

Abstract
Genome editing of human cluster of differentiation 34+ (CD34+) hematopoietic stem and progenitor cells (HSPCs) holds great therapeutic potential. This study aimed to optimize on-target, ex vivo genome editing using the CRISPR-Cas9 system in CD34+ HSPCs and to create a clear workflow for precise identification of off-target effects. Modified synthetic guide RNAs (gRNAs), either 2-part gRNA or single-guide RNA (sgRNA), were delivered to CD34+ HSPCs as part of ribonucleoprotein (RNP) complexes, targeting therapeutically relevant genes. The addition of an Alt-R electroporation enhancer (EE), a short, single-stranded oligodeoxynucleotide (ssODN), significantly increased editing efficiency in CD34+ HSPCs. Notably, similar editing improvement was observed when excess gRNA over Cas9 protein was used, providing a DNA-free alternative suitable for therapeutic applications. Furthermore, we demonstrated that sgRNA may be preferable over 2-part gRNA in a locus-specific manner. Finally, we present a clear experimental framework suitable for the unbiased identification of bona fide off-target sites by Genome-Wide, Unbiased Identification of Double-Strand Breaks (DSBs) Enabled by Sequencing (GUIDE-seq), as well as subsequent editing quantification in CD34+ HSPCs using rhAmpSeq. These findings may facilitate the implementation of genome editing in CD34+ HSPCs for research and therapy and can be adapted for other hematopoietic cells.
Keywords: genome editing, CRISPR-Cas9, CD34+ hematopoietic stem and progenitor cells, chemically modified guide RNAs, off-target sites
Graphical Abstract
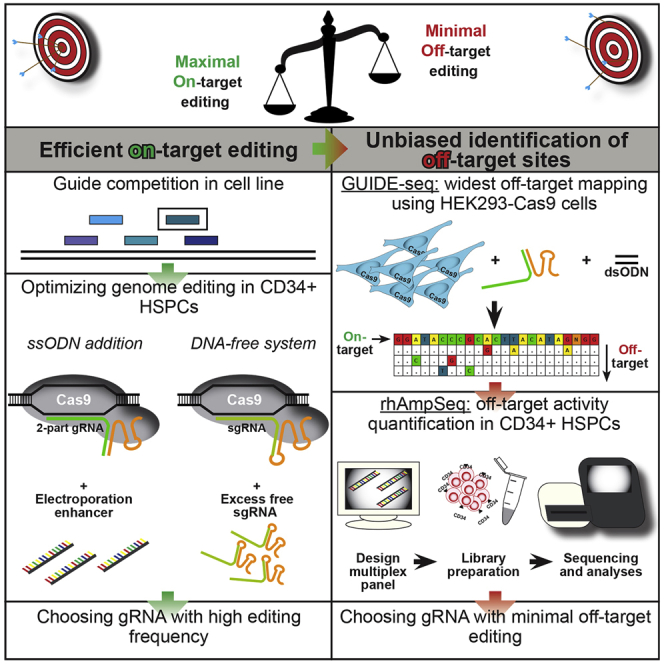
In order to become a universally applied gene-therapy tool, CRISPR-Cas9 must overcome certain pitfalls that lead to ineffective editing or cellular toxicity. Here, we developed a clear workflow to optimize the efficiency and specificity of CRISPR-Cas9 genome editing in hematopoietic stem and progenitor cells for clinical research applications.
Introduction
Human cluster of differentiation 34+ (CD34+) hematopoietic stem and progenitor cells (HSPCs) comprise a heterogeneous population, including a small group of long-term stem cells able to generate de novo the entire hematopoietic system.1,2 Human CD34+ HSPCs are derived from three different sources: umbilical cord blood, bone marrow, and peripheral blood via cytokine mobilization.3,4 The highly expressed cell-surface glycoprotein CD34 provides a marker for the identification and isolation of human CD34+ HSPCs, differentiating them from mature blood cells. HSPCs are crucial for the continued functioning of the hematopoietic system and therefore, present a target for basic, preclinical, and clinical research. Furthermore, CD34+ HSPCs are utilized for hematopoietic stem cell transplantation (HSCT), the only broadly applied clinical stem cell therapy routinely used. HSCT is used to treat patients with primary immunodeficiency diseases, blood disorders, malignant diseases, and certain systemic genetic diseases.5, 6, 7 Whereas allogeneic HSCT can be curative, significant limitations remain, including the availability of matched allogeneic donors, risk of graft-versus-host disease (GVHD), graft rejection, and toxicities related to chemotherapeutic or immunosuppressive agents.8,9 Thus, alternative, safer options are currently being explored, such as gene therapy using viral vectors containing a corrective transgene. However, in disorders in which the corrected gene needs to be expressed in a precise, developmental, and lineage-specific manner, genome-editing techniques may offer an alternative, definitive, and safe treatment approach. Genome editing using engineered nucleases provides a transformative technology with the potential to therapeutically modify any genomic sequence of interest with high efficiency and specificity.
Due to its simplicity and affordability, the CRISPR-Cas9 system is currently the most widely used genome-editing technology that utilizes programmable nucleases. The Cas9 nuclease relies on the direction of a guide RNA (gRNA), complementary to a specific 20-bp-long target site within the genome, for the induction of a site-specific double-strand break (DSB), thus initiating genome editing.10 A DSB activates the cell’s repair machinery, consisting primarily of nonhomologous end-joining (NHEJ) and homology-directed repair (HDR). When the cell repairs a DSB by NHEJ, insertions and deletions (indels) can be created at the site of the break, potentially generating premature stop codons within the targeted gene open-reading frame that may perturb its function. Alternatively, in HDR, the cell uses donor DNA as a template for repair of the break. By using synthetic donor DNA, the new sequence is introduced at the site of the break, thereby creating precise nucleotide genomic changes. Since the first step in genome editing is inducing a site-specific DSB, activity levels of nucleases often need to be optimized for the specific cell type and system used.
