

Abstract
Exposure to chronic hyperglycemia because of diabetes mellitus can lead to development and progression of diabetic kidney disease (DKD). We recently reported that reduced superoxide production is associated with mitochondrial dysfunction in the kidneys of mouse models of type 1 DKD. We also demonstrated that humans with DKD have significantly reduced levels of mitochondrion-derived metabolites in their urine. Here we examined renal superoxide production in a type 2 diabetes animal model, the db/db mouse, and the role of a mitochondrial protectant, MTP-131 (also called elamipretide, SS-31, or Bendavia) in restoring renal superoxide production and ameliorating DKD. We found that 18-week-old db/db mice have reduced renal and cardiac superoxide levels, as measured by dihydroethidium oxidation, and increased levels of albuminuria, mesangial matrix accumulation, and urinary H2O2. Administration of MTP-131 significantly inhibited increases in albuminuria, urinary H2O2, and mesangial matrix accumulation in db/db mice and fully preserved levels of renal superoxide production in these mice. MTP-131 also reduced total renal lysocardiolipin and major lysocardiolipin subspecies and preserved lysocardiolipin acyltransferase 1 expression in db/db mice. These results indicate that, in type 2 diabetes, DKD is associated with reduced renal and cardiac superoxide levels and that MTP-131 protects against DKD and preserves physiological superoxide levels, possibly by regulating cardiolipin remodeling.
Keywords: diabetic nephropathy, kidney, mitochondria, cardiolipin, superoxide ion, cardiac metabolism, elamipretide, MTP-131, reactive oxygen species (ROS), shotgun lipidomics
Introduction
The steadily growing number of patients worldwide with diabetes is projected to increase the prevalence of DKD4 (1). Exposure to chronic hyperglycemia modulates several pathological pathways, including intracellular signaling pathways, transcription factors, cytokines, chemokines, and growth factors, that lead to development and progression of DKD (2). Reactive oxygen species (ROS) play a crucial role in progression of DKD; however, the role of ROS, particularly that of superoxide (O2˙̄), in DKD is controversial. Under healthy conditions, normal levels of O2˙̄ and hydrogen peroxide (H2O2) production have been shown to be critical in regulating several fundamental biological processes, including signal transduction, regulation of immune function, and cell growth (3). In mouse models of DKD, overexpression of cytosolic superoxide dismutase (SOD1) protects against mesangial expansion (4), and genetic ablation of SOD1 accelerates renal matrix deposition (5). Conversely, deletion of mitochondrial SOD (SOD2) does not promote susceptibility to DKD (6). In addition, we recently found that O2˙̄ production is reduced in the kidneys of mice with type 1 diabetes (6) and that enhancement of mitochondrial biogenesis and oxidative phosphorylation lead to increased O2˙̄ production but with beneficial effects on kidney function and structure. However, direct measurement of the O2˙̄ radical is difficult because of its short half-life (7, 8).
Dihydroethidium (DHE; hydroethidine (9)) is a cell-permeable compound that is rapidly oxidized in the presence of O2˙̄ but not H2O2 or peroxynitrite (10). It subsequently interacts with DNA to emit a bright red color (11). An important caveat of this approach is that interpretation of DHE experiments requires special attention, as DHE is particularly prone to auto-oxidation in ambient oxygen (12). Two major oxidation products of DHE are ethidium (E+) and 2-hydroxyethidium (2-OH-E+). Although in vitro studies using HPLC have suggested that the specific oxidation product of DHE by O2˙̄ is 2-OH-E+ (7, 13, 14), a recent report by Hall et al. (9) demonstrated the discrepancy between in vitro and in vivo results for DHE oxidation products. Using a novel in vivo live animal imaging method that utilized the difference of fluorescence lifetimes between the two DHE oxidation products (E+, 6 ns; 2-OH-E+, 12 ns), it was shown that the predominant oxidation product in vivo was E+, not 2-OH-E+ (9).
We recently applied a similar in vivo fluorescence lifetime measurement and confocal imaging to examine renal O2˙̄ production in mouse models of type 1 diabetes (6). Surprisingly, O2˙̄ production was reduced not only in the kidneys but also in heart and liver tissues of type 1 diabetes compared with nondiabetic controls (6). The reduction in renal O2˙̄ production was associated with reduced mitochondrial electron transport chain activity with diabetes or with in vivo inhibition of complex I activity. Interestingly, stimulation of mitochondrial electron transport chain activity led to an increase in renal O2˙̄ production and reduced evidence of DKD. In support of a similar pattern in human DKD, several metabolites related to mitochondrial function were reduced in the urine of patients with DKD, and kidney biopsies exhibited a reduction in mitochondrial biogenesis in patients with chronic kidney disease (15, 16).
In this study, we used a murine model of type 2 diabetes (db/db) and applied confocal imaging of DHE oxidation to examine O2˙̄ production in the kidney. As mitochondrial function is reduced in DKD (6), we also evaluated the role of a novel mitochondrion-targeting peptide agent, MTP-131. MTP-131 is a mitochondrion-targeted tetrapeptide (H-d-Arg-Dmt-Lys-Phe-NH2). It has been shown that MTP-131 has many renoprotective effects in various animal models of kidney disease and cell culture studies, including renal ischemic–reperfusion injury (17–19), unilateral ureteral obstruction (20), and hypoxia/reoxygenation-induced tubular injury (21). The mechanism of these effects has generally been ascribed to reduction of ROS and/or improvement in mitochondrial function and efficiency. We hypothesized that MTP-131 has a protective effect in the kidneys of diabetic db/db mice via optimization of renal O2˙̄ production. We further examined whether MTP-131 had an impact on renal cardiolipins (CLs), a class of key phospholipids that play a major role in restoring normal mitochondrial complex activity (18).
Results
Diabetic db/db mice have reduced renal O2˙̄ production
To evaluate O2˙̄ production in the kidneys, 18-week-old db/db and db/m mice underwent administration of DHE for 16 h prior to euthanasia, and kidneys were harvested (6); at this point, all unreacted DHE has been excreted (9). The DHE oxidation signal was prominent in the kidneys of db/m mice (Fig. 1A), but, as expected, diabetic db/db kidneys showed significantly reduced DHE oxidation in glomeruli and cortical tubules (Fig. 1, B–D), indicating reduced O2˙̄ production.
Figure 1.
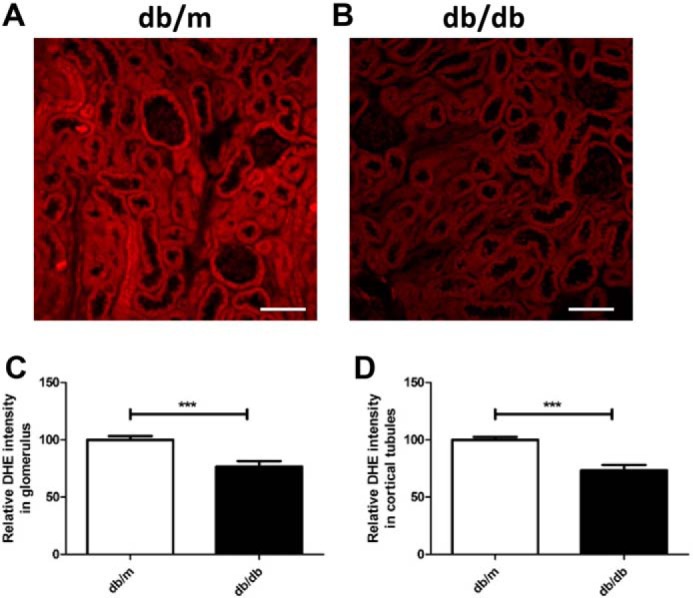
Evaluation of renal superoxide production in db/m and db/db mice. A and B, representative confocal fluorescence images of DHE oxidation indicate that superoxide is reduced in the kidneys of db/db mice compared with db/m mice. Scale bars = 100 μm. C and D, quantification of DHE oxidation in glomerular (C) and cortical tubular (D) lesions. 15 randomly selected glomeruli or tubular lesions per mouse were evaluated (n = 3/group). Values are means ± S.E. ***, p < 0.001.
