

Abstract
In exploration of congenital heart defects produced by TCE, Hepatocyte Nuclear Factor 4 alpha (HNF4a) transcriptional activity was identified as a central component. TCE exposure altered gene transcription in the chick heart in a non-monotonic pattern where only low dose exposure inhibited transcription by HNF4a. As the chick embryo is non-placental, we examine here HNF4a as a target of TCE in developing mouse embryos. Benfluorex and Bi6015, published agonist and antagonist, respectively, of HNF4a were compared to low dose TCE exposure. Pregnant mice were exposed to 10ppb (76 nM) TCE, 5μM Benfuorex, 5μM Bi6015, or a combination of Bi6015 and TCE in drinking water. Litters (E12) were collected during a sensitive window in heart development. Embryonic hearts were collected, pooled for extraction of RNA and marker expression was examined by quantitative PCR. Multiple markers, previously identified as sensitive to TCE exposure in chicks or as published targets of HNF4a transcription were significantly affected by Benfluorex, Bi6015 and TCE. Activity of TCE and both HNF4a-specific reagents on transcription argues that HNF4a is a component of TCE cardiotoxicity and likely a proximal target of low dose exposure during development. The effectiveness of these reagents after delivery in maternal drinking water suggests that neither maternal metabolism, nor placental transport is protective of exposure.
Keywords: Cardiac development, cardiac teratology, Benfluorex, Bi6015
Graphical Abstract
Introduction
Trichloroethylene (TCE) is an industrial solvent with widespread distribution in the environment including Superfund sites, military bases, and manufacturing facilities. TCE is identified as a potential cardiac teratogen by the EPA1 and is designated as a contaminant of concern by the Department of Defense (GAO-07–1042). Reports of TCE cardiac teratogenicity first reported from studies of children in Tucson, Arizona.2 The defects seen in children exposed to up to 270 ppb (parts per billion) TCE in drinking water in utero included aortic stenosis, valvular, septal and muscular defects and the odds ratio (O.R.) for all heart defects was 3.02 and 2.53. Additional links between TCE and human heart defects were investigated in Milwaukee, Wisconsin and Endicott, NY related to vapor exposure.4,5 Although the epidemiological support for cardiac teratogenicity in humans is limited, animal and mechanistic studies establish that TCE exposure can produce cardiac malformations and perturb normal developmental gene expression.1
The mechanisms of cardiac defect formation in exposed embryos appear to be complex. At varying exposure levels, there is either inhibition of the formation of valve precursors6, or proliferation and overgrowth of cardiac valves.7,8 Low dose exposure also perturbs cardiomyocyte contraction.9,10 Low dose TCE exposure affects transcription of a large number of genes including regulators of calcium homeostasis in rat, mouse and chicken heart tissues.9–14 An issue with studies of TCE toxicity is the non-monotonic dose curve observed in vitro and in vivo. Doses in the 8–10 ppb range produce greater effects than seen in the 800ppb- 1 ppm range as measured both by gene transcription an intracellular calcium fluxes.12,15 The reduction in calcium flux corresponds with a deficit in myocyte contraction and a reduction in shear stress markers in the heart.9
Studies show perturbation of gene transcription by TCE exposure in rat, mouse and chick embryonic hearts as well as cell lines in culture.11–15 Based upon a microarray and interactome analysis of chick embryos exposed to 8ppb TCE in ovo, the transcription factor, Hepatocyte Nuclear Factor 4 alpha (HNF4a) and several additional molecules were linked to TCE exposure.16 HNF4α was not previously associated with the developing heart but recent studies in the chick embryo showed that HNF4a is expressed in the heart during the window of sensitivity to TCE and that HNF4a is transiently expressed in human IPSCs during differentiation into cardiomyocytes.10,17
Small molecule screens identified molecules, Benfluorex and BI6015, with specific agonist or antagonist activities towards HNF4a.18,19 In ovo exposure in the chick showed that Benfluorex and TCE both inhibited expression of three genes associated with TCE exposure at low levels of exposure.10 As chick embryo exposures have no link to maternal metabolism nor utilize placental transport during exposure, it is important to evaluate the role of HNF4a in response to both TCE and HNF4a-specific reagents in the context of mammalian heart development. Maternal metabolism and placental transfer may alter exposure of the embryo to TCE or a metabolite to reduce exposure.
To test HNF4a transcriptional activity and TCE exposure during heart development, we treated pregnant mice with TCE, Benfluorex or Bi6015 in drinking water and compared a panel of marker genes known to be sensitive to TCE or as targets of HNF4a transcriptional activity in the hearts of the exposed embryos. TCE, Benfluorex and Bi6015 all perturbed the panels of marker genes associated with HNF4a or previously identified as significant targets of TCE. However, while low dose exposure to TCE, produced an inhibition of markers as seen in vitro, both Benfluorex and Bi6015 produce an upregulation of marker gene expression in vivo. These data show that maternal metabolism and transport across the placenta have little or no influence on TCE-perturbed gene transcription. Further, the data suggest that HNF4a-mediated transcription is a significant target of TCE in the developing heart in this mammalian model.
Materials and Methods
In vitro analysis
Human HepG2 cells (ATCC, HB-8065) were grown in DMEM media with 10% bovine serum and 1% pen/strep. For exposure to HNF4a reagents they were incubated with either Benfluorex Hydrochloride (5μM) (sc-291931, Santa Cruz Biotechnology) or Bi6015 (5μM) (CAS# 93987-28- 2, Tocris Bioscience, Bristol, UK). RNA was collected after a six hour treatment using the E.Z.N.A Total RNA Kit 1 (R6834–02, Omega-BioTek).
In utero exposure and embryo analysis
Wild type 129/Sv mice were obtained from Harlan Sprague Dawley and bred in the University of Arizona Animal Facility. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the University of Arizona and approved by the Animal Ethics Committee of the University of Arizona Institutional Animal Care and Use Committee (IACUC). Presence of a vaginal plug was designated as day E0. Pregnant mice were provided with TCE (10ppb) or Benflorex HCl (5μM) or Bi6015 (5μM) in combination with TCE (10ppb) in their drinking water and provided ad libitum from E0 to E12. To reduce loss of volatile and light sensitive TCE, stainless steel water bottles were used and exposure solutions were prepared and replaced on a daily basis from a 1000x stock solution. The bottles were completely filled each day to minimize head space and loss of TCE. A previous study11 showed no aversion to low dose TCE exposure in drinking water by mice but dehydration was monitored for all groups to verify normal drinking behavior. Three to four independent litters were obtained for each exposure group. Mothers were sacrificed and litters were collected at E12. Litter sizes varied between 7 to11 embryos and there was no observed bias in litter size by treatment. Embryonic hearts were dissected from the body, pooled by litter and stored in RNAlater (ThermoFisher) until RNA extraction. Pooled tissues were extracted for mRNA using an E.Z.N.A Total RNA Kit 1 (R6834–02, Omega-BioTek). If the RNA yield was or quality was poor, hearts from the fourth dam were utilized.
Quantitative real-time RT PCR
Total RNA from mice embryo hearts was treated with DNaseI (E1091–02, Omega-BioTek). cDNA was transcribed using the iScript cDNA synthesis kit (Bio-Rad). Total cDNA was used to normalize quantitative PCR reactions as we have previously published (Mercado-Pimentel; Tavares). cDNA was sensitively measured in each reaction using Quanti-iT Oligreen ssDNA reagent (011492, Invitrogen) with a fluorometer (Turner Biosystems). Equal aliquots of cDNA (10ng) were dispensed in each PCR tube for measurement. Quatitative PCR was performed on a Rotorgene Instrument (Qiagen) using a SensiFAST SYBR buffer (Bio-98002, Bioline). Cycling parameters used a 60°C annealing temperature and 72° C for elongation and data collection. Primers were designed for mouse and human marker genes using NCBI Primer-Blast and selected for pairs that gave melting curves with a single peak. Primers sequences are displayed in Table 1.
Table 1.
