

Abstract
Multiple sclerosis (MS) is a chronic, inflammatory autoimmune disease that affects the central nervous system (CNS) for which there is no cure. In MS, encephalitogenic T cells infiltrate the CNS causing demyelination and neuroinflammation; however, little is known about the role of regulatory T cells (Tregs) in CNS tissue repair. Transplantation of neural stem and progenitor cells (NSCs and NPCs) is a promising therapeutic strategy to promote repair through cell replacement, although recent findings suggest transplanted NSCs also instruct endogenous repair mechanisms. We have recently described that dampened neuroinflammation and increased remyelination is correlated with emergence of Tregs following human NPC transplantation in a murine viral model of immune-mediated demyelination. In the current study we utilized the prototypic murine autoimmune model of demyelination experimental autoimmune encephalomyelitis (EAE) to test the efficacy of hNSC transplantation. Eight-week-old, male EAE mice receiving an intraspinal transplant of hNSCs during the chronic phase of disease displayed remyelination, dampened neuroinflammation, and an increase in CNS CD4+CD25+FoxP3+ regulatory T cells (Tregs). Importantly, ablation of Tregs abrogated histopathological improvement. Tregs are essential for maintenance of T cell homeostasis and prevention of autoimmunity, and an emerging role for Tregs in maintenance of tissue homeostasis through interactions with stem and progenitor cells has recently been suggested. The data presented here provide direct evidence for collaboration between CNS Tregs and hNSCs promoting remyelination.
Keywords: Neural Stem Cells, Neuroinflammation, Remyelination, Regulatory T cells, Multiple Sclerosis
Introduction
Multiple Sclerosis (MS) is an inflammatory disease of the central nervous system (CNS) characterized by immune-mediated demyelination and subsequent axonal loss (Steinman, 1996). Immunosuppressive therapies are directed at the most common clinical form of MS, with efficacy in only 30% of patients (Lassmann et al., 1997), likely due to the fact that these therapies fail to promote sustained remyelination (Torkildsen et al., 2016). Consequently, there is an unmet clinical need for methods to promote remyelination while limiting immune cell infiltration into the CNS.
Studies employing animal models of demyelination demonstrate that transplantation of neural stem and progenitor cells (NSCs and NPCs) is a potential therapeutic strategy to treat diseases affecting white matter integrity. NSCs are capable of generating all three CNS neural lineages; NSCs can differentiate and replace damaged neurons, astrocytes and oligodendrocytes (Gage, 2000). Indeed, multiple studies have demonstrated that surgical implantation of NSCs into the CNS mutes neuroinflammation and enhances remyelination (Ben-Hur et al., 2013; Chen et al., 2014; Greenberg et al., 2014; Mozafari et al., 2015; Plaisted et al., 2016; Pluchino et al., 2009; Salazar et al., 2010). Initially, cell replacement was thought to be the primary mechanism of action; however, subsequent findings suggested transplanted NSCs instruct endogenous repair mechanisms (Pluchino et al., 2009). Much of the work evaluating the efficacy of NSC transplantation has been conducted using grafts from syngeneic donors or transplantation into immune-compromised or immune-suppressed recipients. From a clinical perspective, syngeneic NSC transplants are not feasible for patients with MS because NSCs may have a genetic defect. In addition, major histocompatibility complex (MHC) matched cells are challenging to obtain, and since donor transplants are likely to be allogeneic, life-long systemic immune suppression is required, increasing the risk of infection for the patient. Therefore, it is important to consider the potential for transplanted cells to elicit an immune response as well as the impact of transplanting cells into a chronic inflammatory environment.
Previously, we demonstrated that intraspinal injection of human induced pluripotent stem cell (iPSC) embryoid body (EB)-derived NPCs results in dampened neuroinflammation and remyelination that is correlated with the emergence of regulatory T cells (Tregs) in mice afflicted with murine hepatitis virus (MHV)-induced immune-mediated demyelination (Chen et al., 2014; Mangale et al., 2019; Plaisted et al., 2016). Intracranial inoculation of susceptible mice with the neurotropic JHM strain of MHV results in widespread dissemination of virus throughout the CNS inducing an acute encephalomyelitis evolving into a chronic demyelinating disease (Bergmann et al., 2006; Glass et al., 2004; Hosking and Lane, 2009). MHV infection displays histological and pathological similarities to MS, rendering it an exemplary model to characterize immune mediated demyelinating diseases (Denic et al., 2011; Hosking and Lane, 2009). However, T cells respond to viral epitopes in MHV infection, rather than self-antigens, and thus the model lacks the autoimmune features of MS. Unfortunately, there is no single experimental model that recapitulates the entire spectrum of clinical, pathological or immunological features of MS (Lassmann and Bradl, 2017). Therefore, it is essential to investigate the potential of NSCs to elicit an immune response in multiple animal models of immune-mediated demyelination.
The present study explores the mechanisms through which NSCs affect the pathophysiology of Experimental Autoimmune Encephalomyelitis (EAE), which is the prototypic model of MS. In EAE, mice are immunized with self-antigen Myelin Oligodendrocyte Glycoprotein (MOG35-55) in combination with Complete Freund’s Adjuvant and pertussis toxin results in chronic inflammatory disease in the spinal cord potentiated by autoreactive CD4+ T cells (Mendel et al., 1995). Here, we show that hNSC transplantation leads to an increase in CNS Tregs following transplant that activates endogenous repair pathways and promote remyelination in EAE mice. EAE mice receiving xenogeneic hNSCs displayed remyelination, that correlated with dampened neuroinflammation, and was accompanied by an increase in CNS CD4+CD25+FoxP3+ Tregs. Importantly, ablation of Tregs abrogated histopathological improvement. These data support a vital role for Treg and NSC interactions in promoting tissue repair and remyelination within the CNS.
Materials and Methods
Animal care and EAE immunization
All experiments were approved by the University of California, Irvine Institutional Animal Care and Use Committee. Age-matched (8 weeks) male C57BL/6 mice (H-2b, Charles River) or C57BL/6 B6 FoxP3EGFP mice (B6.Cg-Foxp3tm2(EGFP)Tch/J, Stock No: 006772; Jackson Laboratories) mice were immunized by subcutaneous injections with 100μl of emulsion containing 100μg of Myelin Oligodendrocyte Glycoprotein (MOG35-55) (MEVGWYRSPFSRVVHLYRNGK-COOH; Pierce) in phosphate buffered saline (PBS), with complete Freund’s adjuvant (CFA) containing 200μg Mycobacterium tuberculosis H37Ra (DIFCO Laboratories). The day of NSC transplant is designated as day 0. Mice received intraperitoneal (I.P.) injections with 400ng (C57BL/6) or 200ng (FoxP3EGFP) of Bordetella pertussis toxin (List Biological Laboratories) on day −21 and day −19 prior to transplant. Mice were monitored for clinical symptoms daily. Clinical evaluation was performed double-blind and based on the following scoring system; 0, asymptomatic; 0.5, ruffled fur; 1, flaccid tail; 2, hind limb paresis; 2.5, partial hind limb paralysis; 3, hind limb paralysis; 4, hind limb and forelimb paralysis; 5, moribund. Clinical score data is presented as average ± SEM. No significant difference in locomotor function were observed. Data were analyzed using Prism software (GraphPad).
Mice were given long sipper water bottles and supplemented with 1mL of saline injected subcutaneously when a score of 2 or greater was achieved. Wet food (Envigo 2020X diet) and diet gel (Clear H2O) supplements were also placed at the bottom of the cage. Mice were sacrificed via inhalation of a lethal dose of isoflurane and cardiac perfusion with PBS was performed at defined time points post-transplant (p.t.) for tissue harvesting and analysis.
Transplantation of NSCs
Mice immunized with MOG were injected with 2.5 ×105 enhanced Green Fluorescent Protein (eGFP)-mNSCs, hNSCs, or hDFs in 2.5μl of PBS or PBS alone as a control, at thoracic vertebrae 9 (T9) on day 21 post-immunization (p.i) (Day 0) as previously described (Carbajal et al., 2011).
Derivation and maintenance of mNSCs
eGFP-mNSCs derived from eGFP transgenic C57BL/6 mice were derived and cultured as previously described (Lu et al., 2002). mNSCs were maintained in mNSC medium (DMEM/F12 w/GlutaMax, 250μg/mL Amphotericin B, 100U/mL penicillin, 100μg/mL streptomycin, and 1X N2; all from Thermo Fisher) supplemented with 20μg/mL EGF (Thermo Fisher). 0.05% Trypsin (Thermo Fisher) was used to split cells when the cell density reached 80-90% confluence.