Several properties of the gRNA can influence the outcomes of the editing process. For example, a gRNA for a specific genomic locus can appear in two different types of formulations: 2-part gRNA or single gRNA (sgRNA).11 The 2-part gRNA is comprised of a CRISPR RNA (crRNA), in which the 5′ end is complementary to a 20-nt-long genomic target sequence, and its 3′ end anneals to a trans-activating CRISPR RNA molecule (tracrRNA). Alternatively, the sgRNA can be synthesized as a ∼100-nt-long oligonucleotide in which a stem-loop structure fuses the crRNA and tracrRNA sequences. Additionally, gRNAs can be generated by chemical synthesis, enzymatic synthesis, or expression from DNA vectors. Whereas genome editing using the CRISPR-Cas9 system is highly efficient in human cell lines, CRISPR-Cas9 genome editing in primary human cells, in general, and in CD34+ HSPCs, in particular, is more challenging. Notably, editing efficiency in CD34+ HSPCs can be increased by protecting both sgRNA termini with chemical modifications, probably due to reduced RNase-provoked degradation of exposed sgRNA ends.12 Whereas initial reports indicated that engineered nucleases generated low frequencies of genome editing in CD34+ HSPCs,13 recent approaches revealed that electroporation delivery of the CRISPR-Cas9 system, as either an RNA system or as a ribonucleoprotein (RNP) complex, achieved high frequencies of genome editing.12 Moreover, greater specificity was observed using the RNP rather than the RNA system, making the former an optimal candidate for therapeutic applications. Additional parameters concerning the delivery of the CRISPR-Cas9 system to the cells can contribute to the ensuing editing frequencies. For example, previous reports in cell lines have shown that delivery of the CRISPR-Cas9 RNP by electroporation in the presence of short single-stranded oligodeoxynucleotides (ssODNs) could further increase editing levels.14,15
Genome editing of CD34+ HSPCs has great therapeutic potential, such as in treating immunodeficiency disorders.1,13 In order to translate CD34+ HSPC genome editing to the clinic, it is crucial to find genomic-editing reagents that are both efficient and highly specific. Although the targeting specificity of the CRISPR-Cas9 system is controlled by the gRNA sequence, potential off-target activity has been identified in genomic DNA (gDNA), with up to 5 bp mismatches or with alternative protospacer adjacent motif (PAM) sequences.16, 17, 18, 19, 20, 21 Whereas off-target activity can potentially occur anywhere in the genome, there is still no widely accepted, standardized battery of assays for the identification and measurement of these unwanted outcomes. Traditionally, in silico techniques were used to predict genomic locations susceptible to off-target activity but with limited success.22, 23, 24 More recent methods allow empirical identification of actual off-target activity within edited cells in an unbiased manner.25,26 One such technique is GUIDE-seq, which stands for Genome-Wide, Unbiased Identification of DSBs Enabled by Sequencing.24 This method is dependent on the integration of short, modified double-stranded oligodeoxynucleotides (dsODNs) into the genome and has been executed and evaluated mainly in human cell lines. An additional challenge when measuring off-target activity is to perform an accurate, high-throughput quantification of editing events discovered by the unbiased empirical methods for off-target identification. It was recently shown that the rhAmpSeq technology can simultaneously quantify off-target genome-editing events in a single multiplex reaction (amplifying up to 5,000 genomic loci), thereby enabling comprehensive characterization of off-target activity for the desired gRNAs.27
The current study aimed to optimize the on-target genome editing using the CRISPR-Cas9 system in CD34+ HSPCs and to create a workflow for off-targets characterization. To this end, either a 2-part synthetic gRNA or a sgRNA as part of RNP complexes was electroporated into CD34+ HSPCs. We further demonstrate the locus-specific effect of certain chemical modifications of a gRNA. Moreover, we show that the addition of an Alt-R electroporation enhancer (EE) reagent, a short ssODN with no homology to the human genome, results in a significant increase in genome-editing efficiency in CD34+ HSPCs. Interestingly, in the absence of Alt-R EE, a similar improvement in genome-editing efficiency was observed when an excess of sgRNA over Cas9 protein was used. This DNA-free system is more suitable for therapeutic applications, since there is no risk for random insertional mutagenesis. Lastly, the study presents a clear workflow of an experimental framework for the unbiased identification of off-target sites by GUIDE-seq and quantification of editing levels in CD34+ HSPCs by the rhAmpSeq technology. This framework can facilitate the implementation of genome editing in CD34+ HSPCs for basic, preclinical, and clinical research and can be adapted for other primary hematopoietic cells.
Results
Alt-R EE Significantly Increases Genome-Editing Frequencies in Human CD34+ HSPCs
With the aim to develop a preclinically relevant research workflow for therapeutic genome editing in human CD34+ HSPCs, two targets were chosen for the current study: recombination-activating genes 2 and 1 (RAG2 and RAG1). Diverse mutations in RAG2 and RAG1 prevent the development of CD34+ HSPCs into functional T and B cells and can lead to severe combined immunodeficiency (SCID). The current treatment of RAG2 and RAG1 SCIDs, using allogeneic HSCT,28, 29, 30 presents significant limitations that may be overcome by using CRISPR-Cas9 genome editing as a definitive and safer treatment alternative. In this study, we first designed and screened five different gRNAs in immortalized K562 cells for targeted activity downstream of the translation start codon of the RAG2 and RAG1 genes. Based on previous studies in our lab, the most active gRNA in an immortalized line correlates to high activity in CD34+ HSPCs (unpublished data). Among the gRNAs screened, RAG2 gRNA 2 and RAG1 gRNA 3 were selected, due to their high on-target activity, and were used for all downstream experiments (Table S1; Figure S1).
In order to define the most efficient conditions for on-target editing in CD34+ HSPCs, three different gRNA formulations were synthesized for the selected RAG2 and RAG1 gRNAs, and editing efficiencies were compared. Since the benefit of modified gRNAs in the editing of CD34+ HSPCs has been demonstrated,12 all three of the examined formulations were synthesized with chemical modifications (Figure 1A). The Alt-R 2-part gRNA system was composed of a 36-nt-long crRNA annealed to a 67-nt-long tracrRNA, whereas the Alt-R 2-part XT gRNA system included additional chemical modifications in the crRNA, further increasing its stability. The single molecule Alt-R sgRNA was synthesized as a 100-nt-long sequence that fused crRNA and tracrRNA. In order to form the RNP complexes, the Cas9 nuclease was complexed to each of the above-mentioned formulations, while ensuring a small gRNA excess (a 1:1.2 molar ratio of Cas9:gRNA). Additionally, based on previous reports, we examined the potential enhancement of genome editing in CD34+ HSPCs by adding nonhomologous ssODNs, such as the Alt-R EE, which possibly act by improving RNP delivery.14,15 First, the Alt-R 2-part gRNA, 2-part XT gRNA, and sgRNA RNP complexes were electroporated into CD34+ HSPCs at 1 μM and 4 μM concentrations, with or without the Alt-R EE. After 48 h, cells were collected, and genome editing was quantified by next-generation sequencing (NGS) of PCR amplicons. Delivery of 1 μM RAG2 Alt-R 2-part gRNA, 2-part XT gRNA, and sgRNA without Alt-R EE generated low indel-editing frequencies of 12%, 15%, and 12%, respectively, but raising RNP concentration to 4 μM increased indel-editing frequencies to 33%, 40%, and 62%, respectively (Figure 1B). Notably, the addition of the Alt-R EE significantly increased indel-editing frequencies at 1 μM RNP concentration, bringing the efficiency of Alt-R 2-part gRNA, 2-part XT gRNA, and sgRNA to 68%, 69%, and 69%, respectively, demonstrating its utility. The increase of the RNP concentrations to 4 μM in the presence of Alt-R EE slightly increased indel-editing frequencies of Alt-R 2-part gRNA, 2-part XT gRNA, and sgRNA to 73%, 75%, and 84%, respectively. Under all conditions tested, ssODN did not trigger any observed differences in cell viability.
Figure 1.

