

Abstract
Ovarian cancer is the fifth-leading cause of cancer death among women. The dissemination of ovarian tumors and growth as spheroids accompanies late stage disease. In cell culture, ovarian tumor cell spheroids can exhibit elevated resistance to environmental stressors such as reactive oxygen species. Homeostatic balance of the antioxidant response is a protective mechanism that prevents anoikis, a form of programmed cell death. Signaling pathways activated by integrin receptors suppress anoikis. Rgnef (ARHGEF28/p190RhoGEF) is a guanine nucleotide exchange factor that is activated downstream of integrins. We find that Rgnef protein levels are elevated in late-stage serous ovarian cancer, high Rgnef mRNA levels are associated with decreased progression-free and overall survival, and genomic ARHGEF28 loss is associated with increased patient survival. Using transgenic and transplantable Rgnef knockout mouse models, we find that Rgnef is essential for supporting three-dimensional ovarian spheroid formation in vitro and tumor growth in mice. Using RNA-sequencing and bioinformatic analyses, we identify a conserved Rgnef-supported anti-oxidant gene signature including GPX4, Nqo1, and Gsta4; common targets of the NF-kB transcription factor. Antioxidant treatment enhanced growth of Rgnef-knockout spheroids and Rgnef re-expression facilitated NF-κB-dependent tumorsphere survival. These studies reveal a new role for Rgnef in ovarian cancer to facilitate NF-κB-mediated gene expression protecting cells from oxidative stress.
Keywords: Rgnef, GEF, ovarian cancer, oxidative stress, NF-κB
Introduction
Ovarian cancer is the fifth-leading cause of cancer death among women [1]. The high mortality rate is largely due to the fact that most patients are diagnosed after tumors have metastasized [2]. Ovarian cancer exhibits a unique pattern of metastasis whereby cells detach from the primary tumor into the peritoneal space, which can trigger a form of programmed cell death called anoikis [2]. More aggressive cancer cells resist anoikis, can proliferate anchorage-independently as multicellular spheroids, and can become resistant to chemotherapy [3]. Targeting peritoneal spheroid survival and triggering anoikis may provide therapeutic benefits [4, 5].
Ovarian cancer tumors growing within the peritoneal space must adapt to environmental stresses from hypoxia, anchorage independence, and metabolic challenges [6]. Peritoneal tumor growth must overcome elevated oxidative stress within cells [7, 8]. Oxidative stress occurs when the generation of cellular reactive oxygen species (ROS) cells is not sufficiently balanced by ROS-scavenging pathways [8–10]. Platinum- and taxane-based chemotherapies elevate ROS generation, which is associated with cytotoxicity. Correspondingly, antioxidants that lower cellular ROS levels can enhance tumor initiation and progression [11, 12]. Ovarian cancer cells often exhibit elevated endogenous antioxidant gene expression, driven by transcription factors such as nuclear factor-like 2 (NRF2) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [13, 14]. Although increased NRF2- and NF-κB-driven signaling in ovarian cancer is associated with poor survival [15], regulation of these pathways remains incompletely defined.
Integrins are cell surface receptors that facilitate extracellular matrix attachment and initiate signals that promote cell survival [16]. Loss of matrix attachment can increase ROS levels in ovarian cancer cells [11]. Rgnef (ARHGEF28/ p190RhoGEF) is a Rho-specific guanine nucleotide exchange factor (GEF) that is activated downstream of integrins in a complex with focal adhesion kinase (FAK) [17]. In primary fibroblasts, Rgnef facilitates RhoA GTPase activation, actin stress fiber formation, and paxillin tyrosine phosphorylation needed for the control of cell motility [18, 19]. In advanced-stage colon cancer, Rgnef expression is elevated and promotes tumor growth through its interaction with FAK [20, 21]. In high grade serous ovarian cancer, RhoA pathway activation is associated with poor patient survival [22]. However, a role for Rgnef in ovarian cancer is not known.
Here we show that Rgnef levels are elevated in late-stage serous ovarian cancer and this is associated with decreased progression-free and or overall patient survival. Using transgenic and transplantable Rgnef knockout tumor models and supported by comparative Rgnef re-expression, we find that Rgnef is selectively required for three-dimensional spheroid growth in vitro and growth of tumors in vivo. Rgnef promotes NF-κB associated antioxidant gene expression revealing a novel role for Rgnef in ROS homeostasis and ovarian tumor progression.
Results
Rgnef is elevated in late-stage serous ovarian cancer
Rgnef (ARHGEF28/p190RhoGEF) is a ubiquitously-expressed protein with elevated levels detected in the murine ovary, brain, lung, and spleen [19]. Polyclonal antibodies have been generated against Rgnef and validated using tissues from Rgnef+/+ and Rgnef−/− mice [20]. Human ovarian serous adenocarcinomas tumor microarrays were analyzed by anti-Rgnef immunohistochemical staining (Fig. 1a). In general, Rgnef levels increased as a function of tumor stage. However, there was a wide distribution in staining intensities. Rgnef differences were significant when comparing Stage 3 tumors compared to normal adjacent tissue (P < 0.05, Fig. 1b). Kaplan-Meier analyses of serous OC mRNA array datasets (www.kmplot.com) revealed that elevated Rgnef mRNA levels (>400 patient samples from Stage 3 and above tumors) were associated with a significant decrease in both progression-free and overall survival (Figs. 1c and d). Concordantly, analyses using The Cancer Genome Atlas (TCGA) for serous OC (www.cbioportal.org) revealed that loss of one or more ARHGEF28 gene copies (greater than 200 patient samples) in advanced stage tumors was associated with better progression-free and overall survival (Figs. 1e and f). RNA sequencing data from TCGA showed that elevated ARHGEF28 levels in top 30% was associated with decreased overall survival (P < 0.03). Together, these findings support the notion that Rgnef may function in late-stage serous ovarian cancer.
Figure 1:
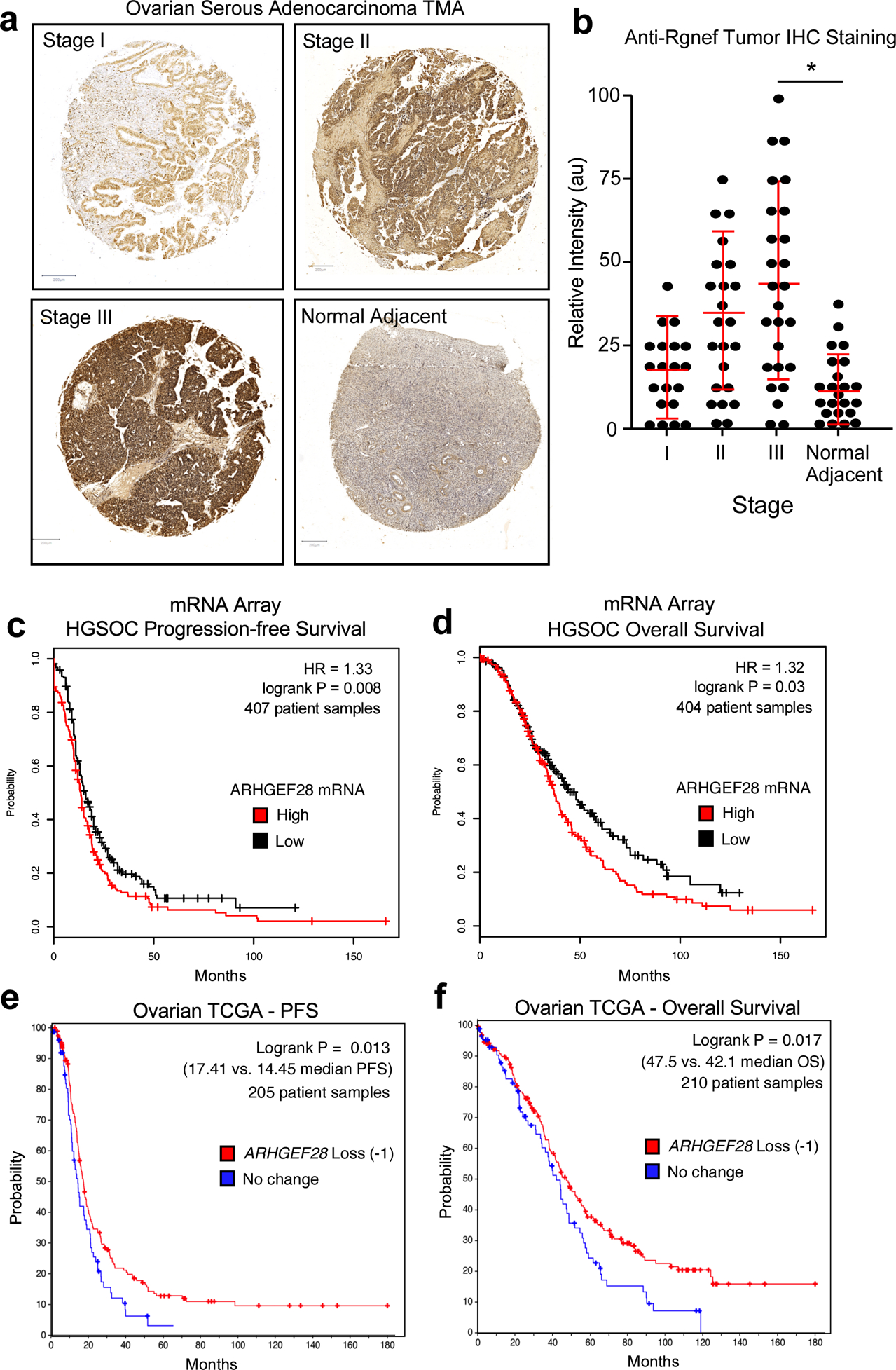
Analysis of Rgnef or ARHGEF28 levels in serous ovarian cancer. (a) Representative high grade serous ovarian cancer tumor micro-array (TMA) cores stained with polyclonal anti-Rgnef antibodies (brown) and nuclei counter-stained with hematoxylin. Stage information was from the manufacturer and normal adjacent is ovarian tissue from tumor-bearing patients. (b) Aperio Image Analysis quantification using the positive pixel count algorithm and relative intensity values (arbitrary units, au) are shown per TMA core. Values are means +/− SD (n=92, P< 0.05, T-Test). (c and d) Kaplan-Meier plotter (http://kmplot.com/ovar) was used to evaluate ARHGEF28 mRNA levels in stage III-IV serous ovarian cancer samples. High ARHGEF28 expression [auto-select cutoff] was significantly associated with (c) progression-free survival (*P = 0.008, n = 407) or (d) overall survival (*P = 0.03, n = 404). Loss of ARHGEF28 at the genomic level (heterozygous loss or homologous deletion vs. maintenance or gain) was similarly evaluated for (e) progression free or overall (f) survival using data available in The Cancer Genome Atlas (TCGA) (*P < 0.02).
