

Abstract
Objective:
To determine the utility of a comprehensive, targeted-capture next-generation sequencing (NGS) assay for the clinical management of children undergoing enucleation for retinoblastoma.
Design:
Cohort study.
Subjects:
32 children with retinoblastoma.
Methods:
We performed targeted NGS using the UCSF500 Cancer Panel on formalin-fixed, paraffin-embedded tumor tissue along with constitutional DNA isolated from peripheral blood, buccal swab, or uninvolved optic nerve. Peripheral blood samples were also sent to a commercial laboratory for germline RB1 mutation testing.
Main Outcome Measures:
Presence or absence of germline RB1 mutation/deletion, tumor genetic profile, and association of genetic alterations with clinicopathologic features.
Results:
Germline mutation or deletion of the RB1 gene was identified in all children with bilateral retinoblastoma (n=12), and these NGS results were 100% concordant with commercial germline RB1 mutation analysis. In tumor tissue tested with NGS, biallelic inactivation of RB1 was identified in 28 tumors and focal MYCN amplification in 4 tumors (two with wildtype RB1 and two with biallelic RB1 inactivation). Additional likely pathogenic alterations beyond RB1 were identified in 13 tumors (41%), several of which have not been previously reported in retinoblastoma. These included focal amplifications of MDM4 and RAF1, as well as damaging mutations involving BCOR, ARID1A, MGA, FAT1, and ATRX. The presence of additional likely pathogenetic mutations beyond RB1 inactivation was associated with aggressive histopathologic features, including higher histologic grade and anaplasia, and also with both unilateral and sporadic disease.
Conclusions:
Comprehensive NGS analysis reliably detects relevant mutations, amplifications, and chromosomal copy number changes in retinoblastoma. The presence of genetic alterations beyond RB1 inactivation correlate with aggressive histopathologic features.
Précis
Comprehensive next-generation sequencing of retinoblastoma shows RB1 inactivation and additional pathogenic alterations have an association with aggressive histological features, a marker for more aggressive disease.
Introduction
Retinoblastoma is an aggressive intraocular cancer, which threatens loss of vision, a painful eye, and death from metastatic disease or direct extension into the brain. In 98% of patients, retinoblastoma is caused by bi-allelic inactivation of the RB1 tumor suppressor gene on chromosome 13q14. This RB1 inactivation can be somatic (tumor-specific), due to a germline mutation or deletion accompanied by somatic inactivation of the remaining allele, or can arise during early embryogenesis leading to post-zygotic mosacism.1 Patients with germline RB1 mutation or constitutional mosaicism are also at risk of pinealoblastoma, osteosarcoma, and other sarcomas, as well as secondary neoplasms following radiation therapy.2,3
A two-step, biallelic inactivation of the RB1 tumor suppressor gene is required for tumor formation in most retinoblastomas1,7,8 However, studies suggest that biallelic RB1 inactivation leads to a non-proliferative retinoma, and progression to retinoblastoma requires additional genetic aberrations.8–10 The most common alterations beyond biallelic RB1 inactivation include recurrent chromosomal copy number alterations, including trisomy 1q, trisomy 2p, trisomy 6p, and monosomy 16q, likely leading to activation of oncogenes or inactivation of tumor suppressor genes at these regions.1,10,11 Similar to many pediatric neoplasms, the overall somatic mutation burden in retinoblastomas is very low, and only a small subset of retinoblastomas have been identified to harbor additional recurrent mutations involving the BCOR and CREBBP transcriptional regulatory genes.1 Approximately 2% of retinoblastomas do not harbor RB1 alterations, and instead are driven by focal high-level amplification of the MYCN oncogene.1,12
Alterations in the RB1 gene are quite heterogeneous including single nucleotide variants and chromosomal deletions and rearrangements spanning the entire gene, and modern techniques such as high-throughput next-generation sequencing (NGS) and array comparative genomic hybridization (aCGH) have significantly improved the sensitivity and specificity of detection. However, the spectrum of cooperating gene mutations is still largely unknown and most studies have been limited to identification of candidate genes at or near the frequent chromosomal copy number variants.11 Herein, we performed comprehensive molecular profiling of retinoblastoma enucleation specimens to identify genetic alterations beyond RB1 inactivation that may correlate with clinical and pathologic features.
Materials and Methods
This study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines, under the approval of the UCSF Institutional Review Board with a waiver of patient consent (CC number 17–23850). The study cohort included 32 consecutive retinoblastoma patients who underwent enucleation at the University of California, San Francisco.
Clinical Methods
Clinical information was obtained through retrospective review of the electronic medical records, and included patient age at diagnosis, sex, laterality, clinical features of the study eye, international intraocular classification of retinoblastoma (IICR) group13, clinical TNM stage,14 treatment modalities, and outcome. Standard clinical evaluation for retinoblastoma included full ophthalmologic examination under anesthesia including fundus photography by RetCam3 (Natus Medical, Pleasanton, CA, USA) and B-scan ultrasound (Eyecubed; Ellex, Adelaide, Australia). Patients with germline retinoblastoma also received magnetic resonance imaging of the brain and orbits, both with and without contrast, performed at diagnosis and every 6 months thereafter. Pre-enucleation chemoreduction included systemic chemotherapy with carboplatin, etoposide, and vincristine or by intra-arterial chemotherapy with melphalan and topotecan, with addition of carboplatin as necessary for insufficient therapeutic response. Some eyes received focal consolidation with laser (Alcon Purepoint, Fort Worth, TX, USA) and/or cryotherapy (Frigitronics, Cooper Surgical, Trumbull, CT, USA).