Marker gene identity and qPCR primer sequences a
| Mouse Genes | Gene Name | Gene ID | FW Sequence | RW Sequence | HNF4a target* |
|---|---|---|---|---|---|
| INSR | Insulin receptor | NM_ 010568.2 | CCCCACCCTTTGAGTCTGAT | CGTTGTGCAGGTAATCCTCG | Y |
| MAP3K3 | Mitogen- activated protein kinase kinase kinase 3 | NM_011947.3 | TGAGTGCTCTGGAGTGTGAG | TGAGTGCTCTGGAGTGTGAG | Y |
| EP300 | E1A binding protein p300 | NM_177821.6 | AACAGCAGCTCAGCCATCTA | CAGGATCCGGAGTGGGAAAT | Y |
| HNF4a | Hepatocyte Nuclear Factor 4, alpha | NM_008261.2 | AGCCAACGATCACCAAGCAA | ACTGGTCCCTCGTGTCACAT | Y |
| TRAF6 | TNF receptor associate factor 6 | NM_009424 | CTCGGAGTGCCGTGTATGTA | ACACACACAACCCGATCGTA | Y |
| SIKE1 | Suppressor of IKBKE | NM_025679.3 | ACAAACCCATTCAGGGGTGG | AGCTTGCTCTACCCTCAGGA | |
| HSPA8 | Heat shock 70kDa Protein 8 | NC_000075.6 | GGGTTGCAGACTTTCTCCAGT | AAGGCTGAGGATGAGAAGC A | Y |
| PRKAB1 | Protein Kinase Activated Beta-1 | NM_031869 | CTTCCTTGTGTCCCTGCAGAT | TTCGGAGTGGAAGATGTCGG | |
| YWHAG | 14-3-3 gamma | NM_018871.3 | AGACGTGGCCCTGTATTTGG | TTGGCGTCATGTATGGCTGT | Y |
| SEPW1 | Selenoprotein W,1 | NM_009156.2 | GTGACAGTAGCCGGGAAGTT | TGATGGCGGTCACCAGTTTC | Y |
| ZMAT2 | Zinc Finger, Matrin-Type 2 | NM_025594.3 | AGTGCGAGTTTTCCCCTCTG | AGCCTGAATAACCCAGTGCC | Y |
| ALDOC | Aldolase C | NM_001303423. 1 | TGGACCGAGCTAATCAGAGGA | TGAGTGGGGCATGATGACA G | Y |
| FXYD6 | FXYD Domain Containing Ion Transport Regulator 6 | NM_022004.6 | GGTTGGTGTTTGCTGTGGTC | CCGCAGCGTTTGTAGTGATG | Y |
| IKBKE | Inhibitor of Kappa Light Polypeptide Gene Enhancer in B Cells, Kinase | NM_019777.3 | CCACTTGGAGTGCAGGAAGA | TTTCGGGCCTTGTACACACT | Y |
| Human Genes | Gene Name | Gene ID | FW Sequence | RW Sequence | |
| EP 300 | E1A binding protein p300 | NM_001429 | GCAGTGTGCCAAACCAGATG | GGGTTTGCCGGGGTACAATA | Y |
| HNF4a | Hepatocyte Nuclear Factor 4-alpha | NM_001287184 | AGGGATTAACTCTGCCCAGC | ACATCCTCCTCCTGCTGCTA | Y |
| CYP1B1 | Cytocrhome P450 family1,subfamil y b 1 | NM_000104 | AACAAGGACCTGACCAGCAG | CCCTGAAATCGCACTGGTGA | Y |
| CYP24A 1 | Cytochrome P450, family 24, subfamily A, polypeptide 1 | NM_001128915 | ACCCAAAGGAATTGTCCGCA | CAAAACGCGATGGGGAGTTC | |
| INSR | Insulin receptor | XM_006722744.1 | GTGACAGACTATTACGTCCCGT | CCATCTGGCTGCCTCTTTCT | Y |
| MAP3K3 | Mitogen- activated protein kinase kinase kinase 3 | NM_203351 | TTGGACAGGTCAGCAGACAG | GGTTCCACCTTTGACCCCTT | Y |
| HBGEF | Heparin binding EGF like growth factor | DQ480720 | TTGAGTGAGAAGAGCCAAGG G | AGGCCTCTTTCATCACAGGA C | Y |
Gene Expression Measurement and Statistical Analysis
Pregnant dams were treated via drinking water, and heart tissue of offspring was excised and pooled by litter. Three technical replicate qPCR measurements were made for each litter, and a minimum of three litters were tested for each chemical exposure and for control treated dams. Expression in technical replicates was normalized relative to the mean of control treated litters and averaged to produce one relative expression value per litter for each gene reported. Hypothesis tests comparing each chemical exposure to control were conducted using Student’s t-test. We evaluate each test at 0.05 significance. Because we are evaluating specific genes identified previously in a different animal model, we do not adjust p-values for multiple comparisons.
Results
In vitro analysis of HNF4a reagents.
Previous studies identified a large number of genes with transcription perturbed in the heart by low dose exposure to TCE in mouse and chick embryos.11,16 Interactome analysis suggested HNF4a as a significant target of TCE toxicity by linkage to a large percentage of identified transcripts.16 Studies in the chick embryo showed that transcription of CYP2C45, HNF4a and NFKBIE were sensitive to both TCE and Benfluorex and that both HNF4a and CYP2C45 showed a switch between inhibition of expression at 10ppb (76nM) or below and induction at ppb (0.76μM) or greater.10 To explore the significance of the observation, we selected additional genes identified as transcriptional targets of TCE toxicity from a microarray analysis of TCE exposure in the mouse embryo heart.11 As cardiomyocytes express HNF4a only transiently during development17, we first tested their expression in a human liver tumor cell line that expresses HNF4a (HepG2).20 Both Benfluorex and BI6015 perturb HNF4a transcriptional activity. Bi6015 was identified as an antagonist of HNF4a activity that binds in its ligand-binding pocket.18 Benfluorex was identified as an agonist of HNF4a by a high throughput screen.19 HepG2 cells were treated for 6 hr with Benfluorex (5μM) or Bi6015 (5μM) and then extracted for RNA. RT-PCR was performed to measure changes in marker transcripts. Figure 1 shows that all of the TCE-sensitive genes tested were responsive to both reagents compared to untreated control cells. The majority of markers were inhibited by both reagents but Cyp2C4A1 transcription was upregulated by Bi6015. Although Benfluorex was identified as an agonist of HNF4a activity in cultures at high concentrations (20 and 80 μM;19), we found it to be an inhibitor of marker gene expression in vitro at 5μM here and in ovo in the chick embryo.10 As Bi6015 showed more variation in response, Benfluorex was chosen for the majority of analysis in mouse embryos after drinking water exposure in pregnant mice.
Figure 1. HepG2 cell marker expression after drug exposure.
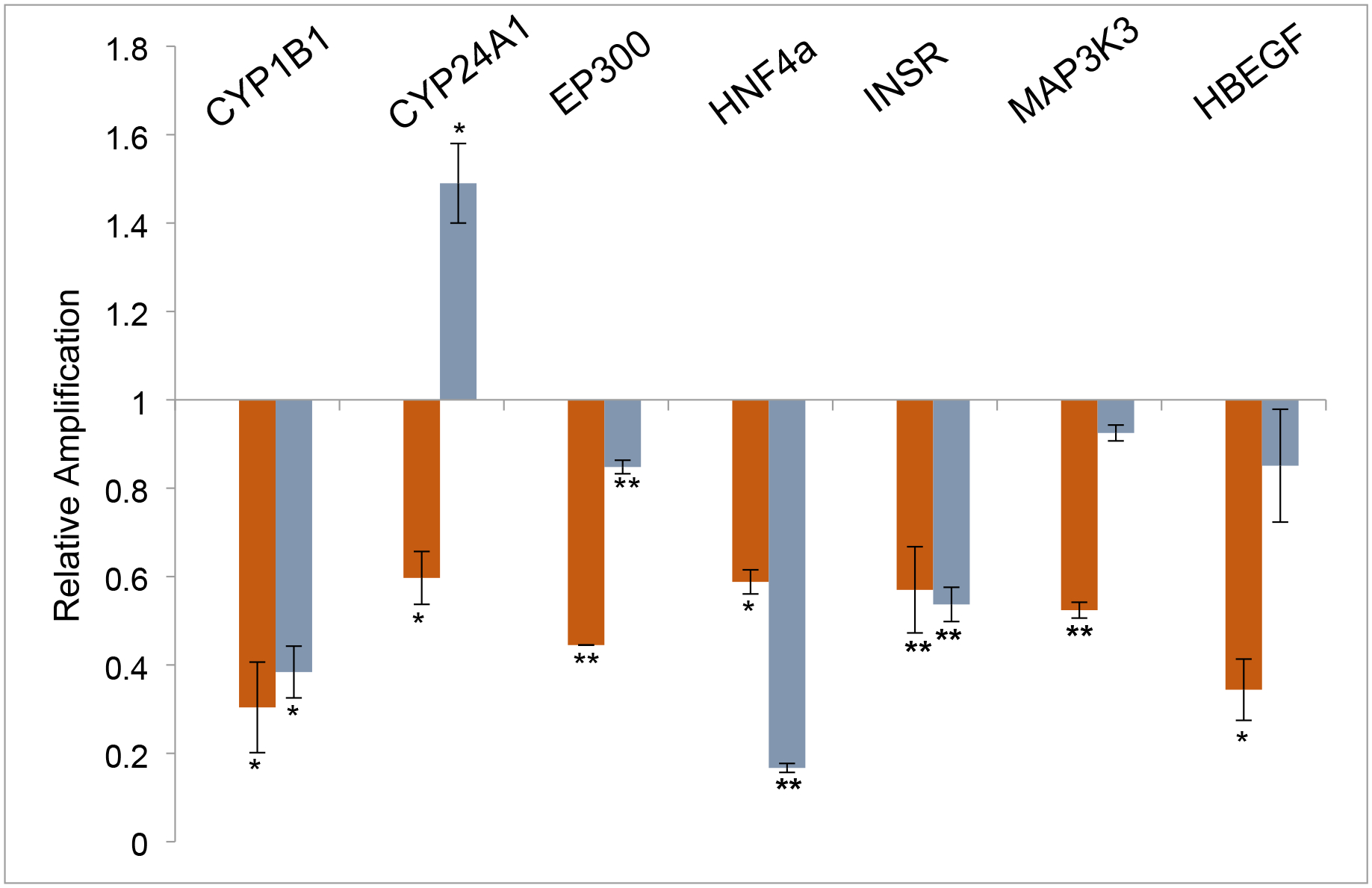
Human HepG2 cells were treated with 5μM Benfluorex or 5μM Bi6015 for 6 hours and RNA was collected and measured by qPCR. The data show the average relative amplification of three independent experiments performed with 3 technical repeats for each experiment and are normalized to untreated HepG2 cells. Benfluorex (red bars) significantly inhibited expression of each of the tested markers compared to untreated cells. Bi6015 showed a significant upregulation of Cyp2C4A1 and non-significant effects on Map3K3 and HBEGF. Significance calculated by Student’s T Test for values obtained with the same primer pair for treatments and controls. *p>.05, **p>.005..