Rgnef knockout inhibits spontaneous ovarian tumor formation
As ARHGEF28 loss is associated with less aggressive disease, we tested the effect of Rgnef knockout in a murine spontaneous ovarian cancer model driven by simian virus 40 T antigen (TAg) under the regulation of the Müllerian inhibiting substance type II receptor (MISIIR) [23] (Fig. 2). In high-grade serous ovarian cancer, the p53 tumor suppressor is mutated or inactivated in ~95% of cases, and the RB1 signaling pathway is deregulated in ~67% of cases [24]. TAg protein binds and inactivates p53 and RB1 in cells [25, 26] and female MISIIR-TAg develop spontaneous poorly differentiated carcinomas with complete penetrance [23]. MISIIR is expressed in the epithelia of Mullerian origin [26, 27] and TAg staining can be detected within cells of the oviduct and ovary (Fig. 2a).
Figure 2:
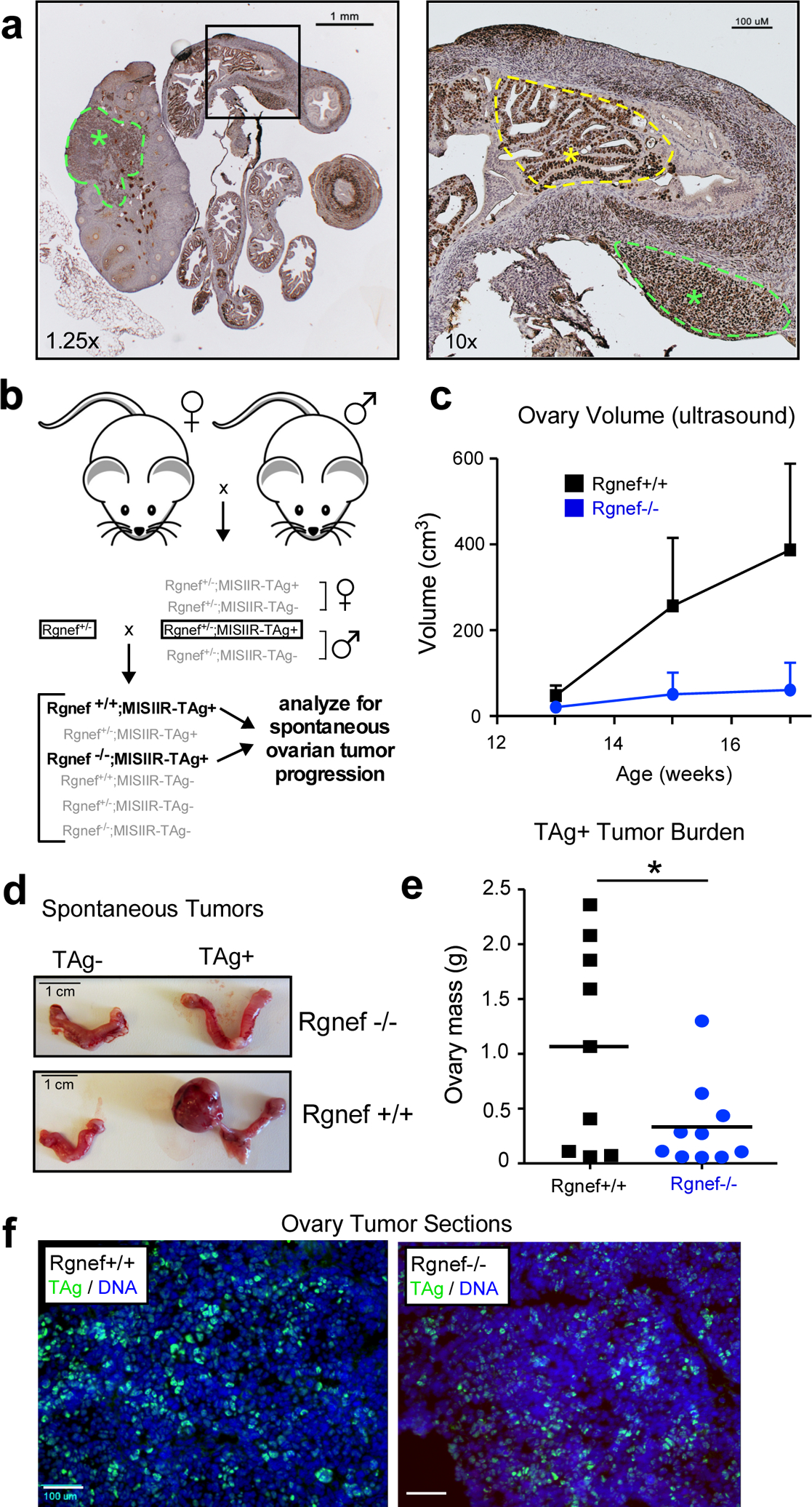
Rgnef knockout prevents spontaneous ovarian tumor formation. (a) Image of a TAg-stained (brown) ovary and oviduct from a MISIIR-TAg+ mouse. TAg is expressed in the murine ovary and fallopian tubal lesions (green asterisks and dotted lines) and oviduct (yellow asterisk and dotted line). Scale is 1 mm (left) or 100 μM (right). (b) Breeding schematic for generation of Rgnef−/− or Rgnef+/+; MISIIR;TAg+ mice. (c) Ovary size of littermates measured by ultrasound. Mice were sacrificed at week 17 or at earlier humane endpoint. Each point represents n ≥ 5 mice with error bars as SEM. (d) Representative pictures of oviducts and ovaries for each group at endpoint. Scale is 1 cm. (e) Mean tumor burden (total ovary mass) was higher in Rgnef+/+ than Rgnef−/− mice (*P < 0.05, SD +/− 0.93 for Rgnef+/+ and 0.39 for Rgnef−/− mice). (f) Immunofluorescent staining for TAg (green) and DNA (Hoechst, blue) in frozen section of ovaries from Rgnef−/− or Rgnef+/+; MISIIR;TAg+ tumor-bearing mice. Scale is 100 μm.
Rgnef knockout (Rgnef−/−) mice are born smaller than wildtype (Rgnef+/+) littermates [19]. However, by 6 to 8 weeks, Rgnef−/− mice are phenotypically normal and fertile. Breeding of Rgnef−/− with MISIIR-TAg+ mice yield Rgnef+/+; MISIIR-TAg+ and Rgnef−/−; MISIIR-TAg+ littermates after two crosses (Fig. 2b) and females were monitored for spontaneous ovarian tumor formation by ultrasound (Fig. 2c). Some Rgnef+/+ mice formed large tumors by week 15, and all mice were euthanized by 18 weeks. Total ovarian tissue mass was used as a measure of tumor burden (Fig. 2d). Rgnef+/+ mice formed significantly larger (P <0.05) tumors than Rgnef−/− mice (Fig. 2e). TAg expression was confirmed by immunofluorescent staining of tumor-bearing ovarian tissue from both Rgnef+/+ and Rgnef−/− mice (Fig. 2f). These results demonstrate that Rgnef expression is required for optimal TAg-driven ovarian tumor growth.
Tumor cell-intrinsic role for Rgnef
In the spontaneous Rgnef−/− MISIIR-TAg+ model, Rgnef loss is in all cells, both tumor and stroma. To determine if Rgnef is required for tumor cell-intrinsic growth, cells were collected from peritoneal wash of either Rgnef+/+ or Rgnef−/− MISIIR-TAg+ tumor-bearing mice at 17 weeks and expanded by growth in adherent culture (Fig. 3a). Pooled populations of murine ovarian carcinoma (MOVCAR) cells were generated. The presence or absence of Rgnef expression was verified by DNA genotyping (Fig. 3b), anti-Rgnef immunoblotting (Fig. 3c) and TAg expression (data not shown). Surprisingly, a pooled population of Rgnef−/− MOVCARs proliferated faster in adherent cell culture than Rgnef+/+ MOVCARs isolated in the same manner (Fig. 3d).