Histopathologic Examination
As part of the routine pathologic examination, all enucleated globes were entirely submitted for microscopic evaluation and the diagnosis of retinoblastoma was confirmed by light microscopy on sections stained with hematoxylin and eosin and, if necessary, by immunohistochemical stains. One of the ophthalmic pathologists re-reviewed all slides for study purposes to evaluate pathologic features including grade, presence of anaplasia, and updated pathologic TNM classification (AJCC 8th edition).14 Tumors with frequent or occasional rosettes (Flexner-Wintersteiner or Homer Wright) were annotated as grade 2 or 3, respectively. Tumor with poorly-differentiated cells without rosettes and/or those with extensive anaplasia involving more than half of the tumor volume were categorized as grade 4.
Commercial Germline RB1 Genetic Testing
As part of the clinical care, all patients underwent germline RB1 mutation and indel testing using peripheral blood samples at the time of initial clinical diagnosis using one of the commercially available laboratories including Ambry Genetics (Aliso Viejo, CA, USA,15), Invitae (San Francisco, CA, USA,16) or Impact Genetics (Toronto, Ontario, Canada,17). These tests utilize Multiplex Ligation-dependent Probe Amplification (MLPA), Sanger sequencing, Quantitative Multiplex Polymerase Chain Reaction (QM-PCR), Allele-Specific PCR (AS-PCR), RB1 promoter methylation, and/or next-generation sequencing limited to the coding exons of the RB1 gene.
Targeted Next-Generation Sequencing of Paired Retinoblastoma Tumor and Normal Specimens
The UCSF500 Cancer Panel is a clinically validated next-generation sequencing assay performed in the UCSF Clinical Cancer Genomics Laboratory, which is accredited by the Collage of American Pathologists (CAP) and certified under the Clinical Laboratory Improvement Amendments (CLIA). Therefore, the laboratory has regulatory approvals to perform UCSF500 testing on cases from any institution and is available to both providers at UCSF Medical Center as well as providers at outside institutions as send-out testing. Sequencing is typically performed on both tumor and normal tissue in order to evaluate for potential germline alterations associated with increased cancer risk and accurately identify the somatic drivers of each patient’s tumor, enabling a precision medicine treatment approach and selection of personalized targeted therapeutics. UCSF500 testing was performed prospectively as part of clinical care in 17 patients, and retrospectively for the purposes of this study in the remaining 15 patients, using tumor-normal pairs whenever available. Genomic DNA was extracted from tumor tissue that had been macrodissected from formalin-fixed, paraffin-embedded (FFPE) blocks of enucleation specimens using the QIAamp DNA FFPE Tissue Kit (Qiagen) according to the manufacturer’s protocol. Genomic DNA for germline testing was extracted from buccal swabs, peripheral blood, or uninvolved optic nerve sections from FFPE specimens. Capture-based next-generation DNA sequencing was performed using an assay that targets all coding exons of 479 cancer-related genes, select introns and upstream regulatory regions of 47 genes to enable detection of structural variants including gene fusions, and DNA segments at regular intervals along each chromosome to enable genome-wide copy number and zygosity analysis, with a total sequencing footprint of 2.8 Mb (UCSF500 Cancer Panel; Supplementary Table 1).18 Specifically, this assay covers all exons of the RB1 gene as well as other genes previously implicated in retinoblastoma (MYCN, MDM4, BCOR, and CREBBP)1.
Multiplex library preparation was performed using the KAPA Hyper Prep Kit (Roche) according to the manufacturer’s specifications using 250 ng of sample DNA. Hybrid capture of pooled libraries was performed using a custom oligonucleotide library (Nimblegen SeqCap EZ Choice). Captured libraries were sequenced as paired-end 100 bp reads on an Illumina HiSeq 2500 instrument. Sequence reads were mapped to the reference human genome build GRCh37 (hg19) using the Burrows-Wheeler aligner (BWA). Recalibration and de-duplication of reads was performed using the Genome Analysis Toolkit (GATK), enabling accurate allele frequency determination and copy number assessment. Coverage and sequencing statistics were determined using Picard CalculateHsMetrics and Picard CollectInsertSizeMetrics. Single nucleotide variant and insertion/deletion mutation calling was performed with Mutect, Unified Genotyper, Pinder and Delly. Variant annotation was performed with Annovar. Single nucleotide variants, insertions/deletions, and structural variants were visualized and verified using Integrated Genome Viewer. Genome-wide copy number analysis based on on-target and off-target reads was performed by CNVkit and visualized using Nexus Copy Number (Biodiscovery).
Statistical analysis
Biostatistical analysis was performed in Stata Version 16 (StataCorp, College Station, Texas, USA). Comparison of clinical and histologic features stratified by molecular alterations and copy number variations were performed using the Mann-Whitney test for continuous variables (diagnosis age) and the Fisher’s exact test for categorical variables (all other variables). Patients were divided into two groups: those with retinoblastomas harboring RB1 inactivation as the sole pathogenic alteration (n=15) and those with retinoblastomas harboring additional likely pathogenic alternations beyond RB1 inactivation (n=13). Only additional somatic variants that were clearly protein-damaging (e.g. truncating mutations) in well-established tumor suppressor genes or focal high-level amplifications of well-established oncogenes were included in this analysis. Somatic variants of uncertain significance (missense variants of uncertain functional significance) were excluded from this subgrouping analysis. A p-value cutoff of 0.05 was used to assess statistical significance, with p values between 0.05 and 0.10 considered borderline significant.
Results
Patient Demographics
The study cohort included 16 males (50%) and 16 females (50%) with a median age of 19 months at diagnosis (range 2–92 months) (Table 1). Seventeen (53%) patients were Hispanic or Latino, nine (28%) were Caucasian/White, five (16%) were Asian and one (3%) was black. Retinoblastoma was unilateral in 20 (63%) and bilateral in 12 (37%) patients, with one patient (3%) presenting with trilateral disease (bilateral retinoblastoma with concurrent pineoblastoma). Patients with unilateral disease were significantly older than those with bilateral disease (28 vs 7.5 months, respectively; p=0.0003).
