

Abstract
An intrinsically disordered neuronal protein α-synuclein (αSyn) is known to cause mitochondrial dysfunction, contributing to loss of dopaminergic neurons in Parkinson’s disease. Through yet poorly defined mechanisms, αSyn crosses mitochondrial outer membrane and targets respiratory complexes leading to bioenergetics defects. Here, using neuronally differentiated human cells overexpressing wild-type αSyn, we show that the major metabolite channel of the outer membrane, the voltage-dependent anion channel (VDAC), is a pathway for αSyn translocation into the mitochondria. Importantly, the neuroprotective cholesterol-like synthetic compound olesoxime inhibits this translocation. By applying complementary electrophysiological and biophysical approaches, we provide mechanistic insights into the interplay between αSyn, VDAC, and olesoxime. Our data suggest that olesoxime interacts with VDAC β-barrel at the lipid–protein interface thus hindering αSyn translocation through the VDAC pore and affecting VDAC voltage gating. We propose that targeting αSyn translocation through VDAC could represent a key mechanism for the development of new neuroprotective strategies.
Electronic supplementary material
The online version of this article (10.1007/s00018-019-03386-w) contains supplementary material, which is available to authorized users.
Keywords: Voltage-dependent anion channel, VDAC-facilitated protein translocation, Mitochondria, SH-SY5Y cells, Proximity ligation assay, Planar lipid membrane, Channel reconstitution, Voltage gating, Fluorescent correlation spectroscopy
Introduction
One of the pathological hallmarks of Parkinson’s disease (PD) is abnormal accumulation of alpha-synuclein (αSyn), a 14.4-kDa, natively unfolded, neuronal protein into Lewy bodies and Lewy neurites and degeneration of dopaminergic neurons within the substantia nigra [1, 2]. Under normal physiological conditions, αSyn is abundant throughout the brain and mainly in presynaptic terminals of neurons. While αSyn is essentially associated with the central nervous system, various reports detect smaller amounts in peripheral tissues (salivary glands, heart, and muscle) and body fluids [3]. A strong body of genetic evidence including point mutations, genomic duplications, triplications, and polymorphisms in regulatory elements of the encoding SNCA gene has nominated αSyn as a causal gene for both familial and sporadic forms of PD [4]. The alterations in αSyn expression levels, associated with its accumulation into misfolded oligomers and larger aggregates, are proposed to be the main reason for its toxicity towards dopaminergic neurons that are lost in PD. However, the detailed mechanisms by which αSyn is involved in neurodegeneration remain enigmatic. Known pathways are diverse and involve interplay with multiple organelles including synaptic vesicles, ER, Golgi apparatus, lysosomes, and mitochondria [5]. A specific relationship between αSyn toxicity and mitochondrial dysfunction is supported by studies showing intramitochondrial accumulation of the protein and impairment of the mitochondrial respiratory complexes I [6–9], IV [10], and the ATP synthase [11]. However, several important aspects of these functional interactions remain undefined, including identification of the mechanism(s) by which the normally cytosolic αSyn protein gains access to mitochondria.
To date, neuroprotective strategies aimed at slowing progression of PD constitute an unmet clinical need. Recently, olesoxime, an investigational neuroprotective compound, was shown to be protective in a neuronally differentiated human cell model of αSyn-mediated toxicity [12]. This low molecular weight, cholesterol-like compound was originally identified in a medium-throughput screen for rescuing motor neurons from neurotrophic factor deprivation or Fas-induced cell death [13–15]. Gouarne et al. showed that olesoxime-mediated neuroprotection was characterized by reduction of αSyn-induced cell death while preserving mitochondrial integrity and function [12]. The authors associated the pro-survival effects of olesoxime with a decrease in mitochondrial cytochrome c release and inhibition of caspase-9 activation. Olesoxime was found associated with two major proteins of the mitochondrial outer membrane (MOM): translocator protein 18 kDa (TSPO), the mitochondrial cholesterol transporter, and the voltage-dependent anion channel (VDAC) [13, 14]. Our interest in the role of VDAC in the olesoxime-promoted neuroprotection is motivated by our in vitro data showing an interaction of VDAC with monomeric αSyn [16]. We found that VDAC, reconstituted into planar lipid membranes, can be reversibly blocked by recombinant monomeric αSyn with nanomolar efficiency. Using electrophysiological experiments in combination with modeling, we demonstrated that αSyn molecules translocate through the VDAC pore [16–18]. These results lead us to conclude that αSyn-membrane binding is the required first step of αSyn–VDAC interaction controlling both VDAC blockage by αSyn and its translocation through the VDAC pore. We proposed a model where monomeric αSyn bound to the MOM from the cytosolic side by its N-terminus domain could disrupt ATP/ADP fluxes through VDAC by dynamically blocking the pore with its anionic C-terminal domain. Thus, in normal cells αSyn could be a functional regulator of mitochondrial bioenergetics. In pathology, αSyn can cross the MOM barrier by translocating through VDAC and target the electron transport chain (ETC) complexes in the mitochondrial inner membrane (MIM) causing their impairment and leading to mitochondrial dysfunction [16]. Experiments on live cells with a yeast strain deficient in VDAC1 (por1Δ) supported our in vitro findings by demonstrating that αSyn toxicity in yeast depends on VDAC. However, whether or not αSyn–VDAC interaction is a physiological means to regulate cellular respiration and energy production in neuronal cells remained unknown.
Here, using neuronally differentiated SH-SY5Y cells overexpressing wild-type (wt) αSyn as a cell model of PD, we studied the mechanism of olesoxime-induced neuroprotection focusing on the potential role of VDAC as one of the olesoxime mitochondrial targets [13, 14]. Using a combination of confocal imaging and molecular biology approaches, we gathered in situ evidence for αSyn translocation through VDAC into the mitochondria and inhibition of this translocation in the presence of olesoxime. Using complementary in vitro electrophysiology method on VDAC reconstituted in planar lipid membranes, we showed that olesoxime hinders αSyn translocation through the VDAC pore and affects VDAC voltage-gating properties. Our data suggest that olesoxime directly interacts with VDAC β-barrel at the lipid–protein interface. These results identify VDAC as a key player in olesoxime neuroprotection against αSyn-induced mitochondrial toxicity.
Materials and methods
Cell-culture conditions and transfection
Human neuroblastoma SH-SY5Y cells were bought from the American Tissue Culture Collection (Manassas, VA) and grown in RPMI 1640 (12-702F, Gibco) medium supplemented with 10% fetal bovine serum (ThermoFisher Scientific). Cultures were maintained at 37 °C in a humidified atmosphere with 5% CO2. Constructs for transient expression of αSyn were built up by insertion of wt human αSyn coding sequence into the pcDNA™ 3.1 vector. SH-SY5Y cells were transiently transfected with either an empty plasmid or plasmid containing wt αSyn using Lipofectamine™ 3000 transfection reagent (Invitrogen) following manufacturer’s instruction and a ratio of 0.8 μg of DNA per 100,000 cells. At the same time, differentiation was induced for all conditions by the addition of 20 μM all-trans retinoic acid (RA; Sigma-Aldrich). Cells were treated with 3 μM olesoxime (TRO 19622, Tocris Bioscience) in 0.5% final DMSO (Sigma-Aldrich) (control cultures received DMSO only). For all experiments, the entire medium was changed every 2 days and all reagents were replaced at the same concentration.
siRNA transfection
Cells were transfected with either the VDAC1 On-Target Plus SMART pool siRNA or siGenome Non-targeting siRNA as a control (Dharmacon, Waltham, MA) at a final concentration of 5 nM, using DharmaFECT™ 1 as per the manufacturer’s instructions (GE Healthcare). Media was renewed 24 h post transfection, with cells typically incubated for a further 24–48 h before harvesting/analyzing.
Trypan-blue exclusion test of cell viability
To evaluate αSyn cytotoxicity following transient transfection, cell viability was measured by the trypan-blue exclusion assay 96 h after transfection. After the addition of 0.4% trypan blue for 2 min (Sigma), the percentage of dead cells (trypan-blue-stained) was counted manually with a hemocytometer, within 5 min.
Live cell imaging of mitochondrial membrane potential
For all confocal imaging, cells were grown on 35 mm MatTek dishes (MatTek corporation). Mitochondrial membrane potential (ΔΨm) was assessed by loading cells with 100 nM MitoTracker™ Red CMXRos (Cell-Signaling Technology) for 25 min. After the incubation, the fluorescent probe was washed out with Hank’s balanced salt solution. The cells were placed in a thermostated chamber on the stage of a Zeiss LSM 710 confocal microscope and images were taken with a 63 × 1.4 N.A. Plan-Apochromat. Image analysis and quantification of the fluorescence intensity were performed with ImageJ (NIH) as described in [19], and averaged for five independent experiments.
Proximity ligation assay (PLA)
Cells were grown, transfected, and differentiated on Nunc™ Lab-Tek™ II CC2™ Chamber Slide System (ThermoFisher scientific). PLA was carried out with Duolink® In Situ Detection Reagents Orange (Sigma-Aldrich) with little modifications from the manufacturer’s instructions, as reported in [20]. The cells were treated with primary antibodies for anti-αSyn (Cell-Signaling Technology, 1:100, rabbit) and anti-VDAC1 (Abcam, 1:100, mouse) or anti-COXIV (Cell-Signaling Technology, 1:100, mouse). Slides were mounted using a minimal volume of Duolink in situ Mounting Medium containing DAPI and images were taken with a Zeiss LSM 710 confocal microscope at 63 × magnification. The microscope settings were kept constant for all images to enable direct comparison. Quantification of signals (number of dots per cell) was obtained from thresholded images using the “analyze particles” feature of ImageJ, which detects isolated continuous objects in the image. Neither PLA puncta size nor circularity was constrained for detection.
