

Abstract
Lucina pectinata live in high concentrations of hydrogen sulfide (H2S) and contains one hemoglobin, Hemoglobin I (HbI), transporting H2S and two hemoglobins, Hemoglobin II (HbII) and Hemoglobin (HbIII), transferring dioxygen to symbionts. HbII and HbIII contain B10 tyrosine (Tyr) and E7 glutamine (Gln) in the heme pocket generating an efficient hydrogen bonding network with the (HbII-HbIII)-O2 species, leading to very low ligand dissociation rates. The results indicate that the oxy-hemeprotein is susceptible to pH from 4 to 9, at acidic conditions, and as a function of the potassium ferricyanide concentration, 100% of the met-aquo derivative is produced. Without a strong oxidant, pH 5 generates a small concentration of the met-aquo complex. The process is accelerated by the presence of salts, as indicated by the crystallization structures and UV-Vis spectra. The results suggest that acidic pH generates conformational changes associated with B10 and E7 heme pocket amino acids, weakening the (HbII-HbIII)-O2 hydrogen bond network. The observation is supported by X-ray crystallography, since at pH 4 and 5, the heme-Fe tends to oxidize, while at pH 7, the oxy-heterodimer is present. Conformational changes also are observed at higher pH by the presence of a 605 nm transition associated with the iron heme-Tyr interaction. Therefore, pH is one crucial factor regulating the (HbII-HbIII)-O2 complex hydrogen-bonding network. Thus, it can be proposed that the hydrogen bonding adjustments between the heme bound O2 and the Tyr and Gln amino acids contribute to oxygen dissociation from the (HbII-HbIII)-O2 system.
Keywords: hemeprotein, tyrosine, glutamine, pH susceptibility, oxygen release
Graphical Abstract
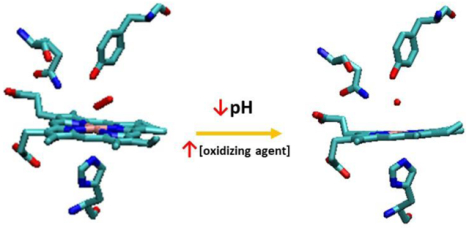
Two factors control the full oxidation of the hemoglobin (Hb) heterodimer (HbII-HbIII)-O2 to the metaquo (HbII-HbIII): an oxidizing agent and a low pH in the 4 to 5 range.
1. Introduction
Hemeproteins comprise a wide variety of proteins specialized in small molecule reactivity, including O2, NO, CO2, and H2S [1–10]. Functionalization is principally governed by the presence of specific amino acid residues in the active site of the protein, which are a result of the organisms’ adaptations to the environment. Hemeproteins with tyrosine (Tyr) and glutamine (Gln) in their heme active site pocket are found in both eukaryotes and bacteria. These hemeproteins are characterized by a high oxygen affinity due to a very low dissociation constant (Table 1) [11,12,13]. In these hemoglobins and analogous systems, a strong hydrogen bond between the ligated O2 molecule and the tyrosine side chain anchors the oxygen in the active center, aided by a weaker hydrogen bond to the distal glutamine [14–18]. The hydrogen bond network accounts for the proteins’ low dissociation constant. Examples include Hemoglobin II (HbII) and Hemoglobin III (HbIII) from the bivalve Lucina pectinata [11,14,19], and several truncated hemoglobins (TrHb) found in all kingdoms of life [20,21], like Mycobacterium tuberculosis TrHb N [13], and Ascaris hemoglobin (AscHb) of the parasitic nematode Ascaris suum [12,17,22]. The high oxygen affinity also depends on other characteristics in these globin families [15,17,21,23], which may reflect the different adaptation of the hemeproteins to the organism’s function.
Table 1:
Rate constants for hemeproteins containing tyrosine (Tyr) at the active site.
The clam L. pectinata is an organism, located in the mangroves coastal zone of Puerto Rico, living in high hydrogen sulfide (H2S) concentration medium with a chemo-symbiotic relationship with bacteria[19]. The symbiosis requires a supply of sulfide and oxygen to support the clam’s nutritional needs. As a function of the mangrove age, sediments, and environment, the pH can be in the range from 2.87 to 6.40, with average bulk soil densities of 1.4 g/cm3 and an estimated H2S concentration ranging from 27 μg to 258 μg per gram of soil. Thus, it can be expected that L. pectinata’s habitat can oscillate from 0.79 mM to 7.4 mM in H2S concentrations [11,24,25]. The H2S concentration in the mangrove habitat of the bivalve is about 1mM [26], equivalent to a pH of 5. Considering the diprotic nature of hydrogen sulfide with a pKa of 7.02 [27], this bivalve lives in predominantly acidic conditions where the H2S dominates.
L. pectinata has three hemeproteins: Hemoglobin I (HbI), HbII, and HbIII [19,28]. Read in 1962, classified HbII, HbIII, and HbII-HbIII as oxygen reactive proteins. Later, Kraus and Wittenberg in 1990 described their O2 function due to the high kon and low koff compared to HbI. Hemoglobin I binds H2S with high affinity and transport it to the bacteria for nutritional sustainability [11,29]. The high H2S affinity is due to fast association and very slow dissociation rate constants controlled by glutamine and one of the phenylalanine residues (Phe) of the “phenylalanine cage” in its active site which provides access and stability to H2S [30]. Two key processes govern the transport of H2S in the clam: (i) the slow dissociation of H2S from the ferric adduct, and (ii) the heme iron reduction followed by H2S liberation [31]. These processes are dependent on H2S concentration and pH. Furthermore, mRNA expression for the three Hb species is also modulated by the amount of H2S to which the clam is exposed [32].
HbII and HbIII differ from HbI in that they associate, and one of the phenylalanine residues in the “phenylalanine cage” in HbI is substituted by tyrosine in HbII and HbIII [11]. It is well established that during the first purification step (Size Exclusion Chromatography), HbII and HbIII elute together [11,30]. The eluate molecular weight and electrophoresis techniques support that a significant component is the (HbII-HbIII)-O2 derivative. The HbII and HbIII protein interactions are so strong that Ion Exchange Chromatography is required to separate oxyHbII from oxyHbIII. This process requires a slow linear salt gradient to separate the associated subunits. Moreover, the (HbII-HbIII)-O2 species can be regenerated by merely mixing equimolar amounts of the isolated subunits. HbII has been extensively described in the homodimer form [14,33,34] and partially as a heterodimer with HbIII [35]. The HbIII homodimer has thus far proven recalcitrant to crystallographic analysis. Under these conditions, the results suggest that the HbII-HbIII heterodimer is an excellent candidate to be a functional oxygen reactive protein present in the gills, which can deliver dioxygen to the L. pectinata symbiont. The presence of tyrosine and glutamine in the heme moiety has been confirmed by X-ray crystallography [14] and absorption spectroscopy studies [33] for HbII. Both residues have a role in the stabilization of the bound O2. These proteins have smaller heme pockets compared to myoglobin, which also contributes to the high O2 affinity [14]. The rate of O2 dissociation for an equimolar mix of HbII and HbIII (the heterodimer) is an average of the oxyHbII and oxyHbIII values [11]. Since chemoautotrophy is very demanding of the oxygen process [36], higher oxygen dissociation rates are needed for the organism’s survival. Therefore, it is crucial to define important factors, like pH, which may help to unravel (HbII-HbIII)-O2 susceptibility, contributing to heme oxygen dissociation.
The (HbII-HbIII)-O2 complex is exposed to environmental conditions that require it to keep the O2 bound until it is necessary to aid the clam’s nutritional needs and survival. A change in pH directly affects the ionization state of amino acids, such as the tyrosine found in hemoglobin’s active site, which is typically protonated at pH < 10.8 [37]. Depending on the local surroundings in the active protein sites, the pKa of amino acids can shift and become functionally important [38–42]. An example of this is the pH-sensitive switch of the histidine residue in human hemoglobin [37,43], where lowering the pH aids in the release of the oxygen. Previous pH-dependent titration experiments on the oxy HbII homodimer showed that a decrease in pH from 7.5 to 5.5 is required to completely change the O2 species Soret band from 414 nm to the met-aquo HbII characteristic wavelength at 403 nm [14,33]. Kraus and Wittenberg determined the experimental values for the reduction potential (Em) of the HbIII homodimer as 165 mV, 117 mV, and 55mV at pH 5.6, 6.6 and 7.8, respectively [11]. For the HbII homodimer, measured values are 149 mV, 118 mV, and 85mV at pH 5.7, 6.7, and 7.4, respectively. The data indicates that increasing the pH leads to a reduced reduction potential. As mentioned before, the physical properties for an equimolar mixture of HbII and HbIII (the heterodimer) are likely to be an average of both homodimers. Consequently, it is suggested that the reduction potential for HbII-HbIII as a function of the pH can be near the average of each reporting point leading to 157 mV, 117.5 mV, and 70 mV at pH’s of 5.65, 6.65, and 7.60, respectively. These values indicate an inverse relationship between the pH and the reduction potential. From the extrapolation of the fitted curve, the Em value at pH 7 corresponds to 95 mV and 105 mV for HbIII and HbII, respectively. Under this scenario, the heterodimer would have a reduction potential of approximately 100 mV.