Table 1:
Patient Demographics
| Number (%), N=32 | |
|---|---|
| Demographics | |
| Laterality, median age at diagnosis (range) | |
| Unilateral: 28 months (5–92) | 19 (59) |
| Bilateral: 7.5 months (2–20) | 12 (37) |
| Trilateral (bilateral retinoblastoma and pineoblastoma): 4 months | 1 (3) |
| Gender | |
| Male | 16 (50) |
| Female | 16 (50) |
| Race/Ethnicity | |
| Hispanic or Latino | 17 (53) |
| White | 9 (28) |
| Asian | 5 (16) |
| Black | 1 (3) |
Study Eye Characteristics
The IIRC at diagnosis was group was D in 5 eyes and E in 27 eyes. Clinical TNM Stage was cT2 in 17 (53%) eyes, cT3 in 6 (19%) eyes, and cT4 in 9(28%) eyes at the time of enucleation (Table 2). Eight patients (25%) presented to our institution with clinical evidence of extraocular retinoblastoma on MRI imaging. One patient (#30) presented with recurrent orbital retinoblastoma 8-years after systemic and intraarterial chemotherapy and plaque brachytherapy elsewhere; one patient (#17) had diffuse, infiltrating retinoblastoma with enhancement/disease to the optic chiasm on MRI at the time of diagnosis. Seven patients (22%) presented with buphthalmos.
Table 2.
Clinical features of the study eye
| Number (%), N=32 | |
|---|---|
| Study eye characteristics | |
| Laterality | |
| Right | 15 (47)* |
| Left | 17 (53) |
| IIRC Group at Diagnosis | |
| Group D | 5 (16) |
| Group E | 27 (84) |
| Enucleation | |
| Primary | 28 (88) |
| Secondary | 4 (12) |
| Buphthalmic | 7 (22) |
| Orbital optic nerve enhancement on MRI (extraocular disease at diagnosis) | 8 (25) |
| Clinical TNM stage (all N0M0) | |
| cT2 | 17 (53) |
| cT2a | 3 |
| cT2b | 14 |
| cT3 | 6 (19) |
| cT3b | 2 |
| cT3c | 4 |
| cT4 | 9 (28) |
| Primary treatment | |
| Enucleation | 28 (88) |
| Systemic Chemotherapy | 14 (44) |
| Intra-arterial chemotherapy | 6 (19) |
Patient #17 had enucleation of the right eye without sufficient viable tumor for genetic sequencing, due to pre-enucleation chemotherapy with disease the chiasm on MRI at presentation. Patient #30 presented with orbital extension of the right eye, and biopsy of the orbital mass was performed at an outside institution. Pre-enucleation chemoreduction was given, with plan for enucleation after chemotherapy is completed.
Pathologic features
Histologic grade was 1, 2, 3 and 4 in 1, 11, 9 and 10 tumors respectively (Table 3). One patient (patient #18) did not have enough viable tumor cells in the enucleated eye for accurate grading. Anaplasia was present in 20 tumors, of which 9 were diffusely anaplastic and 11 were focally anaplastic. Twenty-five tumors were staged as pT1 (78%), 13 of which showed no choroidal or optic nerve invasion (41%), 4 had only minimal choroidal invasion (13%), and 8 had only prelaminar optic nerve invasion (25%). One tumor showed prelaminar optic nerve invasion along with choroidal invasion, resulting in staging as pT2a. One tumor showed massive (>3mm) choroidal invasion (pT3a), and three tumors showed post laminar optic nerve invasion without involvement of the optic nerve margin (pT3b). One tumor showed retinoblastoma cells within the meningeal space (patient #23), and one tumor (patient #30) presented as recurrence with extraocular extension in the orbit on presentation to our center (pT4).
Table 3.
Pathologic features of the enucleated eye
| Number (%),N=31* | |
|---|---|
| Pathologic TNM stage | |
| pT1 | 25 (81) |
| pT2a | 1 (3) |
| pT3 | |
| pT3a | 1 (3) |
| pT3b | 3 (10) |
| pT4 | 2 (6) |
| Optic nerve involvement | |
| None | 19 (61) |
| Prelaminar | 9 (29) |
| Post-laminar | 3 (10) |
| Choroidal invasion | |
| None | 23 (74) |
| Small | 6 (19) |
| Massive (>3mm) | 2 (7) |
| Histologic grade | |
| Grade 1 | 1 (3) |
| Grade 2 | 11 (35) |
| Grade 3 | 9 (29) |
| Grade 4 | 10 (32) |
| Histologic anaplasia | 20 (65) |
Patients #17, 18 and 28 had limited viable eye tumor tissue in the enucleation specimens due to pre-enucleation chemotherapy. Anaplasia and histologic grade could not be determined for patient #18.
Treatment
Enucleation was the primary therapy in 28 (88%) eyes and secondary enucleation was performed for persistent or recurrent tumor following ocular salvage therapy in 3 eyes (10%). Thirteen patients received pre-enucleation systemic chemoreduction, which had been administered for buphthalmos (7), optic nerve involvement (3), recurrent tumor with extraocular spread (1), and diffuse, infiltrating retinoblastoma extending to the chiasm (1), respectively (Figure 1).