Derivation and maintenance of hNSCs
WA09 hESCs were adapted to feeder-free conditions and maintained in Essential 8 medium (Thermo Fisher) on Geltrex™-coated plates (Thermo Fisher). hESCs were differentiated into EB-NSCs according to established methods (Chambers et al., 2009). Briefly, feeder-free hESCs were dissociated using Accutase (Thermo Fisher), transferred to ultra-low adherence 6-well plates, and cultured for 5 days in human ESC medium (DMEM/F12+ GlutaMAX, 20% Knockout Serum Replacement, 1X non-essential amino acids, and 0.1M 2-mercaptoethanol; all from Thermo Fisher) supplemented with 500ng/mL recombinant Noggin (R&D Systems) and 10μM SB431542 (Torcris). On the fifth days of culture, hESCs formed embryoid body-like structures (EBs) and 20-50 EBs were transferred to each well of a Geltrex™-coated 6-well dish. Increasing amounts of N2 medium (DMEM/F12+ GlutaMAX and 1X N2 Supplement; Thermo Fisher) supplemented with 500ng/mL Noggin and 10μM SB431542 were added every other day for 14 days. Resulting columnar rosette structured were collected using Accutase and stained with anti-human CD184, anti-human CD24, anti-human CD44, anti-human, CD271 according to manufacturer recommendations using the BD Stemflow Human Neural Cell Sorting Kit (BD Biosciences). Cells were sorted on a FACSAria Fusion (BD Biosciences) and sorted hNSCs were maintained in hNSC maintenance medium (DMEM/F12+ GlutaMAX, 0.5X N2, 0.5X B27 without vitamin A, 20ng/mL bFGF; all from Thermo Fisher) on Geltrex™-coated dishes using Accutase to split cells when the cell density reached 80-90% confluence.
hNSCs were characterized by flow cytometry analysis. hNSCs were collected using Accutase and stained with mouse anti-human Nestin (1:200; BD Biosciences), mouse anti-human Sox1 (1:200, BD Biosciences), and mouse anti-human Sox2 (1:200; BD Biosciences). For immunofluorescence microscopy differentiated NSCs were seeded on Geltrex-coated chamber slides, fixed with 4% paraformaldehyde, and stained with mouse anti-beta-Ill-tubulin (1:500; Abcam), rabbit anti-GFAP (1:200; Thermo Fisher), or rabbit anti-NG2 (1:200; Chemicon) before addition of respective secondary antibodies (goat anti-rabbit AlexaFluor™ 488, goat anti-rabbit AlexaFluor™ 568; Thermo Fisher). Slides were cover-slipped and mounted using VectaShield, Hard Set Mounting Medium with DAPI (Vector Labs), and all images were captured using a Nikon Eclipse Ti inverted microscope.
Statistical Analyses
Data were analyzed using Prism software (GraphPad). Comparisons were performed using a two-tailed T-test and two-way analysis of variance, where indicated. Clinical scores were analyzed using two-tailed T-tests with a Mann-Whitney post-test. For all statistical models and tests described above, the significance is displayed as follows: NS p>0.05, *p<0.05, **p<0.01, ***p<0.001.
Experimental Design-eGFP-mNSCs transplant
Age-matched (8 weeks) male C57BL/6 mice (H-2b, Charles River) mice were immunized by subcutaneous injections of MOG peptide and Pertussis toxin 21 days prior to NSC transplant as described above. At day 21 p.i. mice received intraspinal transplants of 2.5 ×105 eGFP-mNSCs in 2.5μl of PBS or PBS alone as a control, at thoracic vertebrae 9 (T9).
Immunohistochemistry and analysis of histopathology
For quantification of eGFP-mNSC cell survival and demyelination, mice receiving eGFP-mNSCs (n=12) or PBS (n=13) control transplants were sacrificed 28 days p.t., spinal cords were dissected, fixed overnight in 4% paraformaldehyde, and soaked in 30% sucrose for 7 days before being embedded in OCT compound. Frozen tissues were serially sectioned into 8μM and counterstained with DAPI or stained with Luxol Fast Blue (LFB) and counterstained with hematoxylin and eosin (H&E) to assess the eGFP-mNSC survival and migration or severity of demyelination, respectively. The total area of the white matter was quantified and compared to the area of demyelinated regions using ImageJ software (National Institutes of Health (NIH)). All demyelination measurements were conducted double blind. Both fluorescent and bright field images were obtained on a Nikon TE inverted microscope at 4X magnification and processed using ImageJ software (NIH).
Statistical Analysis
For quantification of demyelination, data is presented as average ± SEM and analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. Data were analyzed using Prism software (GraphPad).
Flow Cytometric Analysis of Tissues
Seven and 14 days p.t. mice receiving eGFP-mNSCs (n=5 at each timepoint) or PBS (n=5 at each timepoint) control transplants were sacrificed and their tissues were analyzed for immune cell populations via flow cytometry. Cervical lymph nodes, spleens or spinal cords from transplanted animals were dissected into single cell suspension, depleted of red blood cells using tris-acetic-acid-chloride (TAC) as previously described (Lane et al., 2000). Cells were filtered, washed, and counted before being blocked and stained with anti-mouse CD16/32 (1:100 CD4-PE, CD8-FITC or PE/Cy7 or APC/Cy7, CD25-PE or APC, and FoxP3-Alexa 488 or APC, and propidium iodide or Zombie Aqua. For intracellular cytokine analysis, cells were fixed and permeabilized according to manufacturer specifications using the eBioscience FoxP3/Transcription Factor Staining Buffer Set (Thermo Fisher). All antibodies were purchased from BD Biosciences, Biolegend, or eBiosciences. Single-stain samples establish PMT voltages, gating, and compensation parameters. Cells were processed using an LSR II flow cytometer (BD)and analyzed using FlowJo software (Tree Star).
Statistical Analysis
For quantification of immune cell populations, data is presented as average ± SEM and analyzed using a two-way ANOVA with a Sidak’s multiple comparisons test.
Experimental Design-hNSC transplant
Age-matched (8 weeks) male C57BL/6 mice (H-2b, Charles River) mice were immunized by subcutaneous injections of MOG peptide and Pertussis toxin 21 days prior to hNSC transplant as described above. At day 21 p.i. mice received intraspinal transplants of 2.5 ×105 hNSCs or hDFs in 2.5μl of PBS or PBS alone as a control, at thoracic vertebrae 9 (T9).
Immunohistochemistry and analysis of histopathology
For quantification of demyelination, mice receiving hNSCs (n=8), hDFs (n=5) or PBS (n=9) control transplants were sacrificed 28 days p.t. spinal cords were dissected, fixed overnight in 4% paraformaldehyde, and soaked in 30% sucrose for 7 days before being embedded in OCT compound. Frozen tissues were serially sectioned into 8μM and stained with Luxol Fast Blue (LFB) and counterstained with hematoxylin and eosin (H&E) to assess the severity of demyelination. The total area of the white matter was quantified and compared to the area of demyelinated regions using ImageJ software (National Institutes of Health (NIH)). All demyelination measurements were conducted double blind. Bright field images were obtained on a Nikon TE inverted microscope at 4X magnification and processed using ImageJ software (NIH).
Statistical Analysis
For quantification of demyelination, data is presented as average ± SEM and analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. Data were analyzed using Prism software (GraphPad).
Flow Cytometric Analysis of Tissues
Seven days p.t. mice receiving hNSCs (n=3) or PBS (n=3) control transplants were sacrificed and their tissues were analyzed for Treg cell populations via flow cytometry. This data is representative of 3 independent experiments. In additional experiments, 7 days p.t. mice receiving hNSCs (n=6), hDFs (n=6), PBS (n=6) or non-transplanted (n=6) animals were sacrificed and their tissues were analyzed for immune cell populations. This data is representative of 3 independent experiments. Cervical lymph nodes, spleens or spinal cords were dissected and processed as described above. Cells were filtered, washed, and counted before being blocked and stained with anti-mouse CD16/32 (1:100), B220-BV510, CD3-BV421, CD4-PE or BV785, CD8-FITC or PE/Cy7 or APC/Cy7, CD11b-Alexa 700, CD25-PE or APC, CD44-PB, CD45-PE/Cy7, CD62L-PE/Cy7, FoxP3-Alexa 488, Gr-1-BV605, Helios-FITC, NK1.1-APC, propidium iodide, Zombie Aqua. For intracellular cytokine analysis, cells were fixed and permeabilized according to manufacturer specifications using the eBioscience FoxP3/Transcription Factor Staining Buffer Set (Thermo Fisher). All antibodies were purchased from BD Biosciences, Biolegend, or eBiosciences. Single-stain samples and FMO controls were used to establish PMT voltages, gating, and compensation parameters. Cells were processed using an LSR II or LSRFortessa flow cytometer (BD) and analyzed using FlowJo software (Tree Star).
Statistical Analysis
For CD4+ and CD4+CD25+FoxP3+ Treg cell analysis, data is presented as average ± SEM and analyzed using an unpaired, two-tailed T-Test. For immune cell populations, data is presented as average ± SEM and analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. Data were analyzed using Prism software (GraphPad).
Experimental Design-hNSC transplant and 2-Photon Imaging
For hNSC transplant and 2-photon imaging experiments Age-matched (8 weeks) male C57BL/6 B6 FoxP3EGFP mice (B6.Cg-Foxp3tm2(EGFP)Tch/J, Stock No: 006772; Jackson Laboratories) were immunized by subcutaneous injections of MOG peptide and Pertussis toxin 21 days prior to hNSC transplant as described above.
2-Photon Imaging
For 2-photon imaging, cell trace yellow (CTY) labeled hNSCs (n=3) or hDFs (n=3) were transplanted into spinal cords of Foxp3EGFP mice undergoing EAE at thoracic vertebrae 9 (T9) on day 21 p.i. An additional group received no transplant (n=3) as a control. Spinal cords were carefully dissected 3 days after p.t, fixed in 4% paraformaldehyde washed in PBS and imaged in PBS at room temperature. Two photon imaging was performed using a custom-built 2-photon system based on an Olympus BX51 upright microscope as previously described (Matheu et al., 2015), fitted with a Ziess W Plan-Apochromat 10x (NA= 0.5) water-immersion objective, and equipped with 3 PMTs and excitation generated by a tunable Chameleon femtosecond laser (Coherent) set to 890nm for optimal excitation of GFP and CTY. 495 nm and 560 nm dichroic filters were arranged in series to separate blue, green and red signals. Several 3D image stacks of x=1000 μm, y=700 μm, and z=750 μm (XYZ voxel size 1 μm x 1 μm x 5 μm) were acquired using image acquisition software Slidebook (Intelligent Imaging Innovations) and motorized Z-Decks stage (Prior Scientific). 3D Image blocks were stitched in Slidebook and Imaris version 9.2.1 (Bitplane) was used for rendering, final image size 3 mm X 32 mm X 0.75 mm.