Metabolic characteristics with MTP-131 treatment
Body weight (Fig. 2A), food intake (Fig. 2B), and glycated hemoglobin (HbA1c) (Fig. 2C) were significantly increased in the db/db groups compared with the nondiabetic (db/m) groups. There were no differences between the db/db and db/db+B (Bendavia/MTP-131) groups during the study period and at the end of the study. Kidney weight per tibial length was increased in the db/db group; however, no difference between treated and untreated diabetic groups was observed (Fig. 2D). Perigonadal fat and liver weights per tibial length were also increased in the db/db group compared with the db/m group (Fig. 2, E and F). There were no significant differences, but a slight trend was noted of a reduction in liver and perigonadal fat weights by MTP-131 treatment. There were no statistical differences in heart weight between groups (Fig. 2G).
Figure 2.

Metabolic characteristics of mice. A, time course of body weights. B, food intake at 13 and 17 weeks of age. C, HbA1c levels before starting treatments (at 6 weeks of age) and at the end of the study (at 18 weeks of age). IFCC, International Federation of Clinical Chemistry and Laboratory Medicine; NGSP, National Glycohemoglobin Standardization Program. D, kidney weight per tibial length. E, perigonadal fat weight per tibial length. F, liver weight per tibial length. G, heart weight per tibial length. n = 15/group. db/m, db/m mice treated with vehicle; db/m+B, db/m mice treated with MTP-131 (Bendavia (B)); db/db, db/db mice treated with vehicle; db/db+B, db/db mice treated with MTP-131. Values are means ± S.E. ***, p < 0.001; **, p < 0.01; *, p < 0.05 versus db/m and db/m+B. ##, p < 0.01; #, p < 0.05.
MTP-131 protects against progression of DKD
Representative findings of the glomeruli in PAS-stained sections are shown in Fig. 3A. Glomerular hypertrophy was observed in both diabetic db/db groups compared with the nondiabetic db/m groups (Fig. 3, A and B). MTP-131 treatment showed a slight improvement in glomerular size, but this was not significant (Fig. 3, A and B). However, mesangial matrix expansion was observed in the db/db group, and MTP-131 treatment significantly reduced mesangial matrix accumulation compared with the db/db group (Fig. 3, A and C). In the db/db group, the urinary albumin/creatinine ratio was significantly increased at 10, 14, and 18 weeks of age and significantly suppressed in the db/db+B group at 14 and 18 weeks of age (Fig. 3D). Urinary hydrogen peroxide/creatinine levels were markedly increased in the db/db group at 10 and 18 weeks age compared with the db/m groups and significantly reduced by MTP-131 treatment at 18 weeks of age (Fig. 3E).
Figure 3.

MTP-131 (Bendavia) suppressed diabetes-induced mesangial matrix expansion, albuminuria, and urine hydrogen peroxide. A, representative images of PAS-stained kidney sections (×400 magnification). Scale bars = 50 μm. B, glomerular hypertrophy was observed in the db/db and db/db+B groups compared with nondiabetic groups. C, PAS-positive mesangial matrix was increased in the db/db group and suppressed ∼50% by MTP-131 treatment. Fifteen randomly selected glomeruli per mouse were examined (n = 5/group). D, urinary albumin/creatinine ratio at 10, 14, and 18 weeks of age. The urinary albumin/creatinine ratio of the db/db group was markedly increased compared with nondiabetic groups and was suppressed in MTP-131-treated db/db mice (n = 8/group). E, urinary H2O2/creatinine ratio at 6, 10, and 18 weeks of age (n = 5–10/group). MTP-131 suppressed urinary hydrogen peroxide in db/db mice at 18 weeks of age. Values are means ± S.E. ***, p < 0.001; **, p < 0.01; *, p < 0.05. ##, p < 0.01 and #, p < 0.05 versus db/db.
MTP-131 reduces perigonadal adipocyte size in db/db mice
We also examined the effect of MTP-131 on adipocyte size to verify whether systemic administration of MTP-131 affected other organs. Representative H&E-stained perigonadal adipose tissue images are shown in Fig. 4A. Interestingly, average adipocyte size was markedly increased in the db/db group and significantly reduced by MTP-131 treatment (Fig. 4, B and C).
Figure 4.

MTP-131 (Bendavia) reduced perigonadal adipocyte size in db/db mice. A, representative images of H&E-stained perigonadal fat sections (×100 magnification). Scale bars = 100 μm. B, average perigonadal adipocyte size. Adipose size was increased in db/db mice and slightly but statistically significantly reduced by MTP-131 treatment. C, adipocyte size in the db/db and db/db+B groups. Forty randomly selected adipocytes per mouse were examined (n = 6/group). Values are means ± S.E. ***, p < 0.001; **, p < 0.01.
MTP-131 preserved renal O2˙̄ production in db/db mice
We then examined the effect of MTP-131 on renal O2˙̄ production (as measured by DHE oxidation using confocal microscopy) in db/db mice. Renal tubules showed intense ethidium staining in db/m mice, whereas glomeruli were quite faint (Fig. 5A). db/db mice showed a significant reduction in renal DHE oxidation compared with the db/m group; however, MTP-131 significantly preserved renal O2˙̄ production in cortical tubules and glomeruli (Fig. 5, A–C). Interestingly, MTP-131 increased DHE oxidation in cortical tubules in the db/m group (Fig. 5C).
Figure 5.
