

This study presents the cell-mediated drug-loaded nanoparticle strategy for selective drugs delivery to treat lung fibrosis.
Abstract
Idiopathic pulmonary fibrosis (IPF) is a highly heterogeneous and fatal disease. However, IPF treatment has been limited by the low drug delivery efficiency to lungs and dysfunctional “injured” type II alveolar epithelial cell (AEC II). Here, we present surface-engineered nanoparticles (PER NPs) loading astaxanthin (AST) and trametinib (TRA) adhered to monocyte-derived multipotent cell (MOMC) forming programmed therapeutics (MOMC/PER). Specifically, the cell surface is designed to backpack plenty of PER NPs that reach directly to the lungs due to the homing characteristic of the MOMC and released PER NPs retarget injured AEC II after responding to the matrix metalloproteinase-2 (MMP-2) in IPF tissues. Then, released AST can enhance synergetic effect of TRA for inhibiting myofibroblast activation, and MOMC can also repair injured AEC II to promote damaged lung regeneration. Our findings provide proof of concept for developing a strategy for cell-mediated lung-targeted delivery platform carrying dual combined therapies to reverse IPF.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a rapidly progressive and fatal interstitial pulmonary disease with a dismal median survival time of just 3 years after diagnosis (1, 2). To date, the IPF therapies depend on blocking myofibroblast activation to inhibit collagen I deposition (3, 4). However, the clinical data showed that these therapies remained far from achieving IPF revision. The main reason is that the IPF therapeutics lack an effectively targeted carrier or ignore some of the other risk factors such as the instability and tolerability of type II alveolar epithelial cell (AEC II) (5, 6). The AEC II, which is considered as “injured” AEC II (7, 8) in the IPF tissues, releases excessive amounts of reactive oxygen species (ROS) that initiate an antifibrinolytic coagulation cascade and promote the overexpression of connective tissue growth factor (CTGF) to provoke myofibroblast overactivation and extracellular matrix (ECM) development and then destroy the lung architecture (9–11). This situation has inspired us to propose that the combination of modulating superoxide in injured AEC II and antimyofibroblast activation as “weeding and uprooting” strategy will be a potential therapeutic strategy for synergistic antifibrosis. Furthermore, another limitation is that current therapies are rarely distributed in the lungs, which cannot achieve full therapeutic effect for treating IPF (12). Thus, the development of an effective lung-targeting drug delivery carrier is highly desirable for IPF therapy.
Recently, local preferred therapeutic agents generated using endogenous cells have served as a strong and promising delivery platform for targeting in situ, achieving considerable progress in several diseases (13–17). In the inflammatory phase of IPF, precursor circulating monocytes (PCMs) have been found to undergo notable proliferation (18). PCMs and injured AEC II release chemotactic factors that specifically recruit chemokine receptor–positive (CR+) cells including monocyte-derived multipotent cell (MOMC) and guide the MOMC to migrate to injured lung tissues through specific binding to chemokine receptors on cell membrane (19, 20). Furthermore, in addition to this migration characteristic, these MOMCs, which originate from hematopoietic stem cells in the bone marrow, still have multipotency to differentiate into a variety of functional cells, including AEC II and endothelial cell (21, 22), which demonstrates that MOMC has the potential to participate in reestablishing lung functions (23). In addition, chronic hypoxic exposure induces the recruitment of MOMC to the pulmonary circulation, and the cell contributes to improving lung functions by producing angiogenic factors (24). It has been reported that monocytes from patients with IPF also show preconditioned prorepair features (25). In general, MOMC as a precise lung-targeting delivery platform will exhibit encouraging therapeutic effects, leading to the repair or regeneration of injured AEC II for IPF treatment.
In this study, we constructed the programmed therapeutics composed of surface-engineered nanoparticles (PER NPs) loading dual drugs adhered to MOMC (named MOMC/PER) to solve the issues in IPF therapy by improving drug accumulation in injured lung sites and completely destroying the fibrotic signaling network in IPF (Fig. 1). The MOMC/PER delivery platform realized efficacy through programmed modules, which consisted of a homing moiety, responsive release moiety, and retargeting moiety. (i) The homing moiety is the native ability of MOMC/PER to migrate to injured lungs due to the homing characteristic of MOMC. (ii) The responsive release moiety of MOMC/PER is activated by matrix metalloproteinase-2 (MMP-2) overexpression in IPF tissues, resulting in pathology-responsive release of PER NPs with exposed cyclic RGDfc (Arg-Gly-Asp) [c(RGDfc)] from the MOMC. (iii) The retargeting moiety is that exposed c(RGDfc) on PER NPs can anchor to injured AEC II via an interaction between αvβ6 and c(RGDfc) (26), allowing the cytoplasm of the injured AEC II internalize PER NPs. Subsequently, astaxanthin (AST) and trametinib (TRA) are released from PER NPs to achieve a weeding and uprooting therapeutic effect. In general, the sustained injury of epithelial cells and highly heterogeneous myofibroblasts is considered as the most critical variable in achieving complete IPF reversion (27). To validate the above hypothesis, in this study, AST was chosen as an antioxidant by neutralizing superoxide to repair injured AEC II (28), and TRA suppressed the activation of myofibroblast by inhibiting CTGF production for IPF therapy (29). MOMC also participates in treating IPF by repairing injured AEC II to promote regeneration of IPF lungs (21). Overall, MOMC/PER, which mimics the features of chimeric antigen receptor T cell immunotherapy, is a precise lung-targeting platform to reverse IPF by improving drug accumulation due to the outstanding homing ability of MOMC to injured lung sites, and the destruction of the fibrotic signaling network by inhibiting the activation of myofibroblast and repairing injured AEC II to promote the damaged lungs regeneration.
Fig. 1. Schematic illustration of lung-targeted programmed MOMC/PER therapeutic designed to reverse IPF.
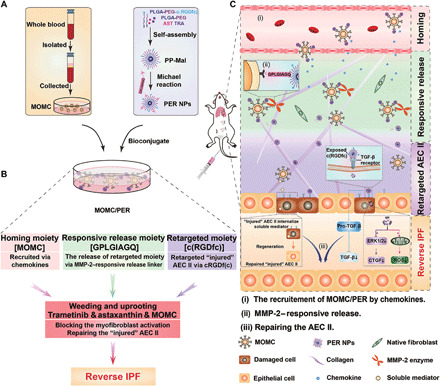
(A) Bioconjugated MOMC/PER was prepared by incubating PER NPs with MOMC. (B) MOMC/PER has multifunctional moieties including a homing moiety, responsive release moiety, and retargeting moiety to reverse IPF. Then, a weeding and uprooting strategy contributes to IPF reversion. (C) Schematic illustration of MOMC/PER for improved drugs accumulation and antifibrotic effect in IPF lung microenvironment.
RESULTS
Potential application and characterization of MOMC/PER in IPF mice
The quantities of MOMC in serum and lung tissues were significantly increased in IPF mice compared with normal mice (Fig. 2A). The proliferation of MOMC was positively related to IPF progression, which might be because increasing numbers of MOMC would be recruited from the bone marrow to the lesion sites when IPF occurred (30). Motivated by the fact that MOMC has a homing ability, we considered MOMC to be a potential delivery carrier to improve delivery efficiency in IPF treatment under pathological conditions.
Fig. 2. The PER NPs can adhere to MOMC and pathology responsively release and then be internalized by A549 in vitro.
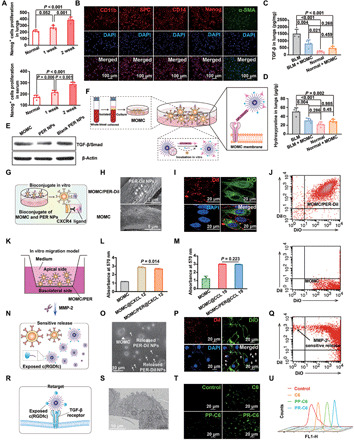
(A) The proliferation of Nanog+ cells in serum and lung tissues by ELISA assay. (B) The MOMC phenotypes. The level of TGF-β (C) and hydroxyproline (D) in IPF lung tissues, respectively. (E) The level of TGF-β/Smad in vitro. (F) Schematic showing the preparation of MOMC/PER. (G) Schematic showing the adhesion of PER NPs to MOMC. (H) SEM images of MOMC and MOMC/PER-DiI. (I) Fluorescent signals of MOMC and PER-DiI NPs by CLSM. (J) The adhesion between MOMC and PER-DiI NPs by flow cytometry. (K) In vitro migration model. The migration ability of MOMC and MOMC/PER in CXCL 12 (L) and CCL 19 (M), respectively. (N) Schematic showing sensitive release of MOMC/PER-DiI triggered by MMP-2. (O) Characterizations by TEM. MOMC is the triangle, and PER-DiI NPs are the arrows. (P) Fluorescent images of MOMC and released PER-DiI NPs by CLSM. (Q) The flow cytometry showed responsive release. (R) Schematic showing the retarget ability of released PER NPs. (S) Characterization of retargeting ability by TEM. (T) The fluorescent images by CLSM. (U) Cellular uptake in A549 by flow cytometry (n = 3). Statistical significance was calculated via one-way analysis of variance (ANOVA).