Western blotting
Proteins were extracted in cell lysis buffer (Cell-Signaling Technology) with additional phosphatase and protease inhibitors (Sigma). Proteins were separated on 4–20% Criterion TGX pre-cast gels (Biorad) in SDS/Tris–glycine running buffer and transferred to PVDF membranes by semi-dry trans-Blot Turbo transfer system (Biorad). The membranes were blocked for 1 h with Odyssey Blocking Buffer (Licor) and then incubated for 1 h at room temperature (RT) with antibodies: anti αSyn (Cell-Signaling Technologies, 1:1000, rabbit), anti-VDAC1 (Abcam, 1:2000, mouse), anti-GAPDH (Sigma, 1:2000, rabbit), and anti-β-actin (Santa Cruz, 1:1000, mouse). The membranes were washed in TBST (3 × 5 min) at RT followed by incubation for 1 h at RT with fluorescently conjugated goat anti-mouse or rabbit IR Dye 680 or 800 antibodies (Licor). The blots were washed in TBST (3 × 5 min) at RT and scanned on an ODYSSEY® CLx (Licor). Quantitation of western blots was performed using Image Studio (Licor), and the intensity of target proteins was standardized with the loading control.
Materials and protein purification
1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), cholesterol (Chol), and a soybean Polar Lipid Extract (PLE) were purchased from Avanti Polar Lipids (Alabaster, AL). Olesoxime was purchased from Tocris. All other chemicals were obtained from Sigma-Aldrich unless noted otherwise.
Recombinant wt αSyn and Alexa-488-labeled αSyn were the generous gifts of Dr. Jennifer Lee (NHLBI, NIH). αSyn was expressed, purified, and characterized as described previously [21] and stored at – 80 °C. αSyn Y136C mutant was labeled with fluorophore Alexa488 at position Y136C as described previously [22]. VDAC was isolated from frozen mitochondrial fractions of rat liver that were a kind gift of Dr. Marco Colombini (University of Maryland, College Park, MD) following the standard method [23] and purified on a hydroxyapatite/Celite (2:1) column as described previously [24]. VDAC purified from mitochondrial fraction of rat liver contains all three isoforms with VDAC1 being the predominant one (~ 80% of total VDACs) [25].
Channel reconstitution and conductance measurements
Planar lipid membranes for single-channel experiments were formed from DOPC/DOPE (1:4 w/w) mixtures with or without 5% (w/w) of olesoxime or cholesterol. For multichannel experiments, planar membranes were prepared from PLE with or without 20% olesoxime (w/w) or from PLE with 4% cholesterol (w/w) and with or without 16% olesoxime (w/w). The mixtures of lipids were prepared from 10 mg/ml aliquots of two lipids, cholesterol or olesoxime solutions in chloroform, followed by drying with nitrogen and then re-dissolving them in pentane to a total lipid concentration of 5 mg/ml. Planar bilayer membranes were formed from two opposing lipid monolayers across ~ 70 μm aperture in the 15-μm-thick Teflon partition separating two ~ 1.2-mL compartments as previously described [26]. VDAC insertion was achieved by adding VDAC isolated from the mitochondria of rat liver in 2.5% triton X-100 buffer to the aqueous phase of 1 M KCl buffered with 5 mM Hepes at pH 7.4 in the cis compartment. Potential is defined as positive when it is greater at the side of VDAC addition (cis). αSyn at a final concentration of 50 nM was added symmetrically to both sides of the membrane. Records for analysis were obtained no less than 15 min after αSyn addition to ensure a steady state. Single-channel conductance measurements were performed as described previously [16] using an Axopatch 200B amplifier (Axon Instruments, Inc., Foster City, CA) in the voltage clamp mode. Data were filtered by a low-pass 8-pole Butterworth filter (Model 900, Frequency Devices, Inc., Haverhill, MA) at 15 kHz and a low-pass Bessel filter at 10 kHz, and directly saved into computer memory with a sampling frequency of 50 kHz. For single-channel data analysis by Clampfit 10.3, a digital 8-pole Bessel low-pass filter set at 5 kHz or 2 kHz was applied to current recordings, and then, individual events of current blockages were discriminated and kinetic parameters were acquired by fitting single exponentials to logarithmically binned histograms [27] as described previously [16, 28]. Four different logarithmic probability fits were generated using different fitting algorithms and the mean of the fitted time constants was used as the mean for the characteristic open and blockage times. Each channel experiment was repeated at least three times on different membranes.
VDAC voltage-dependent properties were assessed on multichannel membranes following the protocol previously devised [29–31] in which gating is inferred from the channels response to a slow symmetrical 5 mHz triangular voltage wave of ± 60 mV amplitude from an Arbitrary Waveform Generator 33220A (Agilent). Data were acquired at a sampling frequency of 2 Hz and analyzed as described previously [30, 31] using pClamp 10.3 software. In each experiment, current records were collected from membranes containing 20–850 channels in response to 5–10 periods of voltage waves. Only the part of the wave during which the channels were reopening was used for the subsequent analysis.
Statistics
Differences between groups were analyzed by a one-way ANOVA using p < 0.05 as the criterion of significance. Data points are the mean ± SD of 3–6 independent experiments or presented as box plots. Images in figures are representative of three or more independent experiments.
Results
Olesoxime attenuates αSyn-induced loss of mitochondrial membrane potential
Olesoxime neuroprotective capacity against αSyn-induced mitochondrial toxicity was studied using retinoic acid differentiated SH-SY5Y cells overexpressing αSyn. Overexpression was confirmed by western blot and immunostaining which also demonstrated the lack of effect of olesoxime on αSyn expression (Supplemental Fig. S1). As seen in Supplemental Fig. S1C, enhanced level of αSyn resulted in a punctate staining of the protein expanded all over the cytoplasm. Under these conditions, no aggregates were detected. The toxicity of αSyn and the neuroprotection of olesoxime were confirmed in this cell model by assessing morphological features and cell survival parameters. Figure 1a shows morphological alterations associated with overexpression of wt αSyn, while olesoxime treatment maintained a normal phenotype, as previously described [12]. By counting the number of trypan-blue excluded cells, we determined that αSyn overexpression resulted in an average of 21.2 ± 4.3% dead cells (Fig. 1b, αSyn), a significant increase of cell death (p < 0.01, here and elsewhere, by one-way ANOVA) when compared with the 1.5 ± 0.7% of dead cells in basal conditions (Fig. 1b, pEV). The addition of 3 µM olesoxime to αSyn overexpressing cells caused a significant decrease, down to 6.9 ± 5.8% (p < 0.01) of dead cells (Fig. 1b, αSyn + Oxime), thus confirming olesoxime’s protective effect. Importantly, 3 µM olesoxime alone is not toxic to the cells (Fig. 1a, b, Oxime).
Fig. 1.

Olesoxime preserves mitochondrial integrity in a cell model overexpressing αSyn. a Phase contrast representative images (× 20 objective, scale bar 100 µm) show morphological alterations such as reduced processes (arrow heads) and rounded floating cells (stars), following transient transfection of wt αSyn in differentiated SH-SY5Y cells. Olesoxime treatment restores cell morphology. b Box plot showing quantification of cell death assessed 96 h after transfection by trypan-blue exclusion assay. Here and in Figs. 2b, c and 3d, the boundary of the box closest to zero indicates the 25th percentile, a line within the box marks the median, and the boundary of the box farthest from zero indicates the 75th percentile. Error bars indicate SD (n = 4). c Representative images of cells loaded with MitoTracker™ CMXRos to evaluate mitochondrial membrane potential for which the relative fluorescence intensity changes are quantified as ratios over pEV control in d (× 63 objective, scale bar 20 µm). In d, data are presented as mean ± SD (n = 4). In b, d, NS (not significant): p > 0.5; **p < 0.01; one-way ANOVA
To assess the effect of olesoxime on mitochondria function in αSyn overexpressing cells, we evaluated mitochondrial membrane potential using the cell permeant MitoTracker™ probe which passively diffuses across the cell membrane and accumulates in energized mitochondria [32]. Analysis of the MitoTracker™ fluorescence showed that the basal fluorescence intensity of αSyn overexpressing cells is significantly lower (46 ± 15%, p < 0.05) than in control cells (Fig. 1c, d, pEV, normalized to 100%). Olesoxime treatment of αSyn overexpressing cells significantly rescued the basal fluorescence intensity (88 ± 5% vs 46 ± 15%, p < 0.05), while cells treated with olesoxime alone had basal fluorescence intensity similar to control cells (103 ± 18%, p > 0.5) (Fig. 1d). These results confirm a key role of mitochondria in olesoxime neuroprotection against αSyn-mediated toxicity.
In cells, olesoxime reduces the αSyn association with mitochondrial membrane proteins
αSyn has been found to associate with both outer and inner mitochondrial membranes [7, 33–35]. However, the pathway by which αSyn enters mitochondria has not been identified. Recent in vitro data suggest that VDAC could be such a pathway for αSyn to cross the MOM [16]. Although αSyn binding to and translocation through VDAC have been extensively studied biophysically [16–18], a visual assessment of this model in situ in cells has never been done. The use of conventional confocal microscopy approaches for such a task is challenging due to high expression levels of both αSyn in the cytosol and VDAC in the MOM. To overcome these limitations, we performed an in situ Proximity Ligation Assay (PLA), a method that allows to visualize the presence of two proteins at close distances with a resolution up to 30 nm [36].