Experimental Design-hNSC transplant and Treg depletion
For hNSC transplant and Treg depletion experiments Age-matched (8 weeks) male C57BL/6 mice (H-2b, Charles River) mice were immunized by subcutaneous injections of MOG peptide and Pertussis toxin 21 days prior to hNSC transplant as described above. Mice were divided into 4 groups and received the following; hNSCs (n=10), hNSCs+PC61.5 (n=7), hNSCs+IgG (n=7) and PBS control (n=8).
In vivo Treg ablation
EAE mice were injected I.P. with 150μg of purified, monoclonal anti-mouse CD25 (rat anti-mouse CD25, clone PC61.5 (Huss et al., 2016) (Biolegend)) or control rat immunoglobulin G (Sigma) at day −2, day 0 and day 2 p.t., as previously described (Chen et al., 2014).
Flow Cytometric Analysis of Peripheral Blood
Efficiency of anti-CD25 treatment was determined by collection of peripheral blood from the peri-orbital sinus of anesthetized animals, followed by quantification of circulating Tregs by flow cytometry analysis as described above.
Transmission Electron Microscopy and G-ratio Analysis
Mice were sacrificed via inhalation of a lethal dose of isoflurane and cardiac perfusion was performed using 1M cacodylate buffer containing 2% paraformaldehyde and 2% glutaraldehyde. Spinal cords (SC) were dissected and fixed in 4% Glutaraldehyde for 24 hours. SC were then washed briefly in 0.1M Sodium Cacodylate (Na Cacodylate) buffer and incubated overnight in 0.1M Na Cacodylate buffer at 4°C. SC were then washed briefly in 0.1M Na Cacodylate buffer and incubated in 1% Osmium tetroxide (0s04; Electron Microscopy Solutions) for 1 hour. SC were rinsed with dH2O and then washed in 50% ethanol (EtOH) for 15 min., 70% EtOH for 15 min., 80% EtOH for 15 min., 95% EtOH for 15 min., 100% EtOH for 15min., 100% EtOH for 15 min. and propylene oxide for 30 min. SC were then placed into resin containing 50% propylene oxide and 50% Spurr resin (Electron Microscopy Solutions) for 25 min. Then 20% propylene oxide and 80% Spurr resin was added for 20min. Samples were then placed into 100% Spurr Resin and degassed overnight at room temperature. SC were then embedded into new 100% Spurr resin and baked for 24 hours at 60°C before being ultra-serially sectioned, stained with uranyl acetate-lead citrate and analyzed as previously described (Liu et al., 2001). G-ratios were determined by measuring axon diameter and comparing it to the total fiber diameter (axon diameter/total fiber diameter) using ImageJ software (NIH). Measurements were performed double blind by two investigators, and more than 200 axons were measured per experimental group.
Data Visualization
All graphs were generated using Prism software (GraphPad). Graphics were created with BioRender.com. Figures were generated using Adobe Illustrator software.
Results
Engraftment of syngeneic neural stem cells promotes remyelination in an autoimmune model of demyelination without altering the T lymphocytes
We have previously shown that transplantation of syngeneic, enhanced Green Fluorescent Protein (eGFP)-expressing mouse NSCs (eGFP-mNSCs) into the spinal cords of JHMV-infected mice with chronic demyelination results in migration, proliferation, and remyelination that correlated with preferential differentiation of engrafted cells into mature oligodendrocytes (Carbajal et al., 2010; Carbajal et al., 2011; Greenberg et al., 2014). To determine if eGFP-mNSCs promoted remyelination in an additional model of immune-mediated demyelination, we utilized the experimental autoimmune encephalomyelitis (EAE) model. Mice were immunized with Myelin Oligodendrocyte Glycoprotein (MOG), and 2.5×105 eGFP-mNSCs were engrafted 21 days post-immunization (p.i.); a timepoint at which clinical disease is present and mice have established demyelinating lesions (Constantinescu et al., 2011; McCarthy et al., 2012). In support of previous findings, there was extensive migration of NSCs from the transplantation site (Figure 1A). Luxol fast blue (LFB) staining of serially-sectioned spinal cord tissue revealed that eGFP-mNSC transplanted animals reduced demyelinated white matter caudal to the injection site compared to phosphate-buffered saline (PBS) injected controls (Figure 1B and 1C), although EAE immunized animals transplanted with eGFP-mNSCs did not display clinical recovery compared to PBS treated controls (data not shown). To investigate whether this muted demyelination was due to cell replacement or a reduction in neuroinflammation, we evaluated the frequency of CD4+ T cells, CD8+ T cells and CD4+CD25+FoxP3+ Tregs in the spinal cord (SC), cervical lymph nodes (cLN), and spleen of mice receiving eGFP-mNSCs or PBS 7, 14, and 21 days post-transplant (p.t.). Flow cytometric analysis revealed that there was a slight, but significant (*p= 0.0182) decrease in frequency of CD4+ T cells within the spinal cord of mice receiving eGFP-mNSCs 14 days p.t., however no significant difference in the frequency of CD4+ T cells, CD8+ T cells and CD4+CD25+FoxP3+ Tregs in the cLN and spleen 7, 14 or 21 days p.t. or the SC 7 and 21 days p.t. (Figure 1D). Therefore, syngeneic eGFP-mNSCs dampen the severity of white matter damage independent of modulating neuroinflammation which is consistent with previous studies from our group (Hardison et al., 2006).
Figure 1. Mouse Neural Stem Cells engraft, migrate and remyelinate in an EAE mouse model of MS with no effect upon CNS immune environment.
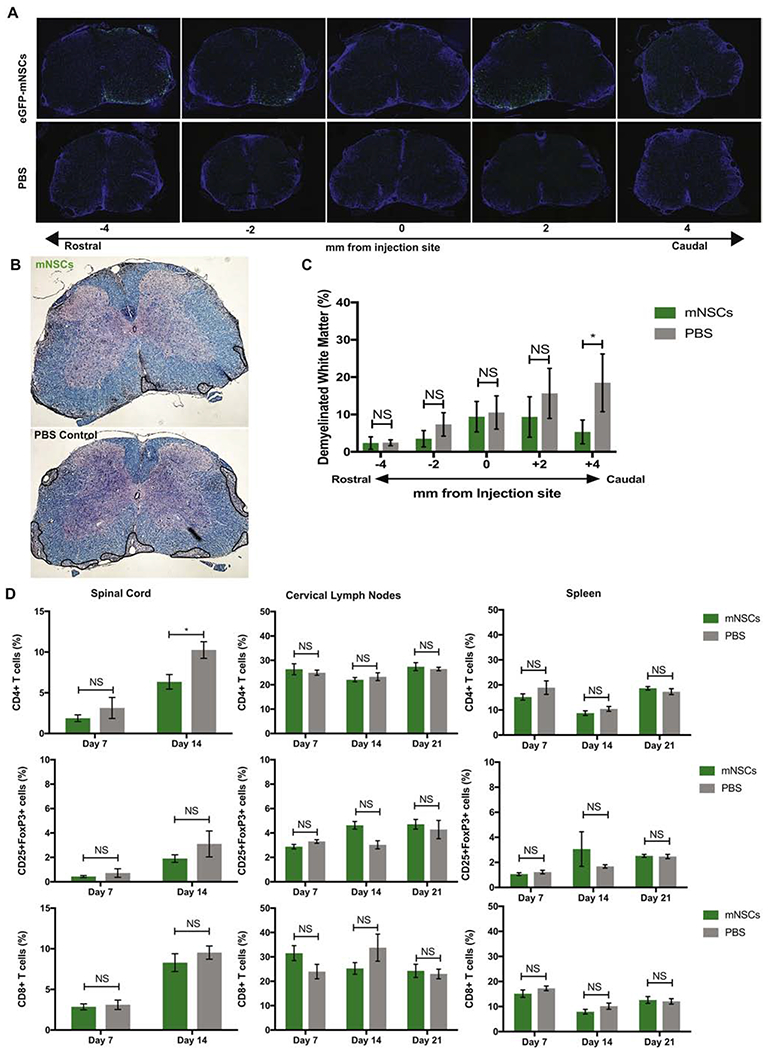
(A) Representative serial sections of spinal cord rostral and caudal to the site of implantation (T9) show that eGFP-mNSCs (green) survive 28 days post-transplantation and migrate towards areas of demyelination compared to PBS treated controls. Nuclei are stained with DAPI (blue) (B) Representative brightfield images of coronal spinal cord sections from eGFP-mNSC (n= 12) or PBS control (n= 13) transplanted animals stained with luxol fast blue (LFB) and counterstained with hematoxylin and eosin (H&E), areas of demyelinated white matter outlined in black. (C) Quantification of demyelination in the ventral white matter of eGFP-mNSC (n= 12) or PBS control (n= 13) transplanted mice revealed significantly (p<0.05) reduced demyelination at sections caudal to the injection site. (D) Quantification of the frequency of adaptive immune cell populations CD4+, CD4+CD25+FoxP3+ and CD8+ T cell subsets within the spinal cord, draining cervical lymph nodes, and spleen of eGFP-mNSC (n= 5) or PBS control (n= 5) injected mice revealed no significant difference in cell populations at day 7 p.t. FACS plots were analyzed and quantified by gating on lymphocytes, excluding forward and side scatter doublets, CD4+ or CD8+ and CD4+CD25+FoxP3+. Sample gating strategy is shown in Supplemental Figure 1. Data represents two independent experiments. 14 days p.t. there was a slight, but significant (*p= 0.0182) decrease in frequency of CD4+ T cells within the spinal cord of mice receiving eGFP-mNSCs 14 days p.t., however no significant difference in the frequency of CD4+ T cells, CD8+ T cells and CD4+CD25+FoxP3+ Tregs in the cLN and spleen 7, 14 or 21 days p.t. or in the SC at day 7 or 21 p.t. For (C), day 28 p.t. demyelinated white matter analysis eGFP-mNSCs (n=12) and control PBS (n=13). Data is presented as average ± SEM and analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. For (D), day 7 p.t. analysis eGFP-mNSCs (n=5) and control PBS (n=5). Data is presented as average ± SEM and analyzed using a two-way ANOVA with a Sidak’s multiple comparisons test.
