

SUMMARY
Androgen deprivation therapy for prostate cancer (PCa) benefits patients with early disease, but becomes ineffective as PCa progresses to a castration-resistant state (CRPC). Initially CRPC remains dependent on androgen receptor (AR) signaling, often through increased expression of full-length AR (ARfl) or expression of dominantly active splice variants such as ARv7. We show in ARv7-dependent CRPC models that ARv7 binds together with ARfl to repress transcription of a set of growth-suppressive genes. Expression of the ARv7-repressed targets and ARv7 protein expression are negatively correlated and predicts for outcome in PCa patients. Our results provide insights into the role of ARv7 in CRPC and define a set of potential bio-markers for tumors dependent on ARv7.
In Brief
Cato et al. utilize cistrome and transcriptome analyses in castration-resistant prostate cancer (CRPC) to reveal that the androgen receptor (AR) splice variant ARv7 functions as a transcriptional repressor and heterodimerizes with full-length AR at a subset of growth-suppressive genes to support CRPC growth.
Graphical Abstract
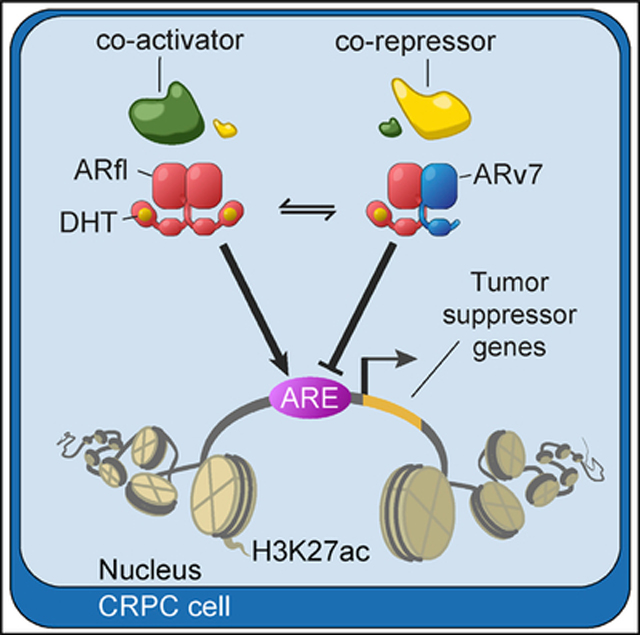
INTRODUCTION
Prostate cancer (PCa) remains one of the most common causes of cancer deaths in men worldwide (Jemal et al., 2011; Siegel et al., 2017). Locally advanced and metastatic PCa is treated with endocrine therapies, aimed at repressing the synthesis of androgens (de Bono et al., 2011; van Poppel and Nilsson, 2008) or at inhibiting androgen receptor (AR) function (Tran et al., 2009). The molecular basis for this therapeutic approach is to block the AR C-terminal, ligand-binding domain (LBD), thereby inhibiting AR-driven oncogenic gene expression programs (Matsumoto et al., 2013; Wang et al., 2009). While this treatment is initially effective, patients frequently develop resistance to endocrine therapy and develop castration-resistant PCa (CRPC). CRPC often continues to rely on AR signaling initially (Watson et al., 2015), but the underlying mechanisms of AR reactivation are poorly understood. Proposed mechanisms include genetic alterations of AR (Taplin et al., 1995; Visakorpi et al., 1995) and the expression of truncated, ligand-independent AR variants (AR-Vs), generated via genomic rearrangements and/or alternative splicing events (Dehm et al., 2008; Guo et al., 2009; Hu et al., 2009; Sun et al., 2010).
Multiple AR-Vs have been described in PCa models, xeno-grafts, and patient tumors (Lu and Luo, 2013). One of the most common variants, AR variant 7 (ARv7), arises through cryptic exon inclusion. This AR isoform consists of a conserved N-terminal activation function-1 (AF-1) domain, a central DNA-binding domain, a partial hinge region and a unique C-terminal 16-amino-acid sequence, in place of the classical LBD. ARv7, unlike the full-length receptor, is continuously localized to the nucleus (Hu et al., 2009), and its expression is correlated with androgen-independent cell proliferation and PCa progression (Dehm et al., 2008). ARv7 levels are increased in metastases of CRPC patients (Hornberg et al., 2011; Qu et al., 2015), and ARv7 is present in over 80% of CRPC patient samples in the SU2C PCa cohort (Robinson et al., 2015). Detection of ARv7 in circulating tumor cells (CTCs) from CRPC patients was associated with resistance to anti-hormonal therapies in a pioneering study (Antonarakis et al., 2014). While some studies confirmed the correlation of ARv7 status with disease progression and overall survival (Antonarakis et al., 2017; Scher et al., 2016; Welti et al., 2016), one follow-up study reported that CTC ARv7 status in patients cannot entirely predict nonresponse to anti-hormonal treatment (Bernemann et al., 2017).
In this article we investigate the genomic function of ARv7 and its potential link with ARfl action by utilizing cistrome and transcriptome studies in CRPC cells.
RESULTS
LNCaP95 and 22Rv1 Cell Growth Is Dependent on ARfl and ARv7
A proposed mechanism underlying the reactivation of AR in CRPC is the increased expression of ARv7 (together with ARfl). Utilizing the CRPC cell line LNCaP95, which endogenously expresses ARfl and ARv7 and is cultured under hormone-starvation conditions, we assessed the dependency of this line for both AR isoforms. We created stable LNCaP95 lines harboring doxycycline (dox)-inducible short hairpin RNAs (shRNAs), selectively silencing the expression of ARfl (shARfl), ARv7 (shARv7), or a GFP control (shGFP) (Figures 1A and S1A). Knockdown (KD) of either AR isoform was sufficient to decrease LNCaP95 cell proliferation in 2D culture (Figure 1B) and in 3D cell culture in a poly(ethylene glycol) diacrylate (PEGda) cryogel matrix (Gö ppert et al., 2016) compared with the shGFP control (Figures 1C, 1D, and S1B). Silencing of either ARfl or ARv7 in 22Rv1 cells, another CRPC model with high endogenous ARv7 expression, also significantly decreased cell growth (Figures S1C and S1D). Therefore, consistent with previous findings (Dehm et al., 2008; Guo et al., 2009; Hu et al., 2009), we conclude that both ARfl and ARv7 are necessary to support proliferation of CRPC cell line models under castration conditions.
Figure 1. LNCaP95 Cell Growth Is Dependent on both ARfl and ARv7.

(A) Top: schematic of the full-length AR (ARfl) and the AR variant 7 (ARv7), with the N-terminal domain (NTD), DNA-binding domain (DBD), hinge region (H), ligand-binding domain (LBD), and cryptic exon (CE3). Bottom: western blot of shGFP, shARv7, or shARfl LNCaP cells using an N-terminal, pan-AR antibody. Actin signals serve as a loading control.
(B) Proliferation assay of indicated cells grown in 2D culture. Data are the mean of three independent experiments ±SEM.
(C) Representative scanning electron microscopy images of LNCaP95 cells after 7 days of growth in 3D/PEGda cryogels. Scale bar, 100 mm.
(D) Quantification of 3D cell growth data in (C). Data are the mean of four independent experiments relative to day 0 ±SEM.
**p % 0.01; ***p % 0.001; ****p % 0.0001 by Student’s t test. See also Figure S1.
ARv7 Functions as a Transcriptional Repressor in CRPC Cells
To better delineate ARv7 from ARfl function, we next profiled gene expression in the inducible shRNA cells by RNA sequencing (RNA-seq). Using differential expression analysis (DEseq; p < 0.05, fold change >1.5), we observed a higher number of upregulated (n = 300) compared with downregulated genes (n = 129) in response to ARv7 depletion (Figure 2A), suggestive of a preferentially repressive ARv7 transcriptional function. In contrast, a slightly higher fraction of genes was downregulated (n = 293) rather than upregulated (n = 242) upon shARfl (Figure 2B). To identify the biological processes associated with these AR isoform-regulated genes, we next employed gene set enrichment analysis (GSEA) (Subramanian et al., 2005). In this analysis some ARfl-activated pathways over-lapped with ARv7-repressed ones, while others were unique, with an unclear discernible pattern (Table S1).
Figure 2. ARv7, Unlike ARfl, Functions as a Transcriptional Repressor in CRPC Cells.

(A and B) Volcano plots of differentially expressed genes in shARv7 (A) or shARfl (B) cells, compared with shGFP control. Significantly altered genes (fold change >±1.5; adjusted p value <0.05) are highlighted in red (activated) or blue (repressed). Select AR targets and significant outliers are labeled.
(C) Top: violin plots of log2 fold changes of ARv7-regulated (blue) and ARfl-regulated (red) genes (relative to the shGFP control) in response to DHT stimulation. Only genes with an adjusted p value of <0.05 are shown. Bottom: bar plots of the mean log2 fold changes of the ARv7-regulated (blue) and ARfl-regulated (red) target genes as above. ****p % 0.0001 by ANOVA and Tukey’s honest significant difference test (HSD).
(D) Comparison of log2 fold changes of significantly dysregulated genes (adjusted p value <0.05) in response to shARv7 or shARfl (as defined in A and B). Colors indicate genes primarily dysregulated by shARfl (red), shARv7 (blue), or both (purple). Select classical AR targets are labeled.
