporting Information).
Figure 1.
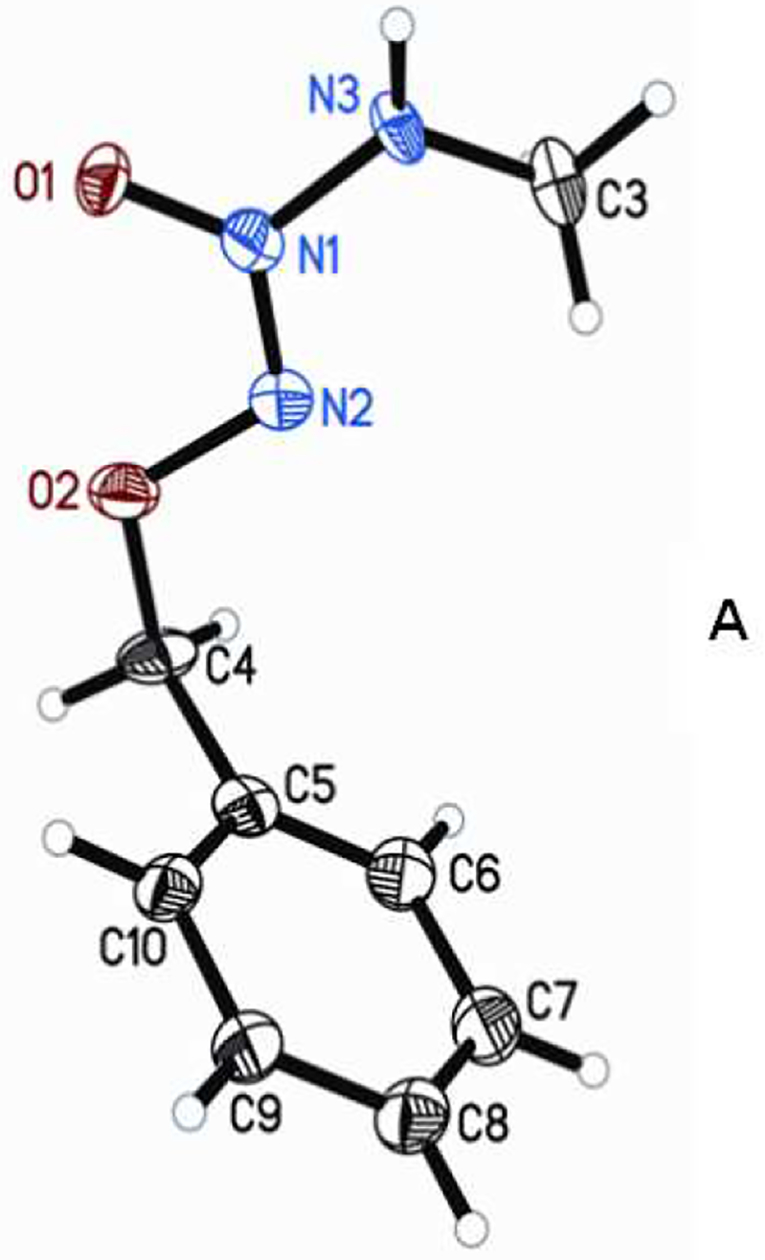
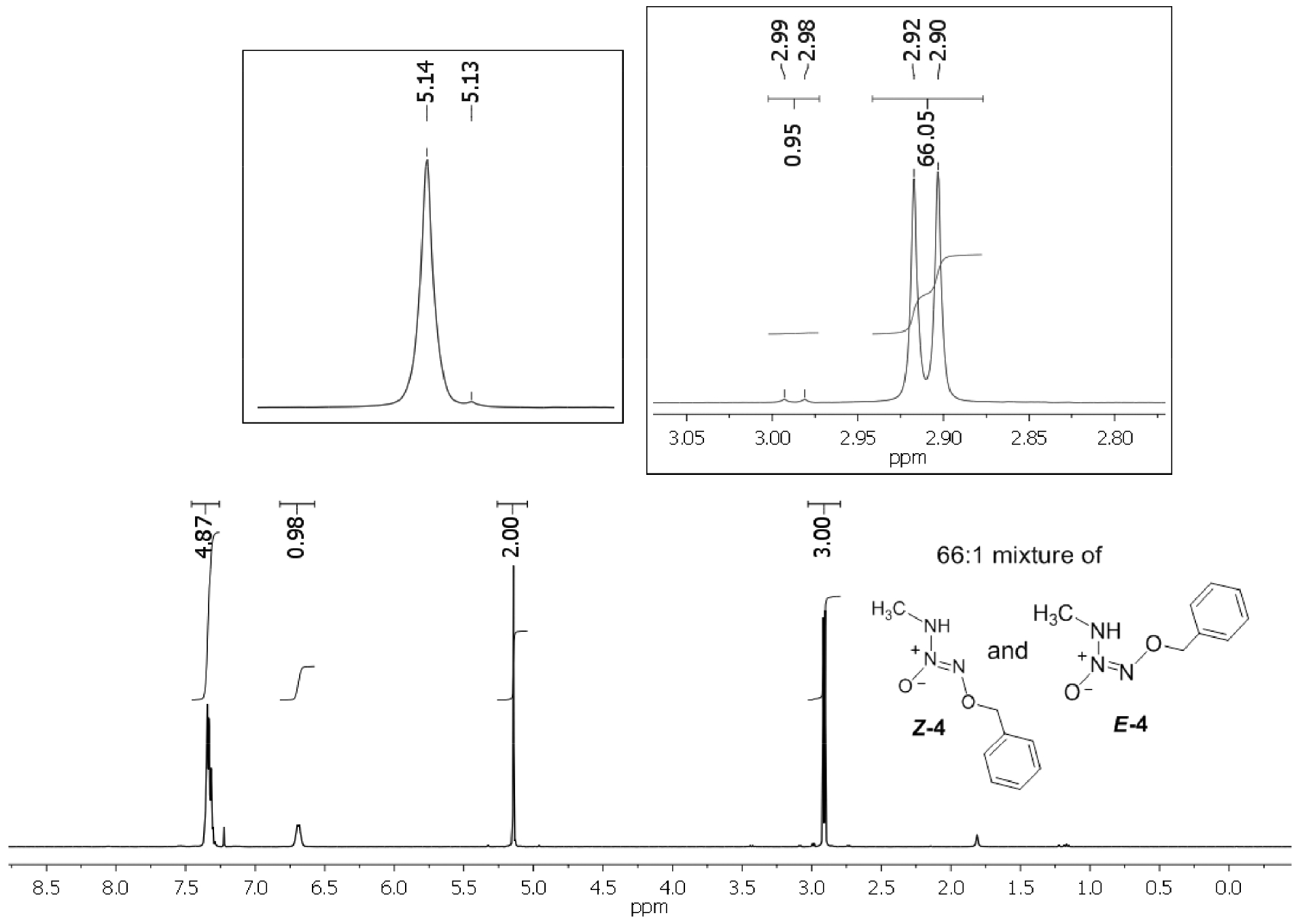
A) Molecular structure and numbering schemes for the crystals of Z-4 (displacement ellipsoids are at the 50% level). B) 1D 1H NMR spectra of recrystallized Z-4 in CDCl3 at −20 °C indicating Z-4 and E-4 isomers to exist in 66:1 ratio in solution phase.