Human ES-derived hNSCs promote remyelination and regulatory T cell expansion in the EAE mouse spinal cord
To evaluate graft survival and migration following intraspinal delivery of embryoid body-NSCs (EB-NSCs) we differentiated human embryonic stem cells (hESCs) into NSCs using dual SMAD inhibition (Figure 2A) as previously described (Chambers et al., 2009), herein referred to as hNSCs. Differentiated cells were enriched for NSCs by FACS sorting for CD184+CD24+CD271−CD44− cells (Figure 2B) (Yuan et al., 2011). Sorted cells strongly expressed Nestin (81.6±2.91), a known marker of neural stem cells, and the NSC markers SOX1 and SOX2 (92.7±0.61) (Figures 2C and 2D). Upon growth factor withdrawal, these cells differentiated and expressed markers associated with all three neural lineages: βIll-tubulin (neurons), GFAP (astrocytes) and NG2 (oligodendrocytes) (Figure 2E).
Figure 2. EB-derived Human Neural Stem Cells are multipotent and express neural lineage markers.
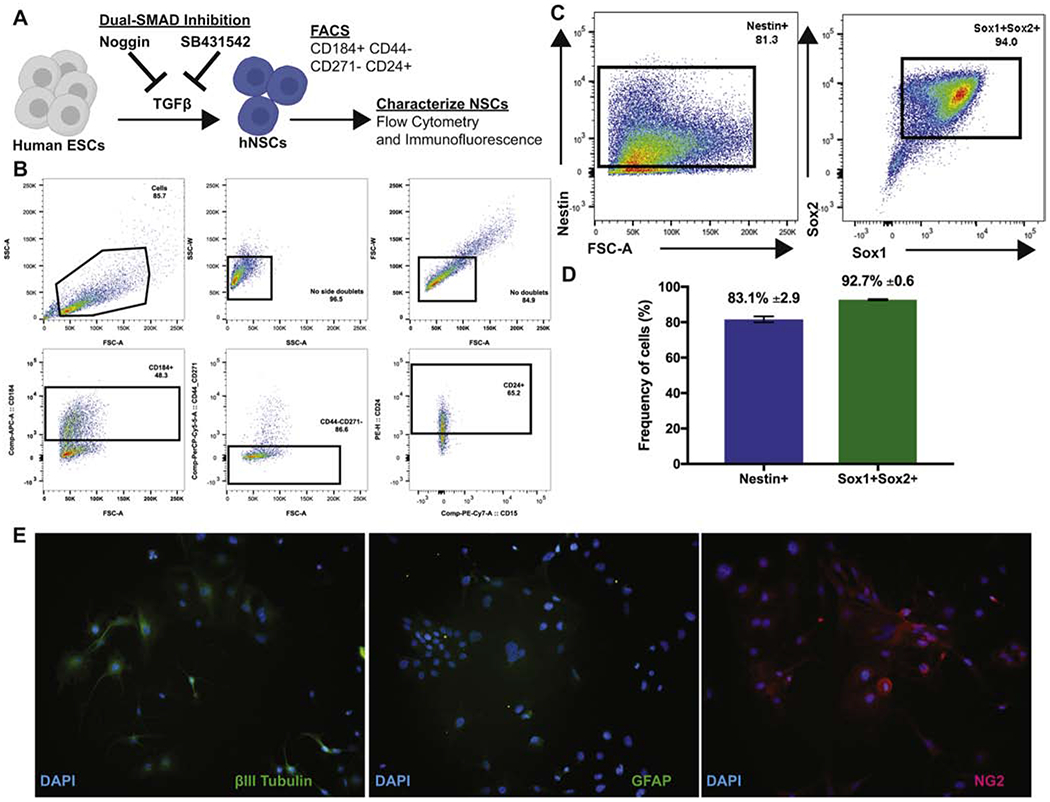
(A) Diagram depicting derivation and differentiation of hNSCs. (B) Representative FACS sort plots for CD184+CD44−CD271− CD24+ hNSCs. Cells were gated sequentially excluding side and forward scatter doublets, CD184+, CD44−CD271−, and CD24+. (C) FACS plots of sorted hNSCs for expression of NSC markers Nestin, Sox1 and Sox2. (D) Frequency of hNSCs expressing NSC makers Nestin (83.1%± 2.9; blue) and Sox1 and Sox2 (92.7% ± 0.6; green). Data is presented as average ± SEM. (E) Microscopy images of differentiated hNSCs that expressed markers of neurons (βIll-tubulin; left), astrocytes (GFAP; middle) and oligodendrocytes (NG2; right). Magnification =20x.
To evaluate the potential of xenogeneic hNSCs to promote remyelination in EAE, we transplanted 2.5 ×105 hESC derived NSCs, human dermal fibroblasts (hDFs) as a xenogeneic control, or administered PBS as a vehicle control, into the spinal cord of EAE immunized mice 21 days p.i. (Figure 3A). Following transplantation, hNSCs and hDFs underwent xenograft rejection and were undetectable 21 days p.t. by immunohistochemical staining (data not shown (Plaisted et al., 2016). However, LFB staining revealed that EAE mice transplanted with hNSCs displayed less severe demyelination compared to the control mice (Figures 3B–3D). Although hNSC transplanted mice did not display a significant clinical response (Figure 3E), we observed a decrease in neuroinflammation in mice receiving hNSCs compared to PBS controls. The reduction in the severity of demyelination was associated with a decrease (p=0.006) in CD4+ T cells and an increase (p=0.004) in CD4+CD25+FoxP3+ Tregs in the spinal cord (SC) and cervical lymph nodes (cLN) of hNSC transplanted mice 7 days p.t. (Figure 4A and 4B). These results are consistent with our previous findings in JHMV infected animals receiving hNSCs (Plaisted et al., 2016). However, this decrease in CD4+ T cells and increase in Tregs appeared to be transient, and corresponded to the time period at which hNSCs were rejected. Accordingly, we did not detect a difference in these populations 21 days p.t. (Supplemental Figure 2) nor did we detect a difference in the frequency in other immune cell populations (CD45lo CD11b+ Microglia, CD45hi CD11b+ Macrophages, CD45+, CD11b+Gr-1+ Neutrophils, CD11b+B220− cDCs, CD11b+B220+ pDCs, B220+ B cells, NK1.1+ NK cells, CD3+NK1.1+ NK T cells, or CD3+ T cells) at day 7 p.t. (Figure 5). Importantly, EAE mice transplanted with hDFs did not display decreased neuroinflammation or decreased demyelination (Figures 3B, 3C and 3D), indicating that the mechanism of hNSC-mediated immune alteration is mediated by hNSCs and not due to a xeno-specific immune response. These results are consistent with the existence of a population of Tregs capable of promoting endogenous repair.
Figure 3. Transplantation of hNSCs results in less demyelination in the EAE mouse spinal cord.
