

A chemical protein-DNA conjugation approach enhances precise genome editing in human cells and mouse zygotes.
Abstract
Site-specific chemical conjugation of proteins can enhance their therapeutic and diagnostic utility but has seldom been applied to CRISPR-Cas9, which is a rapidly growing field with great therapeutic potential. The low efficiency of homology-directed repair remains a major hurdle in CRISPR-Cas9–mediated precise genome editing, which is limited by low concentration of donor DNA template at the cleavage site. In this study, we have developed methodology to site-specifically conjugate oligonucleotides to recombinant Cas9 protein containing a genetically encoded noncanonical amino acid with orthogonal chemical reactivity. The Cas9-oligonucleotide conjugates recruited an unmodified donor DNA template to the target site through base pairing, markedly increasing homology-directed repair efficiency in both human cell culture and mouse zygotes. These chemically modified Cas9 mutants provide an additional tool, one that is complementary to chemically modified nucleic acids, for improving the utility of CRISPR-Cas9–based genome-editing systems.
INTRODUCTION
Chemical conjugation of proteins has emerged as a powerful tool for building protein constructs with potential clinical utility (1, 2), such as antibody-radionuclide conjugates for diagnosis (3, 4), antibody-drug conjugates for cancer therapy (5, 6), and long half-life therapeutic proteins (7). Conjugation was traditionally achieved by random chemical modifications of native amino acid side chains, but the development of various site-specific modification techniques (8) now allows for the generation of homogenous protein conjugates, which are often required for biomedical-related applications to ensure quality control and the consistency of activities (9).
Genome-editing proteins, such as zinc-finger nucleases (10), TALEN (transcription activator–like effector nucleases) (10, 11), and newly developed CRISPR-Cas9 nucleases (12), show great potential for gene therapy. However, these proteins have rarely been chemically modified. The Cas9 protein, which is derived from type II CRISPR bacterial immune systems (13), is becoming a prominent tool for genome editing in diverse organisms (14). The components of CRISPR-Cas9 systems are usually introduced into target cells by means of viruses or plasmid-based approaches. Recently, a new delivery method was developed that uses a recombinantly expressed Cas9 protein and a transcribed or, preferably, chemically synthesized guide RNA (gRNA), which are preassembled to form a ribonucleoprotein (RNP) complex that is delivered directly into target cells to perform the DNA cleavage (15–18).The RNP delivery method is now considered to be the safest and most accurate for therapeutic applications (16, 19–21). In addition, the fact that there is no cleavage delay makes this method superior, particularly for time-dependent editing, such as in dividing zygotes (22, 23). The RNP-delivery method provides the opportunity to improve performance by chemically modifying the gRNA and, potentially, the Cas9 proteins, as demonstrated in this work. Recently, considerable effort has been made to chemically modify gRNA to enhance its stability and reduce off-target effects (24–28). However, there have been only a few studies aimed at chemically modifying Cas9 proteins to make CRISPR a better genome-editing tool (29, 30). We wondered whether a CRISPR-Cas9 system could be improved by site-specifically introducing a bio-orthogonal chemical handle to a recombinantly expressed Cas9 protein to allow bioconjugation of other functional components.
CRISPR-mediated precise genome editing usually relies on homology-directed repair (HDR), which is inefficient because it requires the presence of a donor DNA template at the site of cleavage by Cas9. It has been demonstrated that a key limiting step for HDR may be the local concentration of donor DNA, preferably a single-stranded oligodeoxynucleotide (ssODN) to reduce integration risk, at the genome cleavage site (31). Very recently, several methods for recruiting the ssODN to the Cas9-gRNA cleavage complex to enhance HDR have been reported; these methods take advantage of high-affinity binding between biotin-streptavidin interaction and covalent linkage mediated by fusion domains and substrate interaction (26, 31–35). However, there are several limitations: (i) The fusion domain may influence protein expression and intracellular delivery efficiency, and the linker is susceptible to protease cleavage; (ii) terminally modified long ssODNs [over 100 nucleotides (nt)] are difficult to obtain from commercially sources; and (iii) the ssODN attachment sites are limited in number, usually occur at the N or C terminus of the Cas9 protein, and may not be optimal for the HDR reaction.
Here, we overcame these problems by generating chemically modified Cas9 mutants that were compatible with modified or unmodified oligonucleotides and improved the efficiency of HDR-mediated genome editing. Specifically, we used genetic code–expansion technology to chemically modify a Cas9 protein with an azide-containing noncanonical amino acid (ncAA) at selected positions. These modifications allowed subsequent conjugation of dibenzylcyclooctyne (DBCO)–containing donor ssODN or DNA adaptor by means of strain-promoted alkyne-azide cycloaddition, thereby recruiting a modified or even an unmodified donor DNA template to the cleavage complex. We show that site-specific Cas9-oligonucletide conjugates were not only necessary for the preparation of homogenous Cas9 conjugates but also important for placing the donor DNA near the cleavage site for the HDR reaction. These Cas9 conjugates, especially those prepared with an adaptor oligonucleotide, provide a universal platform for recruiting unmodified ssODNs (which are commercially available in high purity at low cost) to the RNP complex by base pairing, which substantially increases HDR efficiency. To demonstrate the utility of this approach, we applied it to mouse zygotes and showed a marked improvement in HDR-mediated precise genome editing, which demonstrates the great potential of this approach for research and therapeutic applications.