The results presented here indicate that the iron in (HbII-HbIII)-O2 complex partially oxidizes at the acidic condition and requires the presence of a potent oxidizing agent, like potassium ferricyanide (K3Fe(CN)6), to obtain 100% of met aquo (HbII-HbIII) derivative. This hemeprotein oxidation procedure is not associated with the factors that induce oxygen dissociation for the oxy (HbII-HbIII) species. Moreover, lowering only the pH prompt oxy(HbII-HbIII) conformational changes producing a small concentration of met-aquo (HbII-HbIII) complex. The process is accelerated by the presence of salts, as indicated by crystallization structures and UV-Vis data. The results suggest that acid pH generates conformational changes in the Tyr B10 and Gln E7 heme pocket, which would imply that pH is an essential factor regulating the (HbII-HbIII)-O2 active site hydrogen bonding network. Thus, it can be suggested that the hydrogen bonding alteration between the heme bound O2 and the Tyr and Gln groups can contribute to oxygen dissociation in the (HbII-HbIII)-O2 heterodimer of L. pectinata.
2. Materials and Methods
2.2. Protein purification and sample preparation
The L. pectinata was collected in the mangrove swamps of Cabo Rojo, Puerto Rico. The HbII–HbIII complex was isolated from the ctenidia tissue as described previously by Kraus & Wittenberg [11] with the incorporation of an FPLC system [30]. After isolation and purification, the HbII-HbIII species was determined to be (HbII-HbIII)-O2 as verified by UV-Vis spectroscopy, and the characteristic Soret (414 nm) and Q bands (540 nm/576 nm). The concentration of the complex was determined using mean extinction coefficients reported in the literature for HbII (ɛmM = 129) and HbIII (ɛmM = 144) [11] using a SHIMADZU UV- 2600 spectrophotometer.
2.3. Potassium ferricyanide Titrations and Optical absorption spectroscopy
The (HbII-HbIII)-O2 titrations with K3Fe(CN)6 were recorded using a SHIMADZU UV-2600 spectrophotometer with pH and spectral ranges from 4 to 9, and from 350 to 750 nm, respectively. The (HbII-HbIII)-O2 protein sample (20μM) was prepared in PBS (50mM K2HPO4/50mM KH2PO4) with a pH range of 4 to 9. Each sample was then titrated with a 40mM K3Fe(CN)6 solution by adding aliquots of 0.2μL to the samples until no notable change was observed. To study the effect of the crystallization conditions on (HbII-HbIII)-O2 in solution (5.0 M Sodium formate buffered to pH with sodium acetate for pH 4–7 and Tris-HCl for pH 8–9), an aliquot of (HbII-HbIII)-O2 was transferred to a 2.0 M solution of sodium formate at pH 4.6 and monitored through time with a SHIMADZU UV-2600 spectrophotometer. This solution is 60% less concentrated than the crystallization condition to avoid sample precipitation.
2.4. Crystallization Conditions
The (HbII-HbIII)-O2 hemeproteins were crystallized, as reported [35]. In brief, the protein samples were concentrated in distilled water to a final concentration of 30 mg/mL. The crystals were grown employing a two-step capillary counter-diffusion technique [44] at various pH values. A sodium formate pH screen (Triana®) was then used to produce crystals at pH 7, 5, and 4. Crystals showed rounded rather than faceted morphologies, as previously observed [35]. Crystals were kept in the capillary and cryoprotected by equilibrating the capillary with a cryosolution containing 15% glycerol, followed by flash-cooling in liquid N2.
2.5. Crystal structures and refinement
X-ray diffraction data were acquired at the Stanford Synchrotron Radiation Light source (SSRL) beamlines BL12–2 and BL9–2 from crystals grown at pH 4, 5, and 7. The images were processed and scaled using the HKL2000 suite [45]. All crystals belonged to the space group P42212 with unit cell dimensions similar to those reported for HbII and the HbII–HbIII complex (a = b = 74.21 Å and c = 151.32 Å) [14,35,46]. The structures of the (HbII-HbIII)-O2 crystals were solved by molecular replacement methods using the (HbII-HbIII)-O2 model (PDB id: 37PT) [35] with slight modifications. Missing amino acids in the structure were built back into the model based on the HbIII amino acid sequence [47,48]. Refinement of all structures was carried out with REFMAC5 and PHENIX and further improved by incorporation of TLS parameters [49,50]. The models were manually built with COOT [51].
3. Results and Discussion
3.1. Titrations and Optical absorption spectroscopy
The (HbII-HbIII)-O2 complex has a very low koff attributed to the hydrogen bonding network between the nearby tyrosine, glutamine, and the molecular O2 bound to the heme group. However, the factor that contributes to cleave this hydrogen bond network, allowing the heme (FeII) to release the O2 towards the clam’s gill bacteria symbiont, remains unknown. Figure 1 shows the changes in the absorption spectra of (HbII-HbIII)-O2 as a function of pH, without the presence of any oxidant agent (i.e., K3Fe(CN)6). Figure 1a and Figure 1b inserts show an expansion of the Soret (400–430 nm) and Q (580 – 600 nm) regions, respectively. Absorption bands at 412 nm, 540 nm, and 576 nm represent the oxy complex, while the 403 nm, 503 nm, and 633 nm transitions are associated with the met-aquo heme derivative [11,29]. At pH 4, there are transitions at 409 nm, 503 nm, and 633 nm, but the oxy-hemoglobin heterodimer dominates the spectrum as indicated by the 540 nm and 576 nm intensities. Increasing pH to 6 leads to the (HbII-HbIII)-O2 complex with electronic bands at 412 nm, 540 nm, and 576 nm. Upon further increase to pH 7, there is a redshift in the Soret towards 414 nm, a small decrease in the intensity of the characteristic (HbII-HbIII)-O2 Q bands, and a low-intensity transition appears at 605 nm. This new transition in the Q region is maintained up to pH 9. The presence of the 605 nm band was previously attributed to the tyrosine (B10) residue coordinated to the heme (FeIII) moiety [33]. Overall, the data indicate that lowering the pH produces a decrease in the absorption intensity of the oxyheme derivatives suggesting a small population of the met-aquo HbII-HbIII species. However, the Q (540 nm and 576 nm) transitions indicate that the (HbII-HbIII)-O2 derivative dominates.