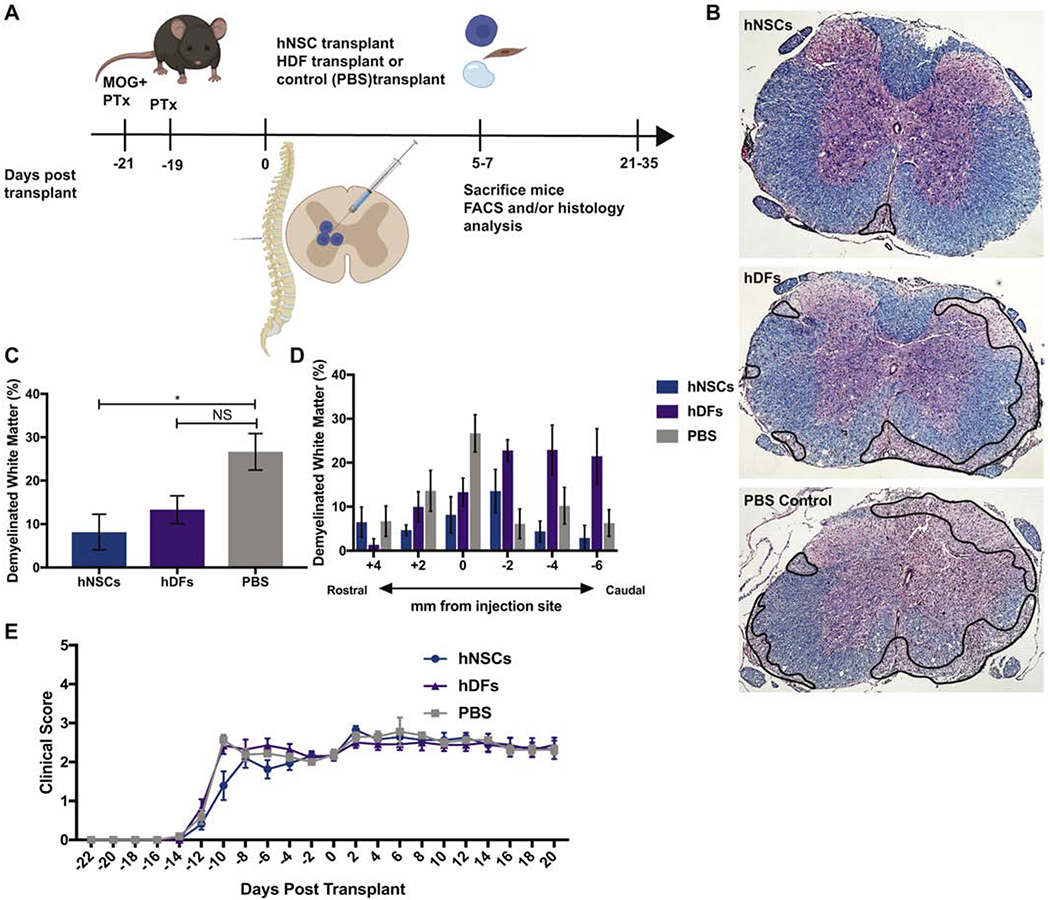
(A) Timeline of EAE induction (MOG35-55 immunization and Pertussis Toxin (PTx) administration), hNSC transplant and analysis. (B) Representative brightfield images of coronal spinal cord sections from hNSC (n=8), hDF (n=5) or PBS (n=9) control transplanted animals stained with luxol fast blue (LFB) and counterstained with hematoxylin and eosin (H&E), areas of demyelinated white matter outlined in black. (C) Quantification of demyelination in the ventral white matter of hNSC (n=8), hDF (n=5) and control PBS (n=9) transplanted mice revealed significantly (p=0.0232) reduced demyelination at the injection site in the spinal cords of hNSC transplanted mice, 28 days p.t. (D) Quantification of demyelination in areas rostral and caudal to the site of injected revealed that reduced demyelination was not sustained throughout the spinal cord. All data is presented as average ± SEM and analyzed using one-way ANOVA followed by Tukeys multiple comparison test (C and D). (E) Graph of clinical scores of mice injected intraspinally with hNSCs (n=8, blue), hDFs (n=7, purple), or control PBS (n=8, grey) at defined timepoints p.t.. Clinical evaluation was performed double-blind and based on the following scoring system; 0, asymptomatic; 0.5, ruffled fur; 1, flaccid tail; 2, hind limb paresis; 2.5, partial hind limb paralysis; 3, hind limb paralysis; 4, hind limb and forelimb paralysis; 5, moribund. Data represents two independent experiments and is presented as average ± SEM. No significant difference in locomotor function were observed. Data was analyzed using a two-way ANOVA.
Figure 4. Increase in CD4+CD25+FoxP3+ regulatory T cells in the spinal cord and cervical lymph nodes following hNSC Transplant.
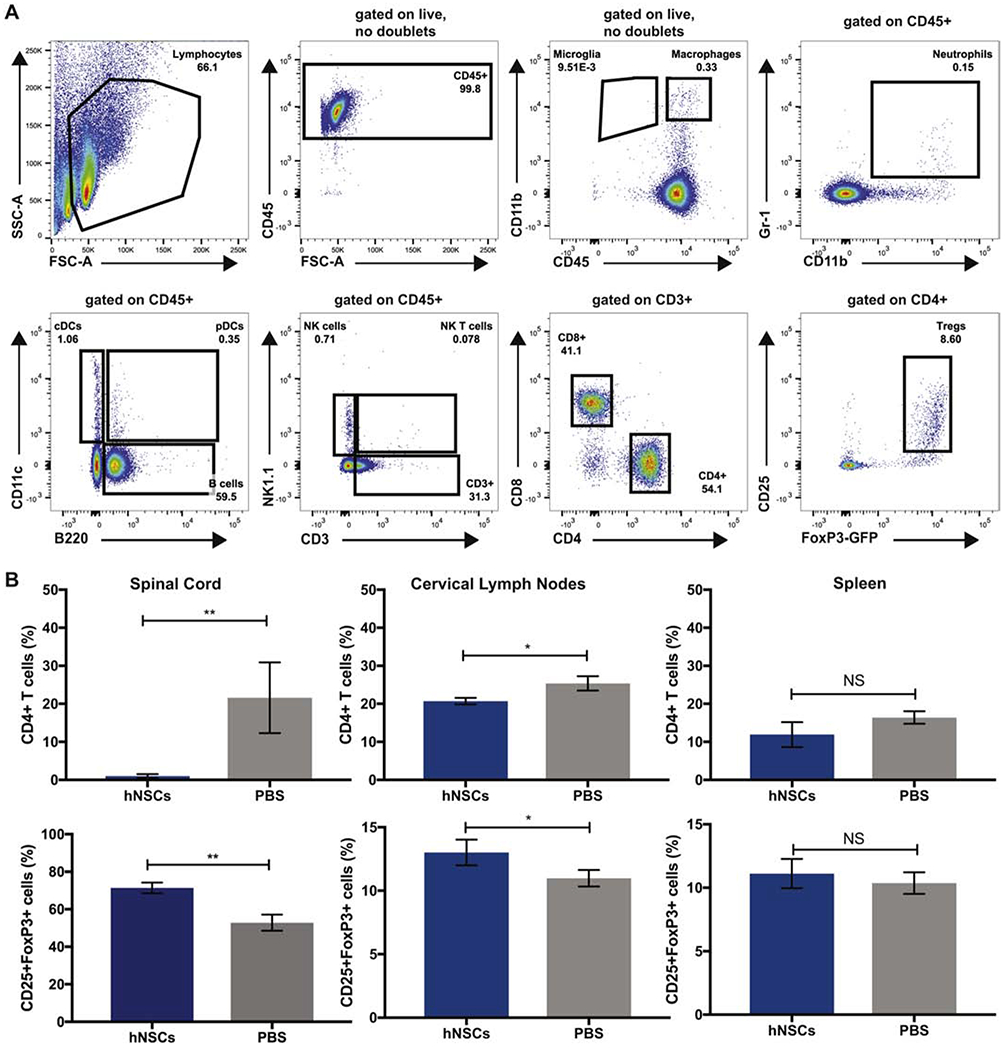
(A) Representative FACS plots showing gating strategy for immune cells. (B) Quantification of the frequency of CD4+, CD4+CD25+FoxP3+ and CD8+ T cell subsets 7 days p.t. from hNSC (n=3) or PBS transplanted (n=3) mice demonstrated a significant (p=0.006) decrease in the percentage of CD4+ T cells within the spinal cord (top left) and draining cervical lymph nodes (top middle). An increase in frequency of CD4+CD25+FoxP3+ Tregs was observed in the spinal cord (lower left) and a significant (p=0.004) increase infrequency of CD4+CD25+FoxP3+ Tregs was observed in the draining cervical lymph nodes (lower middle) of hNSC transplanted mice. A difference in CD4+, CD4+CD25+FoxP3+ cells was not observed in the spleen. Data represents three independent experiments. Data is presented as average ±SEMand analyzed using an unpaired, two-tailed T-Test.
Figure 5. Immune cell populations are unaffected following hNSC Transplant.
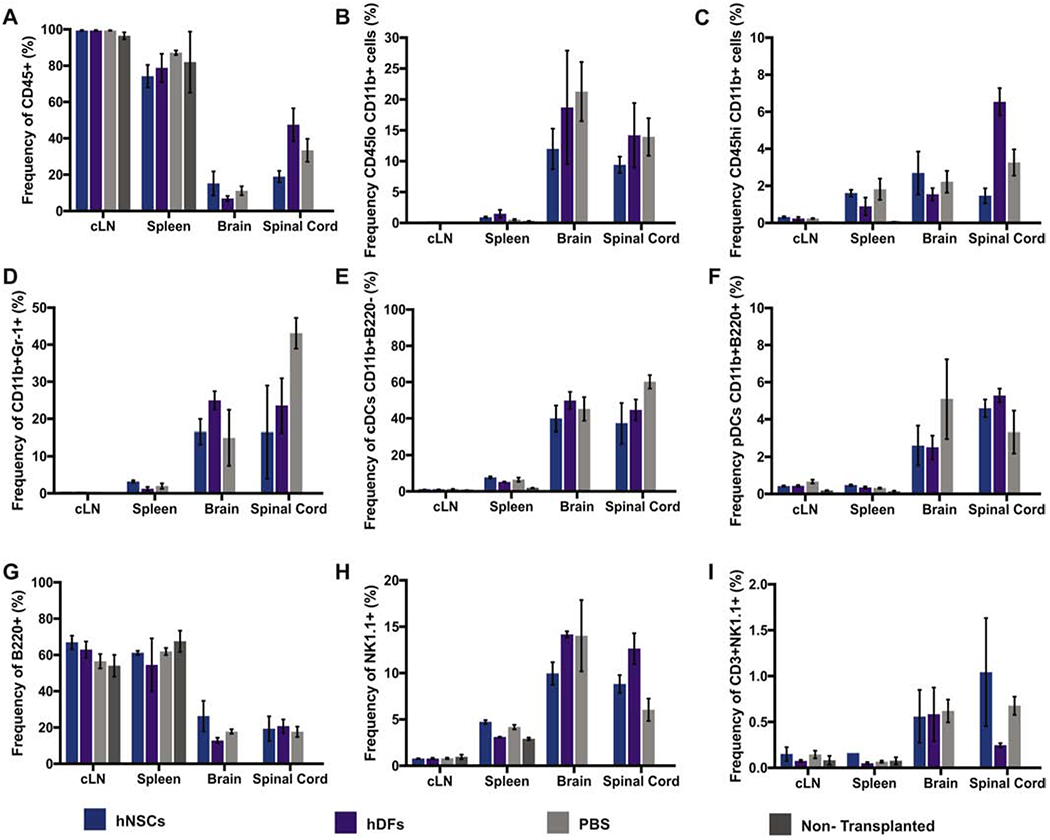
Quantification of the frequency and number of immune cell populations in cervical lymph nodes, spleen, spinal cord and brain of hNSC (n=6), hDF (n=6), PBS (n=6), or non-transplanted (n=6) control mice 7 days p.t. No difference in immune cell populations; CD45+ (A top, left), CD45lo CD11b+ Microglia (B top, middle), CD45hi CD11b+ Macrophages (C top, right), CD11b+Gr-1+ Neutrophils (D middle, left), CD11b+B220− cDCs (E middle, middle), CD11b+B220+ pDCs (F middle, right), B220+ B cells (G bottom, left), NK1.1+ NK cells (H bottom, middle), or CD3+NK1.1+ NK T cells (I bottom, right) were observed. Data represents two independent experiments. Data is presented as average ± SEM and analyzed using one-way ANOVA followed by Tukeys multiple comparison test.