We first isolated MOMC from the peripheral blood of C57BL/6J male mice of IPF. The morphologies of the MOMC were fusiform (fig. S1). To identify the phenotypes of MOMC isolated from IPF mice, we first investigated the presence of specific markers for MOMC by immunofluorescence staining. The results showed that MOMC expressed CD11b and α–smooth muscle actin (α-SMA) (Fig. 2B), which was consistent with the literature (24). In addition, the MOMC also expressed the stem cell markers CD14 and Nanog protein and the injured AEC II’s marker pro-surfactant protein C (SPC), as shown in Fig. 2B. These results indicated that MOMC was pluripotent cells with stem cell– and epithelial cell–like properties. It has been reported that MOMC was recruited to damaged lung areas and participated in recovering injured lung normalization through growth factor release to repair injured AEC II (30). In addition, to inspect the potential risk of injecting MOMC into mice, we further investigated the feasibility of using isolated MOMC as a delivery carrier, including measuring the levels of transforming growth factor–β (TGF-β) and hydroxyproline, which are closely related to the development of IPF in vivo. The results displayed approximately onefold reduction in TGF-β and hydroxyproline levels in IPF mice treated with MOMC compared with untreated bleomycin (BLM)–induced mice, and these indexes were barely changed in normal mice, indicating that MOMC would not induce the occurrence of IPF and partly relieved established IPF (Fig. 2, C and D).
We next prepared PER NPs that contained two target peptides named peptide E5 and c(RGDfc). The poly(lactide-co-glycolide)-block-poly(ethylene glycol) methyl ether maleimide (PLGA-PEG-Mal) and PLGA-PEG-c(RGDfc) (mass ratio, 10:1) were self-assembled by noncovalent interactions of amphiphilic PLGA-PEG copolymer into nanoparticles (31), and then, peptide E5 was bound on the NPs by the Michael reaction (fig. S2A). As determined by 1H nuclear magnetic resonance spectroscopy (fig. S2B) and SDS–polyacrylamide gel electrophoresis (SDS-PAGE) (fig. S2C), we successfully prepared PER NPs, and the grafting rate of peptide E5 in the PER NPs was 43.7%. The PER NPs showed particle sizes of approximately 110 ± 10.39 nm and the zeta potential of −23.37 mV (fig. S2, D and E). In addition, AST and TRA were encapsulated into PER NPs (fig. S2F). The drug loading content of the PER NPs was 1.98 weight % (wt %) for AST and 2.83 wt % for TRA. The sustained release of the loaded AST was 49.5 wt %, and the pH-dependent release of the loaded TRA (weak alkalinity) was 79.6 wt %, which were obtained at pH 5.0 within 72 hours (fig. S2G). Then, we investigated the capacity of MOMC to differentiate into myofibroblast after treatment with PER NPs in vitro. As shown in Fig. 2E, the expression of TGF-β/small mother against decapentaplegic (TGF-β/Smad), which is molecule in the crucial pathway for myofibroblast activation, was decreased, suggesting that the PER NPs could inhibit MOMC differentiation. The possible reason for the inhibition was that the PER NPs partly covered the TGF-β receptor on the MOMC and reduced exogenous TGF-β stimulation within 8 hours, and then, the PER NPs could be gradually internalized. The released drugs could reduce TGF-β expression of MOMC after 8 hours (fig. S3, A and B).
We next constructed MOMC/PER as a delivery platform/therapeutic carrier (Fig. 2F), and PER NPs loaded with both drugs could specifically adhere to MOMC through the interaction between peptide E5 of the PER NPs and the CXCR4 on the MOMC by a temperature-dependent manner (32, 33). The formation of MOMC/PER was positively correlated with the incubation time within 2 hours (fig. S3, A and B). Moreover, the PER NPs could specifically stick to the surface of the MOMC without internalization by the MOMC within 8 hours (Fig. 2G). The reasons may be that peptide E5 conjugated on the surface of the PER NPs is a long-chain peptide that limits internalization into the MOMC and that CXCR4 is not an endocytic receptor (34). The MOMC/PER had a loading capacity of 4.75 μg of TRA and 1.5 μg of AST/1 × 105 cells (fig. S3, C and D). In addition, MOMC cell viability was above 80% with different concentrations of PER NPs and different incubation times (fig. S4, A and B).
To investigate the adhesion of MOMC and PER NPs, the 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) was loaded into blank PER NPs (PER-DiI NPs) to evaluate adhesion behavior. After incubating with MOMC and PER-DiI NPs for 2 hours, the morphologies of the MOMC/PER-DiI were confirmed by scanning electron microscopy (SEM) (Fig. 2H) and confocal laser scanning microscopy (CLSM) (Fig. 2I). Flow cytometry detection also indicated the formation of MOMC/PER-DiI in that the MOMC labeling green and PER-DiI NPs were collected in the double-positive quadrant (Fig. 2J).
Sequentially, migration via the interaction between a receptor and ligand is the vital characteristic that needs to be retained by MOMC/PER to realize efficient delivery (Fig. 2K). The migratory capability of MOMC/PER was detected by a Transwell invasion assay. The results indicated that the migratory ability of the MOMC/PER was unaffected by PER NPs adhering to the surface of MOMC (Fig. 2, L and M) at all studied concentrations (fig. S4, C and D).
To establish the retargeting ability of PER NPs, MMP-2 overexpressed in IPF tissues was used as an activating trigger to release PER NPs from MOMC/PER. As depicted in Fig. 2N, the separation of PER-DiI NPs from MOMC was well evidenced by transmission electron microscopy (TEM) and CLSM (Fig. 2, O and P) and flow cytometry (Fig. 2Q). We also detected the phenomenon by SDS-PAGE and particle sizes changes (fig. S4, E and F). After PER NPs were released from MOMC/PER, the exposed peptide c(RGDfc) of the PR NPs could retarget αvβ6, which is overexpressed on the surface of injured AEC II (Fig. 2R) (35). Then, we investigated the capacity of injured AEC II to uptake PLGA-PEG-c(RGDfc)–coumarin 6 (PR-C6) compared with free C6 and PLGA-PEG–C6 (PP-C6) by TEM and CLSM. The internalization of PR-C6 was better than other forms (C6 and PP-C6) (Fig. 2, S to U). In addition, PR-C6 also underwent lysosomal escape (fig. S4G).
Homing ability and therapeutic efficacy of MOMC/PER in vivo
The homing ability of MOMC/PER was investigated in IPF models in vivo (Fig. 3A). We first examined the lung accumulation of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR)–loaded into blank PER NPs (PER-DiR NPs) adhere to MOMC (MOMC/PER-DiR) after intravenous administration. The DiR fluorescence accumulated in the lungs of the MOMC/PER-DiR group, which indicated that compared with MOMC-loading DiR (MOMC-DiR) and free-DiR, MOMC/PER-DiR had a superior ability to target IPF lungs (Fig. 3B). Then, we quantitatively analyzed the drug distribution in the tissues of each organ. The DiR fluorescence intensity in the lungs was 3.5- and 0.5-fold greater than that in the liver in the MOMC/PER-DiR and MOMC-DiR groups, respectively. In addition, there was little accumulation of free DiR in the lungs than that in the liver (Fig. 3C). MOMC loading of DiR could improve DiR accumulation in IPF lungs due to the homing ability of the MOMC.However, the accumulation of MOMC-DiR was weaker than that of MOMC/PER-DiR. This may be because the free dye carried by the MOMC was limited compared with that carried by the PER NPs, which suggested that MOMC/PER could solve the limitation of conventional drug loading of cells. In addition, we further evaluated the homing capacity of MOMC/PER, the responsive release ability of MOMC/PER mediated by MMP-2, the released PER NPs with exposed c(RGDfc), and the retargeting to injured AEC II by immunofluorescence staining. The DiI was chose to a tracer agent, labeling the PER-DiI NPs with red fluorescence, and then, the PER-DiI NPs adhered to MOMC to form MOMC/PER-DiI. Nanog and SPC, which represented MOMC and injured AEC II, respectively, were labeled in green fluorescence. Then, the MOMC/PER-DiI was administered to IPF mice by intravenous injection. As shown in Fig. 3D, the PER-DiI NPs labeled in red overlapped with the MOMC marked in green, generating a merged yellow signal, which revealed that the MOMC/PER-DiI notably accumulated in the lungs of IPF mice in stage 1 (homing to lungs). In stage 2 (releasing PER-DiI NPs), the PER-DiI NPs labeled in red were separated from the MOMC labeled in green, indicating that the PER-DiI NPs were released from the MOMC membrane surface and exposed c(RGDfc) at fibrotic foci as a result of the overexpression of MMP-2 in the IPF microenvironment. Then, the PER-DiI NPs labeled in red overlapped with injured AEC IISPC+ labeled in green, implying that the PER-DiI NPs retargeted to injured AEC II through the interaction between the exposed c(RGDfc) ligand and the αvβ6 receptor on the surface of the injured AEC II in stage 3 (retarget injured AEC II) (Fig. 3D). Collectively, these results showed that MOMC/PER-DiI had the native ability to home to damaged lungs and then were activated by programmed procedures, confirming that MOMC could function as a vehicle to deliver PER NPs to injured lungs.
Fig. 3. Homing ability and therapeutic efficacy of MOMC/PER in vivo.

(A) Schematic of the targeting performance of MOMC/PER in the blood circulation to IPF lungs. (B) In vivo fluorescence images of IPF mice intravenous injection with MOMC-DiR, MOMC/PER-DiR, and DiR (n = 3). (C) Quantification of the in vivo retention profile (n = 3). (D) The different stages of MOMC/PER-DiI. (E) The whole lungs were imaged and investigated after 28 days. Lung morphologies (i) [Photo credit (i): Xin Chang, China Pharmaceutical University], H&E staining (ii), and Masson staining (iii). The morphologies of mitochondria by TEM (iv). The levels of TGF-β (F), IL-1β (G), and IL-4 (H) by ELISA assay (n = 5). The levels of lymphocytes (I), white blood cells (J), and neutrophils (K) in whole blood (n = 5). The levels of GSH (L) and SOD (M), respectively (n = 5). (N) The expression of SPC. (O) Survival rate curves (n = 10). Statistical significance was calculated via one-way ANOVA.