Figure 1:
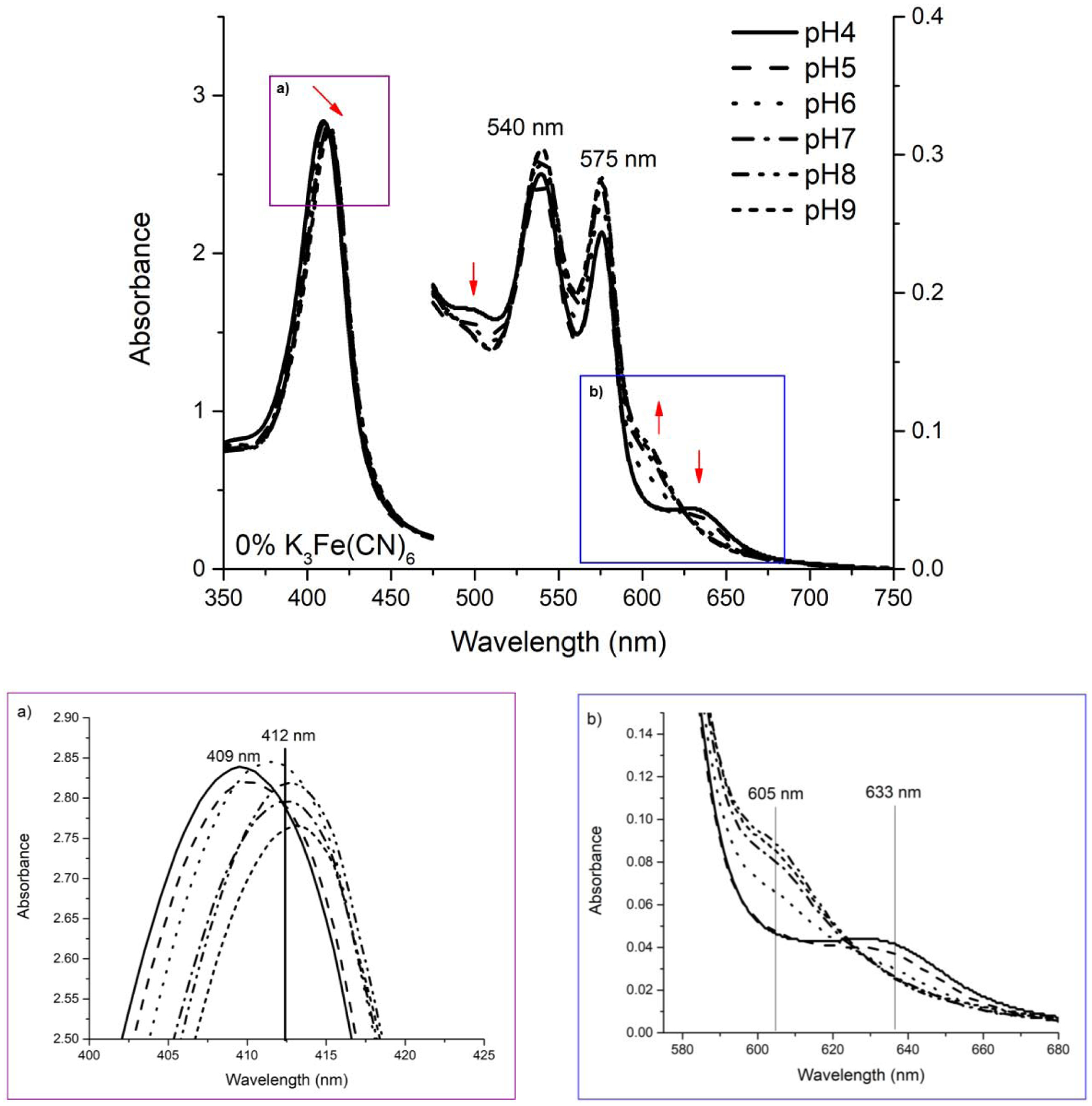
Visible region spectra of the (HbI-HbIII)-O2 complex when transferred to acid and alkaline PBS. Inserts a and b are present to facilitate the changes visualization. Arrows show band shifts going from acid to alkaline environment. The oxy Q bands shift, from pH 4 to around pH 5, forming two new bands at 503 and 633 nm, while the Soret band shifts to 409 nm, towards the 403 nm met-aquo band. At near neutral pH, the oxy complex bands at 540 and 576 nm dominate the Q region and the Soret band at 412 nm. At alkaline pH, the characteristic band of Fe-Tyr appears at ~605 nm.
The data presented in Figure 2a demonstrates changes in the absorption spectra of (HbII-HbIII)-O2 as a function of pH in the presence of 120% K3Fe(CN)6 excess. Once ferrous (HbII-HbIII)-O2 at acidic pH is titrated with the oxidizing agent (K3Fe(CN)6), the ferric aquo (HbII-HbIII) dominates the spectra. At pH 4, the classical absorption spectrum of the met-aquo species (403, 503, and 633 nm) is present, and the Q bands corresponding to (HbII-HbIII)-O2 are absent. Therefore, acidic pH, the presence of a potent oxidizing agent, facilitates the complete oxidation of (HbII-HbIII)-O2. However, from pH 6 to 9, the Q bands associated with the oxy derivative are present along with the 605 nm electronic transition, as previously mentioned, attributed to the interaction between the heme(FeIII) and TyrB10. Nevertheless, at an alkaline limit of pH 7.5, the transitions at 541 and 576 nm are attributed to the hydroxide derivate [29, 33]. These results agree with the observed behavior for the oxyHbII homodimer, where adding an excess of potassium ferricyanide at a pH 5.5 promotes the iron oxidation to the ferric species [14]. Moreover, Figure 2b summarizes the behavior of the 633 nm transition as a function of the pH and the K3Fe(CN)6 concentration. The results show that the band intensity is a minimum in the absence of K3Fe(CN)6, and it increases as the concentration of the oxidizing agent increases, reaching a maximum at pH 4. Clearly, the behavior of the met-aquo-heme 633 nm transition shows a direct relationship to the percentage of K3Fe(CN)6 in solution and an inverse relationship with pH. This hemeprotein oxidation procedure is not associated with oxygen dissociation from the oxy (HbII-HbIII) species.
Figure 2:
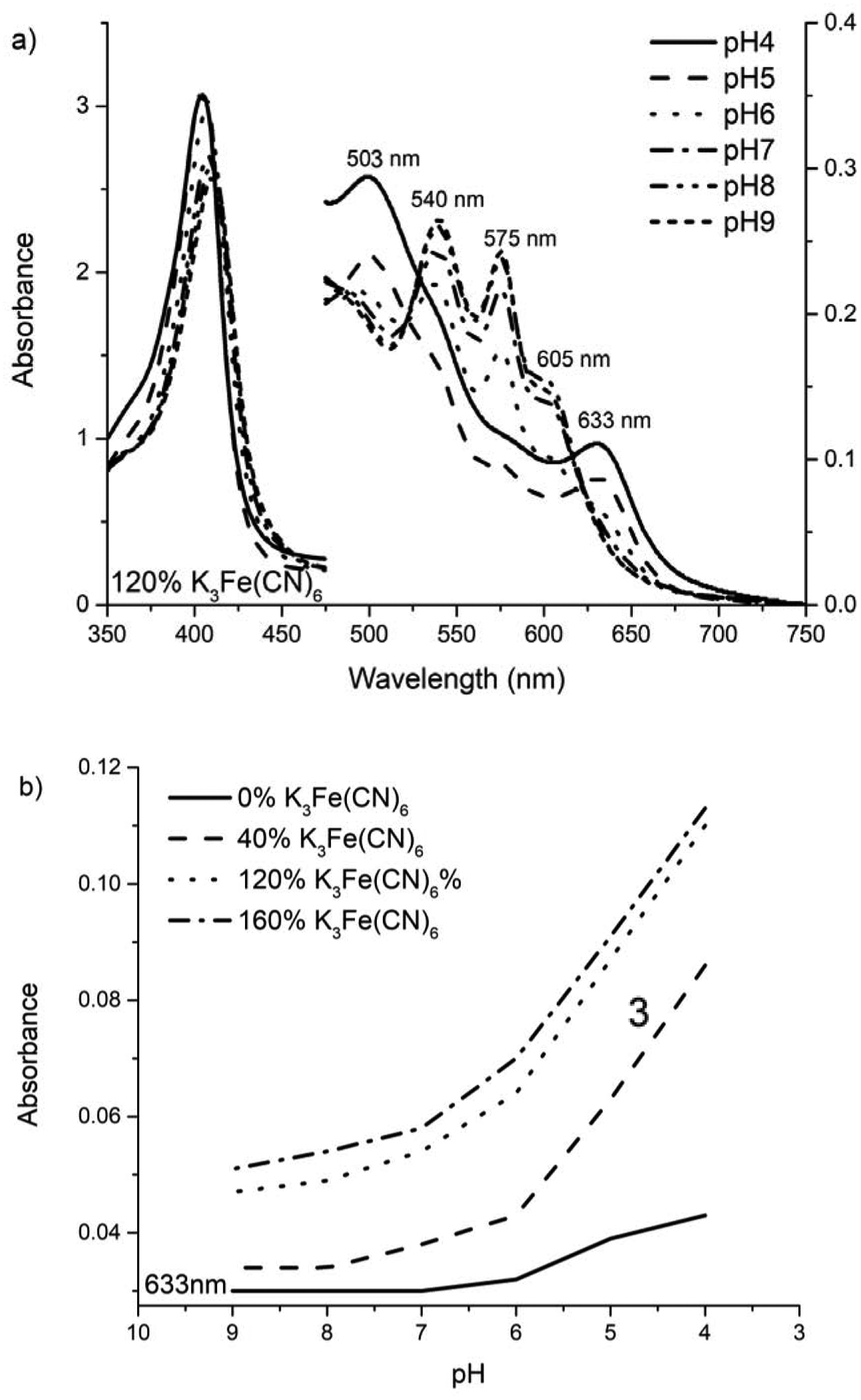
Visible region spectra of the (HbII-HbIII)-O2 complex when transferred to acid/alkaline PBS and titrated with an oxidant agent. (a) Visible region spectrum: At near pH 5, the oxy complex bands shift to the 503 and 633 nm band, and the Soret band shifts to 403 nm, and (b) Ferric characteristic Q band at 633 nm behavior as a function of [K3Fe(CN)6].
3.2. X-ray Structural Analysis of (HbII-HbIII)-O2 species
(HbII-HbIII)-O2 systems were crystallized using the Triana® sodium formate pH variant counter-diffusion kit. Crystals grew to final size in 1–3 days in a spectrum of red shades ranging from brownish (pH 4) to dark red (pH 7). Crystal structures for crystals grown in pH 4, 5, and 7 were solved and deposited in the PDB with PDB IDs 6OTY, 6OTW, and 6OTX, respectively. The experimental set up was done to minimize the radiation damage during data collection. The working resolution during refinement was 2.48 Å, 2.45 Å, and 2.54 Å for the structures at pH 4, 5, and 7, respectively. Table 2 presents the summarized crystallographic parameters and analyses. All data were integrated into the space group P42212, as previously determined for the HbII-HbIII heterodimer [35]. Hemoglobin components in the structure consist of HbII in chain A and HbIII in chain B.