Figure 3:
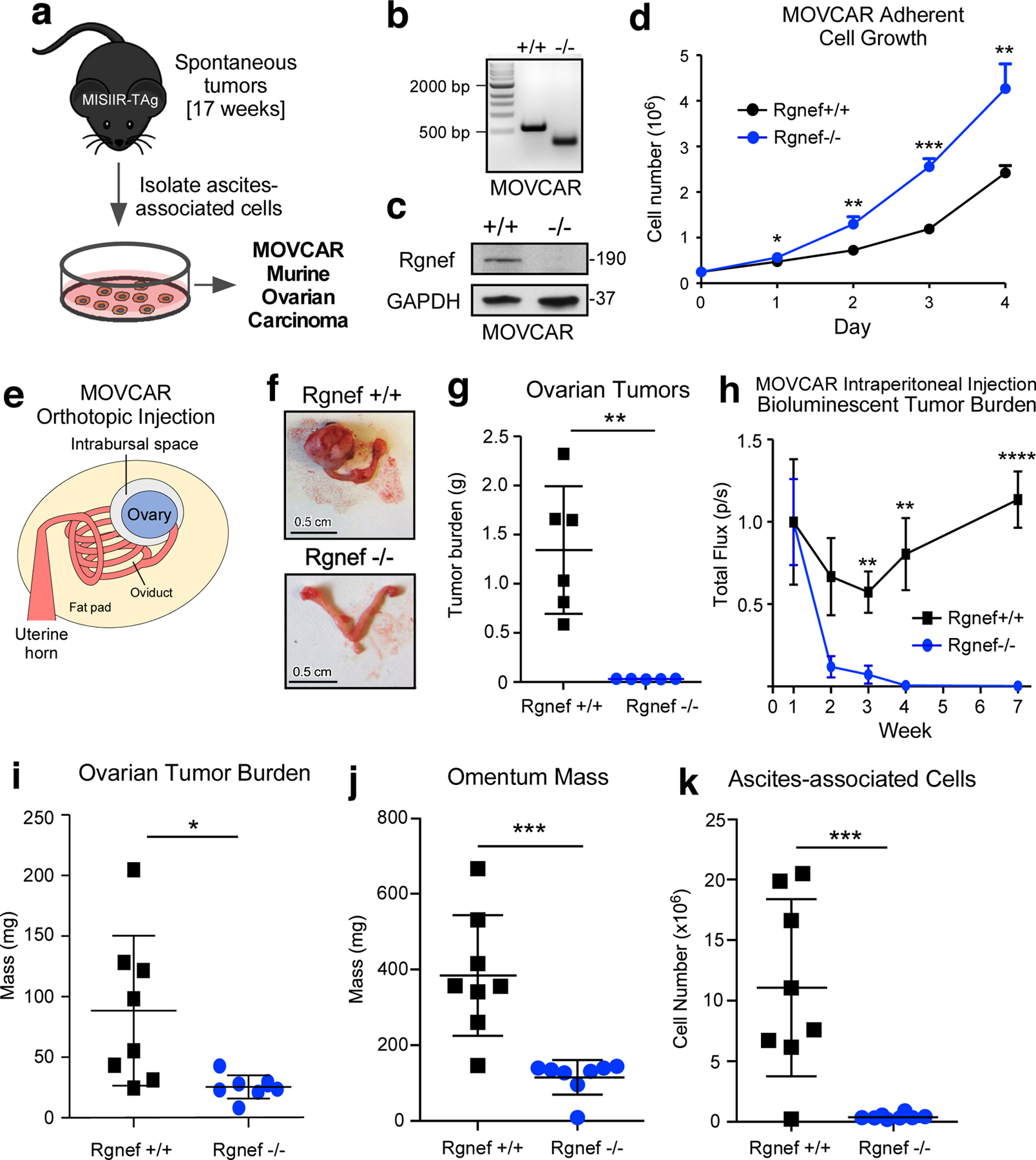
Cell-intrinsic role for Rgnef in promoting murine ovarian cancer (MOVCAR) tumor growth. (a) Schematic of Rgnef+/+ and Rgnef−/− MOVCAR generation. (b) Representative Rgnef exon 24 PCR of MOVCAR cells from Rgnef+/+ and Rgnef−/− TAg+ mice confirming genomic wildtype Rgnef (582 bp) or deletion in Rgnef murine exon 24 (125 bp). (c) Immunoblotting showing Rgnef loss in Rgnef−/− MOVCARs using GAPDH as loading control. (d) Rgnef−/− MOVCARs grow faster in adherent conditions over 4 days (* P ≤ 0.05, ** P ≤ 0.01, ***P ≤0.001, +/−SD, n=3 independent experiments). (e) Schematic of MOVCAR cell orthotopic intrabursal injection into syngeneic MISIIR-TAg-Low; Rgnef+/+ mice. (f) Representative images of oviducts and ovaries from Rgnef+/+ and Rgnef−/− MOVCAR tumor-bearing mice. Scale is 0.5 cm. (g) Quantitation of Rgnef+/+ and Rgnef−/− MOVCAR orthotopic tumor growth (**P ≤ 0.01, +/− SD). (h) Bioluminescent imaging from mice injected intraperitoneally with Rgnef+/+ or Rgnef−/− MOVCARs. Total flux (photons/second) levels are normalized by group to week 1 for each cohort (**P ≤ 0.01, ****P ≤ 0.0001, n=8 for each group, +/− SEM). (i-k) Following intraperitoneal injection, ascites-associated cells were collected at week 14. Rgnef loss impaired metastatic potential as measured by (i) ovarian tumor burden, (j) omentum mass and (k) number of ascites-associated cells (*P ≤ 0.05, ***P ≤ 0.001, +/− SD, n=8 per group).
To test for growth differences between Rgnef+/+ and Rgnef−/− MOVCARs in vivo, cells were injected into the ovarian bursa space (Figs. 3e–g) of syngeneic C57Bl/6 MISIIR-TAg-Low mice [26]. At 15 weeks, ovaries of mice injected with Rgnef+/+ MOVCARs exhibited extensive tumor burden whereas ovarian tumors were very small in mice injected with an equal number of Rgnef−/− MOVCARs (Figs. 3f and g). To monitor potential changes in MOVCAR survival in vivo, Rgnef+/+ and Rgnef−/− MOVCARs were transduced with a luciferase reporter, injected into mice, and in situ luminance was evaluated over time (Fig. 3h). Within 4 weeks, the bioluminescent signal from Rgnef−/− MOVCARs was reduced to background whereas Rgnef+/+ MOVCAR signal continued to increase over time. After 14 weeks, Rgnef+/+ MOVCAR cells had formed large ovarian tumors, infiltrated the omentum, and non-adherent tumor spheroids were collected with ascites (Figs. 3i–k). Rgnef−/− MOVCAR cells failed to form ovarian or omental tumors and few ascites-associated tumor cells were recovered at 14 weeks (Figs. 3i–k). Together, these results support tumor-intrinsic Rgnef importance in promoting ovarian tumor growth.
CRISPR/Cas9 Rgnef knockout in ID8-IP cells
To test the role of Rgnef in a different ovarian murine tumor model, we utilized the aggressive murine ID8-IP cell line, generated by expansion of ID8 cells in C57Bl/6 mice [28]. Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/Cas9-mediated genome editing was used to target ID8-IP Rgnef exon 6, single cells were isolated by dilution cloning, and ID8-IP Rgnef knockout (KO) clones containing insertions or deletions in exon 6 were screened by genomic DNA PCR analyses (Fig. 4a). Interestingly, several Rgnef KO clones spontaneously stopped proliferating and failed to achieve sufficient cell expansion while Rgnef KO-1 and Rgnef KO-11 clones grew significantly slower than parental ID8-IP cells (Fig. 4b).
Figure 4:
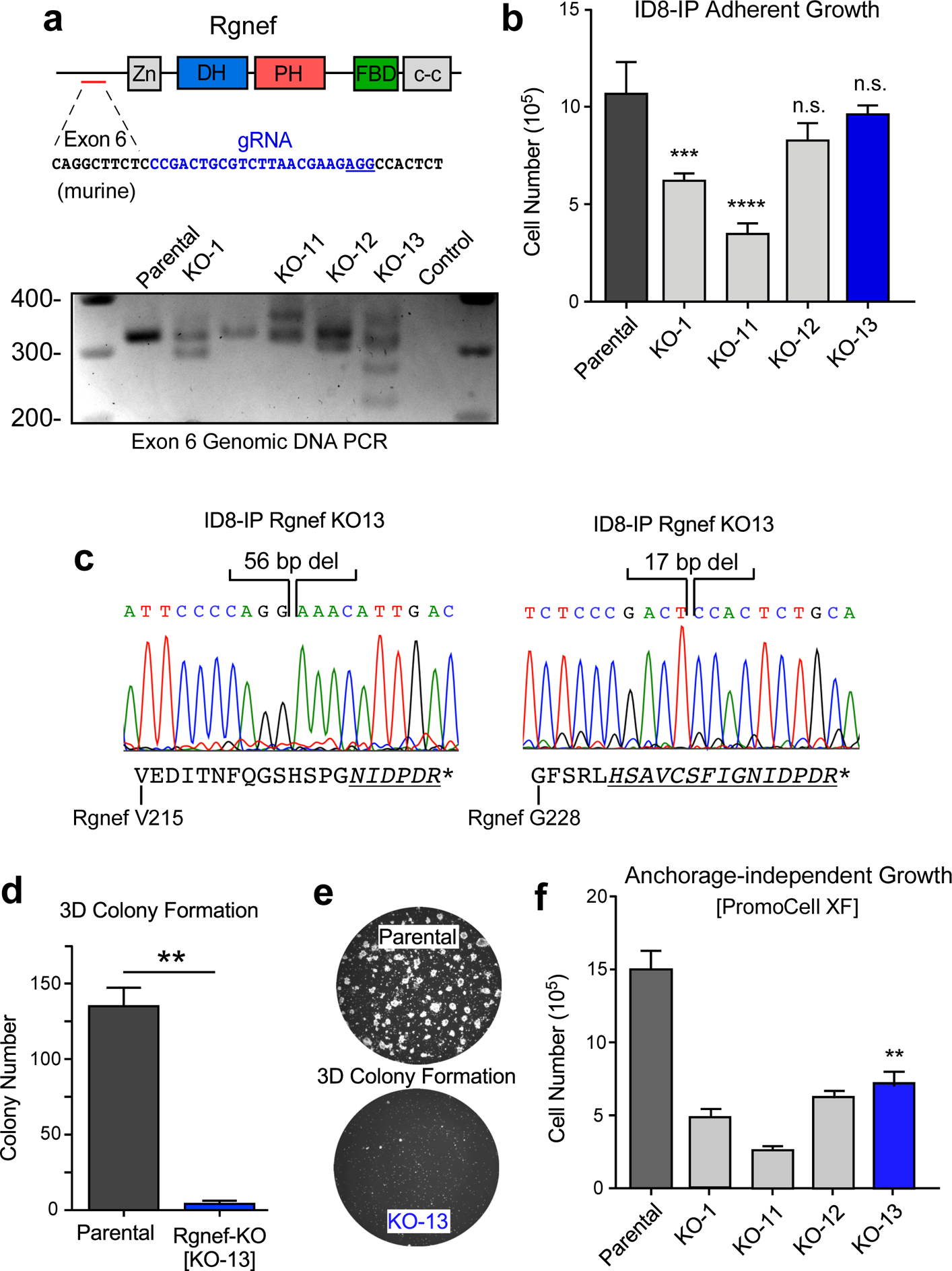
Rgnef loss specifically impairs 3D cell growth. (a) Schematic of CRISPR/Cas9 gRNA targeting of murine Rgnef exon 6 (blue text) within the N-terminal domain. Also shown are Rgnef protein domains: Zn - zinc finger, DH – Dbl homologous, PH - pleckstrin homology, FBD – FAK binding domain, c-c - coiled-coil region. Below: Exon 6 PCR was performed with genomic DNA to identify insertions or deletions in the indicated Rgnef KO clones. Parental ID8-IP cells yield a 393 bp band. (b) Adherent growth of ID8-IP and the indicated Rgnef-KO clones over 4 days (n.s.= no significance, *** P ≤0.001, **** P ≤ 0.0001, n=3 technical replicates, +/− SD). (c) Sanger sequencing of ID8-IP Rgnef KO-13 clone exon 6 identifying two independent deletions (56 and 17 bp) resulting in predicted translational termination. Alternative reading frame is underlined and italicized with position of stop codon (*) denoted (Uniprot protein G5E8P2). (d) ID8-IP and Rgnef KO-13 colony formation (** P ≤ 0.01, n=2 independent experiments, +/− SD). (e) Representative images of colonies in Matrigel. (f) Anchorage-independent growth of ID8-IP and the indicated Rgnef KO clones in serum-free conditions over 5 days (+/− SD, **P ≤ 0.01).