Efficient Editing of RAG2 and RAG1 in CD34+ HSPCs in the Presence of Alt-R EE
(A) RAG2- and RAG1-targeting gRNAs were used in three different synthetic formulations, bearing modifications at all open termini: an Alt-R 2-part gRNA (an annealed complex of chemically modified crRNA and a universal chemically modified tracrRNA), an Alt-R 2-part XT gRNA (similar to the Alt-R 2-part system with additional chemical modifications in the crRNA that further increases its stability), and an Alt-R sgRNA (a chemically synthesized single molecule that fuses the crRNA and tracrRNA). (B and C) Alt-R Streptococcus pyogenes (S.p.) Cas9 nuclease V3 was precomplexed to each of the RAG2 (B) and RAG1 (C) gRNAs at a 1:1.2 Cas9:gRNA molar ratio. CD34+ HSPCs were electroporated with 1 or 4 μM (light and dark bars, respectively) of RNP complexes in the absence (left) or presence (right) of an Alt-R EE. Cells were cultured for 48 h prior to gDNA isolation, and editing frequencies by NGS were quantified at the on-target sites via amplicon sequencing to detect the presence of indels. Bars represent mean indel-editing percentages ± SEM (from three independent human donors), and editing percentages are designated above the bars. Mock-electroporated negative controls were used to subtract background indels when performing the analyses.
Next, we tested the editing frequencies for RAG1 using the same experimental conditions. Similar results were obtained for the RAG1 target when the experiments were performed without the Alt-R EE (Figure 1C). However, whereas for RAG2, all guide RNAs generated similar indel-editing frequencies in the presence of Alt-R EE, for RAG1, the Alt-R 2-part XT gRNA and sgRNA were more efficient than the Alt-R 2-part gRNA. Delivery of 4 μM RAG1 Alt-R 2-part gRNA, 2-part XT gRNA, and sgRNA with Alt-R EE generated frequencies of 23%, 60%, and 80%, respectively. Taken together, this suggests that Alt-R 2-part XT and sgRNA provide more efficient editing when the inherent potency of a given guide is lower.
Although NGS analysis of PCR amplicons for genome-editing experiments is a sensitive tool for indel-editing quantification, NGS can be cost-prohibitive for a small number of experiments, and its interpretation requires instrument availability and bioinformatics expertise. An alternative method for indel-editing quantification is the tracking of indels by decomposition (TIDE) algorithm that was developed to analyze Sanger sequence traces generated from the PCR amplicon using a web-based platform (https://tide.nki.nl/). TIDE analysis is an easy-to-use and cost-effective method, especially when analyzing a small number of samples.31 Amplicons flanking the RAG2 and RAG1 target sites from CD34+ HSPCs were either analyzed by NGS or Sanger sequencing, followed by TIDE analysis. Consistent with prior findings, TIDE and NGS frequencies were highly correlated (Figure S2A), but TIDE frequencies were consistently lower,32 demonstrating its lower sensitivity. The recently developed inference of CRISPR edits (ICE) method, used for analyzing CRISPR data using Sanger sequence traces (https://ice.synthego.com/#/), gave similar results to those of TIDE (Figure S2B). Therefore, these results suggest that whereas indel frequency levels are similar among TIDE, ICE, and NGS, NGS is a more reliable approach due to its higher sensitivity and ability to characterize the specific genomic alterations.
Excess sgRNA Induces High Editing Rates in CD34+ HSPCs, Compensating for the Lack of an Alt-R EE
The Alt-R EE is computationally predicted to be nonhomologous to the human genome and is, therefore, designed to have a low potential for nonspecific genomic integration. This assumption is supported by the finding that ssODNs of similar sizes were shown to have a dramatically reduced tendency to integrate into the genome randomly.14 In order to ensure that this principle holds in CD34+ HSPCs, genomic DNA from RAG2- and RAG1-edited CD34+ HSPCs were analyzed by NGS for Alt-R EE integration at the RAG2 and RAG1 on-target sites. Alt-R EE integration frequencies ranged from 0.01% to 0.05% in both genes (Table 1), demonstrating its low, but detectable, potential insertional mutagenesis.
Table 1.
Genomic Integration Frequencies of Alt-R EE
| Gene | gRNA Formulation | Alt-R EE Insertion (%) | SD |
|---|---|---|---|
| RAG2 | Alt-R sgRNA | 0.009 | 0.01 |
| Alt-R 2-part XT RNA | 0.013 | 0.01 | |
| Alt-R 2-part RNA | 0.035 | 0.03 | |
| RAG1 | Alt-R sgRNA | 0.016 | 0.02 |
| Alt-R 2-part XT RNA | 0.052 | 0.05 | |
| Alt-R 2-part RNA | 0.017 | 0.02 |
The gDNA of CD34+ HSPCs was examined by NGS for the integration of the Alt-R EE, following genome editing with 4 μM RNP complexes, as described in Figure 1. SD, standard deviation.
For CRISPR-Cas9 therapeutic applications, safety is a major concern. High accuracy of CRISPR-Cas9 genome editing is crucial, since even low frequencies of insertional mutagenesis events, such as in Alt-R EE integration, could potentially lead to genotoxicity. This requirement holds especially true in therapeutic procedures, such as HSCT, that require the editing of millions to hundreds of millions of cells. Therefore, to ensure safety and specificity, we explored whether the high frequencies of genome editing in CD34+ HSPCs could be maintained in the absence of the Alt-R EE. Since gRNA is a single-strand nucleic acid molecule, with some similar properties to Alt-R EE, we hypothesized that excess gRNA in the electroporation reaction might enhance editing efficiency. Delivery of 1 μM RAG2 RNPs, composed of a 1:2.5 molar ratio of Cas9:Alt-R 2-part XT gRNA, without Alt-R EE, generated low indel-editing frequencies of 31%, but increasing the RNP concentration to 4 μM raised indel-editing frequencies to 69% (Figure 2A). Interestingly, delivery of 1 μM RNPs, composed of 1:2.5 molar ratio of Cas9:Alt-R sgRNA, without Alt-R EE, generated indel-editing frequencies of 82%, whereas the increase of the RNP concentration to 4 μM further increased the indel-editing frequencies to 90%. These high frequencies were comparable to those obtained by using a 1:1.2 molar ratio of Cas9:gRNA with the Alt-R EE. Similar results were obtained for the RAG1 target (Figure 2B). Notably, in both instances, excess Alt-R sgRNA performed better than an excess of Alt-R 2-part XT gRNA. Therefore, our data demonstrate that highly efficient CRISPR indel-editing frequencies can be induced in CD34+ HSPCs (around 90%) by using either a 1:1.2 molar ratio of Cas9:2-part XT/sgRNA with Alt-R EE or a 1:2.5 molar ratio of Cas9:sgRNA without Alt-R EE. The removal of Alt-R EE from the reaction provides a DNA-free system and prevents the potential risks of harmful insertional mutagenesis.
Figure 2.

Excess sgRNA in RNP Formation Leads to Efficient Editing in CD34+ HSPCs, Compensating for the Lack of an Alt-R EE
(A and B) RAG2- (A) and RAG1 (B)-targeting gRNAs were used as Alt-R 2-part XT gRNA or Alt-R sgRNA in CD34+ HSPCs. RNPs precomplexed at a 1:1.2 Cas9:gRNA molar ratio were electroporated into the cells with an Alt-R EE (left). Alternatively, RNPs were precomplexed at an elevated 1:2.5 Cas9:gRNA molar ratio and delivered to the cells without an Alt-R EE (right). Either 1 μM or 4 μM RNP concentrations (light and dark bars, respectively) were examined, and indel-editing frequencies at the on-target sites were analyzed by NGS. Bars represent mean indel-editing percentages ± SEM (from three independent human donors), and editing percentages are designated above the bars. Mock-electroporated negative controls were used to subtract background indels when performing the analyses.