LC/MS Identification and Partial Chromatographic Separation of Z- and EIsomers.
Following the same synthetic protocol, O2-ethyl protected MA/NO 13 was also prepared in high yield starting from 10 via the formation of the corresponding N-bromoethyl (11) and N-vinyl (12) intermediates (Scheme 4). NMR analysis of this product was consistent with that of the O2-benzyl analog and suggested the formation of 13 as an 11:1 mixture of two isomers, presumably also as Z- and E-conformations. Injection of a purified mixture of these two isomers on an HPLC column allowed a reasonable separation of the two peaks which were fused at the baseline, Figure 2A. Isolation of either form, followed by reinjection, resulted in chromatograms containing both peaks with integration ratios matching those of the original mixture, Figures 2B and 2C. These results were confirmed using HRMS detection. The measured m/z for the [M+H]+ ion of both peaks was 120.07692, which is in excellent agreement, < 1.5 ppm, with the calculated m/z, 120.07675, for the molecular formula, C3H9N3O2 (Figure 3). The m/z for the sodium adduct was extracted and plotted as a function of time. This analysis shows that the taller peak readily forms a sodium adduct while such an adduct was not detected in the smaller peak (Supporting Information). These data are consistent with the view that 13 exists as a mixture of Z- and E-isomers, with the Z-isomer’s cis oxygens uniquely favorably oriented for bidentate coordination to the alkali metal ion (See Supporting Information). This isomerization is presumably mediated through a proton shift from the amine nitrogen (N3) to the diazeniumdiolate nitrogen (N2), and/or direct ionization of the N3-H bond, thus rendering largely single bond character to N1—N2 allowing free rotation about this bond, as indicated by theoretical calculations reported earlier.8,12 (See Figure 1A for numbering scheme).
Scheme 4.
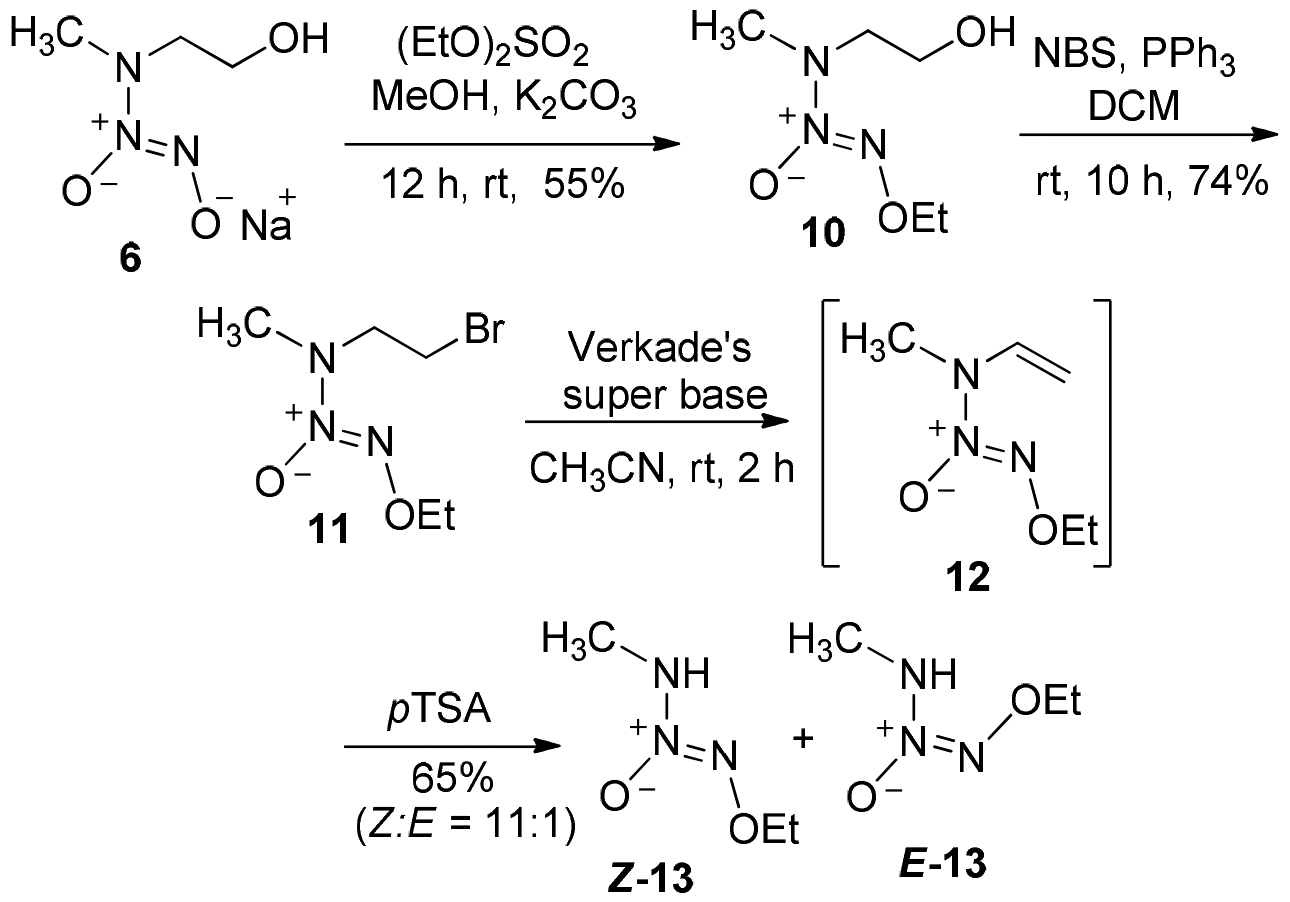
Preparation of a mixture of O2-ethylated MA/NO isomers
Figure 2.