To evaluate the effect of olesoxime on αSyn association with mitochondrial membranes, we used PLA to assess proximities between αSyn and VDAC and between αSyn and complex IV (COX IV), as markers of the MOM and MIM, respectively. Analysis of confocal images of cells after PLA showed that overexpression of αSyn correlates with a significant enrichment of αSyn found in close proximity to VDAC at the MOM (Fig. 2a, upper row). Quantification of PLA signals for pairwise αSyn/VDAC antibodies revealed a ~ 3.5 times increase of the average number of αSyn in close proximity to VDAC relative to the basal conditions (pEV) (Fig. 2b). To provide further evidence of αSyn accumulation at the MOM, we performed an additional PLA for αSyn and the translocase of the outer membrane 20 (TOM20). Using this other marker of the MOM, we found a significant increase (p = 0.01) in PLA signals for αSyn and TOM20 (Supplemental Fig. S2). Olesoxime treatment of cells overexpressing αSyn significantly decreased the αSyn and VDAC co-localization (Fig. 2a, b). However, olesoxime treatment did not affect association of endogenous αSyn with VDAC in non-transfected cells (NS; p > 0.5) (Fig. 2a, b). These results indicate that olesoxime can diminish the co-localization of αSyn and VDAC. Next, using a pair of αSyn/COX IV antibodies, we found a ~ 3 times increase of the average number of αSyn molecules in a close proximity to COX IV at the MIM (Fig. 2a, lower panels and Fig. 2c) in αSyn overexpressing cells. Olesoxime treatment of cells overexpressing αSyn resulted in a significant reduction of αSyn co-localization with COX IV (p < 0.05). Olesoxime per se did not affect association of endogenous αSyn with COX IV (Fig. 2c). Collectively, these results show that olesoxime significantly reduces the association of αSyn with both inner and outer mitochondrial membranes proteins.
Fig. 2.

Effect of olesoxime on αSyn localization at the mitochondrial membranes. a Representative confocal images of PLA showing αSyn in close proximity (~ 30 nm) to VDAC at the MOM and with Complex IV (COX IV) at the MIM in cells overexpressing αSyn before and after olesoxime treatment. The nuclei are stained in blue (DAPI) (× 63 objective, scale bar 20 µm). Controls omitting the primary antibodies are shown for each pairwise combination. b, c Box plots represent the normalized distribution of signals per cell, obtained from an average of 160 cells per experiment with a minimum of 60. Error bars indicate SD. The PLA signals were normalized to the average PLA values in pEV control cells of the corresponding experiment. The dashed lines indicate pEV controls. At least 3 independent experiments were performed for each pairwise of antibodies. Unless indicated by brackets, significance was tested against the corresponding pEV controls (**p < 0.01, *p < 0.05, NS (not significant): p > 0.5; one-way ANOVA)
VDAC1 is involved in αSyn translocation into mitochondria
The finding that αSyn localizes in a close proximity (~ 30–40 nm) to VDAC and COX IV is suggestive of a direct interaction with these two proteins, but does not provide definitive evidence in either case (Fig. 2) and does not show whether VDAC forms a pathway for αSyn translocation across MOM. As the current knowledge indicates VDAC1 as the major docking site at the MOM for misfolded proteins involved in neurodegenerative diseases [37], to further evaluate the role of VDAC in αSyn mitochondrial localization, VDAC1 expression was downregulated in differentiated SH-SY5Y cells using siRNA. Western blot in Fig. 3a, b shows 85% decrease of VDAC1 protein after siRNA treatment. PLA was then used to monitor the ability of αSyn to cross MOM and target COX IV in VDAC1 depleted cells. Under these conditions, we found that overexpression of αSyn did not result in a significant difference (1.3 ± 1.2; p ≥ 0.4) in PLA signal for αSyn/COX IV co-localization (Fig. 3c, d) in contrast with threefold increase of αSyn/COX IV co-localization in cells with endogenous VDAC1 (Fig. 2c, d). Olesoxime treatment of these cells did not affect PLA signals (Fig. 3d). Altogether, in situ results show that in cells overexpressing αSyn, VDAC1 is important for αSyn translocation into mitochondria.
Fig. 3.

αSyn co-localization with COX IV at the inner membrane is lost upon downregulation of VDAC1 expression. a Western blot of VDAC1 protein levels in differentiated SH-SY5Y cells transfected with a non-targeting sequence (NTC) siRNA or VDAC1–siRNA. GAPDH was used as a loading control and maintenance of αSyn overexpression under these conditions has been validated. b Quantification of the Western blot analysis as intensity ratios of VDAC1/GAPDH. Data are presented as mean ± SD (n = 3). In VDAC1–siRNA cells, the VDAC1/GAPDH ratio is significantly less than in NTC control. c Representative confocal images of PLA of VDAC1 and COX IV co-localization in controls and cells depleted of VDAC1, overexpressing αSyn alone or treated with olesoxime. The nuclei are stained in blue (DAPI) (× 63 objective, scale bar 20 µm). d Box plot represents the normalized distribution of PLA signals per cell, obtained from an average of 180 cells per experiment with a minimum of 60. The PLA signals were normalized to averaged PLA value measured in the NTC control cells of the corresponding experiment. The dashed line indicates a normalized PLA signal in NTC cells. Error bars indicate SD. At least 3 independent experiments were performed for each pairwise of antibodies. In b, d, **p < 0.01; NS (not significant): p ≥ 0.2; one-way ANOVA
Olesoxime prevents αSyn translocation through VDAC
The decrease of αSyn co-localization with VDAC and COX IV in cells treated with olesoxime could be explained by various mechanisms. One explanation is based on the previously shown direct interaction between olesoxime and VDAC using competition between olesoxime and photolabeled analog of pregnanolone for VDAC [14]. Therefore, we hypothesized that olesoxime binding to VDAC could interfere with αSyn–VDAC interaction and αSyn translocation through the channel. To assess this hypothesis, we employed electrophysiological measurements on VDAC channels reconstituted into planar lipid membranes. This method enables highly sensitive measurements of αSyn’s dynamic interaction with VDAC at the single-molecule level [16]. Using VDAC electrophysiology, we recently reported that αSyn is able to reversibly block channel conductance in a concentration- and highly voltage-dependent manner. By measuring ionic selectivity of αSyn-blocked state in salt gradient conditions, we showed that the presence of the anionic C-terminal domain of αSyn in the pore shifts VDAC’s selectivity towards more cationic, while the uncharged N-terminal region leaves it relatively unaffected [17]. Using an experimental approach based on this observation, we established a method for direct, model-free, real-time monitoring of the translocation of αSyn polypeptide through the VDAC pore [18]. Therefore, our data imply that αSyn is able to translocate through the channel [16–18]. We capitalized on this earlier established system and compared the effects of olesoxime on VDAC basic channel properties and on the kinetic parameters of αSyn-induced VDAC blockages with the effect of cholesterol as a relevant control for the cholesterol-like olesoxime molecule.
Two zwitterionic lipids, DOPC and DOPE, were chosen to mimic MOM phospholipid composition, where PC and PE make up to 44 and 35% of the total lipid content of the rat liver MOM, respectively [38]. A natural content of cholesterol in mammalian MOM is estimated as ~ 5–7% (w/w) of total phospholipid [39, 40]. VDAC was reconstituted into planar membranes formed from DOPC/DOPE (1:4 w/w) mixture with 5% (w/w) of olesoxime or cholesterol or without them. Figure 4a shows representative current traces through a single VDAC reconstituted into planar membranes formed from PC/PE with olesoxime (i), with the equal amounts of cholesterol (ii), or without either of them (iii). Before the addition of αSyn, a typical ion current through VDAC’s high conducting or “open” state is steady and corresponds to 4 nS conductance in all three lipid compositions (in 1 M KCl solution) [29, 41] (Fig. 4a, left traces), showing that neither cholesterol nor olesoxime affect VDAC open-state conductance. The addition of 50 nM of αSyn to the membrane-bathing solution resulted in rapid and well-resolved millisecond range current fluctuations between the open state and the lower conducting αSyn-blocked state (Fig. 4a, right traces), which is a characteristic effect of αSyn on reconstituted VDAC [16]. Importantly, the current corresponding to the open state did not change in the presence of αSyn indicating the absence of any pore-forming activity of αSyn at this concentration [16]. Each individual blockage event corresponds to a steric blockage of the VDAC pore by individual membrane-bound αSyn molecule [17]. The addition of olesoxime to the lipid membrane did not visibly affect the general pattern of the characteristic current blockage behavior induced by αSyn (i). Therefore, olesoxime does not prevent αSyn from blocking VDAC pore. However, the kinetic analysis of blockage times, the intervals of time when channel is blocked by αSyn, revealed a strikingly different behavior in the presence of olesoxime in comparison with (PC/PE/Chol) or without cholesterol (PC/PE). The blockage times obtained in the presence of olesoxime (PC/PE/Oxime) increased exponentially with the amplitude of the applied voltage up to − 47.5 mV (Fig. 4b). In contrast, the voltage dependences obtained in PC/PE and PC/PE/Chol membranes showed a clear biphasic behavior: blockage time increased exponentially up to ~ − 35 mV (indicated by the vertical dashed line in Fig. 4b) and then markedly decreased at |V| > 35 mV (highlighted in yellow in Fig. 4b).
Fig. 4.