Analysis of TCE and HNF4a reagent exposure to hearts in utero
To explore TCE and HNF4a-specific reagents in vivo, mice were mated and drinking water exposure was initiated when a vaginal plug was observed. Experimental exposures were 10 ppb TCE, 5μM Benfluorex, 5μM Bi6015, or a combination of TCE and Bi6015 delivered in maternal drinking water. Hearts were dissected from each embryo on day E12, pooled by litter and extracted for RNA. For each treatment, three litters were exposed. Quantitative PCR was performed on each litter (7–11 embryos) and averages were compiled for the three litters. Initial qPCR measurements were made of gene transcripts selected from the list of most highly linked genes in the interactome developed from the chick embryo microarray.16 Data was normalized to the average expression and standard error of the mean of an untreated litter after verification that there was no significant difference in marker expression between three control litters. Figure 2 shows significant down-regulation of these genes by TCE exposure (10ppb = 76 nM) in this mouse model. An additional set of markers, selected from known HNF4a targets (HBEGF, INSR) as well as genes strongly down-regulated by TCE in previous mouse or chick microarray data (EP300, SEPW1, ZMAT2, FXYD6, ALDOC) were inhibited by low dose TCE exposure in the developing heart (Figure 3). Gene identification and primer pairs used for each marker are shown in Table 1.
Figure 2. TCE effects on markers identified by the interactome in mouse embryo hearts.
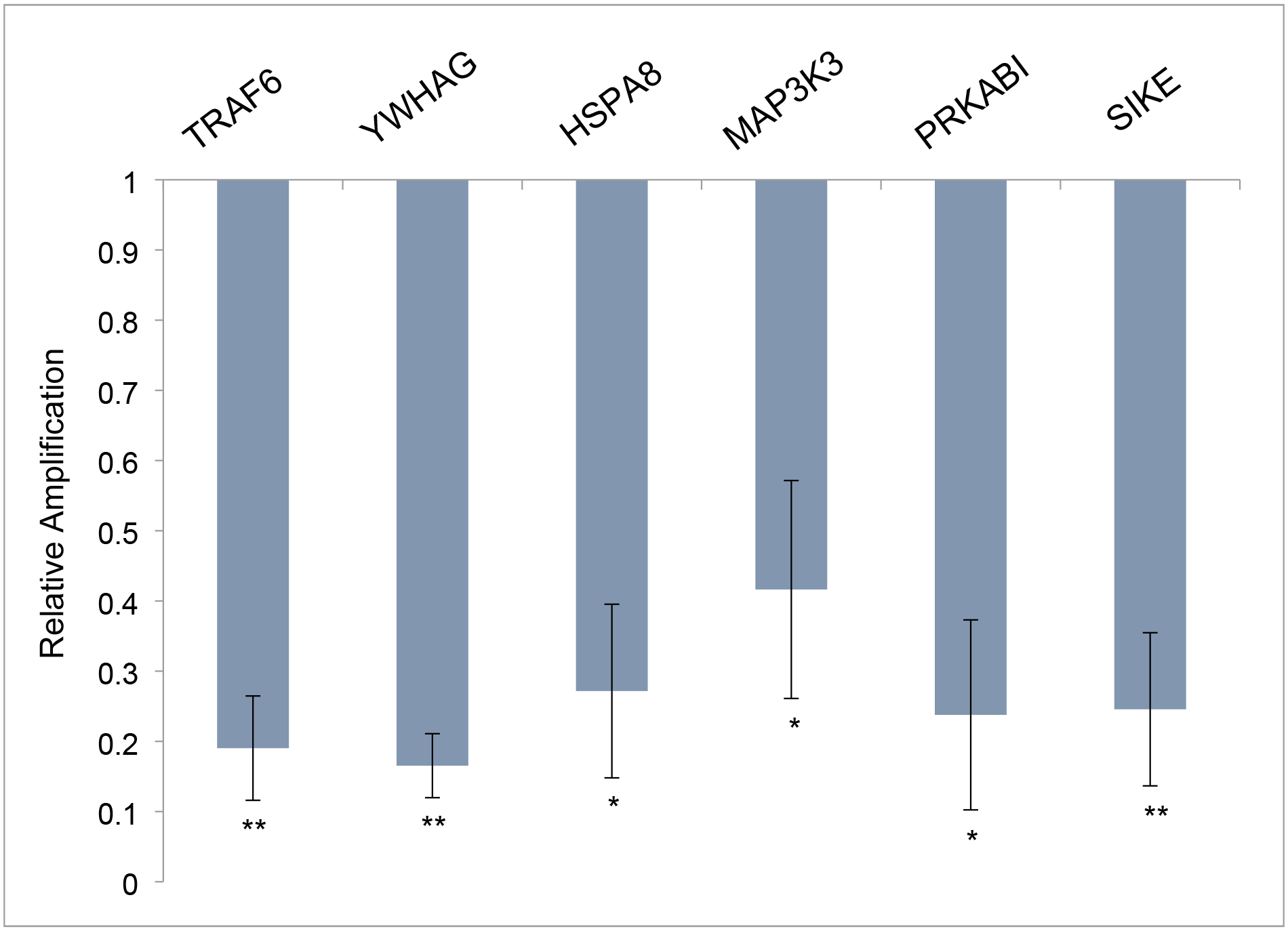
Six of the nine highest linked genes in the TCE interactome other than HNF4a were tested and found to be present in the heart at E12.5. TCE exposure in utero significantly reduced mRNA expression of all of these markers in the heart. Data show the average and SEM of the three litters exposed to 10ppb TCE and are normalized to the average measure of each primer pair with three untreated control litters. All transcriptional changes were significant as evaluated by Student’s T Test (unpaired, two-tailed). *p>.05, **p>.005.
Figure 3. TCE effects on identified HNF4a and TCE markers in mouse embryo hearts.
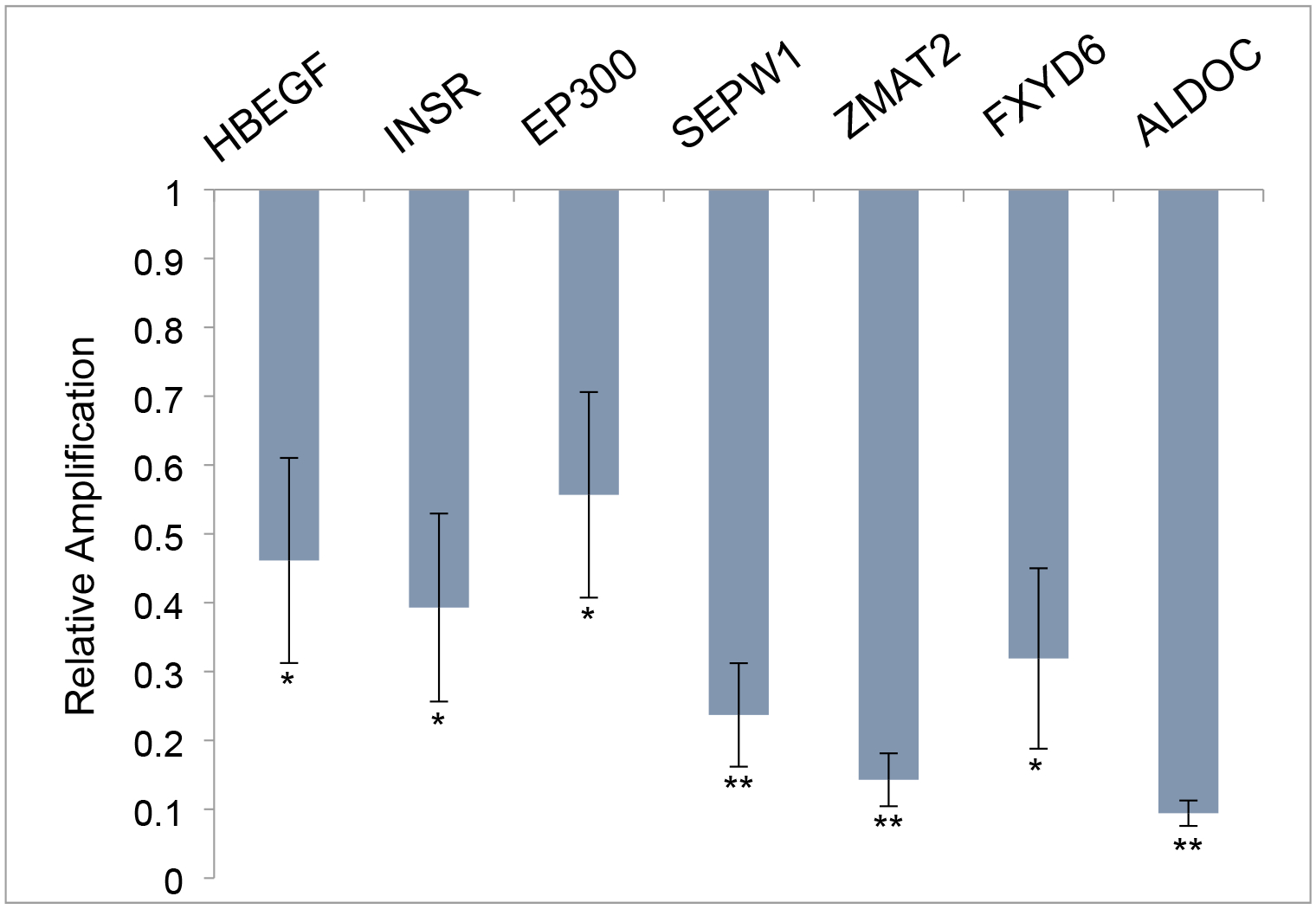
HBEGF and INSR are known targets of HNF4a activity while the remaining markers tested were chosen from among the most significantly affected genes in a microarray analysis of exposure in the developing chick heart. All markers were significantly downregulated by exposure to TCE in utero. Data show the average and SEM of the three litters exposed to 10ppb TCE and are normalized to the average measure of each primer pair with three untreated control litters. All transcriptional changes were significant as evaluated by Student’s T Test (unpaired, two-tailed). *p>.05, **p>.005.