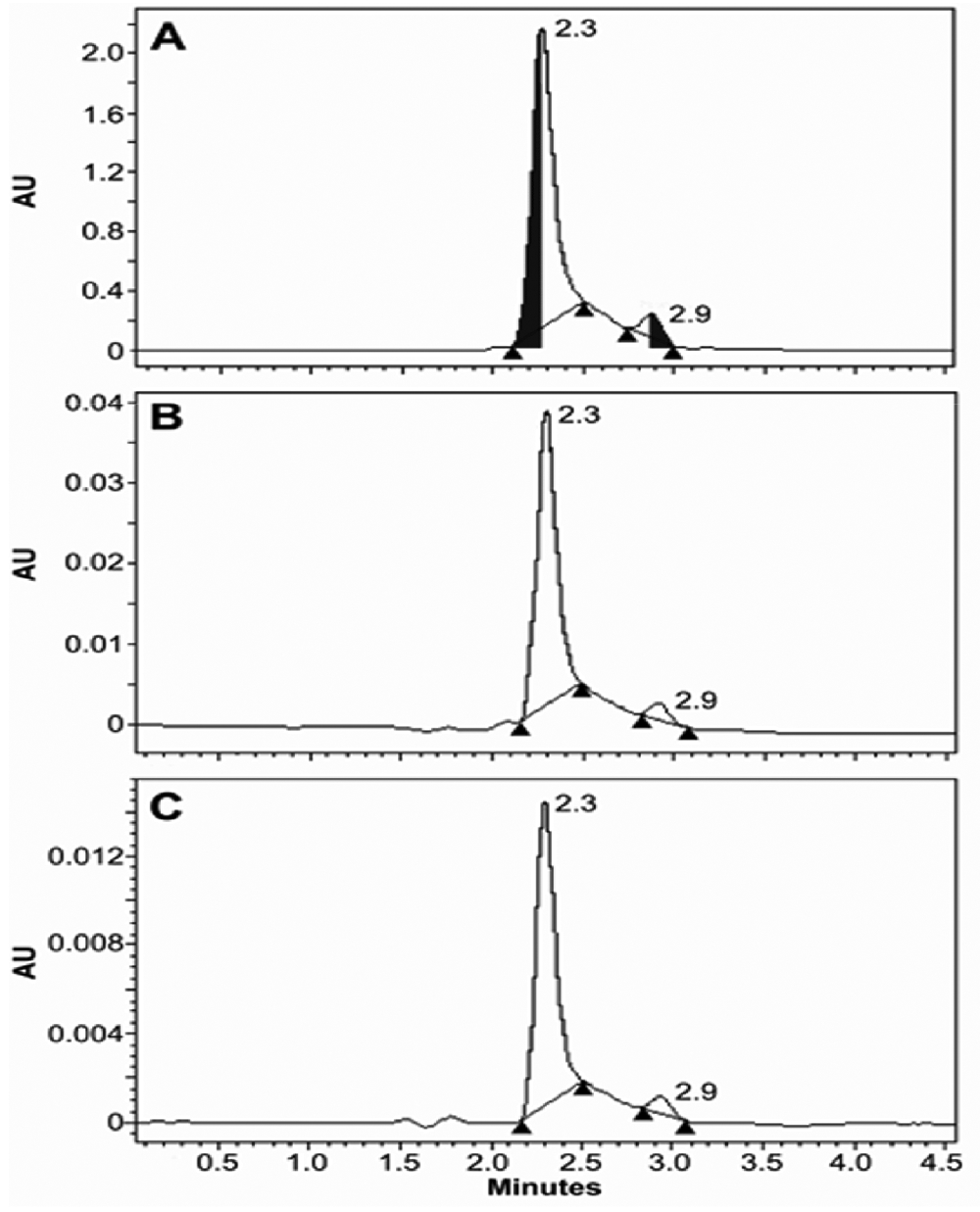
HPLC traces of a mixture of the two 13 isomers observed at 240 nm. A) Chromatogram of the mixture of isomers purified by silica gel chromatography. Both peaks have a λmax at 240 nm. B) The shaded portion of the first peak was collected at 0 °C and immediately injected into the HPLC to obtain the chromatogram of panel B, showing re4equilibration. Panel C is a similar experiment to B showing re-equilibration upon collection of the shaded portion of the second peak.
Figure 3.