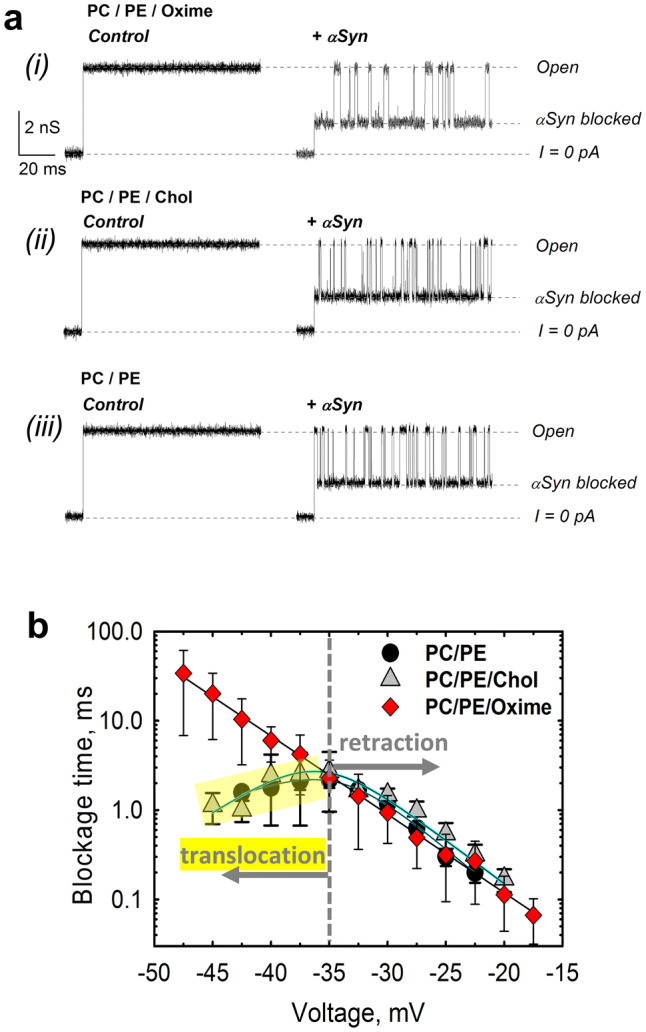
Olesoxime prevents αSyn translocation through reconstituted VDAC. a Representative ion current records of the single VDAC channel reconstituted into a planar lipid bilayer formed from (i) PC/PE (1:4 w/w) mixture with 5% (w/w) of olesoxime (PC/PE/Oxime) before (left column) and after (right column) addition of 50 nM of αSyn to the cis compartment were obtained on the same VDAC channel. The characteristic blockages of VDAC conductance induced by αSyn are seen in trace in the right column. Traces (ii) and (iii) were obtained in the membrane composed of PC/PE (1:4) with 5% (w/w) cholesterol (PC/PE/Chol) (traces ii) and PC/PE (1:4) (traces iii) before (left column) and after addition of 50 nM αSyn (right column). All records were taken at -40 mV applied voltage. The membrane-bathing solutions contained 1 M KCl buffered with 5 mM HEPES at pH 7.4. Dashed lines indicate VDAC open and αSyn-blocked states and zero current. All current records were smoothed with a 5 kHz low-pass Bessel digital filter using pClamp 10.3. b Voltage dependences of mean blockage times of αSyn-induced blockages of VDAC reconstituted into planar membranes made of PC/PE (1:4) with 5% (w/w) of olesoxime or cholesterol or in pure PC/PE (1:4) membranes. The increase of blockage time with absolute amplitude of the applied voltage corresponds to the pore blockage regime; a decrease of blockage time with voltage amplitude (highlighted in yellow for PE/PC and PE/PC/Chol membranes) corresponds to regime of αSyn translocation through the pore. In the presence of olesoxime, translocation of αSyn is not observed at applied voltages up to − 48 mV, while in PC/PE membranes with or without cholesterol, the translocation regime starts at − 35 mV. Data points and error bars represent the mean and SD for 4–6 independent experiments
As shown previously [16–18], the increase of blockage time with the applied voltage corresponds to the regime of reversible VDAC blockage when the acidic C-terminal of αSyn is captured and retracted from the pore, while the αSyn molecule remains at the same side of the channel (Fig. 6). The decrease of dwell time at higher potentials observed in PC/PE or PC/PE/Chol membranes is indicative of the translocation regime when the applied voltage drives the entire αSyn molecule through the pore. This kind of behavior has been demonstrated for polypeptides and DNA translocation through various nanopores (e.g., [42, 43]). Importantly, both voltage dependences obtained on PC/PE membranes with or without cholesterol are overlapping, confirming that cholesterol does not affect either blockage or translocation regimes of αSyn through VDAC. This is drastically different from the exponential increase of blockage times with the voltage amplitude in the olesoxime-containing membranes (Fig. 4b).
Fig. 6.

Proposed model of olesoxime neuroprotective effect. When αSyn is captured by the VDAC pore it disrupts ATP/ADP fluxes through VDAC. Under normal conditions endogenous αSyn regulates these fluxes by reversibly and dynamically blocking VDAC. Under αSyn overexpression-induced stress αSyn translocates across MOM through VDAC and targets ETC in the MIM causing their impairment, mitochondrial dysfunction and eventually neuronal cell death. Olesoxime partitions into the MOM and hinders αSyn translocation through VDAC by interacting with the pore–lipid interface. This model suggests a tentative mechanism of olesoxime protection of mitochondria integrity and promotion of neuronal cell survival
Complementary experiments were performed to confirm the effect of olesoxime on the translocation regimes of αSyn through VDAC. To that end, olesoxime was added to the membrane-bathing solution after recording of αSyn-induced blockages of VDAC inserted into PC/PE/Chol membranes (Supplemental Fig. S3 A, traces a and b). Successive additions of olesoxime (from 10 µM up to a final concentration of 100 µM; Supplemental Fig. S3 A, traces c and d) resulted in a significant increase of blockage events duration (at high applied voltages) following addition of olesoxime, which is indicative of olesoxime interference with αSyn translocation (Supplemental Fig S3B).
Measurable blockage of the VDAC pore occurs when negative potential is applied from the side of αSyn addition [16]. αSyn blocks VDAC in qualitatively similar way from both sides of the channel [16]. Hence, for the sake of clarity, we present results for the blockage from the presumably cytosolic side of the channel which corresponds to the “cis” side of our experimental chamber or to the side of VDAC addition [44]. Considering αSyn’s cytosolic localization in cells, its interaction with cytosolic side of the channel is physiologically most relevant.
Olesoxime affects VDAC voltage gating
These electrophysiological results suggest that olesoxime delays αSyn translocation through the VDAC pore and, consequently, prevents αSyn from targeting the ETC at the MIM. The absence of cholesterol effect on αSyn–VDAC interaction points toward a specific interaction of olesoxime with VDAC that modifies the channel or its immediate lipid environment in a way that prevents αSyn translocation at |V| < 50 mV. To further test this hypothesis using reconstituted VDAC, we examined the effect of olesoxime on VDAC voltage gating, an important VDAC’s property that allows assessing VDAC’s functions [23, 29–31]. Quantification of VDAC gating requires a different experimental approach that involves membranes with multiple reconstituted channels [29, 30, 45].
Voltage gating, which is the voltage-triggered switching between high and low conducting states, the so-called “open” and “closed” states [29], is a characteristic property of VDAC and its namesake. VDAC voltage gating is known to be sensitive to the surrounding lipid environment [30, 46, 47], pH [31, 48], and osmotic stress [49]. To achieve reliable statistical data on VDAC gating which is an inherently stochastic process, we used the well-established experimental approach, where gating is inferred by monitoring the response of multiple channels to triangular voltage waves [29, 30]. The periodic voltage wave is chosen slow enough (5 mHz, corresponding to a period of 200 s) to ensure that the channel reopening is much faster (milliseconds range), so that the obtained results do not depend on the time parameters of the voltage protocol used [45].
Figure 5a shows the normalized conductance plots (G/Gmax, where G is the conductance at a given voltage and Gmax is the maximum conductance at voltages close to 0 mV) vs the applied voltage obtained for the system of multiple VDAC channels reconstituted into the membranes formed from soybean Polar Lipid Extract (PLE), with cholesterol (PLE/Chol), and with olesoxime (PLE/Oxime). PLE is a natural lipid mixture which was chosen for these experiments, as it ensures a pronounced VDAC gating in electrophysiological experiments [47] and also closely mimics MOM phospholipid composition in rat liver mitochondria, including the content of charged lipids and acyl chains [38]. This condition is important, because VDAC gating was shown to be especially sensitive to the lipid headgroup charges [47]. All G/Gmax plots display the characteristic bell-shaped voltage dependences of the normalized conductance (Fig. 5a) with some asymmetry with respect to the polarity of the applied voltage: the minimum conductance Gmin, calculated at |V| > 50 mV, is slightly lower at negative than at positive voltages. The addition of olesoxime (20% w/w in the PLE) enhanced VDAC gating by decreasing Gmin. This decrease was significant at the negative applied voltages (~ 8% decrease of Gmin in comparison with control) (Fig. 5a). The effect of olesoxime on VDAC gating was even more pronounced when olesoxime was added to the PLE membranes with 4% of cholesterol (w/w) (Fig. 5b, showing the data with or without olesoxime). The olesoxime content in experiments with PLE/Chol/Oxime membranes was reduced to 16% w/w to keep the total amount of cholesterol and olesoxime at the same level as in experiments shown in Fig. 5a (20%). In PLE/Chol/Oxime membranes, Gmin decreased by ~ 15% at negative voltages in comparison with Gmin obtained in experiments without olesoxime. Importantly, cholesterol, in contrast to olesoxime, does not affect VDAC gating (Fig. 5a) [47].
Fig. 5.