To compare the responses of the 14 selected TCE-sensitive markers, we examined expression in the Benfluorex-exposed embryonic hearts. Figure 4 shows the interactome-selected markers are all up-regulated by Benfluorex, consistent with its reported activity as an agonist.19 Though the majority of markers are downregulated in culture when exposed at a defined level, they are consistently up-regulated when exposure is provided by maternal drinking water provided ad libitum at the same initial concentration. All of the markers identified as the major targets in the TCE interactome showed an increased expression of 5–11 fold. In addition to the targets identified by the interactome, we examined the set of genes selected as TCE targets (Figure 5). All but one of these genes is significantly upregulated by Benfluorex. The one gene that is more modestly upregulated (less than 2-fold) was the transcript for SEPW1 a ubiquitous selenoprotein of largely unknown function that binds to 14-3-3 proteins.21
Figure 4. Benfluorex effects on interactome markers in mouse embryo hearts.
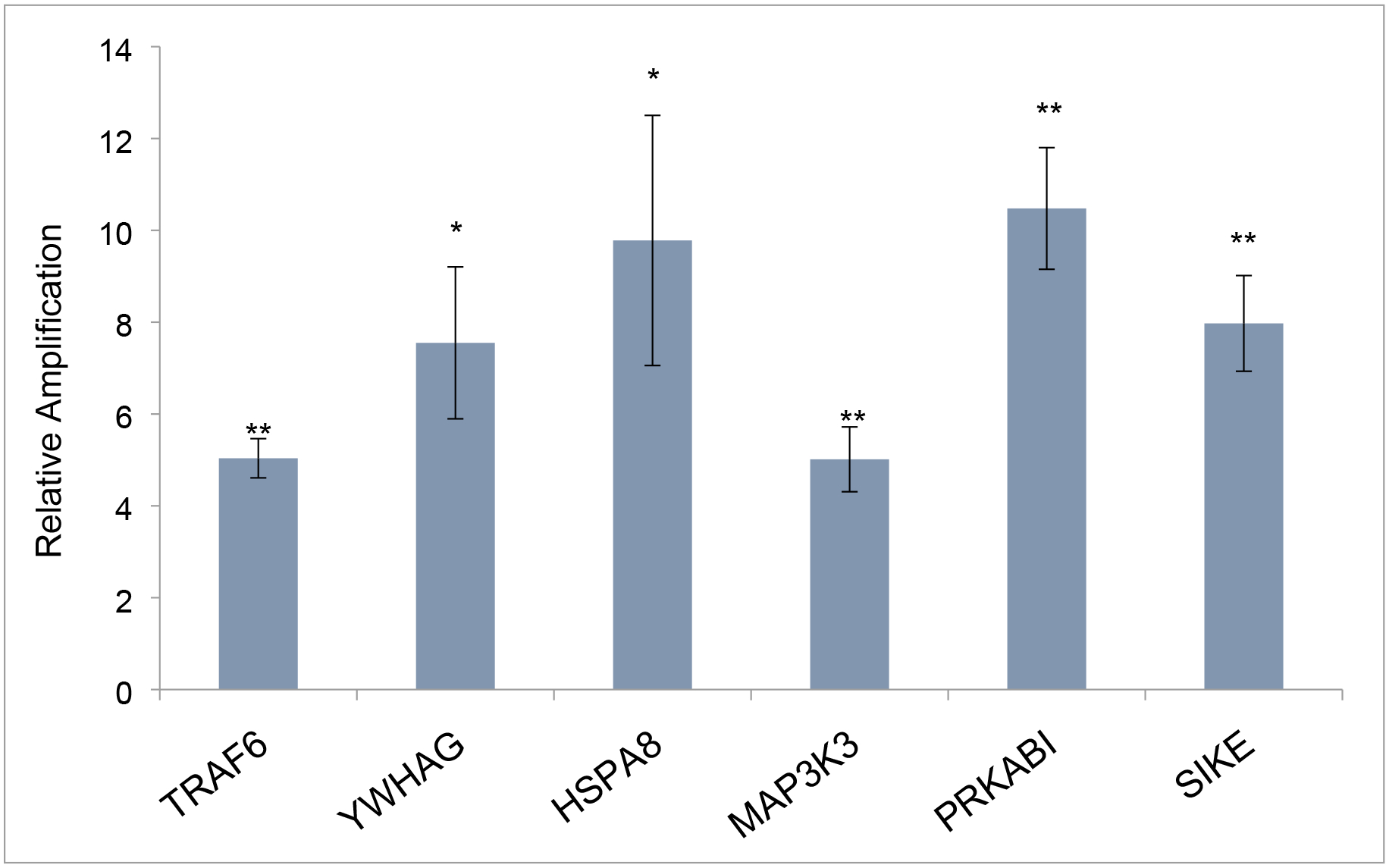
The markers shown in Fig. 2 were examined after in utero treatment with Benfluorex in maternal drinking water. Expression of each marker mRNA was analyzed by qPCR and normalized to its expression in untreated control embryo hearts (set at 1). Data show the average and SEM of the three litters exposed to 5 μM Benfluorex. After ad libitum exposure in drinking water, marker gene expression was elevated in a range of 5–10 fold. All transcriptional changes were significant as evaluated by Student’s T Test (unpaired, two-tailed). *p>.05, **p>.005.
Figure 5. Benfluorex effects on identified HNF4a and TCE markers in mouse embryo hearts.
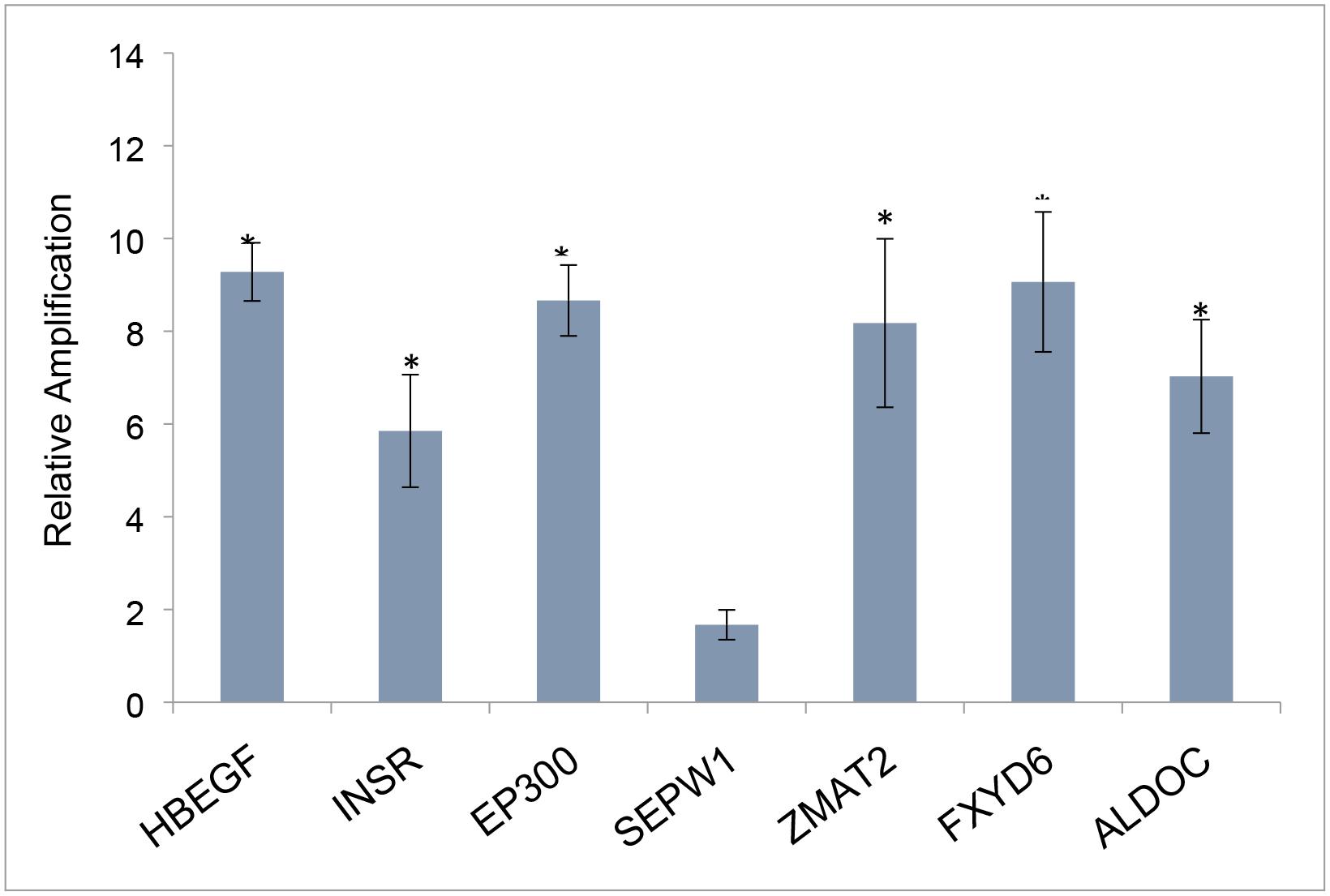
Markers shown in Fig. 3 were examined after exposure to Benflorex in utero. All but 1 marker, the transcript of SelenoproteinW1, were significantly elevated by exposure. Data show the average and SEM of three litters compared to measures in 3 pooled, untreated litters. Significance was evaluated by Student’s T Test (unpaired, two-tailed). *p>.05, **p>.005.