To assess the effect of dihydrotestosterone (DHT) on the AR isoform-regulated transcriptomes, we next performed RNA-seq in the inducible shRNA LNCaP95 lines treated with a range of DHT concentrations for 24 h (Figures 2C, S2A, and S2B; Table S2). We observed that across all DHT concentrations ARv7 retains its repressive effect, with significantly regulated genes (p < 0.05) being mostly upregulated after ARv7 KD (with median fold changes >1). Conversely, ARfl retains its activating effect at all DHT concentrations with dysregulated genes being mostly downregulated after ARfl KD (with median fold changes <1). Moreover, we only observed a limited DHT effect, with a stable amplitude for the ARv7-regulated genes (Figures 2C and S2A). In contrast, the distribution of ARfl-regulated genes increased proportionally to DHT increases (Figures 2C and S2B), which agrees with a model of hormone-dependent, ARfl-mediated transcriptional regulation in PCa cells (Wang et al., 2007).
A direct comparison of ARfl- and ARv7-dependent transcriptomes in the absence of hormone revealed significant differences between the two AR isoform-specific transcriptomes (Table S2), with no significant correlation between them (R [Pearson] = 0.195) (Figure 2D), although expression of some genes, including the canonical AR targets KLK2, KLK3, and IGF1, was activated by ARv7 and ARfl (Figure 2D). This observation conflicts with the hypothesis that ARv7 simply acts as a constitutively active form of ARfl (Li et al., 2013), but instead suggests that ARv7 and ARfl have different transcriptional roles in CRPC (Hu et al., 2012).
ARfl and ARv7 Bind to the Same Sites in Chromatin and Heterodimerize
To further identify the direct targets of ARfl and ARv7, we examined their respective cistromes using chromatin immunoprecipitation sequencing (ChIP-seq) in LNCaP95 cells treated with and without DHT (10 nM) for 4 h. We utilized antibodies specific to ARfl or ARv7 and, as a control, an antibody that recognized both ARfl and ARv7 (Figure S3A). To date, the number of published AR-V cistromes has been limited (Chan et al., 2015; Chen et al., 2018; He et al., 2018; Lu et al., 2015) and there have been no direct comparisons between ARfl and ARv7 cistromes in an endogenous setting. We first confirmed the specificity and ChIP suitability of the ARv7 antibody using coimmunoprecipitation (coIP; Figure S3B) and ChIP-seq following ARv7 KD (Figure S3C). We then carried out ChIP-seq for both AR isoforms and observed 3,497 binding sites for ARv7 and 12,389 binding sites for ARfl, in the absence of hormone (Figures 3A and S3C). DHT treatment increased the ARv7 cistrome 2-fold (n = 6,149), and the ARfl cistrome 5-fold (>60,000 sites). Although most ARv7 sites were contained within the ARfl cistrome, a small number of sites (n = 794 for vehicle and n = 465 for DHT) were exclusive to the ARv7 cistrome, suggesting that ARv7 might function independently of ARfl. To test this hypothesis, we compared the two AR cistromes (without DHT) with a cistrome using an N-terminal AR antibody that recognizes both AR isoforms (Figures S3A and S3B). Peaks that were common between at least two of the three cistromes were defined as “high-confidence” AR-binding sites (n = 2,828) (Figures 3B and 3C). The remaining ARv7-unique peaks (n = 595), unlike the “high-confidence” ARv7 peaks, were only minimally affected by silencing of ARv7 (Figure 3C), despite efficient protein reduction (Figures 1A and S1A). Taken together, these results suggest that most of the exclusive ARv7 peaks are in fact artifacts rather than bona fide ARv7 chromatin-binding sites.
Figure 3. ARfl and ARv7 Binding to Chromatin Is Interdependent.

(A) Euler diagram of the overlap (R1 bp) of ARv7 (blue) and ARfl (red) cistromes, profiled in the absence of hormone (ETOH) or after 4 h of DHT (10 nM) treatment. The numbers of unique and overlapping binding sites are indicated.
(B) Top: Euler diagram of the overlap of ARv7 (blue), ARfl (red), and AR N-terminal cistromes (ARN; purple) in the absence of hormone. Bottom: signal profiles (500-bp interval around the center of ARv7 peaks) of ARv7, ARfl, and ARN cistromes at different ARv7-binding sites. Left panel: 2,629 ARv7, ARfl, and ARN shared peaks. Middle panel: 199 ARv7 and ARN shared peaks. Right panel: 595 “ARv7-unique” peaks.
(C) Signal profiles of ARv7 (blue) and ARfl (red) cistromes in response to ARv7 (shARv7; dashed line) or GFP KD (shGFP; solid lines) at “ARv7-unique” (left) or “high confidence” AR sites (union of ARN, Arv7, and ARfl peaks) (right).
(D) Apparent FRET efficiencies representative of the level of AR isoform interactions, as shown. Values are the mean of 34–48 cells ±SEM.
(E) Signal profiles of ARfl (left) and ARv7 (right) cistromes centered on AR isoform peaks. Cells were induced for 3 days and treated for 4 h with vehicle (ETOH) or 10 nM DHT (DHT).
Given that the majority of ARv7-binding sites overlap with the ARfl cistrome (Figure 3A), we compared sites shared by ARfl and ARv7 (n = 2629) and sites occupied by ARfl only (n = 4,737) (Table S3). This revealed that the AR motif was more prevalent at ARfl/ ARv7 sites than at ARfl-only sites (Figure S3D). Although we did not observe any differences in gene expression in response to ARfl KD for targets in the vicinity of ARfl/ARv7 or ARfl-only sites, shARv7-regulated genes associated with the two sites were significantly differentially expressed (Figure S3E). To investigate whether ARv7 and ARfl co-occupy the same genomic loci, we carried out sequential ARfl/ARv7 ChIP-re-ChIP experiments at select target genes. We detected positive signal enrichment (Figure S3F), suggesting a potential functional interaction (i.e., heterodimerization) of the two receptors. To investigate this further, we next employed acceptor photobleaching fluorescence resonance energy transfer (FRET) (Figure 3D). We observed strong FRET signals for ARfl/ARfl and ARfl/ARv7 or ARv7/ARfl interactions, but not for ARv7/ARv7 homotypic interaction. Although these results reinforce a model of ARfl and ARv7 heterodimerization, they do not establish codependent binding on chromatin, as this is not required for the FRET signal. To further investigate chromatin binding, we performed ChIP-seq of both AR variants in the shGFP, shARv7, and shARfl LNCaP95 cells. Here, KD of ARv7 significantly reduced ARfl chromatin binding in both the vehicle and DHT condition (Figure 3E). Concordantly, loss of ARfl also reduced ARv7 binding in both treatment conditions (Figure 3E). Similar results were obtained in 22Rv1 cells, where codependent binding of ARfl and ARv7 was observed at ARE-containing sites with high levels of both factors (Figures S3G–S3I). Taken together, these results suggest that ARfl and ARv7 form heterodimers and can modulate their respective DNA-binding affinities.
ARv7 Preferentially Interacts with Transcriptional Corepressors
Given the codependent binding of ARfl and ARv7 to chromatin, but divergent transcriptional output, we speculated that other factors might contribute to the divergent genomic function of the two receptors. To address this, we first determined the AR isoform-specific chromatin-binding kinetics using fluorescence recovery after photobleaching (FRAP). The experiment was performed using wild-type (WT; ARfl or ARv7) or DNA-binding domain mutants of AR (ARfl R585K or ARv7 R585K). Previous studies have shown that due to impaired DNA binding, the R585K mutant displays a quicker recovery time after photo-bleaching compared with WT AR, as it does not form long-term interactions with DNA (van Royen et al., 2012). While this was the case for ARfl, ARv7 WT and the R585K mutant FRAP signals were indistinguishable (Figure S4A). This suggests that ARv7, in the absence of ARfl, has few long-term DNA interactions, further supported by the finding that ARfl has a speckled nuclear distribution, which was not observed for ARfl-R585K, ARv7, or ARv7-R585K (Figure S4A).
Next, we examined AR isoform-specific affinities for coregulator binding using a MARCoNI peptide-binding assay (Desmet et al., 2014). In this assay, LXXLL or FXXLF motif-encompassing peptides of known coregulators immobilized on an array are exposed to nuclear receptor-containing cell lysates, and binding is detected using specific antibodies. Using a pan-AR antibody we observed that AR bound coregulator peptides similarly in the shGFP and shARv7 cells (Figure S4B). This suggests that these interactions are primarily mediated by the LBD-containing ARfl and are not significantly altered by the loss of ARv7. In comparison, most interactions were lost in the absence of ARfl, particularly those for classical AR coactivators, such as NCOA1–6 (Figure S4B). However, we also observed increased binding to several corepressor peptides, including those for CNOT1, NCOR1, NCOR2, NRIP1, and PELP1 (Perissi et al., 2010) (Figures 4A and S4B). To test whether this binding was due to the direct interaction of ARv7 with select corepressors, we employed coIP in LNCaP95 cells following ARv7 or ARfl KD. We observed a small but reproducible increase in binding of ARv7 to NCOR1 and NCOR2 upon ARfl depletion (Figures 4B and S4C–S4E), but were unable to validate the ARv7 and NRIP1 interaction (not shown). This supports a model whereby ARv7 preferentially interacts with specific transcriptional corepressors, such as members of the NCOR family.
Figure 4. ARv7 Binds to Transcriptional Co-repressors NCOR1, NCOR2, and NRIP1.

(A) MARCoNI assay (using a pan-AR antibody) of select corepressor peptides and cell lysates from indicated LNCaP95 cells. Results are the mean of three experiments ±SD. *p % 0.05, ***p % 0.001, Student’s t test.