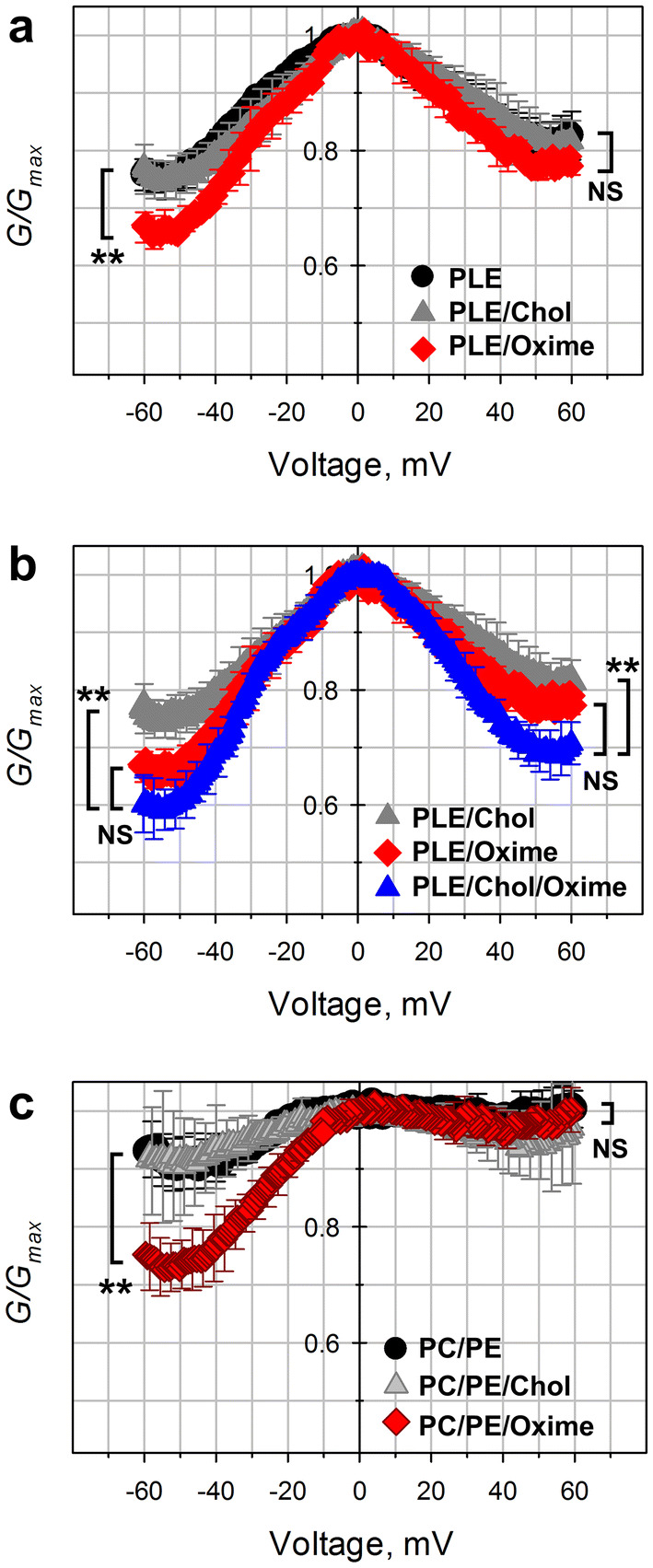
Olesoxime increases VDAC voltage gating. Characteristic bell-shaped plots of normalized average VDAC conductance as function of the applied voltage obtained on planar bilayers of different lipid compositions. a PLE with olesoxime (20% w/w) (PLE/Oxime) or cholesterol (4% w/w) (PLE/Chol) or without either of them (PLE). b PLE with cholesterol (4% w/w) (PLE/Chol) or olesoxime (20% w/w) (PLE/Oxime) or both (4% w/w and 16% w/w, respectively) (PLE/Chol/Oxime). c PC/PE (1:4) with cholesterol (10% w/w) (PC/PE/Chol) or olesoxime (10% w/w) (PC/PE/Oxime) or without either of them (PC/PE). Data were obtained on multichannel membranes with 10–200 reconstituted VDAC channels. Normalized conductance is defined as G/Gmax, where Gmax is the maximum conductance at voltages close to 0 mV. Membrane-bathing solutions consisted of 1 M KCl buffered with 5 mM HEPES at pH 7.4. Data are means of 3–5 experiments ± SD. (error bars shown every 5 points for clarity). To check significance (**p < 0.01, NS (not significant): p > 0.05; one-way ANOVA), the minimum G/Gmax value of each data set was used, corresponding to |V| ~ 52 mV. Each voltage polarity was tested independently
Next, we tested the effect of olesoxime on VDAC gating in the membranes made of the same lipid compositions as those used in single-channel experiments in Fig. 4. In these experiments, the addition of olesoxime (5% w/w) to the DOPC/DOPE (1:4 w/w) mixture significantly decreased closed-state conductance (**p < 0.01; one-way ANOVA) at negative potentials, while cholesterol did not affect gating (Fig. 5c), confirming the results in Fig. 5a, b.
Olesoxime does not measurably change mechanical properties of lipid membranes
The results with reconstituted VDAC suggest that olesoxime modulates VDAC gating either by directly interacting with VDAC or, alternatively, indirectly by changing integral properties of lipid bilayer. Considering a large amount of olesoxime (5–20%) used in the gating experiments, one could expect a change of bilayer properties based on the previously reported data that olesoxime concentrates on mitochondrial membranes and modulates mitochondrial membrane fluidity [50, 51]. Therefore, we tested the possibility that olesoxime changes the integral properties of the lipid bilayer using two independent biophysical approaches. We first employed a channel-forming polypeptide, gramicidin A (gA), as a well-recognized highly sensitive molecular probe of planar lipid bilayer mechanics [28, 30, 52–54]. The advantage of using gA for such a task is that it allows the use of exactly the same system of planar lipid membranes as in the experiments with reconstituted VDAC. Several studies have demonstrated that the lifetime of a gA-conducting dimer is exquisitely sensitive to the bilayer thickness, lipid-packing stress, bilayer curvature, etc. [30, 52, 54, 55]. In particular, it was demonstrated that cholesterol causes a ~ 10 times reduction of gA lifetime due to the increase in the lipid-packing stress [52] or membrane stiffness [55]. In contrast with drastic cholesterol effect, we did not find a significant effect of olesoxime addition (20% w/w) to the DOPE/DOPC membranes on gA lifetime (Supplemental Fig. S4), which suggests that olesoxime does not change appreciably the lipid bilayer mechanical properties under our experimental conditions. Therefore, we conclude that olesoxime most likely affects VDAC gating by directly interacting with the channel.
We then tested a possibility of olesoxime to influence αSyn-membrane binding using Fluorescence Correlation Spectroscopy (FCS). The interaction of αSyn with liposome membranes was assessed using Alexa488-labeled αSyn and large unilamellar vesicles (LUVs) (Supplemental Methods). We found that FCS correlation functions obtained with the same concentration of total lipid in LUVs prepared from DOPC/DOPE and DOPC/DOPE with 10% of olesoxime or cholesterol overlap with each other, indicating that neither olesoxime nor cholesterol affect membrane binding of αSyn (Supplemental Fig. S5 A). To quantify αSyn binding to the membranes with different lipid compositions, we followed the approach described in [22]. The analysis of FCS correlation functions showed that there is no significant difference between LUVs containing olesoxime or the same amount of cholesterol (Supplemental Fig. S5 B). This suggests that olesoxime does not affect αSyn-membrane binding.
Discussion
The relationship between mitochondrial dysfunction and αSyn is central to the understanding of PD pathogenesis and, therefore, could be instructive for the development of neuroprotective compounds. Based on evidence of mitochondrial function protection [13, 50, 51, 56–59], the experimental compound olesoxime has recently been assessed for its potential neuroprotective effect in a human cell model of αSyn toxicity [12]. In this model, olesoxime favored cell survival leading to the hypothesis that its protective effect is exerted through preservation of mitochondrial function. In the current work, we have chosen olesoxime for its protective properties but also as a tool to investigate the mechanism of αSyn-induced mitochondrial toxicity.
Here, we first confirmed olesoxime neuroprotective potential in our cell model and then assayed its effect on mitochondrial function. We found a ~ 60% loss of the MitoTracker™ fluorescence intensity in differentiated SH-SY5Y cells overexpressing αSyn (Fig. 1c, d). This is in line with the previous studies conducted on cell lines or transgenic mice overexpressing αSyn [9, 60–64], as well as in recent models of dopaminergic neurons derived from PD patients [65]. We found that olesoxime significantly protects against αSyn overexpression-induced dissipation of the mitochondrial membrane potential (Fig. 1c, d), thus confirming olesoxime’s protective effects in our cell model. Mechanisms underlying a disturbance of mitochondrial membrane potential include insufficient ETC activity or uncoupled oxidative phosphorylation. As a matter of fact, detrimental effect of αSyn on ETC components, in particular complex I [6, 7, 66] and complex IV [10, 67], and lately ATP synthase [11, 65], has been consistently reported (most recently reviewed in [68]).
Recently, we characterized the interaction between αSyn and VDAC using VDAC reconstituted into planar lipid membranes and proposed a three-step model in which αSyn binding to the membrane is a necessary pre-requisite to interaction with VDAC. According to this model [16–18], VDAC can serve as a gateway for αSyn translocation across the MOM into the mitochondrial intermembrane space. Interaction of αSyn with VDAC has been reported in various cell and animal models of PD (reviewed in [37]) using co-localization or co-immunoprecipitation approaches. In the present work, using VDAC and COX IV as markers for MOM and MIM, respectively, we found that the overexpression of αSyn significantly increases αSyn accumulation in close proximity to VDAC in the MOM and to COX IV in the MIM (Fig. 2). These data confirm, first, αSyn association with both mitochondrial membranes as shown previously [7, 33, 35], and, second, αSyn ability to translocate across MOM, in accordance with our model. We further verified the role of VDAC as a gateway for αSyn by detecting a significant loss of αSyn localization at the MIM in VDAC1 downregulated cells (Fig. 3). The ~ 2 times decrease in normalized αSyn/COX IV PLA signal in VDAC1 SiRNA cells (Fig. 3d) compared to cells with endogenous VDAC1 (Fig. 2c) after olesoxime treatment supports our hypothesis that VDAC is involved in αSyn ability to reach MIM. However, these data play rather supportive than conclusive role in verifying this model. These results are in accord with the proposed model and are in line with our previous study showing that αSyn toxicity in yeast depends on VDAC expression [16].