Additional litters of mice were exposed to the antagonist Bi6015 or to a combination of Bi6015 and TCE. The E12 hearts were pooled from a single litter of each and comparison was made with a subset of markers. As it was seen that the SIKE1 (Suppressor of IKBKE) was altered by both TCE and Benfluorex exposure, primers to examine IKBKE were added. The data in Figure 6 show that both Bi6015 and Benfluorex acted as agonists of marker transcription with the exception of IBKE where the effects of Bi6015 were inhibitory. The combination of TCE and Bi6015 produced a greater upregulation of marker expression than Bi6015 alone except in the example of the Insulin Receptor (INSR).
Figure 6. Comparison of the effects of TCE with Bi6015, Benfluorex or a combination of TCE and Bi6015 on selected marker expression.
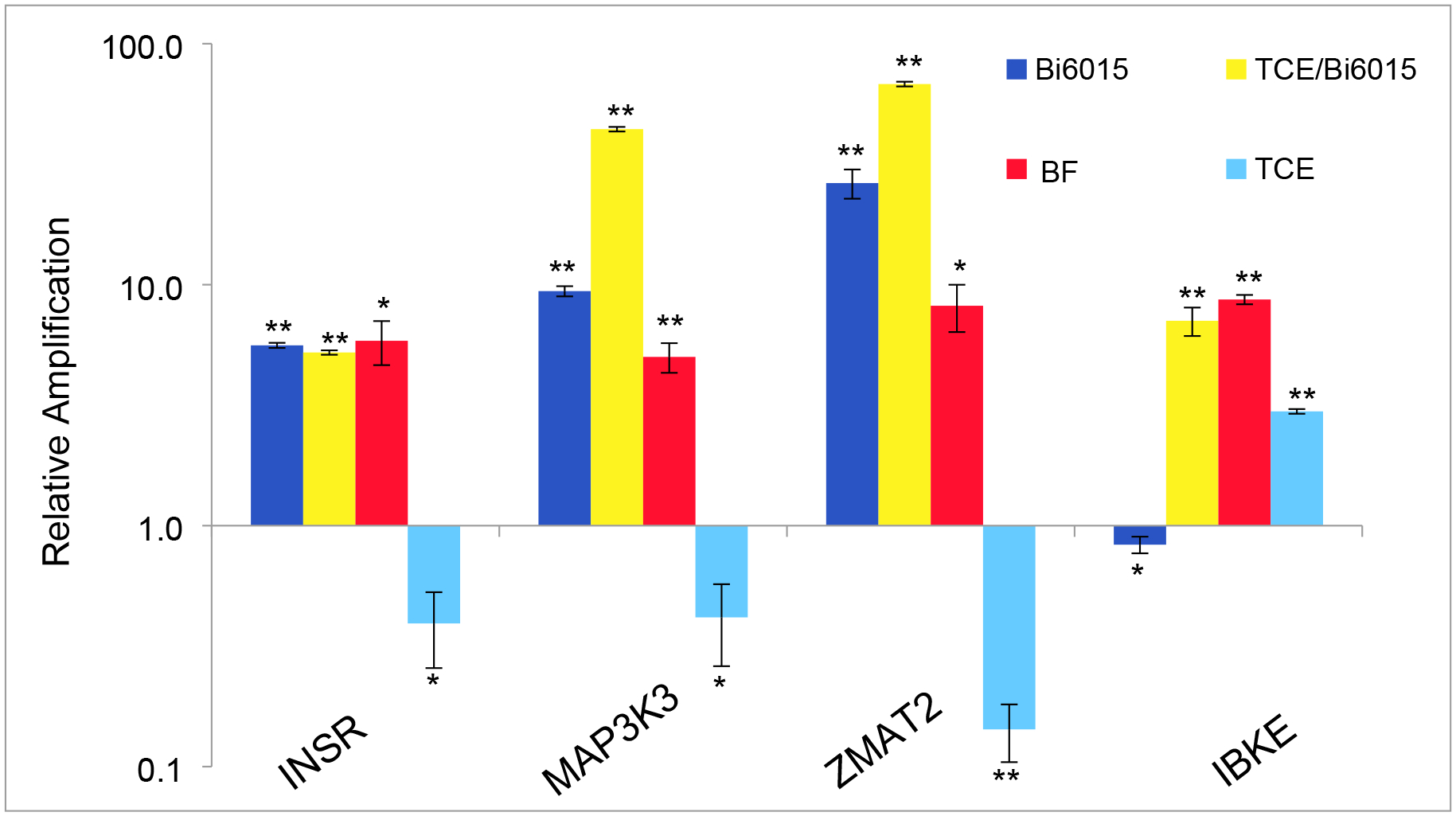
qPCR measurements of selected markers were compared on RNAs extracted from pooled hearts and controls. Measurements were normalized to control litter measurements for each marker (set at 1.0) and are the average of three independent repeats. TCE and Benfluorex data is repeat of previous figures but compared here to Bi6015 and TCE with Bi6015. Scale is logarithmic to capture the large changes seen with the combination of TCE and Bi6015 exposure with MAP3K3 and ZMAT2 markers. While Benfluorex was a consistent agonist of these markers, both Bi6015 and TCE showed agonist or antagonist activity against specific markers. The combination of Bi6015 and TCE showed substantial effects on the expression of some markers but little difference from Benfluorex alone with others. Significance was evaluated by Student’s T Test (unpaired, two-tailed). *p>.05, **p>.005.
HNF4a is known to regulate its own transcription19 but our original microarray data showed no significant change in expression despite its highly linked position in the interactome. HNF4a showed both a change in expression and altered phosphylation in HepG2 cells in vitro.16 Exposure to TCE in the chick embryo was dose sensitive with inhibition of HNF4a expression at exposures of less than 10 ppb and an increase at 100 ppb and 1 ppm. Here, HNF4a transcript levels were up-regulated by both Benfluorex and Bi6015. The combination of TCE and Bi6015 was no more effective than Bi6015 alone. In contrast, TCE, by itself did not produce significant change in expression at the 10ppb dose tested here. (Fig. 7)
Figure 7. Effects of HNF4a reagents on HNF4a mRNA expression.