RESULTS
Chemical modification of Cas9 with an azide-containing ncAA retained in vitro and in cells activities
To prepare chemically modified Cas9 mutants, we first synthesized an ncAA, 4-(2-azidoethoxy)-l-phenylalanine (AeF), which would allow site-specific introduction of an azido group by means of an expanded genetic code (36). On the basis of the structure of Streptococcus pyogenes Cas9 (SpyCas9), a total of 11 amino acids were selected as AeF mutation sites (Fig. 1A): K3, D39, H41, and H754 are located near the N terminus; K1151, K1153, D1361, and G1367 are near the C terminus; and L833, K929, and L1004 are close to the region that binds the target DNA. The codons of these residues were mutated to the TAG codon, and the mutant proteins were expressed in Escherichia coli BL21 cells with a previously developed polyspecific Methanococcus jannaschii tyrosyl-tRNA synthetase (MjPolyRS)/tRNACUA pair (37). The expression yields of the mutants are listed in table S1. Mutations at K3, D39, and L833 resulted either in no expression or in low-quality (unstable or misfolded) proteins. Mutations at H41, H754, K929, and D1361 markedly decreased expression yields relative to that of wild-type (WT) Cas9. Among the 11 mutants, K1151-AeF and G1367-AeF showed the highest expression yields and quality (Fig. 1B) and were therefore selected for further studies. The fidelity of AeF incorporation was demonstrated by tandem mass spectrometry (fig. S1).
Fig. 1. Assessment of the activity of chemically modified Cas9 mutants in vitro and in cells.
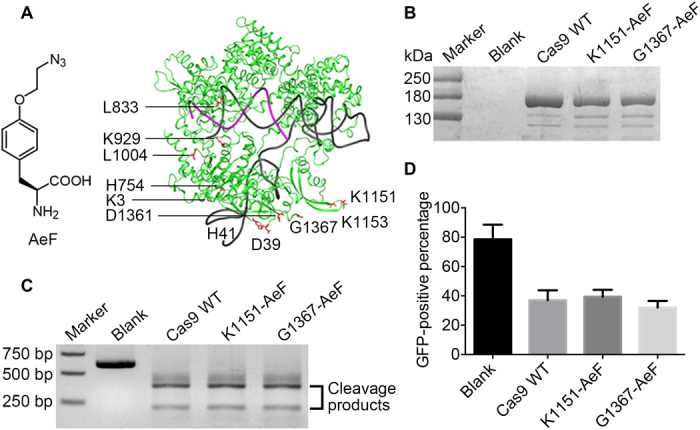
(A) Chemical structure of ncAA AeF and crystal structure of SpyCas9 with gRNA (Protein Data Bank code: 4OO8). The residues selected for AeF mutagenesis are labeled in red. (B) Purified Cas9 mutants analyzed by SDS–polyacrylamide gel electrophoresis. (C) Nuclease activity of WT Cas9 and Cas9 mutants. Double-stranded DNA was digested in the presence of a preassembled ribonucleoprotein complex of Cas9 and gRNA for 1 hour at 37°C. (D) Human embryonic kidney (HEK) 293–Teton–enhanced green fluorescent protein (EGFP) cells were transfected with 3 pmol of AeF-Cas9 or WT Cas9 RNP using RNAiMAX. Genome-editing activity was quantified by flow cytometry 2 days after transfection. Error bars represent the SD in triplicates.
First, we assessed the DNA-cleavage activities of these Cas9 mutants in vitro. A 600–base pair (bp) target DNA template was incubated with Cas9 mutants preassembled with gRNA at 37°C, and the cleavage products were analyzed on an agarose gel (Fig. 1C). Both the K1151-AeF mutant and the G1367-AeF mutant showed cleavage activity comparable to that of the WT protein. To validate their activities in mammalian cells, we constructed a human embryonic kidney (HEK) 293–enhanced green fluorescent protein (EGFP)–Teton cell line, which has an EGFP cassette under the control of a tetracycline-inducible promoter, and we used these cells to evaluate the gene disruption efficiency of the mutants. Both mutants showed an EGFP-quenching percentage of 50 to 60% (as indicated by flow cytometry analysis), which was identical to that of the WT Cas9 protein (Fig. 1D). These results demonstrate that chemical modification of Cas9 at the two selected positions did not affect cleavage activity either in vitro or in cells.
Covalent ssODN-Cas9 conjugates enhanced Cas9 RNP-mediated HDR efficiency
It has been demonstrated that increasing the local concentration of donor DNA near the cleavage complex may increase HDR efficiency (33). To effectively increase the concentration of donor DNA around the RNP complex, we first attempted to covalently attach a DBCO-modified donor ssODN to an azido-Cas9 protein by means of a strain-promoted alkyne-azide cycloaddition reaction (Fig. 2A). For gene knock-in, we designed a 186-nt ssODN donor that contained a 36-nt insertion encoding for a C-terminal HiBiT reporter tag (38) and two 75-nt homology arms corresponding to the flanking regions of the GAPDH gene (all DNA and primer sequences used in this work are listed in table S2). A successful HDR reaction would result in an in-frame insertion of the HiBiT tag gene at the end of the endogenous GAPDH gene. The HiBiT tag is an 11–amino acid split-nanoluciferase peptide tag that allows for sensitive quantification of HDR efficiency. The DBCO-modified ssODN was prepared by treating the corresponding NH2-ssODN with excess DBCO-N-hydroxysuccinimidyl ester and purified by high-performance liquid chromatography.
Fig. 2. Cas9-ssODN conjugation strategy to increase Cas9-mediated HDR efficiency.