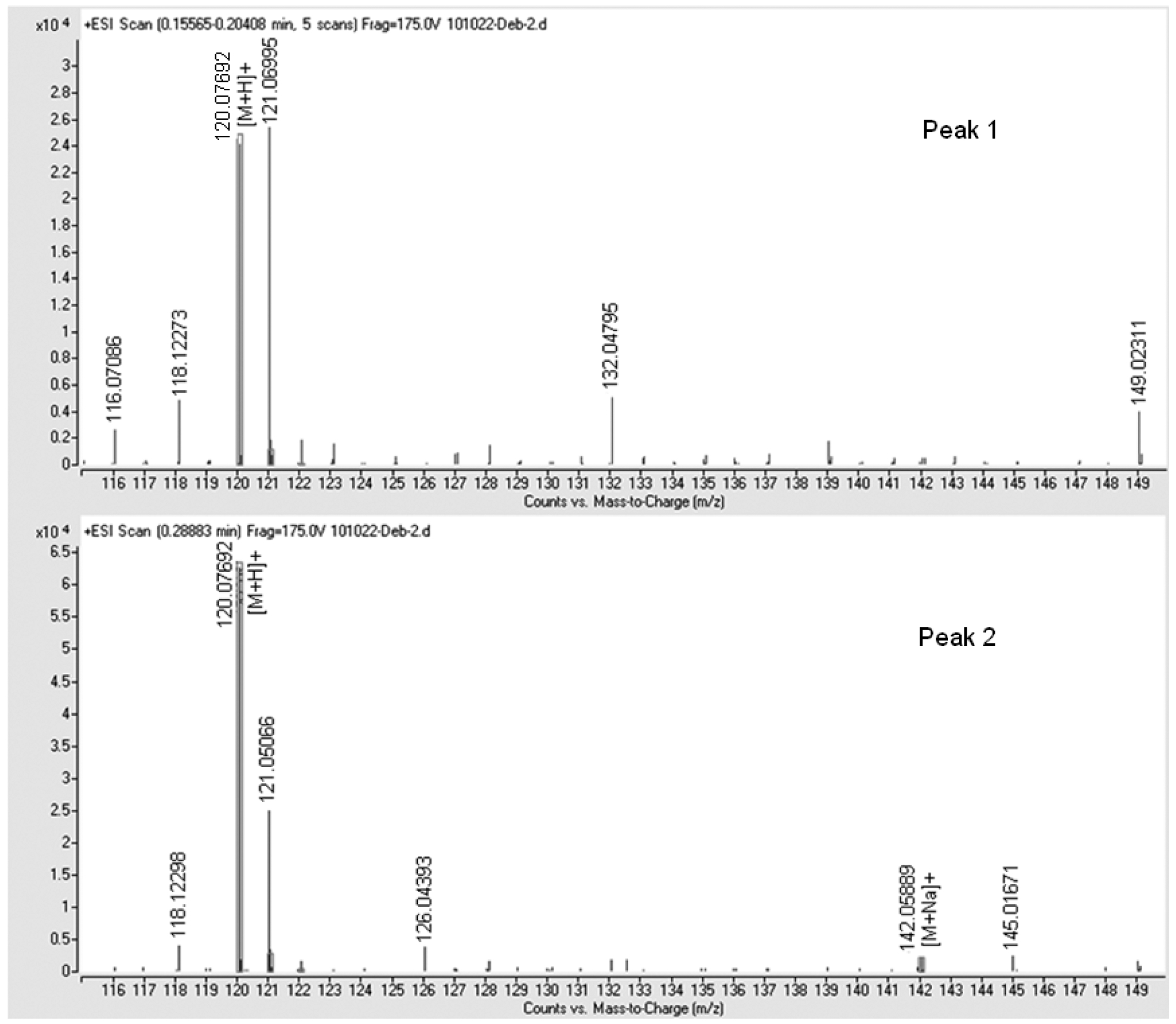
High resolution mass spectra for the two peaks of compound 13 in the extracted ion chromatogram presented in the supporting information. Peak 1 has an [M+H]+ = 120.07682. Peak 2 has an [M+H]+ = 120.07692 and an [M+Na]+ = 142.05889. Note, the ion with an m/z = 121.051 is due to purine, which was added as an internal reference ion to obtain high resolution spectra of our analytes. All other ions are attributable to background interference.
UV-Vis Spectroscopic Determination of pKa.
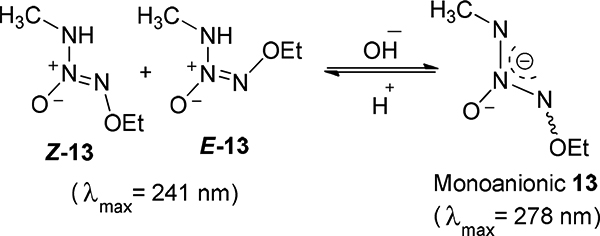
Potential acidity of the NH bond of a purified mixture of Z- and E-13 was examined by UV spectroscopy using buffers of different pH following the similar procedure as reported for that of O2-methyl IPA/NO.12 The extinction coefficients were determined by dissolving a known amount of 13 in a given quantity of ethanol, diluting with a large excess of 0.01 M phosphate buffer for runs at pH 7.4 – 12 and then recording the spectrum within 1 – 2 s of dilution at room temperature. A single peak was found for 13 at λmax = 241 nm at all the pHs from 1 to 11. As the pH was increased above 11, the peak intensity decreased and a new one emerged at λmax = 278 nm due to the ionization of the NH proton. Re-acidification of the alkaline solution restored the 241-nm peak at its original intensity.
The pKa of the process was measured by adjusting the pH in small increments between 11.0 and 12.0 until the 241- and 278-nm peaks were each at half-maximal intensity. As the extinction coefficients for the un-ionized and anionic forms of 13 are 11.0 and 10.0 mM−1 cm−1, respectively, the pH was adjusted so that the apparent extinction coefficients were approximately 5.5 and 5.0 mM−1 cm−1, respectively. Half-maximal absorption intensities for both forms were observed approximately at pH 11.70 and this pH value was taken as the pKa for 13.
Kinetic Studies.
Ionization of the N-H bond of a mixture of these two isomers at room temperature in D2O was also examined by 1H NMR spectroscopy. Samples were run in D2O adjusted to pD values13 of about either 8.5, 10.5, or 13.0 by adding NaOD to solutions in D2O. Prominent exchange line broadening of N-methyl and O-methylene signals was observed for both the isomers at the pD value of 10.5 (Figure 4C). The observed spectrum at the pD value of 13.0 indicates ionization of the acidic primary amine proton with formation of the corresponding charge-delocalized monoanionic species with a line-broadened singlet for the N-methyl and quartet for the O-methylene signals (Figure 4D). Kinetic data for the exchange between the anion and its conjugate acid were determined from the signal broadening of the peak centered at 2.95 ppm using saturation transfer and dynamic NMR spectroscopic methods described previously.8,14 The rate constant for exchange, ~1.2 × 103 s−1, is similar to that of a structurally analogous primary amine diazeniumdiolate shown to have an acidic NH bond.12 The interconversion barrier was found to be 17.4 kcal/mol, which is in close agreement with the theoretical calculations for a similar molecule reported earlier.8 Re-acidification of this solution with DCl back to the pD value of 3.5 revealed a re-equilibration with Z/E ratio and spectral patterns identical to those of the parent mixture at a pD value of 7.5. Analysis of this sample after 14 h at room temperature indicated no significant change in the isomer ratio.
Figure 4.