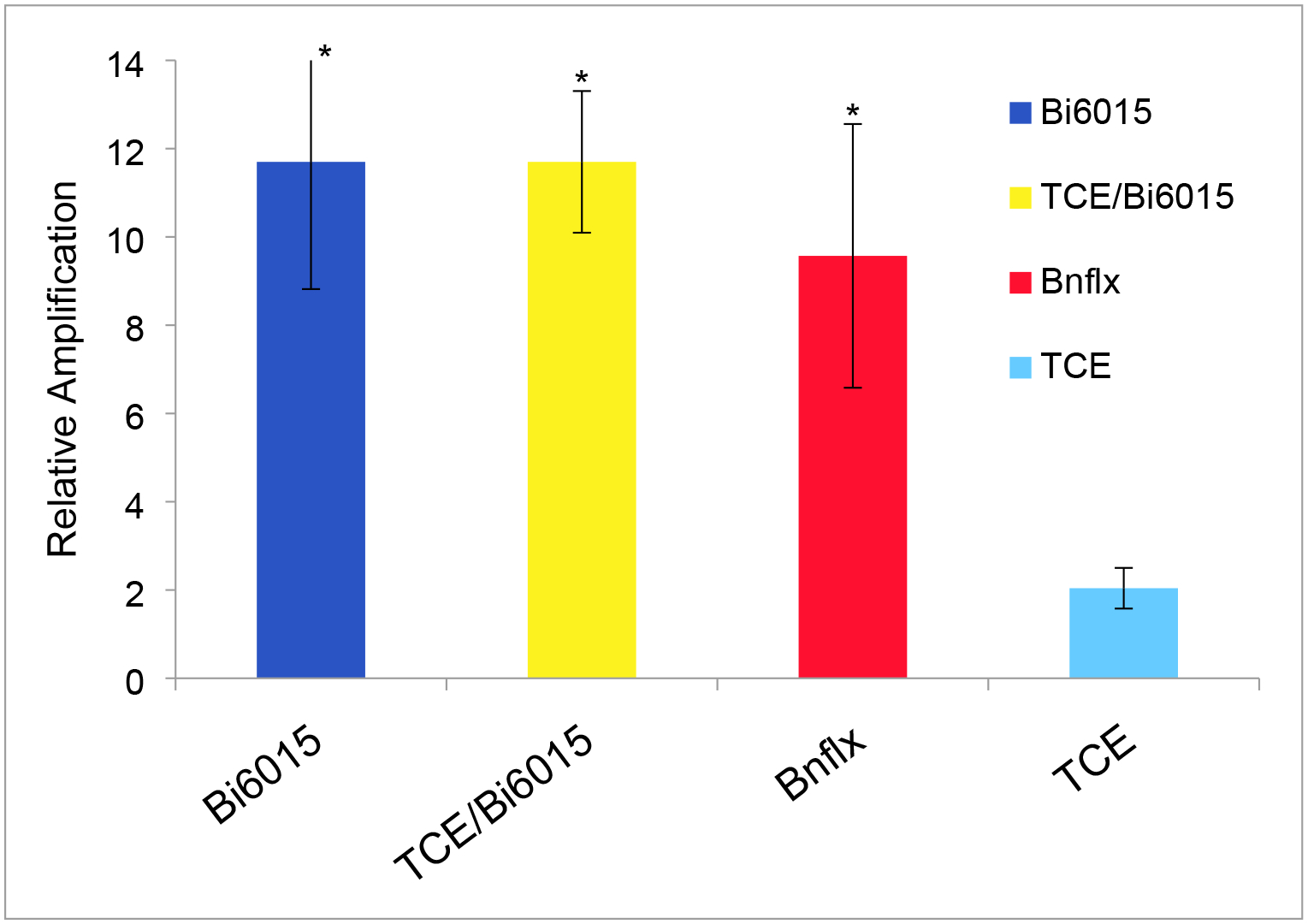
HNF4a is potentially autoregulatory although our original microarray data showed no direct effect of TCE on HNF4a RNA expression. Measurement of HNF4a by qPCR showed that both Bi6015 and Benfluorex are significant agonists of HNF4a expression while TCE had no significant effect. Significance was evaluated by Student’s T Test (unpaired, two-tailed). *p>.05, **p>.005.
Discussion
The link between congenital heart defects and TCE exposure is an area of controversy.22–26 The controversy persists, in large part, due to the lack of established adverse outcome pathways (AOP) and the complexity of heart development. The early studies of Dawson et al.27 and Johnson et al.28 focused on high dose exposures and the gross examination of more than 2000 exposed embryos for morphologically detectable defects. While their studies drew attention to an apparent cardiac-specific response to TCE, the exposures they examined (1100 ppm-1.5ppm) were 2–4 orders of magnitude greater than the first reported environmental exposure.2 More recent work has explored environmentally relevant exposure levels to TCE in the parts per billion (ppb) range.7,11, 29. Together, these studies show non-monotonic dose responses as measured by marker gene transcription, thousands of transcripts sensitive to low dose exposure and functional effects on calcium homeostasis and muscle contraction in response to TCE (reviewed in Selmin et al.16). The data shown here and in Harris et al.10 support an interaction between TCE and HNF4a as a molecular interaction consistent with an initiation of a novel AOP leading to cardiac defects. We do not exclude that there are likely additional AOPs involved in development of the valves or the proliferation of valvular fibroblasts with increased exposure levels.6–8
TCE exposure produces a significant alteration of gene transcription in both animal embryos and cells at low doses.11–13,16 Although formation of valves is sensitive to TCE6,7, comparison of transcript identities and numbers between cell types suggested that myocytes were more significantly perturbed by low dose TCE exposure.12,13 As perturbation of myocardial function is capable of producing cardiac malformation31, we focused on several genes that mediate calcium homeostasis and regulation of contraction. Serca2 and Ryr2 were among the molecules whose expression was altered by TCE exposure in embryonic hearts.13, 31 Loss of these molecules alters regulated release and uptake of calcium in myocytes. Using H9C2 myoblasts, exposure to TCE at levels down to 10 ppb showed a 40% reduction in the calcium flux produced by vasopressin.12 This was followed by studies that showed a corresponding reduction in the rate of contraction in cardiomyocytes from exposed embryos and a reduction of shear stress markers in exposed hearts.9 Though other pathways of significance in the developing heart are altered by TCE exposure6,34, the effects of TCE on calcium homeostasis may be sufficient for teratogenesis due to altered hemodynamics.30
Studies in several laboratories and with different species and models found that low dose exposures of TCE produced greater effects on morphology and gene expression than at elevated exposures.8.9.33.34 This suggests that the biphasic response is evolutionarily conserved. At 10ppb TCE, H2C9 cells show a larger reduction calcium homeostasis that at 100 ppb or 10 ppm exposures12. We first speculated that low dose TCE exposure might be more toxic because phase I and II enzymes might not be induced at lower exposure levels. Higher exposures could be less toxic through induction of metabolizing enzymes. However, an examination of cytochrome p450 expression in the early chick embryo revealed that cytochromes were exclusively expressed in the embryonic heart during a sensitive window to TCE exposure. Surprisingly, induction of several cytochromes was greater at 8–10ppb than at 800 ppb.15 The data confirmed the non-monotonic dose response by another sensitive measure of gene transcription but did not explain the basis for this effect.
Microarray analysis of chick embryo hearts exposed to 8ppb TCE injected in ovo showed changes in approximately 4000 genes. The data were examined against a chick interactome database to explore linkage.16 Interactome analysis of the dataset found that the highest linkage was to the transcription factor, HNF4a.16 Though HNF4a, itself, was not significantly perturbed, it was linked to 75% of the nodes (genes). An examination of HNF4a expression and its most highly linked targets in the developing chick embryo showed that HNF4a was transiently expressed in the heart during the developmental window that coincides with sensitivity to TCE exposure.10,33 A nonmonotonic dose response was observed in expression of TCE-sensitive markers in the chick along with a functional effect on cardiac contraction.10
Though an established model of heart development, the chick is not a conventional toxicology model and this was raised in the controversy related to TCE.35 Here, we investigated selected genes identified in the TCE interactome as well as identified targets of HNF4a.11,16 A substantial overlap between TCE-sensitive and HNF4a target genes is consistently observed, suggesting that HNF4a is a central target of gene transcription in the heart. A similar examination of TCE effects on larval zebrafish hearts exposed to 10 ppb TCE was recently published.32 They identified 70 genes with significantly altered transcription. Though they focused on the effects of TCE of the FAK signaling pathway, we observe that human homologues of 53 of the 70 genes are targets of HNF4a as judged by the presence of 4 or more HNF4a binding motifs in their promoter region.20
HNF4a is a transcription factor that is abundantly found in adult liver and pancreatic islets, where almost 40% and 11% of genes, respectively, have an HNF4a response element.36 HNF4a has not been widely identified in the heart. The Mouse Genome and the Mouse Gene Expression databases (www.informatics.jax.org) show no expression in E14 or older fetal or adult hearts although an HNF4a reporter shows prominent expression in the E13.5 heart.37 In addition to microarray data, HNF4a was confirmed in mouse (E11) and stage 15–22 chick embryonic heart tissue by PCR analysis, western blot and immunostaining.10,16 HNF4a is transiently expressed during development of human IPS cells into cardiomyocytes.17 The impression is that HNF4a expression in the heart is limited to stages prior to embryonic liver development when the heart is developing and cytochrome p450 enzymes are expressed. As the HNF4a null mouse is embryonic lethal by E10.5 with a loss of mesoderm and increased apoptosis in the primitive streak, no direct role for HNF4a in heart development has been explored.39
HNF4a is a transcription factor that binds fatty acids, primarily linoleic acid, as its natural ligand.39 HNF4a is linked to regulation of cytochrome p450 expression, hepatic differentiation, hepatic cancer and maturity onset of diabetes.40–42 By interaction with insulin regulation, Bi6015 and Benfluorex were identified as having antagonist or agonist activity, respectively, towards HNF4a. Our data suggest that both Bi6015 and Benfluorex act similarly and as agonists or antagonists depending on concentration. HepG2 cells treated with 5μM Benfluorex showed inhibition of all marker transcripts while 5μM Bi6015 showed both agonist and antagonist activity. Since Benfluorex inhibits marker transcription acutely in vitro and is stimulatory when provided in drinking water, the difference could be due to chronic exposure or to a differing final concentration.