(B) Quantification of ARv7:NCOR1,2 coIP signals from nuclear lysates of shGFP, shARv7, and shARfl cells. Values are the means ±SEM of 5–6 replicates (n). n.s., not significant; *p % 0.05,**p % 0.01, ***p % 0.001, paired one-tailed t test.
(C) Heatmap of the union of dysregulated genes (adjusted p value <0.05) in response to NCOR1 or NCOR2 KD (siNCOR1 or siNCOR2) relative to control (siCtrl) in the background of shGFP, shARv7, and shARfl cells.
See also Figure S4.
To interrogate the functional link between NCOR- and ARv7-dependent transcription, we investigated the effect of small interfering RNA (siRNA)-mediated corepressor depletion on gene expression in each individual KD cell line. We could only evaluate transcriptional effects mediated by siRNA corepressor depletion within the background of a specific shRNA line (i.e., shGFP, shARv7, and shARfl), because the shRNA-mediated effects were much stronger than the transient siRNA effects. When this was done, we observed that the transcriptional responses to siNCOR1 or siNCOR2 were strongly attenuated within the shARv7 and shARfl cell lines, whereas this was not the case in the control cell line (shGFP) containing both ARv7 and ARfl (Figures 4C, S4F, and S4G). This suggests that NCOR-mediated transcription is, at least in part, dependent on the presence of ARv7 and ARfl. Consistently, KD of NRIP1, which could not be validated convincingly as an interactor of ARv7, did not produce this effect (Figure S4H). Taken together, these findings suggest that ARv7 mediates its repressive function by preferentially interacting with corepressors NCOR1 and NCOR2.
ARv7 Negatively Regulates H3K27ac
To further characterize the mechanism whereby ARv7 negatively affects transcription, we utilized ChIP-seq to assess levels of histone H3K27 acetylation (H3K27ac), a mark of active enhancers and transcriptional activity (Nord et al., 2013; Rada-Iglesias et al., 2011; Wang et al., 2016) H3K27ac cistromes were determined in the AR KD cell lines, and signals centered at “high-confidence” AR-binding sites (union of ARN, ARv7, and ARfl peaks) were analyzed (Figure 5A). To correlate H3K27ac levels with AR function, we stratified the averaged H3K27ac signals at the AR sites using Pearson correlation. This allowed us to distinguish two clusters with distinct H3K27ac signals (Figures 5A and S5A). Cluster I (n = 3,284) showed decreased H3K27ac levels after ARv7 and ARfl KD, indicative of ARfl and ARv7 positively regulating H3K27ac at these sites. In contrast, H3K27ac levels in cluster II (n = 4,268) were significantly increased upon ARv7 KD, yet decreased upon ARfl KD. This suggests that genes in cluster II sites are mostly differentially regulated by the two AR iso-forms. To test this hypothesis, we examined the ARv7- and ARfl-binding sites (Figures 5B and 5C). Although no substantial difference in ARv7 or ARfl binding was apparent between the two clusters, we observed diminished signal intensities in response to KD of either AR isoform. This indicates an interdependent binding of ARv7 and ARfl, in agreement with our previous finding (Figure 3E). We next correlated the cluster-specific AR cistromes with the previously determined AR transcriptomes (Figures 2A and 2B). As this analysis depends on a stringent peak-to-gene association, we only considered targets significantly dysregulated (DEseq; p <0.05) upon AR KD, localized within 10 kb of an AR-binding site (Table S4). For cluster I targets, the fold changes for the majority of genes was <0 (Figure 5D), consistent with these genes being activated by either AR isoform. In contrast, cluster II was biased toward shARv7 upregulated (fold change >0) and shARfl downregulated genes (fold change <0) (Figure 5D), which suggests that cluster II is predominantly associated with ARv7-repressed genes. These findings indicate that ARv7-dependent gene repression is a consequence of ARv7-mediated inhibition of H3K27ac.
Figure 5. ARv7 Negatively Regulates H3K27ac and FOXA1 Chromatin Binding.

(A–C) Top: heatmaps of H3K27ac (A), ARv7 (B), and ARfl ChIP-seq (C) in indicated cells, depicting only signals for AR “high-confidence” binding sites (union of ARN, Arv7, and ARfl peaks). Clusters (I and II) are based on the H3K27ac data. Bottom: bar graphs of the average normalized ChIP-seq counts, as indicated. Data are the mean of each cell line data within each cluster ±SEM (cluster I, n = 3,284 peaks; cluster II, n = 4,268 peaks). ****p < 0.0001 by ANOVA and Tukey’s HSD.
(D) Violin plots of the differentially expressed (adjusted p value <0.05) ARv7-target (blue) and ARfl-target (red) genes (within 10 kb of an AR site). The number of up- and downregulated genes and median values (black dot) are shown. The Fisher’s exact test between clusters has a p = 0.074 for shARv7 and p = 1 for shARfl.
(E) Histogram of the fraction of ChIP-seq peaks in clusters I and II from (A) that contain an AR-binding (left) or FOXA1-binding motif (right).
(F) Heatmaps (left) and bar graphs (right) for FOXA1 ChIP-seq in indicated cell lines and clusters, as in (A) to (C).
To identify additional genomic factors involved in ARv7-dependent repression, we examined the underlying DNA sequences in each cluster. Although we identified the AR-binding motif as the top sequence in both clusters, its enrichment was much higher in cluster I than in cluster II (Figure 5E). This suggests that the differences in the isoform transcriptomes may be influenced by the strength of the AR-binding motif. The second most common binding motif identified was that of FOXA1, a known determinant of AR action (Jin et al., 2014; Sahu et al., 2011). In contrast to the AR motif, enrichment of the FOXA1 motif was identical across the two clusters (Figure 5E). FOXA1 binding increased across both clusters in response to AR isoform depletion, albeit significantly more for ARv7 compared with ARfl KD (Figure 5F), with no apparent changes in FOXA1 level (Figure S5B). In addition, significantly higher FOXA1 levels were also observed at ARfl and ARv7 shared sites (n = 2,629) compared with ARfl-only sites (n = 4,737) (Figures S3D, S3E, and S5C). Combined, these results suggest that ARv7 (and to a lesser extent ARfl) reprogram the FOXA1 cistrome. Since this phenotype is observed at both positively and negatively regulated ARv7 sites (i.e., clusters I and II), it is likely an important downstream effect of AR isoform depletion. In summary, these data suggest that the ARv7 repressive function stems from binding NCOR corepressors, which results in negative regulation of H3K27ac.
ARv7 Represses Negative Regulators of Tumor Cell Proliferation
To better understand the clinical relevance of the ARv7 repressor function in PCa, we compared the LNCaP95-derived ARv7 gene signature with gene expression data from CRPC biopsies. The ARv7 gene signature was defined as shARv7-dysregulated genes (DEseq; p < 0.05) with an ARv7-binding site within 50 kb of their transcription start site; this resulted in 118 ARv7-activated and 157 ARv7-repressed genes (Table S5). ARv7 status in patient biopsies was determined by immunohistochemistry (IHC) (Figures S6A–S6C) and classified into low or high ARv7 expression. However, only the highest and lowest quartiles, based on ARv7 IHC score, were considered for subsequent analyses. Significance analysis of microarrays (SAM) (Tusher et al., 2001) was used to define gene expression differences between patient groups (using unpaired, two-sample t tests) and controlled for multiple testing by q value estimation, using the false discovery rate (FDR) method. Genes were then ranked according to their t test score and compared with the cell line-derived ARv7 gene signatures using GSEA (Subramanian et al., 2005). We detected a significant negative distribution of the ARv7-repressed signature within the entire dataset (p = 0.003), but no significant correlation with the ARv7-activated signature (p = 0.193; Figures 6A and 6B; Table S5). This suggests that ARv7-dependent repression observed in cells is also present in CRPC cases (with high levels of ARv7). As a control, we also performed a GSEA-based comparison between the ARfl gene signatures (shARfl-dysregulated genes within 50 kb of an ARfl-binding site; DEseq: p < 0.05; fold change >1.5) and the patient gene expression profiles. As expected, only ARfl-activated (n = 82; p = 0), but not ARfl-repressed (n = 55; p = 0.705), genes were significantly associated with gene expression in CRPC (Figure 6A and Table S5). Taken together, these analyses suggest that both ARfl-dependent gene activation and ARv7-dependent gene repression are prominent features of CRPC.
Figure 6. ARv7 Represses Genes with a Tumor-Suppressive Function.

(A) GSEA of ARfl- and ARv7-specific gene signatures (Table S5), compared with genes ranked by transcriptome data from CRPC tumors with IHC-defined ARv7 expression. The number of genes in each signature is indicated in parentheses. The normalized enrichment score (NES), the nominal p value (NOM p), and the FDR q value (FDR q) are shown.
(B) GSEA-determined enrichment profile of the ARv7-repressed gene signature (ARv7 rep) as shown in (A). Genes are ranked by their expression in IHC-defined ARv7 high (red, left, ARv7 pos. corr.) versus low patient tumors (blue, right, ARv7 neg. corr.).
(C) Heatmap of relative expression of 57 target genes defined by the leading edge in (B), in shGFP and shARv7 LNCaP95 cells. Genes in red were also identified in (D).
(D) Hockey-stick plot of positively selected genes identified in a genome-wide CRISPR KO screen. Genes were ranked according to their r score and log10 p value. Target genes from the ARv7-repressed gene signature in (A) are indicated, and genes highlighted in red overlap with the leading-edge analysis in (C).
(E) Hierarchical clustering of the 4 ARv7-target genes from (D) on gene expression data from patient samples (Decipher-GRID). Three clusters, based on the average expression of the four genes, are shown: low (blue), mixed (orange), and high (red).
