

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal late-onset neurodegenerative disease that specifically affects the function and survival of spinal and cortical motor neurons. ALS shares many genetic, clinical, and pathological characteristics with frontotemporal dementia (FTD), and these diseases are now recognized as presentations of a disease spectrum known as ALS/FTD. The molecular determinants of neuronal loss in ALS/FTD are still debated, but the recent discovery of nucleocytoplasmic transport defects as a common denominator of most if not all forms of ALS/FTD has dramatically changed our understanding of the pathogenic mechanisms of this disease. Loss of nuclear pores and nucleoporin aggregation, altered nuclear morphology, and impaired nuclear transport are some of the most prominent features that have been identified using a variety of animal, cellular, and human models of disease. Here, we review the experimental evidence linking nucleocytoplasmic transport defects to the pathogenesis of ALS/FTD and propose a unifying view on how these defects may lead to a vicious cycle that eventually causes neuronal death.
Keywords: Nucleocytoplasmic transport, Amyotrophic lateral sclerosis, Frontotemporal dementia, ALS/FTD
1. Introduction
Nucleocytoplasmic transport (NCT) is a tightly regulated process that controls the exchange and cellular distribution of proteins and RNAs between the nucleus and the cytoplasm. Three main groups of proteins are involved in the regulation of NCT, including 1) the nucleoporins that make up the nuclear pore complex (NPC), which serves as a gateway connecting the nucleoplasm and cytoplasm, 2) the nuclear transport receptors (NTRs), which chaperone RNA and protein cargo through the selective permeability barrier of the NPC, and 3) the small GTPase Ran with associated GTPase-activating protein RanGAP1 and Ran guanine nucleotide exchange factor (RanGEF/RCC1), which regulate NTR-specific cargo-loading and release in the nucleoplasm and cytoplasm, thus governing directionality of the transport (Jamali et al., 2011; Schmidt and Görlich, 2016). NPCs are large multiprotein complexes, composed of more than 30 different nucleoporins arranged in an 8-fold symmetry (Hampoelz et al., 2019; Stoffler et al., 2003). They provide a tightly controlled barrier for the shuttling of high molecular weight (MW) cargo (i.e.~ >40 kDa) in and out of the nucleus, thanks to the presence of phenylalanine glycine-rich nucleoporins (FG-Nups) in the central channel (Figure 1A). These highly dynamic components of the NPC are characterized by the presence of intrinsically disordered regions that form a permeability barrier, acting as a size filter (Hayama et al., 2017; Hülsmann et al., 2012; Li et al., 2016). Current models suggest either the formation of a hydrogel-like molecular sieve formed via attractive interactions between hydrophobic FG-repeats, or the formation of polymer ‘brushes’ dominated by repulsive interactions, with both types of FG-Nup polymers serving to repel large macromolecules from NPC passage (Lemke, 2016; Zilman, 2018). Only large proteins that are endowed with nuclear localization (NLS) or nuclear export signals (NES) can pass through the pore by binding to NTRs, the most common of which are karyopherins that act as either importins or exportins. The best characterized NLS types are the classical NLS (cNLS), consisting of basic amino acid clusters which is recognized by an importin-α/β heterodimer, and the proline-tyrosine NLS (PY-NLS) which is bound by importin-β2/transportin-1 (Soniat and Chook, 2015). Karyopherins facilitate the transport by forming transient interactions with the FG-Nups in the pore to chaperone their protein and/or RNA cargos across the permeability barrier (Aramburu and Lemke, 2017). The directionality for this process is provided through a gradient formed by the small Ras-related nuclear protein, Ran, which is highly enriched in its GTP-bound form in the nucleus. Here, Ran-GTP forms a trimeric complex with exportin-bound cargos that are shuttled to the cytoplasm, and is then hydrolyzed to Ran-GDP by RanGAP1, triggering the release of the cargo. Ran-GDP is imported by nuclear transport factor 2 (NTF2) back into the nucleus, where the nucleotide exchange factor RCC1 catalyzes the conversion to the GTP-bound state. In the nucleus, binding of Ran-GTP to importins triggers the release of their cargo and their translocation back to the cytoplasm, restarting the cycle (Figure 1B) (Matsuura, 2016; Steggerda and Paschal, 2002).
Figure 1: The NPC and associated factors.
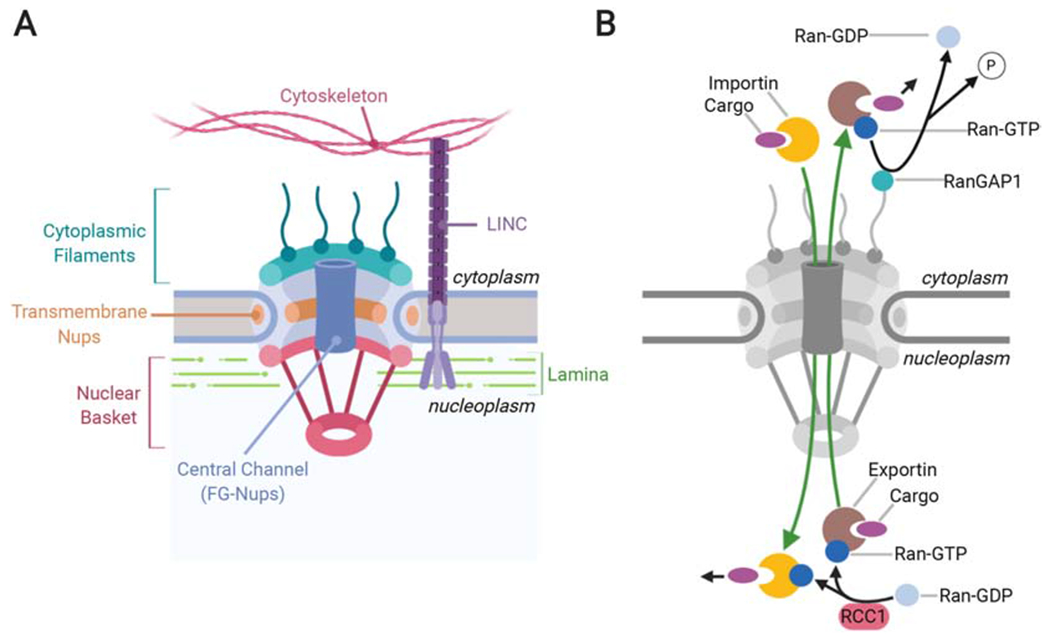
A. Depiction of the basic structure of the NPC. Cytoplasmic filaments and the nuclear basket extend into the cytoplasm and nucleoplasm respectively, while transmembrane Nups anchor the NPC to the nuclear envelope. The central channel is rich in FG-Nups, that form a selectivity filter to prevent the unregulated passage of large proteins (i.e. ~>40kDa) though the pore. The LINC complex is tightly associated with the nuclear pore and connects the nuclear lamina with the cytoskeleton. B. Representation of the NCT pathway. Importins bind to their cargo and facilitate movement through the NPC. In the nucleoplasm, Ran-GTP either binds to importins to trigger the release of the cargo, or to the exportin-cargo complex, facilitating its transport through the nuclear pore to the cytoplasm. RanGAP1 then hydrolyzes Ran-GTP to Ran-GDP and triggers the release of the cargo into the cytoplasm. RCC1 restores the GTP-bound state of Ran in the nucleoplasm, resetting the cycle.