The data here support the idea that HNF4a is an early or proximal target of TCE toxicity. Low dose (8–10ppb) TCE exposure, via maternal drinking water, perturbs the transcription of more than 1000 genes in mouse while injection in ovo perturbs more than 4000 in the chick.11,16 Gene ontological analysis identifies a wide variety of pathways and processes including stress proteins, nucleic acid repair as well as calcium homeostasis, differentiation and cell migration.11,13 The diversity of effects is complex and while we focused on a calcium homeostatic process in relation to cardiac function it is clear that additional mechanisms are operating. The present data confirm TCE-sensitivity of targets such as SepW1, Zmat2 as well as established targets of HNF4a including INSR and HBEGF. SEPW1 was previously identified in chick heart microarrays as the most strongly down-regulated transcript after TCE exposure (16 and unpublished data). Benfluorex and Bi6015 have complete overlap with TCE- sensitive genes in perturbing transcription although TCE appears to be consistently an antagonist while Bi6015 and Benfluorex show both agonist and antagonist activity. Importantly, the most highly linked genes in the TCE interactome are linked to HNF4a by sensitivity to HNF4a-specific reagents and by identification of HNF4a response elements in their promoters.20 While Benfluorex and Bi6015 may have additional effects other than acting on HNF4a, the similarity of their effects produced and the overlap with TCE targets is consistent with HNF4a as a target of TCE exposure. This view is supported by the data showing that both Benfluorex and TCE directly perturb HNF4a-mediated transcription in HepG2 cells. Strikingly, TCE exposure produced a non-monotonic dose response in HNF4a reporter transcription.10
In published chick embryo experiments, TCE and Benfluorex show a similar, concentration dependent, shift between inhibition and induction of marker expression.10 Data here show that TCE is inhibitory and that both Benfluorex and BI6015 are stimulatory of marker expression. As the effect on expression is concentration dependent and higher concentrations are stimulatory in the chick, we speculate that the persistence of exposure in this model is important. Our data reinforce the observation that 8–10ppb are more toxic than 100–800 ppb.12,15 Exposure in the chick embryo model was for one day with a single injection of either TCE or Benfluorex. In thie present study, pregnant mice were exposed for twelve days, with consistent drinking water exposure. Due to its volatility and potential metabolism, we suggest that the concentration of TCE in the tissues does not accumulate to sufficient quantities to induce gene expression. In contrast, the non-volatile reagents, Benfluorex and BI6015, may accumulate over the 12 day exposure to reach a concentration that is inductive of HNF4a transcription rather than inhibitory. Alternatively, Benfluorex or Bi6015 may interact differently with mammalian HNF4a or its endogenous endogenous ligand, linoleic acid to alter the activity of the HNF4a transcription complex.39
During the window of TCE toxicity in the heart, HNF4a both regulates expression of cytochromes and mediates calcium homeostasis.41,43 A mis-regulated cascade of gene transcription from HNF4a in the heart could lead to a variety of altered pathways involving TCE metabolism and altered calcium homeostasis with impaired contraction. Impaired contraction cardiac output is sufficient to produce heart defects.30 Concomitant with liver development and the loss of HNF4a expression in the heart during embryonic development, metabolism of TCE in the embryo would shift from the heart to the liver and the window of cardiac sensitivity to TCE would close. Though TCE may alter regulation of HNF4a in other tissues, misregulation of this molecule may not lead to permanent defects in differentiation as seen in the developing heart.
In summary, a body of literature shows that TCE can alter cardiac differentiation in several animal models consistent with reports in exposed human populations. Controversy exists due to an inconsistency in effects but may be entirely due to the nonmonotonic-like dose curve. The data here show that exposure in embryonic mouse hearts, to reported agonists or antagonists of HNF4a produces a perturbation of marker expression. Markers sensitive to HNF4a perturbation are also sensitive to TCE. We also show that exposure of HepG2 cells (expressing HNF4a) to either Benfluorex or Bi6015 similarly inhibits the same markers. Together these data support both a previous bioinformatic analysis identifying HNF4a as an early or proximal target of TCE exposure and studies in the chick embryo showing the temporal expression of HNF4a during the window of sensitivity to TCE. Importantly, the data describe HNF4a as a target of TCE exposure in the mammalian embryo and a novel component in the formation of cardiac defects. This appears to be a novel AOP unrelated to a TCE-mediated effect on valve and septal formation but may be upstream of the altered gene transcription associated with defects in calcium homeostasis. As responses in the chick and mouse embryo models are similar, maternal metabolism and placental transfer do not appear to be significant barriers to cardiac teratogenicity in mammals although it remains possible that placental metabolism is involved in the final exposure levels.44
Acknowledgements
We thank Clark Lantz, Richard Vaillancourt and Nathan Cherrington for useful comments and discussions during the conduct of this study. We thank Dean Billheimer and Elmira Torabzadeh for their assistance with the statistics.
Funding Information
This work was supported by the National Institute for Environmental Health Sciences of the National Institutes of Health through the Southwest Environmental Health Sciences Center at the University of Arizona [P30 ES006694].
Footnotes
Conflicts of Interest
There are no conflicts of interest to declare.
References
- 1.Chiu WA, Caldwell JC, Keshava N, and Scott CS, Key scientific issues in the health risk assessment of trichloroethylene. Environmental health perspectives 2006, 114, 1445–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldberg SJ, Lebowitz MD, Graver EJ, and Hicks S, An association of human congenital cardiac malformations and drinking water contaminants. Journal of the American College of Cardiology, 1990,16, 155–64. [DOI] [PubMed] [Google Scholar]
- 3.Bove F, Shim Y, and Zeitz P, Drinking water contaminants and adverse pregnancy outcomes: A review. Environmental Health Perspectives, 2002. 110, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forand SP, Lewis-Michl EL, and Gomez MI, Adverse Birth Outcomes and Maternal Exposure to Trichloroethylene and Tetrachloroethylene through Soil Vapor Intrusion in New York State. Environmental Health Perspectives, 2012, 120, 616–621, Doi 10.1289/Ehp.1103884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yauck JS, Malloy ME, Blair K, Simpson PM and McCarver DG, Proximity of residence to trichloroethylene-emitting sites and increased risk of offspring congenital heart defects among older women. Birth Defects Research Part a-Clinical and Molecular Teratology, 2004, 70, 808–814, Doi 10.1002/Bdra.20060. [DOI] [PubMed] [Google Scholar]
- 6.Boyer A, Finch W and Runyan R, Trichloroethylene inhibits development of embryonic heart valve precursors in vitro. Toxicological Sciences 2000, 53,109–117, Doi 10.1093/toxsci/53.1.109. [DOI] [PubMed] [Google Scholar]
- 7.Drake VJ, Koprowski SL, Hu N, Smith SM, and Lough J, Cardiogenic effects of trichloroethylene and trichloroacetic acid following exposure during heart specification of avian development. Toxicological Sciences, 2006, 94,153–162, 10.1093/toxsci/kfl083. [DOI] [PubMed] [Google Scholar]
- 8.Mishima N, N., Hoffman S, Hill EG and Krug EL, Chick embryos exposed to trichloroethylene in an ex ovo culture model show selective defects in early endocardial cushion tissue formation. Birth Defects Research Part a-Clinical and Molecular Teratology, 2006, 76, 517–527, 10.100 [DOI] [PubMed] [Google Scholar]
- 9.Makwana O, King NM, Ahles L, Selmin O, Granzier HL, and Runyan RB, Exposure to low-dose trichloroethylene alters shear stress gene expression and function in the developing chick heart. Cardiovascular toxicology 2010, 10, 100–7, 10.1007/s12012-010-9066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris AP, Ismail KA, Nunez M, Martopullo I, Lencinas A, Selmin OI and Runyan RB, Trichloroethylene perturbs HNF4a expression and activity in the developing chick heart. Toxicol Lett. 2018, 285, 113–120. doi: 10.1016/j.toxlet.2017.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caldwell PT, Manziello A, Howard J, Palbykin B, Runyan RB, and Selmin O, Gene Expression Profiling in the Fetal Cardiac Tissue after Folate and Low-Dose Trichloroethylene Exposure. Birth Defects Research Part a-Clinical and Molecular Teratology, 2010. 88, 111–127, Doi 10.1002/Bdra.20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caldwell PT, Thorne PA, Johnson PD, Boitano S, Runyan RB and Selmin O, Trichloroethylene disrupts cardiac gene expression and calcium homeostasis in rat myocytes. Toxicological Sciences 2008. 104, 135–143, 10.1093/toxsci/kfn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collier JM, Selmin O, Johnson PD and Runyan RB, Trichloroethylene effects on gene expression during cardiac development. Birth Defects Research Part A-Clinical and Molecular Teratology, 2003, 67, 488–495. [DOI] [PubMed] [Google Scholar]