Tregs localize to the site of hNSC injection in EAE Spinal cord
To investigate the in situ organization of Tregs in the vicinity of transplanted hNSCs in vivo, we utilized two-photon (2P) microscopy and image stitching to generate organ wide distribution maps. We transplanted cell trace yellow (CTY) labeled hNSCs or hDFs into FoxP3EGFP EAE mice 21 days p.i.; non-transplanted mice were also used as imaging controls. 3 days p.t., spinal cords were carefully dissected, embedded in 5% agarose, fixed and then imaged by 2P microscopy (Weinger et al., 2015). Montage images revealed few FoxP3EGFP+ Tregs (green) localized to sparse clusters along the entire length of spinal cord in the control non-transplanted mice (Figure 6A). Collagen fibers are detected as blue (second-harmonic generation, SHG). Dramatic increase in both numbers and size of Treg-clusters was observed at the site of hNSC injection (red) (Figure 6B). hDFs transplanted spinal cords did not show any increase in number or density of Tregs (Figure 6C), further confirming hNSC-specific local expansion of Tregs. Notably, Tregs from hNSC spinal cords were brighter than both controls (NT and hDFs), indicating a stronger activation of Foxp3 gene. Altogether, spatial mapping data demonstrate that Tregs preferentially localize to the sites injection and directly interact with hNSCs in during EAE.
Figure 6. Tregs accumulate at the site of hNSC injection in the EAE Spinal cord.
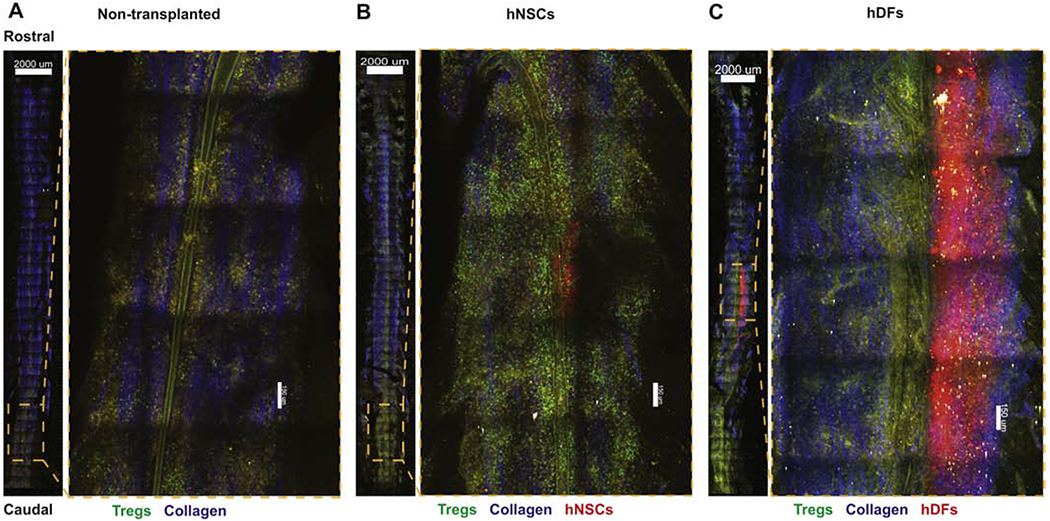
Representative 2-photon-microscopy-derived spinal cord montage and magnified images showing distribution of FoxP3EGFP Tregs (green), transplanted cell trace yellow (CTY) labeled hNSCs or hDFs (red), second-harmonic signal from collagen (blue), and autofluorescent structures (yellow) in the ventral side of the spinal cord, 3 days p.t. of mice that were non-transplanted (n=3) (A), received hNSCs (n=3) (B), or hDFs (n=3) (C). Images are maximal intensity projections through the z-axis, ventral-dorsal (750 μm), scale bar (overview) = 2000 μm; scale bar (magnification) = 150 μm.
Regulatory T cells induced following hNSC transplantation are required for remyelination
Previous studies from our group have demonstrated that spinal cord engraftment of hNSCs promotes remyelination in a viral model of neurological disease, in part, through expansion of Tregs (Chen et al., 2014; Plaisted et al., 2016).To determine whether Tregs were necessary for remyelination in mice with EAE that were engrafted with hNSCs, experimental mice were depleted of Tregs using an anti-CD25 antibody (PC61.5) (Huss et al., 2016; Setiady et al., 2010). EAE mice received no antibody treatment, PC61.5 or an IgG isotype control, and were then transplanted with hNSCs or PBS 21 days p.i. (Supplemental Figure 3A). Ablation of Tregs abrogated histopathological improvement. PC61.5 treated mice displayed a decreased frequency and number of CD4+CD25+FoxP3+ cells in peripheral blood 3 and 9 days p.t. (Supplemental Figures 3B and 3C) with no effect upon the frequency and number of CD4+ T cells (Supplemental Figures 3D and 3E) or clinical outcome (Supplemental Figure 3F). Assessment of the g-ratio, the ratio of the inner axonal diameter of the total outer fiber diameter, is commonly employed as a structural index of remyelination; lower ratios indicate more extensive myelination (Chen et al., 2014; Marro et al., 2019).To determine whether Tregs were necessary for remyelination following hNSC transplant, we compared the g-ratio’s between EAE mice transplanted with hNSCs, hNSCs+ PC61.5, hNSCs+ IgG control, or PBS control mice. Mice transplanted with hNSCs had extensive signs of remyelination at the transplant site (Figure 7A) and subsequent g-ratio analyses confirmed a significant increase (p<0.001) in myelin at the transplant site; hNSC and hNSC+IgG transplanted mice had significantly lower calculated g-ratios (0.7414±0.0063 and 0.748±0.0049, respectively) compared to those transplanted with hNSCs but depleted of Tregs (hNSCs+PC61.5) (0.7808±0.0057) (Figure 7B). Mice receiving hNSCs and hNSCs+IgG also had more axons with a smaller diameter compared to mice transplanted with hNSCs but depleted of Tregs (hNSCs+PC61.5), suggesting less damage (Figure 7C and 7D). Mice receiving hNSCs hNSCs+IgG, or PBS had less demyelinated axons (Figure 7E) and more remyelinated axons than mice receiving hNSCs+PC61.5 (Figure 7F and 7G). Mice treated with PC61.5 also had slightly fewer axons compared to the three other treatment groups (Figure 7H). These results confirm that Tregs are necessary for remyelination; depletion of Tregs abrogates histopathological improvement.
Figure 7. Regulatory T cells are necessary for remyelination following hNSC transplant.