Our findings are of particular interest because of the current lack of consensus regarding the molecular identity of the MOM pathway for αSyn. αSyn has been shown to bind to different subunits of the translocases of the outer membrane complex (TOM20, TOM22, TIM23, TOM40), among which TOM40 has been reported to participate in αSyn import to the matrix [7]. To test this possibility, we performed experiments with the recombinant TOM40 channel reconstituted into planar membranes and found that αSyn, up to 100 nM, does not affect TOM40 conductance (Supplemental Fig. S6). This result is in striking contrast with established αSyn–VDAC interaction [16, 22], but is not surprising, because TOM40 is a highly cation selective channel [69–71]. Besides, TOM complex recognizes specific mitochondrial sequences that are not present on αSyn. Therefore, VDAC remains as the most plausible candidate for MOM translocator of αSyn.
Since its identification, olesoxime has been described as a mitochondrial-targeting compound, because it selectively binds to two important components of the MOM, TSPO, and VDAC. While phenotypical observation of mitochondrial protection was confirmed in various models of neurodegeneration [50, 51, 56], a VDAC-related molecular mechanism has not been explored. We took advantage of VDAC being a potential target to both αSyn and olesoxime to understand the role of VDAC in olesoxime neuroprotection. The PLA experiments showed a significant decrease of αSyn co-localization with VDAC and COX IV in cells treated with olesoxime (Fig. 2). Whether αSyn specifically binds to both VDAC and COX IV, nonspecifically targets mitochondrial membranes, interacts with other membrane proteins, or all of the above, is not certain considering the crowded protein environment in both mitochondrial membranes and the limitation of PLA resolution to 30 nm between a pair of proteins of interest. Based on the earlier report that olesoxime could modulate mitochondrial membrane properties, such as membrane fluidity [50, 51], and the well-described effects of membrane lipid composition on αSyn-membrane-binding efficiency [72–74], we considered the possibility for olesoxime to affect αSyn affinity to MOM. Olesoxime, upon partitioning into MOM and thus modifying membrane properties, could cause a depletion of αSyn from MOM, and consequently, abolish its intramitochondrial targeting (Fig. 2). Under this scenario, olesoxime should nonspecifically modulate all cellular organelle membranes affecting mitochondrial and cell integrity. On the contrary, our data show that olesoxime is not toxic for cells and mitochondria (Fig. 1) and does not significantly affect association of endogenous αSyn with VDAC or COX IV (Fig. 2) or αSyn expression level (Supplemental Fig. S1A, B). The results of FCS experiments support this conclusion, because 10% of olesoxime in liposome membranes does not measurably affect αSyn binding to liposomes (Supplemental Fig. S5). Considering that extracellular addition of 3 μM olesoxime should result in a significantly lower than 10% doping of the mitochondrial membrane with olesoxime, we thus concluded that olesoxime does not induce αSyn detachment from mitochondrial membranes in situ. The complementary experiments on planar membranes, using a gA channel as a sensitive molecular probe of lipid bilayer mechanics, further confirmed that olesoxime does not change integral properties of lipid membranes even at a high content of the compound in the membrane (20% w/w of the total lipid) that is not physiologically achievable (Supplemental Fig. S4). However, olesoxime affects VDAC voltage gating (Fig. 5) pointing to direct interaction between olesoxime and VDAC in in vitro experiments. The effect on gating is even stronger in the presence of a physiologically relevant amount of cholesterol in planar membranes.
To address a question of how olesoxime could impair αSyn ability to reach MIM complexes by translocating through VDAC, we used single channels experiments with VDAC reconstituted into planar lipid membranes. Notably, while olesoxime does not qualitatively change the pattern of the reversible VDAC blockage by αSyn, it hinders αSyn translocation through the channel (Fig. 4b and Supplemental Fig. S3).
The cartoon in Fig. 6 illustrates a proposed mechanism of olesoxime action, by affecting αSyn translocation through and most likely binding to VDAC. The absence of the olesoxime effect on VDAC conductance (Fig. 4a) indicates that highly hydrophobic olesoxime does not enter the water-filled pore and, therefore, most likely interacts with VDAC at the protein–lipid interface. In the proposed model (Fig. 6), olesoxime interacts with hydrophobic exterior of the VDAC’s β-barrel, targeting it from the lipid medium and leaving the hydrophilic interior of the pore—and consequently the channel conductance—unaffected. It should be noted that if olesoxime were to interact with channel interior, it inevitably should change the dynamics of αSyn molecule capture by the VDAC pore, i.e., the blockage parameters at all applied voltages. The results (Fig. 4b) show that this is not the case and that olesoxime does not affect the regime of VDAC blockage at low voltages (at |V| < 35 mV), but prevents or delays αSyn translocation through the pore (VDAC blockage at high voltages, |V| > 35 mV). Olesoxime could interact with VDAC at lipid–pore interface between the lipid shell consisting of one or two lipid layers [75] and the β-barrel while leaving membrane integral properties unchanged. The ability of VDAC to modify its surrounding lipid shell to compensate for a hydrophobic mismatching between its β-barrel and an adjusted lipid has been proposed recently for the VDAC2 isoform [76] and should be applicable for other VDAC isoforms. We further speculate that olesoxime may also induce subtle structural changes in the flexible loops of VDAC, which connect β-strands and form channel entrances, by interfering with the specific interactions between VDAC loops and lipid headgroups as proposed by Mlayeh et al. for plant VDAC [46]. A similar mechanism was recently suggested to explain interaction between VDAC2 and MOM-located pro-apoptotic Bak and Bax proteins [77, 78]. Even subtle changes in channel structure could cause the increase of the energy barrier for αSyn translocation through the pore [17, 18] without altering the VDAC β-barrel integrity. Taken together, our in vitro data suggest that olesoxime could affect αSyn translocation through VDAC by interacting with the lipid–pore interface. To the best of our knowledge, such a mechanism, where a neuroprotective compound inhibits transport of a neurodegenerative-related protein into mitochondria through interaction with another mitochondrial membrane protein, has never been previously described.
We previously proposed that under physiological conditions, αSyn may play a role as a regulator of VDAC (and, consequently, the MOM) permeability by modulating ATP/ADP and other respiratory substrates fluxes through it (Fig. 6). Under stress conditions, such as αSyn overexpression, αSyn translocates through VDAC, targeting proteins in the MIM and potentially impairing ETC. This leads to the loss of mitochondrial membrane potential and finally to mitochondrial dysfunction. This model is in accord with the current trend positing VDAC as the main mitochondrial docking site for various neurodegenerative disease-related misfolded proteins [37]. In particular, similar to αSyn, some of these proteins—i.e., amyloid β associated with Alzheimer disease—could have the ability to translocate through the VDAC pore to reach and impair the ETC [37, 79]. We speculate that by partitioning to the MOM, olesoxime interferes with αSyn–VDAC interaction, thus hindering αSyn translocation into mitochondria, where it targets ETC at the MIM (Fig. 6). As a result, olesoxime maintains mitochondria integrity and eventually promotes neuronal cell survival.
To conclude, the findings presented here suggest αSyn interaction with VDAC as a new target for the development of anti-αSyn toxicity drugs for treating PD. In particular, the use of molecules interacting with the αSyn–VDAC complex could be a promising and effective pharmacological treatment of neurodegenerative diseases, aimed to decrease mitochondrial deficiencies in affected neurons.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institutes of Health (NIH), Eunice Kennedy Shriver National Institute of Child Health Human Development (NICHD) and National Institute of Aging. Authors are grateful to Megha Rajendran (NICHD, NIH) for fruitful discussion.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
1/9/2020
In the published article, an error was noticed and this has been corrected with this erratum publication.