Growth of Rgnef KO clones KO-12 and KO-13 was equivalent to parental ID8-IP cells, consistent with normal growth of Rgnef−/− MOVCARs in standard culture conditions. ID8-IP Rgnef KO-13 cells contained two deletions in Rgnef exon 6, predicted to result in translational reading frame shifts and stop codon introduction (Fig. 4c). Interestingly, despite equivalent growth in adherent cell culture conditions, ID8-IP KO-13 cells (hereafter termed Rgnef-KO) did not grow as three-dimensional colonies embedded in Matrigel (Figs. 4d and e) and exhibited limited growth as spheroids in serum-free anchorage-independent culture conditions (Fig. 4f).
Rgnef promotes ID8-IP spheroid growth in vitro and in vivo
To confirm that Rgnef inactivation was causal for anchorage-independent ID8-IP growth defects, GFP-Rgnef was stably re-expressed in ID8-IP Rgnef-KO cells (Fig. 5). Transduced cells were enriched by fluorescence-activated cell sorting, full-length GFP-Rgnef expression was confirmed by immunoblotting (Fig. 5a), and GFP-Rgnef was localized to the cytoplasm and plasma membrane (Fig. 5b). Previous studies showed that GFP-Rgnef rescues signaling defects of murine Rgnef−/− fibroblasts [19, 29]. Notably, whereas no adherent growth differences were observed between Rgnef-KO and GFP-Rgnef re-expression (Fig. 5c), 3D colony growth of ID8-IP Rgnef-KO cells in Matrigel was significantly increased by GFP-Rgnef re-expression (Figs. 5d and e).
Figure 5:
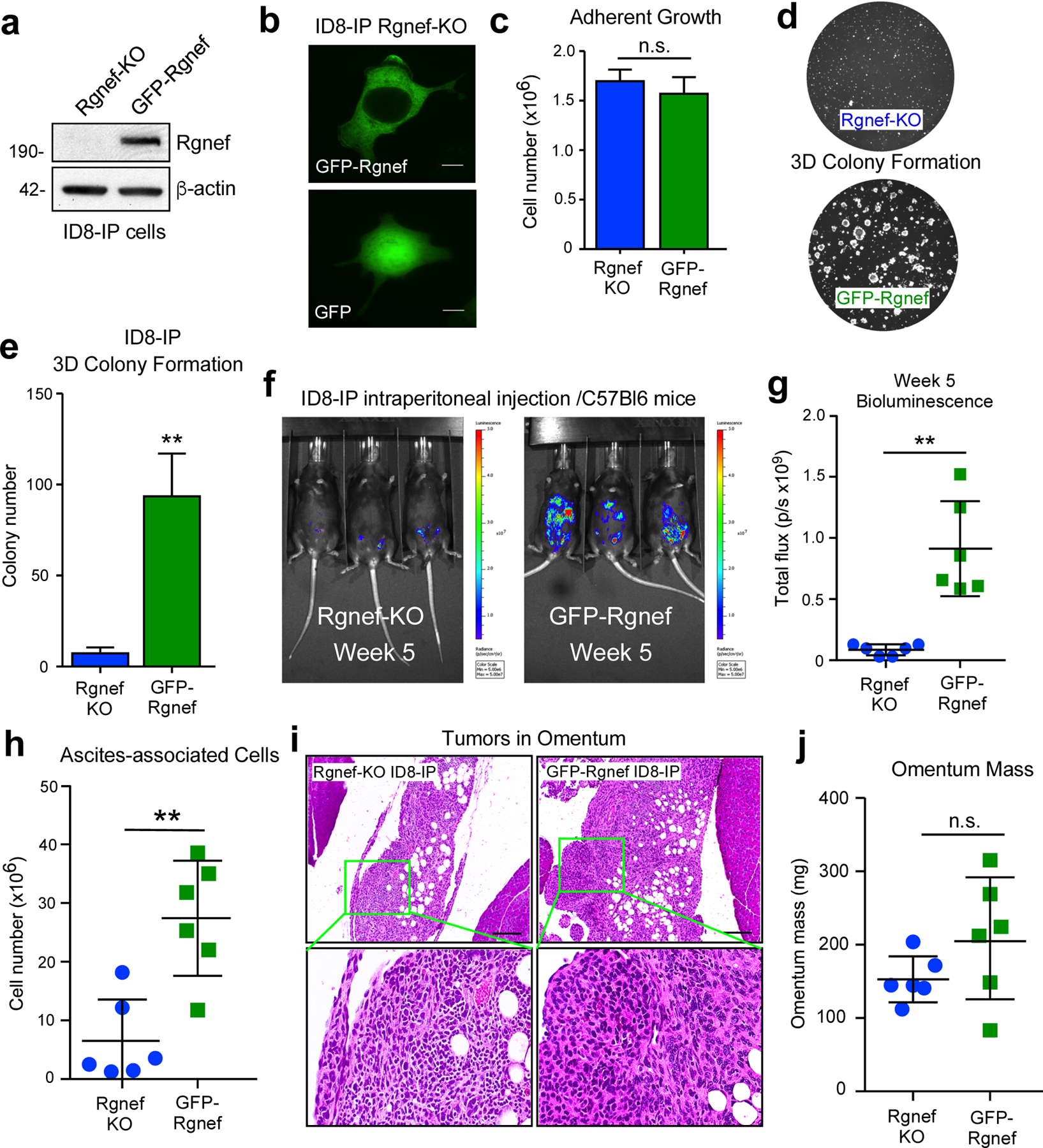
Rgnef promotes anchorage-independent growth in vitro and in vivo. (a) Anti-Rgnef immunoblotting showing GFP-Rgnef re-expression in ID8-IP Rgnef-KO cells. Actin is a control. (b) GFP-Rgnef or GFP distribution in ID8-IP Rgnef-KO cells visualized by confocal microscopy. Scale is 2 μm. (c) ID8-IP Rgnef-KO and GFP-Rgnef re-expressing cells exhibit no adherent growth difference (n=3 technical replicates, n.s.= no significance, +/− SD). (d) Colony growth is enhanced by GFP-Rgnef re-expression. Representative images (d) and quantification (e) are shown (**P ≤ 0.01, n=3 biological replicates, +/− SD). (f) Representative bioluminescent imaging of ID8-IP Rgnef-KO or GFP-Rgnef re-expressing cells after 5 weeks. (g) Bioluminescent flux quantified on experimental Day 34 (**P ≤ 0.01, n = 6, +/− SD). (h) Quantitation of ascites-associated cells recovered at Day 39 (**P ≤ 0.01, n = 6, +/− SD). (i) Representative H&E stained images of ID8-IP Rgnef-KO or GFP-Rgnef tumor implants within the omentum. Scale is 50 μm. Inset, high magnification. (j) Final omentum mass from ID8-IP Rgnef-KO or GFP-Rgnef tumor-bearing mice. Differences are not significant (n.s.)
Upon intraperitoneal injection of luciferase-labeled Rgnef-KO into C57Bl6 mice, only GFP-Rgnef expressing cells produced a significantly higher bioluminescent signal by IVIS imaging after 5 weeks (Figs. 5f and g). Combined Vi-Cell and flow cytometry analyses confirmed elevated number of cells recovered from GFP-Rgnef compared to Rgnef KO tumor-bearing mice after 39 days (Fig. 5h). However, small Rgnef-KO tumors were detected within omental fat tissue by sectioning and H&E staining (Fig. 5i). These tumors exhibited a dense staining pattern compared to GFP-Rgnef tumor implants. Although there was a trend of increased total omental tissue-tumor mass in GFP-Rgnef tumor bearing mice, there was no significant difference (Fig. 5j). Together, these results support the notion that Rgnef expression contributes to anchorage-independent ID8-IP cell growth, and this may underlie the enhanced tumor-promoting role for Rgnef in vivo.
Rgnef enhances an antioxidant gene signature
To explore whether Rgnef supports changes in gene expression that may enhance the anchorage-independent growth phenotype observed in MOVCARs and ID8-IP models, microarray gene expression profiling was performed with Rgnef+/+ and Rgnef−/− MOVCARs (Figs. 6a and b) and RNA sequencing was performed with Rgnef-KO and GFP-Rgnef re-expressing ID8-IP cells (Figs. 6c–f). In MOVCARs, 394 genes were upregulated and 774 genes were downregulated between Rgnef+/+ vs Rgnef−/− cells. Gene set enrichment analysis (GSEA) revealed elevated levels of targets associated with oxidoreductase activity (Fig. 6a, Supplemental Table S1) and Kyoto Encyclopedia of Genes and Genomes (KEGG) revealed enrichment of pathways including glutathione metabolism in Rgnef+/+ MOVCARs (Fig. 6b, Supplemental Table S1).