Each individual nucleus contains several hundreds to thousands of NPCs, but their precise number and density changes depending on phases of the cell cycle and cell types (Weberruss and Antonin, 2016). For instance, the number of NPCs almost doubles during interphase in dividing cells, a process regulated by cyclin-dependent kinases (Maeshima et al., 2010). The insertion, anchoring, and positioning of NPCs in metazoans is mediated by their interaction with the nuclear lamina, a meshwork of intranuclear intermediate filaments that include A- and B-type Lamins (de Leeuw et al., 2018; Fiserova and Goldberg, 2010). The nuclear lamina also anchors the whole nucleus to the cell cytoskeleton through its interaction with the linker of nucleoskeleton and cytoskeleton (LINC) complex (Figure 1A). This multiprotein complex spans both the inner and outer nuclear membrane, transmitting tensile force generated by the cytoskeleton onto the nucleus to regulate nuclear morphology, positioning, and migration (Burke, 2019; Goldberg, 2017; Hieda, 2017; Jahed et al., 2016). Although much is known about the NPC lifecycle in dividing cells, the mechanisms controlling the formation, anchoring, and especially maintenance and repair of the nuclear pores in postmitotic cells are still largely unknown. Indeed, contrary to dividing cells that refresh their pool of NPCs at the end of mitosis when the nuclear envelope is newly formed, postmitotic neurons lack this replacement mechanism (Hetzer, 2010). Some nucleoporins are among the longest lived proteins in the cell, and they are never or very slowly replaced during the life time of a cell once the NPC is formed (D’Angelo et al., 2009; Savas et al., 2012; Toyama et al., 2013). It is thus likely that even subtle alterations that damage the function and integrity of the NPC would accumulate over time, leading to late onset defects in the import and export of cargo across the pore, accumulation of nuclear proteins in the cytoplasm, and eventually cell death. These considerations point to the NPC as a prime suspect to explain the late-onset neuron-specific cell death observed in neurodegenerative diseases. In fact, defects to the NCT pathway have been recently identified as a common pathogenic event in several neurodegenerative diseases, including Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD), Alzheimer’s disease (AD), and Huntington’s disease (HD) (Hutten and Dormann, 2019; Kim and Taylor, 2017). Cytoskeletal dysregulation, protein aggregation, and oxidative stress, common phenomena in neurodegenerative diseases, have been shown to induce NCT defects such as loss of NPCs at the nuclear envelope, protein mislocalization, nuclear RNA retention, and disrupted nuclear lamina. Here we will focus on the accumulated evidence linking NCT defects to the pathobiology of ALS and FTD, and propose a mechanistic view on how such defects may originate in the first place.
2. NCT defects as a common feature of ALS/FTD
ALS and FTD form a disease spectrum based on shared clinical, genetic, and neuropathological features (Lattante et al., 2015; Talbot and Ansorge, 2006; Weishaupt et al., 2016). A defining characteristic of ALS/FTD is the loss of specific RNA-binding proteins (RBPs) from the nucleus and their mislocalization into cytoplasmic aggregates (Ito et al., 2017; Mandrioli et al., 2019). TDP-43 pathology is present in ~ 97% of ALS, and~ 45% of FTD cases, whereas FUS pathology is less common and only found in <1% of ALS and~ 9% of FTD cases (Ling et al., 2013). Genetic studies demonstrate that TDP-43 and FUS mutations can directly cause ALS, suggesting a direct functional connection of these mislocalized proteins to the disease process (Lagier-Tourenne and Cleveland, 2009). In the past few years, there has been accumulating evidence that beyond the mislocalization of these proteins, general NCT defects in protein import and RNA export are also a common feature of ALS/FTD and other age-related neurodegenerative diseases, suggesting a possible link between these two phenomena. The first evidence for potential defects in the NCT machinery itself in ALS comes from the observation of irregularly shaped nuclear membranes and abnormal NTR distribution in spinal motor neurons from sporadic ALS and familial ALS cases with SOD1 mutations (mSOD1). Nuclear staining of the FG-rich Nup62 was irregular or completely absent in some cases (Aizawa et al., 2019; Kinoshita et al., 2009; Nagara et al., 2013; Yamashita et al., 2017). This was also confirmed in mSOD1 mice where Nup62 staining progressively weakened with age and became discontinuous (Kinoshita et al., 2009; Nagara et al., 2013). These early observations were followed by a series of studies based on genetic modifier screens in flies and yeast models of C9ORF72 GGGGCC (G4C2) repeat expansion pathology, one of the most common genetic causes of ALS/FTD (DeJesus-Hernandez et al., 2011). These studies reported a direct connection between C9ORF72 G4C2 pathology and morphological and functional defects to the NCT pathway (Freibaum et al., 2015; Jovičić et al., 2015; Zhang et al., 2015). Similar defects were observed in models of TDP-43 proteinopathy and sporadic ALS patient cells, suggesting that NCT dysfunction may be a common hallmark not only for C9ORF72-linked disease (C9-ALS/FTD), but for most if not all ALS/FTD cases (Chou et al., 2018). While these studies identified co-aggregation of components of the NCT machinery with pathological protein aggregates or RNA foci as potential pathomechanisms, newer studies have implicated cytoskeletal defects as additional mechanisms that may cause NCT defects in ALS/FTD and AD (Eftekharzadeh et al., 2018; Giampetruzzi et al., 2019; Paonessa et al., 2019). A compendium and schematic of all NCT-related defects identified thus far in ALS/FTD models as well as patient tissue are provided in Table 1 and Figure 2, respectively.