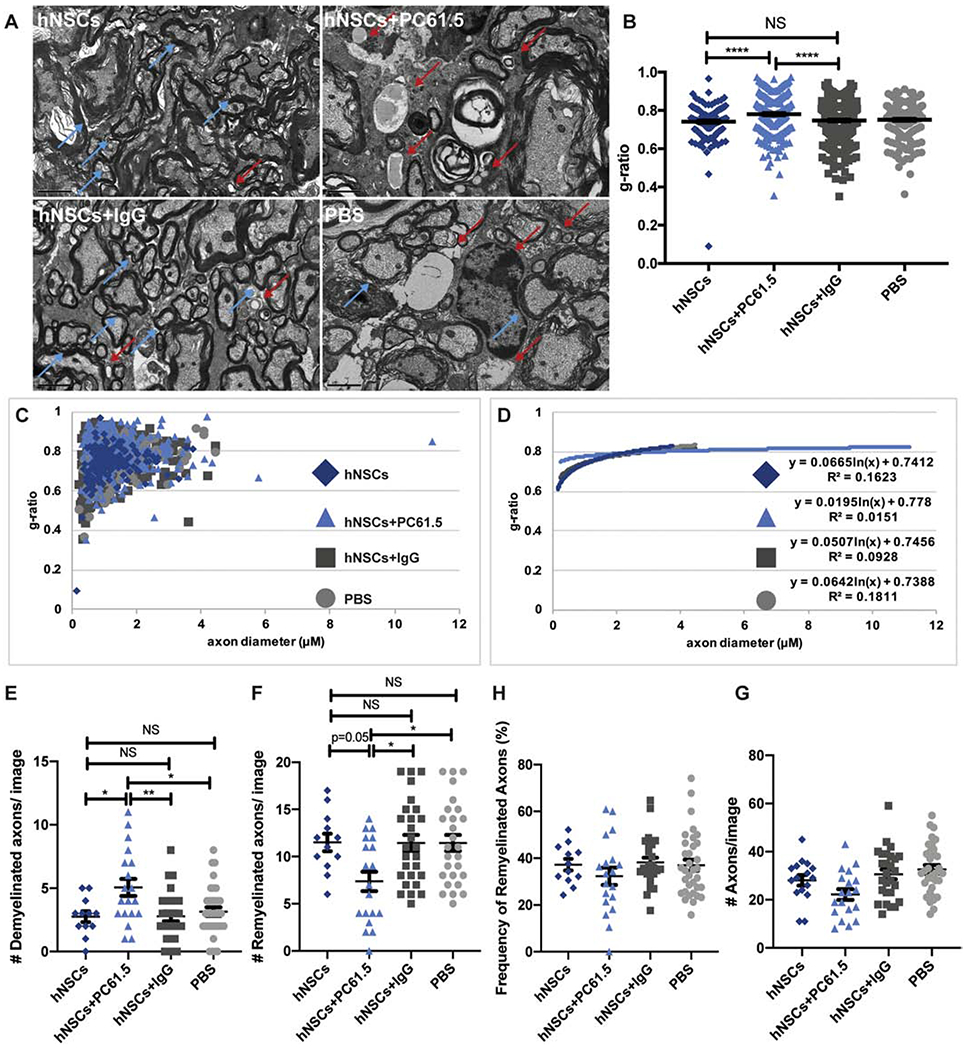
(A) Representative electron micrographs of coronal spinal cord sections from hNSCs (n=10; top, left), hNSCs+PC61.5 (n= 7; top, right), hNSCs+IgG (n= 7; bottom, left), and control PBS (n=8; bottom, right) injected mice. Demyelinated axons indicated by red arrows. Remyelinated axons indicated by blue arrows. (B) Quantification of ratio of inner axon diameter versus total myelinated fiber (g-ratio) G-ratio of hNSCs (0.7414±0.0063), hNSCs+PC61.5 (0.7808±0.0057), hNSCs+IgG (0.748±0.0049), and PBS (0.7512±0.0054). More than 200 axons were measured per group. Data is presented as average ± SEM and analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. (C) Scatter plot displaying g-ratio of individual axons as a function of axonal diameter from hNSC (dark blue, diamonds), hNSC+PC61.5 (light blue, triangles), hNSC+IgG (dark grey, squares), and control PBS (light grey, circles) injected mice. More than 200 axons were measured per group. (D) Logarithmic trend lines for scatter plot displayed in C. Quantification of the number of (E) demyelinated axons, (F) remyelinated axons, (G) percentage of remyelinated axons and (H) number of total axons per EM image from mice receiving hNSCs, hNSCs+ PC61.5, hNSCs+IgG, and control PBS. For E-H, more than 200 axons were counted per group. Data is presented as average ± SEM and analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. (*p<0.05**p<0.01; **p<0.001; NS is not significant).
Discussion
In this study, we provide direct evidence that interactions between NSCs and Tregs results in remyelination during EAE. Pre-clinical studies of NSC transplantation in animal models of human neurological disorders, including MS, have demonstrated significant improvements in disease pathology and clinical outcome (Pluchino et al., 2009). In this manuscript, we have shown that EAE mice receiving syngeneic mNSC transplants displayed less demyelination, compared to non-transplanted animals due to cell replacement. However, the mechanism by which xenogeneic hNSC transplantation promotes remyelination remains unclear as remyelination in these mice was found to be dependent upon Tregs as opposed to cell replacement by donor hNSCs.
Consistent with our lab’s previous results in MHV (Tirotta et al., 2010), mice with established MOG35-55 induced EAE receiving intraspinal injections of syngeneic eGFP-mNSCs displayed less demyelination at sites caudal to the transplantation site compared to control animals receiving PBS. Transplanted eGFP-mNSCs engrafted and migrated extensively from the transplant site. Reduction in demyelination is likely due to cell replacement, with little impact on immune cells. While these results are promising, syngeneic NSC transplants may not be feasible for patients with MS because the etiology of MS is unknown, but is considered to have a genetic component and therefore, MS patient-derived NSCs may have genetic/epigenetic defects leading to inefficient tissue repair. Therefore, much attention has focused on the use of hNSCs for promoting repair through potential immunomodulatory mechanisms.
Following hNSC transplantation into the spinal cords of mice with established EAE, we observed an increase in CD4+CD25+FoxP3+ Tregs in the CNS and cervical lymph nodes of MOG35-55 immunized animals, but not in PBS controls or animals that received hDFs. Utilizing, 2P microscopy we observed Tregs accumulating at the site of hNSC transplant within the CNS at early timepoints p.t. An increased frequency of Tregs was associated with a decrease in CD4+ T cells. In addition, g-ratio analyses revealed that mice receiving hNSC transplants had significantly more myelin compared to controls. Importantly, in vivo depletion of Tregs with a monoclonal antibody abrogated histopathological improvement, suggesting that Tregs augment remyelination. Remyelination was observed after transplanted hNSCs could no longer be observed, suggesting that these results were not due to cell replacement, as hNSCs underwent xenograft rejection. Nor was remyelination and expansion of Tregs due to a xenogeneic response, as transplanted hDFs were also rejected and did not elicit an expansion of Tregs. Together these results demonstrate that transplantation of hNSCs elicits a specific Treg response resulting in dampened neuroinflammation and increased remyelination.
Consistent with our previous results using iPSC EB-derived hNPCs, transplantation into JMHV infected mice did not significantly improve clinical recovery, measured by clinical score (Plaisted et al., 2016). In this study, transplantation of NSCs was performed in the chronic stage of EAE (21 days p.i.), whereas all other cell transplantation studies resulting in clinical improvement inject cells prior to or at the onset of disease. Since demyelination is widespread at the time of transplant, lack of clinical improvement is likely due to local Treg recruitment to the transplantation site, whereas other areas of the spinal cord remain damaged. Future studies should focus on methods to expand Treg recruitment and repair throughout the spinal cord.
These results support a novel role for NSC and Treg cooperation within the CNS in promoting tissue repair. While NSCs are traditionally thought to replace cells, recent reports have suggested transplanted hNSCs do not readily differentiate into neural lineages in murine models, but rather exert their immunomodulatory effects via trophic factors (Blurton-Jones et al., 2009; Muller et al., 2006; Neirinckx et al., 2013). hNSCs have also been reported to convert T conv cells to a Treg phenotype (Gao et al., 2014; Nazmi et al., 2014). Although Tregs have been traditionally implicated in suppressing inflammatory T cell responses, an emerging role for Tregs as potentiators of tissue repair through interactions with stem and progenitor cells has been suggested (Josefowicz et al., 2012; Li et al., 2018). Tregs have been reported to facilitate stem and progenitor cell maturation in visceral adipose tissue (Feuerer et al., 2009; Tontonoz and Spiegelman, 2008), bone (Glowacki et al., 2013), skin hair follicles (Ali et al., 2017), lung (Arpaia et al., 2015; Mock et al., 2014), skeletal muscle (Burzyn et al., 2013) and recently the CNS (Dombrowski et al., 2017; Ito et al., 2019). Therefore, it is possible that CNS Tregs promote remyelination indirectly through maturation of immature oligodendrocyte progenitor cells to mature, myelin producing oligodendrocytes. While the role for adaptive immune cells within the CNS remains controversial, benefits of Tregs in the CNS is likely dependent on spatial and temporal factors (Kleinschnitz et al., 2013; Walsh et al., 2014).
Conclusions
Collectively, our results demonstrate that Tregs directly interact with stem and progenitor cells within tissues, including the CNS to promote tissue regeneration and repair. Significantly, these results support a role for Tregs in tissue regeneration, in addition to their well-established function in immune modulation.
Supplementary Material
Supplemental Figure 1. T cell FACS gating strategy. Representative dot plots of CD4+, CD8+ and CD25+FoxP3+ Tregs from Figure 4D sequentially gated on lymphocytes, excluding forward and side scatter doublets, CD4+ or CD8+ cells, CD4+CD25+FoxP3+. Gates are indicated above plots.
Supplemental Figure 2. T cell populations are unaffected 28 days post hNSC Transplant. Quantification of the frequency of adaptive immune cell populations CD4+, CD4+CD25+FoxP3+ and CD8+ T cell subsets within the spinal cord, draining cervical lymph node, and spleen of hNSC (n=3) or control PBS (n=3) injected mice revealed no significant difference in cell populations at day 28 p.t. Data represents two independent experiments.
Supplemental Figure 3. PC61.5 treatment of mice depleted Tregs without affecting T conv cell population or EAE clinical score. (A) Timeline of experiment. EAE mice received intraspinal transplant of hNSCs and intraperitoneal (i.p.) PBS (n=10), hNSCs and anti-CD25 (clone PC61.5) i.p. (n=7), hNSCs and IgG i.p. (n=7), or PBS (n=8) as a control. Anti-CD25 antibody selectively depletes Tregs, IgG was given as an isotype control. EAE mice were administered anti-CD25 and IgG antibodies −2, 0, and 2 days p.t. of hNSCs. Quantification of frequency (B) and number (C) of CD25+FoxP3+ Tregs in peripheral blood 3, 9, and 21 days p.t. Quantification of frequency (D) and number (E) of CD4+ Tconv cells in peripheral blood 3, 9, and 21 days p.t. (F) Graph of clinical scores of mice injected intraspinally with hNSCs (dark blue), hNSCs+PC61.5 (light blue), hNSCs+IgG (dark grey), or PBS (light grey) at defined timepoints post-transplant. PC61.5 treatment does not impact clinical score. Data is presented as average ± SEM and analyzed using two-way ANOVA followed by Tukey’s multiple comparison test (*p=0.01; ****p<0.0001).
Highlights:
Neural Stem Cell transplantation increases regulatory T cells.
Neural Stem Cells interact with regulatory T cells to promote remyelination.
Cell transplantation is a promising therapeutic strategy for repair in Multiple Sclerosis.
Acknowledgements:
We would like to thank Warren, Plaisted, Edna Hingco, Angel Zavala, Louis Parker, Jackie Mitchell and Collene Worne for technical support.