Figure 6:
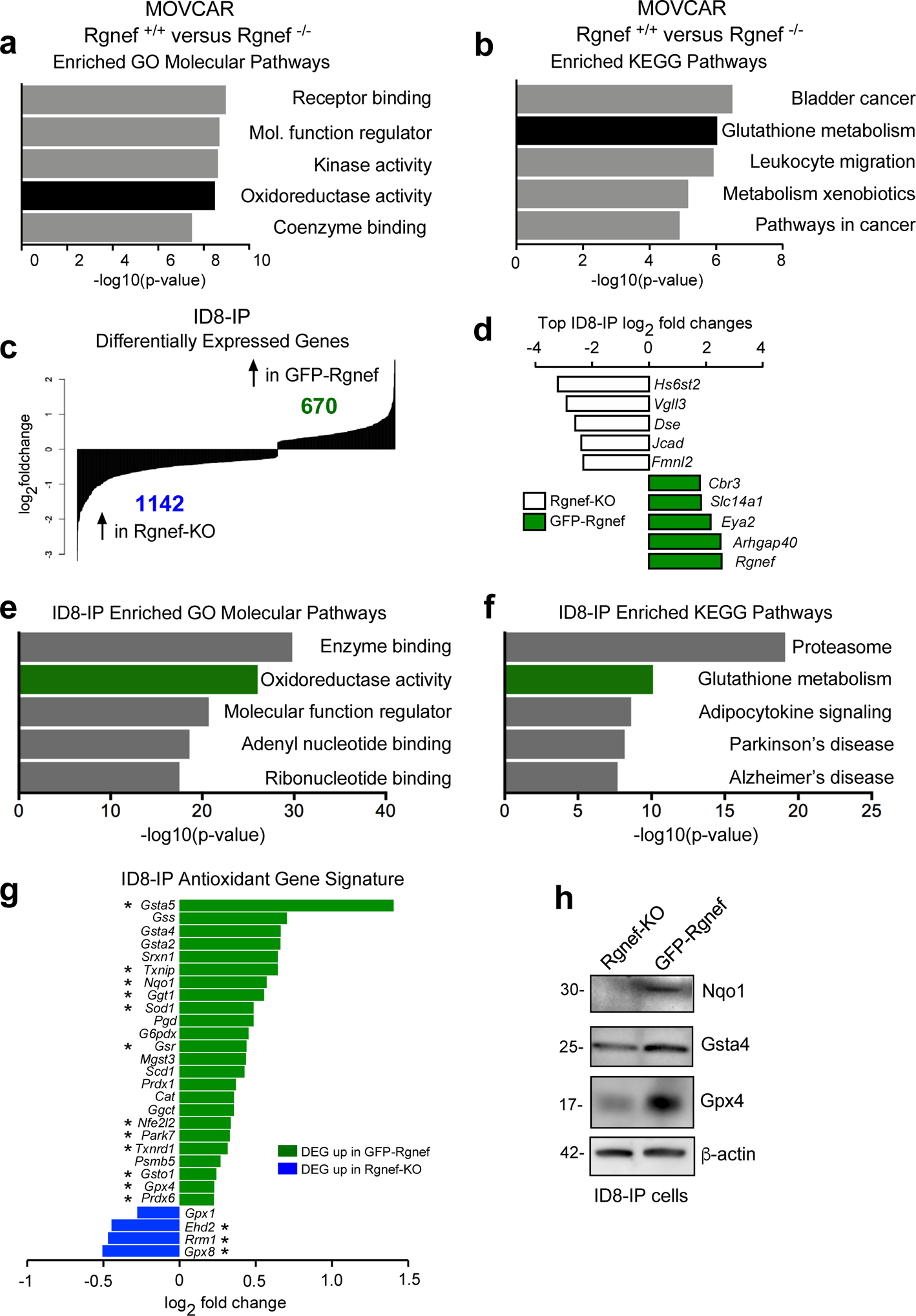
Rgnef promotes an antioxidant gene signature. (a) Differentially upregulated mRNAs in MOVCAR Rgnef+/+ as compared to Rgnef−/− were determined using Illumina BeadChip Array and processed by Gene Set Enrichment Analysis (GSEA). Top 5 most enriched GO molecular function (a) or KEGG pathways (b) in the set of 313 upregulated transcripts are shown. (c) RNA sequencing was used to determine differential mRNA levels in Rgnef-KO and GFP-Rgnef re-expressing ID8-IP cells. 670 targets were upregulated in the GFP-Rgnef re-expressing, and 1142 targets were upregulated in Rgnef-KO cells. (d) The top log2 fold changes in the ID8-IP Rgnef-KO or GFP-Rgnef re-expressing cells are shown. (e) Top 5 most enriched GO molecular function or (f) KEGG pathways in the set of 670 upregulated genes in the ID8-IP GFP-Rgnef re-expressing cells. Enrichment score was determined using GSEA. (g) Differentially expressed genes were compared to a curated list of antioxidant genes (Supplemental Table 2). Genes differentially upregulated (green) or downregulated (blue) in ID8-IP GFP-Rgnef cells vs Rgnef-KO cells are shown. (h) Immunoblotting shows increased levels of Nqo1, Gsta4, and Gpx4 in lysates of anchorage-independent (5 days) ID8-IP GFP-Rgnef cells with β-actin as a loading control.
In ID8-IP cells, 670 genes were upregulated and 1142 genes were downregulated in GFP-Rgnef re-expressing compared to Rgnef-KO cells (Fig. 6c, GSE126560). Rgnef had the highest global top fold-change in ID8-IP cells, as expected for cells engineered to re-express GFP-Rgnef (Fig. 6d). Notably, oxidoreductase activity and glutathione metabolism were also enriched in GFP-Rgnef cells by GO and KEGG pathway analyses, respectively (Figs. 6e–f and Supplemental Table S1). The shared alterations between MOVCAR and ID8-IP cells with or without Rgnef reinforce the potential importance of Rgnef in regulating these pathways.
Glutathione is the largest pool of antioxidant reducing equivalents in the cell, and reduced glutathione is used as a substrate for antioxidant enzymes that protect against oxidative stress [8]. Therefore, we investigated differential expression of antioxidant genes in ID8-IP GFP-Rgnef versus Rgnef-KO cells using a curated list that includes glutathione peroxidases, thioredoxins, peroxiredoxins, superoxide dismutases, and members of the glutathione metabolism pathway (Supplementary Table S2). Twenty-four of these transcripts were upregulated and four were downregulated in response to GFP-Rgnef re-expression (Fig. 6g). Elevated protein levels of targets such as Nqo1 (reductase), Gsta4 (glutathione transferase), and Gpx4 (glutathione peroxidase) were confirmed by immunoblotting (Fig. 6h). Together, these results demonstrate that Rgnef supports the expression of common targets within an antioxidant gene set.
Increased ROS limits anchorage-independent Rgnef-KO growth
Loss of matrix attachment can increase ROS levels in ovarian carcinoma cells, which in turn can trigger anoikis [11, 30]. As Rgnef loss prevents ID8-IP growth as spheroids and alters antioxidant-associated transcripts, ROS levels in suspended Rgnef-KO and GFP-Rgnef cells were measured using a redox-sensitive fluorescent probe (Fig. 7a). Elevated ROS levels were present in Rgnef-KO cells compared to cells expressing Rgnef. Addition of the antioxidant Trolox, a water-soluble vitamin E analog, to the culture media reduced ROS levels in suspended Rgnef-KO cells (Fig. 7a). Treatment with Trolox also promoted anchorage-independent Rgnef-KO growth under defined serum-free media conditions (Fig. 7b). However, Trolox did not alter anchorage-independent GFP-Rgnef cell growth, supporting the notion that these cells do not derive additional benefit from antioxidant treatment. These results show that oxidative stress is a limiting factor for anchorage-independent ID8-IP Rgnef-KO growth.
Figure 7:
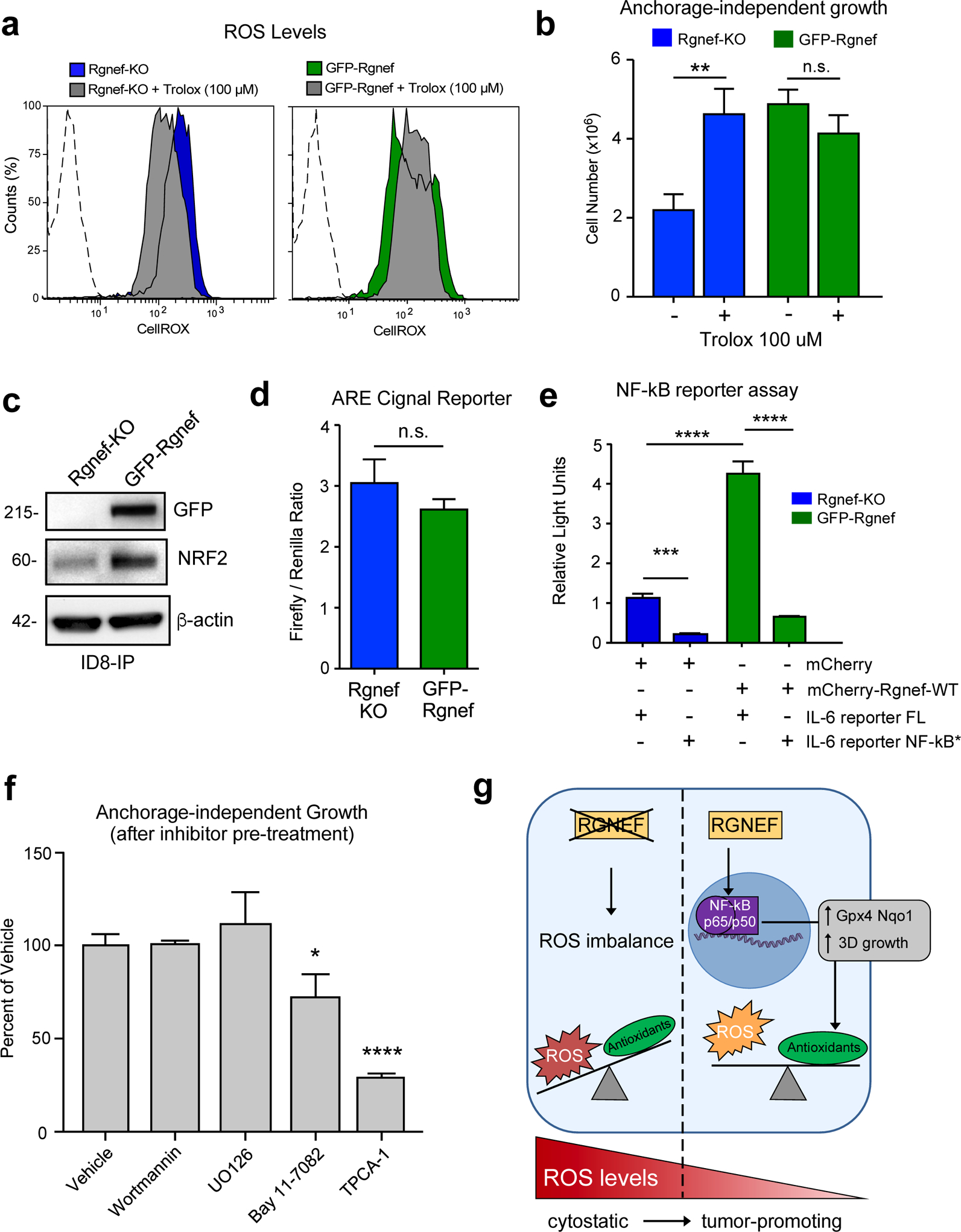
Rgnef supports NF-κB activation needed for ID8-IP anchorage-independent cell growth. (a) Cellular reactive oxygen species (ROS) levels in ID8-IP Rgnef-KO and GFP-Rgnef re-expressing cells as measured by CellROX staining with or without antioxidant Trolox (100 μM, 1 h) addition. Flow cytometry histograms are shown. Negative control (dotted line). (b) Trolox increases ID8-IP Rgnef-KO growth in suspension (** P ≤ 0.01, n=3 independent experiments, +/− SD). (c) Immunoblot of ID8-IP Rgnef KO and GFP-Rgnef cell lysates for GFP, NRF2, and actin protein levels. (d) GFP-Rgnef expression did not enhance ARE (antioxidant response element) reporter for NRF2 transcriptional activity in human 293T cells (n.s.= no significance, +/− SD, n=3 technical replicates). (e) mCherry-Rgnef transfection activates an IL-6 luciferase reporter in human 293T cells dependent on NF-κB DNA binding site integrity (NF-κB*). Dual-luciferase activity was measured and normalized (***P ≤ 0.001, ****P ≤ 0.0001, +/− SD, n=3 technical replicates). (f) Adherent ID8-IP GFP-Rgnef cells were pre-treated with DMSO (vehicle), 1 μM wortmannin, 10 μM U0126, 10 μM Bay 11–7082, or 10 μM TPCA-1 for 24h, washed, and then evaluated for anchorage-independent growth (n.s.=no significance, *P ≤ 0.05, ****P ≤ 0.0001, +/− SD, n=3 technical replicates). (g) Model of Rgnef in mediating cell response to oxidative stress. Upon Rgnef loss, the cell is not able to balance increased ROS resulting from growth in suspension, resulting in oxidative stress. When Rgnef is re-expressed, NF-κB-mediated transcription downstream of Rgnef promotes the expression of an antioxidant gene signature, resulting in redox homeostasis.