(F) Kaplan-Meier graphs of prostate-specific antigen recurrence-free survival for the three patient clusters defined in (E). Log-rank test: n.s, not significant; ****p < 0.0001.
(G and H) Expression of the 4 ARv7-regulated genes in (D) in patient subgroups (Decipher-GRID). Samples are grouped by patients that did (n = 492) or did not (n = 1,134) develop metastasis (G), or did (n = 236) or did not (n = 1,211) succumb to the tumor (PCa-specific mortality; H). Standard t test: ***p % 0.001, ****p % 0.0001. Box plots show the median, and the first and third quartile. Whiskers extend to 1.5 the interquartile range and data beyond that are shown as individual points.
To elucidate the function of ARv7 repression, we focused on the subset of 57 genes at the core of the enrichment in our GSEA analysis (“leading-edge”; Figure S6D). As expected, ARv7 KD in the cells led to an upregulation of all “leading-edge” genes (Figure 6C), consistent with an ARv7-repressive function. To elucidate the function of these genes, we next compared them with positively selected genes from a genome-wide CRISPR knockout (KO) screen in LNCaP95 cells (MAGeCK; p < 0.05, Figure 6D and Table S6). We detected four ARv7-repressed genes with a negative effect on CRPC cell proliferation (SLC30A7, B4GALT1, HIF1A, and SNX14) (Figure 6D), indicating that these genes may have anti-tumor functions. However, no unequivocal correlation between the expression of the four genes and genetic aberrations in AR or PTEN, as visualized with cbioportal (Cerami et al., 2012; Kumar et al., 2016), could be identified (Figure S6E). In support of the potential clinical importance of this finding, we observed in the Taylor (Taylor et al., 2010) and expanded Decipher-GRID cohorts (Benzon et al., 2017; Boormans et al., 2013; Den et al., 2014; Erho et al., 2013; Karnes et al., 2013; Klein et al., 2015; Ross et al., 2016; Taylor et al., 2010) that PCa patients with low expression of the four genes (Figures 6E and S6F) are at greater risk of disease recurrence than patients with high expression of all four genes (Figures 6F and S6G). Moreover, in the Taylor dataset, this finding was observed for patients with primary and metastatic disease (Figure S6G), and patients with primary disease only (Figure S6H). In addition, expression of the four genes was lower in metastatic than in primary disease (Figure S6I). Similarly, B4GALT1, SLC30A7, SNX14, and HIF1A expression was negatively correlated with metastasis development and PCa-specific mortality (Figures 6G and 6H). Taken together, these results suggest that ARv7 promotes CRPC progression by repressing genes that negatively regulate tumor growth, and are associated with poor PCa prognosis.
DISCUSSION
The goal of this study was to uncover how ARv7 contributes to CRPC development. In this article we show that ARv7 primarily acts as a transcriptional repressor, despite colocalization, inter-dependence, and potential heterodimerization of ARv7 with ARfl. This implies that ARv7 and ARfl have divergent transcriptional properties. These differences can be explained, in part, by the structural differences between the two isoforms. Although the N-terminal AF-1 domain harbors substantial transcriptional activity (Bevan et al., 1999; Ma et al., 1999), the C-terminal LBD, which ARv7 lacks, is necessary for optimal receptor activation. Besides its ability to be bound and activated by hormones, the LBD is also required for the intra- and intermolecular AR N/C-terminal interaction, which stabilizes the receptor, regulates coregulatory interactions (van Royen et al., 2007), and enhances its transactivation function (He et al., 2002). Moreover, the LBD also harbors the capacity to bind LXXLL- and FXXLF-containing coregulators, which in turn modulate the activity of the receptor (Huang et al., 1998; Matias et al., 2000).
In this work we observed that ARv7 preferentially associates with the NCOR transcriptional corepressors, whereas ARfl associates with both coactivators and corepressors, in agreement with the idea that ARfl and ARv7 display differential coregulatory binding repertoires. Moreover, increases in ARv7 and NCOR1,2 binding upon ARfl KD suggest that ARfl partially inhibits the ARv7/NCOR interaction, thereby limiting a repressive transcriptional response. We find that the difference in ARfl and ARv7 transcriptional activity is correlated with AR isoform-specific differences in H3K27 acetylation. Therefore, ARv7 likely functions by recruiting corepressors, such as NCOR1 and NCOR2, which in turn control the genomic recruitment of histone deacetylases, such as HDAC3 (Perissi et al., 2010), which negatively regulate H3K27 acetylation. Reprogramming of the FOXA1 cistrome following ARv7 depletion is probably an important consequence of AR inhibition, but may not be directly linked to ARv7-dependent repression, since it is also observed at ARv7-activated sites. This observation is further supported by the finding that ARv7 and FOXA1 are unable to interact by coIP (He et al., 2018; Wang et al., 2011). Moreover, additional ARv7 cooperating factors may also exist; HOXB13 was recently indicated as an important mediator of ARv7 function (Chen et al., 2018).
Despite having distinct functions, our findings suggest that ARv7 and ARfl preferentially heterodimerize on chromatin. In contrast to previous studies (Xu et al., 2015), we were unable to verify the presence of ARv7 homodimers by either ChIP-seq or FRET. There are a number of explanations for this discrepancy, including the difference between ectopically and endogenous expression of ARv7, as found in LNCaP95 cells. We did confirm that ectopically expressed ARv7 alone was able to induce transcription in an AR reporter assay (data not shown). This suggests that the function of ARv7 may differ depending on the abundance of ARfl protein. In general, ARv7 is expressed at much lower levels than ARfl in CRPC (Guo et al., 2009; Robinson et al., 2015), and no naturally occurring cell lines or tumor models solely expressing ARv7 have been identified. In CRPC cells, ARv7 production is tightly coupled to the transcription rate of the AR gene (Liu et al., 2014), and level of AR gene amplification (Henzler et al., 2016). The presence of large structural rearrangements of the AR locus can uncouple AR-V and ARfl production; it has been previously demonstrated that ARv567es is sufficient to drive CRPC proliferation in the absence of ARfl (Dehm et al., 2008; Nyquist et al., 2013). Therefore, differences in the genetic structure of the AR locus, as well as the total amount and relative ratios of all AR isoforms, will dictate the prevalence of each AR dimer.
In this study, we propose that the repressive function of ARv7 acts by negatively regulating genes that limit CRPC proliferation. Specifically, we found that SLC30A7, B4GALT1, HIF1A, and SNX14 have a negative impact on cell growth, while being repressed by ARv7. Interestingly, three of these genes have been previously associated with tumor-suppressor activities. Specifically, SLC30A7 deletion accelerates prostate tumor formation (Singh et al., 2016; Tepaamorndech et al., 2011) while SNX14 is commonly deleted in human cancers (Dong et al., 2008). Moreover, HIF1A, although primarily studied as an oncogene, also has tumor-suppressive activity (Chiavarina et al., 2010; Velasco-Hernandez et al., 2014). In PCa patients, expression of the four genes is negatively correlated with ARv7 protein levels and time to recurrence. This is consistent with the concept that ARv7 acts by repressing tumor-suppressive genes during PCa progression, and that expression of ARv7-repressed genes may serve as a biomarker to assess ARv7 inhibition.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be direct to and will be fulfilled by the Lead Contact, Myles Brown (myles_brown@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
LNCaP95 cells have been previously described (Hu et al., 2012) and were maintained in phenol red-free RPMI 1640 medium, supplemented with 10% charcoal/dextran-treated fetal bovine serum (FBS), 1% L-Glutamine and 1% penicillin/streptomycin. 22Rv1 cells were obtained from the American Type Culture Collection (ATCC) and were grown in regular RPMI 1640 medium, supplemented with 10% FBS, 1% L-Glutamine and 1% penicillin/streptomycin. For experiments requiring hormone starvation, 22Rv1 cells were grown in phenol red-free RPMI 1640 medium supplemented with 10% charcoal/dextran-treated FBS, 1% L-Glutamine and 1% penicillin/streptomycin for 72 hr prior to any subsequent vehicle or hormone addition. All cell lines were tested for mycoplasma using the MycoAlert mycoplasma detection kit (Lonza, Portsmouth, NH) and their identities were confirmed by short tandem repeat profiling (BioSynthesis, Lewisville, TX).
Clinical Patient Samples
A set of 59 metastatic tumors from 37 men with CRPC were obtained with informed consent through the University of Washington Prostate Cancer Donor Autopsy Program (Morrissey et al., 2013) and used for transcript profiling by microarray, as described (Kumar et al., 2016) using frozen tissues. Soft tissue tumors were laser capture-microdissected and bone metastases were sampled using a 1 mm diameter tissue punch. All procedures involving human subjects were approved by the Institutional Review Board (IRB) of the University of Washington and of the Fred Hutchinson Cancer Research Center.