Figure 1:
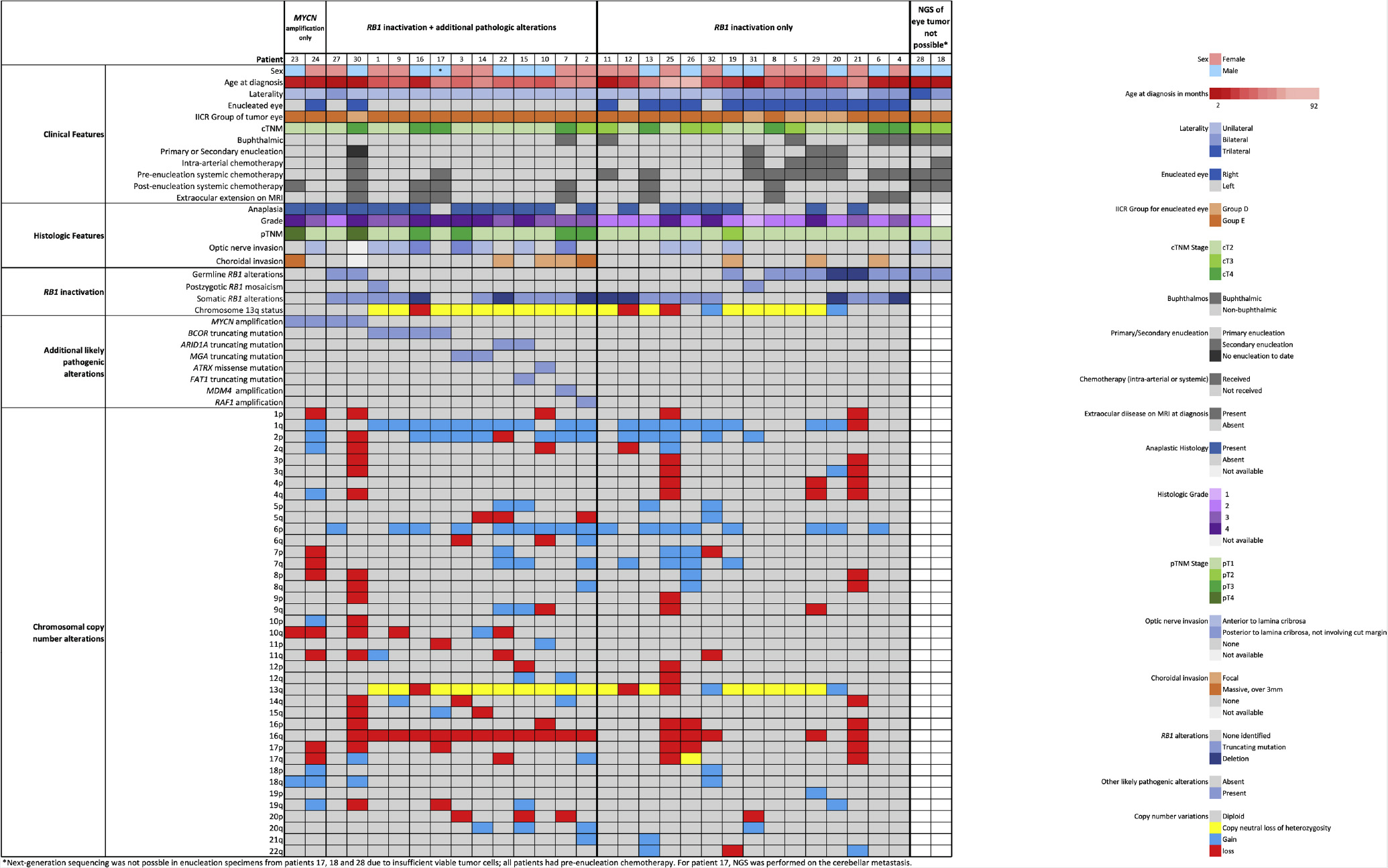
Oncoprint summary table of the clinical, pathologic, and genetic features of the 32 retinoblastoma patients.
Germline RB1 Sequencing Analysis with the UCSF500 and Commercial Assays
Targeted NGS using the UCSF500 Cancer Panel was performed on 30 constitutional DNA samples to assess for germline alterations (Supplementary table 2). A normal sample was not available for 2 patients (#7, #29), and their germline data were obtained from the commercial laboratory results. Overall, twelve patients (38%) were found to have inactivating germline mutation or deletion in RB1, eleven of whom had bilateral retinoblastoma (including one child with imaging evidence of pineoblastoma). No germline alterations in the RB1 gene were found in the remaining 20 patients. However, in two of these children (#1 and #31), RB1 mutations were found in the constitutional DNA samples (from peripheral blood and uninvolved optic nerve, respectively) with variant allele frequencies of 13% and 19%, indicative of post-zygotic mosaicism with acquisition of an RB1 mutation during embryogenesis rather than in a germ cell transmitted from one of the parents. The variant allele frequencies of these RB1 mutations in the respective tumor samples were 99% and 83%, indicating that the retinoblastomas in these two children arose from a retinal progenitor cell harboring the post-zygotic RB1 mutation following loss of heterozygosity of chromosome 13q that eliminated the remaining wildtype allele. In all cases, the germline and post-zygotic mosaicism results from the UCSF500 panel were perfectly concordant with the results of the commercial germline RB1 testing.
Retinoblastoma Tumor Genetic Analysis with the UCSF500
Genetic analysis of tumor tissue with NGS was successful in 30 (94%) patients: 28 enucleation specimens, one cerebellar metastasis (patient #17), and one recurrence with orbital extraocular extension (patient #30). Detailed results are reported in Table 4, Figure 1 and Supplementary Tables 2 and 3. Three patients (17, 18 and 28) had insufficient viable tumor tissue in the enucleated globe for genetic analysis, due to pre-enucleation chemotherapy.
Table 4.
Tumor Genetics by UCSF500 Next-Generation Sequencing Panel
| Germline: Number (%), N=32 | |
|---|---|
| Tumor: Number (%), N=30* | |
| Germline or Mosaic Alterations in Normal Sample: | |
| Germline RB1 mutation or deletion | 12 (38) |
| Post-zygotic mosaicism for RB1 mutation | 2 (6) |
| Somatic Alterations in Tumor: | |
| Known Genetic Alternations in Retinoblastoma | |
| RB1 mutation or deletion | 28 (93) |
| MYCN amplification | 4 (13) |
| BCOR mutation (truncating) | 4 (13) |
| MDM4 amplification | 1 (3) |
| Novel Likely Pathogenic Alternations | |
| MGA mutation (truncating) | 2 (7) |
| ARID1A mutation (truncating) | 2 (7) |
| FAT1 mutation (splice site) | 1 (3) |
| ATRX mutation (missense) | 1 (3) |
| RAF1 amplification | 1 (3) |
| Frequent Chromosomal Copy Number Variations | |
| Chromosome 13q loss or loss of heterozygosity | 20 (67) |
| Chromosome 1q gain | 19 (63) |
| Chromosome 6p gain | 17 (57) |
| Chromosome 16q loss | 17 (57) |
| Chromosome 2p gain | 13 (43) |
Due to limited or no viable eye tumor in enucleation specimens in patients #17, 18 and 28, all of whom had pre-enucleation chemotherapy, Next-Generation Sequencing of the eye tumor was not possible. Patient #16 had genetic testing of a cerebellar metastatic lesion.