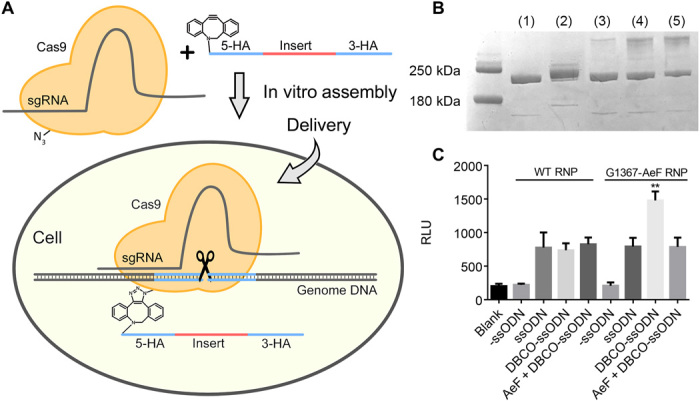
(A) Schematic representation of Cas9-ssODN conjugate formation and rationale for improving HDR efficiency. Azido-modified Cas9 underwent bio-orthogonal strain-promoted alkyne-azide cycloaddition with DBCO-modified ssODN, and the resulting conjugate was delivered into cells, which effectively increased the local concentration of the donor ssODN near the target region. (B) Cas9 G1367-AeF (500 ng) was allowed to react with the DBCO-adaptor and DBCO-ssODN at various molar ratios, and the conjugation efficiency was evaluated by SDS–polyacrylamide gel electrophoresis: (1) Cas9 G1367; (2) Cas9 G1367-AeF:DBCO-adaptor, 1:1; (3) Cas9 G1367-AeF:DBCO-ssODN, 1:1; (4) Cas9 G1367-AeF:DBCO-ssODN, 1:3; (5) Cas9 G1367-AeF:DBCO-ssODN, 1:5. (C) The RNP-ssODN complex (3 pmol) was transfected into HEK293 cells, and a HiBiT assay was conducted 48 hours after transfection. Data are means (n = 3), and error bars indicate SDs. Significance was calculated by means of a two-tailed Student’s t test. **P < 0.01. sgRNA, single guide RNA; HA, homology arms; RLU, relative luminescence unit.
Next, to investigate the conjugation reaction, we carried out in vitro reactions of the G1367-AeF mutant and the 186-nt DBCO-ssODN at various stoichiometries (Fig. 2B). Reaction of equimolar protein and ssODN resulted in less than 10% conjugation and increasing the amount of ssODN to five equivalents could improve the conjugation efficiency to near 50%. In contrast, we found that the reaction between the G1367-AeF mutant and DBCO-modified ssODN under 100 nt was nearly quantitative (fig. S2). These results suggest that direct conjugation of the ssODN to the Cas9 protein may have been impeded by the high molecular weight of the DBCO-oligonucleotide. Nevertheless, we proceeded to determine whether direct conjugation of the donor DNA to the Cas9 nuclease could indeed enhance HDR efficiency. First, we show that the Cas9-ssDON complex does not interfere with the Cas9 cleavage activity both in vitro and in cell (fig. S3). Next, HEK293 cells were cotransfected with the WT RNP or G1367-AeF RNP assembling with either a DBCO or a NH2-modified ssODN, and the HDR efficiency was evaluated using HiBiT assay. As shown in Fig. 2C, cells cotransfected with the G1367-AeF RNP and DBCO-modified ssODN had a 1.6 times HDR increase as compared to other groups (Fig. 2C), indicating that covalent conjugation of ssODN donor DNA to Cas9 can enhance HDR efficiency.
Cas9-adaptor conjugate recruited an unmodified donor ssODN and increased HDR efficiency
Although we have shown that direct conjugation of a DBCO-modified ssODN to Cas9 mutants increased HDR efficiency, chemically modifying gRNA and ssODNs can be expensive, and the conjugation efficiency is compromised for longer ssODNs. In contrast, modification of the Cas9 protein alone would permit the development of a universal strategy for recruiting unmodified ssODNs, which are commercially available at low cost and high purity, for efficient HDR. Therefore, we attempted to create a Cas9-oligonucleotide conjugate bearing a short DNA adaptor attached to the azido-Cas9 mutants to recruit donor DNA to the cleavage complex by base pairing (Fig. 3A). First, we designed a 25-nt 3′-DBCO–modified adaptor oligonucleotide (CAAATTCGTTGTCATACCTAGAAGA), the sequence of which was reverse complementary to the 5′ homologous arm of GAPDH. To determine the best conjugation conditions, we incubated the DBCO-modified adaptor with the Cas9 G1367-AeF mutant under various reaction conditions. We found that incubation of a 1:1 mixture of the azido-Cas9 mutants and the DBCO-modified adaptor at 4°C resulted in nearly complete reaction in 3 hours (Fig. 3B).
Fig. 3. Cas9-adaptor conjugation strategy to increase Cas9-mediated HDR efficiency.