To confirm the curative effect of MOMC/PER, we investigated lung morphologies after the administration of MOMC/PER or other treatments. As showed in Fig. 3E, MOMC/PER could greatly relieve IPF according to hematoxylin and eosin (H&E) and Masson staining. Images of lung morphologies showed obvious normalization after treatment with MOMC or MOMC/PER compared with no treatment (Fig. 3E, i). H&E staining showed that lung tissues in the MOMC/PER group were not destroyed and that the alveolar sizes were same as normal lung tissues (Fig. 3E, ii). In addition, compared with no treatment, MOMC also partly protected the lung architecture; however, there was a gap between the MOMC/PER and normal groups. Similarly, Masson staining also showed that the MOMC/PER group exhibited an excellent reduction in collagen I deposition (Fig. 3E, iii). IPF is also induced by mitochondrial oxidative stress in injured AEC II. Hence, we examined the capability of MOMC/PER to repair injured AEC II by maintaining mitochondrial morphologies (Fig. 3E, iv). The morphologies of mitochondria were close to normal in the MOMC/PER group compared with the MOMC group and BLM group, suggesting that MOMC/PER could repair injured AEC II to maintain normal lungs by improving mitochondrial function. Furthermore, we tested the expression of proinflammatory cytokines [TGF-β, interleukin-1β (IL-1β), and IL-4], which play major roles in excessive ECM formation during IPF progression. As shown in Fig. 3 (F to H), the expression of TGF-β in the MOMC/PER treatment group was nearly threefold lower than that in the BLM group, and the expression of IL-1β and IL-4 also decreased by nearly 0.5- and 1-fold, respectively, in the MOMC/PER group compared with the BLM group, suggesting that MOMC/PER could block IPF progression by inhibiting the secretion of proinflammatory cytokines. In addition, the formulations of MOMC and MOMC/PER showed well biocompatibility in a hemolysis test (fig. S5). In addition, inflammatory cells were quantified in whole blood in these groups after treatment. Compared with the BLM group, the MOMC/PER group showed inhibited inflammatory cell proliferation (Fig. 3, I to K), which indicated that MOMC/PER had the ability to alleviate IPF progression in the inflammatory phase. In addition, the results implied that MOMC had a certain ability to inhibit the proliferation of inflammatory cells. Next, glutathione (GSH) and superoxide dismutase (SOD), which are significant inhibitors of ROS, were used to balance the ROS content of injured AEC II. Compared with no treatment, treatment with MOMC/PER increased the GSH level nearly onefold (Fig. 3L), and MOMC also enhanced the GSH level. Similarly, MOMC/PER increased the SOD level to a certain extent in lung tissues (Fig. 3M). We further explored the repair mechanism for injured AEC II in IPF lungs treated with MOMC or MOMC/PER. The expression of SPC was markedly increased in the MOMC/PER group compared with the BLM group; there was also an augmentation in the expression of SPC in the MOMC group, which showed that MOMC/PER could up-regulate AEC II proliferation or recover injured AEC II to normalize the lungs in IPF and demonstrated that MOMC/PER could promote IPF lungs regeneration (Fig. 3N). The survival time of the MOMC/PER group exceeded 60 days, which was longer than the survival time of the BLM group (Fig. 3O), and the MOMC/PER group did not exhibit any changes in body weight (fig. S6).
Reprogramming of MOMC/PER in vivo
To investigate the targeting ability of PER NPs through reprogramming to form MOMC/PER in the blood circulation, we conducted the following experiments. The E5-mediated targeting ability of PER-DiI NPs was first evaluated in IPF mice (Fig. 4A). CLSM showed the adhesion of PER-DiI NPs to the surface of MOMC (Fig. 4B, bottom). The confocal images produced the same result as Fig. 2I. Furthermore, PER-DiI NPs were administered to IPF mice model by intravenous injection. The results demonstrated that the PER-DiI NPs adhered to the surface of MOMC (Fig. 4B, middle). More detailed results revealed that the PER-DiI NPs could bind to the MOMC surface and reprogram the MOMC to form MOMC/PER in the peripheral blood by SEM (Fig. 4B, top). In addition, immunofluorescence staining confirmed that the PER-DiI NPs homed to IPF lungs and accumulated in the injured AEC II area after intravenous injection (Fig. 4C). In addition, Nanog-labeled MOMC (green fluorescence) accumulated in higher numbers in IPF lungs than normal lungs, which was similar to previous results (Fig. 2I). We also investigated the targeting capacity of PER-DiR NPs at different time points by in vivo imaging system following intravenous injection, and PLGA-PEG-DiR (PP-DiR NPs), PLGA-PEG-c(RGDfc)–DiR (PR-DiR NPs), and PLGA-PEG-E5-DiR (PE-DiR NPs) were used as controls. The PP-DiR NPs and PR-DiR NPs were mainly found in the liver, while the PER-DiR NPs and PE-DiR NPs mainly accumulated in IPF lungs (Fig. 4D and fig. S7A). The accumulation of the PER-DiR NPs in the lungs peaked at 8 hours, while the lung accumulation of the PE-DiR NPs quickly decreased. The primary reason may be that the PE NPs were delivered to the lungs via MOMC; however, they could not anchor on injured AEC II because they lacked c(RGDfc) and were therefore more rapidly cleared from the circulation than the PER-DiR NPs. Compared with the PE-DiR NPs, the PER-DiR NPs accumulated in IPF lungs for a long time (more than 8 hours), which is important for treating lung disease. A quantitative region of interest (ROI) analysis of PER-DiR NPs accumulation was performed by detecting DiR signal variation in the lungs and other organs (Fig. 4E). Moreover, we evaluated PER-DiI NPs behavior in lung tissues after administration at different times (Fig. 4F). After administration at 0.5 hours, increasing levels of overlapping yellow fluorescence in lung blood vessels were observed for PER-DiI NPs labeled in red and MOMC marked in green, indicating that the PER-DiI NPs arrived at IPF lung tissues through reprogramming to form MOMC/PER-DiI in the blood circulation. Then, the red and green signals were separated at the time point of 2 hours, indicating that the PER-DiI NPs were released from the reprogrammed MOMC/PER-DiI due to the overexpression of MMP-2 in the IPF mice. Furthermore, the released PER-DiI NPs showed a wide distribution in the lung tissues at 4 hours after intravenous injection, which is powerful for treating diseases. These data demonstrated that PER NPs could target IPF lungs by means of attaching to circulating MOMC quickly and could accumulate in lung tissues for a long time to achieve therapeutic efficacy.
Fig. 4. Schematic representation of the performance of PER NPs.
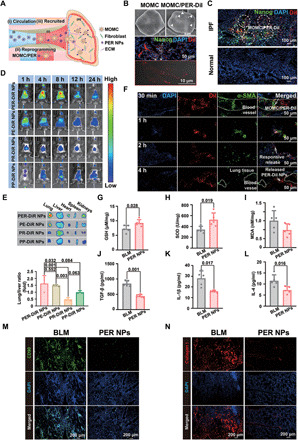
(A) Schematic of PER NPs circulation in vivo, reprogramming of MOMC/PER, and recruitment to IPF tissue. (B) The targeting ability of PER-DiI NPs. (C) The accumulation of PER-DiI NPs in normal and IPF lungs. (D) Fluorescence IVIS imaging (n = 3). (E) Ex vivo fluorescence imaging and quantification of major organs (n = 3). (F) The accumulation PER-DiI NPs in the lungs at different times. Lung function indexes of GSH (G), SOD (H), and MDA (I). TGF-β (J), IL-1β (K), and IL-4 (L) by ELISA assay (n = 5). (M) Proliferation of fibroblasts. (N) Expression of collagen I. Statistical significance was calculated via one-way ANOVA.
We further investigated the antifibrotic efficacy of PER NPs in vivo. With IPF progression, injured AEC II gradually died out due to oxidative stress, which leads to mouse suffocation. Hence, restoring normal lung function has important significance. Compared with the BLM group, the PER NPs group had the promising abilities to repair injured lungs and keep them normal. GSH and SOD levels in the PER NPs group were obviously improved with 0.3- and 1-fold, respectively, which could relieve the oxidative stress in injured AEC II to some extent. The level of malondialdehyde (MDA), a key indicator of oxidative stress, was reduced 0.3-fold in the PER NPs group compared with the BLM group (Fig. 4, G to I). Compared with controls, treatment with PER NPs reduced the production of the three cytokines (TGF-β, IL-1β, and IL-4) in the lungs by 1-, 0.85-, and 0.7-fold, respectively, which showed that the PER NPs effectively inhibited the inflammatory response in IPF lungs (Fig. 4, J to L). We also examined the levels of TGF-β, IL-1β, and IL-4 in the spleen tissues (fig. S7, B to D), which showed consistent results. These results indicated that PER NPs could treat IPF by inhibiting inflammatory responses in IPF lungs. As seen in Fig. 4M, immunofluorescence staining results revealed that the population levels of fibroblasts CD90+ labeled in green remained in a relatively stable range, while the population levels of activated fibroblasts indicated great proliferation in the BLM group, supporting the conclusion that compared with no treatment group, the PER NPs had an efficient ability to reverse IPF by inhibiting the activation of fibroblasts. Figure 4N showed that the expression of collagen I was notably decreased in the PER NPs group, which confirmed that PER NPs could achieve therapeutic effects by inhibiting ECM deposition.