Table 2:
Data Collection and Refinement Statistics
| Collection | pH 4 | pH 5 | pH 7 |
|---|---|---|---|
| Space group | P42212 | P42212 | P42212 |
| Unit cell dimensions (Å) | a = b = 74.43 c= 152.10 | a = b = 74.30 c= 152.30 | a = b = 74.74 c= 152.57 |
| Resolution range (Å) | 50.00 – 2.60 (2.64 – 2.60)a | 50.00 – 2.45 (2.49 – 2.45)a | 50.00 – 2.54 (2.58 – 2.54)a |
| Total reflections (observed / unique) | 784334 / 13907 | 262087 / 16508 | 592952 / 15012 |
| Rmeasb (%) | 8.6 (90.1) | 9.2 (141.2) | 6.4 (72.9) |
| Rpimc (%) | 3.0 (39.4) | 3.5 (54.1) | 1.9 (22.3) |
| I/σI | 22.8 (1.4) | 26.7 (1.2) | 36.6 (2.0) |
| Completeness (%) | 99.5 (95.1) | 97.5 (96.6) | 99.6 (95.8) |
| CC½d | 96.2 (79.5) | 93.2 (69.1) | 98.4 (93.9) |
| Multiplicity | 7.7 (4.4) | 6.9 (6.6) | 10.0 (8.7) |
| Wilson B (Å2) | 58.0 | 58.6 | 58.7 |
| Refinement | |||
| R-factor / Rfree (%)e | 18.07/23.49(H2O) | 18.99/24.81(H2O) | 19.22/24.88 |
| 20.49/27.08(O2) | 18.88/24.04(O2) | ||
| Total atoms - protein / solvent | 2520 | 2537 | 2548 |
| Atomic displacement parameters (Å2) | |||
| - protein | 78.0 | 76.8 | 83.1 |
| - solvent | 75.0 | 67.7 | 79.6 |
| - hemef | 76.1 / 69.3 | 66.8 / 67.6 | 71.2 / 73.5 |
| - ligandf | 75.2 / 46.5 | 52.5 / 64.4 | 106.3 / 81.6 |
| rms deviation from ideality | |||
| - bond distances (Å) | 0.008(H2O) | 0.009(H2O) | 0.008 |
| 0.009(O2) | 0.008(O2) | ||
| - bond angles (°) | 0.995(H2O) | 1.080(H2O) | 0.971 |
| 1.012(O2) | 0.919(O2) | ||
| Ramachandran plotg | |||
| - residues in favored regions (%) | 95.6(H2O) | 95.0(H2O) | 96.0 |
| 94.3(O2) | 94.6(O2) | ||
| - outliers | 0.0(H2O) | 0.67(H2O) | 1.00 |
| 0.34(O2) | 1.34(O2) | ||
| Molprobity score | 1.92(H2O) | 1.93(H2O) | 2.03 |
| 1.96(O2) | 1.90(O2) | ||
| Fe-O distance | 2.85/2.59(H2O) | 2.34/2.49(H2O) | 1.86/1.79(O2) |
| 1.84/1.78(O2) | 1.89/1.82(O2) | ||
Numbers in parentheses relate to the highest resolution shell.
Rmeas is the redundancy-independent merging R factor [61].
Rpim is the precision-indicating merging R factor [62].
Percentage of correlation between intensities from random half-sets of data [63].
Rfree was calculated with 5% of the reflections, chosen randomly.
Atomic displacement parameters for Chain A/Chain B for HbII-HbIII with H2O as a ligand in the protein at pH 4 and pH 5.
Calculated with the program MOLPROBITY [64].
Figure 3 shows the superposition of the HbII-HbIII tertiary structure heterodimer at pH 4 (blue), pH 5 (green), and pH 7 (pink). The visual data shows that there is no significant displacement in the overall position of the main chain of the helices for chain A (HbII, left) and chain B (HbIII, right). Nevertheless, there are some small geometrical changes in the connecting loops. These relatively small changes are minimal overall as a function of pH; however, under these perturbation environments, the heme moiety responds unequally in the two monomers. Figure 4 shows the electron density map for the heme pocket at pH 4, pH 5, and pH 7, respectively. In the pH 7 structure, continuous electron density is observed for the Fe-ligand-tyrosine-glutamine hydrogen bond network, as suggested in the literature [14]. The electron density around the Tyr and Gln oxygen heme system is slightly missing at the HbII subunit relative to the HbIII analog. In the HbIII monomer, O2 is anchored to the heme by the network of the tyrosine and glutamine residues, as was previously reported for the oxy HbII homodimer [46]. In general, the HbII-HbIII species remains oxygenated as expected from the solution experiment described above, and as previously reported [11,19]. The partial density loss at pH 7 can be correlated with the experimental set up accounting for radiation damage, because there is a displacement of the hydrogen bonding network between the heme pocket amino acid, Gln and Tyr, and the oxygen ligand.
Figure 3:
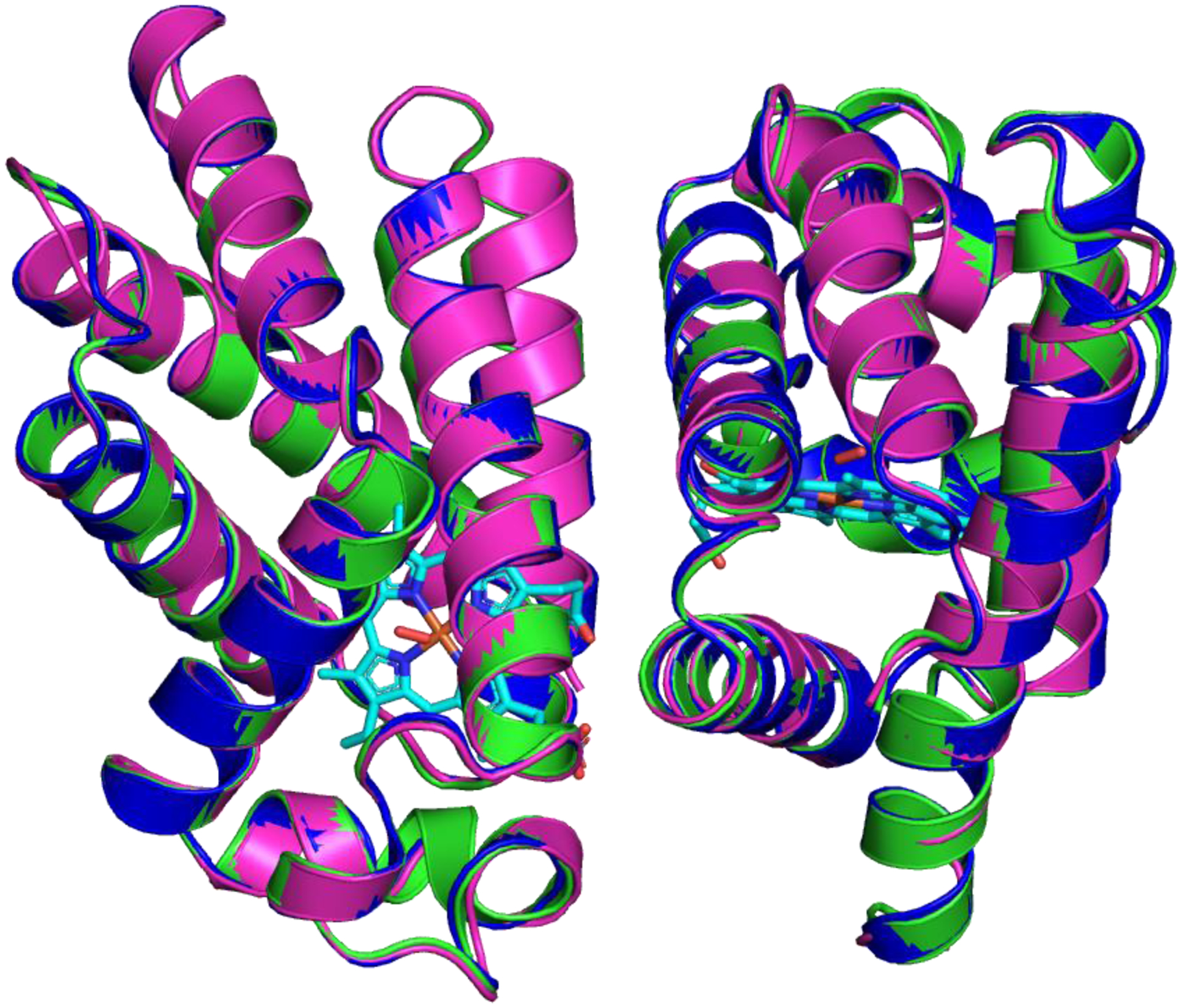
Global tertiary structure of L. pectinata (HbII-HbIII)-O2 at pH 4 (blue), PDB: 6OTY, pH 5 (green), PDB: 6OTW, and pH 7 (pink), PDB: 6OTX.