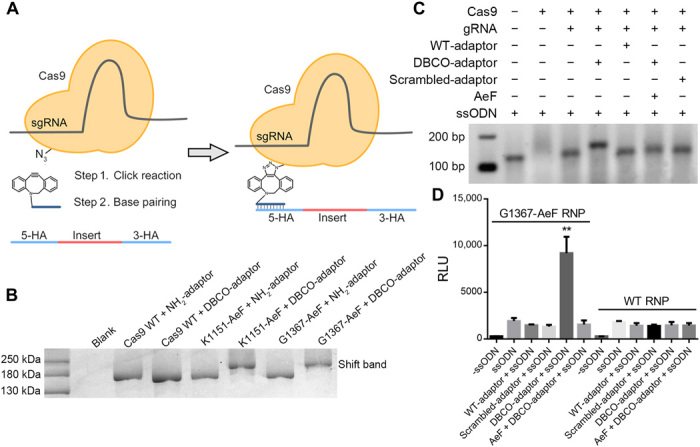
(A) Schematic representation of Cas9-adaptor conjugate formation and rationale for improving HDR efficiency. Azido-modified Cas9 was conjugated with a DBCO-modified DNA adaptor, which tethered the donor ssODN by base pairing. (B) SDS–polyacrylamide gel electrophoresis analysis showing complete reaction between equimolar azido-modified Cas9 (160 kDa) and the DBCO-modified adaptor (8 kDa). (C) Cas9 or Cas9-adatpor conjugate was incubated with ssODN at equal molar and analyzed by for electrophoretic mobility shift on a 2.5% agarose gel. (D) HiBiT assay to evaluate HDR mediated by Cas9-adaptor conjugates. Means ± SE, n = 3. **P < 0.01 by Student’s t test compared to RNP-adaptor-ssODN group.
An in vitro activity assay confirmed that the conjugates retained the DNA-cleavage activity of the Cas9 protein. To further explore the influence of the adaptor on the cleavage activity of the RNP in cells, we performed an EGFP reporter gene disruption assay as described above (fig. S4). A slight increase in gene-editing efficiency was observed for the adaptor-conjugated RNP complexes of both the K1151-AeF mutant and the G1367-AeF mutant. Increasing the overall negative charge of the RNP complex has been reported to increase chemical transfection efficiency (39). Therefore, the increase in efficiency that we observed may have been a result of better intracellular delivery due to conjugation of a negatively charged oligonucleotide. Together, our data support our hypothesis that conjugation of an adaptor oligonucleotide had little impact on Cas9 activity either in vitro or in cells.
Next, we determined whether the Cas9-adaptor conjugates could recruit a donor DNA template to promote HDR. First, base pairing between the adaptor oligonucleotide and the donor ssODN was evaluated. Specifically, an increasing amount of adaptor was mixed with the ssODN, and the assembled complexes were analyzed on an agarose gel (fig. S5). These experiments revealed that hybridization of the adaptor to the ssODN was quantitative at a 1:1 stoichiometry. Next, an equimolar amount of the ssODN was added to the preassembled G1367-AeF RNP-adaptor conjugate or WT RNP control group under the same conditions. A band shift can be observed only for the adaptor conjugated complex in an electrophoretic mobility shift assay (Fig. 3C). Subsequently, the entire complex was transfected into HEK293 cells for assessment of HDR efficiency by HiBiT luminescence assay. As shown in Fig. 3D, HDR efficiency was 10-fold enhanced for Cas9-adaptor conjugates. In contrast, a sequence-scrambled adaptor oligonucleotide had no influence in HDR efficiency. In addition, the WT RNP with either DBCO adaptor or scrambled adaptor showed no influence in HDR improvement. To validate the efficiency of HiBiT, integration was also confirmed and quantified with deep sequencing. The result showed that HDR efficiency notably increased compared with the unconjugated group, which is consistent with the HiBiT luminescence assay. It is noted that the chemical modification and conjugation increase the absolute HDR efficiency without influencing the HDR/non-homologous end joining ratio (fig. S6). These results suggest that HDR enhancement was due to the donor ssODN recruiting to the cleavage site by base pairing. To further investigate whether attaching more ssODN at the Cas9 protein could further increase the HDR efficiency, we prepared and assessed two AeF-modified Cas9 D576-AeF–G1367-AeF in cell with double-adaptor conjugates. A twofold HDR increase can be observed for double-mutant RNP conjugates as compared to single conjugate (fig. S7). These results confirm that the Cas9-oligonucleotide conjugates could act as a plug-and-play system to recruit unmodified, commercially available ssODNs and could therefore serve as a universal platform for highly efficient precise genome editing.
RNP conjugates and a ssODN-binding complex enhanced HDR efficiency in mouse zygotes
The generation of targeted animal mutants is a key technology in biomedical research and may have potential utility for gene therapy. To determine whether our chemical conjugation approach could be used for animal research, we delivered an RNP-adaptor-ssODN complex into mouse zygotes by microinjection and evaluated the efficiency of HDR-mediated genome editing. Specifically, we designed a 42-nt gene encoding a 14–amino acid V5-tag for insertion at the 3′ end of the SOX2 gene as a model system for evaluating HDR efficiency. A gRNA targeting a region before the stop codon of SOX2 and a 162-nt donor ssODN consisting of a 42-nt V5-tag coding sequence with a 60-nt homolog arm on each side were designed and chemically synthesized in high purity. Successful HDR would result in insertion of the V5-tag gene at the 3′ end of SOX2 (Fig. 4A). To recruit donor DNA to the target site, we designed a 25-nt DBCO-modified DNA with a sequence that was reverse complementary to the 5′ homologous arm of the SOX2 gene. On the basis of our previous results, we chose the Cas9 G1367-AeF mutant to assemble the RNP-adaptor-ssODN complex, which was then delivered into mouse zygotes by microinjection.
Fig. 4. Precise genome editing in mouse zygotes using chemically modified Cas9-adaptor conjugates.