GUIDE-Seq Off-Target Identification is Less Robust in CD34+ HSPCs Than in Cas9-Expressing HEK293 Cells
Since genome-editing safety for therapeutic applications of CD34+ HSPCs is a major concern, we next investigated the off-target effects of the synthetic gRNAs. To discover empirically, through an unbiased approach, the potential off-target sites of the synthetic RAG2 and RAG1 gRNAs, we have used the GUIDE-seq methodology.24 Delivery of RAG2 and RAG1 RNPs, composed of a 1:2.5 molar ratio of Cas9:Alt-R sgRNA (conditions that generated the highest editing in CD34+ HSPCs), led to identification of only 4 and 8 off-target sites, respectively (Figure 3A). GUIDE-seq discovery of CRISPR off-targets has successfully been demonstrated mainly in cell lines and not in primary cells.33 Therefore, we also performed the GUIDE-seq experiments in HEK293 cells using either CRISPR RNP complexes or cells with stable expression of Cas9 (HEK293-Cas9). Delivery of the RNP complexes into the HEK293 cells also led to identification of a few off-target sites but using the HEK293-Cas9 cells, dramatically increased the number of off-target sites identified for both RAG2 and RAG1 gRNAs. Notably, GUIDE-seq, using Alt-R 2-part XT gRNA with the HEK293-Cas9 cells, identified the highest number of sites for both targets, revealing 50 sites for RAG2 and 84 sites for RAG1 (Figure 3B; Table S2). Consistently, off-target sites identified in HEK293-Cas9 cells included those uncovered in both CD34+ HSPCs and HEK293 cells using the RNP system. Genomic mapping of all identified off-target sites revealed that these sites were located in introns, exons, non-coding RNA, and non-transcribed DNA, demonstrating the possible location of off-target sites in functional genomic areas (Figure S3). When compared to the target sequences, these off-target sites contained as many as eight mismatches or gaps with a descending trend, with a low correlation between the number of mismatches and GUIDE-seq read counts (Figure S4). Therefore, our data demonstrated that performing GUIDE-seq analysis in HEK293-Cas9 cells uncovers a wider landscape of the off-target site risks compared with transient Cas9 expression.
Figure 3.

Experimental Identification of Potential Off-Target Sites of RAG2 and RAG1 gRNAs by GUIDE-Seq
RAG2- and RAG1-targeting Alt-R 2-part XT gRNAs or Alt-R sgRNAs were delivered either directly to HEK293-Cas9 stable cells or HEK293 cells as part of RNP complexes. RAG2- and RAG1-targeting Alt-R sgRNAs were delivered to CD34+ HSPCs as part of RNP complexes. The gRNAs or RNP complexes were delivered to the cells together with a short dsODN. Genomic sites, which incorporated the dsODN sequence, were mapped by NGS. (A) Venn diagrams showing the number of overlapping off-target sites identified under the different experimental conditions. (B) Representative off-target sites identified in HEK293-Cas9 cells following editing with Alt-R 2-part XT gRNAs. The total number of mismatches for each off-target site and the number of sequencing reads are indicated. On-target sites are highlighted in yellow.
Quantification of CRISPR Off-Target Editing Events in CD34+ HSPCs by rhAmpSeq-Targeted Sequencing Technology
In order to quantify the off-target activity at the sites uncovered by the GUIDE-seq method, off-target indel frequencies were measured by Illumina sequencing using the rhAmpSeq library preparation technology. First, the same HEK293 gDNA samples used for the GUIDE-seq analysis underwent rhAmpSeq multiplexed amplification (Figure S5). The rhAmpSeq indel-editing measurements of every off-target site were plotted against the GUIDE-seq measurements, which are presented as a percentage, normalized to 100% of the on-target site. Positive correlations between rhAmpSeq and GUIDE-seq measurements for RAG2 and RAG1 off-target sites, in all samples, confirm the power of GUIDE-seq to identify real off-target sites. Next, to test the specificity of the conditions that generated the highest on-target editing in CD34+ HSPCs, either 4 μM RNPs composed of a 1:2.5 molar ratio of Cas9:Alt-R sgRNA without Alt-R EE or 4 μM RNPs composed of a 1:1.2 molar ratio of Cas9:Alt-R 2-part XT gRNA with Alt-R EE were delivered into the cells, and off-target frequencies were measured by rhAmpSeq. On- and off-target activity was quantified in CD34+ HSPCs at all 50 RAG2 sites and 84 RAG1 sites uncovered by the wider GUIDE-seq map generated in the HEK293-Cas9 cells described above. We first focused on the RAG2 gRNA, which had shown high on-target editing levels for both Alt-R sgRNA and Alt-R 2-part XT gRNA conditions (87% and 85%, respectively). Out of the 49 off-target sites tested in the CD34+ HSPCs using the Alt-R sgRNA and 2-part XT gRNA RAG2 formulations, only six and seven sites showed indel-editing frequencies above 0.1%, respectively (Figure 4A). These results demonstrate that both high activity and relatively high specificity can be achieved when CRISPR-Cas9 is delivered as an RNP system into CD34+ HSPCs.
Figure 4.

rhAmpSeq-Quantified Editing in RAG2 and RAG1 Off-Target Sites in CD34+ HSPCs
(A and B) RAG2 (A) and RAG1 (B) on- and off-target editing levels were determined by rhAmpSeq in CD34+ HSPCs. WT Cas9 was complexed with either Alt-R sgRNA at a 1:2.5 Cas9:gRNA molar ratio (top plots) or with Alt-R 2-part XT gRNA at a 1:1.2 Cas9:gRNA (middle plots). Only 2-part XT RNPs were delivered in the presence of Alt-R EE. Alt-R HiFi Cas9 was complexed with sgRNA at a 1:2.5 Cas9:gRNA molar ratio (bottom plots). rhAmpSeq data were analyzed using a custom-built pipeline (IDT). Sites with editing levels higher than 0.1% in at least one of the experimental set-ups are presented (0.1% threshold designated by red dashed lines). Editing percentages are designated above the bars, and the numbers of the identified off-target sites appear below the graphs. On-target bars are colored in orange, and off-target bars are colored in gray. Error bars represent a 95% confidence interval (see Materials and Methods).
For the RAG1 guides, delivery of the Alt-R sgRNA formulation generated a high level of 83% on-target editing frequencies, and out of 83 off-target sites tested, only six sites showed indel-editing frequencies above 0.1% (Figure 4B). As described above, the Alt-R 2-part XT gRNA formulation generated a lower level of 55% on-target editing frequency. Consistent with this relatively low on-target editing level, only one site had an off-target activity above 0.1%.