References
- 1.Kim WS, Kagedal K, Halliday GM. Alpha-synuclein biology in Lewy body diseases. Alzheimers Res Ther. 2014;6(5):73. doi: 10.1186/s13195-014-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goedert M, Jakes R, Spillantini MG. The synucleinopathies: twenty years on. J Parkinsons Dis. 2017;7(s1):S53–S71. doi: 10.3233/JPD-179005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma LY, Liu GL, Wang DX, Zhang MM, Kou WY, Feng T. Alpha-synuclein in peripheral tissues in Parkinson’s disease. ACS Chem Neurosci. 2019;10(2):812–823. doi: 10.1021/acschemneuro.8b00383. [DOI] [PubMed] [Google Scholar]
- 4.Nussbaum RL. Genetics of synucleinopathies. Cold Spring Harb Perspect Med. 2018;8:a024109. doi: 10.1101/cshperspect.a024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23(2):1–13. doi: 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chinta SJ, Mallajosyula JK, Rane A, Andersen JK. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett. 2010;486(3):235–239. doi: 10.1016/j.neulet.2010.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283(14):9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loeb V, Yakunin E, Saada A, Sharon R. The transgenic overexpression of alpha-synuclein and not its related pathology associates with complex I inhibition. J Biol Chem. 2010;285(10):7334–7343. doi: 10.1074/jbc.M109.061051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reeve AK, Ludtmann MH, Angelova PR, Simcox EM, Horrocks MH, Klenerman D, Gandhi S, Turnbull DM, Abramov AY. Aggregated alpha-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis. 2015;6:e1820. doi: 10.1038/cddis.2015.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elkon H, Don J, Melamed E, Ziv I, Shirvan A, Offen D. Mutant and wild-type alpha-synuclein interact with mitochondrial cytochrome C oxidase. J Mol Neurosci. 2002;18(3):229–238. doi: 10.1385/JMN:18:3:229. [DOI] [PubMed] [Google Scholar]
- 11.Ludtmann MH, Angelova PR, Ninkina NN, Gandhi S, Buchman VL, Abramov AY. Monomeric alpha-synuclein exerts a physiological role on brain ATP synthase. J Neurosci. 2016;36(41):10510–10521. doi: 10.1523/JNEUROSCI.1659-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gouarne C, Tracz J, Paoli MG, Deluca V, Seimandi M, Tardif G, Xilouri M, Stefanis L, Bordet T, Pruss RM. Protective role of olesoxime against wild-type alpha-synuclein-induced toxicity in human neuronally differentiated SHSY-5Y cells. Br J Pharmacol. 2015;172(1):235–245. doi: 10.1111/bph.12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bordet T, Berna P, Abitbol JL, Pruss RM. Olesoxime (TRO19622): a novel mitochondrial-targeted neuroprotective compound. Pharmaceuticals (Basel) 2010;3(2):345–368. doi: 10.3390/ph3020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bordet T, Buisson B, Michaud M, Drouot C, Galea P, Delaage P, Akentieva NP, Evers AS, Covey DF, Ostuni MA, Lacapere JJ, Massaad C, Schumacher M, Steidl EM, Maux D, Delaage M, Henderson CE, Pruss RM. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2007;322(2):709–720. doi: 10.1124/jpet.107.123000. [DOI] [PubMed] [Google Scholar]
- 15.Sunyach C, Michaud M, Arnoux T, Bernard-Marissal N, Aebischer J, Latyszenok V, Gouarne C, Raoul C, Pruss RM, Bordet T, Pettmann B. Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology. 2012;62(7):2346–2352. doi: 10.1016/j.neuropharm.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Rostovtseva TK, Gurnev PA, Protchenko O, Hoogerheide DP, Yap TL, Philpott CC, Lee JC, Bezrukov SM. Alpha-synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J Biol Chem. 2015;290(30):18467–18477. doi: 10.1074/jbc.M115.641746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoogerheide DP, Gurnev PA, Rostovtseva TK, Bezrukov SM. Mechanism of alpha-synuclein translocation through a VDAC nanopore revealed by energy landscape modeling of escape time distributions. Nanoscale. 2017;9(1):183–192. doi: 10.1039/c6nr08145b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoogerheide DP, Gurnev PA, Rostovtseva TK, Bezrukov SM. Real-time nanopore-based recognition of protein translocation success. Biophys J. 2018;114(4):772–776. doi: 10.1016/j.bpj.2017.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maldonado EN, Patnaik J, Mullins MR, Lemasters JJ. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 2010;70(24):10192–10201. doi: 10.1158/0008-5472.CAN-10-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rostovtseva TK, Gurnev PA, Hoogerheide DP, Rovini A, Sirajuddin M, Bezrukov SM. Sequence diversity of tubulin isotypes in regulation of the mitochondrial voltage-dependent anion channel. J Biol Chem. 2018 doi: 10.1074/jbc.RA117.001569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfefferkorn CM, Lee JC. Tryptophan probes at the alpha-synuclein and membrane interface. J Phys Chem B. 2010;114(13):4615–4622. doi: 10.1021/jp908092e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobs D, Hoogerheide DP, Rovini A, Zhiping J, Lee JC, Rostovtseva TK, Bezrukov SM. Probing membrane association of α-synuclein domains with VDAC nanopore reveals unexpected binding pattern. Sci Rep. 2019;9(1):4580. doi: 10.1038/s41598-019-40979-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blachly-Dyson E, Peng SZ, Colombini M, Forte M. Selectivity changes in site-directed mutants of the VDAC ion channel—structural implications. Science. 1990;247(4947):1233–1236. doi: 10.1126/science.1690454. [DOI] [PubMed] [Google Scholar]
- 24.Palmieri F, De Pinto V. Purification and properties of the voltage-dependent anion channel of the outer mitochondrial membrane. J Bioenerg Biomembr. 1989;21(4):417–425. doi: 10.1007/BF00762514. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto T, Yamada A, Watanabe M, Yoshimura Y, Yamazaki N, Yoshimura Y, Yamauchi T, Kataoka M, Nagata T, Terada H, Shinohara Y. VDAC1, having a shorter N-terminus than VDAC2 but showing the same migration in an SDS-polyacrylamide gel, is the predominant form expressed in mitochondria of various tissues. J Proteome Res. 2006;5(12):3336–3344. doi: 10.1021/pr060291w. [DOI] [PubMed] [Google Scholar]
- 26.Rostovtseva TK, Gurnev PA, Chen MY, Bezrukov SM. Membrane lipid composition regulates tubulin interaction with mitochondrial voltage-dependent anion channel. J Biol Chem. 2012;287(35):29589–29598. doi: 10.1074/jbc.M112.378778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52(6):1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weinrich M, Worcester DL, Bezrukov SM. Lipid nanodomains change ion channel function. Nanoscale. 2017;9(35):13291–13297. doi: 10.1039/c7nr03926c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colombini M. Voltage gating in the mitochondrial channel, VDAC. J Membr Biol. 1989;111(2):103–111. doi: 10.1007/BF01871775. [DOI] [PubMed] [Google Scholar]
- 30.Rostovtseva TK, Kazemi N, Weinrich M, Bezrukov SM. Voltage gating of VDAC is regulated by nonlamellar lipids of mitochondrial membranes. J Biol Chem. 2006;281(49):37496–37506. doi: 10.1074/jbc.M602548200. [DOI] [PubMed] [Google Scholar]
- 31.Teijido O, Rappaport SM, Chamberlin A, Noskov SY, Aguilella VM, Rostovtseva TK, Bezrukov SM. Acidification asymmetrically affects voltage-dependent anion channel implicating the involvement of salt bridges. J Biol Chem. 2014;289(34):23670–23682. doi: 10.1074/jbc.M114.576314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson SM, Yellon D, Duchen MR. Assessing mitochondrial potential, calcium, and redox state in isolated mammalian cells using confocal microscopy. Methods Mol Biol. 2007;372:421–430. doi: 10.1007/978-1-59745-365-3_30. [DOI] [PubMed] [Google Scholar]
- 33.Li WW, Yang R, Guo JC, Ren HM, Zha XL, Cheng JS, Cai DF. Localization of alpha-synuclein to mitochondria within midbrain of mice. NeuroReport. 2007;18(15):1543–1546. doi: 10.1097/WNR.0b013e3282f03db4. [DOI] [PubMed] [Google Scholar]
- 34.Cole NB, Dieuliis D, Leo P, Mitchell DC, Nussbaum RL. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp Cell Res. 2008;314(10):2076–2089. doi: 10.1016/j.yexcr.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robotta M, Gerding HR, Vogel A, Hauser K, Schildknecht S, Karreman C, Leist M, Subramaniam V, Drescher M. Alpha-synuclein binds to the inner membrane of mitochondria in an alpha-helical conformation. ChemBioChem. 2014;15(17):2499–2502. doi: 10.1002/cbic.201402281. [DOI] [PubMed] [Google Scholar]
- 36.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3(12):995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 37.Magri A, Messina A. Interactions of VDAC with proteins involved in neurodegenerative aggregation: an opportunity for advancement on therapeutic molecules. Curr Med Chem. 2017;24(40):4470–4487. doi: 10.2174/0929867324666170601073920. [DOI] [PubMed] [Google Scholar]
- 38.de Kroon AI, Dolis D, Mayer A, Lill R, de Kruijff B. Phospholipid composition of highly purified mitochondrial outer membranes of rat liver and Neurospora crassa. Is cardiolipin present in the mitochondrial outer membrane? Biochim Biophys Acta. 1997;1325(1):108–116. doi: 10.1016/S0005-2736(96)00240-4. [DOI] [PubMed] [Google Scholar]
- 39.Ardail D, Lerme F, Louisot P. Further characterization of mitochondrial contact sites: effect of short-chain alcohols on membrane fluidity and activity. Biochem Biophys Res Commun. 1990;173(3):878–885. doi: 10.1016/S0006-291X(05)80868-X. [DOI] [PubMed] [Google Scholar]
- 40.Horvath SE, Daum G. Lipids of mitochondria. Prog Lipid Res. 2013;52(4):590–614. doi: 10.1016/j.plipres.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 41.Rostovtseva TK, Bezrukov SM. Function and regulation of mitochondrial voltage-dependent anion channel. Electrophysiology of unconventional channels and pores. Switzerland: Springer; 2015. [Google Scholar]