Antifibrosis mechanism with dual-drug synergy
We next investigated the antifibrosis mechanism based on the synergistic effect of TRA and AST. We firstly established the different formulations, including PLGA-PEG-TRA-AST (PPTA), PLGA-PEG-TRA (PPT), and PLGA-PEG-AST (PPA), and the morphologies of PPA, PPT, and PPTA were evaluated by TEM (fig S8). As shown in Fig. 5A, the results of immunofluorescence staining showed that the expression of the vimentin as cytoskeletal protein was increased after treatment with different formulations in human lung epithelial cell carcinoma (A549). In particular, compared with other treatments, the PER NPs significantly increased the expression of vimentin, indicating that PER NPs had the capacity to keep injured AEC II normal. To assess the repair mechanism induced by the drugs combination, the ROS were detected using ROS probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA) via inverted fluorescence microscopy and flow cytometry. The ROS content was significant decreased in the PPTA group compared with the untreated and single-drug groups (PPT and PPA) (Fig. 5B). Although the PPA group exhibited some changes than PPT, this effect was not as strong as that in PPTA group, because the PPTA groups exhibited synergistic effect that relieved oxidative stress in injured AEC II than other control formulations. Furthermore, the PPTA group also showed a reduced mitochondrial membrane potential in TGF-β–induced cells (Fig. 5C), which supported the conclusion that the efficacy in PPTA group was the result of repairing mitochondrial function with relief of oxidative stress in the mitochondria. In the microenvironment of IPF lungs, myofibroblast can be derived from injured AEC II undergoing epithelial-mesenchymal transition (EMT), which aggravates the progression of IPF. As observed in a wound healing assay and invasion assay (Fig. 5D), PPTA effectively inhibited the occurrence of EMT. Furthermore, fibronectin is a structural protein in the ECM, which is a crucial indicator of IPF progression. The expression of fibronectin was obviously decreased in PPTA group than PPT and PPA groups, thus inhibiting the differentiation of injured AEC II into myofibroblast (Fig. 5E). Next, we also tested IPF-reversing efficacy by monitoring the recovery of the lung architecture and improvement in lung functions in vivo. As shown in Fig. 5F, collagen I deposition in the PPTA group returned to normal levels, as determined by H&E and Masson staining, demonstrating that the PPTA could recover the architecture of injured lungs compared with no treatment or single-drug groups (PPT and PPA). Similarly, the expressions of α-SMA and collagen I were tested by immunohistochemistry (IHC), which also obtained the same results that the synergistic effect of PPTA could effectively inhibit myofibroblast activation and ECM deposition. In addition, the level of hydroxyproline, the main component of the ECM, was also decreased after treatment with PPTA compared with other treatments (Fig. 5G), suggesting that PPTA had the ability to diminish ECM deposition and retard IPF progression. Furthermore, we detected the expression of α-SMA to evaluate the myofibroblasts activation by Western blotting. The lungs were collected after treatment with PPTA, PPT, or PPA for 28 days. The results showed that α-SMA expression, as the major evaluation index for IPF, was significantly reduced in PPTA group (Fig. 5H). In addition, the results of real-time quantitative polymerase chain reaction (qPCR) showed that the relative mRNA expressions of CTGF (Ctgf) and α-SMA (Acta2) significantly decreased in PPTA group, which indicated that the combination of AST and TRA can achieve efficient therapeutic efficacy by inhibiting myofibroblasts overactivation (Fig. 5, I and J). Moreover, MDA expression decreased, and SOD and GSH levels increased after treatment in PPTA group compared with the PPT and PPA groups. Together, these results implied that the combination of AST and TRA could recover IPF lung function through synergistic effect that was not observed with the other treatments (Fig. 5, K to M). The various formulations as mentioned above were safe by intravenous injection through H&E staining (fig. S9).
Fig. 5. Antifibrosis mechanism with dual drugs synergy.
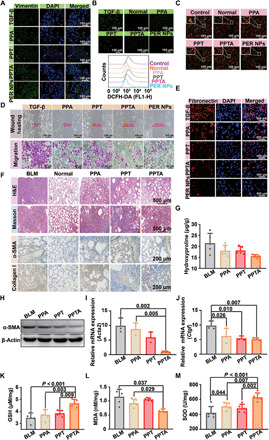
(A) Expression of the vimentin in vitro. (B) The ROS level in vitro. (C) The changes of mitochondrial membrane potential. (D) Invasion assay. (E) Fibronectin expression. (F) H&E, Masson, and IHC staining. (G) The level of hydroxyproline. (H) The α-SMA and β-actin by Western blotting. The mRNA expression of Acta2 (I) and Ctgf (J) by qPCR (n = 3). Contents of GSH (K), MDA (L), and SOD (M) (n = 5). Statistical significance was calculated via one-way ANOVA.
Comparison of the antifibrotic effect between MOMC/PER and traditional treatment
To further investigate the antifibrotic efficacy of MOMC/PER and pirfenidone as a conventional therapeutics for IPF, we evaluated the ability of these treatments to repair lung tissue and inhibit collagen I deposition through H&E and Masson staining, respectively, after 28 days of administration. As shown in Fig. 6A, the alveolar structure in the BLM group collapsed, and alveolar wall thickness increased notably, indicating that collagen I was accumulated and that alveolar heterogeneity was aggravated. Similarly, the alveolar morphologies in the pirfenidone group also showed collapse via H&E staining. In contrast, MOMC/PER could obviously repair the collapsed part of the alveolar space, narrow the spaces between the alveoli, and produce a thinner alveolar wall that tended to appear normal by H&E staining, which demonstrated that MOMC/PER had greater reparative effect on alveolar structure than pirfenidone. In addition, MOMC/PER group showed notable decrease compared with the pirfenidone group in inflammatory cell infiltration. The PER NPs were also more competent in restoring alveolar structure than the clinical drug pirfenidone. This effect was observed because the PER NPs could undergo reprogramming to form MOMC/PER in the blood circulation and then reach their destination, which was consistent with the above results. We further confirmed therapeutic efficacy in regard to ECM deposition by Masson staining (Fig. 6B). Compared with that in the pirfenidone group, the ECM accumulation in the lungs, which appeared as blue staining, was notably reduced in the MOMC/PER group. The results for Masson staining showed that MOMC/PER had greater power than pirfenidone to prevent IPF progression by inhibiting ECM deposition. The main reason for the limited therapeutic effect of pirfenidone was its low bioavailability as an oral drug, and onefold treatment target is the second therapeutic limitation of pirfenidone. Overall, the antifibrotic efficacy in the MOMC/PER group was the best efficacy observed through H&E and Masson staining, and PER NPs had better efficacy than pirfenidone or MOMC. In addition, the fibrosis score of different formulations showed the same trend in fig. S10. These data demonstrated that MOMC/PER showed a preferable combination efficacy over the U.S. Food and Drug Administration (FDA)–approved therapeutic pirfenidone or using MOMC or PER NPs alone.
Fig. 6. The whole lungs were investigated after treatment on day 28.
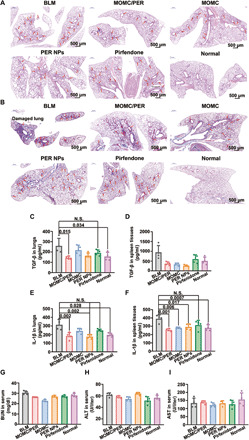
(A) H&E staining. (B) Masson staining. The levels of TGF-β in the lungs (C) and spleen tissues (D). N.S., not significant. The levels of IL-1β in the lungs (E) and spleen tissues (F). BUN (G), ALT (H), and aspartate aminotransferase (I) in serum (n = 4). Statistical significance was calculated via one-way ANOVA.
Then, we further evaluated the antifibrotic effect in various groups by examining biochemical indexes of IPF. We first examined the expressions of TGF-β and IL-1β in the lungs and spleen tissues, respectively. The results in the lungs showed that TGF-β expression was reduced onefold in the MOMC/PER group (P = 0.015) compared with the BLM group and became close to normal (Fig. 6, C and D). However, there was no significant difference between the pirfenidone group and the BLM group, and the level of TGF-β in the PER NPs group was lower than that in the pirfenidone group. In addition, the TGF-β level in the spleen tissues was significantly decreased in the MOMC/PER group (P = 0.002) compared with the BLM group. However, the level of TGF-β in the pirfenidone group was similar to that in the BLM group. The main reason is that pirfenidone is used to treat IPF by inhibiting the accumulation of collagen I, but it has no therapeutic effect on the simultaneous inflammatory response or cytokine expression. The results demonstrated that MOMC/PER had the best antifibrotic efficacy, which was superior to the efficacy achieved by pirfenidone and was mediated by inhibiting the expression of cytokines in the lungs. Furthermore, we detected the expression of IL-1β in the lungs and spleen tissues to investigate the antifibrotic effects of different formulations (Fig. 6, E and F). The trends in IL-1β expression were similar to TGF-β; all the treatments could reduce the expression of IL-1β, and MOMC/PER showed the best therapeutic efficacy (P = 0.001) in all the treatments. In addition, the IL-1β level in the pirfenidone group was maximal, indicating that compared with the other treatment groups, including the MOMC/PER and PER NPs alone groups, the pirfenidone group showed minimal anti-inflammatory effects. These results indicated that MOMC/PER could achieve a greater treatment effect on IPF than pirfenidone by inhibiting the expression of cytokines in the inflammatory phase; the efficacy of PER NPs was second only to MOMC/PER, and pirfenidone and MOMC were weaker than the PER NPs.
To assess the safety of the treatments in vivo, we then evaluated biological indexes for each formulation after treatment. The levels of blood urea nitrogen (BUN), alanine transaminase (ALT), and aspartate aminotransferase were detected to evaluate the function of the kidneys, liver, and heart, respectively (Fig. 6, G to I). The levels of BUN, ALT, and aspartate aminotransferase were not significantly different between the pirfenidone and other groups (the MOMC/PER, PER NPs, and MOMC groups). As an oral drug approved by the FDA for the treatment of IPF, pirfenidone is highly recognized for its safety in application. Similarly, our different formulations obtained results of safety equivalent to pirfenidone for a certain period of time, indicating that MOMC/PER, MOMC, and PER NPs could also be safely administered by intravenous injection and could be used clinically.