Figure 4:
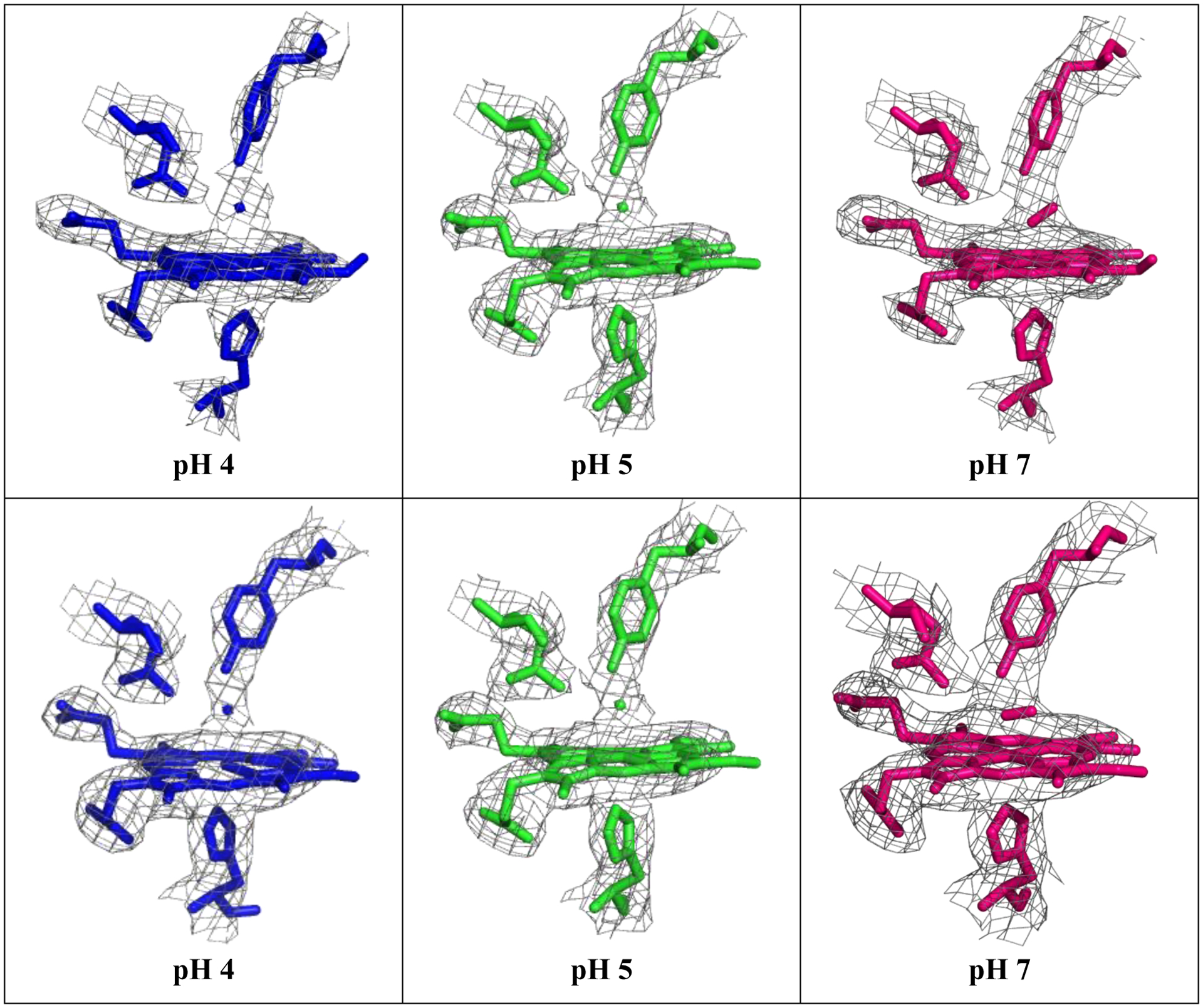
Electron density map of the heme pocket of L. pectinata (HbII-HbIII)-O2 at pH 4(blue), pH 5 (green), and pH 7(pink), chain A (top) and chain B(bottom). The 2Fo-Fc contours for the images presented here are 1σ within 2 Å of the selected atoms.
The hydrogen bonding networking in the crystal structures obtained at pH 4 and pH 5 is the most susceptible to conformational changes. They were refined in tandem with O2 and H2O near the porphyrin iron. The electron density for the heme ligand could be refined either as H2O (full occupancy of 1.00) or as O2 (partial occupancy of 0.58). However, as shown in Table 2, the overall statistics for refinement for HbII-HbIII at pH 4 with H2O as the ligand are slightly better than for O2, while for the structure at pH 5, statistics are almost the same. For both structures, the electron density in the Gln and Tyr hydrogen bond network with the heme group is not continuous, suggesting a broken hydrogen network between the heme and its ligand. Therefore, the structure in Figure 4 correlates with the solution experiments and allows the visualization of the (HbII-HbIII)-O2 structures as a function of pH, the heme moiety, and the hydrogen bond networking. It is important to note that there is a known structure of the (HbII-HbIII)-O2 heterodimer determined at pH 5 (PDB id 3PT7) [35] which is very similar to our pH 5 structure, with the exception that based on our solution experiments, we fit our X-ray data with H2O as the ligand. Nevertheless, the previously known structure can be directly compared with O2 as ligand, as presented in Figure 5, and the structure reported here with H2O as the ligand.
Figure 5:
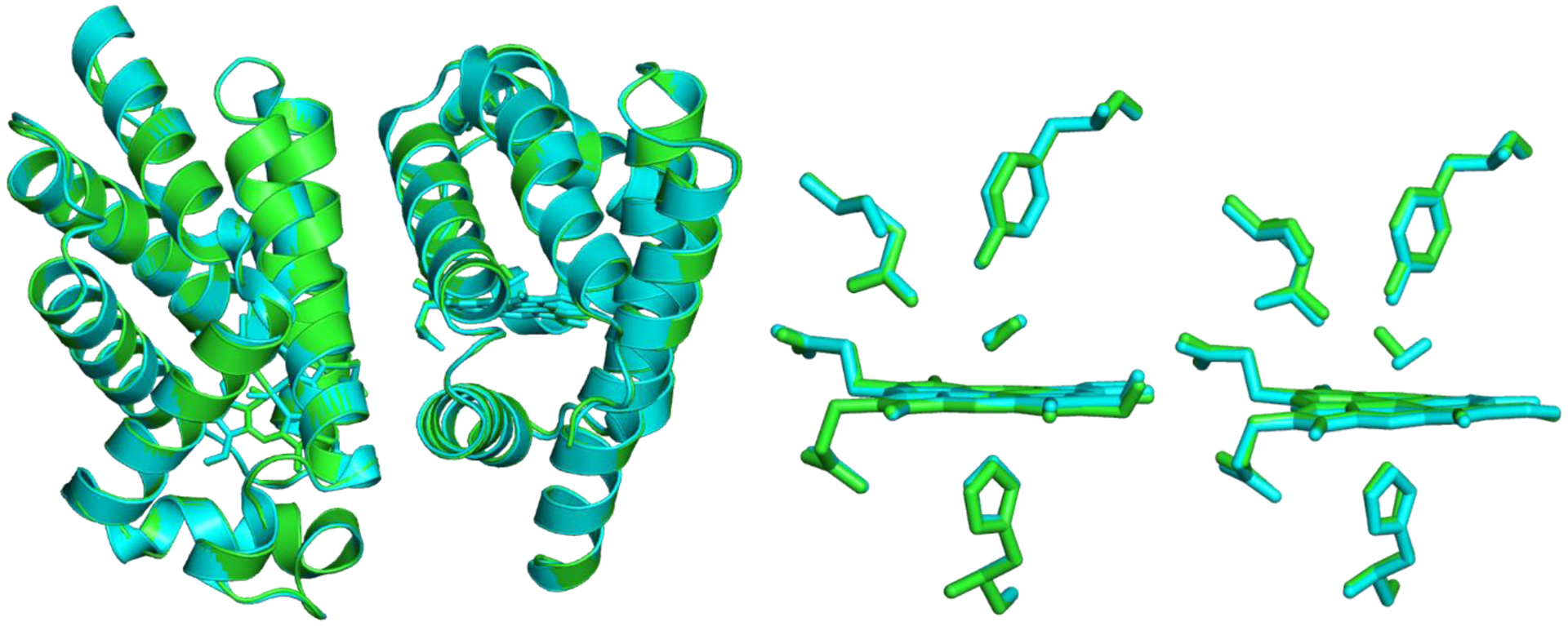
Superposition of 3PT7(cyan) onto the pH 5 heterodimer structure(green) reported here. There has been no relative movement of the HbII and HbIII chains in the current structure compared to the published structure.