METHOD DETAILS
Generation of Doxycycline-Inducible shARfl or shARv7 Cell Lines
Doxycycline (dox)-inducible cell lines were generated using lentiviral vectors in pLKO-Tet-On backbones (Wiederschain et al., 2009) targeting GFP, ARfl (exon 8) or ARv7 (cryptic exon 3). Oligonucleotides containing the shRNA sequences, and AgeI and EcoRI restriction enzyme sites compatible with cloning into the pLKO-Tet-On vector, were designed using following primers (shRNA sequences are shown in bold): shGFP forward:
5’-CCGGGCAAGCTGACCCTGAAGTTCACTCGAGTGAACTTCAGGGTCAGCTTGCTTTTTG-3’shGFP reverse:
5’-AATTCAAAAAGCAAGCTGACCCTGAAGTTCACTCGAGTGAACTTCAGGGTCAGCTTGC-3’shARfl forward:
5’-CCGGCCTGCTAATCAAGTCACACATCTCGAGATGTGTGACTTGATTAGCAGGTTTTTG-3’shARfl reverse:
5’-AATTCAAAAACCTGCTAATCAAGTCACACATCTCGAGATGTGTGACTTGATTAGCAGG-3’shARv7 forward:
5’-CCGGGTAGTTGTGAGTATCATGACTCGAGTCATGATACTCACAACTACTTTTTG-3’shARv7 reverse:
5’-AATTCAAAAAGTAGTTGTGAGTATCATGACTCGAGTCATGATACTCACAACTAC-3’
Oligonucleotides were annealed in 1x annealing buffer (1 M NaCl, 100 mM Tris-HCl, pH 7.4) and heated for 5 min at 95°C. Annealed oligos were ligated into pre-digested (EcoRI and AgeI) pLKO-Tet-On lentiviral plasmid and subsequently transformed into Stbl3 competent bacteria. Lentiviral particles were generated using calcium phosphate, and transfer, VSV-G envelope (pMD2G, Addgene #12259) and packaging vectors (pCMVR8.74, Addgene #22036) (Barde et al., 2010). Prostate cancer cells were infected with unconcentrated virus and selected with 1 mg/ml puromycin. shRNA expression was induced by treating cells with 1 mg/ml dox for at least 72 hr.
siRNA Transfection
ON-TARGETplus siRNA for non-targeting siCtrl, siNCOR1, siNCOR2, and siNRIP1 was purchased from GE Dharmacon.
| Name | Catalog | Sequence |
|---|---|---|
| siCtrl_1 | D-001810-01-20 | 5’-UGGUUUACAUGUCGACUAA-3’ |
| siCtrl_2 | D-001810-02-20 | 5’-UGGUUUACAUGUUGUGUGA-3’ |
| siNCOR1_1 | J-003518-06-0002 | 5’-GCUGAGGGCUUCUGCAGAU-3’ |
| siNCOR1_2 | J-003518-07-0002 | 5’-GGAAAGUCCUCCCAUACGA-3’ |
| siNCOR2_1 | J-020145-10-0002 | 5’-CAGCCAGGGAAGACGCAAA-3’ |
| siNCOR2_2 | J-020145-11-0002 | 5’-AGGCAUCCCAGGACCGAAA-3’ |
| siNRIP1_1 | J-006686-05-0002 | 5’-GAAGCGUGCUAACGAUAAA-3’ |
| siNRIP1_2 | J-006686-06-0002 | 5’-AGAAGGAUGUUGGCAGUUA-3’ |
Transfection of siRNA was performed using Lipofectamine RNAiMax (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Cell Proliferation Assays
For the proliferation assays, indicated cell lines were plated in a 24-well plate format at 2×104 cells/well. Unless otherwise stated, cells were induced for 3 days with dox prior to cell growth assessment. Cell growth was determined for indicated times by trypan blue exclusion using a hemocytometer, or by using the direct cell count function on a Celigo Imaging Cytometer (Nexcelom Bioscience). 3D cell proliferation assay was performed using poly(ethylene glycol) diacrylate 575 cryogels (Gö ppert et al., 2016), incubated with 2.5 × 105 cells in a 12-well plate format. Dox was added one day after seeding. Cryogels were removed at indicated time points and cells lysed using 10 mM Tris-HCl pH 7.5, 1 mM EDTA, 1% sodium dodecyl sulfate and 200 mg/ml Proteinase K (Sigma-Aldrich). DNA was extracted and quantified and cell growth visualization was carried out using scanning electron microscopy.
Tissue Microarray
Tissue microarray was performed on whole human genome Agilent microarrays at the University of Washington (TMA 55). IHC was performed using the rabbit ARv7 monoclonal antibody clone RM7 (RevMab). Antigen retrieval was achieved by microwaving slides in citrate buffer (pH 6.0) for 18 min at 800 W. Endogenous peroxidase was blocked using 3% H2O2 solution. Blocking was performed using the protein block solution from the Novolink polymer detection system (Leica, Wetzlar, Germany). EP343 was diluted 1:200 and the tissue was incubated for 1 hr. The reaction was visualized using the Novolink polymer and DAB chromogen. Nuclear ARv7 on TMA slides were scanned with an Aperio ScanScope AT2 (Leica Biosystems Pathology Imaging, Vista, CA) at 40x (0.25 microns/ pixel), and stored on a server running Aperio eSlide Manager digital slide repository and database software.
Protein Assays
Proteins for Western blotting were isolated using TIVE lysis buffer (50 mM Tris-HCl pH 7.8, 2 mM EDTA, 150 mM NaCl, 1% NP-40, protease inhibitors), or lysis buffer A (10 mM HEPES pH 7.5, 10 mM KCl, 0.1 mM EGTA, 0.1 mM EDTA, 1 mM DTT, protease inhibitors) plus 0.5% NP-40 and lysis buffer C (20 mM HEPES pH 7.5, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% Glycerol, 1 mM DTT, protease inhibitors) for total, cytoplasmic and nuclear proteins respectively. Co-immunoprecipitation (co-IP) was carried using the nuclear protein fraction, similarly to previously described (Jehle et al., 2014) and ARv7 antibody-coupled Protein A Dynabeads (Thermo Fisher Scientific). Co-IP experiments with formaldehyde crosslinked material was carried out using isolated chromatin samples, as described below. Western blotting was carried out using following antibodies: AR (N20 and 441, Santa-Cruz Biotechnologies; PG21, Millipore), AR C-terminus (SP242, Spring Bioscience; C-19, Santa-Cruz Biotechnologies), ARv7 (RM7, RevMAb; H6 253, Epitomics), NCOR1 (PA1–844A, Invitrogen), NCOR2 (ab24551, Abcam), FOXA1 (ab5089, Abcam), b-Actin (Abcam) and Lamin B1 (EPR8985, Abcam), and secondary antibody IgG fraction monoclonal mouse anti-rabbit IgG, light chain specific (211–032-171, Jackson ImmunoResearch).
RNA-Sequencing (RNA-seq)
Unless otherwise stated, cells were induced for 3 days with dox and treated with vehicle (ETOH) or DHT for 4 hr prior to RNA isolation. Total genomic RNA was isolated using TRIzol (Thermo Fisher Scientific) and the RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. mRNA libraries were generated by the Center for Functional Cancer Epigenetics (Dana-Farber Cancer Institute (DFCI)) using 1 mg of total RNA and the Illumina TruSeq stranded mRNA sample kit. Libraries were sequenced on the Illumina NextSeq 500 platform at the Molecular Biology Core Facility (DFCI).
ChIP-Sequencing (ChIP-seq)
Unless otherwise stated, cells were induced for 3 days with dox and were cultured without hormone prior to ChIP. Cells were then crosslinked with 1% formaldehyde and chromatin sonicated to 300–500 bp. ChIP was carried out using Protein A/G Dynabeads (Thermo Fisher Scientific) coupled to antibodies against N-terminal AR (N-20; Santa-Cruz Biotechnology), C-terminal AR (Clone SP242; Spring Bioscience), ARv7 (H6 253, Epitomics), H3K27ac (C15410196, Diagenode) or FOXA1 (Mix of ab23738 and ab5089; Abcam). ChIP DNA was purified using the PCR purification kit (Qiagen). ChIP-re-ChIP was carried out as described for ChIP, but releasing the ChIP DNA with 10 nM DTT (30 min incubation at 37°C) prior to subsequent ChIP with a different antibody. ChIP-seq libraries were generated using the ThruPLEX DNA-seq Kit (Rubicon Genomics) according to the manufacturer’s instructions and standard 8 bp Illumina primers. Libraries were pooled and sequenced on the Illumina NextSeq 500 platform at the Molecular Biology Core Facility (DFCI).
FRET and FRAP
N-terminal EYFP-AR and C-terminal ECFP-AR were previously generated (van Royen et al., 2012). C-terminal ECFP-ARv7 was generated by cloning CFP into pCerulean-(GA)6-ARv7. N-terminal ARv7-EYFP was generated by cloning YFP into pARv7-(GA)6-Venus. The AR R585K mutation was generated by site-directed mutagenesis of pEYFP-AR-ECFP, using the following primers:
5’-GTCTTCTTCAAAAAAGCCGCTGAAGGG-3’ (forward)
5’-CCCTTCAGCGGCTTTTTTGAAGAAGAC-3’ (reverse)
To obtain single-tagged DBD mutant AR, the DBD of pYFP-(GA)6-AR was replaced with R58K-DBD from pEYFP-R585K_AR-ECFP. The ARv7-R585K mutant was generated by site-directed mutagenesis of pYFP-(GA)6-ARv7 using primers:
5’-AGCAAAAATGATTGCACTATTGAT-3’ (forward)
5’-GGCGCACAGGTACTTCTGTTT-3’(reverse)
Plasmids were transfected in the presence of 1 nM DHT into Hep3B cells using FuGENE6 (Promega). Co-expression of ARfl and ARv7 was confirmed for each cell measurement, based on CFP signal intensities. Confocal live cell imaging was performed using a Zeiss LSM510 confocal laser scanning microscope equipped with a Plan-Neofluar 40x/1.3 NA oil objective (Carl Zeiss). In acceptor photobleaching experiments (abFRET), CFP and YFP were scanned once before photobleaching (CFP ex: 458 nm, em: 470–500 nm BP; YFP ex: 514 nm, 560 nm LP). YFP was then bleached 25 times at 100% laser power at 514 nm. New images were collected after photobleaching to calculate apparent abFRET eficiency. For FRAP, YFP was scanned 1.9 msec/line of 512 pixels. A 10-pixel wide strip was selected in the middle of the nucleus for photobleaching. The fluorescent signal was monitored at 21 msec time intervals by scanning the region of interest for 40 secs at low excitation. When 200 iterations were completed, the strip was bleached for 2 iterations at maximum intensity.