Among the 10 children with heterozygous germline RB1 mutation or deletion with viable tumor for sequencing analysis, 6 had tumors with an additional somatic RB1 mutation or deletion, whereas 4 had tumors with loss of heterozygosity involving chromosome 13q that eliminated the remaining wildtype RB1 allele. Sixteen sporadic retinoblastomas (including the two cases arising due to post-zygotic mosaicism) harbored biallelic somatic mutation or deletion of the RB1 gene. Additionally, two patients with unilateral retinoblastoma had tumors demonstrating loss of heterozygosity of chromosome 13q but without a detectable RB1 mutation or deletion on the remaining allele, likely representing a cryptic RB1 alteration not detected by this NGS assay. These tumors were considered to have RB1 inactivation for study purposes, as both cases were found to have loss of Rb protein expression by immunohistochemistry. Overall, 28 (93%) of the 30 evaluated tumors harbored biallelic RB1 inactivation, and the two remaining tumors without RB1 germline or somatic alterations both contained focal high-level amplification of the MYCN oncogene (with greater than 50 extra copies anticipated to be present in the majority of tumor cells based on the NGS results).
Among the 28 tumors with RB1 inactivation, 15 (54%) had RB1 inactivation as the solitary pathogenic alteration identified, while the remaining 13 (46%) harbored additional likely pathogenic alterations (Table 4, Figure 1 and Supplementary table 2). These included MYCN focal amplification (n=2), RAF1 focal amplification (n=1), MDM4 focal amplification (n=1), BCOR truncating mutation (n=4), ARID1A truncating mutation (n=2), MGA truncating mutation (n=2), ATRX missense mutation (n=1), and FAT1 splice site mutation (n=1). These additional likely pathogenic alterations were mutually exclusive among the 13 tumors (i.e. each retinoblastoma harbored biallelic RB1 inactivation plus one additional genetic alteration), except for patient #15 whose tumor harbored biallelic RB1 inactivation plus ARID1A nonsense mutation and FAT1 splice site mutation. Notably, the ARID1A and FAT1 mutations were present at subclonal allele frequencies in this tumor, indicating they were only present in a subset of the tumor cells, perhaps each within different subclones.
Among the 28 retinoblastomas with viable tumor for sequencing analysis that harbored RB1 biallelic inactivation, the presence of additional likely pathogenic alterations was associated with higher histologic grade (Fisher’s exact test, p=0.002), histologic anaplasia (Fisher’s exact test, p=0.05), optic nerve involvement at time of enucleation (Fisher’s exact test, p=0.06), higher pathologic stage (Fisher’s exact test, p=0.07), unilateral disease (Fisher’s exact test, p=0.05), and was less common in patients with germline RB1 alterations (Fisher’s exact test, p=0.05) (Table 5).
Table 5.
The presence of additional pathogenic alterations beyond RB1 inactivation correlates with specific clinicopathologic features in retinoblastoma
| RB1 inactivation only (n=15) | RB1 + additional pathogenic alteration# (n=13) | p-value | ||
|---|---|---|---|---|
| Age at diagnosis, median (range) | 14 months (3–92) | 27 months (2–19) | p=0.37 | |
| Germline RB1 alteration | 8 (54%) | 2 (15%) | p=0.05 | |
| Laterality | p=0.05 | |||
| Bilateral | 8 (53%) | 2 (15%) | ||
| IICR tumor group | p=0.33 | |||
| Group E | 11 (73%) | 12 (92%) | ||
| p=0.48 | ||||
| stage | cT3 or cT4 | 8 (53%) | 5 (38%) | |
| Extraocular extension on MRI | 4 (27%) | 4 (31%) | p=1.00 | |
| p=0.07 | ||||
| stage | pT2 - pT4 | 1 (7%) | 5 (38%) | |
| Optic nerve involvement | 3 (20%) | 7 (58%) | p=0.06 | |
| Choroidal invasion | 3 (20%) | 4 (33%) | p=0.66 | |
| Histologic grade | P<0.01 | |||
| Grade 3 or 4 | 5 (33%) | 12 (92%) | ||
| Anaplastic histology | 7 (47%) | 11 (85%) | p=0.05 | |
Additional likely pathogenic alterations beyond RB1 inactivation that were present in these 13 tumors included: focal high-level MYCN amplification (2), MDM2 amplification (1), RAF1 amplification (1), truncating BCOR mutation (4), truncating MGA mutation (2), truncating ARID1A mutation (2), and ATRX missense mutation (1). Somatic variants of unknown significance (e.g. missense mutations not recurrently found in the COSMIC database and involving genes not known to be recurrently mutated in retinoblastoma) were not considered as likely pathogenic alterations beyond RB1 inactivation for this analysis.
Chromosomal copy number aberrations were frequently seen including loss of chromosome 13q (n=3, 10%) or copy-neutral loss of heterozygosity involving chromosome 13q (n=17, 57%). Other common copy number variations included gains of chromosomes 1q (n=19, 63%), 2p, (n=13, 43%) and 6p (n=17, 57%), as well as loss of chromosome 16q (n=17, 57%). Details of the chromosomal copy number variations are reported in Table 4, Figure 1, and Supplementary Table 3. The mean number of chromosomal copy number changes per tumor was 8 (range 0–20, median=8). Representative genome-wide copy number plots from a retinoblastoma with focal homozygous/biallelic deletion of the RB1 gene on chromosome 13q14 and a retinoblastoma with focal high-level amplification of the MYCN gene on chromosome 2p24 are shown in Figure 2.