Interestingly, for RAG2, only three out of seven off-target sites that demonstrated bona fide activity above 0.1% in CD34+ HSPCs via rhAmpSeq were nominated by GUIDE-seq in CD34+ HSPCs, whereas four were nominated in HEK293 using RNP complexes (Table 2). Similarly, for RAG1, only four out of seven verified off-target sites were nominated by GUIDE-seq in CD34+ HSPCs, and five were nominated by GUIDE-seq in HEK293 using RNP complexes. These results highlight the importance of performing the GUIDE-seq method in HEK293-Cas9 cells in order to uncover the widest potential off-target risks.
Table 2.
Guide-Seq in HEK293-Cas9 Cells Offers the Widest Identification Potential for Off-Target Risks in CD34+ HSPCs
|
RAG2 Sites Detected by rhAmpSeq in HSPCs (Editing > 0.1%) |
RAG2 Sites Detected by GUIDE-seq (Using Different Cell Types and gRNA Formulations) |
|||||
|---|---|---|---|---|---|---|
| Site | Genomic Position | Alt-R XT 2-Part RNA HEK293-Cas9 | Alt-R sgRNA HEK293-Cas9 | Alt-R XT 2-Part RNA HEK293 RNP | Alt-R sgRNA HEK293 RNP | Alt-R sgRNA HSPCs RNP |
| 2–on | Chr11:36594108 | + | + | + | + | + |
| 1 | Chr3:182671721 | + | + | + | + | + |
| 4 | Chr4:102437906 | + | + | + | + | + |
| 5 | Chr3:171315260 | + | + | + | + | − |
| 7 | Chr13:85131231 | + | + | + | + | − |
| 9 | Chr1:86729397 | + | + | − | − | − |
| 12 | Chr3:136434089 | + | − | − | − | − |
| 13 | Chr6:154714216 | + | + | − | − | + |
|
RAG1 Sites Detected by rhAmpSeq in HSPCs (Editing > 0.1%) |
RAG1 Sites Detected by GUIDE-seq (Using Different Cell Types and gRNA Formulations) |
|||||
|---|---|---|---|---|---|---|
| Site | Genomic Position | Alt-R XT 2-Part RNA HEK293-Cas9 | Alt-R sgRNA HEK293-Cas9 | Alt-R XT 2-Part RNA HEK293 RNP | Alt-R sgRNA HEK293 RNP | Alt-R sgRNA HSPCs RNP |
| 1–on | Chr11:36573321 | + | + | + | + | + |
| 2 | Chr1:16415506 | + | + | + | + | + |
| 4 | Chr11:57348913 | + | + | + | − | − |
| 5 | Chr3:140708416 | + | + | − | + | − |
| 8 | Chr1:34144141 | + | + | + | + | + |
| 19 | Chr19:4360240 | + | + | + | + | + |
| 35 | Chr12:90948235 | + | + | + | + | + |
| 37 | Chr6: 26286921 | + | + | − | − | − |
This table presents a comparison of the GUIDE-seq identification potential under different experimental conditions for confirmed off-target sites that had the rhAmpSeq activity level above 0.1% in CD34+ HSPCs. Chr, chromosome.
To explore whether the specificity of the RAG2 editing could be further improved, we delivered a high-fidelity Cas9 variant (Alt-R high-fidelity [HiFi] Cas9)34 using 4 μM RNPs, composed of a 1:2.5 molar ratio of Cas9:Alt-R sgRNA without Alt-R EE. The Alt-R HiFi Cas9 induced similar, high-level, on-target indel editing (88%), while reducing the off-target indel-editing frequencies below 0.1% (Figure 4A). For RAG1, when the wild-type (WT) Cas9 was compared with the Alt-R HiFi Cas9 complexed with the sgRNA, on-target editing was reduced from 83% to 60%, whereas the off-target activity was only partially reduced. Importantly, for both RAG2 and RAG1 gRNAs, no off-target sites with activity above 0.1% were observed within protein-coding regions (Table S2). Altogether, the data emphasize the need to optimize and quantify genome-editing outcomes when working with new targets, especially for therapeutic applications where high activity and specificity are required.
Discussion
Although the CRISPR-Cas9 system is widely used in genome-editing applications, there are still significant challenges regarding best practice strategies for achieving highly efficient and specific genome editing in human CD34+ HSPCs. In this study, we show that Alt-R EE significantly increases indel-editing frequencies in human CD34+ HSPCs, delivered with RNP complexes composed of a 1:1.2 molar ratio of Cas9:Alt-R gRNA. Interestingly, we show that in certain cases, when the CRISPR-Cas9 system is delivered by electroporation of RNP complexes, such as with the RAG1 target, additional synthetic chemical modifications of the gRNAs (Alt-R 2-part XT gRNA and sgRNA) can further increase editing levels. Moreover, the high indel-editing frequencies that were achieved in this study using conditions that are clinically relevant might eliminate the need for selection strategies, such as in the case of gene knockout of the C-C chemokine receptor type 5 (CCR5) gene, which encodes a co-receptor of HIV and is currently being investigated as a target for therapeutic genome editing in anti-HIV clinical trials.35
The Alt-R EE is a ssODN with no sequence homology to the human genome. Previous works suggest that adding ssODN increases genomic indel frequencies due to the stimulation of error-prone DNA repair.15 However, later reports suggest that ssODN induces higher levels of editing due to improved delivery of the RNP complexes but only when electroporation is employed.14 Here, we demonstrate that excess sgRNA over Cas9 protein can improve the indel-editing frequencies without the need to include any ssODN within the reaction. The positive effect of sgRNA may be due to the structural similarity between gRNAs and ssODNs, with both being single-strand nucleic acids. The provision of such a DNA-free alternative strategy reduces insertional mutagenesis risks and is more suitable for therapeutic genome-editing applications. Our results in CD34+ HSPCs are consistent with a recent study performed in primary human CD4+ T cells that showed that excess gRNA, or addition of ssODN, could stabilize the RNP complexes within the electroporation solution. This added stability may underlie the higher editing efficiency observed.36 Understanding the mechanism by which ssODNs, such as Alt-R EE or short RNA molecules, improve genome-editing efficiency is an important area for further research.
Our data also demonstrate that chemical modifications of specific gRNAs (Alt-R 2-part XT gRNA and sgRNA) further increase editing levels. This finding is in line with previous studies reporting that chemically synthesized sgRNA, as well as modified crRNA and tracrRNA, improves genome-editing efficiency when electroporated with Cas9 mRNA.11,12 Additionally, it was reported that end modifications of sgRNA improved genome-editing efficiencies with Cas9 protein.12 Furthermore, Basila et al.11 compared the modified 2-part system against the modified sgRNA with Cas9 protein and did not notice any difference between the two systems. Interestingly, in the current study, when using RAG2 RNP complexes without Alt-R EE, the modified Alt-R sgRNA induced higher indel-editing frequencies compared with modified Alt-R 2-part XT gRNA. However, when Alt-R EE was added, editing efficiencies significantly increased, and there was no difference between the two formulations. Importantly, for the RAG1 target, modified Alt-R sgRNA induced higher indel-editing frequencies compared with modified Alt-R 2-part XT gRNA, both with and without the Alt-R EE. The relative preference of the Alt-R sgRNA over the Alt-R 2-part XT gRNA in certain instances may arise from reduced exposure to exonuclease activity of the two single-molecule ends relative to the four ends of the 2-part molecule system. Furthermore, each of the Alt-R sgRNA molecule ends is more heavily chemically modified than any of the four exposed ends of the Alt-R 2-part XT gRNA, conferring higher protection from exonuclease activity. The differences in editing frequencies between genes might be derived from the sequence properties or epigenetic properties of the targeted locus. Broadening the investigation to include additional targets may shed more light on the locus-dependent properties that affect genome editing and may reveal the genetic and epigenetic parameters underlying low editing frequencies, such as in the case of RAG1. Future studies based on machine and deep-learning approaches might allow predicting difficult targeting sites that can benefit from higher modified guides. Until such tools are developed, finding the most effective formulations should be addressed experimentally, as done in the current study.