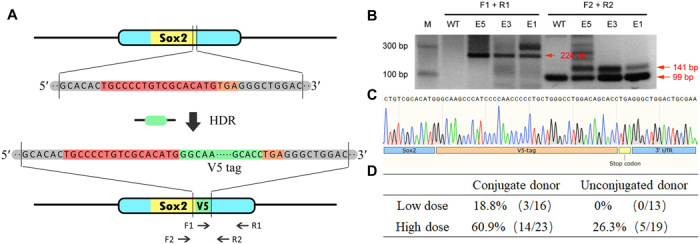
(A) Schematic representation of targeted SOX2 gene editing with insertion of a V5-tag. The gRNA sequence and stop codon are highlighted in red and orange, respectively. The V5 donor ssODN contains a V5 insertion sequence (highlighted in green) and a 60-nt homologous arm on each side. Polymerase chain reaction (PCR) primers (F1, R1, F2, and R2) used for genotyping are indicated by the arrows. (B) For validation of V5 gene insertion, V5-specific primer F1 was used with primer R1 to amplify a unique 224-bp fragment from the edited sequence. In addition, a 141-bp fragment containing a 42-bp V5-tag could be specifically detected in edited zygotes by using primers F2 and R2, whereas unedited zygotes gave a 99-bp amplified band. (C) DNA sequencing results confirmed integration of the V5-tag at the end of the SOX2 gene. 3′ UTR, 3′ untranslated region. (D) RNP-ssODN complexes were microinjected in mouse zygotes at doses of 200 and 600 ng, and HDR efficiency was determined by genotyping PCR.
Sixteen mouse embryos [embryonic day (E) 1 to E16] were microinjected with the preassembled RNP-adaptor-ssODN complex (200 ng), and 13 control group 1-13 (C1-C13) were treated with a mixture of unconjugated RNP and an ssODN under the same conditions. The resulting zygotes were cultured to the morula stage for genotyping analysis and sequencing. A 224-bp polymerase chain reaction (PCR) band was amplified with V5-specific primers (F1 and R1), which could be detected only in E1, E3, and E5. Confirming these results, further PCR analysis showed an extra band (141 bp) containing the 42-bp V5-tag gene amplified by primers F2 and R2 in the three knock-in embryos, whereas only a 99-bp band corresponding to the unmodified genome was amplified in the WT embryos (Fig. 4B). Last, the presence of the V5-tag at the 3′ end of SOX2 was detected by DNA sequencing, which confirmed knock-in at the desired location (Fig. 4C). In summary, 3 of 16 zygotes were successfully edited using a Cas9-adaptor conjugate paired with donor DNA, whereas the unconjugated control group failed to produce any knock-in mice. In addition, higher concentrations (600 ng) of RNP-adaptor-ssODN complex were microinjected into zygotes and analyzed, as described above. As shown in Fig. 4D and fig. S8, 14 of 23 embryos showed precise SOX2 editing, for an HDR efficiency of 60.9%. On the contrary, the HDR efficiency was only 26.3% (5 of 19) in the unconjugated group under the same conditions. Together, these results confirm that the chemically conjugated RNP-adaptor-ssODN complex could increase HDR efficiency in mouse zygotes.
DISCUSSION
The use of CRISPR-Cas9–mediated HDR for precise genome editing has great therapeutic potential. However, its utility is limited by the low efficiency of HDR, which requires the presence of a donor DNA template near the site of cleavage. Here, we described the preparation of site-specific chemical Cas9 mutants containing reactive ncAAs, which allows a direct oligonucleotide conjugation with a precise control of geometry and stoichiometry. We showed that these mutant proteins could tether modified or unmodified donor DNA either directly via chemical conjugation or indirectly via base pairing. In particular, with a Cas9-adaptor conjugate, modification of the donor ssODN was unnecessary, so it could be conveniently synthesized at low cost and in high purity from commercial sources. By means of our approach, the donor ssODN was effectively brought to the genome cleavage site, where it was available for the HDR reaction immediately after cleavage, therefore resulting in a notable improvement in HDR efficiency. We also demonstrated that our approach worked efficiently both in human cell–based assays and in mouse zygotes. In summary, we have developed a novel, effective platform based on chemical conjugation of Cas9 proteins to improve HDR efficiency; this platform can be used to construct animal models and may have clinical applications. These chemically modified Cas9 proteins can supplement and are compatible with chemically modified nucleic acids for improving the utility of CRISPR-Cas9–based genome editing. With the development of genetic code expansion technology (40), multiple ncAAs could be genetically encoded in Cas9 proteins. The geometry and stoichiometry of conjugation could be further control by the positions, numbers, and types of ncAAs of choice, and the resulting mutants could recruit more than two ssODN and might further improve the HDR efficiency, therefore extending the applicability of our method.
MATERIALS AND METHODS
General information
For all chemical synthesis, solvents and reagents were purchased from Sigma-Aldrich, TCI (Shanghai), or Bide Pharmatech and used directly without further purification. Analytical thin-layer chromatography was performed on 0.25-mm silica gel 60-F254. Visualization was carried out with ultraviolet light and vanillin or ninhydrin staining. All the chemical-modified primers (DBCO or phosphorylation-modified oligos) were purchased from General Biosystems (Anhui) Co. Ltd. Unmodified ssODN targeting SOX2 and 5-amine–modified ssODN targeting GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were purchased from Integrated DNA Technologies Inc. and labeled with DBCO in General Biosystems (Anhui) Co. Ltd.
Plasmid construction
All plasmids were constructed using Gibson assembly [New England Biolabs (NEB)]. Mutagenesis PCR was conducted using KOD-One PCR Master Mix (TOYOBO), followed by DpnI (NEB) digestion.