DISCUSSION
IPF is characterized by injured AEC II and activated myofibroblast, resulting in ECM deposition. To date, the FDA has approved only two drugs (pirfenidone and nintedanib) for IPF treatment. Unfortunately, curing end-stage IPF is inefficient due to the narrow therapeutic spectrums and insufficient accumulation of these drugs in the lungs (3). As a result, traditional therapies have done little to reverse IPF (4). To address this problem, we developed programmed therapeutics MOMC/PER to reverse IPF by efficient lung delivery, programmed modules, and double synergetic strategies.
Two synergetic strategies including drug/drug and cell/drug involved in reversing IPF were shown for the MOMC/PER here. First, the drug/drug as weeding and uprooting strategy could repair injured AEC II and inhibit myofibroblast activation, achieving first synergetic antifibrosis effect. In particular, one drug (AST) acted as the “uprooting” part of the treatment strategy, repairing injured AEC II by neutralizing oxidative stress. The other drug (TRA) acted as the “weeding” portion of the strategy, inhibiting the differentiation of fibroblasts into myofibroblast by suppressing CTGF production. Second, some studies have demonstrated that MOMC is multipotent cell that can be specifically recruited to injured lung tissues through interactions between chemokine receptors and chemotactic factors (36–38) and contribute to lung tissue normalization and regeneration (39). In addition, MOMC also plays a vital role in regulating the population of immune cells during the inflammatory phase of disease progression (40). Similarly, our results also showed that MOMC could inhibit the proliferation of inflammatory cells, such as lymphocytes and white blood cells. This is another synergetic effect called cell/drug. PER NPs also exhibited greater antifibrotic effects than pirfenidone due to their efficient lung-targeting ability and combination of AST and TRA, as these PER NPs could target MOMC in the circulation, accumulate in the lungs effectively, and then reverse IPF collaboratively. The limited treatment efficacy of pirfenidone is mainly due to its low bioavailability, narrow therapeutic spectrum, and functions by inhibiting myofibroblast activation only.
In addition, MOMC/PER is strategically distinct from nanodelivery carrier (41) and drug-loaded cells carrier (14). The traditional nanodelivery system for IPF always presents dissatisfactory accumulation and unexpected drug release at the lesion site. Even these defects can be avoided for IPF therapy, the therapeutic efficacy is also limited to onefold treating target, and these shortcomings make IPF hard to reverse. In addition to nanodelivery systems, cell-mediated drug delivery has also received more attention in disease treatment. The classic cell-based delivery strategy for treating disease is reliant on drugs being loaded into cells by endocytosis (15). However, cells are difficult to load with large quantities of drugs, and chemotherapeutics may be highly toxic to cells undergoing loading. Hopefully, the PER NPs adhere to the MOMC surface in our study could surmount this challenge in conventional drug loading of cells. PER NPs were firstly attached to MOMC surface and then precisely delivered to the lungs via the homing ability of MOMC and activated for IPF reversion. However, there is still an unresolved point in our research, which is that the treatment mechanism of MOMC remains unclear. Our results indicated that MOMC might up-regulate AEC II proliferation or recover injured AEC II to normalize the lungs. The mechanism of MOMC differentiation for IPF treatment requires further exploration. In addition, some studies have indicated that MOMC could partly treat early IPF through regulating the immune response by inhibiting the proliferation of immune cells in vivo (42). It is unclear whether MOMC is effective for IPF therapy during different periods.
Compared with conventional antifibrotic strategies, our previous unknown programmed therapeutics MOMC/PER has showed accurate lung targeting and excellent therapeutic effects. The excellent antifibrotic efficacy of the MOMC/PER was achieved through the following features. (i) MOMC has the ability to backpack PER NPs, constructing programmed therapeutics MOMC/PER. (ii) MOMC/PER can precisely accumulate in IPF lung tissues due to the homing ability of the MOMC. (iii) PER NPs are sensitively released from MOMC/PER due to the overexpression of MMP-2 in the IPF microenvironment. (iv) Released PER NPs are able to retarget injured AEC II through c(RGDfc). (v) PER NPs can reduce the secretion of TGF-β by occupying TGF-latent sites. (vi) Two drugs loaded into PER NPs are the key factors in achieving IPF reversion of drug/drug as weeding and uprooting. In addition, MOMC also participates in AEC II regeneration using cell/drug strategy. Specifically, MOMC-mediated delivery therapeutics is convenient, and the materials used in our PER NPs have all been approved by the FDA, which indicates certain advantages for further clinical development. Overall, we have proposed an innovative concept to cure IPF through using native cells as a delivery carrier and a dual-drug combination as therapeutic agents, and this strategy is likely to be applicable to other major diseases.
MATERIALS AND METHODS
Materials
MMP-2, α-SMA rabbit anti-mouse antibody, and DCFH-DA were purchased from Sigma-Aldrich (St. Louis, USA). SPC rabbit anti-mouse antibody was purchased from Millipore (St. Louis, USA). Lymphocyte isolation kit was purchased from Solarbio Science & Technology Co. Ltd. (Beijing, China). PLGA-PEG-Mal and PLGA-PEG-c(RGDfc) were purchased from Jinan Daigan Biomaterial Co. Ltd. (Jinan, China). RPMI 1640, fetal bovine serum (FBS), and bicinchoninic acid (BCA) protein assay kit were purchased from Jiangsu KeyGEN BioTECH Co. Ltd. (Nanjing, China). The peptide E5 (CGPLGIAGQCGGRSFFLLRRIQGCRFRNTVDD) was synthesized by Top Peptide Biotechnology Co. Ltd. (Shanghai, China). AST was purchased from Yuanye Bio-Technology Co. Ltd. (Shanghai, China). TRA was purchased from J&K Scientific Co. Ltd. (Beijing, China). DiI, 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), and DiR were purchased from Fanbo Biochemicals Co. Ltd. (Beijing, China). DAPI (4,6-diamino-2-phenylindole) and mitochondrial membrane potential kit of JC-1 were purchased from Beyotime Biotechnology Co. Ltd. (Shanghai, China). CXC chemokine ligand 12 (CXCL 12) and CC chemokine ligand 19 (CCL 19) were purchased from Zoonbio Biotechnology Co. Ltd. (Beijing, China). BLM was purchased from Zhejiang Huahai Pharmaceutical Co. Ltd. (Linhai, China). TGF-β was purchased from Multi Sciences Biotech Co. Ltd. (Hangzhou, China). C6 was purchased from Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan). LysoTracker Red DND-99 kit was purchased from Thermo Fisher Scientific (Waltham, USA). Nanog, TGF-β/Smad and collagen I rabbit anti-mouse antibodies, and H&E and Masson staining kits were purchased from Servicebio Co. Ltd. (Nanjing, China). GSH, MDA, SOD, and hydroxyproline were purchased from Jiancheng Biotech Co. Ltd. (Nanjing, China). IL-1β and IL-4 detection kits were purchased from eBioscience (Waltham, USA). TGF-β detection kit was purchased from BioLegend (CA, USA). Polyvinylidene difluoride (PVDF) was purchased from PALL (NY, USA). Electrogenerated chemiluminescence (ECL) was purchased from Tanon Science & Technology Co. Ltd. (Shanghai, China).
Isolation and culture of primary MOMC
MOMC was isolated from peripheral blood by a mouse lymphocyte isolation kit. To obtain the MOMC, peripheral blood was collected from the C57BL/6J mice of IPF with EDTA and diluted three times with phosphate-buffered saline (PBS). The isolated protocol of MOMC was as follows: The mouse Percoll was added into diluted peripheral blood solution, and MOMC was isolated from peripheral blood by centrifugation at 600 rpm for 30 min. The solution was divided into upper, middle, and lower layers, and MOMC existed in the middle layer. Then, the MOMC was taken into centrifuge tube of 15 ml, and cell washing buffer of 5 ml was added into the tube. The cell suspension was then centrifuged for another 30 min, and it needed to be repeated three times to obtain MOMC. Last, the cell deposits were resuspended, and the suspension was put into the culture dish in RPMI 1640 medium with 10% FBS at 37°C and 5% CO2 (22, 23). The adherence time for dishes of MOMC was about 2 weeks. The MOMC was a kind of adherent cells, and the morphologies of cells were fusiform. After the cells adhere to the dish, the medium was changed once every 3 days.
Preparation of PER NPs
PER NPs were prepared by antisolvent precipitation method. Peptide E5 modification was prepared as follows. Specifically, the PLGA-PEG-Mal and PLGA-PEG-c(RGDfc) (mass ratio, 10:1) and dual drugs of AST and/or TRA were dissolved in dimethyl sulfoxide (50 mg/ml, 2 ml), added dropwise into deionized water with 100 ml, and then stirred with 300 rpm for 2 hours. Next, the prepared nanoparticle solution (NPs) was centrifuged at 2800 rpm for 15 min to discard the large particles and free drug. The NPs were then condensed to concentration of 2 ml by ultrafiltration device for further use. Last, the peptide E5 was added into the solution of NPs to form PER NPs. The preparation process of other groups including PLGA-PEG-c(RGDfc) (PR NPs), the preparation of PLGA-PEG-E5 (PE NPs), and the preparation of PLGA-PEG (PP NPs) were similar to PER NPs.
Preparation of MOMC/PER
The preparation of MOMC/PER was carried out by incubating MOMC with PER NPs. Briefly, the MOMC (2 × 105 cells/ml) was cultured in a petri dishes with a diameter of 100 mm. After incubated with the FBS-free media for 1 hour, PER NPs at a TRA concentration of 40 μg/ml were added into MOMC medium and incubated for 2 hours at 37°C and 5% CO2. At the same time, the CXCR4 receptor and ligand peptide E5 would undergo bioconjugate reaction.