Table 1:
Summary of functional and morphological NCT defects in ALS/FTD.
| Defect | ALS/FTD subtype | Models | References |
|---|---|---|---|
| Irregular nuclear morphology | C9ORF72 | Drosophila, mouse | Freibaum et al., 2015; Chew et al., 2019 |
| PFN1 | cell lines | Giampetruzzi et al., 2019 | |
| SetX | patient tissue, mouse | Bennett et al., 2018 | |
| SOD1 | patient tissue, mouse | Kinoshita et al., 2009; Nagara et al., 2013 | |
| Tau | Drosophila, cell lines, patient tissue | Frost et al., 2016; Eftekharzadeh et al., 2018; Cornelison et al., 2019; Paonessa et al., 2019 | |
| TDP-43 | cell lines | Chou et al., 2018; Roczniak-Ferguson et al., 2019 | |
| VAPB | cell lines | Tran et al., 2012 | |
| sALS | patient tissue | Kinoshita et al., 2009; Nagara et al., 2013; Aizawa et al., 2018 | |
| Loss of Ran gradient | C9ORF72 | cell lines | Zhang et al., 2015; Zhang et al., 2018 |
| GRN | mouse | Ward et al., 2014 | |
| PFN1 | cell lines | Giampetruzzi et al., 2019 | |
| Tau | patient tissue, mouse | Eftekharzadeh et al., 2018 | |
| TDP-43 | cell lines | Zhang et al., 2018; Gasset-Rosa et al., 2019; | |
| Protein import defect | C9ORF72 | cell lines | Zhang et al., 2015; Khosravi et al., 2017; Shi et al., 2017; Vanneste et al., 2019 |
| PFN1 | cell lines | Giampetruzzi et al., 2019 | |
| SetX | mouse | Bennett et al., 2018 | |
| Tau | cell lines | Eftekharzadeh et al., 2018; Paonessa et al., 2019; | |
| TDP-43 | cell lines | Chou et al., 2018 | |
| RNA export defect | C9ORF72 | Drosophila | Freibaum et al., 2015 |
| Tau | cell lines | Cornelison et al., 2019 | |
| TDP-43 | cell lines | Woerner et al., 2016; Chou et al., 2018 | |
| Mislocalization/aggregation of NCT proteins | ADAR-2 | mouse | Yamashita et al., 2018 |
| C9ORF72 | mouse | Zhang et al., 2016 | |
| SOD1 | mouse | Shang et al., 2017 | |
| Tau | cell lines, patient tissue | Eftekharzadeh et al., 2018 | |
| TDP-43 | cell lines | Chou et al., 2018; Gasset-Rosa et al., 2019 | |
| sALS | patient tissue | Shang et al., 2017; Chou et al., 2018, Aizawa et al., 2019 | |
| Mislocalization of importins | C9ORF72 | Drosophila, patient tissue | Xao et al., 2015; Somolon et al., 2018 |
| SOD1 | mouse | Nagara et al., 2013 | |
| TDP-43 | cell lines, Drosophila | Gasset-Rosa et al., 2019; Solomon et al., 2018 | |
| sALS | patient tissue | Kinoshita et al., 2009; Nishimura et al., 2010; Yamashita et al., 2017; Aizawa et al., 2019 | |
| Loss of nuclear integrity | Tau/AD | patient tissue | Eftekharzadeh et al., 2018 |
| Aging | C. elegans, rat | D’Angelo et al., 2009 | |
| Blocked nuclear pores | C9ORF72 | cell lines | Shi et al., 2017 |
Figure 2: ALS/FTD-linked defects in the NPC and NCT.
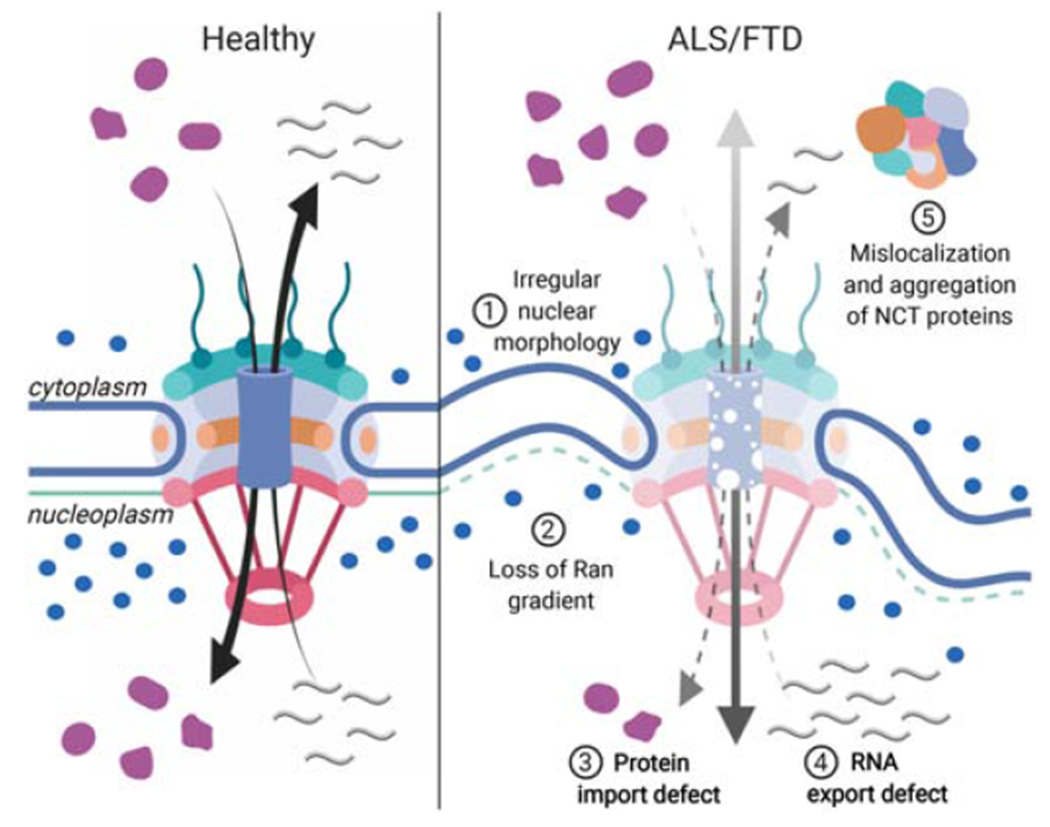
(left panel) In healthy cells, RNA export and protein shuttling through the nuclear pore are tightly regulated processes that depend on the nucleocytoplasmic distribution of GTP-bound Ran (blue circles). (right panel) In ALS/FTD cells, the nuclear pore integrity is compromised and the shuttling of RNAs and proteins is no longer controlled. Irregular nuclear morphology is often observed with deep membrane invaginations and disruption of the lamina (1). The Ran gradient is impaired, leading to increased levels of Ran in the cytoplasm (2). This impairs the import of proteins (3), as well as the export of mature RNAs from the nucleus (4). The mislocalization and aggregation of shuttling proteins sequesters NCT factors such as importins and nucleoporins into the cytoplasm (5), further disrupting the process.