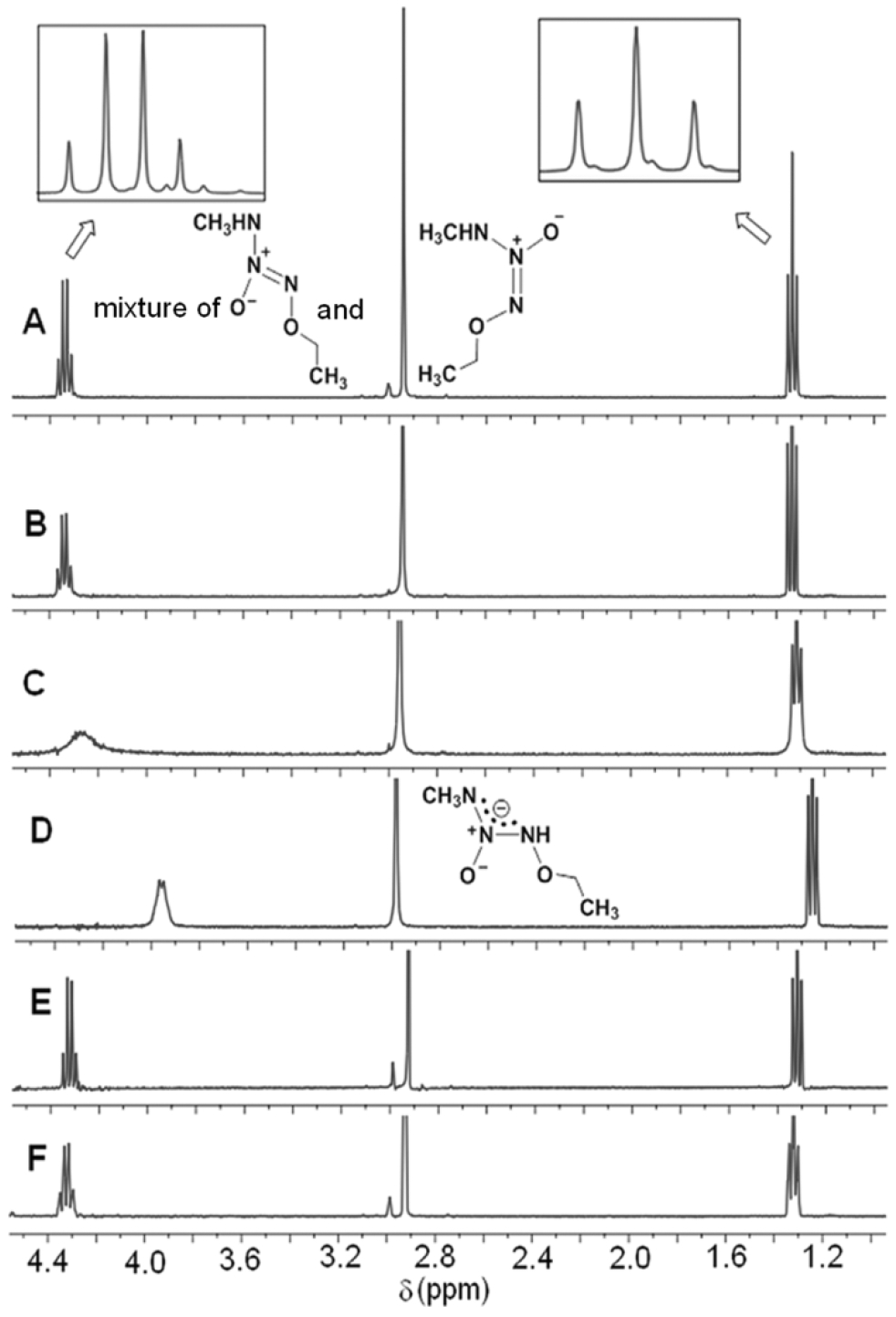
1H NMR spectra of the mixture of Z-13 and E-13 at 400 MHz revealing the slow exchange process accompanying ionization of the N-H bond at room temperature. Spectra A, B, C and D were run in D2O adjusted to measured pD values of about 7.5, 8.5, 10.5 and 13.0, respectively, by adding NaOD. Spectrum E was run after adjusting the pD value of the sample used for D back to 3.5 from 13.0 by adding DCl. Spectrum F shows the status of the pD 3.5 solution in D2O run after 14 h.
CONCLUSIONS
Our results reveal important similarities as well as differences between the well characterized iPrHN-N(O)=NOR derivatives and their methylamine analogs. While the sodium salt of the former (R = Na+) can be stored indefinitely without incident,15 salts of its analog MA/NO have thus far defied isolation in pure form, even when the methylamine/NO reaction was carried out at cryogenic temperatures. Anions of both IPA/NO and MA/NO can be O-alkylated to produce stable derivatives, but because MA/NO’s instability has precluded isolation of the salts in reasonable yield we had to devise an indirect dealkylative synthesis in order to produce enough MeHN-N(O)=NOR (R = benzyl or ethyl) for efficient characterization. The O-alkylated IPA/NO and MA/NO derivatives were very similar to each other in their ultraviolet properties, the pKas for ionization of their N-H bonds, and the rate constants for protonation/deprotonation at pHs near the pKas. Additionally, there was evidence that the O-alkylation products of both IPA/NO and MA/NO can isomerize from the favored Z configuration (oxygens cis) to the E form, but with one critical difference: the E isomer of IPA/NO had to be trapped in an intramolecular cyclization reaction to confirm its existence, while that of MeHN-N(O)=NOR proved directly observable as a constituent of the equilibrium mixture. The latter’s isomers were partially separable. It may be that conditions can be found that will permit one MA/NO derivative or another to be isolated as a pure E form, facilitating further insights into the chemistry (and possibly the biology) of diazeniumdiolates having this thus far little-studied configuration.
EXPERIMENTAL SECTION
Instrumentation.
All reactions were performed under an inert atmosphere. All chemicals were purchased from commercial sources and used without further purification. Ultraviolet (UV) spectra were recorded on a diode array spectrophotometer. Nuclear magnetic resonance (NMR) spectra were collected with a 400-MHz spectrometer using appropriate deuterated solvent with chemical shifts reported in parts per million (ppm) downfield from tetramethylsilane. The analytical HPLC/MS analyses were performed using systems with photodiode array UV-visible detector and accurate-mass quadrupole time-of-flight (Q-TOF) mass spectrometer.
Reaction of Methylamine with NO.