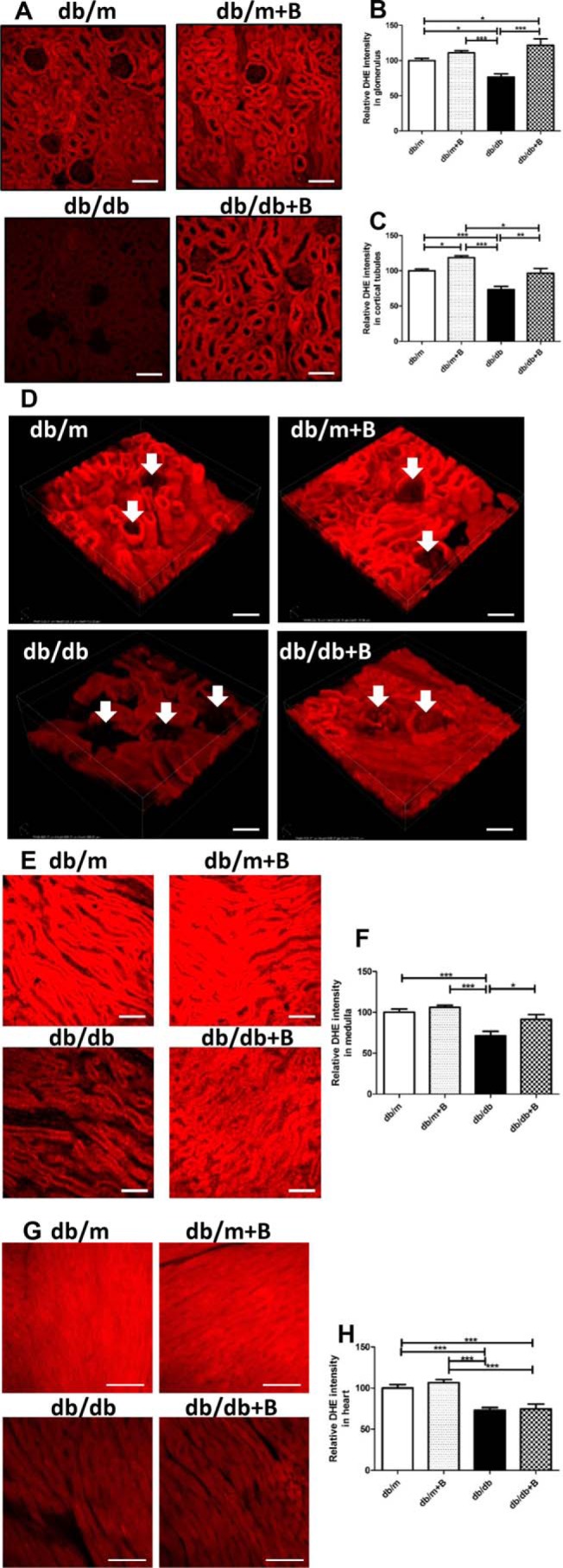
MTP-131 (Bendavia) preserves DHE oxidation in the renal cortex, renal medulla, and heart tissue of db/db mice. A, representative confocal fluorescence images of DHE oxidation. Scale bars = 100 μm. B and C, quantification of DHE oxidation in glomerular (B) and cortical tubular (C) tissue. 15 randomly selected glomeruli or tubular lesions per mouse were evaluated (n = 3/group). D, representative multiphoton 3D fluorescence images of DHE oxidation. Arrows, glomeruli. Scale bars = 100 μm. E, representative confocal fluorescence images of DHE oxidation in the renal medulla. Scale bars = 100 μm. F, quantification of DHE oxidation in renal medullary lesions. 15 randomly selected medullary lesions per mouse were evaluated (n = 3/group). G, representative confocal fluorescence images of DHE oxidation in the heart. Scale bars = 50 μm. H, quantification of DHE oxidation in heart tissue. 15 randomly selected lesions per mouse were evaluated (n = 3/group). Values are means ± S.E. ***, p < 0.001; *, p < 0.05.
We further examined DHE oxidation using multiphoton microscopy to construct renal 3D images. 3D DHE imaging demonstrated DHE fluorescence primarily in the cytosolic compartment of tubular cells, with reduction of DHE oxidation in db/db mice and restoration by MTP-131 treatment (Fig. 5D). As with the renal cortex, DHE oxidation was also significantly decreased in medullary regions of the db/db group and fully preserved by MTP-131 treatment (Fig. 5, E and F). We also measured the protein levels of the five oxidative phosphorylation system complexes I–V in renal cortical tissues; however, none of them showed significant differences among four groups of mice (Fig. S1).
As it has been reported that MTP-131 plays protective roles in the heart (22, 23), we also examined O2˙̄ production in the heart. Although the db/db group showed a significant reduction in DHE oxidation, MTP-131 did not maintain O2˙̄ production in heart tissues (Fig. 5, G and H).
MTP-131 regulates lysocardiolipin, Pla2, and LCLAT1 expression in the kidneys of db/db mice
As it has been reported that MTP-131 interacts with CL to protect mitochondrial function (24), we quantified total CL and lysoCL contents in the kidney cortex. Unexpectedly, there was no difference in total CL content between the groups (Fig. 6A). However, total lysoCL levels were markedly increased in the db/db group and significantly reduced by treatment with MTP-131 (Fig. 6B). We examined whether the protein content in the kidney cortex affected the measurement of CL or lysoCL, but there was no difference between the groups (Fig. 6C).
Figure 6.

MTP-131 (Bendavia) preserves lysoCL, Pla2, and LCLAT1 in the kidneys of db/db mice. A and B, total CL and total lysoCL content in the renal cortex by shotgun lipidomics quantification. C, total protein content/tissue weight in the renal cortex. D–P, quantitative results of lysoCL species by shotgun lipidomics analysis. Q, relative mRNA expression of Pla2 detected by RT-qPCR. Data were normalized to actin (n = 4/group). R, representative Western blot images of LCLAT1 and β-actin (n = 3–4/group). S, quantitative Western blot result of LCLAT1 (n = 3–4/group). Values are means ± S.E. ***, p < 0.001; **, p < 0.01.
Specific lysoCL species were also measured to verify which lysoCL species were related to the protective effects of MTP-131 (Fig. 6, D–P). It is noteworthy that the major lysoCL species, including 18:2–18:2–18:1, 18:2–18:2–18:2, and combined 18:2–18:1–20:3 and 18:2–18:2–20:2, were significantly increased in the db/db group and inhibited by MTP-131 treatment (Fig. 6, I, J, and M). As lysoCL is involved in CL remodeling, we examined the expression of phospholipase A2 (Pla2), an enzyme for deacylation of immature CL to lysoCL (25, 26), and lysocardiolipin acyltransferase 1 (LCLAT1), a key enzyme for CL remodeling and reacylation of lysoCL to CL (27, 28). quantitative RT-PCR (RT-qPCR) data showed that expression of Pla2 was enhanced in cortical tissues of db/db mice and suppressed by MTP-131 treatment (Fig. 6Q). Interestingly, expression of LCLAT1 was markedly decreased in the kidneys of db/db mice and significantly preserved by MTP-131 treatment (Fig. 6, R and S).
MTP-131 regulates immature cardiolipins and long-chain mature cardiolipins in the kidneys of the db/db mice
Diabetic mice had lower levels of immature CL species (those containing short fatty acid chains such as C16:1 and C16:0), including 18:1–18:1–16:0–16:1 (Fig. 7A) and 18:1–18:1–16:1–16:1 (Fig. 7B), in the renal cortex. MTP-131 treatment preserved immature CL levels significantly (e.g. 16:1–18:1–18:1–18:0/16:0–18:1–18:1–18:1, Fig. 7C) or tendentiously (18:1–18:1–16:0–16:1 and 18:1–18:1–16:1–16:1; Fig. 7, A and B). However, long-chain mature CL (Fig. 7, D and E) were greater in the renal cortex of diabetic mice. The accumulation of long-chain CL species was reduced by MTP-131 treatment. Specifically, 18:2–18:2–18:2–20:2 was lower in the renal cortex of the db/db +B group compared with the db/db group (Fig. 7E). In addition, another long-chain mature CL, 18:2–18:2–18:2–20:3, was decreased in the kidney cortex of MTP-131–treated db/db mice compared with db/m mice with or without MTP-131 (Fig. 7F). These observations clearly indicate that CL species are remodeled to those containing longer fatty acyl chains, as reported previously for diabetic mouse hearts (29), and that MTP-131 treatment protects normal regulation of CL synthesis and remodeling.
Figure 7.