3. NCT defects linked to protein aggregation
Abnormal protein aggregation is considered a central disease mechanism in ALS/FTD. Affected neurons in postmortem tissue as well as in animal models of ALS are most often characterized by the presence of large cytoplasmic inclusions positive for proteasome and autophagy-associated markers such as ubiquitin and p62 (Blokhuis et al., 2013). The main protein component of these aggregates was TDP-43, a mostly nuclear RBP that abundantly translocates to the cytoplasm and is lost from the nucleus under pathological conditions now referred to as TDP-43 proteinopathies (Neumann et al., 2006). Nearly all sporadic and familial ALS cases test positive for TDP-43 pathology in postmortem tissue, with the notable exceptions of FUS- and SOD1-associated ALS. The role of aggregates in ALS as well as other neurodegenerative diseases is still controversial, with conflicting hypotheses describing the accumulation of misfolded proteins in insoluble aggregates as either a protective mechanism or toxic initiator of neuronal death (Bolognesi et al., 2019; Hergesheimer et al., 2019; Zhu et al., 2018). Of note, regulators of autophagy and protein homeostasis represent a major class of ALS/FTD disease genes, suggesting that a diminished ability of the cell to clear misfolded and aggregating proteins causes increased toxicity. Taken together, these observations point to misfolded proteins and aggregates as being more of a foe than a friend (Hergesheimer et al., 2019; Ramesh and Pandey, 2017). Furthermore, repeat-associated non-ATG (RAN) translation of aggregation-prone dipeptide-repeat proteins (DPRs) from C9ORF72 repeat expansions has been shown to be a prominent cause of toxicity in several animal and cellular model of disease, strengthening the case for protein aggregation as a cause of cellular toxicity. However, the mechanisms by which DPR protein aggregation induces cell death and its potential contribution to C9ORF72-related ALS/FTD phenotypes is not fully understood.
Recent studies have demonstrated that cytoplasmic protein aggregation, regardless of the misfolded protein species, can lead to NCT defects and neuronal degeneration. Amyloidogenic β-sheet proteins have been shown to undergo fibrillary aggregation when they accumulate in the cytoplasm, and disrupt the nucleocytoplasmic shuttling of proteins and RNAs (Woerner et al., 2016). Formation of nuclear RNA foci and cytoplasmic DPRs in C9-ALS/FTD has been shown to cause NCT dysfunction (K. Zhang et al., 2016). NCT defects can also be caused by the formation of pathological TDP-43 inclusions that can abnormally sequester nucleoporins into aggregates and affect the localization of other NCT factors in ALS/FTD patients (Chou et al., 2018). While mechanisms of cytoplasmic mislocalization of FUS in ALS/FTD have been well studied, in contrast to TDP-43 there is currently little evidence for FUS pathology directly causing NCT defects. A targeted screen for modifiers of FUS toxicity in Drosophila found that downregulation of Nup154 and exportin-1 prevented neurotoxicity induced by overexpression of human FUS in motor neurons. However, knock-down of exportin-1 also significantly reduced the propensity of FUS to form inclusions upon stress (Steyaert et al., 2018), suggesting that NCT proteins modulated FUS toxicity by directly or indirectly acting on the mislocalization and aggregation of FUS (see also section 6). In this section, we will review the evidence linking NCT disruption and aggregation caused by different ALS/FTD disease genes.
3.1. C9ORF72 repeat expansion
The most common genetic cause of ALS and FTD is the presence of expanded G4C2 hexanucleotide repeats (HRE) in intron 1 of the C9ORF72 gene (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Potential disease mechanisms include loss of function due to haploinsufficiency and a toxic gain of function of either the intronic RNA repeats or DPRs generated via RAN-translation (Gendron and Petrucelli, 2018). While the cellular function of the two long and short C9ORF72 protein isoforms is not fully defined, the short C9 isoform strongly localizes to the nuclear membrane but relocates to the plasma membrane in ALS motor neurons. Both long and short C9 isoforms also interact with Ran and importin-β1, suggesting that reduced levels of these proteins could interfere with NCT, thus contributing to pathology (Xiao et al., 2015). Currently substantial more evidence exists linking repeat RNA and DPR toxicity to NCT dysfunction in several cell and animal models of ALS/FTD (Rodriguez and Todd, 2019).