- 14.Selmin O, Thorne P, Caldwell P, Johnson P and Runyan R, Effects of trichloroethylene and its metabolite trichloroacetic acid on the expression of vimentin in the H9c2 cell line. Cell Biol Toxicol, 2005, 21, 83–95. [DOI] [PubMed] [Google Scholar]
- 15.Makwana O, Ahles L, Lencinas A, Selmin OI and Runyan RB. Low-Dose Trichloroethylene Alters Cytochrome P450–2C Subfamily Expression in the Developing Chick Heart. Cardiovascular Toxicology 2013, 13, 77–84, 10.1007/s12012-012-9180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selmin O, O., Makwana O, and Runyan R, Environmental Sensitivity to Trichloroethylene (TCE) in the Developing Heart In Trichloroethylene: Toxicity and Health Risks (Gilbert KM, and Blossom SJ, Eds.), pp. 153–169. Springer; London: Doi: 10.1007/978-1-4471-6311-4_8,2003 [DOI] [Google Scholar]
- 17.Zeidler S, Meckbach C, Tacke R, Raad FS, Roa A, Uchida S, Zimmermann WH, Wingender E and Gültas M, Computational Detection of Stage-Specific Transcription Factor Clusters during Heart Development. Frontiers in Genetics, 2016, 7, 33 Doi: 10.3389/fgene.2016.00033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiselyuk A, Lee SH, Farber-Katz S, Zhang M, Athavankar S, Cohen T, Pinkerton AB, Ye M, Bushway P, Richardson AD, Hostetler HA, Rodriguez-Lee M, Huang L, Spangler B, Smith L, Higginbotham J, Cashman J, Freeze H, Itkin-Ansari P, Dawson MI, Schroeder F, Cang Y, Mercola M and Levine F, HNF4alpha antagonists discovered by a high-throughput screen for modulators of the human insulin promoter. Chem Biol, 2012, 19, 806–18, 10.1016/j.chembiol.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SH, Athavankar S, Cohen T, Piran R, Kiselyuk A and Levine F, Identification of Alverine and Benfluorex as HNF4 alpha Activators. ACS Chem Biol, 2013, 8, 1730–1736, Doi. 10.1021/cb4000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolotin E, Liao H, Ta TC, Yang C, Hwang-Verslues W, Evans JR, Jiang T and Sladek FM, Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays. Hepatology, 2010, 51, 642–53, 10.1002/hep.23357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dikiy A, Novoselov SV, Fomenko DE, Sengupta A, Carlson BA, Cerny RL, Ginalski K, Grishin NV, Hatfield DL and Gladyshev VN,. SelT, SelW, SelH, and Rdx12: Genomics and molecular insights into the functions of selenoproteins of a novel thioredoxin-like family. Biochemistry 2007, 46, 6871–6882, Doi. 10.1021/bi602462q. [DOI] [PubMed] [Google Scholar]
- 22.Bukowski J, Critical review of the epidemiologic literature regarding the association between congenital heart defects and exposure to trichloroethylene. Critical reviews in toxicology, 2014, 44, 581–9, Doi. 10.3109/10408444.2014.910755. [DOI] [PubMed] [Google Scholar]
- 23.Makris SL, Scott CS, Fox J, Knudsen TB, K Hotchkiss A, Arzuaga X, Euling SY, Powers CM, Jinot J, Hogan KA, Abbott BD, Hunter ES 3rd and Narotsky MG, A systematic evaluation of the potential effects of trichloroethylene exposure on cardiac development. Reprod. Toxicol, 2016, 65, 321–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeSesso JM and Risotto SP, Review of TCE cardiac defects data by Makris et al. is not systematic. Reprod. Toxicol, 2017, 71, 134 Doi. 10.1016/j.reprotox.2017.05.012 [DOI] [PubMed] [Google Scholar]
- 25.DeSesso JM, Coder PS, York RG, Budinsky RA, Pottenger LH, Shiladitya S, Lucarell JM, Bevan C and Bus JS, Trichloroethylene in drinking water throughout gestation did not produce congenital heart defects in Sprague Dawley rats. Birth Defects Res, 2019, 111, 1217–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Runyan RB, R.B., Selmin OI, Smith SM and Freeman JL, J.L., Letter to the Editor. Birth Defects Res, 2019, 111, 1234–1236. [DOI] [PubMed] [Google Scholar]
- 27.Dawson BV, Johnson PD, Goldberg SJ, and Ulreich JB, Cardiac teratogenesis of trichloroethylene and dichloroethylene in a mammalian model. Journal of the American College of Cardiology, 1990, 16, 1304–1309. [DOI] [PubMed] [Google Scholar]
- 28.Johnson P, Goldberg S, Mays M and Dawson B Threshold of trichloroethylene contamination in maternal drinking waters affecting fetal heart development in the rat. Environ. Health Perspect, 2003, 111, 289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hogers B, DeRuiter MC, GittenbergerdeGroot AC and Poelmann RE, Unilateral vitelline vein ligation alters intracardiac blood flow patterns and morphogenesis in the chick embryo. Circulation Research, 1997, 80, 473–481. [DOI] [PubMed] [Google Scholar]
- 30.Selmin OI, Thorne PA, Caldwell PT and Taylor MR, Trichloroethylene and trichloroacetic acid regulate calcium signaling pathways in murine embryonal carcinoma cells p19. Cardiovascular Toxicology, 2008, 8, 47–56. Doi. 10.1007/s12012-008-9014-2. [DOI] [PubMed] [Google Scholar]
- 31.Wirbisky SE, Damayanti NP, Mahapatra CT, Sepulveda MS, Irudayaraj J and Freeman JL, Mitochondrial Dysfunction, Disruption of F-Actin Polymerization, and Transcriptomic Alterations in Zebrafish Larvae Exposed to Trichloroethylene. Chem Res Toxicol, 2016, 29, 169–179. Doi. 10.1021/acs.chemrestox.5b00402. [DOI] [PubMed] [Google Scholar]
- 32.Drake VJ, Koprowski SL, Lough J, Hu N and Smith SM, Trichloroethylene exposure during cardiac valvuloseptal morphogenesis alters cushion formation and cardiac hemodynamics in the avian embryo. Environmental Health Perspectives, 2006, 114, 842–847, Doi. 10.1289/ehp.8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rufer ES, Hacker TA, Flentke GR, Drake VJ, Brody MJ, Lough J and Smith SM, Altered cardiac function and ventricular septal defect in avian embryos exposed to low-dose trichloroethylene. Toxicol Sci, 2010,113, 444–52, Doi. 10.1093/toxsci/kfp269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hardin BD, Kelman BJ and Brent RL, Trichloroethylene and dichloroethylene: a critical review of teratogenicity. Birth defects research. Part A, Clinical and molecular teratology, 2005, 73, 931–55. Doi. 10.1002/bdra.20192. [DOI] [PubMed] [Google Scholar]
- 35.Chandra V, V., Huang P, Potluri N, Wu D, Kim Y and Rastinejad F, Multidomain integration in the structure of the HNF-4alpha nuclear receptor complex. Nature, 2013, 495, 394–8, Doi. 10.1038/nature11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vincent SD and Robertson EJ, Targeted insertion of an IRES Cre into the Hnf4 alpha locus: Cre-mediated recombination in the liver, kidney, and gut epithelium. Genesis, 2004, 39, 206–211, Doi 10.1002/Gene.20047. [DOI] [PubMed] [Google Scholar]
- 37.Chen WS, Manova K, Weinstein DC, Duncan SA, Plump AS, Prezioso VR, Bachvarova RF and Darnell JE Jr., Disruption of the HNF-4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes Dev, 1994, 8, 2466–77. [DOI] [PubMed] [Google Scholar]
- 38.Yuan XH, Ta TC, Lin M, Evans JR, Dong YC, Bolotin E, Sherman MA, Forman BM and Sladek FM, Identification of an Endogenous Ligand Bound to a Native Orphan Nuclear Receptor. Plos One, 2009, 4, ARTN e5609, Doi. 10.1371/journal.pone.0005609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hwang-Verslues WW and Sladek FM, HNF4a-role in drug metabolism and potential drug target?, Curr. Opin. Pharmacol, 2010, 10, 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayhurst GP, Lee YH, Lambert G, Ward JM, and Gonzalez FJ, Hepatocyte nuclear factor 4 alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Molecular and Cellular Biology, 2001, 21(4), 1393–1403, Doi 10.1128/Mcb.21.4.1393-1403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jover R, Moya M, and Gomez-Lechon MJ, Transcriptional regulation of cytochrome p450 genes by the nuclear receptor hepatocyte nuclear factor 4-alpha. Curr Drug Metab, 2009, 10(5), 508–19. [DOI] [PubMed] [Google Scholar]
- 42.Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA, Gifford DK, Fraenkel E, Bell GI, and Young RA, Control of pancreas and liver gene expression by HNF transcription factors. Science, 2004, 303(5662), 1378–81, 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moore BD, Jin RU, Lo H, Jung M, Wang H, Battle MA, Wollheim CB, Urano F, & Mills JC, Transcriptional Regulation of X-Box-binding Protein One (XBP1) by Hepatocyte Nuclear Factor 4α (HNF4A) Is Vital to Beta-cell Function. The Journal Of Biological Chemistry, 2016, 291(12), 6146–6157. 10.1074/jbc.M115.685750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elkin ER, Harris SM,Su AL,Lash LH and Loch-Caruso R, Placenta as a target of trichloroethylene toxicity. Environmental Science: Processes and Impacts, 2020, 2020 Doi: 10.1039/C9EM00537D [DOI] [PMC free article] [PubMed] [Google Scholar]