The equipment used for conducting reactions with NO gas under anaerobic conditions has been described previously.16,17 Nitric oxide was obtained in UHP grade and allowed to stand in a ballast tank at about five atmospheres of pressure over potassium hydroxide pellets for a minimum of several hours before use. A 2 M solution of methylamine in THF (20 mL) was diluted with 20 mL of diethyl ether and 10 mL of acetonitrile. The resulting solution was placed in a standard thick-walled glass hydrogenation bottle, cooled to −80 °C, flushed with nitrogen, charged with 50 psi NO, and stirred for 2 h. After completion of the reaction, about 500 mg (10% yield) of the methylammonium salt of MA/NO (2) was collected by filtration using a pre-cooled Büchner funnel. While 2 was still moist and cold, it was promptly suspended in ether, cooled to −80 °C and treated with 750 μL of 4.6 M sodium methoxide solution in methanol; care was taken to use less than one equivalent of sodium methoxide as estimated from the weight of crude compound 2. The resulting product was filtered while cold to afford about 140 mg (3% yield) of the sodium salt of MA/NO (3) as a white powder which was quickly stored under nitrogen at −20 °C: UV (0.01 M NaOH) λmax (ε) 249 nm (8.3 mM−1cm−1). [Special Note: Compound 3 and its methylammonium analog are unstable and decompose, often explosively, at ambient temperature.]
O2-Benzyl [N-(2-Hydroxyethyl)-N-methylamino]diazen-1-ium-1,2-diolate (7).
Compound 610 (3.00 g, 19.1 mmol) was dissolved in 15 mL of anhydrous DMF and cooled to 0 °C with constant stirring. A solution of benzyl bromide (3.89 g, 22.8 mmol) in 5 mL of anhydrous DMF was slowly added. The reaction mixture was then allowed to warm to room temperature and stirred for an additional 24 h. The reaction mixture was then quenched with H2O and extracted with ether (3×20 mL). The combined organic layers were washed with water and dried over anhydrous sodium sulfate. After concentration under reduced pressure, the crude product was purified on a silica gel column (hexanes:ethyl acetate 15:85) to afford 2.62 g (61% yield) of 7 as a hygroscopic, pale yellow oil: 1H NMR δ (CDCl3, 400 MHz) 7.40–7.32 (m, 5H), 5.22 (s, 2H), 3.66–3.62 (m, 2H), 3.34 (t, J=5.2 Hz, 2H), 2.97 (s, 3H), 2.36 (br s, 1H); 13C NMR δ (CDCl3, 100 MHz) 135.5, 128.6, 128.5, 128.5, 75.7, 59.4, 57.1, 42.0; UV (MeOH) λmax (ε) 244 nm (10.0 mM−1cm−1).
Anal. Calcd for C10H15N3O3. (0.3 H2O): C, 52.07; H, 6.82; N, 18.22. Found: C, 52.09; H, 6.66; N, 18.08.
O2-Benzyl [N-(2-Bromoethyl)-N-methylamino]diazen-1-ium-1,2-diolate (8).
Compound 7 (1.37 g, 6.08 mmol) was dissolved in 15 mL of dichloromethane and cooled to 0 °C with constant stirring. Triphenylphosphine (1.91 g, 7.30 mmol) was dissolved in 5 mL of dichloromethane and added to the reaction mixture and stirred for 15 min. N-Bromosuccinimide (1.29 g, 7.30 mmol) was added in portions to the reaction mixture over a period of 10 min and allowed to warm to room temperature and stirred overnight. Solvent was removed on a rotary evaporator and the crude product was purified on a silica gel column (hexanes:ethyl acetate 80:20) to afford 1.20 g (69% yield) of 8 as a dark yellow oil: 1H NMR δ (CDCl3, 400 MHz) 7.40–7.34 (m, 5H), 5.23 (s, 2H), 3.60 (t, J=6.8 Hz, 2H), 3.35 (t, J=6.8 Hz, 2H), 3.02 (s, 3H); 13C NMR δ (CDCl3, 100 MHz) 135.6, 128.7, 128.7, 128.6, 75.8, 55.9, 41.8, 27.3; UV (EtOH) λmax (ε) 246 nm (9.17 mM−1cm−1).
Anal. Calcd for C10H14N3O2Br: C, 41.68; H, 4.90; N, 14.58. Found: C, 41.44; H, 4.98; N, 14.29.
Isomeric Mixture of O2-Benzyl (N-Methylamino)diazen-1-ium-1,2-diolate (Z-4 and E-4).
Compound 8 (0.50 g, 1.7 mmol) was dissolved in 4 mL of anhydrous acetonitrile and slowly added to a stirring solution of 2,8,9-trimethyl-2,5,8,9-tetraaza-1-phosphabicyclo[3.3.3]undecane (0.41 g, 1.91 mmol) in 3 mL of anhydrous acetonitrile pre-cooled to 0 °C. Subsequently, the reaction mixture was warmed to room temperature and stirred for another 2 h while the reaction was monitored by TLC. After the completion of the reaction, the reaction mixture was re-cooled to 0 °C and a solution of p-toluenesulfonic acid (0.33 g, 1.74 mmol) in 1 mL of H2O was added and stirred for 1 h. The reaction mixture was concentrated under reduced pressure and extracted with ethyl acetate (3×10 mL). The combined organic layers were washed with saturated brine solution and dried over anhydrous sodium sulfate. After concentration under reduced pressure, the crude product was purified on a silica gel column (hexanes:ethyl acetate 40:60) to afford 0.22 g (69% yield) of a mixture of Z- and E-isomers of 4 in 11:1 ratio as a white solid: mp 48–50 °C; 1H NMR of Z-isomer δ (CDCl3, 400 MHz) 7.40–7.10 (m, 5H), 6.37–6.33 (m, 1H) 5.18 (s, 2H), 2.98 (d, J=5.6 Hz, 3H); 13C NMR δ (CDCl3, 100 MHz) 135.7, 128.6, 128.6, 128.5, 75.4, 34.3; UV (MeOH) λ max(ε) 245 nm (12.1 mM−1cm−1)
Anal. Calcd for C8H11N3O2: C, 53.03; H, 6.12; N, 23.19. Found: C, 53.06; H, 6.02; N, 23.14.
O2-Ethyl [N-(2-Hydroxyethyl)-N-methylamino]diazen-1-ium-1,2-diolate (10).