Evidence for expanded RNA repeats disrupting NCT comes from a candidate-based genetic screen in a Drosophila model expressing (G4C2)30 repeat constructs, which identified Drosophila RanGAP as a potent suppressor of neurodegeneration (Zhang et al., 2015). A previous study using a proteome array to identify G4C2–binding proteins had found that HREs can physically bind to RanGAP1 (Donnelly et al., 2013), which formed large perinuclear puncta in G4C2-expressing Drosophila cells, as well as in iPSC-derived neurons and in the motor cortex of C9ALS/FTD patients (Zhang et al., 2015). It has also been reported that RanGAP1 abnormally accumulates in nuclear invaginations in the cortex of (G4C2)149 mice (Chew et al., 2019). Disrupting the interaction between RanGAP1 and HREs rescued NCT defects, suggesting that RanGAP1 sequestration by G4C2 RNA is upstream of nuclear import dysfunction (Zhang et al., 2015). Since RanGAP1 is an essential regulator of the Ran cycle, its sequestration and loss from the nuclear envelope is expected to alter the nuclear-cytoplasmic distribution of Ran. Indeed, overexpression of RanGAP1 could rescue Ran cytoplasmic accumulation, and further rescue photoreceptor and motor neuron degeneration in (G4C2)30–expressing flies, indicating that disruption of the Ran gradient contributes to G4C2 RNA toxicity. Accordingly, overexpression of RCC1 enhanced the degenerative phenotype, while its knock-down rescued it. HREs also caused nuclear import deficits in Drosophila cells and iPSC neurons, including an abnormal cytoplasmic accumulation of TDP-43. Modulation of NCT via either the exportin-1 inhibitor KPT-276, RNAi-mediated downregulation of exportin-1, or overexpression of importin-α rescued photoreceptor neuron degeneration in these flies. Although the exact mechanisms responsible for this rescue are not fully understood, these data suggest that NCT defects are central to disease pathogenesis (Zhang et al., 2015). Interestingly, the eye phenotype in these flies appeared to be linked to a G4C2 RNA-mediated mechanism, since DPRs were either absent or below the detection level in the fly eye at the time of photoreceptor neuron degeneration.
Very similar observations were also made in a parallel study that employed (G4C2)58 flies in which poly(GP) and poly(GR) expression via RAN-translation was evident (Freibaum et al., 2015). Ran was identified as a strong suppressor of the rough eye phenotype, highlighting the Ran gradient again as an important target of the G4C2 toxicity. Expanded G4C2 disrupted the nuclear envelope in Drosophila salivary glands and induced Nup107 mislocalization, which has also been reported in patient tissue (Freibaum et al., 2015; Zhang et al., 2015). Knock-down of the NTRs transportin-1, exportin-1, and of nuclear basket nucleoporins Nup50 and Nup153 – nucleoporins involved in the initiation of NPC assembly (Schwartz et al., 2015) - enhanced retinal degeneration, further strengthening the link between NCT and ALS/FTD pathology. Interestingly, several RNA nuclear export factors were identified as genetic modifiers in this disease model. These included the mRNA export factors ALYREF and GLE1 (Freibaum et al., 2015), the latter having been previously associated with recessive fetal motor neuron diseases and familial forms of ALS (Kaneb et al., 2015; Nousiainen et al., 2008). In support of these genetic data, nuclear retention of RNA was observed in flies and in different cell models of G4C2 toxicity, and downregulation of ALYREF partially rescued nuclear RNA accumulation in flies, presumably by permitting RNA degradation via the nuclear exosome (Freibaum et al., 2015). Although this study did not directly explore the role of DPRs vs HRE in inducing NCT defects, this question has been addressed by using DPR-only models of disease as discussed below.
A clear connection between DPR toxicity and NCT defects has been established by a study based on unbiased genetic screens for modifiers of poly(PR)50 toxicity in yeast (Jovičić et al., 2015). Potent modifiers included karyopherins and other components of the NCT machinery, a category also represented in genetic screens for modifiers of poly(GR)100 toxicity in yeast (Chai and Gitler, 2018). Interestingly, several NCT factors were also found to modify the degenerated eye phenotype in poly(GR)50-expressing flies. These proteins only interact with arginine-containing poly(PR) and poly(GR), which appear to be especially harmful in comparison with other DPRs (Lee et al., 2016). Moreover, poly(PR) have been shown to increase the permeability of NPCs by direct binding to FG-Nups (Shi et al., 2017). These findings suggest that NCT dysfunction may be a key disease mechanism in poly(PR)/(GR) pathology. On the other hand, co-aggregation of RanGAP1 and transmembrane nucleoporin Pom121 with poly(GA) inclusions was reported in poly(GA)-expressing mice. Surprisingly, these aberrations were not observed in mice expressing a soluble form of poly(GA), suggesting that poly(GA)-induced NCT defects are caused by GA aggregation (Y.-J. Zhang et al., 2016). Interestingly, a study measuring nuclear import and export in DPR-expressing cell lines found that poly(GA), but not poly(GR) and poly(PR), induced NCT deficits (Vanneste et al., 2019), specifically altering the importin-α/β-dependent import pathway (Khosravi et al., 2017). Finally, in flies TDP-43 was shown to accumulate in the cytoplasm in the presence of DPRs but not HREs, which favors the role of DPRs in NCT dysfunction (Solomon et al., 2018). Taken together, these studies demonstrate that RNA repeat as well as DPR-mediated toxicity can cause NCT defects in cellular and animal models, and may both contribute to the disease phenotype in patients where these pathologies are found together.
3.2. TDP-43 proteinopathy
An effort to characterize the proteome associated with pathological TDP-43 inclusions has led to the discovery that aggregates formed by the 25 kDa C-terminal fragment of TDP-43 (TDP-CTF) did not resemble stress granules (SG) but were highly enriched in components of the endomembrane system that are involved in intracellular transport, including components of the NCT machinery (Chou et al., 2018). TDP-CTF is a major component of cytoplasmic inclusions in ALS/FTD brain but not spinal cord tissue, and its expression in cellular models shows hallmarks of human TDP-43 proteinopathy, such as ubiquitination and hyperphosphorylation (Chou et al., 2018; Fallini et al., 2012; Igaz et al., 2008). The TDP-CTF associated proteome showed particular enrichment in nucleoporins, and co-expression studies in cells with mCherry-tagged TDP-CTF and GFP-tagged nucleoporins caused a cytosolic mislocalization and co-aggregation with TDP-CTF aggregates of most nucleoporins, including Nup62 and other FG-Nups (Chou et al., 2018). In a seeding model of TDP-43 proteinopathy, exposing mitotically arrested neuroblastoma cells to recombinant TDP-43 amyloid fibrils caused the loss of nuclear endogenous TDP-43 and the formation of cytoplasmic TDP-43 aggregates. Over time, these cytosolic TDP-43 assemblies sequestered Nup62 and induced mislocalization of RanGAP1, Ran, and Nup107, causing NCT defects and cell death (Gasset-Rosa et al., 2019). Ran and RanGAP1 have also been reported to form cytoplasmic puncta in motor neurons of ALS patients (Shang et al., 2017; Xiao et al., 2015). Defects in the localization of other nucleoporins have also been reported in spinal motor neurons of ALS patients, including nuclear aggregation of Nup50 and cytoplasmic accumulation of gp210 and Nup205, with the latter strongly colocalizing with TDP-43 aggregates (Shang et al., 2017). Cytoplasmic inclusions of Nup205 were also detected in different brain regions of sporadic ALS, familial TDP-ALS/FTD and familial C9-ALS/FTD cases, and strong co-localization of phospho-TDP-43 and Nup205 in large cytoplasmic inclusions was demonstrated in the motor cortex and hippocampus of a TDP-ALS case. Interestingly, in these cases Nup205 aggregation was not observed in the cerebellum, which remains unaffected by TDP-43 pathology, and it was also absent in the motor cortex of a familial SOD1-ALS patient without TDP-43 pathology, highlighting a distinct role of TDP-43 in NCT pathology (Chou et al., 2018). The role of TDP-43 pathology in C9-ALS/FTD and how it may contribute to the observed NCT defects is currently unclear.