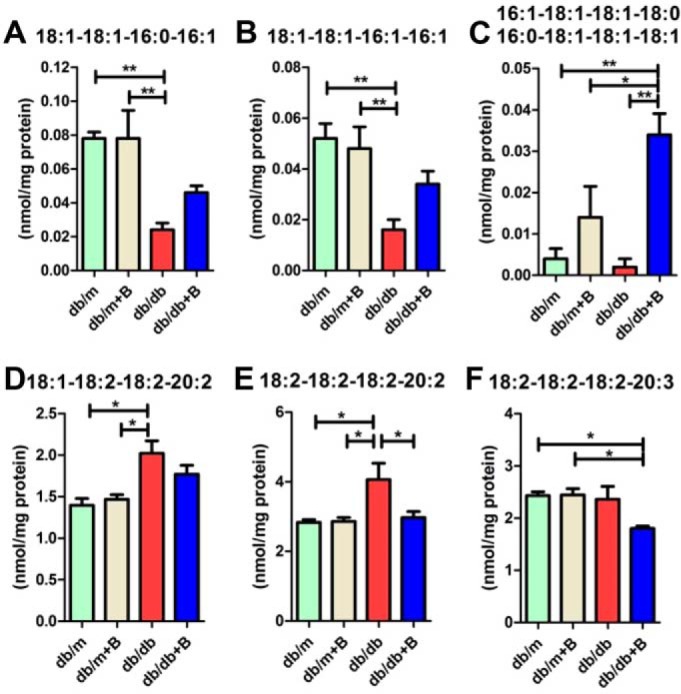
MTP-131 regulates immature CL and long-chain mature CL in the kidneys of db/db mice. A–C, levels of immature CL species in the renal cortex by shotgun lipidomics quantification. D–F, quantitative results of long-chain mature CL in the renal cortex by shotgun lipidomics analysis (n = 5/group). Values are means ± S.E. **, p < 0.01; *, p < 0.05.
MTP-131 regulates mitochondrial fusion machinery in the kidneys of db/db mice
As CL plays important roles in mitochondrial dynamics (30), we measured relative expression of genes regulating mitochondrial fusion and fission. The expression of optic astrophy 1 (Opa1), which regulates mitochondrial fusion and crista structure in the inner mitochondrial membrane and contributes to ATP synthesis and apoptosis (31, 32), was decreased in db/db mice versus the db/m control (Fig. S2A). In addition, the expression of mitofusin 1 (Mfn1), a mediator of mitochondrial fusion in the outer mitochondrial membrane (33, 34), was up-regulated after MTP-131 treatment in db/db mice compared with the db/db mice with vehicle treatment (Fig. S2B). Another dynamin-related GTPase, mitofusion 2 (Mfn2), did not show a difference among the four groups of mice (Fig. S2C. No significant changes were observed in the level of mediator (e.g. dynamin 1 (Dnm1)) or its receptors (e.g. mitochondrial fission 1 protein (Fis1)), involved in mitochondrial fission (Fig. S2, D and E). These results further confirm that diabetic kidneys have reduced mitochondrial fusion and that MTP-1 treatment may protect mitochondrial fusion. We also assessed the mRNA abundance of several mitochondrial biogenesis transcription factors (mitochondrial transcription factor A (Tfam), peroxisome proliferator–activated receptor α (Ppara), nuclear respiratory factor 1 (Nrf1), and nuclear respiratory factor 2 (Nrf2); Fig. S2, F–I) and found it to be unchanged.
Discussion
Our findings reveal that db/db mice, a well-accepted model of type 2 diabetes, experienced a significant reduction of renal O2˙̄ in association with increased albuminuria, elevated urine H2O2, and mesangial matrix expansion. With a new mitochondrial protective agent, MTP-131, renal O2˙̄ levels were preserved, and there was an improvement in urine albumin, H2O2, and mesangial matrix expansion. The beneficial effect of MTP-131 is likely due to reduced lysoCL and an increase in LCLAT1.
Two prior studies have examined DHE oxidation using kidneys of db/db mice and found an increased DHE oxidation signal in db/db mice compared with db/m mice (35, 36). However, in both studies, frozen kidney sections were used, and DHE was applied under ambient oxygen conditions, where auto-oxidation may occur rapidly. Therefore, in this study, DHE was prepared under anaerobic conditions and immediately delivered intraperitoneally for in vivo measurement of O2˙̄ production. Under these conditions, DHE oxidation was markedly reduced in the kidneys of db/db mice, indicating a reduction in renal O2˙̄ production. We further found that MTP-131 increased the levels of renal O2˙̄ in db/db mice similar to control levels without affecting hyperglycemia or body weight. The basis for the preserved renal O2˙̄ production may be improved mitochondrial function, although specific aspects of mitochondrial function that are altered in db/db kidneys and protected by MTP-131 remain to be identified. Restoring a normal CL profile could be one of the underlying mechanisms of preserved mitochondrial function and renoprotection by MTP-131.
Similar to diabetic kidneys, we found that the hearts of db/db mice also exhibited reduced O2˙̄. A similar pattern was noted in type 1 diabetes as well, suggesting that hyperglycemia may be the driving force to reduce O2˙̄ production in multiple organs, independent of insulin levels. On the other hand, MTP-131 was unable to preserve O2˙̄ production in the heart. A recent report has shown that radioisotope-labeled MTP-131 ([125I]SS-31) is rapidly absorbed and selectively concentrated in the kidney (15–100 times higher concentration than in heart tissues) after subcutaneous injection (18). The difference in restoration of DHE oxidation between kidney and heart tissues may be attributed to the difference in MTP-131 distribution.
In this study, in contrast to the reduction in renal O2˙̄ production in db/db mice, urine H2O2 levels were significantly increased in db/db mice and partly inhibited by MTP-131 treatment. The increased H2O2 in the urine may be due to increased renal NADPH oxidase–dependent ROS production (37). In fact, it has been reported that generation of NADPH oxidase (Nox)–derived ROS is increased in the kidneys of multiple models of DKD, with Nox4 likely the most important isoform that is up-regulated in the diabetic kidney (38). In addition, Nox4 can be a potent source of H2O2 production, independent of O2˙̄ production (39). The increased H2O2 is unlikely to originate from mitochondria, as we have reported that isolated mitochondria from diabetic mice have reduced H2O2 production (6).
Interestingly, it has been reported that MTP-131 can directly scavenge ROS, including H2O2 and free radicals; in addition, the Tyr and 2,6-dimethyltyrosine residues of the peptide play critical roles in its ROS-scavenging ability (19). However, a recent study was unable to detect any scavenging activity of MTP-131 (40). Thus, further work will be needed to determine the mechanism of reduction of urinary H2O2 by MTP-131. MTP-131 can also inhibit high-fat diet–induced insulin resistance in muscle tissue (41) and islet cell apoptosis (42). In this study, although there was no difference in HbA1c levels or overall body weight between the db/db and db/db+B groups, MTP-131 treatment partially reduced visceral adipocyte size, which may reduce inflammation and insulin resistance.
The effects of MTP-131 are thought to be partly mediated by interaction with CL and inhibition of cytochrome c–mediated peroxidation (18). In this study, there was no detectable difference in total renal CL content between the control and db/db groups (Fig. 6A), indicating that total CL remains sustained despite severe hyperglycemia. In contrast to total CL content, there was a clear increase in renal total lysoCL content in db/db mice, which was significantly inhibited by MTP-131 treatment (Fig. 6B). It has been reported that an increase in lysoCL is associated with CL oxidation and suppression of CL oxidation by a mitochondrion-targeted nitroxide (XJB-5-131) prevented CL oxidation and hydrolysis (43), suggesting that the much smaller lysoCL pool is a more sensitive end point than the total CL pool (Fig. 6, A versus B).