Characterization of MOMC/PER
The hydrodynamic diameters and ζ potentials for PER NPs suspended in 1 × PBS were measured by Brookhaven Instruments (NY, USA). The morphologies of PER NPs were characterized by TEM (Hitachi TEM system, Japan).
For MOMC/PER-DiI, characterization of adhesion between MOMC and PER-DiI NPs was imaged by CLSM (Carl Zeiss 700, Germany). Specifically, MOMC was cultured in 35-mm culture dishes and incubated with PER-DiI NPs for 2 hours, and the preparation of PER-DiI NPs was the same as mentioned above. The nucleus of MOMC was labeled with DAPI and MOMC membrane was labeled by DiO. For SEM characterization, MOMC/PER-DiI was coated with gold/palladium and examined by Hitachi-SU8020 (Japan).
Release of PER NPs from MOMC/PER at the condition of MMP-2 in vitro
The sensitive release properties of PER NPs from MOMC/PER in vitro was evaluated under the microenvironment of MMP-2 in vitro. MMP-2 enzyme was applied to release PER NPs by degrading the linker of GPLGIAGQ between PER NPs and MOMC. The characterization of released activity was investigated in vitro. First, MMP-2 (2 μg/ml, 1 ml) was added into MOMC/PER-DiI medium for 30 min. Then, MOMC and released PER-DiI NPs were detected by flow cytometry (BD FACSCalibur, USA) or fixed by 4% paraformaldehyde (w/v) for CLSM and 2.5% glutaral for TEM. Besides, the nucleus of MOMC was labeled with DAPI for 30 min at 37°C, and the MOMC membrane was labeled with DiO for 15 min at 37°C. MMP-2 enzyme was dissolved in deionized water and free RPMI 1640 (volume ratio, 1:20) at a concentration of 2 μg/ml. After that, the released PER NPs and MOMC in solution were prepared for the other testing assay and image.
Loading ability of MOMC
The loading ability of MOMC was detected by the concentration of TRA and AST. First, MOMC was cultured in dishes with 100 mm. After MOMC adhered on the dishes, PER NPs were added into the culture dishes and incubated with MOMC for 2 hours in RPMI 1640 with free FBS, and then, MOMC/PER was washed thrice with PBS and digested with 0.25% trypsin-EDTA solution. Next, cells were harvested by centrifugation at 1000 rpm for 3 min and counted with a hemocytometer. Last, the cells were resuspended with PBS of 1 ml, and the absorbance was determined at 326 nm for TRA and 491 nm for AST by Multiskan GO (Thermo Fisher Scientific, USA).
Migration assay of MOMC/PER in vitro
The migration capacity of MOMC/PER was investigated by a Transwell device. First, the MOMC were cultured into the upper chambers with pore sizes of 8.0 μm with 2 × 104 in 400 μl of RPMI 1640 with FBS-free media for 24 hours. The cells were divided into three groups, including CXCL 12 (−) or CCL 19 (−) of MOMC, CXCL 12 (+) or CCL 19 (+) of MOMC, and CXCL 12 (+) or CCL 19 (+) of MOMC/PER. RPMI 1640 with 10% FBS and CXCL 12 (10 μg/ml) or CCL 19 of 600 μl was added to the lower chamber into the 24-well plates. Then, the MOMC in MOMC/PER group was added PER NPs at concentration of 40 μg/ml and incubated for another 24 hours. At last, the Transwell chambers were stained with crystal violet and were dissolved with 33% acetic acid, and the absorbance of solution was tested at 570 nm.
Animals and cell culture
C57BL/6J male mice were obtained from East China Normal University Laboratory Animal Technology Co. Ltd. (Shanghai, China) and housed with a 12-hour light/12-hour dark cycle at 25°C. All the animal protocols and procedures were performed under the guidelines for human and responsible use of animals in research approved by the regional ethics committee of China Pharmaceutical University. After acclimatization for 7 days, mice were subjected to IPF model experiments. IPF mice models were established by inhalation of BLM through endotracheal intubation (2 U/kg, 40 μl; Braintree Scientific, USA). Next, the mice were randomly assigned to the treatment. For the cell culture, A549 and MOMC were cultured in RPMI 1640 media containing 10% FBS and 1% penicillin and streptomycin at 37°C and 5% CO2.
Antifibrosis mechanism of dual drugs in vitro
A549 cells were cultured in six-well plates at 37°C and 5% CO2 for 24 hours and then incubated for another 24 hours with pure PPA, PPT, or PPTA at TRA concentration of 20 nM. The expression of vimentin and fibronectin was investigated by immunofluorescence staining.
Wound healing assay in vitro
The invasion ability of TGF-β–induced A549 cells was evaluated by wound healing tests after treated with various formulations. First, A549 cells were seeded in six-well plates at 15 × 104 cells per dish and incubated for 24 hours. Next, a 10-μl pipette tip was used to scratch wells in the middle of the dishes, and then, A549 cells were washed three times with PBS to remove suspended cells. The cells from each group were imaged by inverted fluorescence microscope (Nikon, Japan) to observe the extents of wound healing after treated with PPT, PPA, PPTA, and PER NPs at 24 hours.
Cell migration assay in vitro
The migration capability of TGF-β–induced A549 cells was investigated by a Transwell device. The A549 cells of 5 × 104 in 400 μl of RPMI 1640 with FBS-free media were added to the upper chambers with pore sizes of 8.0 μm for 24 hours, and RPMI 1640 with 10% FBS media of 600 μl was added to the lower chamber into the 24-well plates. Then, the cells were incubated with PPT, PPA, PPTA, and PER NPs (20 nM of TRA concentration) for another 24 hours. After incubation, the Transwell chambers were stained with crystal violet and were dissolved with 33% acetic acid, and the absorbance of solution was tested at 570 nm.
ROS detection in vitro
A549 cells were seeded on six-well plates (15 × 104 per well) and incubated overnight. First, the cells were activated by TGF-β, and then, various treatment groups were incubated with A549 cells for 24 hours (TGF-β, normal, PPT, PPA, PPTA, and PER NPs). Next, the ROS probe DCFH-DA (5 μM) was added into the dishes and incubated with cells in RPMI 1640 media with free FBS at 37°C under 5% CO2 in the dark for 15 min. Last, cells were digested, and the content of ROS was analyzed by flow cytometry (BD Accuri C6, USA) and imaged by inverted fluorescence microscope, respectively.
Changes of mitochondrial membrane potential in vitro
A549 cells were cultured on six-well plates (15 × 104 per well) overnight at 37°C and 5% CO2. After treated with different formulations for 24 hours, 500 μl of mitochondrial membrane potential reagent of JC-1 (1×) solution was added into the dishes for 20 min. Then, the cells were stained by 4% paraformaldehyde (w/v) and imaged by inverted fluorescence microscope.
Intracellular uptake in vitro
First, A549 cells were seeded on a 35-mm sterile glass bottom culture dishes (2 × 105 cells) and cultured overnight in RPMI 1640 with 10% FBS. The preparation of PR-C6 and PP-C6 was same as mentioned above for PER NPs. The PR-C6, PP-C6, and C6 were then incubated with A549 cells for 4 hours at 5 μg/ml. Next, the cells were washed three times by PBS, and the nucleus was stained with DAPI. Images and data were acquired with CLSM and flow cytometry (BD Accuri C6, USA).
Lysosome escape in vitro
The A549 cells of 5 × 104 were cultured in a 35-mm sterile glass bottom culture dishes. After the cells were cultured overnight in RPMI 1640 with 10% FBS for 24 hours, PR-C6 and PP-C6 were incubated with A549 cells for 1 and 4 hours at 5 μg/ml. Then, the cell dishes were washed three times by PBS and fix by 4% paraformaldehyde (w/v). Lysosome was stained with LysoTracker Red DND-99 kit (100 nM) for 15 min, and the dishes were washed three times with PBS. Then, the cell nucleus was stained by DAPI like above method. Images were acquired by CLSM.
Lung target capacity and biodistribution study of MOMC/PER-DiR in vivo
MOMC/PER-DiR was prepared by the method as mentioned above for PER NPs. The homing ability of MOMC was detected in vivo by IVIS imaging system (Kodak, USA). C57BL/6J mice were induced IPF by inhalation of BLM. Then, the IPF mice were injected with MOMC/PER-DiR, MOMC-DiR, and free DiR by intravenous injection and tested by IVIS living system at different point times. Then, the mice are sacrificed, and the lungs and other organs were harvested for ex vivo imaging after 8 hours of intravenous injection. ROI was circled around the lungs and the other organs (liver, heart, spleen, and kidneys). The fluorescence intensity of the DiR was determined by living image software.
Behavior of MOMC/PER-DiR in vivo
The PER NPs can be released from MOMC/PER-DiI by MMP-2, owing to responsive blocking the linker between PER NPs and MOMC. We have investigated the homing capability, responsive release, and retarget ability. We first administrated MOMC/PER-DiI by intravenous injection, and the preparation of MOMC/PER-DiI was applied by the methods as mentioned above. The mice were sacrificed, and lung tissues were harvested at different point time of 30 min, 1 hour, and 2 hours in a dark place. First, the lung tissues were fixed with 4% paraformaldehyde (w/v), and then, the tissues were embedded and dewaxed before slicing. Next, the lung slices were labeled by Nanog and SPC at 4°C for 1 hour and washed with 0.2% Triton X-100 for three times. Then, they were incubated with relevant secondary antibodies for 2 hours. Thereafter, the slices were stained with DAPI and viewed under fluorescence microscope.