Targeted CRISPR genome editing in CD34+ HSPCs offers promising potential for definitively curing a significant number of disorders that affect the blood and the immune system.10 To deliver CRISPR to the clinic, gRNAs should have high activity at the on-target site, whereas minimizing off-target editing. One of the important challenges in evaluating the specificity and safety of genome editing is that there is no universal agreed-upon assay or battery of assays that can accurately predict these outcomes for therapeutic genome-editing applications.37,38 It is important to take into account that assessment of CRISPR genome-editing specificity must also be put into the context that cells acquire many mutations during their lifetime. Since scanning the entire genome of millions of edited cells is neither practical nor cost-effective, unbiased prediction techniques are needed to highlight the loci prone to off-target activity. Currently, there are three main approaches to identify off-target sites. In the first, computational methods are used to identify off-target sites based on a homology search approach; however, these tools might miss important sites and overpredict others.39 In the second, in vitro biochemical methods, such as In Vitro Nuclease-Digested Whole-Genome Sequencing (Digenome-seq),40 Selective Enrichment and Identification of Adapter-Tagged DNA Ends by Sequencing (SITE-seq),25 and Circularization In Vitro Reporting of Cleavage Effects by Sequencing (CIRCLE-seq),41 are used to identify off-target sites; however, like the computational tools, they are over-predictive.25 In the third, cell-based methods, such as GUIDE-seq,24 Breaks Labeling In Situ and Sequencing (BLISS),42 and Discovery of In Situ Cas Off-Targets and Verification by Sequencing (DISCOVER-Seq),33 are used to identify off-target sites in living human cells. In this study, we used the GUIDE-seq method to identify bona fide off-targets for the RAG2 and RAG1 guides. Interestingly, delivery of the CRISPR system into CD34+ HSPCs or HEK293 as an RNP system identified a low number of off-targets using the GUIDE-seq but using HEK293-Cas9 cells, greatly increased the number of off-target sites for both RAG2 and RAG1 guides. Importantly, some of the off-target sites identified only by using the HEK293-Cas9 cells were indeed confirmed to have off-target editing in the CD34+ HSPCs when specifically investigated by targeted sequencing. Therefore, our study highlights the importance of performing the GUIDE-seq method in cells with stable expression of Cas9 to identify the widest potential off-target risk.
The unbiased detection of off-target sites, enabled by a method like GUIDE-seq, is reliably being used to empirically nominate off-target sites in cells; however, this method often yields inconsistent quantification of off-target editing frequencies. Therefore, we used the rhAmpSeq-targeted sequencing technology to measure off-target editing frequencies. Interestingly, for the 49 RAG2 and 83 RAG1 off-target sites identified by GUIDE-seq in HEK293-Cas9 cells, only six sites showed indel-editing frequencies above 0.1%, in CD34+ HSPCs electroporated with sgRNA RNP complexes. Moreover, whereas for RAG2, use of the HiFi Cas9 maintained high on-target activity and significantly reduced off-target activity, for RAG1, there was some reduction in on-target activity, and the off-target activity was only partially reduced. Our data demonstrate the need to empirically quantify and optimize genome-editing parameters with new targets developed toward therapeutic applications.
In summary, this study shows that chemically synthesized gRNAs induce high on-target activity when delivered as RNP complexes into CD34+ HSPCs, whereas in a subset of targets, editing can benefit from higher modifications as the Alt-R 2-part XT gRNA and sgRNA. Moreover, the combination of optimized GUIDE-seq in HEK293-Cas9 for off-target nomination and the orthogonal rhAmpSeq technology in CD34+ HSPCs for off-target quantification can be used as a streamlined workflow for comprehensively assessing the off-target potential of the CRISPR-Cas9 system in preclinical studies.
Materials and Methods
gRNA Synthesis
Chemically modified gRNA oligomers were synthesized by Integrated DNA Technologies (IDT; Coralville, IA, USA). The 20-nt-long specific sequence for targeting the RAG2 gene was: 5′-TGAGAAGCCTGGCTGAATTA-3′ and for targeting the RAG1 gene was: 5′-AACTGAGTCCCAAGGTGGGT-3′. Alt-R 2-part gRNAs were synthesized as a 36-nt-long, sequence-specific crRNA and a 67-nt-long, nonspecific tracrRNA. The sgRNA was synthesized as a 100-nt-long molecule. Reagents were resuspended in 1× Tris-EDTA (TE), PH 8, solution (IDT, Coralville, IA, USA) to the desired concentrations: 200 μM 2-part gRNAs and 100 μM sgRNAs.
RNP Complex Preparation
Alt-R crRNAs and Alt-R tracrRNA (IDT, Coralville, IA, USA) were mixed in an equimolar ratio and heated at 95°C for 5 min. Next, annealed Alt-R 2-part gRNAs were allowed to form at room temperature (RT) for 10 min. For each electroporation reaction, 104 pmol of Alt-R Cas9 protein (IDT, Coralville, IA, USA) was complexed with either Alt-R sgRNAs or annealed Alt-R 2-part gRNAs in a 1:1.2 or 1:2.5 molar ratio (120 or 260 pmol per reaction, respectively) in a PBS solution (5 μL total volume). Complexes were allowed to form for 10–20 min at room temperature before electroporation.
Genome Editing by RNP Electroporation in CD34+ HSPCs
Mobilized human CD34+ HSPCs (AllCells, Alameda, CA, USA) were thawed and cultured for 48 h at a density of 2.5 × 105 cells/mL in StemSpan SFEM II (StemCell Technologies, Vancouver, Canada), supplemented with 100 U/mL penicillin and 0.1 mg/mL streptomycin (Biological Industries, Beit Haemek, Israel), stem cell factor (SCF), thrombopoietin (TPO), Fms-related tyrosine kinase 3 ligand (Flt3), and interleukin (IL)-6 (100 ng/mL each; PeproTech, Rocky Hill, NJ, USA). Cells were cultured at 37°C, 5% CO2, and 5% O2. 0.5 × 105 CD34+ HSPCs were reconstituted in P3 Primary Cell electroporation solution, according to the manufacturer’s instructions (Lonza, Basel, Switzerland) and mixed with RNP complexes at a final concentration of 1 or 4 μM. Cells were then supplemented with either 3.85 μM Alt-R EE (IDT, Coralville, IA, USA)14 or an equivalent volume of PBS. The supplemented cell solution (final cell concentration of 2 × 106 /mL) was transferred into the Lonza 4D-Nucleofector and electroporated using the DZ-100 program. Recovered cells were cultured for 48–72 h prior to gDNA extraction with QuickExtract (Lucigen, Middleton, WI, USA). RNP complexes were formed and delivered in the same manner with the Alt-R HiFi Cas9 variant (IDT, Coralville, IA, USA).