Compound 610 (3.00 g, 19.1 mmol) was dissolved with constant stirring in 20 mL of pre-cooled anhydrous methanol containing 1.00 g of finely powdered potassium carbonate suspended in it. To this mixture, a solution of diethyl sulfate (4.11 g, 28.7 mmol) in 10 mL of anhydrous methanol was slowly added. The reaction mixture was allowed to warm to room temperature and stirred for an additional 10 h. The reaction mixture was filtered, the methanol was removed on a rotary evaporator, and the residue was extracted with ethyl acetate (3×25 mL). The combined organic layers were washed with saturated brine solution and dried over anhydrous sodium sulfate. After concentration under reduced pressure, the crude product was purified on a silica gel column (hexanes:ethyl acetate 20:80) to afford 1.71 g (55% yield) of 10 as a hygroscopic, pale yellow oil: 1H NMR δ (CDCl3, 400 MHz) 4.17 (q, J=7.2 Hz, 2H), 3.72–3.57 (m, 2H), 3.26 (t, J=5.2 Hz, 2H), 2.89 (s, 3H), 2.71 (br s, 1H), 1.25 (t, J=7.2 Hz, 3H); 13C NMR δ (CDCl 100 MHz) 69.7, 59.3, 57.0, 42.0, 14.2; UV (MeOH) λmax (ε) 242 nm (11.7 mM−1 −1 cm−1)
Anal. Calcd for C5H13N3O3. 0.2H2O: C, 36.01; H, 8.10; N, 25.20. Found: C, 35.77; H, 7.82; N, 24.95.
O2-Ethyl [N-(2-Bromoethyl)-N-methylamino]diazen-1-ium-1,2-diolate (11).
Compound 10 (3.00 g, 18.4 mmol) was dissolved in 15 mL of dichloromethane and cooled to 0 °C with constant stirring. Triphenylphosphine (9.64 g, 36.8 mmol) was dissolved in 5 mL of dichloromethane and added to the reaction mixture and stirred for 15 min. N-Bromosuccinimide (6.51g, 36.80 mmol) was added in portions to the reaction mixture over a period of 10 min and allowed to warm to room temperature and stirred overnight. Solvent was removed on a rotary evaporator and the crude product was purified on a silica gel column (hexanes:ethyl acetate 80:20) to afford 2.75 g (74% yield) of 11 as a yellow oil: 1H NMR δ (CDCl3, 400 MHz) 4.20 (q, J=7.2 Hz, 2H), 3.53 (t, J=7.2 Hz, 2H), 3.35 (t, J=6.8 Hz, 2H), 2.95 (s, 3H), 1.28 (t, J=7.2 Hz, 3H); 13C NMR δ (CDCl3, 100 MHz) 69.8, 55.8, 42.0, 27.3, 14.3; UV (EtOH) λmax (ε) 244 nm (10.6 mM−1cm−1).
Anal. Calcd for C5H12N3O2Br: C, 26.56; H, 5.35; N, 18.59. Found: C, 26.81; H, 5.43; N, 18.25.
Isomeric Mixture of O2-Ethyl (N-Methylamino)diazen-1-ium-1,2-diolate (Z-13 and E-13).
Compound 11 (0.50 g, 2.21 mmol) was dissolved in 4 mL of anhydrous acetonitrile and slowly added to a stirring solution of 2,8,9-trimethyl-2,5,8,9-tetraaza-1-phosphabicyclo[3.3.3]- undecane (0.57 g, 2.65 mmol) in 3 mL of anhydrous acetonitrile pre-cooled to 0 °C. Subsequently, the reaction mixture was warmed to room temperature and stirred for another 2 h while the reaction was monitored by TLC. After the completion of the reaction, the reaction mixture was re-cooled to 0 °C and a solution of p-toluenesulfonic acid (0.42 g, 2.21 mmol) in 1 mL of H2O was added and stirred for 1 h. The reaction mixture was concentrated under reduced pressure and extracted with ethyl acetate (3×10 mL). The combined organic layers were washed with saturated brine solution and dried over anhydrous sodium sulfate. After concentration under reduced pressure, the crude product was purified on a silica gel column (hexanes:ethyl acetate 40:60) to afford 0.17 g (65% yield) of a mixture of Z- and E-isomers of 13 in 11:1 ratio as a pale yellow oil: 1H NMR of Z-isomer δ (CDCl3, 400 MHz) 4.25 (q, J=8.4 Hz, 2H), 3.02 (d, J=5.6 Hz, 3H), 1.37 (t, J=7.2 Hz, 3H); 13C NMR δ (CDCl3, 100 MHz) 69.4, 34.3, 14.3; UV (MeOH) λ max (ε) 245 nm (11.1 mM−1cm−1); HRMS (ESI) m/z calculated for C3H9N3O [M+H]+ 120.07675, found 120.07692, Δ ppm = 1.4
Anal. Calcd for C3H9N3O2. (0.076 EtOAc): C, 31.54; H, 7.70; N, 33.40. Found: C, 31.14; H, 7.65; N, 33.00.
HPLC Analysis.
HPLC separations of a mixture of the two isomeric forms of 13 (Figure 2) were performed using a liquid chromatograph instrument connected to a photodiode array UV-visible detector. A reverse phase C18 column (150×4.6 mm) with 5-μm particle size with a guard column of the same material, both at 25 °C, was used for the stationary phase. The mobile phase used for the separation was comprised of an isocratic solution of 30% acetonitrile in water containing 0.1% formic acid. The eluate was monitored at the wavelength of 250 nm. The fractions were collected manually at room temperature and immediately cooled down to 0 °C and preserved at the same temperature until further analysis.
Supplementary Material
Table 1.
UV absorbances of the un-ionized and the monoanionic forms of 13 at 241 and 278 nm.
 | |||
|---|---|---|---|
| Entry | pH | λ241 (AU) | λ278 (AU) |
| 1. | 7.40 | 0.94 (ε = 11.0 mM−1 cm−1) |
0 |
| 2. | 11.00 | 0.88 | 0.35 |
| 3. | 11.20 | 0.72 | 0.36 |
| 4. | 11.40 | 0.52 | 0.39 |
| 5. | 11.60 | 0.48 | 0.41 |
| 6. | 11.70 (pKa) | 0.47 | 0.42 |
| 7. | 11.75 | 0.42 | 0.44 |
| 8. | 11.80 | 0.29 | 0.51 |
| 9. | 11.90 | 0.23 | 0.68 |
| 10. | 12.00 | 0.17 | 0.80 |
| 11. | 14.00 | 0 | 0.85 (ε = 10.0 mM−1 cm−1) |
ACKNOWLEDGEMENTS
We thank Mr. John Klose, LPAT-ATP, SAIC-Frederick Inc., Frederick National Laboratory for Cancer Research, for assistance in NMR determination of the exchange rate constant and Dr. Sergey Tarasov and Ms. Marzena A. Dyba of the Biophysics Resource in the Structural Biophysics Laboratory, Frederick National Laboratory for Cancer Research, for assistance with the high resolution mass spectrometer. This project has been funded with Federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. Crystallographic studies were supported by the National Institute on Drug Abuse (NIDA) under contract Y1-DA1101 and by the Naval Research Laboratory.