Funding: This work was supported by the California Institute for Regenerative Medicine [TR05603]; the National Multiple Sclerosis Society [CA1058]; the National Institute of Neurological Disorders and Stroke [1T32NS082174, 5R01NS092042]; and the National Institute of Allergy and Infectious Diseases [5R01AI121945].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests statement: The authors have no competing interests to disclose.
References
- Ali N, et al. , 2017. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell. 169, 1119–1129 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, et al. , 2015. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell. 162, 1078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Hur T, et al. , 2013. Cell-based reparative therapies for multiple sclerosis. Curr Neurol Neurosci Rep. 13, 397. [DOI] [PubMed] [Google Scholar]
- Bergmann CC, et al. , 2006. Coronavirus infection of the central nervous system: host-virus stand-off. Nat Rev Microbiol. 4, 121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blurton-Jones M, et al. , 2009. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 106, 13594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D, et al. , 2013. A special population of regulatory T cells potentiates muscle repair. Cell. 155, 1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbajal KS, et al. , 2010. Migration of engrafted neural stem cells is mediated by CXCL12 signaling through CXCR4 in a viral model of multiple sclerosis. Proc Natl Acad Sci U S A. 107, 11068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbajal KS, et al. , 2011. Surgical transplantation of mouse neural stem cells into the spinal cords of mice infected with neurotropic mouse hepatitis virus. J Vis Exp. e2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, et al. , 2009. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 27, 275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, et al. , 2014. Human neural precursor cells promote neurologic recovery in a viral model of multiple sclerosis. Stem Cell Reports. 2, 825–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu CS, et al. , 2011. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 164, 1079–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denic A, et al. , 2011. The relevance of animal models in multiple sclerosis research. Pathophysiology. 18, 21–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrowski Y, et al. , 2017. Regulatory T cells promote myelin regeneration in the central nervous system. Nat Neurosci. 20, 674–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M, et al. , 2009. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 15, 930–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH, 2000. Mammalian neural stem cells. Science. 287, 1433–8. [DOI] [PubMed] [Google Scholar]
- Gao L, et al. , 2014. Transplanted neural stem cells modulate regulatory T, gammadelta T cells and corresponding cytokines after intracerebral hemorrhage in rats. Int J Mol Sci. 15, 4431–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass WG, et al. , 2004. Antibody targeting of the CC chemokine ligand 5 results in diminished leukocyte infiltration into the central nervous system and reduced neurologic disease in a viral model of multiple sclerosis. J Immunol. 172, 4018–25. [DOI] [PubMed] [Google Scholar]
- Glowacki AJ, et al. , 2013. Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes. Proc Natl Acad Sci U S A. 110, 18525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ML, et al. , 2014. Two-photon imaging of remyelination of spinal cord axons by engrafted neural precursor cells in a viral model of multiple sclerosis. Proc Natl Acad Sci U S A. 111, E2349–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison JL, et al. , 2006. Transplantation of glial-committed progenitor cells into a viral model of multiple sclerosis induces remyelination in the absence of an attenuated inflammatory response. Exp Neurol. 197, 420–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosking MP, Lane TE, 2009. The Biology of Persistent Infection: Inflammation and Demyelination following Murine Coronavirus Infection of the Central Nervous System. Curr Immunol Rev. 5, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss DJ, et al. , 2016. Anti-CD25 monoclonal antibody Fc variants differentially impact regulatory T cells and immune homeostasis. Immunology. 148, 276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, et al. , 2019. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. 565, 246–250. [DOI] [PubMed] [Google Scholar]
- Josefowicz SZ, et al. , 2012. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 30, 531–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschnitz C, et al. , 2013. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood. 121, 679–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane TE, et al. , 2000. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J Virol. 74, 1415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H, Bradl M, 2017. Multiple sclerosis: experimental models and reality. Acta Neuropathol. 133, 223–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H, et al. , 1997. Remyelination in multiple sclerosis. Mult Scler. 3, 133–6. [DOI] [PubMed] [Google Scholar]
- Li J, et al. , 2018. Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front Immunol. 9, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MT, et al. , 2001. Neutralization of the chemokine CXCL10 reduces inflammatory cell invasion and demyelination and improves neurological function in a viral model of multiple sclerosis. J Immunol. 167, 4091–7. [DOI] [PubMed] [Google Scholar]
- Lu B, et al. , 2002. Transplantation of EGF-responsive neurospheres from GFP transgenic mice into the eyes of rd mice. Brain Res. 943, 292–300. [DOI] [PubMed] [Google Scholar]
- Mangale V, et al. , 2019. Promoting remyelination through cell transplantation therapies in a model of viral-induced neurodegenerative disease. Dev Dyn. 248, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marro BS, et al. , 2019. Disrupted CXCR2 Signaling in Oligodendroglia Lineage Cells Enhances Myelin Repair in a Viral Model of Multiple Sclerosis. J Virol. 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheu MP, et al. , 2015. Imaging regulatory T cell dynamics and CTLA4-mediated suppression of T cell priming. Nat Commun. 6, 6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DP, et al. , 2012. Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods Mol Biol. 900, 381–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel I, et al. , 1995. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. 25, 1951–9. [DOI] [PubMed] [Google Scholar]
- Mock JR, et al. , 2014. Foxp3+ regulatory T cells promote lung epithelial proliferation. Mucosal Immunol. 7, 1440–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozafari S, et al. , 2015. Skin-derived neural precursors competitively generate functional myelin in adult demyelinated mice. J Clin Invest. 125, 3642–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FJ, et al. , 2006. Gene therapy: can neural stem cells deliver? Nat Rev Neurosci. 7, 75–84. [DOI] [PubMed] [Google Scholar]
- Nazmi A, et al. , 2014. Neural stem/progenitor cells induce conversion of encephalitogenic T cells into CD4+-CD25+-FOXP3+ regulatory T cells. Viral Immunol. 27, 48–59. [DOI] [PubMed] [Google Scholar]
- Neirinckx V, et al. , 2013. Concise review: adult mesenchymal stem cells, adult neural crest stem cells, and therapy of neurological pathologies: a state of play. Stem Cells Transl Med. 2, 284–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisted WC, et al. , 2016. Remyelination Is Correlated with Regulatory T Cell Induction Following Human Embryoid Body-Derived Neural Precursor Cell Transplantation in a Viral Model of Multiple Sclerosis. PLoS One. 11, e0157620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluchino S, et al. , 2009. Human neural stem cells ameliorate autoimmune encephalomyelitis in non-human primates. Ann Neurol. 66, 343–54. [DOI] [PubMed] [Google Scholar]
- Salazar DL, et al. , 2010. Human neural stem cells differentiate and promote locomotor recovery in an early chronic spinal cord injury NOD-scid mouse model. PLoS One. 5, e12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setiady Y Y., et al. , 2010. In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcgammaRIII+ phagocytes. Eur J Immunol. 40, 780–6. [DOI] [PubMed] [Google Scholar]
- Steinman L, 1996. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 85, 299–302. [DOI] [PubMed] [Google Scholar]
- Tirotta E, et al. , 2010. Cell replacement therapies to promote remyelination in a viral model of demyelination. J Neuroimmunol. 224, 101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM, 2008. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 77, 289–312. [DOI] [PubMed] [Google Scholar]
- Torkildsen O, et al. , 2016. Disease-modifying treatments for multiple sclerosis - a review of approved medications. Eur J Neurol. 23 Suppl 1, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JT, et al. , 2014. Regulatory T cells in central nervous system injury: a double-edged sword. J Immunol. 193, 5013–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinger JG, et al. , 2015. Two-photon imaging of cellular dynamics in the mouse spinal cord. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SH, et al. , 2011. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One. 6, e17540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. T cell FACS gating strategy. Representative dot plots of CD4+, CD8+ and CD25+FoxP3+ Tregs from Figure 4D sequentially gated on lymphocytes, excluding forward and side scatter doublets, CD4+ or CD8+ cells, CD4+CD25+FoxP3+. Gates are indicated above plots.
Supplemental Figure 2. T cell populations are unaffected 28 days post hNSC Transplant. Quantification of the frequency of adaptive immune cell populations CD4+, CD4+CD25+FoxP3+ and CD8+ T cell subsets within the spinal cord, draining cervical lymph node, and spleen of hNSC (n=3) or control PBS (n=3) injected mice revealed no significant difference in cell populations at day 28 p.t. Data represents two independent experiments.
Supplemental Figure 3. PC61.5 treatment of mice depleted Tregs without affecting T conv cell population or EAE clinical score. (A) Timeline of experiment. EAE mice received intraspinal transplant of hNSCs and intraperitoneal (i.p.) PBS (n=10), hNSCs and anti-CD25 (clone PC61.5) i.p. (n=7), hNSCs and IgG i.p. (n=7), or PBS (n=8) as a control. Anti-CD25 antibody selectively depletes Tregs, IgG was given as an isotype control. EAE mice were administered anti-CD25 and IgG antibodies −2, 0, and 2 days p.t. of hNSCs. Quantification of frequency (B) and number (C) of CD25+FoxP3+ Tregs in peripheral blood 3, 9, and 21 days p.t. Quantification of frequency (D) and number (E) of CD4+ Tconv cells in peripheral blood 3, 9, and 21 days p.t. (F) Graph of clinical scores of mice injected intraspinally with hNSCs (dark blue), hNSCs+PC61.5 (light blue), hNSCs+IgG (dark grey), or PBS (light grey) at defined timepoints post-transplant. PC61.5 treatment does not impact clinical score. Data is presented as average ± SEM and analyzed using two-way ANOVA followed by Tukey’s multiple comparison test (*p=0.01; ****p<0.0001).