MARCoNI Assay
The MARCoNI assay was performed on PamChIP #88101 arrays (PamGene International), using whole cell lysates and AR antibody 441 (sc-7305, Santa-Cruz Biotechnology) for detection, and FITC-conjugated swine anti-rabbit (F0205, DAKO) for visualization. Cells were grown with dox and induced for 30 min with 10 nM DHT prior to cell lysis.
CRISPR Screen
Pooled genome-wide CRISPR screens were performed using ~2×108 LNCaP95 cells infected with pooled lentiviral GeCKO v2 library (Fei et al., 2017). After three days of puromycin (2 mg) selection, the surviving cells were divided into three groups (0 day control, vehicle, and 10 nM DHT treatment) and cultured for four weeks prior to genomic DNA extraction. The genomic DNA was subsequently amplified using two rounds of PCR and each library was sequenced at 30–40 million reads to achieve ~300× coverage of the CRISPR library.
QUANTIFICATION AND STATISTICAL ANALYSIS
Tissue Microarray Analysis
Using Aperio ImageScope software, ARv7-stained TMA slides were annotated by outlining each TMA spot on a separate annotation layer to create regions of interest for analysis. Quantitative image analysis of the annotated regions of interest was performed using Aperio Brightfield Image Analysis Toolbox software (Leica Biosystems Pathology Imaging, Vista, CA). The data for each TMA spot was extracted into Microsoft Excel for further analysis. The quantitative analysis data for each TMA spot included total numbers and percentages of nuclei (positive and negative), average positive intensity, average positive optical density, and area of analysis (Krajewska et al., 2009; Rizzardi et al., 2012). The TMA 55 cores were then categorized into low and high ARv7 groups by selecting those with the highest and lowest quartiles of ARv7 IHC score. The Significance Analysis of Microarrays (SAM) program (Tusher et al., 2001) was used to analyze expression differences between groups using unpaired, two-sample t-tests and controlled for multiple testing by estimation of q-values using the false discovery rate (FDR) method.
Patient Data Clustering and Analysis of Recurrence-free Survival
Patients from the Decipher-GRID cohorts (Benzon et al., 2017; Boormans et al., 2013; Den et al., 2014; Erho et al., 2013; Karnes et al., 2013; Klein et al., 2015; Ross et al., 2016; Taylor et al., 2010) were clustered according to the expression (mRNA; Z-score) of the four tumor suppressive genes (SLC30A7, B4GALT1, HIF1A and SNX14). Recurrence-free survival was assessed using the Kaplan-Meier method. Patients were censored at the time of their last clinical, tumor-free, follow-up visit. Time to PSA recurrence (as defined by each study individually) was selected as the clinical endpoint.
Western Blot Quantification
Western blots were quantified calculating the average grey level for each band after background subtraction, using a custom script in ImageJ (Schneider et al., 2012).
RNA-seq Analysis
Differential gene expression was determined using the Visualization Pipeline for RNA-seq (VIPER)(Cornwell et al., 2018) and DEseq (Anders and Huber, 2010).
ChIP-seq Analysis
ChIP-seq data was aligned to the hg19 genome and ChIP-seq peaks determined using the ChIP-seq data quality and analysis pipe-line 2 (Qin et al., 2016). Read counts for cluster analyses were quantified using the bamliquidator tool (Loven et al., 2013; Whyte et al., 2013), with read counts being subsampled to match the lowest read count for each antibody. While evaluating the expression of the genes associated with the clusters only genes that could be associated to a unique cluster were considered. DNA motif analysis was performed using SeqPos (He et al., 2010).
FRET and FRAP Analysis
Apparent abFRET efficiencies were calculated using the equation: abFRET=((CFPafter-CFPbefore) *YFPbefore)/((CFPafter*YFPbefore)-(YFPafter*CFPbefore)). Apparent abFRET efficiencies were normalized to the negative (CFP and YFP single fluorescent plasmid) and positive controls (CFP-YFP fusion protein). FRAP efficiencies were calculated according to this formula: Inorm, t = (It-Ibackground)/ (Iprebleach-Ibackground).
MARCoNI Assay Analysis
Binding was determined through array image analysis, consisting of automated spot finding, quantification and background subtraction, using the BioNavigator software (PamGene International).
CRISPR Screen Analysis
CRISPR screen sequencing data was analyzed using MAGeCK (Li et al., 2014).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-AR (N-20) | Santa Cruz Biotechnologies | Cat# sc-816, RRID:AB_1563391 |
| Mouse monoclonal anti-AR (441) | Santa-Cruz Biotechnologies | Cat# sc-7305, RRID:AB_626671 |
| Rabbit polyclonal anti-AR (PG21) | Millipore | Cat# 50-172-110 |
| Rabbit polyclonal C-terminal AR (Cone SP242) | Spring Bioscience | Cat# M5422 |
| Rabbit polyclonal C-terminal AR (C-19) | Santa Cruz Biotechnologies | Cat# sc-815, RRID:AB_630864 |
| Rabbit monoclonal ARv7 (H6 253) | Epitomics (now Abcam) | Cat# ab198394 |
| Rabbit monoclonal anti-ARv7 (clone RM7) | RevMab | Cat# 31-1109-00, RRID:AB_2716436 |
| Rabbit polyclonal NCOR1 (PA1-844A) | Thermo Fisher Scientific | Cat# PA1-844A, RRID:AB_2149004 |
| Rabbit polyclonal NCOR2 (ab24551) | Abcam Abcam | Cat# ab24551, RRID:AB_2149134 |
| Rabbit polyclonal anti-H3K27ac (C15410196) | Diagenode | 15410196;RRID:AB_2637079 |
| Rabbit polyclonal FOXA1 (ab23738) | Abcam | Cat# ab23738, RRID:AB_2104842 |
| Goat polyclonal FOXA1 (ab5089) | Abcam | Cat# ab5089, RRID:AB_304744 |
| Mouse monoclonal b-Actin | Abcam | Cat# ab8226 |
| Rabbit monoclonal Lamin B1 (EPR8985) | Abcam | Cat# ab194109 |
| Monoclonal mouse anti-rabbit IgG, light chain specific | Jackson ImmunoResearch | Cat# 211-032-171 |
| FITC-conjugated swine anti-rabbit | DAKO | Cat# F0205 |
| Biological Samples | ||
| CRPC tumor metastases samples | University of Washington and Fred Hutchinson Cancer Research Center | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DHT (5α-androstan-17β-ol-3-one) | Sigma | Cat# A8380 |
| Critical Commercial Assays | ||
| TruSeq stranded mRNA sample kit | Illumina | Ref# 20020594 |
| ThruPLEX DNA-seq kit | Rubicon Genomics | Ref# R400427 |
| Deposited Data | ||
| RNA-seq | This paper | GEO: GSE106560 |
| ChIP-seq | This paper | GEO: GSE106559 |
| CRPC tumor metastases samples | Kumar et al., 2016 | GEO: GSE77930 |
| Genome-wide expression profiling of radical prostatectomies | Den et al., 2014 | GEO: GSE72291 |
| Genome-wide expression profiling of radical prostatectomies | Ross et al., 2016 | GEO: GSE79957 |
| Genome-wide expression profiling of radical prostatectomies | Benzon et al., 2017 | GEO: GSE79956 |
| Genome-wide expression profiling of radical prostatectomies | Karnes et al., 2013 | GEO: GSE62116 |
| Genome-wide expression profiling of radical prostatectomies | Erho et al., 2013 | GEO: GSE46691 |
| Genome-wide expression profiling of radical prostatectomies | Klein et al., 2015 | GEO: GSE62667 |
| Genome-wide expression profiling of radical prostatectomies | Boormans et al., 2013 | GEO: GSE41408 |
| Genome-wide expression profiling of radical prostatectomies | Taylor et al., 2010 | GEO: GSE21032 |
| Taylor dataset | Taylor et al., 2010 | N/A |
| Experimental Models: Cell Lines | ||
| Human: LNCaP95 | Hu et al., 2012 | N/A |
| Human: LNCaP95 shGFP | This paper | N/A |
| Human: LNCaP95 shARfl | This paper | N/A |
| Human: LNCaP95 shARv7 | This paper | N/A |
| Human: 22Rv1 | ATCC | Cat# CRL-2505 |
| Human: 22Rv1 shGFP | This paper | N/A |
| Human: 22Rv1 shARfl | This paper | N/A |
| Human: 22Rv1 shARv7 | This paper | N/A |
| Human: Hep3B | ATCC | Cat# HB-8064 |
| Oligonucleotides | ||
| shGFP-forward: 5’-CCGGGCAAGCTGACCCTGAAGTTCACTCG AGTGAA CTTCAGGGTCAGCTTGCTTTTTG-3’ | This paper | N/A |
| shGFP-reverse: 5’-AATTCAAAAAGCAAGCTGACCCTGAAGTT CACTCGAGTGAACTTCAGGGTCAGCTTGC-3’ | This paper | N/A |
| shARfl-forward: 5’-CCGGCCTGCTAATCAAGTCACACATCTCG AGATGTGTGACTTGATTAGCAGGTTTTTG-3’ | This paper | N/A |
| shARfl-reverse: 5’-AATTCAAAAACCTGCTAATCAAGTCACAC ATCTCGAGATGTGTGACTTGATTAGCAGG-3’ | This paper | N/A |
| shARv7-forward: 5’-CCGGGTAGTTGTGAGTA TCATGACTCGAGTCATGATACTCACAACTACTTTTTG-3’ | This paper | N/A |
| shARv7-reverse: 5’-AATTCAAAAAGTAGTTGTG AGTATCATGACTCGAGTCATGATACTCACAACTAC-3’ | This paper | N/A |
| siCtr_1: 5’-UGGUUUACAUGUCGACUAA-3’ | GE Dharmacon | Cat# D-001810-01-20 |
| siCtr_2: 5’-UGGUUUACAUGUUGUGUGA-3’ | GE Dharmacon | Cat# D-001810-02-20 |
| siNCOR1_1: 5’-GCUGAGGGCUUCUGCAGAU-3’ | GE Dharmacon | Cat# J-003518-06-0002 |
| siNCOR1_2: 5’-GGAAAGUCCUCCCAUACGA-3’ | GE Dharmacon | Cat# J-003518-07-0002 |
| siNCOR2_1: 5’-CAGCCAGGGAAGACGCAAA-3’ | GE Dharmacon | Cat# J-020145-10-0002 |
| siNCOR2_2: 5’-AGGCAUCCCAGGACCGAAA-3’ | GE Dharmacon | Cat# J-020145-11-0002 |
| siNRIP1_1: 5’-GAAGCGUGCUAACGAUAAA-3’ | GE Dharmacon | Cat# J-006686-05-0002 |
| siNRIP1_2: 5’-AGAAGGAUGUUGGCAGUUA-3’ | GE Dharmacon | Cat# J-006686-06-0002 |
| Primers for site-directed mutagenesis of pEYFP-(GA)6-ARv7 (forward): 5’-AGCAAAAATGATTGCA CTATTGAT-3’ | This paper | N/A |
| Primers for site-directed mutagenesis of pEYFP-(GA)6-ARv7 (reverse): 5’-GGCGCACAGGTACT TCTGTTT-3’ | This paper | N/A |
| Recombinant DNA | ||
| pLKO-TET-ON | Addgene | Cat# 21915 |
| LentiCRISPR v2 | Fei et al., 2017 | N/A |
| pEYFP-(GA)6-AR R585K | This paper | N/A |