Footnotes
Supporting Information
Full 1H and 13C NMR spectra for new compounds, crystallographic data for compound Z-4, details of the dynamic NMR experiment, pKa studies, and chromatographic isomer separation. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES:
- 1).(a) Keefer LK ACS Chem. Biol 2011, 6, 1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hrabie JA; Keefer LK Chem. Rev 2002, 102, 1135–1154. [DOI] [PubMed] [Google Scholar]; (c) Biswas D; Deschamps JR; Keefer LK; Hrabie JA Chem. Commun 2010, 46, 5799–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).(a) Shami PJ; Saavedra JE; Wang LY; Bonifant CL; Diwan BA; Singh SV; Gu Y; Fox SD; Buzard GS; Citro ML; Waterhouse DJ; Davies KM; Ji X; Keefer LK Mol. Cancer Ther 2003, 2, 409–417. [PubMed] [Google Scholar]; (b) Saavedra JE; Southan GJ; Davies KM; Lundell A; Markou C; Hanson SR; Adrie C; Hurford WE; Zapol WM; Keefer LK J. Med. Chem 1996, 39, 4361–4365. [DOI] [PubMed] [Google Scholar]; (c) Murad FN Engl. J. Med 2006, 355, 2003–2011. [DOI] [PubMed] [Google Scholar]; (d) Brune B; Zhou J Cardiovasc. Res 2007, 75, 275–282. [DOI] [PubMed] [Google Scholar]
- 3).(a) Irvine JC; Ritchie RH; Favaloro JL; Andrews KL; Widdop RE; Kemp- Harper BK Trends in Pharmacol. Sci 2008, 29, 601–608. [DOI] [PubMed] [Google Scholar]; (b) Miranda KM Coord. Chem. Rev 2005, 249, 433–455. [Google Scholar]; (c) Fukuto JM; Bianco CL; Chavez TA Free Radic. Biol. Med 2009, 47, 1318–1324. [DOI] [PubMed] [Google Scholar]; (d) Paolocci N; Jackson MI; Lopez BE; Miranda KM; Tocchetti CG; Wink DA; Hobbs AJ; Fukuto JM Pharmacol. Ther 2007, 113, 442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).(a) Mason MG; Nicholls P; Wilson MT; Cooper CE Proc. Natl. Acad. Sci. USA 2006, 103, 708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Espey MG; Miranda KM; Thomas DD; Xavier S; Citrin D; Vitek MP; Wink DA Ann. N. Y. Acad. Sci 2002, 962, 195–206. [DOI] [PubMed] [Google Scholar]
- 5).Arulsamy N; Bohle DS; Holman CL; Perepichka IJ Org. Chem 2012, 77, 7313–7318. [DOI] [PubMed] [Google Scholar]
- 6).(a) Arulsamy N; Bohle DS; Perepichka I Can. J. Chem 2007, 85, 105–117. [Google Scholar]; (b) Arulsamy N; Bohle DS Angew. Chem. Int. Ed 2002, 41, 2089–2091. [PubMed] [Google Scholar]
- 7).(a) Drago RS; Karstetter BR J. Am. Chem. Soc 1961, 83, 1819–1822. [Google Scholar]; (b) Miranda KM; Katori T; Torres de Holding CL; Thomas L; Ridnour LA; McLendon WJ; Cologna SM; Dutton AS; Champion HC; Mancardi D; Tocchetti CG; Saavedra JE; Keefer LK; Houk KN; Fukuto JM; Kass DA; Paolocci N; Wink DA J. Med. Chem 2005, 48, 8220–8228. [DOI] [PubMed] [Google Scholar]; (c) Andrei D; Salmon DJ; Donzelli S; Wahab A; Klose JR; Citro ML; Saavedra JE; Wink DA; Miranda KM; Keefer LK J. Am. Chem. Soc 2010, 132, 16526–16532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Wang Y–N; Bohle DS; Bonifant CL; Chmurny GN; Collins JR; Davies KM; Deschamps J; Flippen-Anderson JL; Keefer LK; Klose JR; Saavedra JE; Waterhouse DJ; Ivanic JJ Am. Chem. Soc 2005, 127, 5388–5395. [DOI] [PubMed] [Google Scholar]
- 9).Dutton AS; Suhrada CP; Miranda KM; Wink DA; Fukuto JM; Houk KN Inorg. Chem 2006, 45, 2448–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Velázquez CA; Chen Q; Citro ML; Keefer LK; Knaus EE J. Med. Chem 2008, 51, 1954–1961. [DOI] [PubMed] [Google Scholar]
- 11).Hong SY; Saavedra JE; Keefer LK; Chakrapani H Tetrahedron Lett. 2009, 50, 2069–2071. [Google Scholar]
- 12).Saavedra JE; Bohle DS; Smith KN; George C; Deschamps JR; Parrish D; Ivanic J; Wang Y; Citro ML; Keefer LK J. Am. Chem. Soc 2004, 126, 12880–12888. [DOI] [PubMed] [Google Scholar]
- 13). The pD value of each solution was taken as pD = pH + 0.4.Keefer LK; Hrabie JA; Hilton BD; Wilbur DJ Am. Chem. Soc 1988, 110, 7459–7462 and Ref. 28 therein.
- 14).Sandstrӧm J Dynamic NMR Spectroscopy; Academic Press: London, 1982; p 78. [Google Scholar]
- 15).Salmon DJ; Torres de Holding CL Thomas L; Peterson KV; Goodman GP; Saavedra JE; Srinivasan A; Davies K,M; Keefer LK; Miranda KM Inorg. Chem 2011, 50, 3262–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Hrabie JA; Klose JR; Wink DA; Keefer LK J. Org. Chem 1993, 58, 1472–1478. [Google Scholar]
- 17).Keefer LK; Nims RW; Davies KM; Wink DA Methods Enzymol. 1996, 268, 281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.