Rgnef supports NFκB transcription factor activation
To explore possible regulatory mechanisms underlying the Rgnef-associated antioxidant mRNA changes, we queried the ChEA (Chip Enrichment Analysis) database using the 670 transcripts upregulated in ID8-IP GFP-Rgnef re-expressing cells. ChEA calculates overrepresented transcription factor targets in a gene set against a database of mammalian Chip-X experiments [31]. Transcription factors with the most significant target overlap with genes upregulated by GFP-Rgnef included NRF2, KLF1, RELA, and SOX2 (Table 1). NRF2 is a known master regulator of the antioxidant response [32] and ID8-IP GFP-Rgnef cells express elevated NRF2 levels compared to Rgnef-KO cells by immunoblotting (Fig. 7c). However, the activity of NRF2 transcriptional reporter was similar in ID8-IP Rgnef-KO and GFP-Rgnef cells (Fig 7d).
Table 1:
Chromatin immunoprecipitation (ChIP) Enrichment Analysis (ChEA) of transcription factor binding sites of differentially-expressed genes in GFP-Rgnef re-expressing ID8-IP cells as compared to Rgnef-KO.
| Transcription Factor | Overlap | Adjusted P-value | Combined Score | PMID |
|---|---|---|---|---|
| NRF2 | 104/1331 | 1.91E-13 | 66.7 | 26677805 |
| NRF2 | 83/1055 | 6.60E-11 | 54.2 | 20460467 |
| KLF1 | 90/1239 | 4.06E-10 | 47.2 | 21900194 |
| RELA | 82/1182 | 2.37E-08 | 37.6 | 24523406 |
| SOX2 | 121/2000 | 8.80E-09 | 34.6 | 27498859 |
RELA, a component of the NF-κB transcription factor complex, is activated by ROS and can protect cells from oxidative stress [13]. Notably, twelve of the twenty-four antioxidant gene targets identified as differentially upregulated in ID8-IP GFP-Rgnef cells are targets of NF-κB (Fig. 6g, starred), including NRF2. Additionally, RELA was one of the transcription factors identified by ChEA analysis (Table 1). NF-κB can promote interleukin-6 (IL-6) gene expression [33] and GFP-Rgnef over-expression activated an IL-6 transcriptional reporter (Fig. 7e). IL-6 reporter activation was dependent on NF-κB DNA binding site integrity (Fig. 7e) and over-expression of RhoGEF-inactive point-mutant GFP-Rgnef Y1003A did not elevate IL-6 reporter activity (Supplemental Figure S1a). Together, these results show that Rgnef signaling supports NF-κB-mediated gene expression in a GEF-dependent manner [34].
Importance of NF-κB in ID8-IP spheroid growth
ID8-IP are aggressive cells that exhibit up-regulated FAK expression and FAK activation in anchorage-independent conditions [28, 35]. As Rgnef can function upstream to promote FAK activation [29], and FAK inhibitor treatment prevents ID8-IP anchorage-independent growth [28], signaling inhibition downstream of Rgnef-FAK was tested for effects on ID8-IP anchorage-independent spheroid growth (Fig. 7f). ID8-IP three-dimensional spheroid growth over 5 days remain unchanged after 24-hour pre-treatment of cells with wortmannin or UO126, inhibitors of phosphatidylinositol kinase and mitogen-activated protein kinases, respectively. However, ID8-IP growth was inhibited by pre-treatment with inhibitors of NF-κB signaling, Bay 11–7082 and TPCA-1 (Fig. 7e). Inhibitor action in ID8-IP cells was confirmed by immunoblotting (U0126 and wortmannin) and by using an NF-κB-dependent transcriptional reporter (Supplemental Fig. S1b–d). Together, these results support the importance of NF-κB activation downstream of Rgnef in the regulation of oxidative stress and anchorage-independent growth.
Discussion
Signaling pathways that confer protection from oxidative stress can become activated in ovarian cancer and sustain cell survival in anchorage-independent conditions. Rgnef (ARHGEF28) is a large protein that canonically functions downstream of integrin receptors to control cell adhesion and RhoA GTPase activity upon cell engagement with matrix proteins [17]. Interestingly, proteomic and transcriptomic analyses have identified RhoA regulatory and integrin-linked kinase pathways as elevated in short versus long-term OC survivors [22]. Previously, elevated Rgnef expression has been shown to activate FAK activation, paxillin tyrosine phosphorylation, cell motility, matrix degradation and colon carcinoma tumor metastasis in a mouse model [20]. Although it is hypothesized that OC patient survival differences may be linked to increased cell motility and tumor metastasis [22], we find that elevated Rgnef levels confer additional phenotypes to OC – reduction in oxidative stress and enhancement of anchorage-independent cell growth.
Rgnef protein levels are increased in late-stage serous OC, high Rgnef mRNA levels are associated with decreased progression-free and overall survival, and genomic ARHGEF28 loss is associated with increased patient survival. Using transgenic and transplantable Rgnef KO mouse models, we find that Rgnef is essential for supporting three-dimensional ovarian spheroid formation in vitro and tumor growth in mice. Using RNA-sequencing and bioinformatic analyses, we identify a conserved Rgnef-supported anti-oxidant gene signature including GPX4, Nqo1, and Gsta4; common targets of the NF-kB transcription factor. Our results support a model in which Rgnef KO disrupts cellular ROS equilibrium, leading to enhanced cellular stress, and acting to limit tumor spheroid growth (Fig. 7g). Notably, addition of the antioxidant Trolox to Rgnef KO cells decreased ROS levels and increased anchorage-independent cell growth. These results support a novel connection between Rgnef, the regulation of oxidative stress, and ovarian tumorigenesis.
Anchorage-independent survival is a hallmark of oncogenic cell transformation. Progression and malignancy in advanced stage OC proceeds as a function of the survival and dissemination of peritoneal spheroids. One of the barriers to achieving anchorage-independent survival is the mitigation of elevated ROS levels after matrix detachment [11]. By comparisons of Rgnef KO and GFP-Rgnef re-expressing ID8-IP cells, we show that Rgnef protects cells from increased ROS in anchorage-independent conditions. This role for Rgnef is consistent with previous findings in other models whereby Rgnef promotes cell survival after elevation of oxidative or other stress stimuli [36, 37]. Although we find that a common set of antioxidant genes were regulated by Rgnef by comparisons of MOVCAR and ID8-IP knockout mouse models, the promoters of these genes also share common transcription factor binding sites for NRF2, KLF1, RELA (the 65 kDa subunit of NF-κB), and SOX2. We did not detect direct regulation of a NRF2 reporter. However, Rgnef expression and Rgnef GEF activity were required to activate an NF-κB reporter. Although it is interesting that SOX2 is also elevated in ovarian cancer [38], analyses of potential signaling connections between Rgnef and KLF1 or SOX2 remain under investigation.
In addition to inducing antioxidant gene expression, NF-kB activation downstream of Rgnef may also confer cellular stress protection via regulation of anti-apoptotic genes [13]. Nevertheless, as Rgnef knockout reduced primary and metastatic tumor burden, increased cellular ROS, and impaired anchorage-independent growth, targeting spheroid survival and triggering anoikis via Rgnef inhibition may provide OC patients with therapeutic benefits. To this end, small molecule inhibitors blocking other RhoGEF and RhoA interactions are in early development [39] and this holds potential for future studies aimed at inhibiting tumor progression and metastasis.
Materials and Methods
Antibodies, Plasmids, and Reagents
Rabbit polyclonal antibodies to Rgnef (#3647 and #1397) were affinity-purified and used as described [20]. Antibody to β-actin (AC-17) was from Sigma. GAPDH (GT239) antibody was from GeneTex. NRF2 (A-10) and SV40 TAg (Pab 101) antibodies were from Santa Cruz Biotechnology. GFP (B34) was from Biolegend. NQO1 (A180), GPX4 (EPNCIR144), and Ki67 (SP6) were from Abcam. GSTA4 (ABS1652) was from Millipore. Akt (pan) (C67E7, #4961) and p-Akt (Ser473) (#9271) were from Cell Signaling. Phospho-ERK1/ERK2 (Thr185, Tyr187) (44–680G), ERK1/ERK2 (44–654G), AlexaFluor-488, and AlexaFluor-647 labeled antibodies were from ThermoFisher. Growth factor reduced Matrigel (GFR, 356231) was from Corning. D-luciferin for IVIS imaging was from PerkinElmer. Trolox, wortmannin, U0126, and Bay 11–7082 were from Sigma-Aldrich. TPCA-1 was from Abcam.
Cre-driven Rgnef recombination in Rgnef−/− MOVCARs was determined by PCR using forward (5’-ACTGCAGATCAGCATGTCTTG-3’ and reverse (5’-GCTGCTATCTCCAAACGCTAT-3’) primers. PCR for TOPO-TA cloning in ID8-IP Rgnef-KO clones was performed using forward (5’-CTGGGAAGCTGTGTGGATTT-3’) and reverse (5’-AGGACATGGGGTTAGAGCCT-3’) primers. The dTomato-luciferase (pUltra-Chili-Luc) vector was a gift from Malcolm Moore (Addgene #48688). dTomato-luciferase and pCDH-CMV-MCS1-eGFP were used to produce lentivirus as described [18]. Cells were transduced using standard methods (System Biosciences) and enriched by fluorescence-activated cell sorting. High-titer lentivirus using pCDH-CMV-MCS1-eGFP-Rgnef [18] was produced by Systems Biosciences. CRISPR gRNA construct was from GenScript (5’-CCGACTGCGTCTTAACGAAG-3’ in pSpCas9 BB-2A Puro PX459). The pmIL-6 FL and pmIL-6 mutant NF-kB plasmids were gifts from Gail Bishop (Addgene p#61286 and #61293, respectively). pTK-Green Renilla Luc Vector was from ThermoFisher.