On-Target Indel Editing Frequency Quantification Using NGS
The following primers were used to amplify the gDNA sequences flanking the on-target sites of the genes of interest: RAG2_forward (Fw): 5′-GCCTTTTTGTCCAAAGAAGAAAA-3′, RAG2_reverse (Re): 5′-CAGAAACTATGTCTCTGCAGATG-3′; RAG1_Fw: 5′-TCATTGTTCTCAGGTACCTCAG-3′, RAG1_Re: 5′-CAGGTGTCTTTTCAAAGGATCT-3′. Primers also contained universal 5′ tails with second PCR adaptors. Next, the amplicons were introduced with Illumina sequencing adaptors and sample barcodes by a second round of PCR. Samples were prepared for Illumina MiSeq according to the manufacturer’s instructions and sequenced using 2 × 150 bp paired-end reads. Data were analyzed using a custom-built pipeline (IDT, Coralville, IA, USA) as described previously.34 Editing frequency was calculated as the percentage of reads containing an indel out of total reads at each target site. Mock-electroporated negative controls were used to subtract background indels when performing the analyses.
Detection of Alt-R EE Integration
The on-target insertions, detected by NGS, were processed to evaluate Alt-R EE integration events within the genome of CD34+ HSPCs, following editing in the presence of 3.85 μM Alt-R EE.
Experimental Off-Target Site Identification by GUIDE-Seq
The GUIDE-seq method was used for each of the guides to identify global editing events in an unbiased fashion.24 The procedure was performed with HEK293-Cas9 cells, generated by IDT, as previously described.34 The stable cell line has a single copy of the Cas9 gene and since being neomycin resistant, was cultured in DMEM, supplemented with 10% fetal bovine serum (FBS; Gibco, Invitrogen, Grand Island, NY, USA), 1% penicillin/streptomycin (Gibco, Invitrogen, Grand Island, NY, USA), and 500 μg mL−1 G418 (Gibco, Invitrogen, Grand Island, NY, USA). The Alt-R sgRNA or Alt-R 2-part XT gRNAs (10 μM) were electroporated into HEK293-Cas9 cells along with dsODNs, a 34-bp dsDNA donor fragment (for sequence, see Tsai et al.24). The GUIDE-seq method was also conducted in HEK293 cells and in CD34+ HSPCs, which were codelivered with 4 μM RNP complexes with dsODNs. HEK293 cells were cultured using identical conditions as HEK293-Cas9 cells, except for the G418 addition. Both cell lines were electroporated by the Lonza 96-Well Shuttle System (Lonza, Basel, Switzerland). 72 h after electroporation, gDNA was extracted from the cells by column-purification. NGS library preparation, sequencing, and operation of the GUIDE-seq software were performed, as described previously.24 The genomic locations of the GUIDE-seq-identified off-target sites (OTEs) were analyzed via the University of California, Santa Cruz (UCSC), genome browser version GRCh38/hg38. The sequencing data were deposited to the Sequence Read Archive (SRA), under accession number: PRJNA628100.
On- and Off-Target Editing Frequency Quantification by rhAmpSeq
rhAmpSeq primer amplification is dependent upon perfect-match hybridization to the target DNA and subsequent recognition of the RNA/DNA hybrid duplex by RNase H2 to cleave the target-matched primers that contain an RNA base and polymerase-blocking 3′ modification and allow for locus-specific amplification.27 Primers were designed to flank the on-target and all off-target sites, nominated by GUIDE-seq of HEK293-Cas9 cells, following editing with Alt-R 2-part XT gRNAs. Primers were pooled for locus-specific RNase H2-dependent multiplex assay amplification, followed by a universal PCR to add adaptor ends for NGS. PCR amplicons were sequenced on an Illumina MiSeq instrument (v.2 chemistry, 150 bp paired-end reads; Illumina, San Diego, CA, USA). Read depth was at least 3,000 reads per locus. The average read depth was around 40,000 reads for each gRNA. Data were analyzed using a custom-built pipeline (IDT, Coralville, IA, USA), as described previously.34 The sequencing data were deposited to the SRA, under accession number: PRJNA628100. Editing frequency was calculated as the percentage of reads containing an indel out of total reads at each target site. Mock-electroporated negative controls were used to compute an estimate of the activity rate by subtraction. A 95% confidence interval (CI) was calculated for each target site using the standard equation for the difference between the following two proportions:
where and denote the inferred editing frequency in treatment and mock, respectively. and denote the total number of reads in treatment and mock, respectively. is the desired confidence level, which is 0.05 for the figures presented here.
Author Contributions
J.S., O.I., A.M.J., R.T., and G.R.R. designed and conducted the experiments and interpreted and analyzed the data. M.S.M., I.A., and Z.Y. performed bioinformatics analyses. A.T.-R. helped with protocol design. A.H. and M.A.B. conceived the study and designed the experiments and the analysis approaches. A.H., A.T.-R., and J.S. wrote the manuscript, which was edited by O.I., A.M.J., M.S.M., R.T., G.R.R., I.A., A.T.-R., Z.Y., and M.A.B.
Conflicts of Interest
A.M.J., M.S.M., R.T., G.R.R., and M.A.B. are employees of Integrated DNA Technologies (IDT), which sells reagents similar to some described in the manuscript. All other authors declare no competing interests.
Acknowledgments
We thank Dr. Leon Anavy and Ms. Alona Levy from the Technion–Israel Institute of Technology for their useful support with the computational analyses. We thank Mr. Daniel Allen for critically reading the manuscript and providing practical advice. We gratefully acknowledge the funding support from the European Research Council (ERC) under the Horizon 2020 Research and Innovation Program (grant agreement number 755758).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.04.027.