4. NCT defects linked to cytoskeletal alterations
The role of cytoskeletal disruption in neurodegenerative diseases in general and ALS/FTD in particular has become increasingly evident in recent years (Hensel and Claus, 2018; Kounakis and Tavernarakis, 2019). Mutations in several cytoskeletal-related genes are causative or associated with disease, including microtubule-associated protein tau (MAPT) in FTD, and actin-binding Profilin-1 (PFN1), tubulin alpha-4A (TUBA4A), and the motor protein kinesin heavy chain isoform 5A (KIF5A) in ALS/FTD (Iqbal et al., 2016; Nicolas et al., 2018; Smith et al., 2014; Wu et al., 2012). Pathological forms of TDP-43 and FUS interfere with normal cytoskeletal function such as axonal outgrowth and intracellular transport (Baskaran et al., 2018; Fallini et al., 2012; Klim et al., 2019; Melamed et al., 2019; Oberstadt et al., 2018; Sama et al., 2017; Sugiura et al., 2016; Yasuda et al., 2017), and alterations to the cytoskeleton have been observed as early events in mSOD1G93A mice as well as mSOD1 iPSCs (Bellouze et al., 2016; Chen et al., 2014; Fanara et al., 2007). But how can these defects lead to specific (moto)neuron degeneration and cell death? Evidence from different genetic models of ALS and dementia suggest a direct link between the cytoskeleton and the disruption of nuclear pore stability and function. Several studies have shown that in human and animal models of AD and FTD associated with pathological forms of tau, neuronal nuclei are misshapen, presenting with deep and frequent invaginations and folds of the nuclear membrane (Cornelison et al., 2019; Eftekharzadeh et al., 2018; Frost et al., 2016; Paonessa et al., 2019). Nuclei isolated from mild and severe AD brains were also found to be leaky, as large MW dextran molecules are no longer excluded from the nucleoplasm, suggesting that the diffusion-barrier function of the nuclear membrane and/or the nuclear pores was severely compromised (Eftekharzadeh et al., 2018). Indeed, nuclear aberrant morphologies were accompanied by loss of nucleoporins, particularly the FG-rich Nup62 and Nup98, possibly due to their interaction with soluble and oligomeric forms of phospho-tau accumulating in the perinuclear region. While the tau–nucleoporin interaction and co-aggregation could explain some phenotypes observed in these tauopathy models, a different mechanism has been proposed to be responsible for the distortion of the nuclear morphology and nuclear lamina disruption observed. Using iPSC-derived neurons carrying two different FTD-associated tau mutations, it was shown that the presence of hyperphosphorylated tau qualitatively altered microtubule dynamics in the neuron’s cell body (Paonessa et al., 2019). Microtubule plus ends labeled with the GFP-EB3 marker were frequently observed projecting into the nucleus of human FTD neurons, severely deforming the nuclear envelope with deep lamin-positive invaginations. Microtubule depolymerization via nocodazole treatment restored normal nuclear morphology and reversed NCT defects, demonstrating a direct link between abnormal microtubule dynamics and nuclear morphology (Paonessa et al., 2019).
A disease-relevant association between cytoskeltal structures and NPCs has been further supported by studies in an ALS cellular model characterized by mutant PFN1. Mutations in this actin binding protein cause subtle changes to actin polymerization, leading to reduced F-actin levels in motor neuron growth cones and reduced axonal outgrowth in vitro (Wu et al., 2012). A combination of genetic and pharmacological tools demonstrated that the modulation of actin homeostasis, either positive or negative, impacts the structural integrity and function of the NPC (Giampetruzzi et al., 2019). While mutant forms of PFN1 caused changes to the NPC composition and/or density on the nuclear envelope and severely decreased nuclear import rates, even slight increases in actin polymerization via the overexpression of wild type PFN1 or the constitutively active form of the formin mDia1, which nucleates and elongates actin filaments (Breitsprecher and Goode, 2013; Wallar and Alberts, 2003), reduced nuclear import rates. Similarly, actin depolymerization via Latrunculin A treatment in normal neurons disrupted the normal nucleocytoplasmic Ran gradient and RanGAP1 localization to the nuclear envelope, suggesting that actin homeostasis is critical to the maintenance of functional NPCs. Strikingly, restoring the right balance of actin polymerization in mutant PFN1 neurons led to a full rescue of both structural and functional defects of the NPC, restoring a normal staining pattern of NPC markers and RanGAP1. Nuclear import defects were also rescued, which led to the reversal of TDP-43 cytoplasmic mislocalization and a normalization of TDP-43-dependent mRNA post-transcriptional regulation. One surprising finding was that this approach was effective in rescuing NPC defects not only in PFN1-linked ALS, but also in neurons expressing the C9ORF72 repeat expansion and in C9-ALS/FTD patient fibroblasts. Although there is currently no strong evidence linking C9ORF72 repeat expansions to actin disruption, decreased levels of the C9ORF72 protein have been found associated with reduced actin dynamics and cofilin activity in cellular models of C9-ALS/FTD, including iPSC-derived neurons (Sivadasan et al., 2016).