CL is one of the most unsaturated lipids in the body, and the degree of unsaturation is maintained by means of a constant remodeling process (44). Pla2 is a key enzyme that catalyzes hydrolysis of immature CL to lysoCL, which is involved in CL remodeling (25, 26, 28). In the remodeling process of CL, LCLAT1, also known as ALCAT1 or LYCAT, catalyzes acylation of lysoCL to produce CL (27, 28, 44). In this study, up-regulation of Pla2 and reduction of LCLAT1 expression in the kidneys of db/db mice may lead to accumulation of lysoCL in db/db mice. In addition, our study uniquely demonstrates that MTP-131 can suppress Pla2 expression, preserve LCLAT1 expression, and normalize lysoCL content in the kidneys of db/db mice. This may prove to be a key mechanism by which MTP-131 provides renoprotection via regulation of CL remodeling pathway.
However, the roles of LCLAT1 in various animal disease models remain controversial. Li et al. (27) reported that LCLAT1-overexpressing mouse myoblast cells show a reduction of total CL contents compared with the vector control. In addition, they showed that LCLAT1 deficiency prevents onset of diet-induced obesity and improves mitochondrial complex I activity and lipid oxidation in the liver. Interestingly, LCLAT1 whole-body-deficient mice also show hyperactivity, hyperphagia, and hypermetabolism (27). However, these changes in phenotype may also directly affect mitochondrial function and systemic metabolic changes. It has been reported that LCLAT1 mRNA and protein expression are significantly decreased in heart tissues of patients with tetralogy of Fallot, a common form of cyanotic congenital heart defect; however, the mechanism underlying the regulation of LCLAT1 remains unknown (45). On the other hand, LCLAT1 expression is increased in lungs from patients with idiopathic pulmonary fibrosis and in murine models of lung fibrosis (46). It is noteworthy that LCLAT1 mRNA expression levels in peripheral blood mononuclear cells (PBMCs) positively correlates with improved lung function and survival rate in patients with idiopathic pulmonary fibrosis. In addition, overexpression of LCLAT1 attenuates fibrogenesis, and knockdown of LCLAT1 accentuates inflammation and injury in murine models of lung fibrosis, suggesting protective roles of lysocardiolipin acyltransferase 1. The latter data are consistent with the findings in this study, as there was a reduction in matrix accumulation in association with an increase in LCLAT1. The protective role of MTP-131 may be partly explained by stimulation of renal LCLAT1 expression.
Reduced content of immature CL and increased levels of lysoCL species in diabetic kidneys indicate enhanced CL hydrolysis (i.e. up-regulated Pla2) and loss of CL remodeling activity, such as LCLAT1 activity. However, long-chain mature unsaturated CLs were increased in diabetic kidneys and reduced by MTP-131 treatment. Accumulation of CL species containing longer fatty acyl chains (greater than 18 carbon atoms) has been reported as a detrimental factor in diabetic conditions (e.g. diabetic myocardium) (29). It has been reported that tetra 18:2 CL is a fully functional CL species, and increases in long chain CLs are associated with mitochondrial dysfunction (29, 47, 48). Although LCLAT1 has no acyl-chain specificity, we postulate that LCLAT1 tends to remodel lysoCL to more long-chain unsaturated mature CL in diabetic kidneys. We also found that MTP-131 might protect mitochondrial function by suppressing CL hydrolysis and reducing levels of long-chain (>18C) unsaturated CLs.
The profile of CL and lysoCL after remodeling in diabetic kidneys leads to mitochondrial dysfunction, likely because of increased basal uncoupling and H+ leakage. CL play an important role in regulating the uncoupling activity of uncoupling proteins (UCPs) and H+ translocation through the membrane (49). Modest depolarization of the mitochondrial inner membrane is known to attenuate mitochondrial ROS, such as O2˙̄ generation (50). Normal CL could maintain proper activation of UCPs for mitochondrial depolarization to attenuate electron transport chain ROS production without disturbing ATP production (50). The mechanism underlying the role of lysoCL and long-chain unsaturated mature CL in diabetic kidneys might be related to enhanced activation of UCPs and leaking of H+ across the mitochondrial inner membrane, dissipating the H+ gradient generated by the respiratory chain. This reduced respiration could lead to reduced O2˙̄ production compared with normal conditions. MTP-131 treatment could reverse the CL remodeling in diabetic kidneys and preserve normal O2˙̄ levels. The mechanisms of disordered CL remodeling and mitochondrial dysfunction in kidneys as well as MTP-131's potential roles in renoprotection through preserved CL remodeling and mitochondrial O2˙̄ levels in diabetes are summarized in Fig. 8.
Figure 8.

Mechanisms of CL remodeling and mitochondrial dysfunction in diabetic kidneys and MTP-131's potential roles in renoprotection. Diabetic renal cortical tissues had reduced content of immature CL and increased level of lysoCL, indicating enhanced CL hydrolysis, dysregulated CL remodeling, and loss of LCLAT1 activity. Accumulation of bad CL (e.g. long-chain CL) leads to mitochondrial dysfunction, likely because of enhanced basal uncoupling and H+ leak and reduced electron transport chain (ETC) activity. Reduced respiration leads to decreased mitochondrial O2˙̄ production, which further contributes to mitochondrial dysfunction and DKD phenotypes. The renoprotective mechanism of MTP-131 might be related to its roles in restoring normal CL remodeling by reducing lysoCL and “bad” CL levels. Furthermore, MTP-131 preserves mitochondrial O2˙̄ production and alleviates DKD phenotypes. Red represents effects of diabetes. Under the condition of diabetes, (+) means up-regulated, and (−) means down-regulated. Blue represents effects of MTP-131 treatment to prevent or inhibit diabetes-induced changes.
In conclusion, we found, for the first time, that renal O2˙̄ production is reduced in the kidney and heart tissues of db/db mice and that MTP-131 (Bendavia) can partly preserve renal O2˙̄ levels in the kidneys, lower elevated urinary albumin, and, importantly, delay expansion of the mesangial matrix, arguably the central lesion of DKD. MTP-131's effect to normalize lysoCL via restoration of LCLAT1 expression may contribute to renoprotective roles of MTP-131 in progression of DKD in db/db mice. These findings provide a better understanding of the pathogenesis of DKD in type 2 diabetes and support the concept that mitochondrion-protective agents may be developed into effective treatments for chronic kidney disease and DKD.