Supplemental Information
References
- 1.Booth C., Gaspar H.B., Thrasher A.J. Treating Immunodeficiency through HSC Gene Therapy. Trends Mol. Med. 2016;22:317–327. doi: 10.1016/j.molmed.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Mohty R., Brissot E., Battipaglia G., Ruggeri A., Sestili S., Mediavilla C., Belhocine R., Dulery R., Mohty M., Malard F. CD34+-selected stem cell “Boost” for poor graft function after allogeneic hematopoietic stem cell transplantation. Curr. Res. Transl. Med. 2019;67:112–114. doi: 10.1016/j.retram.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Pafumi C., Leanza V., Carbonaro A., Leanza G., Iemmola A., Abate G., Stracquadanio M.G., D’Agati A. CD34(+) stem cells from umbilical cord blood. Clin. Pract. 2011;1:e79. doi: 10.4081/cp.2011.e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lidonnici M.R., Aprile A., Frittoli M., Mandelli G., Paleari Y., Spinelli A., Gentner B., Zambelli M., Parisi C., Bellio L. Human CD34+ Cells from Different Sources Disclose a Specific Stemness Signature. Blood. 2016;128:4709. [Google Scholar]
- 5.Kühl J.S., Suarez F., Gillett G.T., Hemmati P.G., Snowden J.A., Stadler M., Vuong G.L., Aubourg P., Köhler W., Arnold R. Long-term outcomes of allogeneic haematopoietic stem cell transplantation for adult cerebral X-linked adrenoleukodystrophy. Brain. 2017;140:953–966. doi: 10.1093/brain/awx016. [DOI] [PubMed] [Google Scholar]
- 6.Aldenhoven M., Wynn R.F., Orchard P.J., O’Meara A., Veys P., Fischer A., Valayannopoulos V., Neven B., Rovelli A., Prasad V.K. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125:2164–2172. doi: 10.1182/blood-2014-11-608075. [DOI] [PubMed] [Google Scholar]
- 7.Vellodi A., Young E.P., Cooper A., Wraith J.E., Winchester B., Meaney C., Ramaswami U., Will A. Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch. Dis. Child. 1997;76:92–99. doi: 10.1136/adc.76.2.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohty B., Mohty M. Long-term complications and side effects after allogeneic hematopoietic stem cell transplantation: an update. Blood Cancer J. 2011;1:e16. doi: 10.1038/bcj.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hierlmeier S., Eyrich M., Wölfl M., Schlegel P.G., Wiegering V. Early and late complications following hematopoietic stem cell transplantation in pediatric patients - A retrospective analysis over 11 years. PLoS ONE. 2018;13:e0204914. doi: 10.1371/journal.pone.0204914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porteus M.H. A New Class of Medicines through DNA Editing. N. Engl. J. Med. 2019;380:947–959. doi: 10.1056/NEJMra1800729. [DOI] [PubMed] [Google Scholar]
- 11.Basila M., Kelley M.L., Smith A.V.B. Minimal 2′-O-methyl phosphorothioate linkage modification pattern of synthetic guide RNAs for increased stability and efficient CRISPR-Cas9 gene editing avoiding cellular toxicity. PLoS ONE. 2017;12:e0188593. doi: 10.1371/journal.pone.0188593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dever D.P., Porteus M.H. The changing landscape of gene editing in hematopoietic stem cells: a step towards Cas9 clinical translation. Curr. Opin. Hematol. 2017;24:481–488. doi: 10.1097/MOH.0000000000000385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobi A.M., Rettig G.R., Turk R., Collingwood M.A., Zeiner S.A., Quadros R.M., Harms D.W., Bonthuis P.J., Gregg C., Ohtsuka M. Simplified CRISPR tools for efficient genome editing and streamlined protocols for their delivery into mammalian cells and mouse zygotes. Methods. 2017;121-122:16–28. doi: 10.1016/j.ymeth.2017.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richardson C.D., Ray G.J., Bray N.L., Corn J.E. Non-homologous DNA increases gene disruption efficiency by altering DNA repair outcomes. Nat. Commun. 2016;7:12463. doi: 10.1038/ncomms12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X.H., Tee L.Y., Wang X.G., Huang Q.S., Yang S.H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids. 2015;4:e264. doi: 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S., Kim J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cradick T.J., Fine E.J., Antico C.J., Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hendel R.C., Frost L. Off-label, but on-target: use of regadenoson with exercise. J. Nucl. Cardiol. 2013;20:179–181. doi: 10.1007/s12350-012-9643-5. [DOI] [PubMed] [Google Scholar]
- 23.Singh R., Kuscu C., Quinlan A., Qi Y., Adli M. Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 2015;43:e118. doi: 10.1093/nar/gkv575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cameron P., Fuller C.K., Donohoue P.D., Jones B.N., Thompson M.S., Carter M.M., Gradia S., Vidal B., Garner E., Slorach E.M. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat. Methods. 2017;14:600–606. doi: 10.1038/nmeth.4284. [DOI] [PubMed] [Google Scholar]
- 26.Gabriel R., von Kalle C., Schmidt M. Mapping the precision of genome editing. Nat. Biotechnol. 2015;33:150–152. doi: 10.1038/nbt.3142. [DOI] [PubMed] [Google Scholar]
- 27.Dobosy J.R., Rose S.D., Beltz K.R., Rupp S.M., Powers K.M., Behlke M.A., Walder J.A. RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 2011;11:80. doi: 10.1186/1472-6750-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadofsky M.J. The RAG proteins in V(D)J recombination: more than just a nuclease. Nucleic Acids Res. 2001;29:1399–1409. doi: 10.1093/nar/29.7.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meshaal S.S., El Hawary R.E., Abd Elaziz D.S., Eldash A., Alkady R., Lotfy S., Mauracher A.A., Opitz L., Pachlopnik Schmid J., van der Burg M. Phenotypical heterogeneity in RAG-deficient patients from a highly consanguineous population. Clin. Exp. Immunol. 2019;195:202–212. doi: 10.1111/cei.13222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cirillo E., Giardino G., Gallo V., D’Assante R., Grasso F., Romano R., Di Lillo C., Galasso G., Pignata C. Severe combined immunodeficiency--an update. Ann. N Y Acad. Sci. 2015;1356:90–106. doi: 10.1111/nyas.12849. [DOI] [PubMed] [Google Scholar]
- 31.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sentmanat M.F., Peters S.T., Florian C.P., Connelly J.P., Pruett-Miller S.M. A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci. Rep. 2018;8:888. doi: 10.1038/s41598-018-19441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wienert B., Wyman S.K., Richardson C.D., Yeh C.D., Akcakaya P., Porritt M.J., Morlock M., Vu J.T., Kazane K.R., Watry H.L. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science. 2019;364:286–289. doi: 10.1126/science.aav9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018;24:1216–1224. doi: 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu L., Wang J., Liu Y., Xie L., Su B., Mou D., Wang L., Liu T., Wang X., Zhang B. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 2019;381:1240–1247. doi: 10.1056/NEJMoa1817426. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen D.N., Roth T.L., Li P.J., Chen P.A., Apathy R., Mamedov M.R., Vo L.T., Tobin V.R., Goodman D., Shifrut E. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 2020;38:44–49. doi: 10.1038/s41587-019-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hendel A., Fine E.J., Bao G., Porteus M.H. Quantifying on- and off-target genome editing. Trends Biotechnol. 2015;33:132–140. doi: 10.1016/j.tibtech.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zischewski J., Fischer R., Bortesi L. Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol. Adv. 2017;35:95–104. doi: 10.1016/j.biotechadv.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 39.Tsai S.Q., Joung J.K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet. 2016;17:300–312. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim D., Bae S., Park J., Kim E., Kim S., Yu H.R., Hwang J., Kim J.-I., Kim J.-S. Digenome-seq: Genome-Wide Profiling of CRISPR-Cas9 Off-Target Effects in Human Cells. Nat. Methods. 2015;12:237–243. doi: 10.1038/nmeth.3284. 1 p following 243. [DOI] [PubMed] [Google Scholar]
- 41.Tsai S.Q., Nguyen N.T., Malagon-Lopez J., Topkar V.V., Aryee M.J., Joung J.K. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan W.X., Mirzazadeh R., Garnerone S., Scott D., Schneider M.W., Kallas T., Custodio J., Wernersson E., Li Y., Gao L. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017;8:15058. doi: 10.1038/ncomms15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.