Two interesting observations emerge from these studies. First, changes in cytoskeletal dynamics and stability are key events in leading to the break-down of the NCT pathway in multiple genetic models of ALS/FTD. Disruption of nuclear morphology and lamina stability appears to be the first insult, which leads to a cascade of currently not well-defined events that terminates with the functional demise of the NPC. While mild overexpression of wild-type PFN1 had no negative impact on nucleoporin or Ran localization, disrupted Lamin A/C staining as well as reduced import rates were already evident (Giampetruzzi et al., 2019). Second, NPC defects observed in these cytoskeletal protein-related models of disease do not seem to be dependent on protein aggregation. In both cultured neurons and postmortem tissue, the mislocalization of nucleoporins and the dissolution of the Ran gradient were occurring in the presence of oligomeric and/or soluble forms of both tau and PFN1 (Eftekharzadeh et al., 2018; Giampetruzzi et al., 2019), suggesting that these defects are directly linked to tau and PFN1 function as regulators of cytoskeletal dynamics. How are defects in actin or microtubule dynamics disrupting the nuclear lamina and NPC function? One possible mechanism is via their association with the LINC complex. Previous research has demonstrated that changes to perinuclear actin homeostasis negatively regulate nuclear integrity (Kanellos et al., 2015; Wiggan et al., 2017). Knock-out of ADF/cofilin in keratinocytes and mouse embryonic fibroblasts led to an actin- and Arp2/3-dependent increase in nuclear deformation and breaks in the continuity of the nuclear lamina. Interestingly, knock-down or expression of dominant negative forms of Nesprin2 – a component of the cytoplasmic side of the LINC complex – completely rescued that phenotype. LINC does not only associate with actin, but it can bind either directly or indirectly via molecular motors to microtubules (Burke, 2019; Yang et al., 2018). This interaction is essential for correct positioning of the nucleus, especially in migrating cells and myotubes, possibly via favoring microtubule nucleation (Burke, 2019; Gimpel et al., 2017). However, no clear role has been outlined thus far for the LINC-cytoskeleton association in postmitotic neurons. Of note, mislocalization of LINC proteins was also observed in neurons containing TDP-43 aggregates (Chou et al., 2018), and mutations in LINC proteins are associated with cerebellar ataxia (Gros-Louis et al., 2007; Wang et al., 2015), suggesting that this complex may play an essential role in maintaining neuronal function and survival. It will be interesting to further explore the role of this complex in mediating NPC/NCT disruption during aging and in disease.
5. NCT defects linked to cellular stress
Together with protein aggregation and cytoskeletal disruption, increased oxidative stress is a hallmark of disease in ALS and FTD (Bozzo et al., 2017; Singh et al., 2019). Mutations in genes involved in the stress response such as ATXN2, TIA1, and hnRNPA2B1 are associated with ALS (Elden et al., 2010; Mackenzie et al., 2017; Martinez et al., 2016; Murakami et al., 2015; Ostrowski et al., 2017), and pathogenic forms of TDP-43 and FUS, as well as DPRs from mutant C9ORF72, have been shown to alter the dynamics of SG formation and dissolution (Boeynaems et al., 2017; Bosco et al., 2010; Khalfallah et al., 2018; Lee et al., 2016). Similar to protein aggregation and cytoskeletal disruption, also cellular stress has been linked to changes to the NCT pathway. Several labs using a wide range of models from yeast to human iPSC-derived neurons have demonstrated that upon cellular stress many NCT factors, including NTRs and THOC2, localize to SGs (Chang and Tarn, 2009; Fujimura et al., 2010; Jain et al., 2016; Mahboubi et al., 2013; Markmiller et al., 2018; Zhang et al., 2018). Severe stress from oxidative, hyperosmotic, or heat insults have been shown to cause the cytoplasmic mislocalization of nuclear proteins including Ran, and the nuclear retention of importins (Kodiha et al., 2008, 2004; Miyamoto et al., 2004; Stochaj et al., 2000; Zhang et al., 2018). Several nucleoporins, including Nup50, Nup88, and Nup205, have also been shown to localize to SGs (Jain et al., 2016; Kodiha et al., 2008, 2004; Zhang et al., 2018), as well as Ran, although only minimally (Fujimura et al., 2010; Zhang et al., 2018).
From these studies, it appears clear that stress induces changes to the NCT pathway, but whether these changes are a physiological and protective response, or a cytotoxic consequence to the stress is not yet fully understood, at least in healthy cells. For instance, damaging mutations in yeast orthologs of Ran and RanGAP1 reduce SG formation upon nutrient starvation, reducing survival rates after stress removal, and overall lead to a hyperactivation of gene expression programs in response to stress. This would suggest that changes to NCT happen as an important protective response to stress (Yang et al., 2014). This hypothesis is further supported by the observation that transportin-1 localization to SGs is required to shuttle the RBP tristetraprolin (TTP) and its bound mRNAs between processing (P) bodies and SGs, aiding in the cellular response to stress (Chang and Tarn, 2009). However, chronic stress such as during ageing or in disease may disrupt this balance, leading to the breakdown of physiological stress response. In fact, pharmacological inhibition of SG formation was shown to normalize Ran localization and NCT, and rescued neurodegeneration in fly models of ALS expressing the G4C2 hexanucleotide repeat, although whether this effect was directly linked to the modulation of NCT factors or other mechanisms is not clear (Zhang et al., 2018). Some nucleoporins are extremely long lived (Savas et al., 2012; Toyama et al., 2013) and are prone to accumulate oxidative stress-related injury, especially in postmitotic cells. Oxidative stress was shown to inhibit exportin-1-dependent nuclear export, and to increase phosphorylation and O-glycosylation of several FG-Nups (i.e. Nup358, Nup214, Nup98, and Nup62) (Crampton et al., 2009). Nup107, Nup153, Nup205, and Nup214 have also been identified as substrates of thioredoxinl, a redox protein essential for the removal of specific disulfide bonds and other cysteine post-translational modifications (Wu et al., 2014), suggesting that the NPC is subject to increased and age-dependent oxidative damage. Nuclei of ageing mice become progressively leaky, allowing large dextrans and other cytoplasmic proteins to permeate the nucleoplasm (D’Angelo et al., 2009). This phenotype may be directly caused by a loss of the NPC permeability barrier integrity due to carbonylation of nucleoporins such as Nup62, Nup93, and Nup153. Exposing worms to increased oxidative stress dramatically accelerated this process, directly linking aging, oxidative stress and NPC function (D’Angelo et al. 2009). In a comparison of neurons trans-differentiated from fibroblasts isolated from young and old individuals, it was found that NCT is specifically affected in aging neurons, perhaps because of reduced levels of RanBP17, a member of the karyopherin β family NTR family. Interestingly, cell reprogramming into rejuvenated induced pluripotent stem cells followed by differentiation into neurons erased such changes, suggesting that aging leads to an accumulation of epigenetic and metabolic defects that affect the NPCs (Mertens et al., 2015). Aging and oxidative stress also lead to the accumulation of protein aggregates that, as we discussed above, can cause NCT defects (Nowotny et al., 2014). However, TDP-CTF aggregate formation inhibits stress granule formation (Chou et al., 2018) and TDP-43 ubiquitylation and insolubility can be triggered via distinct pathways and independent of stress granules (Hans et al., 2019). Thus, stress-dependent aggregates are most likely different from the aggregation of disease-related proteins due to their distinct protein composition (Chou et al., 2018; Gasset-Rosa et al., 2019; Mann et al., 2019), and the relationship between SGs as direct precursors of TDP-43 aggregates in ALS remains controversial.