Experimental procedures
Animal study
Male db/db mice (BKS.Cg-Dock7m +/+ Leprdb/J strain) were purchased from The Jackson Laboratory (Bar Harbor, ME), and the corresponding heterozygote lean db/m mice were used as controls. Mice were given standard rodent chow and water ad libitum. MTP-131 (Bendavia) was provided by Stealth BioTherapeutics Inc. (Newton, MA). Six-week-old male nondiabetic db/m mice and diabetic db/db mice were randomly assigned to four groups (n = 15/group): db/m mice treated with vehicle (db/m), db/m mice treated with MTP-131 (Bendavia) (db/m+B), db/db mice treated with vehicle (db/db), and db/db mice treated with MTP-131 (db/db+B). Vehicle (PBS) or MTP-131 (3 mg/kg of body weight) was injected subcutaneously daily for 4 weeks and then infused subcutaneously via Alzet osmotic minipumps (Durect Corp., Cupertino, CA; 3 mg/kg/day) over an 8-week period. Osmotic minipumps were implanted subcutaneously in the back of all mice. As the life expectancy of the osmotic pump was 4 weeks, all pumps were replaced with new filled pumps when mice reached the age of 14 weeks. HbA1c was measured with a DCA Vantage Analyzer (Siemens, Malvern, PA). Urine albumin and creatinine were measured with the Mouse Albumin ELISA Quantitation Set (Bethyl Laboratories, Inc., Montgomery, TX) and the Creatinine Companion Kit (Exocell, Philadelphia, PA) according to the manufacturers' protocols. Urinary hydrogen peroxide levels were measured using a 2900D Biochemistry Analyzer (YSI Inc., Yellow Springs, OH). At 18 weeks of age, all mice were euthanized, and the organs were harvested as described previously (51). All animal studies were approved by the Institutional Animal Care and Use Committee of the University of California, San Diego.
Histology
Periodic acid-Schiff (PAS)–stained kidney sections were analyzed as described previously with slight modifications (51). To evaluate the glomerular size and mesangial matrix area, 15 randomly selected glomeruli per mouse were analyzed using i-solution software (Advanced Imaging Concepts, Princeton, NJ). To quantify perigonadal adipocyte size, H&E-stained sections were analyzed using ImageJ (National Institutes of Health; RRID: SCR_003070). Quantitative analysis was performed in a blinded manner.
Superoxide measurement
Superoxide generation was examined as described previously (6). DHE was purchased from Sigma-Aldrich (St. Louis, MO). Briefly, mice were administered two intraperitoneal doses of DHE (each 27 mg/kg) given 30 min apart. Sixteen hours after DHE injection, mice were anesthetized and perfused intracardially with cold PBS followed by 4% paraformaldehyde, and the kidneys were harvested. For confocal imaging, the right kidney was sectioned using a vibrating microtome (Leica Biosystems Inc., Buffalo Grove, IL), mounted on glass slides using Vectashield Antifade Mounting Medium (Vector Laboratories, Burlingame, CA), and imaged by A1R+ confocal microscopy (excitation, 561 nm; emission, 595 nm; Nikon Instruments Inc., Melville, NY). For multiphoton imaging, the kidney was cut into 3-mm thickness and placed onto a glass slide (Fisher Scientific, Pittsburgh, PA), washed gently with PBS overnight, and imaged using an A1R MP+ multiphoton confocal microscope (excitation, 800 nm; emission, 629 nm; Nikon). 3D images were reconstructed using NIS-Elements software (Nikon). DHE signal intensity was quantified by ImageJ. Briefly, average pixel intensity was quantified in 15 randomly selected cortical tubular lesions or glomerular lesions from each sample (n = 3/group). The results were expressed as relative intensity compared with the db/m group. Quantitative analysis was performed in a blinded manner.
Cardiolipin and monolysocardiolipin quantification
For cardiolipin and monolysocardiolipin (lysoCL) analysis, kidney cortex tissues were snap-frozen in liquid nitrogen immediately after harvest. Lipids were extracted from frozen kidneys, and cardiolipins were analyzed by shotgun lipidomics as described previously (52).
RT-qPCR
Gene expression was assessed by RT-qPCR. Briefly, total RNA was isolated from kidney tissue using RNeasy Plus Universal Mini Kits (Qiagen, Germantown, MD) in accordance with the manufacturer's instructions. Total RNA (1.5 μg) was reverse-transcribed to complementary DNA using the SuperScriptTM VILOTM Kit (Thermo Fisher Scientific, Grand Island, NY). Real-time qPCR was performed with the gene-specific TaqMan qPCR primers (Thermo Fisher Scientific) according to the manufacturer's instructions in a real-time PCR machine (QuantStudio 3, Applied Biosystems by Thermo Fisher Scientific).
Immunoblotting
Kidney cortex lysates were prepared using radioimmunoprecipitation assay buffer (Cell Signaling Technology, Danvers, MA) according to the manufacturer's instructions. Total protein concentration was determined using the BCA Protein Assay Kit (Life Technologies). 30 μg of protein per sample was loaded onto a 4%–12% BisTris gel and transferred to a nitrocellulose membrane. The blot was blocked with 5% nonfat dry milk and incubated with anti-LCLAT1 antibody (1:500, Aviva Systems Biology, San Diego, CA) at 4 °C overnight. Following immunoblotting, ECL rabbit IgG and HRP-linked whole antibody (GE Healthcare Life Sciences, Chicago, IL) were applied, and the signals were developed with SuperSignalTM West Pico Chemiluminescent Substrate (Life Technologies).
Statistics
Data are expressed as mean ± S.E., with n values as indicated in the text or figure legends. Statistical significance was determined by Student's t test or one-way analysis of variance, with p < 0.05 considered significant.
Data availability
All data described are contained within the manuscript and Figs S1 and S2. Raw data used for generation of graphs are available upon request.
Author contributions
S. Miyamoto, G. Z., X. H., and K. S. data curation; S. Miyamoto and G. Z. formal analysis; S. Miyamoto, G. Z., and K. S. investigation; S. Miyamoto, G. Z., S. Maity, and X. H. methodology; S. Miyamoto and G. Z. writing-original draft; S. Miyamoto and K. S. project administration; S. Miyamoto, G. Z., D. H., P. J. O., S. Maity, M. M., X. H., and K. S. writing-review and editing; G. Z. and K. S. software; G. Z., S. Maity, and K. S. validation; G. Z. and K. S. visualization; K. S. conceptualization; K. S. resources; K. S. supervision; K. S. funding acquisition.
Supplementary Material
This work was supported by a Veterans Affairs MERIT Award 1I01BX003234 (to K. S.) and NIDDK, National Institutes of Health Grants U01 DK060995, DP3 DK094352-01, and DK083142 (to K. S.). P. J. O. consults with Stealth Biotherapeutics Inc. and has a financial interest with the company. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- DKD
- diabetic kidney disease
- ROS
- reactive oxygen species
- DHE
- dihydroethidium
- CL
- cardiolipin
- PAS
- periodic acid-Schiff
- RT-qPCR
- quantitative RT-PCR
- Nox
- NADPH oxidase
- UCP
- uncoupling protein
- H2O2
- hydrogen peroxide
- HbA1c
- glycated hemoglobin
- H&E
- hematoxylin and eosin
- LCLAT1
- lysocardiolipin acyltransferase 1
- lysoCL
- lysocardiolipin
- Mfn1
- mitofusin 1
- MTP-131
- Bendavia
- O2˙̄
- superoxide
- Pla2
- phospholipase A2.