Expression and purification of SpyCas9
The pET28a vector containing SpyCas9 with hexa-histidine and 2xNLS was a gift from C. Gao’s lab at Institute of Genetics and Developmental Biology in Chinese Academy of Sciences. The plasmid was transformed into the E. coli BL21 (DE3) strain for protein production. Briefly, cells were grown at 37°C to optical density at 600 nm (OD600) of 0.6 to 0.8, 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Sigma-Aldrich) was then added, and culture temperature was lowered to 18°C. Cells were grown overnight and harvested by centrifugation at 6000 rpm at 4°C. The protein was purified first by Ni2+ affinity chromatography and then by cation exchange (HitrapTM SP HP, GE Healthcare), followed by size-exclusion chromatography (Superdex 200, GE Healthcare). Fractions containing Cas9 protein were collected and concentrated using 50K Amicon Ultra column (Millipore). Purity of Cas9 protein was analyzed by SDS–polyacrylamide gel electrophoresis. Catalytic activities of purified Cas9 were comparable to those of purchased standard SpyCas9 (NEB) under our experimental condition.
Expression and purification of azido-Cas9 mutants
E. coli BL21 cells transformed with pET28a-Cas9-mutants and pUltra-MjPolyRS were cultivated in 2YT medium. Cells were grown at 37°C, and 1 mM AeF was added at OD600 0.3. The cells were continuing until the OD600 reached 0.6 to 0.8, and then 0.2 mM IPTG (Sigma-Aldrich) was added. The culture temperature was lowered to 18°C for overnight culture. The purification procedure is the same as the WT protein.
In vitro transcription of gRNA
DNA templates for gRNA transcription were obtained by PCR from a vector containing a T7 promoter and a gRNA scaffold. The amplified PCR product was extracted using gel extraction kit (OMEGA) and severed as DNA template for gRNA transcription reaction. Transcription reactions (20 μl) were conducted in 1× reaction buffer along with 2 μl of adenosine 5′-triphosphate, guanosine 5′-triphosphate, cytidine 5′-triphosphate, and uridine 5′-triphosphate (100 mM each), 2 μl of T7 RNA polymerase (HiScribe T7 High Yield RNA Synthesis Kit, NEB), and 1 μg of DNA template. Reactions proceeded at 37°C for 4 hours, and the gRNA was subsequently purified with RNA Clean & Concentrator-5 Kit (Zymo Research).
In vitro Cas9 cleavage assay
Target DNA template used for cleavage activity was PCR amplified from plasmid pcDNA3.1-EGFP. One-hundred fifty nanograms of DNA template was added to preassembled RNP complexes (3 pmol) and incubated for 1 hour at 37°C in NEB 10× Cas9 reaction buffer. The cleavage complex was inactivated at 65°C for 10 min. The cleavage product was separated on a 1% agarose gel running at 150 V for 25 min. Relative intensities of full-length and Cas9-cleavage DNA fragments were determined on a Tanon 1600R Gel Imaging System.
Human cell culture and transfection
Both HEK293 cells and HEK293 Teton EGFP cells were cultured at 37°C with 5% CO2 in advanced Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum. Cell culture reagents were purchased from Macgene (Beijing) Co. Ltd., and cell culture supernatant was tested twice a month for mycoplasma. Transfections were performed using RNAiMAX (Thermo Fisher Scientific, Germany). On the day of transfection, WT Cas9 or Cas9 mutants (1.5 pmol) were assembled with gRNA (4.5 pmol) at room temperature for 10 min to form RNP complex. Donor DNA was added immediately into RNPs and incubated at 4°C for 3 hours before mixing with 1.2 μl of RNAiMAX in Opti-MEM (25 μl). The resulting mixture was incubated for 10 min at room temperature and dropped into 96-well plate. One hundred microliters of diluted cells (400,000 cells/ml) was added to the transfection complexes per well (final concentration of RNP will be 10 nM). Twenty-four hours after transfection, cells were transferred to 24-well tissue culture plate. Forty-eight hours after transfection, cells were digested for flow cytometry analysis.
Electroporation of RNP-ssODN complex
The electroporation machine (catalog no. CTX-1500A LE), tubes (20 μl; catalog no. 12-0107), and the buffer (catalog no. 13-0104) were provided by Celetrix LLC, Manassas VA. Cells were resuspended in electroporation buffer to 1 × 106 cells/ml. For Cas9 RNP-ssODN transfection, 18 pmol of Cas9 mutant protein was premixed with 24 pmol of gRNA at room temperature for 5 min first, and then, the ssODN (18 pmol) or the adaptor (18 pmol) was added to the formed RNP complex for 3 hours at 4°C. After that, the complex was mixed with the cells and transferred to 20-μl electroporation tube. The electroporation condition was 420 V for 30 ms at one pulse. After electroporation, the cells were immediately transferred back to warm medium to continue culture.
Flow cytometry
Doxycycline (Sigma-Aldrich) was added to HEK293 Teton EGFP reporter cells 24 hours after transfection. After another 24 hours, cells were trypsinized and resuspended in fluorescence-activated cell sorting buffer containing phosphate-buffered saline having 1% fetal bovine serum. Data were acquired on a Beckman CytoFLEX Flow Cytometer and were further analyzed using CytExpert software. In all experiments, a minimum of 10,000 cells were analyzed. Live cells were gated on the basis of forward scatter area (FSC-A) and side scatter area (SSC-A). Live-gated cells were further used to quantify the percentage of EGFP-positive populations.