PER NPs are reprogramming of PER NPs in vivo by IVIS imaging system
The IPF mice were injected with PER-DiR NPs, PE-DiR NPs, PR-DiR NPs, and PP-DiR NPs by intravenous injection at different point times for 1, 4, 8, 12, and 24 hours and tested by IVIS imaging system. The preparation of PER-DiR NPs, PE-DiR NPs, PR-DiR NPs, and PP-DiR NPs were the same method as mentioned above, and then, the mice were sacrificed, and the lungs and other organs were harvested to detect ex vivo imaging after intravenous injection at 24 hours. ROI was tested on the lungs and the other organs (liver, heart spleen, and kidneys). The fluorescence intensity of the DiR was determined by living image software.
Treatment efficacy of treatments in vivo
The antifibrotic efficacy in MOMC/PER, MOMC, PER NPs, and pirfenidone for IPF treatment in vivo was evaluated on IPF male mice (C57BL/6J, age of 6 to 8 weeks). The preparations of MOMC/PER and PER NPs were the same method as mentioned above. Pirfenidone was administered by gastrointestinal because it is an oral medication, and other formulations were administered via intravenous injection.
Biochemical indexes
The contents of MDA, SOD, and GSH were detected after treated with various treatments in lung tissues. The protocols are as follows: Solution 1 of 1.5 ml was added into the lung tissues solution (0.5 ml) and mixed thoroughly. Then, the samples were centrifuged for 10 min at 3500 to 4000 rpm. The sample supernatant was added into 3,3′,5,5′-tetramethyl benaidine (TMB substrate), and the absorbance of GSH was detected after 5 to 10 min at 420 nm. The contents of SOD and MDA were tested at 550 and 532 nm, respectively.
Content of TGF-β, IL-1β, and IL-4 in vivo
The lungs and spleen tissues were collected and diluted in precooled solution. The IL-1β, TGF-β, and IL-4 in lung tissues were assayed using enzyme-linked immunosorbent assay (ELISA) method as instructed by the manufacturer. First, the wells were washed three times with a washing buffer for 3 min each time. Next, the blocking solution of 200 μl was added into each well and incubated for 1 to 2 hours at 37°C. Then, the sealing film was removed carefully and putted it into the washing machine and washed three to five times. Furthermore, the sample of 100 μl was added and should be tested diluted appropriately to the above coated reaction wells, and the diluted biotinylated antibody working solution was added with 100 μl into each well. Then, the samples were sealed with a sealing membrane and incubated at 37°C for 1 hour. The following step was that 100 μl of diluted enzyme conjugate working solution was added into each well. Next, TMB substrate solution with 100 μl was added into each well and should be avoided reaction with light for 10 to 30 min at 37°C until a notable color gradient appears in the diluted standard well. Within 10 min, the absorbance of each well was measured on a microplate reader at 450 nm with zero adjustment of the blank control well.
Content of hydroxyproline in vivo
The content of hydroxyproline was detected by a hydroxyproline detection kit. The lung tissues of 30 to 100 mg were mixed with hydrolysate to 1 ml and hydrolyzed in boiling water for 20 min. The sample solution was pH 6.0 to 6.8. Then, the serum hydrolysate was added activated carbon and mixed at 60°C for 15 min. After cooling, the serum samples were centrifuged at 3000 rpm for 20 min, and the supernatant was detected by microplate reader at 550 nm.
Proliferation of inflammatory cells in vivo
The whole blood from mice was collected by anticoagulant tube with EDTA, and white blood cell counts (including lymphocytes and monocytes) were assayed using a standard blood analyzer (Mindray, China).
HE, Masson, and IHC in vivo
After treatment with different formulation, the lungs, heart, liver, spleen, and kidneys were harvested, and lungs were investigated by H&E, Masson, and IHC staining. Other organs were detected by H&E staining to evaluate the application security. First, the lung tissues were fixed with 4% paraformaldehyde (w/v) for more than 48 hours and embedded in paraffin. Then, the lung tissues were cut into 4-μm sections for H&E staining and Masson staining. The levels of collagen I and α-SMA were evaluated by IHC, and protocols are as follows: The slices were incubated with primary antibodies (collagen I and α-SMA or fluorescently labeled CD90 and collagen I, Servicebio, China) and then incubated with corresponding secondary antibodies to detect the expression of relative proteins.
mRNA expression of Ctgf and Acta2 in vivo
The qPCR analysis was evaluated RNA expression of Ctgf and Acta2. qPCR was conducted in ABI StepOnePlus (Thermo Fisher Scientific, USA). Lung tissues (100 mg) were homogenized to extract the total RNA according to the protocols. Complementary DNA (2 μg) was prepared using the Reverse Transcription System, and then, the expression of related genes was determined using q-PCR. The primers used are Acta2 (NM_007392.3), Ctgf (43), and Gapdh (NM_008084.2).
Western blotting
The harvested lungs were homogenized in PBS buffer and then centrifuged at 3000 rpm for 30 min. The supernatant of total protein was taken for further experiments. Total protein concentration in the solution was determined with a BCA protein assay kit. After detecting in SDS-PAGE with protein samples in different treatment groups, the bands were transferred onto PVDF membrane. Next, the PVDF membranes were blocked with 5% milk at room temperature for 2 hours and incubated with primary antibodies (α-SMA, TGF-β/Smad, and β-actin rabbit anti-mouse antibodies) at 4°C overnight and then incubated with corresponding secondary antibodies for 2 hours at room temperature. Last, the bands were detected using ECL (Tanon, China) Western blotting substrate (Thermo Fisher Scientific, USA). The β-actin was used as an endogenous control.
Safety evaluation
The mice were sacrificed, and the serum samples were detected in different treatment groups. The contents of ALT, aspartate aminotransferase, and BUN in the serum were determined using the relevant assay kits (Servicebio, China).
Statistical analysis
Statistical analyses were performed using GraphPad Prism software (GraphPad Software, USA). All error bars were means ± SEM; differences detection index between the treated groups and control groups were determined via one-way analysis of variance (ANOVA). P < 0.05 was considered significantly different.
Supplementary Material
Acknowledgments
We thank the Cellular and Molecular Biology Center of China Pharmaceutical University for assistance with confocal microscopy work. Funding: This work was supported by the National Key R&D Program of China (2017YFA0205400). We thank the National Natural Science Foundation of China (NSFC; grant nos. 81773667, 81573369, and 81430082) and NSFC Projects of International Cooperation and Exchanges (81811540416). This work was also supported by the Fundamental Research Funds for the Central Universities (2632018PT01 and 2632018ZD12), the “111” Project from the Ministry of Education of China and the State Administration of Foreign Experts Affairs of China (B16046), the Double First-Class Project (CPU2018GY06), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. Author contributions: H.-L.J., H.-P.H., X.C., and L.X. conceived the project, designed all the experiments, analyzed the data, and wrote the manuscript. X.C. and L.X. conducted the experiments. X.C. and Y.W. analyzed the data. X.C., Y.W., C.-X.Y., Y.-J.H., T.-J.Z., X.-D.G., and L.L. wrote the manuscript. All authors edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba3167/DC1
REFERENCES AND NOTES
- 1.Raghu G., Flaherty K. R., Lederer D. J., Lynch D. A., Colby T. V., Myers J. L., Groshong S. D., Larsen B. T., Chung J. H., Steele M. P., Benzaquen S., Calero K., Case A. H., Criner G. J., Nathan S. D., Rai N. S., Ramaswamy M., Hagmeyer L., Davis J. R., Gauhar U. A., Pankratz D. G., Choi Y., Huang J., Walsh P. S., Neville H., Lofaro L. R., Barth N. M., Kennedy G. C., Brown K. K., Martinez F. J., Use of a molecular classifier to identify usual interstitial pneumonia in conventional transbronchial lung biopsy samples: A prospective validation study. Lancet Respir. Med. 7, 487–496 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Schwartz D. A., Van Fossen D. S., Davis C. S., Helmers R. A., Dayton C. S., Burmeister L. F., Hunninghake G. W., Determinants of progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 149, 444–449 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Lederer D. J., Martinez F. J., Idiopathic pulmonary fibrosis. N. Engl. J. Med. 378, 1811–1823 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Martinez F. J., Collard H. R., Pardo A., Raghu G., Richeldi L., Selman M., Swigris J. J., Taniguchi H., Wells A. U., Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Prim. 3, 17074 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Gu B. H., Madison M. C., Corry D., Kheradmand F., Matrix remodeling in chronic lung diseases. Matrix Biol. 73, 52–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shetty S. K., Tiwari N., Marudamuthu A. S., Puthusseri B., Bhandary Y. P., Fu J., Levin J., Idell S., Shetty S., p53 and miR-34a feedback promotes lung epithelial injury and pulmonary fibrosis. Am. J. of Pathol. 187, 1016–1034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolters P. J., Blackwell T. S., Eickelberg O., Loyd J. E., Kaminski N., Jenkins G., Maher T. M., Molina-Molina M., Noble P. W., Raghu G., Richeldi L., Schwarz M. I., Selman M., Wuyts W. A., Schwartz D. A., Time for a change: Is idiopathic pulmonary fibrosis still idiopathic and only fibrotic. Lancet Respir. Med. 6, 154–160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weng T., Ko J., Masamha C. P., Xia Z., Xiang Y., Chen N. Y., Molina J. G., Collum S., Mertens T. C., Luo F., Philip K., Davies J., Huang J., Wilson C., Thandavarayan R. A., Bruckner B. A., Jyothula S. S., Volcik K. A., Li L., Han L., Cleavage factor 25 deregulation contributes to pulmonary fibrosis through alternative polyadenylation. J. Clin. Invest. 