Figure 5 is the superposition of 3PT7 onto the pH 5 heterodimer structure reported here. This superposition gives a rmsd of 0.24 Å for all 303 matching Cα atoms in both chains. When the monomers are superimposed individually, the rmsd values are similar (0.25 Å for HbII and 0.22 Å for HbIII), which indicates that there has been no relative movement of the HbII and HbIII chains in the current structure compared to the published structure. Moreover, an inspection of the superimposed structures shows that they are virtually indistinguishable. The Fe-heme moieties in the two structures overlay almost precisely, with only minor structural deviations in the propionyl side chains. In the HbIII molecule (chain B) modeled with O2, the oxygen species has rotated approximately 180° from the orientation observed in 3PT7. In the current pH 5 structure, the O2 points towards the side chain of Gln66, and one of the oxygen atoms is within hydrogen-bonding distance of the Nε2 atom. In this orientation, there is a close contact (2.0 Å) to the Oη atom of Tyr31. In the HbII molecule (chain A), the O2 species is in almost the same orientation as in 3PT7, pointing away from the Gln66 side chain and interacting with the Oη atom of Tyr31. These results do not contradict the reported structure nor invalidate the structure presented here with H2O as a ligand.
Nieves et al. [46] reported the crystal structures for the HbII-O2 homodimer as a function of pH (PDB IDs 3PI1, 3PI2, 3PI3, and 3PI4 for pH 9, 8, 5 and 4, respectively). They proposed that only HbII-O2 at pH 4 undergoes oxidation, refining the structure with water as the ligand suggesting the metaquo complex formation. Nevertheless, the structures at pH 4 and pH 5 can both be refined with water as the ligand. The observed partial oxidation in the solution experiments without an oxidizing agent suggests that the crystals either underwent autoxidation, or that the crystallization condition accelerates the process. The crystallization condition used to grow the crystals contains sodium formate as the primary component, with sodium acetate for pH 4 and 5, and Tris-HCl for pH 6, 7, 8, and 9. The pKa for each is 3.74, 4.74, and 8.1, respectively. At low pH, this means that sodium formate tends to predominate in the protonated state as formic acid and has an oxidizing capability. Figure 6 shows the oxidation of the (HbII-HbIII)-O2 dimer when an aliquot was transferred to a 2.0 M solution of sodium formate at pH 4.6. The metaquo transitions for HbII-HbIII at 405 nm, 503 nm, and 633 nm are evident after 10 minutes. This demonstrates that the precipitant agent in the crystallization conditions speeds up autoxidation of the (HbII-HbIII)-O2 complex. To avoid the precipitation of the sample, a 60% less concentrated solution of the crystallization conditions was used. Under these conditions, the sample transformation at a particular pH explains the observed behavior where the percentage of the oxy and met-aquo heme complex in the solution experiments tend to be higher than the analogous derivatives of the crystallographic structure.
Figure 6:
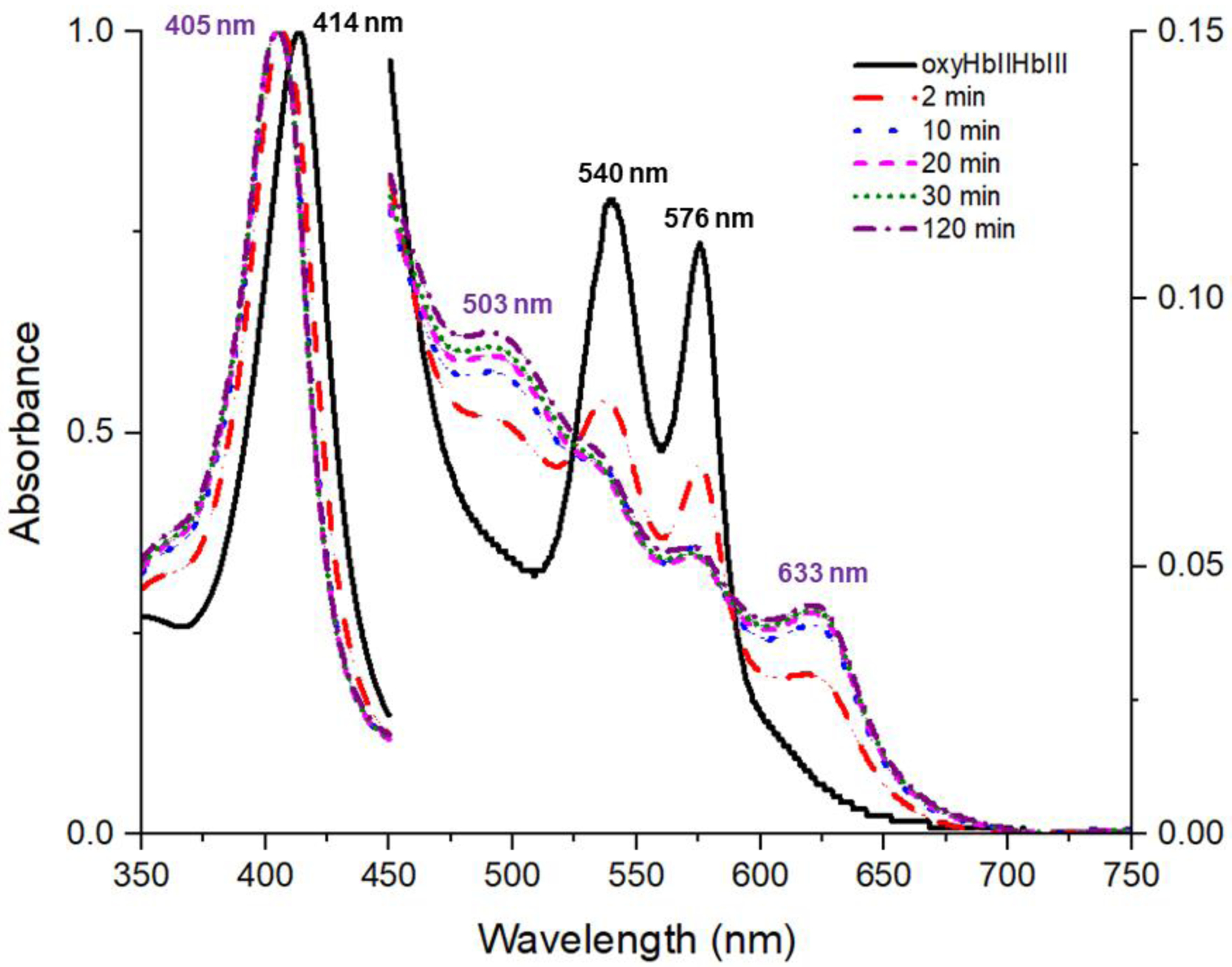
UV-vis spectra of an aliquot of (HbII-HbIII)-O2 transferred into a 2.0 M solution of sodium formate at pH 4.6. Spectra were normalized. This solution is 60% less concentrated than the crystallization condition to avoid precipitation of the sample and observe the effect of the crystallization solution on the oxidation state of L. pectinata (HbII-HbIII)-O2 at acidic pH.
3.3. What does this translate to L. pectinata?
The solution experiment results show two scenarios that are relevant to (HbII-HbIII)-O2. Scenario 1 presents that pH, in combination with ferricyanide, is necessary to obtain 100% of the met-aquo (HbII-HbIII) derivative from (HbII-HbIII)-O2 state (Figure 2). Similar to myoglobin [52–55], the process only is an experimental procedure to obtain the met-aquo (HbII-HbIII) derivative. The conditions have not been reported previously for (HbII-HbIII)-O2 or analogous systems. Therefore, this hemeprotein oxidation process is not associated with the factors that induce oxygen dissociation for the (HbII-HbIII)-O2 species. It is important to note that for further studies of the met-aquo species, the excess of K3Fe(CN)6 must be removed to avoid secondary heme protein reactions.