RNP assembly for HDR reaction
The azido-modified Cas9 protein was mixed with gRNA, DBCO adaptor, and ssODN at a 1:3:1.2:1.2 molar ratio with gentle mixing at room temperature for 5 min and then incubated at 4°C for another 3 hours before delivery into cells.
HiBiT assay
Luminescence measurements were performed using the Nano-Glo HiBiT lytic assay system (Promega). Briefly, confluent cells were detached and lysed in lysis buffer, which contained the recombinant N terminus of nanoluciferase (LgBiT) and nanoluciferase substrate furimazine in 96-well plates. Lysates were incubated for 10 min rotating at room temperature, and luminescence intensities were recorded at a 1-s integration time using Infinite M200 PRO instrument (TECAN).
Genomic analysis
DNA was isolated from cells using FastPure Cell/Tissue DNA Isolation Mini Kit (Vazyme). Genomic PCR was performed following the manufacturer’s instructions using Phanta polymerase (Vazyme), and the PCR products were then purified using a cycle pure purification kit (Omega) and quantified using a NanoDrop. Primer sequences are listed in table S2. Deep sequencing was performed with an Illumina MiSeq with 2 × 150–bp paired-end reads (GENEWIZ, Amplicon-EZ). Sequencing reads were analyzed using CRISPResso.
Zygotes microinjection
B6D2F1 (C57BL/6 DBA2) female mice was used as embryo donors. Superovulated female B6D2F1 mice (7 to 8 weeks old) were mated to B6D2F1 stud males, and fertilized embryos were collected from oviducts. Azido-Cas9 RNP-adaptor-ssODN complex at different concentrations were microinjected into pronucleus of fertilized eggs with well-recognized pronuclei in M2 medium (Sigma-Aldrich). The injected zygotes were cultured in kalium (K+) simplex optimized medium (KSOM) with amino acids at 37°C under 5% CO2 in air until blastocyst stage by 3.5 days.
Genotype validation
Single blastocyst was lysed in 50 μl of lysis buffer [0.005% SDS and protein K (0.4 mg/ml) in water]. The mixture was further incubated at 50°C for 1 hour and then 99°C for 30 min. After cooled to room temperature, genotyping PCR was performed using Phanta polymerase (Vazyme), and the PCR products were cloned in T-vector for further sequencing.
Supplementary Material
Acknowledgments
We acknowledge L. Su at Peking University Medical and Health Analysis Center for the assistance in flow cytometry experiments and X. Shi and X. Zhang at the State Key Laboratory of Natural and Biomimetic Drugs for assistance in high-resolution protein mass spectrometry. Funding: This work was financially supported by the National Natural Science Foundation of China (91853111, 21922701, 81871160, and 21778005), the National Key Research and Development Program of China (no. 2016YFA0201400), National Major Scientific and Technological Special Project for “Significant New Drugs Development” (2019ZX09739001), and Clinical Medicine Plus X–Young Scholars Project (PKU2019LCXQ002 and PKU2020LCXQ029), Peking University. Author contributions: X.L., X.G., M.L., and T.L. designed the research. X.L., X.G., L.C., and L.T. performed the plasmid construction, protein expression, purification, and activity assay in vitro. W.Z. and H.C. performed the chemical synthesis. X.L., X.G., and L.C. performed the transfection and activity assay in cell. X.L. and B.X. performed the mouse zygotes microinjection. X.L., X.G., H.C., Y.H., M.L., and T.L. analyzed the data and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/15/eaaz0051/DC1
REFERENCES AND NOTES
- 1.Spicer C. D., Davis B. G., Selective chemical protein modification. Nat. Commun. 5, 4740 (2014). [DOI] [PubMed] [Google Scholar]
- 2.van Vught R., Pieters R. J., Breukink E., Site-specific functionalization of proteins and their applications to therapeutic antibodies. Comput. Struct. Biotechnol. J. 9, e201402001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morais M., Ma M. T., Site-specific chelator-antibody conjugation for PET and SPECT imaging with radiometals. Drug Discov. Today Technol. 30, 91–104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hallam T. J., Smider V. V., Unnatural amino acids in novel antibody conjugates. Future Med. Chem. 6, 1309–1324 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Beck A., Goetsch L., Dumontet C., Corvaïa N., Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 16, 315–337 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Tsuchikama K., An Z., Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 9, 33–46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turecek P. L., Bossard M. J., Schoetens F., Ivens I. A., PEGylation of biopharmaceuticals: A review of chemistry and nonclinical safety information of approved drugs. J. Pharm. Sci. 105, 460–475 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Boutureira O., Bernardes G. J. L., Advances in chemical protein modification. Chem. Rev. 115, 2174–2195 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Liu T., Du J., Luo X., Schultz P. G., Wang F., Homogeneously modified immunoglobulin domains for therapeutic application. Curr. Opin. Chem. Biol. 28, 66–74 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Gaj T., Gersbach C. A., Barbas C. F. III, ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joung J. K., Sander J. D., TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 14, 49–55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F., Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang F., Doudna J. A., CRISPR–Cas9 structures and mechanisms. Annu. Rev. Biophys. 46, 505–529 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Wang H., La Russa M., Qi L. S., CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem. 85, 227–264 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Cho S. W., Lee J., Carroll D., Kim J.-S., Lee J., Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9–sgRNA ribonucleoproteins. Genetics 195, 1177–1180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuris J. A., Thompson D. B., Shu Y., Guilinger J. P., Bessen J. L., Hu J. H., Maeder M. L., Joung J. K., Chen Z.