| pECFP-(GA)6-AR | van Royen et al., 2012 | N/A |
| pEYFP-(GA)6-AR | van Royen et al., 2012 | N/A |
| pEYFP-(GA)6-ARv7 R585K | This paper | N/A |
| pECFP-(GA)6-ARv7 | This paper | N/A |
| pEYFP-(GA)6-ARv7 | This paper | N/A |
| Software and Algorithms | ||
| Aperio ImageScope | Leica Biosystems | N/A |
| Aperio Brightfield Image Analysis Toolbox | Leica Biosystems | N/A |
| SAM | Tusher et al., 2001 | N/A |
| ImageJ | Schneider et al., 2012 | N/A |
| VIPER | Cornwell et al., 2018 | https://bitbucket.org/cfce/viper/ |
| DEseq | Anders and Huber, 2010 | N/A |
| CHIPS 2 | Qin et al., 2016 | N/A |
| Bamliquidator | Loven et al., 2013; Whyte et al., 2013 | N/A |
| SeqPos | He et al., 2010 | N/A |
| MAGeCK | Li et al., 2014 | N/A |
Highlights.
ARfl and ARv7 genomic binding is interdependent and colocalized
ARv7, unlike ARfl, preferentially represses transcription
Expression of ARv7-repressed genes negatively correlates with recurrence
Re-expression of ARv7-repressed genes may serve as a biomarker of ARv7 inhibition
Significance.
Development of resistance to androgen receptor (AR)-targeted therapy remains a challenge in treating advanced prostate cancer. Our work reveals that the hormone-independent AR splice variant 7 (ARv7) contributes to this process by repressing the transcription of genes with tumor-suppressive activity. Thus, targeting ARv7 may improve currently available prostate cancer therapies by restoring expression of these genes that can serve as biomarkers of ARv7 inhibition.
ACKNOWLEDGMENTS
We thank David Labbé , Teng Fei, Jin Zhao, Didier Trono, Renato Paro, and members of his group for helpful discussions, advice, and technical help. We thank Jutta Stober and Alban Muslija for help with 3D cell culture. We also thank the MBCF (DFCI), particularly Zach Herbert, for help with Illumina sequencing. This work was supported by the Claudia Adams Barr Foundation (L.C.), the Prostate Cancer Foundation (L.C., A.C.B.C., P.S.N., S.R.P., and M.B.) and NIH grant P01CA163227 (L.C., S.P.B., P.S.N., S.R.P., and M.B.). S.R.P. and P.S.N. are funded by the Pacific NW Prostate Cancer SPORE (P50 CA97186), and S.R.P. is additionally funded by DOD W81XWH-13-1-0093 and the Veterans Affairs Research Service. P.S.N. is funded by DOD W81XWH-15-1-0430 and W81XWH-15-1-0562, A.C.G. was funded by the Swiss National Science Foundation (Ambizione grant PZ00P3_167938). X.S.L. is funded by NIH R01HG008927 and DOD PC140817P1 (to MB and XSL). S.P.B. and M.B. were funded by the DF/HCC Prostate SPORE (P50 CA090381).
M.B. has been a consultant for GTx and Novartis. He receives sponsored research support from Novartis and serves on the Scientific Advisory Board of Kronos Bio. L.C. and A.C.G. are employees of Sanofi and Basilea Pharmaceutica, respectively.
Footnotes
DECLARATION OF INTERESTS
The other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and six tables and can be found with this article online at https://doi.org/10.1016/j.ccell.2019.01.008.
REFERENCES
- Anders S, and Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Zhu Y, Silberstein JL, Taylor MN, Maughan BL, Denmeade SR, et al. (2017). Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J. Clin. Oncol 35, 2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al. (2014). AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med 371, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde I, Salmon P, and Trono D. (2010). Production and titration of lentiviral vectors. Curr. Protoc. Neurosci. Chapter 4, Unit 4.21 10.1002/0471142905.hg1210s54. [DOI] [PubMed] [Google Scholar]
- Benzon B, Zhao SG, Haffner MC, Takhar M, Erho N, Yousefi K, Hurley P, Bishop JL, Tosoian J, Ghabili K, et al. (2017). Correlation of B7-H3 with androgen receptor, immune pathways and poor outcome in prostate cancer: an expression-based analysis. Prostate Cancer Prostatic Dis. 20, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernemann C, Schnoeller TJ, Luedeke M, Steinestel K, Boegemann M, Schrader AJ, and Steinestel J. (2017). Expression of AR-V7 in circulating tumour cells does not preclude response to next generation androgen deprivation therapy in patients with castration resistant prostate cancer. Eur. Urol 71, 1–3. [DOI] [PubMed] [Google Scholar]
- Bevan CL, Hoare S, Claessens F, Heery DM, and Parker MG (1999). The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol. Cell. Biol 19, 8383–8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boormans JL, Korsten H, Ziel-van der Made AJ, van Leenders GJ, de Vos CV, Jenster G, and Trapman J. (2013). Identification of TDRD1 as a direct target gene of ERG in primary prostate cancer. Int. J. Cancer 133, 335–345. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SC, Selth LA, Li Y, Nyquist MD, Miao L, Bradner JE, Raj GV, Tilley WD, and Dehm SM (2015). Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. 43, 5880–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Wu D, Thomas-Ahner JM, Lu C, Zhao P, Zhang Q, Geraghty C, Yan PS, Hankey W, Sunkel B, et al. (2018). Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc. Natl. Acad. Sci. U S A 115, 6810–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, Tanowitz HB, Casimiro MC, Wang C, Pestell RG, et al. (2010). HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: autophagy drives compartment-specific oncogenesis. Cell Cycle 9, 3534–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornwell M, Vangala M, Taing L, Herbert Z, Koster J, Li B, Sun H, Li T, Zhang J, Qiu X, et al. (2018). VIPER: visualization pipeline for RNA-seq, a Snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinformatics 19, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB Jr., Saad F, et al. (2011). Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med 364, 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, and Tindall DJ (2008). Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 68, 5469–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den RB, Feng FY, Showalter TN, Mishra MV, Trabulsi EJ, Lallas CD, Gomella LG, Kelly WK, Birbe RC, McCue PA, et al. (2014). Genomic prostate cancer classifier predicts biochemical failure and metastases in patients after postoperative radiation therapy. Int. J. Radiat. Oncol. Biol. Phys 89, 1038–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet SJ, Dejager L, Clarisse D, Thommis J, Melchers D, Bastiaensen N, Ruijtenbeek R, Beck IM, Libert C, Houtman R, et al. (2014). Cofactor profiling of the glucocorticoid receptor from a cellular environment. Methods Mol. Biol 1204, 83–94. [DOI] [PubMed] [Google Scholar]
- Dong X-Y, Rodriguez C, Guo P, Sun X, Talbot JT, Zhou W, Petros J, Li Q, Vessella RL, Kibel AS, et al. (2008). SnoRNA U50 is a candidate tumor suppressor gene at 6q14.3 with a mutation associated with clinically significant prostate cancer. Hum. Mol. Genet 17, 1031–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, Bergstralh EJ, Kollmeyer T, Fink S, Haddad Z, et al. (2013). Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS One 8, e66855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei T, Chen Y, Xiao T, Li W, Cato L, Zhang P, Cotter MB, Bowden M, Lis RT, Zhao SG, et al. (2017). Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc. Natl. Acad. Sci. U S A 114, E5207–E5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gö ppert B, Sollich T, Abaffy P, Cecilia A, Heckmann J, Neeb A, Backer A, Baumbach T, Gruhl FJ, and Cato AC (2016). Superporous Poly(ethylene glycol) diacrylate cryogel with a defined elastic modulus for prostate cancer cell research. Small 12, 3985–3994. [DOI] [PubMed] [Google Scholar]
- Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper CG, et al. (2009). A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 69, 2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Lee LW, Minges JT, and Wilson EM (2002). Dependence of selective gene activation on the androgen receptor NH2- and COOH-terminal inter-action. J. Biol. Chem 277, 25631–25639. [DOI] [PubMed] [Google Scholar]
- He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, et al. (2010). Nucleosome dynamics define transcriptional enhancers. Nat. Genet 42, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Lu J, Ye Z, Hao S, Wang L, Kohli M, Tindall DJ, Li B, Zhu R, Wang L, et al. (2018). Androgen receptor splice variants bind to constitutively open chromatin and promote abiraterone-resistant growth of prostate cancer. Nucleic Acids Res. 46, 1895–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, Liu G, Coleman I, Lakely B, Li R, et al. (2016). Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat. Commun 7, 13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A, and Wikstrom P. (2011). Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One 6, e19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al. (2009). Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 69, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, Edwards J, Isaacs WB, Nelson PS, Bluemn E, et al. (2012). Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 72, 3457–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, vom Baur E, Garnier JM, Lerouge T, Vonesch JL, Lutz Y, Chambon P, and Losson R. (1998). Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. EMBO J. 17, 3398–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehle K, Cato L, Neeb A, Muhle-Goll C, Jung N, Smith EW, Buzon V, Carbo LR, Estebanez-Perpina E, Schmitz K, et al. (2014). Coregulator control of androgen receptor action by a novel nuclear receptor-binding motif. J. Biol. Chem 289, 8839–8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, and Forman D. (2011). Global cancer statistics. CA Cancer J. Clin 61, 69–90. [DOI] [PubMed] [Google Scholar]