Figure 2:
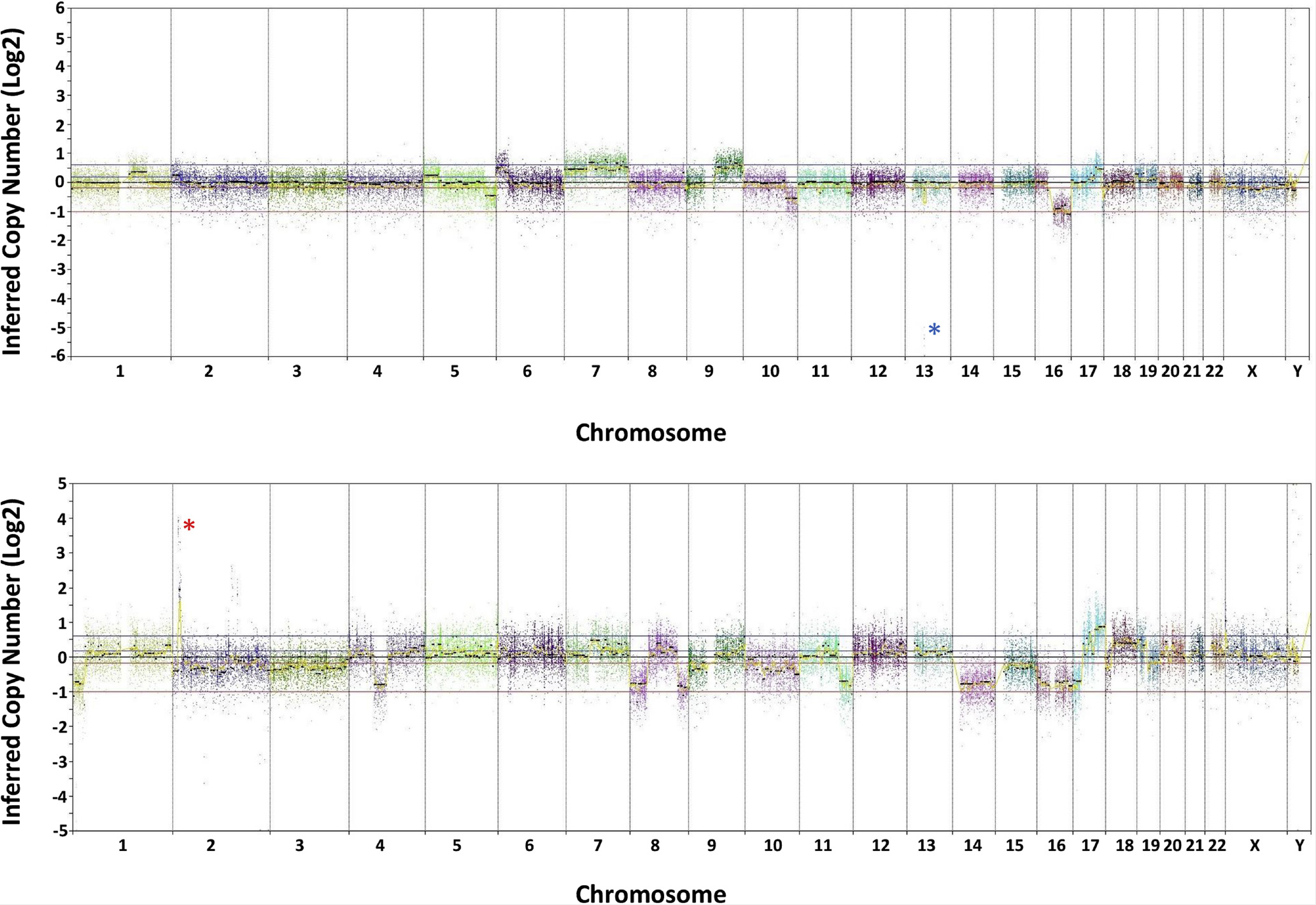
Genome-wide copy number profiles for two representative retinoblastoma cases. Shown is inferred copy number calls (log2 scale) along each of the chromosomes. Top: Retinoblastoma case with focal homozygous/biallelic deletion on chromosome 13q14 that encompasses the 5’ portion of the RB1 tumor suppressor gene (blue asterisk). Additional chromosomal copy number variations include gains of proximal 1q, 5p, distal 6p, 7, 9q, portions of 17q, and distal Xp, as well as losses of distal 2p, distal 5q, distal 10q, distal 11q, 16q, and interstitial 17q. Bottom: Retinoblastoma case with focal high-level amplification of the MYCN oncogene on chromosome 2p24 (red asterisk). Additional chromosomal copy number variations include gains of 17q and 18q, as well as losses of distal 1p, 2, 3, interstitial 4q, 8p, distal 8q, 9p, 10, distal 11q, 14q, 15q, 16, 17p, and 19q.
Discussion
Retinoblastoma was previously thought to principally be a monogenic disease driven only by biallelic inactivation of the RB1 tumor suppressor gene. Other groups reported that somatic alterations beyond RB1 are rare and mostly limited to copy number changes.1 Through targeted NGS analysis on 30 retinoblastomas, we identified a high frequency (46%) of additional somatic likely pathogenic alterations beyond RB1 biallelic inactivation. These included focal high-level amplification of well-described oncogenes including MYCN, MDM4, and RAF1, as well as truncating (nonsense, frameshift, or splice site) mutations in well-described tumor suppressor genes including BCOR, ARID1A, MGA, and FAT1. The presence of these additional likely pathogenic alterations correlated with aggressive histopathologic features including higher histologic grade and anaplasia, and we found a borderline significant association (p=0.06) with optic nerve involvement seen on pathology. Of note, while the size of this cohort is limited, these results support a role for comprehensive, paired tumor-normal NGS analysis in the clinical management of children with retinoblastoma treated with enucleation.