Scenario 2 presents (Figure 1) that pH changes prompt (HbII-HbIII)-O2 conformational changes, which lead to the presence of a small concentration of the met-aquo (HbII-HbIII) complex. In agreement with the autoxidation process of sperm whale oxy-myoglobin [52–55], the induced oxidation of the oxy (HbII-HbIII) complex is extremely slow at neutral pH values at room temperature. The data show that the process is accelerated by lowering pH (Figure 1) as well as by the presence of the ions as indicated by the crystallization structures and the UV-Vis data (Figures 5 and 6). The results indicate that acid pH generates conformational changes in the heme pocket amino acids (Gln E7 and Tyr B10) that affects the hydrogen bond network of the (HbII-HbIII)-O2 complex. The observation is supported by the appearance of the 605 nm transition associated with the Tyr interaction with the iron in the heme (Figure 1) and associated with charge transfer transition [33]. The reported presence of two bands at 1,924 cm−1 and 1,964 cm−1 in the (HbII-HbII)-CO confirms the existence of conformational changes due to the orientation of the TyrB10 concerning the active heme center.[33,56,57]. Consequently, the (HbII-HbIII)-O2 species hydrogen bond network created by Gln and Tyr amino acids stabilizes the heme oxygen complex, and it minimizes dissociation of the bound O2 as well as the heme complex auto-oxidation. Although heme autooxidation and oxygen dissociation are not the same phenomena but have the same early hydrogen bond network structures, it can be suggested that the weakening of the hydrogen bonding network in the distal pocket is common in both processes. Therefore, analogous to oxy-myoglobin [52–55], it can be suggested that the opening of the (HbII-HbIII)-O2 hydrogen bonding network by a pH decrease may play a role in regulating the heme pocket conformations changes as well as oxygen dissociation.
A pH 7 solution contains a hydrogen sulfide equilibrium of 18–28% as H2S, and 72–85% in the deprotonated form [27,58]. The balance moves toward the H2S by lowering the pH at pH 6.5 or lower. Hence, having an acidic physiological microenvironment promotes a higher H2S concentration available for the clam symbiosis. Literature reports establish that for other invertebrates living in H2S environments can lower internal pH under particular conditions [6,59,60], as well as other cells such as lysosomes [61], that could have lower intracellular pH [62]. In the natural habitat of L. pectinata, a pH decrease could be attributed to the H2S metabolism in the ctenidia tissue, specifically in the bacteriocytes [63], which tends to increase its H+ concentration. The bacteriocytes harbor the HbI, HbII, and HbIII proteins in addition to the symbionts that use the H2S and O2 for CO2 fixation to generate sugars. It is still not clear how these proteins engage in the bacteria since the specific symbiont has not been isolated from the clam [64]. The oxyheme proteins from Lucina pectinata are known to withstand low pH without denaturing [11] as also shown in the work presented here. The behavior is supported by the information indicating that increasing the pH leads to a reduced reduction potential [11]. Besides, Figure S1 (Supplemental data) shows that the dioxygen release from the (HbII-HbIII)-O2 species increases by 1.6-fold from pH 7 to pH 5. Therefore, it is suggested that under these environments, the oxygen dissociation constant from the (HbII-HbIII)-O2 derivative can be influenced by the pH as well as by the hydrogen sulfide concentration. Future work is going in this direction.
4. Conclusion
The solution experiment results show that pH, in combination with ferricyanide, is necessary to obtain 100% of the met-aquo (HbII-HbIII) derivative form (HbII-HbIII)-O2 state, but the process is not associated with the factors that induce oxygen dissociation for the (HbII-HbIII)-O2 species. Titration and X-ray structural data for (HbII-HbIII)-O2 species as a function of the pH demonstrates its susceptibility to conformational changes influencing hydrogen bonding networking between the heme oxygen complex and the pocket Tyr and Gln amino acids. The (HbII-HbIII)-O2 derivative dominates at pH 6 to 7 but forms small amounts of the oxidized met-aquo derivative at lower pH. The presence of glutamine and tyrosine in these hemoglobins is crucial for their functionality, and the structures presented show that their hydrogen-bonding interactions with the ligand are debilitated by lowering the pH. Thus, it can be suggested that H2S, in the natural environment of L. pectinata, could provide acidic conditions that contribute weakening the oxy (HbII-HbIII)-O2 hydrogen bonding network that could contribute to dioxygen deliver to the clam symbiont.
Supplementary Material
Highlights:
Lucina pectinata possess an oxygen reactive hemoglobin (Hb) heterodimer, HbII-HbIII.
In vitro, the hydrogen bond network in (HbII-HbIII)-O2 is susceptible to changes in pH.
The crystal structures at pH 4, 5, and 7 behave the same as the complex in solution.
Acidic pH and an oxidizing agent control the oxidation process of (HbII-HbIII)-O2.
In vivo, pH could be the driving force for the cleavage of the heme Fe-O2 bond.
Acknowledgments
This work was supported by the National Science Foundation (NSF)-Science and Technology Center (STC) “BioXFEL” through award STC-1231306 and the SLOAN Minority Ph. D. Program. Part of this work was completed at the Stanford Synchrotron Radiation Lightsource. Use of SSRL, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). National Institute of Health-INBRE PR (P20GM103475 to J.L.G.), The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. and the Alfred P. Sloan (NACME Grant 2010-3-02).
Abbreviation
- Hb
Hemoglobin
- HbI
Hemoglobin I
- HbII
Hemoglobin II
- HbIII
Hemoglobin III
- H2S
Hydrogen sulfide
- Tyr
Tyrosine
- Gln
Glutamine
- Phe
phenylalanine
- AscHb
Ascaris hemoglobin
- TrHb
truncated hemoglobins
- koff
Dissociation Constant
- (K3Fe(CN)6)
Potassium ferricyanide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none.
References
- [1].Powell MA, Somero GN, Biol. Bull 171 (1986) 274–290. [Google Scholar]
- [2].Li T, Bonkovsky HL, Guo J, BMC Struct. Biol 11 (2011) 13 10.1186/1472-6807-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kraus DW, Integr. Comp. Biol 35 (1995) 112–120. 10.1093/icb/35.2.112. [DOI] [Google Scholar]
- [4].Bonaventura J, Lance VP, Am. Zool 41 (2001) 346–359. 10.1668/0003-1569(2001)041. [DOI] [Google Scholar]
- [5].Bonaventura C, Henkens R, Alayash AI, Banerjee S, Crumbliss AL, Antioxid. Redox Signal 18 (2013) 2298–2313. 10.1089/ars.2012.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hourdez S, Weber RE, J. Inorg. Biochem 99 (2005) 130–141. 10.1016/j.jinorgbio.2004.09.017. [DOI] [PubMed] [Google Scholar]
- [7].Feis A, Howes BD, Milazzo L, Coppola D, Smulevich G, Biopolymers. (2018) 1–11. 10.1002/bip.23114. [DOI] [PubMed]
- [8].Kundu S, Blouin GC, Premer SA, Sarath G, Olson JS, Hargrove MS, Biochemistry. 43 (2004) 6241–6252. 10.1021/bi049848g. [DOI] [PubMed] [Google Scholar]
- [9].Smagghe BJ, Hoy JA, Percifield R, Kundu S, Hargrove MS, Sarath G, Hilbert JL, Watts RA, Dennis ES, Peacock WJ, Dewilde S, Moens L, Blouin GC, Olson JS, Appleby CA, Biopolym. - Pept. Sci. Sect 91 (2009) 1083–1096. 10.1002/bip.21256. [DOI] [PubMed] [Google Scholar]
- [10].Terwilliger NB, J. Exp. Biol 201 (1998) 1085–1098. [DOI] [PubMed] [Google Scholar]
- [11].Kraus D, Wittenberg JB, J. Biol. Chem 265 (1990) 16043–16053. [PubMed] [Google Scholar]
- [12].Das TK, Friedman JM, Kloek AP, Goldberg DE, Rousseau DL, Biochemistry. 39 (2000) 837–842. 10.1021/bi9922087. [DOI] [PubMed] [Google Scholar]