References
- 1. Tuttle K. R., Bakris G. L., Bilous R. W., Chiang J. L., de Boer I. H., Goldstein-Fuchs J., Hirsch I. B., Kalantar-Zadeh K., Narva A. S., Navaneethan S. D., Neumiller J. J., Patel U. D., Ratner R. E., Whaley-Connell A. T., and Molitch M. E.. 2014) Diabetic kidney disease: a report from an ADA Consensus Conference. Am. J. Kidney Dis. 64, 510–533 10.1053/j.ajkd.2014.08.001 [DOI] [PubMed] [Google Scholar]
- 2. Sharma K. 2015) Mitochondrial hormesis and diabetic complications. Diabetes 64, 663–672 10.2337/db14-0874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brieger K., Schiavone S., Miller F. J. Jr, and Krause K. H.. 2012) Reactive oxygen species: from health to disease. Swiss. Med. Wkly. 142, w13659 [DOI] [PubMed] [Google Scholar]
- 4. Craven P. A., Melhem M. F., Phillips S. L., and DeRubertis F. R.. 2001) Overexpression of Cu2+/Zn2+ superoxide dismutase protects against early diabetic glomerular injury in transgenic mice. Diabetes 50, 2114–2125 10.2337/diabetes.50.9.2114 [DOI] [PubMed] [Google Scholar]
- 5. Fujita H., Fujishima H., Takahashi K., Sato T., Shimizu T., Morii T., Shimizu T., Shirasawa T., Qi Z., Breyer M. D., Harris R. C., Yamada Y., and Takahashi T.. 2012) SOD1, but not SOD3, deficiency accelerates diabetic renal injury in C57BL/6-Ins2(Akita) diabetic mice. Metabolism 61, 1714–1724 10.1016/j.metabol.2012.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dugan L. L., You Y. H., Ali S. S., Diamond-Stanic M., Miyamoto S., DeCleves A. E., Andreyev A., Quach T., Ly S., Shekhtman G., Nguyen W., Chepetan A., Le T. P., Wang L., Xu M., et al. (2013) AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Invest. 123, 4888–4899 10.1172/JCI66218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maghzal G. J., Krause K. H., Stocker R., and Jaquet V.. 2012) Detection of reactive oxygen species derived from the family of NOX NADPH oxidases. Free Radic. Biol. Med. 53, 1903–1918 10.1016/j.freeradbiomed.2012.09.002 [DOI] [PubMed] [Google Scholar]
- 8. Bordt E. A., and Polster B. M.. 2014) NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: a bipartisan affair? Free Radic. Biol. Med. 76, 34–46 10.1016/j.freeradbiomed.2014.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hall D. J., Han S. H., Chepetan A., Inui E. G., Rogers M., and Dugan L. L.. 2012) Dynamic optical imaging of metabolic and NADPH oxidase-derived superoxide in live mouse brain using fluorescence lifetime unmixing. J. Cereb. Blood Flow Metab. 32, 23–32 10.1038/jcbfm.2011.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fernandes D. C., Wosniak J. Jr, Pescatore L. A., Bertoline M. A., Liberman M., Laurindo F. R., and Santos C. X.. 2007) Analysis of DHE-derived oxidation products by HPLC in the assessment of superoxide production and NADPH oxidase activity in vascular systems. Am. J. Physiol. Cell Physiol. 292, C413–C422 10.1152/ajpcell.00188.2006 [DOI] [PubMed] [Google Scholar]
- 11. Tarpey M. M., Wink D. A., and Grisham M. B.. 2004) Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R431–R444 10.1152/ajpregu.00361.2003 [DOI] [PubMed] [Google Scholar]
- 12. Dikalov S. I., Kirilyuk I. A., Voinov M., and Grigor'ev I. A.. 2011) EPR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radic. Res. 45, 417–430 10.3109/10715762.2010.540242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao H., Kalivendi S., Zhang H., Joseph J., Nithipatikom K., Vásquez-Vivar J., and Kalyanaraman B.. 2003) Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic. Biol. Med. 34, 1359–1368 10.1016/S0891-5849(03)00142-4 [DOI] [PubMed] [Google Scholar]
- 14. Dikalov S. I., and Harrison D. G.. 2014) Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 20, 372–382 10.1089/ars.2012.4886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sharma K., Karl B., Mathew A. V., Gangoiti J. A., Wassel C. L., Saito R., Pu M., Sharma S., You Y. H., Wang L., Diamond-Stanic M., Lindenmeyer M. T., Forsblom C., Wu W., Ix J. H., et al. (2013) Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 24, 1901–1912 10.1681/ASN.2013020126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kang H. M., Ahn S. H., Choi P., Ko Y. A., Han S. H., Chinga F., Park A. S., Tao J., Sharma K., Pullman J., Bottinger E. P., Goldberg I. J., and Susztak K.. 2015) Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21, 37–46 10.1038/nm.3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Szeto H. H., Liu S., Soong Y., Wu D., Darrah S. F., Cheng F. Y., Zhao Z., Ganger M., Tow C. Y., and Seshan S. V.. 2011) Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 22, 1041–1052 10.1681/ASN.2010080808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Birk A. V., Liu S., Soong Y., Mills W., Singh P., Warren J. D., Seshan S. V., Pardee J. D., and Szeto H. H.. 2013) The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 24, 1250–1261 10.1681/ASN.2012121216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao K., Zhao G. M., Wu D., Soong Y., Birk A. V., Schiller P. W., and Szeto H. H.. 2004) Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 279, 34682–34690 10.1074/jbc.M402999200 [DOI] [PubMed] [Google Scholar]
- 20. Mizuguchi Y., Chen J., Seshan S. V., Poppas D. P., Szeto H. H., and Felsen D.. 2008) A novel cell-permeable antioxidant peptide decreases renal tubular apoptosis and damage in unilateral ureteral obstruction. Am. J. Physiol. Renal Physiol. 295, F1545–F1553 10.1152/ajprenal.00395.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao W. Y., Han S., Zhang L., Zhu Y. H., Wang L. M., and Zeng L.. 2013) Mitochondria-targeted antioxidant peptide SS31 prevents hypoxia/reoxygenation-induced apoptosis by down-regulating p66Shc in renal tubular epithelial cells. Cell Physiol. Biochem. 32, 591–600 10.1159/000354463 [DOI] [PubMed] [Google Scholar]
- 22. Dai D. F., Hsieh E. J., Chen T., Menendez L. G., Basisty N. B., Tsai L., Beyer R. P., Crispin D. A., Shulman N. J., Szeto H. H., Tian R., MacCoss M. J., and Rabinovitch P. S.. 2013) Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 6, 1067–1076 10.1161/CIRCHEARTFAILURE.113.000406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sloan R. C., Moukdar F., Frasier C. R., Patel H. D., Bostian P. A., Lust R. M., and Brown D. A.. 2012) Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J. Mol. Cell Cardiol. 52, 1009–1018 10.1016/j.yjmcc.2012.02.009 [DOI] [PubMed] [Google Scholar]
- 24. Birk A. V., Chao W. M., Bracken C., Warren J. D., and Szeto H. H.. 2014) Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 171, 2017–2028 10.1111/bph.12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hsu Y. H., Dumlao D. S., Cao J., and Dennis E. A.. 2013) Assessing phospholipase A2 activity toward cardiolipin by mass spectrometry. PLoS ONE 8, e59267 10.1371/journal.pone.0059267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kiebish M. A., Yang K., Liu X., Mancuso D. J., Guan S., Zhao Z., Sims H. F., Cerqua R., Cade W. T., Han X., and Gross R. W.. 2013) Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 54, 1312–1325 10.1194/jlr.M034728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li J., Romestaing C., Han X., Li Y., Hao X., Wu Y., Sun C., Liu X., Jefferson L. S., Xiong J., Lanoue K. F., Chang Z., Lynch C. J., Wang H., and Shi Y.. 2010) Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 12, 154–165 10.1016/j.cmet.2010.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He Q., and Han X.. 2014) Cardiolipin remodeling in diabetic heart. Chem. Phys. Lipids 179, 75–81 10.1016/j.chemphyslip.2013.10.007 [DOI] [PubMed] [Google Scholar]