Notably, four of the 30 evaluated tumors in this cohort harbored focal high-level amplification of the MYCN oncogene, two of which were RB1 wild-type and two of which harbored biallelic RB1 inactivation. Amplification of the MYCN oncogene has been reported in a small subset (~2–5%) of retinoblastomas, typically those with wild-type RB1 alleles.1,12,19 Such MYCN amplified, RB1 wild-type retinoblastomas are reported to have aggressive histologic features and young age at diagnosis compared with sporadic retinoblastoma harboring somatic RB1 inactivation.1,12 The patients with retinoblastomas harboring MYCN amplification in our cohort (cases 23, 24, 27 and 30) were 8,11, 4 and 2 months old at diagnosis, respectively. The two children whose tumors harbored dual MYCN amplification and RB1 biallelic inactivation were: patient 30, who experienced late disease recurrence after intra-arterial chemotherapy and had histologic anaplasia and extraocular extension (stage pT4); and patient 27, with a Group E eye who underwent enucleation at 4 months of age (stage pT1). We speculate that such retinoblastomas with combined RB1 inactivation and MYCN amplification are more likely to be associated with worse outcome than those harboring only one of these alterations.
Truncating mutation in the BCOR gene is the most frequent additional genetic alteration beyond RB1 biallelic inactivation in our cohort (4/28 RB1 inactivated tumors, 14%). This is consistent with the previously reported rate of BCOR mutations (10–13%) among RB1-inactivated retinoblastomas.1,20 The BCOR gene encodes a transcriptional co-repressor protein that interacts with BCL6, as well as class I and II histone deacetylases.21 BCOR is an important epigenetic regulatory gene during embryogenesis, with inactivating germline mutations being the cause of an X-linked oculofaciocardiodental syndrome (Online Mendelian Inheritance in Man #300166) characterized by microphthalmia, congenital cataracts, long narrow face, dental radiculomegaly with persistent primary teeth, and cardiac septal defects. Studies in osteodentinogenic mesenchymal stem cells from a patient with oculofaciocardiodental syndrome found that BCOR mutation disrupted homeostasis by resulting in increased methylation of lysine 4 and lysine 36 on the tail of histone H3, thereby reactivating transcription of silenced target genes.22 In addition to retinoblastoma, somatic mutations or rearrangements in the BCOR gene have been identified in a wide spectrum of human tumors including acute myeloid leukemia, medulloblastoma, Ewing-like round cell sarcoma, endometrial stromal sarcoma, clear cell sarcoma of kidney, and primitive mesenchymal myxoid tumor of infancy. All four of the retinoblastomas in our cohort with dual RB1 and BCOR mutations were unilateral Group E tumors. Three of these cases had anaplastic histology with optic nerve involvement at time of enucleation, while the fourth case later developed intracranial metastasis.
Additional likely pathogenic alterations that we identified in our cohort included focal high-level amplifications of the MDM4 and RAF1 oncogenes. The MDM4 gene on chromosome 1q32 has been previously implicated as one of the potential oncogenes driving the frequent gains of chromosome 1q in retinoblastoma, although focal MDM4 amplification is a rare event.1,23 MDM4 encodes an E3 ubiquitin ligase that functions as a negative regulator of p53 activity. RAF1 amplification has not been previously reported in retinoblastomas, but it is a well-studied oncogene and its amplification and overexpression are expected to be activating the MAP kinase signaling pathway in this retinoblastoma. Other novel genetic alterations in this cohort include recurrent truncating mutations in the MGA and ARID1A tumor suppressor genes (2 tumors each, both 7%). MGA encodes a DNA-binding protein that regulates c-Myc and n-Myc transcriptional activity and is known to harbor recurrent loss-of-function mutations in lung and colon adenocarcinomas.24,25 ARID1A encodes a subunit of the Swi/Snf chromatin remodeling complex involved in transcriptional regulation and chromatin architecture. Inactivating ARID1A mutations are known to be present in a wide range of human cancers including neuroblastoma,26 but this is the first report of recurrent ARID1A mutations in retinoblastoma to the best of our knowledge. Based on our study, the additional genetic alterations acquired in retinoblastomas beyond RB1 inactivation appear to involve a diverse array of genes that function in transcriptional regulation (MYCN, MGA, BCOR, and ARID1A), p53 regulation (MDM4), and MAP kinase pathway signaling (RAF1).
Clinical Implications
Our study suggests a clinical role for comprehensive, paired tumor-normal NGS analysis in the clinical management of patients with retinoblastoma treated with enucleation. First, this method may detect genetic aberrations beyond RB1 inactivation that indicate an increased risk of recurrent or metastatic disease and hence a greater need for close monitoring and perhaps systemic adjuvant therapy. Second, NGS may reveal RB1 germline mutations or consitutional mosaicism in patients otherwise thought to have sporadic disease. Simultaneous paired tumor-normal sequencing analysis using a targeted NGS panel enables more reliable determination of sporadic versus heritable disease, as sporadic tumor development in children with negative germline RB1 results in confirmed by the finding of somatic RB1 biallelic inactivation (or occasionally MYCN amplification). Third, the ability of modern NGS technology to successfully interrogate formalin-fixed, paraffin-embedded tumor tissue makes genetic tumor analysis possible in situations where storage and transport of fresh-frozen samples is not possible, also reducing costs. Additionally, the ability of NGS to successfully study FFPE tumor specimens obfuscates the need to disrupt the globe for fresh tumor sampling with the intent of enabling genetic analysis, thereby preserving globe architecture for optimal histopathologic analysis.While use of FFPE samples has been routine in many other tumor types for the past several years, both clinically and for research purposes, to the best of our knowledge, this is the first study to use FFPE samples for NGS analysis of retinoblastoma. In many institutions, fresh frozen samples are commonly obtained from enucleated retinoblastoma globes, as well as other childhood solid tumors. Multiple studies have documented that genetic analysis of FFPE tissue performs equivalently to fresh snap-frozen tissue for identification of high-confidence genetic alterations across multiple human cancer types.27,28 This carries considerable clinical benefits, especially in heavily necrotic tumors such as retinoblastoma, since the molecular tests can be directed specifically to the areas of viable tumor via microdissection, avoiding necrotic samples. In addition, archival samples, which are routinely FFPE, can be used for molecular testing not only in later stages of clinical management as indicated, but also for larger research efforts.