Cells
Mouse ovarian carcinoma (MOVCAR) cells were isolated from ascites of tumor-bearing MISIIR-TAg+; Rgnef+/+ or Rgnef−/− mice and expanded by adherent growth, as described [23]. ID8-IP cells were isolated from ID8 peritoneal ascites and expanded in suspension, as described [28]. 293T Lenti-X cells were from Clontech. Cell lines were periodically tested for Mycoplasma. For adherent growth, ID8-IP and 293T Lenti-X cells were maintained in DMEM (Corning) supplemented with 10% FBS, 100 μM non-essential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin on tissue culture-treated plastic plates (Corning). MOVCAR cells were maintained in DMEM (Corning) supplemented with 4% FBS, 100 μM non-essential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1x insulin-transferrin-selenium (Mediatech). CRISPR/Cas9-mediated knockout was performed as previously described [40] using a guide RNA in the PX459 vector. Confirmation of Rgnef exon 6 deletions in ID8-IP Rgnef-KO clones was performed using TOPO-TA cloning (Thermo Fisher), followed by Sanger sequencing and analyses using MacVector (v16.0.8).
Cell Growth
For 2D growth, cells were seeded (3×105 cells per well) in tissue culture-treated 6-well plates (Costar). At the indicated time, cells were enumerated and stained with Trypan blue (ViCell XR, Beckman). For 3D anchorage-independent growth, cells were seeded at 10,000 cells/ml equivalent in poly-HEMA-coated 6-, or 24-well plates (Costar). After 5 days, cells were phase-contrast imaged (Olympus CKX41), enumerated (ViCell XR), or collected by centrifugation. Colony growth was evaluated in a dense layer of Matrigel diluted 1:1 with culture media solidified for 30 mins at 37°C. A single-cell suspension (3000 cells/cm2 in a 1:50 dilution of Matrigel and culture medium) was added on top of the base layer at twice the volume of the base layer and cultured at 37°C. Colony growth over 10 days was monitored by phase-contrast imaging (Olympus) and colony number and size were determined using ImageJ.
Protein Analyses
Cell protein extracts were prepared using a lysis buffer containing (25 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 10 mM MgCl2, 1 mM EDTA, 10 mM NaF, 1 mM Na3VO4) with 1% NP-40, 0.25% sodium deoxycholate, 0.1% SDS and protease inhibitors (Roche Diagnostics). Total protein levels in lysates were determined through a bicinchoninic acid assay (Pierce), proteins were resolved by SDS-PAGE (NuPAGE 4–12 % Tris-Bis gels, Thermo), and transferred to polyvinylidene difluoride membranes (Immobilon-FL, Millipore) for immunoblotting. Levels of protein expression and/or phosphorylation were detected with specific primary antibodies and HRP-conjugated secondary antibodies with chemiluminescent detection (ChemiDoc, BioRad).
Mice and tumor experiments
All animal experiments were performed in accordance with The Association for the Assessment and Accreditation for Laboratory Animal Care (AALAC) guidelines and were approved by the University of California San Diego Institutional Animal Care and Use Committee (IACUC). Rgnef−/− mice were developed and genotyped as described [19]. MISIIR-TAg+ and MISIIR-TAg-Low mice were developed as described [23, 26]. High resolution ultrasound imaging of MISIIR-TAg+ mice was performed using a Vevo 2100. For spontaneous tumor experiments, mice were euthanized when tumors reached standards for humane endpoint, or by 18 weeks.
ID8-IP and MOVCAR cells were transduced with a lentiviral vector expressing dTomato and luciferase (pUltra-Chili-Luc) and cells were enriched by FACS. Orthotopic bursa injections were performed as described [28]. Briefly, MOVCAR cells were mixed with Matrigel (GFR, Corning), at a concentration of 0.5 × 106 cells per 7 μL and injected into the ovarian bursal space using a Hamilton syringe and 29.5-gauge needle. Incisions were closed with surgical stables and mice were evaluated for health daily. For intraperitoneal injection, cells were mixed with PBS + 25% Matrigel (GFR, Corning) for a final concentration of 7.5 × 106 (MOVCAR) or 5 × 106 (ID8-IP) cells per 250 μL. Ascites-associated cells were recovered by peritoneal washings by injection and immediate removal of PBS (5 ml), followed by erythrocyte lysis (RBC lysis buffer, eBioscience), trypsinization, and total cell enumeration with trypan blue viability staining (ViCell XR, Beckman). Tumor growth was monitored via luciferase bioluminescent imaging (IVIS, Perkin Elmer).
Kaplan-Meier analyses
Kaplan-Meier survival analyses using patient data were performed using the integrated datasets from three major research centers (Berlin, Bethesda and Melbourne datasets), accessed at (http://kmplot.com/analysis/) [41]. Parameter selections were: Affymetrix probe ARHGEF28 (1560348_at), auto-select cutoff, stages III-IV, serous histology, exclude outlier arrays, debulk all, chemotherapy all, and follow-up threshold all. TCGA copy number variation data was accessed using the cBioPortal online tool (http://www.cbioportal.org) to interrogate the TCGA high grade serous ovarian cancer provisional dataset [42, 43].
RNA and bioinformatics analyses
For differential RNA expression from MOVCAR cells, total RNA was isolated from adherently grown Rgnef+/+ or Rgnef−/− MOVCAR cells (n=1) grown using PureLink RNA Mini Kit (ThermoFisher). Sample preparation (Ambion/Illumina TotalPrep RNA amplification kit) and runs (Illumina BeadChip Microarray MouseRef-8 v2.0) were performed at the UCSD Veterans Medical Research Foundation Gene Core. Differentially-expressed genes had at least two-fold differences (P < 0.05).
For RNA-sequencing, total RNA was isolated from ID8-IP Rgnef-KO or GFP-Rgnef cells grown on a 0.2% Matrigel 3D matrix (n=3 per group) using PureLink RNA Mini Kit (Thermo Fisher). RNA library preparation and sequencing were performed by Novogene Inc. (Beijing, China). RNA samples were prepared using NEB Next Ultra RNA Library Prep Kit (New England Biolabs) as per manufacturer recommendations. Each transcriptome was sequenced using a 150 bp paired-end protocol on the Illumina HiSeq 4000 platform, generating at least 37 million reads for all samples. Reads were mapped to the reference genome (>87% mapped) using TopHat2 [44]. Differential expression analysis was performed using the DESeq2 R package [45]. To test for enriched pathways, we used the online version of GSEA [46]. ChEA analysis was performed using Enrichr [31, 47]. The RNA-Seq FASTQ files used in this paper have been deposited to the NCBI Gene Expression Omnibus for under the accession number GSE126560.
Immunohistochemistry
For paraffin-embedded staining, ovary or omentum was removed and fixed in formalin for staining using TAg antibody or hematoxylin and eosin (H&E). Tissue deparaffinization, rehydration, antigen unmasking, and peroxidase quenching was performed as described [28]. Tissues were blocked (10% goat serum, 0,1% TritonX-100 in PBS) for 45 minutes and incubated with anti-TAg (1:200) or anti-Rgnef antibody (#1397, 1:100) overnight in blocking buffer. Biotinylated goat-anti-mouse/rabbit IgG (1:300 in PBS), Vectastain Elite ABC, and diaminobenzidine (Vector Labs) were used to visualize antibody binding. Slides were counter-stained with hematoxylin or methyl green. Images were captured using an upright microscope (Olympus BX43) with a color camera (Olympus SC100). Human tumor microarrays OV481, OV803, OV807, OV811, OV1001, and OV8011 containing grade and stage information were from US Biomax (Derwood, MD). High resolution digital scans were acquired (Aperio CS2 scanner) using Image Scope software (Leica Biosystems). Quantification was performed using Aperio Image Analysis software (v12.3.0.5056) using the positive pixel count (v9) algorithm. At least 20 tumor cores from Stage I, Stage II, Stage III from serous ovarian adenocarcinoma were digitally analyzed. Normal adjacent is cancer-adjacent normal ovarian tissue.
For cryo-staining, ovaries were snap-frozen in Optimal cutting temperature (OCT) compound (Tissue Tek) and thin-sectioned, (Leica CM1950) and mounted onto glass slides. For TAg and DNA visualization, slides were fixed in acetone, rehydrated (PBS, 0.5% BSA), blocked (1:50 normal goat serum, 0.5% BSA in PBS), and incubated overnight with TAg antibody (1:100). Slides were incubated with secondary antibody (AlexaFluor 488, 1:300) and Hoechst (1:50,000), and coverslips were mounted with Vectashield Hardset (Vector Labs). For GFP visualization, cells were plated onto glass-bottom dishes (MatTek). All immunofluorescence was visualized using an inverted microscope (IX81, Olympus) and Slidebook software (v5.0, Intelligent Imaging). Images were pseudocolored using ImageJ software.
Cellular ROS levels
ID8-IP Rgnef-KO or GFP-Rgnef re-expressing cells were grown adherently, trypsinized, enumerated (ViCell XR) and equal numbers of cells were seeded in poly-HEMA-coated low-attachment plates (Costar) in serum-free define media (PromoCell XF). After 24 h, cells were treated with vehicle or 100 μM Trolox (1 hr) prior to staining with 750 nM CellROX Deep Red Reagent (Invitrogen) for 30 mins followed by flow cytometry analyses (FACScalibur, BD).
Reporter assays
293T Lenti-X cells were seeded in triplicate at 2.25×105 cells/well in 24 well plates. For the NF-kB reporter assay, cells transfected with either pCDH-MCS1-mCherry-Rgnef, or pCDH-MCS1-GFP-p190RhoGEF Y1003A and either pMIL-6 FL or pmIL-6 mut NF-kB using Fugene HD (Promega) or jetPRIME (Polyplus). For inhibitor treatments, 30 min before transfection, cells were treated with vehicle or 10 μM Bay 11–7082, or 10 μM TPCA-1 and further incubated for 24 h. The ARE (antioxidant response element) Cignal™ Reporter Assay (Qiagen) was performed according to standard manufacturer instructions. Dual-Luciferase Reporter Assay system (Promega) was carried out 24h after transfection according to the manufacturer’s protocol using a Synergy HTX multi-mode plate reader (BioTek). As an internal control of transfection efficiency, the renilla luciferase encoding plasmid (pRL-TK, Promega) was co-transfected and for each sample, firefly luciferase activity was normalized to the renilla luciferase activity.
Statistical Methods
No statistical methods were used to predetermine sample size. Mouse cages were randomized between treatments in all in vivo allograft experiments. For MOVCAR bursa and intraperitoneal injection, mice were randomly chosen from pool of available female MISIIR-TAg-Low mice aged 8–10 or 10–12 weeks, respectively, at the time of injection. No blinding was used during in vivo experiments. In bursa injection experiments, MISIIR-TAg-Low mice with injection site leaks were removed from the experiment. Statistical difference between experimental groups was determined using one-way ANOVA with Bonferroni post-hoc analysis using a Tukey post-hoc analysis. Differences between pairs of data were determined using an unpaired two-tailed Student’s t-test. All statistical analyses were performed using Prism (GraphPad Software, v7). P-values of <0.05 were considered significant.