- 29. Han X., Yang J., Yang K., Zhao Z., Abendschein D. R., and Gross R. W.. 2007) Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study. Biochemistry 46, 6417–6428 10.1021/bi7004015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kameoka S., Adachi Y., Okamoto K., Iijima M., and Sesaki H.. 2018) Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. 28, 67–76 10.1016/j.tcb.2017.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Patten D. A., Wong J., Khacho M., Soubannier V., Mailloux R. J., Pilon-Larose K., MacLaurin J. G., Park D. S., McBride H. M., Trinkle-Mulcahy L., Harper M. E., Germain M., and Slack R. S.. 2014) OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 33, 2676–2691 10.15252/embj.201488349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Santarelli R., Rossi R., Scimemi P., Cama E., Valentino M. L., La Morgia C., Caporali L., Liguori R., Magnavita V., Monteleone A., Biscaro A., Arslan E., and Carelli V.. 2015) OPA1-related auditory neuropathy: site of lesion and outcome of cochlear implantation. Brain 138, 563–576 10.1093/brain/awu378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adams M. D., Kerlavage A. R., Fields C., and Venter J. C.. 1993) 3,400 new expressed sequence tags identify diversity of transcripts in human brain. Nat. Genet. 4, 256–267 10.1038/ng0793-256 [DOI] [PubMed] [Google Scholar]
- 34. Santel A., and Fuller M. T.. 2001) Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114, 867–874 [DOI] [PubMed] [Google Scholar]
- 35. Zhang Y., Yuen D. A., Advani A., Thai K., Advani S. L., Kepecs D., Kabir M. G., Connelly K. A., and Gilbert R. E.. 2012) Early-outgrowth bone marrow cells attenuate renal injury and dysfunction via an antioxidant effect in a mouse model of type 2 diabetes. Diabetes 61, 2114–2125 10.2337/db11-1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y., Wada J., Hashimoto I., Eguchi J., Yasuhara A., Kanwar Y. S., Shikata K., and Makino H.. 2006) Therapeutic approach for diabetic nephropathy using gene delivery of translocase of inner mitochondrial membrane 44 by reducing mitochondrial superoxide production. J. Am. Soc. Nephrol. 17, 1090–1101 10.1681/ASN.2005111148 [DOI] [PubMed] [Google Scholar]
- 37. Towler D. A. 2013) Mitochondrial ROS deficiency and diabetic complications: AMP[K]-lifying the adaptation to hyperglycemia. J. Clin. Invest. 123, 4573–4576 10.1172/JCI72326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eid A. A., Ford B. M., Block K., Kasinath B. S., Gorin Y., Ghosh-Choudhury G., Barnes J. L., and Abboud H. E.. 2010) AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J. Biol. Chem. 285, 37503–37512 10.1074/jbc.M110.136796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takac I., Schröder K., Zhang L., Lardy B., Anilkumar N., Lambeth J. D., Shah A. M., Morel F., and Brandes R. P.. 2011) The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 286, 13304–13313 10.1074/jbc.M110.192138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brown D. A., Hale S. L., Baines C. P., del Rio C. L., Hamlin R. L., Yueyama Y., Kijtawornrat A., Yeh S. T., Frasier C. R., Stewart L. M., Moukdar F., Shaikh S. R., Fisher-Wellman K. H., Neufer P. D., and Kloner R. A.. 2014) Reduction of early reperfusion injury with the mitochondria-targeting peptide Bendavia. J. Cardiovasc. Pharmacol. Ther. 19, 121–132 10.1177/1074248413508003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Anderson E. J., Lustig M. E., Boyle K. E., Woodlief T. L., Kane D. A., Lin C. T., Price J. W. 3rd, Kang L., Rabinovitch P. S., Szeto H. H., Houmard J. A., Cortright R. N., Wasserman D. H., and Neufer P. D.. 2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 119, 573–581 10.1172/JCI37048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thomas D. A., Stauffer C., Zhao K., Yang H., Sharma V. K., Szeto H. H., and Suthanthiran M.. 2007) Mitochondrial targeting with antioxidant peptide SS-31 prevents mitochondrial depolarization, reduces islet cell apoptosis, increases islet cell yield, and improves posttransplantation function. J. Am. Soc. Nephrol. 18, 213–222 10.1681/ASN.2006080825 [DOI] [PubMed] [Google Scholar]
- 43. Ji J., Baart S., Vikulina A. S., Clark R. S., Anthonymuthu T. S., Tyurin V. A., Du L., St Croix C. M., Tyurina Y. Y., Lewis J., Skoda E. M., Kline A. E., Kochanek P. M., Wipf P., Kagan V. E., and Bayır H.. 2015) Deciphering of mitochondrial cardiolipin oxidative signaling in cerebral ischemia-reperfusion. J. Cereb. Blood Flow Metab. 35, 319–328 10.1038/jcbfm.2014.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Esposti M. D. 2002) Lipids, cardiolipin and apoptosis: a greasy licence to kill. Cell Death Differ. 9, 234–236 10.1038/sj.cdd.4400997 [DOI] [PubMed] [Google Scholar]
- 45. Xia Y., Hong H., Ye L., Wang Y., Chen H., and Liu J.. 2013) Label-free quantitative proteomic analysis of right ventricular remodeling in infant tetralogy of Fallot patients. J. Proteomics 84, 78–91 10.1016/j.jprot.2013.03.032 [DOI] [PubMed] [Google Scholar]
- 46. Huang L. S., Mathew B., Li H., Zhao Y., Ma S. F., Noth I., Reddy S. P., Harijith A., Usatyuk P. V., Berdyshev E. V., Kaminski N., Zhou T., Zhang W., Zhang Y., Rehman J., et al. (2014) The mitochondrial cardiolipin remodeling enzyme lysocardiolipin acyltransferase is a novel target in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 189, 1402–1415 10.1164/rccm.201310-1917OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Han X., Yang J., Cheng H., Yang K., Abendschein D. R., and Gross R. W.. 2005) Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry 44, 16684–16694 10.1021/bi051908a [DOI] [PubMed] [Google Scholar]
- 48. Zachman D. K., Chicco A. J., McCune S. A., Murphy R. C., Moore R. L., and Sparagna G. C.. 2010) The role of calcium-independent phospholipase A2 in cardiolipin remodeling in the spontaneously hypertensive heart failure rat heart. J. Lipid Res. 51, 525–534 10.1194/jlr.M000646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Klingenberg M. 2009) Cardiolipin and mitochondrial carriers. Biochim. Biophys. Acta 1788, 2048–2058 10.1016/j.bbamem.2009.06.007 [DOI] [PubMed] [Google Scholar]
- 50. Sack M. N. 2006) Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc. Res. 72, 210–219 10.1016/j.cardiores.2006.07.010 [DOI] [PubMed] [Google Scholar]
- 51. Zhao J., Miyamoto S., You Y. H., and Sharma K.. 2015) AMP-activated protein kinase (AMPK) activation inhibits nuclear translocation of Smad4 in mesangial cells and diabetic kidneys. Am. J. Physiol. Renal Physiol. 308, F1167–F1177 10.1152/ajprenal.00234.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Han X., Yang K., Yang J., Cheng H., and Gross R. W.. 2006) Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. J. Lipid Res. 47, 864–879 10.1194/jlr.D500044-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data described are contained within the manuscript and Figs S1 and S2. Raw data used for generation of graphs are available upon request.