Conclusions
Overall, our results demonstrate the utility of using a targeted sequencing panel that covers a wide spectrum of cancer-associated genes for the genetic characterization of enucleated retinoblastoma specimens. This approach enabled the identification of gene alterations of known relevance in retinoblastoma, as well as multiple novel and likely pathogenic alterations in our cohort. Our results do not support the view that somatic genomic alterations in retinoblastoma beyond RB1 are rare events and mainly limited to chromosome copy number changes, as has been suggested by other studies.1 Instead, the results of this study suggest retinoblastomas with RB1 inactivation and additional pathogenic alterations have an association with higher histologic grade, anaplasia, and higher pathologic stage, all of which are known predictors of more aggressive disease. Future studies with larger patient cohorts are needed to corroborate these findings and firmly establish an association between tumor genotype and clinical outcomes for children with retinoblastoma.
Supplementary Material
Acknowledgements
We thank the staff of the UCSF Clinical Cancer Genomics Laboratory for assistance with genetic profiling.
Financial support: This study was supported in part by That Man May See, Inc. (Seed Grant to A.R.A.), San Francisco, CA, an unrestricted grant from Research to Prevent Blindness, New York, NY, The National Eye Institute (EY002162, Core Grant for Vision Research and 1K23EY02746, Career Development Award to A.R.A.), Bethesda, MD, and the National Institutes of Health (NIH Director’s Early Independence Award, DP5 OD021403 to D.A.S.), Bethesda, MD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kooi IE, Mol BM, Massink MP, et al. Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci Rep. 2016;6:25264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleinerman RA, Yu CL, Little MP, et al. Variation of second cancer risk by family history of retinoblastoma among long-term survivors. J Clin Concol. 2012;30(9):950–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong JR, Morton LM, Tucker MA, et al. Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J Clin Oncol. 2014;32(29):3284–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li WL, Buckley J, Sanchez-Lara PA, et al. A Rapid and Sensitive Next-Generation Sequencing Method to Detect RB1 Mutations Improves Care for Retinoblastoma Patients and Their Families. J Mol Diagn. 2016;18: 480e493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sippel KC, Fraioli RE, Smith GD, et al. Frequency of Somatic and Germ-Line Mosaicism in Retinoblastoma: Implications for Genetic Counseling. Am J Human Genet. 1998;62:610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Z, Moran K, Richards-Yutz J, et al. Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat. 2014;35(3):384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68: 820–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallie BL, Campbell C, Devlin H, et al. Developmental basis of retinal specific induction of cancer by RB mutation. Cancer Res. 1999;59: 1731s–1735s. [PubMed] [Google Scholar]
- 9.Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes, chromosomes & cancer. 2007;46(7):617–634. [DOI] [PubMed] [Google Scholar]
- 10.Bowles E, Corson TW, Bayani J, et al. Profiling genomic copy number changes in retinoblastoma beyond loss of RB1. Genes, chromosomes & cancer. 2007; 6(2):118–129. [DOI] [PubMed] [Google Scholar]
- 11.Kooi IE, Mol BM, Massink MP, et al. A meta-analysis of retinoblastoma copy numbers refines the list of possible driver genes involved in tumor progression. PLoS One. 2016;11(4):e0153323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rushlow DE, Mol BM, Kennett JY, et al. Characterization of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. 2013;14:327e334. [DOI] [PubMed] [Google Scholar]
- 13.Murphree LA. Intraocular retinoblastoma: the case for a new group classification. Ophthalmol Clin North Am. 2005;18(1):41–53, viii. [DOI] [PubMed] [Google Scholar]
- 14.Mallipatna A, Gallie BL, Chévez-Barrios P, et al. Retinoblastoma In: Amin MB, Edge SB, Greene FL, eds. AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer; 2017:819–831. [Google Scholar]
- 15. https://www.ambrygen.com/clinician/genetic-testing/138/oncology/hereditary-retinoblastoma.
- 16. https://www.invitae.com/en/physician/tests/01738/#info-panel-resources.
- 17. https://impactgenetics.com/testing-services/retinoblastoma/info-for-rb-clinicians/
- 18.Kline CN, Joseph NM, Grenert JP, et al. Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro Oncol. 2017;19:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McEvoy J, Nagahawatte P, Finkelstein D, et al. RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget. 2014;5(2):438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, Benaventa CA, McEvoy J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481(7381):329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huynh KD, Fischle W, Verdin E, et al. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 2000;14: 1810–1823. [PMC free article] [PubMed] [Google Scholar]
- 22.Fan Z, Yamaza T, Lee SJ, et al. BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat Cell Biol. 2000;11:1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dimaras H, Khetan V, Halliday W, et al. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17(10):1363–1372. [DOI] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jo YS, Kim MS, Yoo NJ et al. Somatic mutation of a candidate tumour suppressor MGA gene and its mutational heterogeneity in colorectal cancers. Pathology. 2016;48(5):525–7. [DOI] [PubMed] [Google Scholar]
- 26.Sausen M, Leaery RJ, Jones S, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013;45(1):12–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spencer DH, Sehn JK, Abel HJ, et al. Comparison of clinical targeted next-generation sequence data from formalin-fixed and fresh-frozen tissue specimens. J Mol Diagn. 2013. September;15(5):623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oh E, Choi YL, Kwon MJ, et al. Comparison of Accuracy of Whole-Exome Sequencing with Formalin-Fixed Paraffin-Embedded and Fresh Frozen Tissue Samples. PLoS One. 015 Dec 7;10(12):e0144162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.