- 42.Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Characterization of individual polynucleotide molecules using a membrane channel. Proc Natl Acad Sci USA. 1996;93(24):13770–13773. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Movileanu L, Schmittschmitt JP, Scholtz JM, Bayley H. Interactions of peptides with a protein pore. Biophys J. 2005;89(2):1030–1045. doi: 10.1529/biophysj.104.057406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rostovtseva TK, Bezrukov SM. VDAC inhibition by tubulin and its physiological implications. Biochim Biophys Acta. 2012;1818(6):1526–1535. doi: 10.1016/j.bbamem.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rappaport SM, Teijido O, Hoogerheide DP, Rostovtseva TK, Berezhkovskii AM, Bezrukov SM. Conductance hysteresis in the voltage-dependent anion channel. Eur Biophys J. 2015;44(6):465–472. doi: 10.1007/s00249-015-1049-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mlayeh L, Krammer EM, Leonetti M, Prevost M, Homble F. The mitochondrial VDAC of bean seeds recruits phosphatidylethanolamine lipids for its proper functioning. Biochem Biophys Acta. 2017;1858(9):786–794. doi: 10.1016/j.bbabio.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 47.Queralt-Martin M, Bergdoll L, Jacobs D, Bezrukov SM, Abramson J, Rostovtseva TK. Assessing the role of residue E73 and lipid headgroup charge in VDAC1 voltage gating. Biochim Biophys Acta Bioenerg. 2019;1860(1):22–29. doi: 10.1016/j.bbabio.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bowen KA, Tam K, Colombini M. Evidence for titratable gating charges controlling the voltage dependence of the outer mitochondrial-membrane channel, VDAC. J Membr Biol. 1985;86(1):51–59. doi: 10.1007/Bf01871610. [DOI] [PubMed] [Google Scholar]
- 49.Zimmerberg J, Parsegian VA. Polymer inaccessible volume changes during opening and closing of a voltage-dependent ionic channel. Nature. 1986;323(6083):36–39. doi: 10.1038/323036a0. [DOI] [PubMed] [Google Scholar]
- 50.Eckmann J, Clemens LE, Eckert SH, Hagl S, Yu-Taeger L, Bordet T, Pruss RM, Muller WE, Leuner K, Nguyen HP, Eckert GP. Mitochondrial membrane fluidity is consistently increased in different models of Huntington disease: restorative effects of olesoxime. Mol Neurobiol. 2014;50(1):107–118. doi: 10.1007/s12035-014-8663-3. [DOI] [PubMed] [Google Scholar]
- 51.Magalon K, Le Grand M, El Waly B, Moulis M, Pruss R, Bordet T, Cayre M, Belenguer P, Carre M, Durbec P. Olesoxime favors oligodendrocyte differentiation through a functional interplay between mitochondria and microtubules. Neuropharmacology. 2016;111:293–303. doi: 10.1016/j.neuropharm.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 52.Weinrich M, Rostovtseva TK, Bezrukov SM. Lipid-dependent effects of halothane on gramicidin channel kinetics: a new role for lipid packing stress. Biochemistry. 2009;48(24):5501–5503. doi: 10.1021/bi900494y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andersen OS, Nielsen C, Maer AM, Lundbaek JA, Goulian M, Koeppe RE., 2nd Ion channels as tools to monitor lipid bilayer-membrane protein interactions: gramicidin channels as molecular force transducers. Methods Enzymol. 1999;294:208–224. doi: 10.1016/S0076-6879(99)94013-2. [DOI] [PubMed] [Google Scholar]
- 54.Lundbaek JA, Andersen OS. Spring constants for channel-induced lipid bilayer deformations. Estimates using gramicidin channels. Biophys J. 1999;76(2):889–895. doi: 10.1016/S0006-3495(99)77252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lundbaek JA, Birn P, Girshman J, Hansen AJ, Andersen OS. Membrane stiffness and channel function. Biochemistry. 1996;35(12):3825–3830. doi: 10.1021/bi952250b. [DOI] [PubMed] [Google Scholar]
- 56.Rovini A, Carre M, Bordet T, Pruss RM, Braguer D. Olesoxime prevents microtubule-targeting drug neurotoxicity: selective preservation of EB comets in differentiated neuronal cells. Biochem Pharmacol. 2010;80(6):884–894. doi: 10.1016/j.bcp.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 57.Martin LJ. Olesoxime, a cholesterol-like neuroprotectant for the potential treatment of amyotrophic lateral sclerosis. IDrugs. 2010;13(8):568–580. [PMC free article] [PubMed] [Google Scholar]
- 58.Gonzalez S, Berthelot J, Jiner J, Perrin-Tricaud C, Fernando R, Chrast R, Lenaers G, Tricaud N. Blocking mitochondrial calcium release in Schwann cells prevents demyelinating neuropathies. J Clin Invest. 2016;126(7):2773. doi: 10.1172/JCI88179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiao WH, Zheng FY, Bennett GJ, Bordet T, Pruss RM. Olesoxime (cholest-4-en-3-one, oxime): analgesic and neuroprotective effects in a rat model of painful peripheral neuropathy produced by the chemotherapeutic agent, paclitaxel. Pain. 2009;147(1–3):202–209. doi: 10.1016/j.pain.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int J Biochem Cell Biol. 2009;41(10):2015–2024. doi: 10.1016/j.biocel.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 61.Vekrellis K, Xilouri M, Emmanouilidou E, Stefanis L. Inducible over-expression of wild type alpha-synuclein in human neuronal cells leads to caspase-dependent non-apoptotic death. J Neurochem. 2009;109(5):1348–1362. doi: 10.1111/j.1471-4159.2009.06054.x. [DOI] [PubMed] [Google Scholar]
- 62.Banerjee K, Sinha M, Pham Cle L, Jana S, Chanda D, Cappai R, Chakrabarti S. Alpha-synuclein induced membrane depolarization and loss of phosphorylation capacity of isolated rat brain mitochondria: implications in Parkinson’s disease. FEBS Lett. 2010;584(8):1571–1576. doi: 10.1016/j.febslet.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 63.Bir A, Sen O, Anand S, Khemka VK, Banerjee P, Cappai R, Sahoo A, Chakrabarti S. alpha-Synuclein-induced mitochondrial dysfunction in isolated preparation and intact cells: implications in the pathogenesis of Parkinson’s disease. J Neurochem. 2014;131(6):868–877. doi: 10.1111/jnc.12966. [DOI] [PubMed] [Google Scholar]
- 64.Esteves AR, Gozes I, Cardoso SM. The rescue of microtubule-dependent traffic recovers mitochondrial function in Parkinson’s disease. Biochem Biophys Acta. 2014;1842(1):7–21. doi: 10.1016/j.bbadis.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 65.Ludtmann MHR, Angelova PR, Horrocks MH, Choi ML, Rodrigues M, Baev AY, Berezhnov AV, Yao Z, Little D, Banushi B, Al-Menhali AS, Ranasinghe RT, Whiten DR, Yapom R, Dolt KS, Devine MJ, Gissen P, Kunath T, Jaganjac M, Pavlov EV, Klenerman D, Abramov AY, Gandhi S. alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat Commun. 2018;9(1):2293. doi: 10.1038/s41467-018-04422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu J, Wang X, Lu Y, Duan C, Gao G, Lu L, Yang H. Pink1 interacts with alpha-synuclein and abrogates alpha-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis. 2017;8(9):e3056. doi: 10.1038/cddis.2017.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26(1):41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vicario M, Cieri D, Brini M, Cali T. The close encounter between alpha-synuclein and mitochondria. Front Neurosci. 2018;12:388. doi: 10.3389/fnins.2018.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hill K, Model K, Ryan MT, Dietmeier K, Martin F, Wagner R, Pfanner N. Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins [see comment] Nature. 1998;395(6701):516–521. doi: 10.1038/26780. [DOI] [PubMed] [Google Scholar]
- 70.Suzuki H, Kadowaki T, Maeda M, Sasaki H, Nabekura J, Sakaguchi M, Mihara K. Membrane-embedded C-terminal segment of rat mitochondrial TOM40 constitutes protein-conducting pore with enriched beta-structure. J Biol Chem. 2004;279(48):50619–50629. doi: 10.1074/jbc.M408604200. [DOI] [PubMed] [Google Scholar]
- 71.Kuszak AJ, Jacobs D, Gurnev PA, Shiota T, Louis JM, Lithgow T, Bezrukov SM, Rostovtseva TK, Buchanan SK. Evidence of distinct channel conformations and substrate binding affinities for the mitochondrial outer membrane protein translocase pore Tom40. J Biol Chem. 2015;290(43):26204–26217. doi: 10.1074/jbc.M115.642173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.O’Leary EI, Jiang Z, Strub MP, Lee JC. Effects of phosphatidylcholine membrane fluidity on the conformation and aggregation of N-terminally acetylated alpha-synuclein. J Biol Chem. 2018;293(28):11195–11205. doi: 10.1074/jbc.RA118.002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pfefferkorn CM, Jiang Z, Lee JC. Biophysics of alpha-synuclein membrane interactions. Biochem Biophys Acta. 2012;1818(2):162–171. doi: 10.1016/j.bbamem.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rhoades E, Ramlall TF, Webb WW, Eliezer D. Quantification of alpha-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys J. 2006;90(12):4692–4700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eddy MT, Ong TC, Clark L, Teijido O, van der Wel PC, Garces R, Wagner G, Rostovtseva TK, Griffin RG. Lipid dynamics and protein-lipid interactions in 2D crystals formed with the beta-barrel integral membrane protein VDAC1. J Am Chem Soc. 2012;134(14):6375–6387. doi: 10.1021/ja300347v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Srivastava SR, Zadafiya P, Mahalakshmi R. Hydrophobic mismatch modulates stability and plasticity of human mitochondrial VDAC2. Biophys J. 2018 doi: 10.1016/j.bpj.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Naghdi S, Varnai P, Hajnoczky G. Motifs of VDAC2 required for mitochondrial Bak import and tBid-induced apoptosis. Proc Natl Acad Sci USA. 2015;112(41):E5590–E5599. doi: 10.1073/pnas.1510574112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chin HS, Li MX, Tan IKL, Ninnis RL, Reljic B, Scicluna K, Dagley LF, Sandow JJ, Kelly GL, Samson AL, Chappaz S, Khaw SL, Chang C, Morokoff A, Brinkmann K, Webb A, Hockings C, Hall CM, Kueh AJ, Ryan MT, Kluck RM, Bouillet P, Herold MJ, Gray DHD, Huang DCS, van Delft MF, Dewson G. VDAC2 enables BAX to mediate apoptosis and limit tumor development. Nat Commun. 2018;9(1):4976. doi: 10.1038/s41467-018-07309-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smilansky A, Dangoor L, Nakdimon I, Ben-Hail D, Mizrachi D, Shoshan-Barmatz V. The voltage-dependent anion channel 1 mediates amyloid beta toxicity and represents a potential target for Alzheimer disease therapy. J Biol Chem. 2015;290(52):30670–30683. doi: 10.1074/jbc.M115.691493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.