-Y., Liu D. R., Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 33, 73–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeWitt M. A., Corn J. E., Carroll D., Genome editing via delivery of Cas9 ribonucleoprotein. Methods 121–122, 9–15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang J., Chen X., Glass Z., Gao F., Mao L., Wang M., Xu Q., Integrating combinatorial lipid nanoparticle and chemically modified protein for intracellular delivery and genome editing. Acc. Chem. Res. 52, 665–675 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim S., Kim D., Cho S. W., Kim J., Kim J.-S., Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 24, 1012–1019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang X., Potter J., Kumar S., Zou Y., Quintanilla R., Sridharan M., Carte J., Chen W., Roark N., Ranganathan S., Ravinder N., Chesnut J. D., Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 208, 44–53 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Wang M., Zuris J. A., Meng F., Rees H., Sun S., Deng P., Han Y., Gao X., Pouli D., Wu Q., Georgakoudi I., Liu D. R., Xu Q., Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 113, 2868–2873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Modzelewski A. J., Chen S., Willis B. J., Lloyd K. C. K., Wood J. A., He L., Efficient mouse genome engineering by CRISPR-EZ technology. Nat. Protoc. 13, 1253–1274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao X., Zhang M., Wang X., Ying W., Hu X., Dai P., Meng F., Shi L., Sun Y., Yao N., Zhong W., Li Y., Wu K., Li W., Chen Z. J., Yang H., Tild-CRISPR allows for efficient and precise gene knockin in mouse and human cells. Dev. Cell 45, 526–536.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Hendel A., Bak R. O., Clark J. T., Kennedy A. B., Ryan D. E., Roy S., Steinfeld I., Lunstad B. D., Kaiser R. J., Wilkens A. B., Bacchetta R., Tsalenko A., Dellinger D., Bruhn L., Porteus M. H., Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 33, 985–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelley M. L., Strezoska Ž., He K., Vermeulen A., van B. Smith A., Versatility of chemically synthesized guide RNAs for CRISPR-Cas9 genome editing. J. Biotechnol. 233, 74–83 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Lee K., Mackley V. A., Rao A., Chong A. T., Dewitt M. A., Corn J. E., Murthy N., Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife 6, e25312 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basila M., Kelley M. L., van B. Smith A., Minimal 2′-O-methyl phosphorothioate linkage modification pattern of synthetic guide RNAs for increased stability and efficient CRISPR-Cas9 gene editing avoiding cellular toxicity. PLOS ONE 12, e0188593 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryan D. E., Taussig D., Steinfeld I., Phadnis S. M., Lunstad B. D., Singh M., Vuong X., Okochi K. D., McCaffrey R., Olesiak M., Roy S., Yung C. W., Curry B., Sampson J. R., Bruhn L., Dellinger D. J., Improving CRISPR–Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res. 46, 792–803 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hemphill J., Borchardt E. K., Brown K., Asokan A., Deiters A., Optical control of CRISPR/Cas9 gene editing. J. Am. Chem. Soc. 137, 5642–5645 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki T., Asami M., Patel S. G., Luk L. Y. P., Tsai Y.-H., Perry A. C. F., Switchable genome editing via genetic code expansion. Sci. Rep. 8, 10051 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma M., Zhuang F., Hu X., Wang B., Wen X.-Z., Ji J.-F., Xi J. J., Efficient generation of mice carrying homozygous double-floxp alleles using the Cas9-Avidin/Biotin-donor DNA system. Cell Res. 27, 578–581 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carlson-Stevermer J., Abdeen A. A., Kohlenberg L., Goedland M., Molugu K., Lou M., Saha K., Assembly of CRISPR ribonucleoproteins with biotinylated oligonucleotides via an RNA aptamer for precise gene editing. Nat. Commun. 8, 1711 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gu B., Posfai E., Rossant J., Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 36, 632–637 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Savic N., Ringnalda F. C., Lindsay H., Berk C., Bargsten K., Li Y., Neri D., Robinson M. D., Ciaudo C., Hall J., Jinek M., Schwank G., Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. eLife 7, e33761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aird E. J., Lovendahl K. N., Martin A., Harris R. S., Gordon W. R., Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun. Biol. 1, 54 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maza J. C., McKenna J. R., Raliski B. K., Freedman M. T., Young D. D., Synthesis and incorporation of unnatural amino acids to probe and optimize protein bioconjugations. Bioconjug. Chem. 26, 1884–1889 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young D. D., Young T. S., Jahnz M., Ahmad I., Spraggon G., Schultz P. G., An evolved aminoacyl-tRNA synthetase with atypical polysubstrate specificity. Biochemistry 50, 1894–1900 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwinn M. K., Machleidt T., Zimmerman K., Eggers C. T., Dixon A. S., Hurst R., Hall M. P., Encell L. P., Binkowski B. F., Wood K. V., CRISPR-mediated tagging of endogenous proteins with a luminescent peptide. ACS Chem. Biol. 13, 467–474 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Park H. M., Liu H., Wu J., Chong A., Mackley V., Fellmann C., Rao A., Jiang F., Chu H., Murthy N., Lee K., Extension of the crRNA enhances Cpf1 gene editing in vitro and in vivo. Nat. Commun. 9, 3313 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lajoie M. J., Rovner A. J., Goodman D. B., Aerni H.-R., Haimovich A. D., Kuznetsov G., Mercer J. A., Wang H. H., Carr P. A., Mosberg J. A., Rohland N., Schultz P. G., Jacobson J. M., Rinehart J., Church G. M., Isaacs F. J., Genomically recoded organisms expand biological functions. Science 342, 357–360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/15/eaaz0051/DC1