- Jin HJ, Zhao JC, Wu L, Kim J, and Yu J. (2014). Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat. Commun 5, 3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnes RJ, Bergstralh EJ, Davicioni E, Ghadessi M, Buerki C, Mitra AP, Crisan A, Erho N, Vergara IA, Lam LL, et al. (2013). Validation of a genomic classifier that predicts metastasis following radical prostatectomy in an at risk patient population. J. Urol 190, 2047–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein EA, Yousefi K, Haddad Z, Choeurng V, Buerki C, Stephenson AJ, Li J, Kattan MW, Magi-Galluzzi C, and Davicioni E. (2015). A genomic classifier improves prediction of metastatic disease within 5 years after surgery in node-negative high-risk prostate cancer patients managed by radical prostatectomy without adjuvant therapy. Eur. Urol 67, 778–786. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Smith LH, Rong J, Huang X, Hyer ML, Zeps N, Iacopetta B, Linke SP, Olson AH, Reed JC, et al. (2009). Image analysis algorithms for immunohistochemical assessment of cell death events and fibrosis in tissue sections. J. Histochem. Cytochem 57, 649–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, et al. (2016). Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med 22, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, and Dehm SM (2013). Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 73, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, and Dong X. (2014). Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 33, 3140–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, and Luo J. (2013). Decoding the androgen receptor splice variants. Transl. Androl. Urol 2, 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Lonergan PE, Nacusi LP, Wang L, Schmidt LJ, Sun Z, Van der Steen T, Boorjian SA, Kosari F, Vasmatzis G, et al. (2015). The cistrome and gene signature of androgen receptor splice variants in castration resistant prostate cancer cells. J. Urol 193, 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Hong H, Huang SM, Irvine RA, Webb P, Kushner PJ, Coetzee GA, and Stallcup MR (1999). Multiple signal input and output domains of the 160-kilodalton nuclear receptor coactivator proteins. Mol. Cell. Biol 19, 6164–6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, Otto N, Joschko S, Scholz P, Wegg A, et al. (2000). Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J. Biol. Chem 275, 26164–26171. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Sakari M, Okada M, Yokoyama A, Takahashi S, Kouzmenko A, and Kato S. (2013). The androgen receptor in health and disease. Annu. Rev. Physiol 75, 201–224. [DOI] [PubMed] [Google Scholar]
- Morrissey C, Roudier MP, Dowell A, True LD, Ketchanji M, Welty C, Corey E, Lange PH, Higano CS, and Vessella RL (2013). Effects of androgen deprivation therapy and bisphosphonate treatment on bone in patients with metastatic castration-resistant prostate cancer: results from the University of Washington Rapid Autopsy Series. J. Bone Mineral Res 28, 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, Phouanenavong S, Plajzer-Frick I, Shoukry M, Afzal V, et al. (2013). Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyquist MD, Li Y, Hwang TH, Manlove LS, Vessella RL, Silverstein KA, Voytas DF, and Dehm SM (2013). TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc. Natl. Acad. Sci. U S A 110, 17492–17497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissi V, Jepsen K, Glass CK, and Rosenfeld MG (2010). Deconstructing repression: evolving models of co-repressor action. Nat. Rev. Genet 11, 109–123. [DOI] [PubMed] [Google Scholar]
- Qin Q, Mei S, Wu Q, Sun H, Li L, Taing L, Chen S, Li F, Liu T, Zang C, et al. (2016). ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC Bioinformatics 17, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Dai B, Ye D, Kong Y, Chang K, Jia Z, Yang X, Zhang H, Zhu Y, and Shi G. (2015). Constitutively active AR-V7 plays an essential role in the development and progression of castration-resistant prostate cancer. Sci. Rep 5, 7654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J. (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzardi AE, Johnson AT, Vogel RI, Pambuccian SE, Henriksen J, Skubitz AP, Metzger GJ, and Schmechel SC (2012). Quantitative comparison of immunohistochemical staining measured by digital image analysis versus pathologist visual scoring. Diagn. Pathol 7, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. (2015). Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AE, Johnson MH, Yousefi K, Davicioni E, Netto GJ, Marchionni L, Fedor HL, Glavaris S, Choeurng V, Buerki C, et al. (2016). Tissue-based genomics augments post-prostatectomy risk stratification in a natural history cohort of intermediate- and high-risk men. Eur. Urol 69, 157–165. [DOI] [PubMed] [Google Scholar]
- Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, Sankila A, Turunen JP, Lundin M, Konsti J, et al. (2011). Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J 30, 3962–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, Johnson A, Jendrisak A, Bambury R, Danila D, et al. (2016). Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol. 2, 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A. (2017). Cancer statistics, 2017. CA Cancer J. Clin 67, 7–30. [DOI] [PubMed] [Google Scholar]
- Singh CK, Malas KM, Tydrick C, Siddiqui IA, Iczkowski KA, and Ahmad N. (2016). Analysis of zinc-exporters expression in prostate cancer. Sci. Rep 6, 36772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for inter-preting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, et al. (2010). Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest 120, 2715–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, and Balk SP (1995). Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med 332, 1393–1398. [DOI] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepaamorndech S, Huang L, and Kirschke CP (2011). A null-mutation in the Znt7 gene accelerates prostate tumor formation in a transgenic adenocarcinoma mouse prostate model. Cancer Lett. 308, 33–42. [DOI] [PubMed] [Google Scholar]
- Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, et al. (2009). Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, and Chu G. (2001). Significance analysis of micro-arrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U S A 98, 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Poppel H, and Nilsson S. (2008). Testosterone surge: rationale for gonadotropin-releasing hormone blockers? Urology 71, 1001–1006. [DOI] [PubMed] [Google Scholar]
- van Royen ME, Cunha SM, Brink MC, Mattern KA, Nigg AL, Dubbink HJ, Verschure PJ, Trapman J, and Houtsmuller AB (2007). Compartmentalization of androgen receptor protein-protein interactions in living cells. J. Cell Biol 177, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Royen ME, van Cappellen WA, de Vos C, Houtsmuller AB, and Trapman J. (2012). Stepwise androgen receptor dimerization. J. Cell Sci 125, 1970–1979. [DOI] [PubMed] [Google Scholar]
- Velasco-Hernandez T, Hyrenius-Wittsten A, Rehn M, Bryder D, and Cammenga J. (2014). HIF-1alpha can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood 124, 3597–3607. [DOI] [PubMed] [Google Scholar]
- Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, and Kallioniemi OP (1995). In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet 9, 401–406. [DOI] [PubMed] [Google Scholar]
- Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. (2011). Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474, 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, and Brown M. (2007). A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell 27, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, et al. (2009). Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zang C, Xiao T, Fan J, Mei S, Qin Q, Wu Q, Li X, Xu K, He HH, et al. (2016). Modeling cis-regulation with a compendium of genome-wide histone H3K27ac profiles. Genome Res. 26, 1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Arora VK, and Sawyers CL (2015). Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welti J, Rodrigues DN, Sharp A, Sun S, Lorente D, Riisnaes R, Figueiredo I, Zafeiriou Z, Rescigno P, de Bono JS, et al. (2016). Analytical validation and clinical qualification of a new immunohistochemical assay for androgen receptor splice variant-7 protein expression in metastatic castration-resistant prostate cancer. Eur. Urol 70, 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao YM, Lengauer C, et al. (2009). Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8, 498–504. [DOI] [PubMed] [Google Scholar]
- Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, Gambhir SS, Lee P, Sartor O, Flemington EK, et al. (2015). Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res. 75, 3663–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.