Supplementary Material
Supplemental Figure S1. Rgnef signaling and NF-κB reporter activation. (a) Human 293T transfection with mCherry-tagged Rgnef wildtype, Rgenf Y1003A (GDP exchange defective) and an IL-6 luciferase reporter with or without mutated NF-κB binding sites. (b and c) Adherent ID8-IP GFP-Rgnef cells were serum starved overnight, pre-treated with DMSO (vehicle), 10 μM U0126, or 1 μM wortmannin for 2h, and then stimulated with 5% fetal bovine serum plus 5% insulin-transferrin-selenium supplement for the indicated times prior to cell lysis. (b) Immunoblotting for phosphorylated ERK1/2 (T185/Y187) or total ERK1/2. (c) Immunoblotting for phosphorylated Akt (S473) or total Akt. (d) Human 293T cells were transfected as above and incubated with 10 μM Bay 11–7082 or 10 μM TPCA-1 for 24 h prior to analysis of luciferase activity. (a and d) Mean luciferase values are normalized from three independent analyses (*** P <0.001 and **** P < 0.0001, unpaired T test).
Acknowledgements
Supported by grants RO1CA180769, RO1CA102310, and UCSD Cancer Center Support grant P30CA023100, from UCSD Altman Clinical and Translational Research grant NIH UL1TR001442, and from charitable donations from Nine Girls Ask. D. Stupack was supported by NIH RO1CA107263. K. Taylor and A. Barrie are fellows of the UCSD Reproductive Medicine Gynecologic Oncology Program and were supported by the Gaines Family fellowship. Carlos Diaz-Osterman was supported by NIH training grant (T32-CA121938). D. Connolly was supported by NCI P30 CA006927, NCI CA195723, DOD W81XWH-16-1-0142 and charitable donations from The Roberta Dubrow Fund.
Abbreviations
- 3D
3-dimensional
- ARE
antioxidant response element
- ChEA
chip enrichment analysis
- CRISPR
clustered regularly interspaced short palindromic repeats
- FAK
focal adhesion kinase
- GEF
guanine nucleotide exchange factor
- GFP
green fluorescent protein
- GO
gene ontology
- GSEA
gene set enrichment analysis
- IP
intraperitoneal
- KEGG
Kyoto encyclopedia of genes and genomes
- KO
knockout
- MOVCAR
murine ovarian carcinoma
- NF-κB
nuclear factors kappa b
- OC
Ovarian Cancer
- PBS
phosphate buffered saline
- Rgnef
rho guanine nucleotide exchange factor, also known as Arhgef28
- SV40 TAg
simian virus 40 T-antigen
Footnotes
Conflicts of Interest: All authors declare no conflicts of interest.
Electronic supplementary material.
The online version of this article contains supplemental files.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018; 68: 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Lengyel E. Ovarian cancer development and metastasis. Am J Pathol 2010; 177: 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shield K, Ackland ML, Ahmed N, Rice GE. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol Oncol 2009; 113: 143–148. [DOI] [PubMed] [Google Scholar]
- 4.Kenny HA, Dogan S, Zillhardt M, A KM, Yamada SD, Krausz T et al. Organotypic models of metastasis: A three-dimensional culture mimicking the human peritoneum and omentum for the study of the early steps of ovarian cancer metastasis. Cancer Treat Res 2009; 149: 335–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al Habyan S, Kalos C, Szymborski J, McCaffrey L. Multicellular detachment generates metastatic spheroids during intra-abdominal dissemination in epithelial ovarian cancer. Oncogene 2018; 37: 5127–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roy L, Cowden Dahl KD. Can Stemness and Chemoresistance Be Therapeutically Targeted via Signaling Pathways in Ovarian Cancer? Cancers (Basel) 2018; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11: 85–95. [DOI] [PubMed] [Google Scholar]
- 8.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013; 12: 931–947. [DOI] [PubMed] [Google Scholar]
- 9.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 2009; 8: 579–591. [DOI] [PubMed] [Google Scholar]
- 10.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol 2014; 24: R453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009; 461: 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015; 27: 211–222. [DOI] [PubMed] [Google Scholar]
- 13.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res 2011; 21: 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Wijst MG, Brown R, Rots MG. Nrf2, the master redox switch: the Achilles’ heel of ovarian cancer? Biochim Biophys Acta 2014; 1846: 494–509. [DOI] [PubMed] [Google Scholar]
- 15.Konstantinopoulos PA, Fountzilas E, Pillay K, Zerbini LF, Libermann TA, Cannistra SA et al. Carboplatin-induced gene expression changes in vitro are prognostic of survival in epithelial ovarian cancer. BMC Med Genomics 2008; 1: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol 2015; 25: 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller NL, Kleinschmidt EG, Schlaepfer DD. RhoGEFs in cell motility: novel links between Rgnef and focal adhesion kinase. Curr Mol Med 2014; 14: 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim Y, Lim ST, Tomar A, Gardel M, Bernard-Trifilo JA, Chen XL et al. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol 2008; 180: 187–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller NL, Lawson C, Chen XL, Lim ST, Schlaepfer DD. Rgnef (p190RhoGEF) knockout inhibits RhoA activity, focal adhesion establishment, and cell motility downstream of integrins. PLoS One 2012; 7: e37830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu HG, Nam JO, Miller NL, Tanjoni I, Walsh C, Shi L et al. p190RhoGEF (Rgnef) promotes colon carcinoma tumor progression via interaction with focal adhesion kinase. Cancer Res 2011; 71: 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masia-Balague M, Izquierdo I, Garrido G, Cordomi A, Perez-Benito L, Miller NL et al. Gastrin-stimulated Galpha13 Activation of Rgnef Protein (ArhGEF28) in DLD-1 Colon Carcinoma Cells. J Biol Chem 2015; 290: 15197–15209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016; 166: 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X et al. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res 2003; 63: 1389–1397. [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474: 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bargonetti J, Reynisdottir I, Friedman PN, Prives C. Site-specific binding of wild-type p53 to cellular DNA is inhibited by SV40 T antigen and mutant p53. Genes Dev 1992; 6: 1886–1898. [DOI] [PubMed] [Google Scholar]
- 26.Quinn BA, Xiao F, Bickel L, Martin L, Hua X, Klein-Szanto A et al. Development of a syngeneic mouse model of epithelial ovarian cancer. J Ovarian Res 2010; 3: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gabbasov R, Xiao F, Howe CG, Bickel LE, O’Brien SW, Benrubi D et al. NEDD9 promotes oncogenic signaling, a stem/mesenchymal gene signature, and aggressive ovarian cancer growth in mice. Oncogene 2018; 37: 4854–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ward KK, Tancioni I, Lawson C, Miller NL, Jean C, Chen XL et al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin Exp Metastasis 2013; 30: 579–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller NL, Lawson C, Kleinschmidt EG, Tancioni I, Uryu S, Schlaepfer DD. A non-canonical role for Rgnef in promoting integrin-stimulated focal adhesion kinase activation. J Cell Sci 2013; 126: 5074–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev 2005; 24: 425–439. [DOI] [PubMed] [Google Scholar]
- 31.Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010; 26: 2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018; 34: 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baccam M, Woo SY, Vinson C, Bishop GA. CD40-mediated transcriptional regulation of the IL-6 gene in B lymphocytes: involvement of NF-kappa B, AP-1, and C/EBP. J Immunol 2003; 170: 3099–3108. [DOI] [PubMed] [Google Scholar]
- 34.Lee JR, Ha YJ, Kim HJ. Cutting edge: induced expression of a RhoA-specific guanine nucleotide exchange factor, p190RhoGEF, following CD40 stimulation and WEHI 231 B cell activation. J Immunol 2003; 170: 19–23. [DOI] [PubMed] [Google Scholar]
- 35.Tancioni I, Miller NL, Uryu S, Lawson C, Jean C, Chen XL et al. FAK activity protects nucleostemin in facilitating breast cancer spheroid and tumor growth. Breast Cancer Res 2015; 17: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheung K, Droppelmann CA, MacLellan A, Cameron I, Withers B, Campos-Melo D et al. Rho guanine nucleotide exchange factor (RGNEF) is a prosurvival factor under stress conditions. Mol Cell Neurosci 2017; 82: 88–95. [DOI] [PubMed] [Google Scholar]
- 37.Wu J, Zhai J, Lin H, Nie Z, Ge WW, Garcia-Bermejo L et al. Cytoplasmic retention sites in p190RhoGEF confer anti-apoptotic activity to an EGFP-tagged protein. Brain Res Mol Brain Res 2003; 117: 27–38. [DOI] [PubMed] [Google Scholar]
- 38.Hellner K, Miranda F, Fotso Chedom D, Herrero-Gonzalez S, Hayden DM, Tearle R et al. Premalignant SOX2 overexpression in the fallopian tubes of ovarian cancer patients: Discovery and validation studies. EBioMedicine 2016; 10: 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diviani D, Raimondi F, Del Vescovo CD, Dreyer E, Reggi E, Osman H et al. Small-Molecule Protein-Protein Interaction Inhibitor of Oncogenic Rho Signaling. Cell Chem Biol 2016; 23: 1135–1146. [DOI] [PubMed] [Google Scholar]
- 40.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lanczky A, Nagy A, Bottai G, Munkacsy G, Szabo A, Santarpia L et al. miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat 2016; 160: 439–446. [DOI] [PubMed] [Google Scholar]
- 42.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013; 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016; 44: W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Rgnef signaling and NF-κB reporter activation. (a) Human 293T transfection with mCherry-tagged Rgnef wildtype, Rgenf Y1003A (GDP exchange defective) and an IL-6 luciferase reporter with or without mutated NF-κB binding sites. (b and c) Adherent ID8-IP GFP-Rgnef cells were serum starved overnight, pre-treated with DMSO (vehicle), 10 μM U0126, or 1 μM wortmannin for 2h, and then stimulated with 5% fetal bovine serum plus 5% insulin-transferrin-selenium supplement for the indicated times prior to cell lysis. (b) Immunoblotting for phosphorylated ERK1/2 (T185/Y187) or total ERK1/2. (c) Immunoblotting for phosphorylated Akt (S473) or total Akt. (d) Human 293T cells were transfected as above and incubated with 10 μM Bay 11–7082 or 10 μM TPCA-1 for 24 h prior to analysis of luciferase activity. (a and d) Mean luciferase values are normalized from three independent analyses (*** P <0.001 and **** P < 0.0001, unpaired T test).