129, 1984–1999 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xaubet A., Marin-Arguedas A., Lario S., Ancochea J., Morell F., Ruiz-Manzano J., Rodriguez-Becerra E., Rodriguez-Arias J. M., Inigo P., Sanz S., Campistol J. M., Mullol J., Picado C., Transforming growth factor-β1 gene polymorphisms are associated with disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 168, 431–435 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Mora A. L., Rojas M., Pardo A., Selman M., Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 16, 755–772 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Guo L., Karoubi G., Duchesneau P., Aoki F. G., Shutova M. V., Rogers I., Nagy A., Waddell T. K., Interrupted reprogramming of alveolar type II cells induces progenitor-like cells that ameliorate pulmonary fibrosis. NPJ Regen. Med. 14, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newman S. P., Delivering drugs to the lungs: The history of repurposing in the treatment of respiratory diseases. Adv. Drug Deliv. Rev. 133, 5–18 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Xue J., Zhao Z., Zhang L., Xue L., Shen S., Wen Y., Wei Z., Wang L., Kong L., Sun H., Ping Q., Mo R., Zhang C., Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotech. 12, 692–700 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Zhang W. Z., Wang M. Z., Tang W., Wen R., Zhou S. Y., Lee C. B., Wang H., Jiang W., Delahunty I. M., Zhen Z. P., Chen H., Chapman M., Wu Z. H., Howerth E. W., Cai H., Li Z. B., Xie J., Nanoparticle-laden macrophages for tumor-tropic drug delivery. Adv. Mater. 30, 1805557 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Q., Sun W., Wang J., Ruan H., Zhang X., Ye Y., Shen S., Wang C., Lu W., Cheng K., Dotti G., Zeidner J. F., Wang J., Gu Z., Conjugation of haematopoietic stem cells and platelets decorated with anti-PD-1 antibodies augments anti-leukaemia efficacy. Nat. Biomed. Eng. 2, 831–840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aghajanian H., Kimura T., Rurik J. G., Hancock A. S., Leibowitz M. S., Li L., Scholler J., Monslow J., Lo A., Han W., Wang T., Bedi K., Morley M. P., Linares-Saldana R. A., Bolar N. A., McDaid K., Assenmacher C. A., Smith C. L., Wirth D., June C. H., Margulies K. B., Jain R., Puré E., Albelda S. M., Epstein J. A., Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ling C., Nishimoto K., Rolfs Z., Smith L. M., Frey B. L., Welham N. V., Differentiated fibrocytes assume a functional mesenchymal phenotype with regenerative potential. Sci. Adv. 5, eaav7384 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y., Peng H., Sun H. X., Peng X. Y., Tang C. Y., Gan Y., Chen X. S., Mathur A., Hu B. Q., Slade M. D., Chitinase 3-like 1 suppresses injury and promotes fibroproliferative responses in mammalian lung fibrosis. Sci. Transl. Med. 6, 240ra276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strieter R. M., Gomperts B. N., Keane M. P., The role of CXC chemokines in pulmonary fibrosis. J. Clin. Invest. 117, 549–556 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elias J. A., Zurier R. B., Schreiber A. D., Leff J. A., Daniele R. P., Monocyte inhibition of lung fibroblast growth: Relationship to fibroblast prostaglandin production and density-defined monocyte subpopulations. J. Leukoc. Biol. 37, 15–28 (1985). [DOI] [PubMed] [Google Scholar]
- 21.Seta N., Kuwana M., Derivation of multipotent progenitors from human circulating CD14+ monocytes. Exp. Hematol. 38, 557–563 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Kuwana M., Okazaki Y., Kodama H., Izumi K., Yasuoka H., Ogawa Y., Kawakami Y., Ikeda Y., Human circulating CD14+ monocytes as a source of progenitors that exhibit mesenchymal cell differentiation. J. Leukoc. Biol. 74, 833–845 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Noriyuki S., Masataka K., Potential involvement of human circulating CD14+ monocytes in tissue repair and regeneration. Inflamm. Regen. 32, 1–7 (2012). [Google Scholar]
- 24.Maria G. F., Jacqueline A. B., Danielle L. B., Todd C. C., Neil J. D., John T. R., Mark T. R., Nico V. R., Kurt R. S., Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am. J. Pathol. 168, 659–669 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fraser E., Blirando L., Noble V. S., Benamore R., Hoyles R., Benlahrech A., Ho L. P., S50 monocytes from IPF patients show pre-conditioned pro-repair features. Thorax 71, A30–A31 (2016). [Google Scholar]
- 26.Maltsev O. V., Marelli U. K., Kapp T. G., Di Leva F. S., Di Maro S., Nieberler M., Reuning U., Schwaiger M., Novellino E., Marinelli L., Kessler H., Stable peptides instead of stapled peptides: Highly potent αvβ6-selective integrin ligands. Angew. Chem. Int. Ed. Engl. 55, 1535–1539 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Agha E. E., Kramann R., Schneider R. K., Li X. K., Seeger W., Humphreys B. D., Bellusci S., Mesenchymal stem cells in fibrotic disease. Cell Stem Cell 21, 166–177 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Markou G., Nerantzis E., Microalgae for high-value compounds and biofuels production: A review with focus on cultivation under stress conditions. Biotechnol. Adv. 31, 1532–1542 (2013). [DOI] [PubMed] [Google Scholar]
- 29.McQuade J. L., Daniel C. R., Hess K. R., Mak C., Wang D. Y., Rai R. R., Park J. J., Haydu L. E., Spencer C., Wongchenko M., Lane S., Lee D. Y., Kaper M., McKean M., Beckermann K. E., Rubinstein S. M., Rooney I., Musib L., Budha N., Hsu J., Nowicki T. S., Avila A., Haas T., Puligandla M., Lee S., Fang S., Wargo J. A., Gershenwald J. E., Lee J. E., Hwu P., Chapman P. B., Sosman J. A., Schadendorf D., Grob J. J., Flaherty K. T., Walker D., Yan Y., McKenna E., Legos J. J., Carlino M. S., Ribas A., Kirkwood J. M., Long G. V., Johnson D. B., Menzies A. M., Davies M. A., Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: A retrospective, multicohort analysis. Lancet Oncol. 19, 310–322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toonkel R. L., Hare J. M., Matthay M. A., Glassberg M. K., Mesenchymal stem cells and idiopathic pulmonary fibrosis potential for clinical testing. Am. J. Respir. Crit. Care Med. 188, 133–140 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Jimena G. D., Aniket S. W., Nina P. C., Rebeca G., Heather M. A., Sen P., Nhan L. T., Olga G. G., Graeme F. W., Jeffrey A. W., Anthony J. K., Decreased nonspecific adhesivity, receptor-targeted therapeutic nanoparticles for primary and metastatic breast cancer. Sci. Adv. 6, eaax3931 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hennrick K. T., Keeton A. G., Nanua S., Kijek T. G., Goldsmith A. M., Sajjan U. S., Bentley J. K., Lama V. N., Moore B. B., Schumacher R. E., Thannickal V. J., Hershenson M. B., Lung cells from neonates show a mesenchymal stem cell phenotype. Am. J. Respir. Crit. Care Med. 175, 1158–1164 (2007). [DOI] [PubMed] [Google Scholar]
- 33.de la Rica R., Matsui H., Applications of peptide and protein-based materials in bionanotechnology. Chem. Soc. Rev. 39, 3499–3509 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X. J., Guo H., Duan H. Y., Yang Y. L., Meng J., Liu J., Wang C., Xu H. Y., Targeting CXCR 4/CXCL 12 axis to improve chemotherapeutics efficiency in acute myeloid leukemia treatments by synthesized peptide E5. Nanomedicine 14, 1764–1765 (2018). [Google Scholar]
- 35.Takahashi M., Ishiko T., Kamohara H., Hidaka H., Ikeda O., Ogawa M., Baba H., Curcumin (1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione) blocks the chemotaxis of neutrophils by inhibiting signal transduction through IL-8 receptors. Mediators Inflamm. 2007, 10767 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costabel U., Inoue Y., Richeldi L., Collard H. R., Tschoepe I., Stowasser S., Azuma A., Efficacy of nintedanib in idiopathic pulmonary fibrosis across prespecified subgroups in inpulsis. Am. J. Respir. Crit. Care Med. 193, 178–185 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Gallo R., Gambelli F., Gava B., Sasdelli F., Tellone V., Masini M., Marchetti P., Dotta F., Sorrentino V., Generation and expansion of multipotent mesenchymal progenitor cells from cultured human pancreatic islets. Cell Death Differ. 14, 1860–1871 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Förster R., Davalos-Misslitz A. C., Rot A., CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol. 8, 362–371 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Serbina N. V., Pamer E. G., Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR 2. Nat. Immunol. 7, 311–317 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Takada S., Kambe N., Kawasaki Y., Niwa A., Honda-Ozaki F., Kobayashi K., Osawa M., Nagahashi A., Semi K., Hotta A., Asaka I., Yamada Y., Nishikomori R., Heike T., Matsue H., Nakahata T., Saito M. K., Pluripotent stem cell models of Blau syndrome reveal an IFN-γ-dependent inflammatory response in macrophages. J. Allergy Clin. Immunol. 141, 339–349.e11 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Ding L., Zhu C., Yu F., Wu P., Chen G., Ullah A., Wang K., Sun M., Li J., Oupický D., Pulmonary delivery of polyplexes for combined PAI-1 gene silencing and CXCR 4 inhibition to treat lung fibrosis. Nanomedicine 14, 1765–1776 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Wong E., Xu R.-H., Rubio D., Lev A., Stotesbury C., Fang M., Sigal L. J., Migratory dendritic cells, group 1 innate lymphoid cells, and inflammatory monocytes collaborate to recruit NK cells to the virus-infected lymph node. Cell Rep. 24, 142–154 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lo L., McLennan S., Song W., Tan J., Twigg S., Williams P., Bonner J., Connective tissue growth factor inhibits adipocyte differentiation. Am. J. Physiol. 295, C740–C751 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba3167/DC1