- [13].Couture M, Yeh SR, a Wittenberg B, Wittenberg JB, Ouellet Y, Rousseau DL, Guertin M, Proc. Natl. Acad. Sci. U. S. A 96 (1999) 11223–8. 10.1073/pnas.96.20.11223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gavira JA, Camara-Artigas A, De Jesús-Bonilla W, López-Garriga J, Lewis A, Pietri R, Yeh SR, Cadilla CL, García-Ruiz JM, J. Biol. Chem 283 (2008) 9414–9423. 10.1074/jbc.M705026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Igarashi J, Kobayashi K, Matsuoka A, J. Biol. Inorg. Chem 16 (2011) 599–609. 10.1007/s00775-011-0761-3. [DOI] [PubMed] [Google Scholar]
- [16].Ouellet Y, Milani M, Couture M, Bolognesi M, Guertin M, Biochemistry. 45 (2006) 8770–8781. 10.1021/bi060112o. [DOI] [PubMed] [Google Scholar]
- [17].Peterson ES, Huang S, Wang J, Miller LM, Vidugiris G, Kloek AP, Goldberg DE, Chance MR, Wittenberg JB, Friedman JM, Biochemistry. 36 (1997) 13110–13121. 10.1021/bi971156n. [DOI] [PubMed] [Google Scholar]
- [18].Huang S, Huang J, Kloek AP, Coldberg DE, Friedman JM, J. Biol. Chem 271 (1996) 958–962. [DOI] [PubMed] [Google Scholar]
- [19].Read KRH, Comp. Biochem. Physiol 15 (1965) 137–158. [DOI] [PubMed] [Google Scholar]
- [20].Pesce A, Couture M, Dewilde S, Guertin M, Yamauchi K, Ascenzi P, Moens L, Bolognesi M, EMBO J. 19 (2000) 2424–34. 10.1093/emboj/19.11.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bustamante JP, Radusky L, Boechi L, Estrin DA, ten Have A, Martí MA, PLoS Comput. Biol 12 (2016) 1–26. 10.1371/journal.pcbi.1004701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dasgupta J, Sen U, Choudhury D, Datta P, Chakrabarti A, Chakrabarty SB, Chakrabarty A, Dattagupta JK, Biochem. Biophys. Res. Commun 303 (2003) 619–623. 10.1016/S0006-291X(03)00379-6. [DOI] [PubMed] [Google Scholar]
- [23].Ascenzi P, Masi A, Tundo GR, Pesce A, Visca P, Coletta M, PLoS One. 9 (2014) 1–15. 10.1371/journal.pone.0102811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kryger L, Lee SK, Biogeochemistry. 35 (1996) 367–375. 10.1007/BF02179960. [DOI] [Google Scholar]
- [25].Hossain MD, Nuruddin AA, J. Environ. Sci. Technol 9 (2016) 198–207. 10.3923/jest.2016.198.207. [DOI] [Google Scholar]
- [26].Laurent MCZ, Gros O, Brulport JP, Gaill F, Le Bris N, Mar. Environ. Res 67 (2009) 83–88. 10.1016/j.marenvres.2008.11.006. [DOI] [PubMed] [Google Scholar]
- [27].Olson KR, Biochim. Biophys. Acta 1787 (2009) 856–863. 10.1016/j.bbabio.2009.03.019. [DOI] [PubMed] [Google Scholar]
- [28].Read KRH, Biol. Bull 123 (1962) 605–617. [Google Scholar]
- [29].Kraus D, Wittenberg JB, Jing-Fen L, Peisach J, J. Biol. Chem 265 (1990) 16043–16059. [PubMed] [Google Scholar]
- [30].Pietri R, León RG, Kiger L, Marden MC, Granell LB, Cadilla CL, López-Garriga J, Biochim. Biophys. Acta - Proteins Proteomics 1764 (2006) 758–765. 10.1016/j.bbapap.2005.11.006. [DOI] [PubMed] [Google Scholar]
- [31].Pietri R, Lewis A, León RG, Casabona G, Kiger L, Yeh SR, Fernandez-Alberti S, Marden MC, Cadilla CL, López-Garriga J, Biochemistry. 48 (2009) 4881–4894. 10.1021/bi801738j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Montes-Rodríguez IM, Rivera LE, López-Garriga J, Cadilla CL, PLoS One. 11 (2016) 1–31. 10.1371/journal.pone.0147977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pietri R, Granell L, Cruz A, De Jesús W, Lewis A, Leon R, Cadilla CL, Garriga JL, Biochim. Biophys. Acta - Proteins Proteomics 1747 (2005) 195–203. 10.1016/j.bbapap.2004.11.005. [DOI] [PubMed] [Google Scholar]
- [34].Bonaventura C, Henkens R, De Jesus-Bonilla W, Lopez-Garriga J, Jia Y, Alayash AI, Siburt CJP, Crumbliss AL, Biochim. Biophys. Acta - Proteins Proteomics 1804 (2010) 1988–1995. 10.1016/j.bbapap.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ruiz-Martínez CR, Nieves-Marrero CA, Estremera-Andújar RA, Gavira JA, González-Ramírez LA, López-Garriga J, García-Ruiz JM, Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun 65 (2009) 25–28. 10.1107/S1744309108038542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Childress JJ, Girguis PR, J. Exp. Biol 214 (2011) 312–325. 10.1242/jeb.049023. [DOI] [PubMed] [Google Scholar]
- [37].Berg JM, Tymoczko JL, Stryer L, Biochemistry 5th Editition, W. H. Free. Co. (2002). [Google Scholar]
- [38].Di Russo NV, Estrin DA, Martí MA, Roitberg AE, PLoS Comput. Biol 8 (2012). 10.1371/journal.pcbi.1002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pace CN, Grimsley GR, Scholtz JM, J. Biol. Chem 284 (2009) 13285–13289. 10.1074/jbc.R800080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Harris TK, Turner GJ, IUBMB Life. 53 (2002) 85–98. 10.1080/10399710290038972. [DOI] [PubMed] [Google Scholar]
- [41].Fitch CA, Karp DA, Lee KK, Stites WE, Lattman EE, Bertrand García-Moreno E, Biophys. J 82 (2002) 3289–3304. 10.1016/S0006-3495(02)75670-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Isom DG, Castaneda CA, Cannon BR, Garcia-Moreno EB., Proc. Natl. Acad. Sci 108 (2011) 5260–5265. 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zheng G, Schaefer M, Karplus M, Biochemistry. 52 (2013) 8539–8555. 10.1021/bi401126z. [DOI] [PubMed] [Google Scholar]
- [44].Ng JD, Gavira JA, García-Ruíz JM, J. Struct. Biol 142 (2003) 218–231. 10.1016/S1047-8477(03)00052-2. [DOI] [PubMed] [Google Scholar]
- [45].Otwinowski Z, Minor W, Methods Enzymol. 276 (1997) 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- [46].Nieves-Marrero CA, Ruiz-Martínez CR, Estremera-Andjar RA, González-Ramírez LA, López-Garriga J, Gavira JA, Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun 66 (2010) 264–268. 10.1107/S1744309109053081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hockenhull Johnson JD, Stern MS, Martin P, Dass C, Desiderio DM, Wittenberg JB, Vinogradov SN, Walz DA, J. Protein Chem 10 (1991) 609–622. [DOI] [PubMed] [Google Scholar]
- [48].Rivera L, López-Garriga J, Cadilla CL, Gene. 410 (2008) 122–128. 10.1016/j.gene.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN, Acta Crystallogr. Sect. D Biol. Crystallogr 60 (2004) 2184–2195. 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- [50].Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, Acta Crystallogr. Sect. D Biol. Crystallogr 66 (2010) 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Emsley P, Cowtan K, Acta Crystallogr. Sect. D Biol. Crystallogr 60 (2004) 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- [52].Antonini M, Brunori E, Hemoglobin and Myoglobin in Their Reactions with Ligands, Front Biology, Amsterdam-London: (1971). [Google Scholar]
- [53].Shikama K, Chem. Rev 98 (1998) 1357–1373. 10.1021/cr970042e. [DOI] [PubMed] [Google Scholar]
- [54].Brantley RE, Smerdonll SJ, Wilkinsonllll AJ, Singleton EW, Olson JS, 268 (1993) 6995–7010. [PubMed] [Google Scholar]
- [55].Postnikova GB, Shekhovtsova EA, Biochem. 83 (2018) 168–183. 10.1134/S0006297918020098. [DOI] [PubMed] [Google Scholar]
- [56].De Jesús-Bonilla W, Jia Y, Alayash AI, López-Garriga J, Biochemistry. 46 (2007) 10451–10460. 10.1021/bi7003262. [DOI] [PubMed] [Google Scholar]
- [57].De Jesús-Bonilla W, Cruz A, Lewis A, Cerda J, Bacelo DE, Cadilla CL, López-Garriga J, J. Biol. Inorg. Chem 11 (2006) 334–342. 10.1007/s00775-006-0082-0. [DOI] [PubMed] [Google Scholar]
- [58].Li Q, Lancaster JRJ, Nitric Oxide. (2013) 1–32. 10.1016/j.niox.2013.07.001.Chemical. [DOI] [Google Scholar]
- [59].Sommer S, Jahn A, Funke F, Brenke N, Naturwissenschaften. 87 (2000) 283–287. [DOI] [PubMed] [Google Scholar]
- [60].Volkel S, Amer. Zool 35 (1995) 145–153. [Google Scholar]
- [61].Asokan A, Cho MJ, J. Pharm. Sci 91 (2002) 903–913. [DOI] [PubMed] [Google Scholar]
- [62].Boron WF, Adv. Physiol. Educ 28 (2004) 160–179. 10.1152/advan.00045.2004. [DOI] [PubMed] [Google Scholar]
- [63].Liberge M, Gros O, Frenkiel L, Mar. Biol (2001) 401–409. 10.1007/s002270000526. [DOI]
- [64].Lechaire J-P, Frebourg G, Gaill F, Gros O, I Biol. Cell 98 (2006) 163–170. 10.1042/BC20040502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.