6. Mislocalization of importins in ALS/FTD
Aside from the sequestration of nucleoporins into aggregates and the disruption of the Ran gradient, NCT defects in ALS/FTD are also associated with reduced levels or mislocalization of importins. Nuclear depletion and cytoplasmic accumulation of importin-β1 have been reported in patient motor neurons by several groups, with no obvious sequestration into TDP-43-positive aggregates (Aizawa et al., 2019; Kinoshita et al., 2009; Nagara et al., 2013; Xiao et al., 2015). However, no changes in importin-β1 protein levels were observed in the cortex of TDP-ALS/FTD patients or in spinal cord of TDP-43-positive sporadic ALS patients (Nishimura et al., 2010), despite importin-β1 being a direct RNA target of TDP-43 (Sephton et al., 2011). Expression of amyloidogenic β-sheet proteins in HEK293 cells caused the cytoplasmic sequestration of importin-α1 and importin-α3 into cytoplasmic aggregates, thus interfering with protein nuclear import (Woerner et al., 2016). Cytoplasmic mislocalization and reduced protein levels of importin-α3 were also found in the frontal cortex of sporadic FTD and C9-ALS/FTD patients with or without TDP-43/DPR pathology (Solomon et al., 2018). Importin-α3 cytoplasmic accumulation has also been observed in spinal motor neurons of mSOD1G93A mice (Nagara et al., 2013), suggesting that importin-α3 mislocalization does not necessarily correlate with TDP-43/DPR pathology. Nuclear depletion of both importin-α1 and importin-α3 was also reported in TDP-43 proteinopathy fly models, even in the absence of a marked disruption of the NPCs (Solomon et al., 2018). Of interest, knock-down of importin-α3 enhanced the rough eye phenotype in a fly model of poly(GR) pathology (Lee et al., 2016). Immunostaining of exportin-2, which recycles the importin-α/Ran-GTP complex back to the cytoplasm, was reduced both in the nucleus and in the cytoplasm of FTD and ALS patients, while importin-α1 was depleted from the nucleus (Nishimura et al., 2010). These results correlate with reduced levels of exportin-2 and importin-α1 versus no change in importin-β1 levels in aged fibroblasts (Pujol et al., 2002). Interestingly, exportin-2 immunoreactivity was also absent in motor neurons of mice with knock-out of ADAR2, a protein downregulated in motor neurons of sporadic ALS patients (Yamashita et al., 2017). These observations are of particular interest, since the importin-α/β1 NTR complex mediates the nuclear import of many cargo including TDP-43, which could explain why TDP-43 accumulates in the cytoplasm of ALS/FTD patients (Kim et al., 2012; Nishimura et al., 2010). However, future studies will be required to define the mechanisms and disease relevance of these defects, and how they relate to a non-canonical role of importins as molecular chaperones to prevent the aggregation of their cargo proteins, as discussed below.
7. Importins as molecular chaperones to prevent cargo aggregation
Beside their canonical role as import factors, karyopherin β family importins have been shown to prevent the cytoplasmic aggregation of histones and ribosomal proteins (Jäkel et al., 2002) as well as FG-Nups (Milles et al., 2013), and the inappropriate non-nucleosomal interactions of histones (Padavannil et al., 2019). These observations suggest that this class of NTRs may have a non-canonical chaperone-like activity. At least for FUS protein, importins appear to govern not only its nuclear import but also its liquid-liquid phase separation (LLPS) and aggregation in the cytoplasm (Mikhaleva and Lemke, 2018). Similarly to TDP-43, FUS pathology has been reported in ALS and FTD patients (Ling et al., 2013), while mutations in FUS have been linked to familial ALS (Shang and Huang, 2016). Four independent studies have elegantly demonstrated that transportin-1/importin-β2 can bind FUS and prevent it from undergoing LLPS and aberrant liquid-to-solid phase transition (LSPT) into pathological fibrils, both in vitro and in vivo (Guo et al., 2018; Hofweber et al., 2018; Qamar et al., 2018; Yoshizawa et al., 2018). Transportin-1 regulates nuclear import of FUS by binding to its C-terminal PY-NLS, and defective nuclear import of FUS has been shown to cause its cytoplasmic accumulation and aggregation in disease. FUS can phase-separate in vitro into spherical liquid droplets which can undergo fusion events, as well as LSPT (Patel et al., 2015; Qamar et al., 2018). Transportin-1 prevented and reversed phase separation of FUS into droplets and stable hydrogels (Guo et al., 2018; Hofweber et al., 2018; Qamar et al., 2018; Yoshizawa et al., 2018). These results indicate that transportin-1 can both chaperone and disaggregate FUS. This dual activity is inhibited by Ran-GTP which dissociates transportin-1 from its cargo, and it highly depends on the presence of the PY-NLS (Guo et al., 2018; Hofweber et al., 2018; Yoshizawa et al., 2018). Interestingly, LLPS of other RBPs with a PY-NLS can also be prevented by transportin-1 (Guo et al., 2018). However, transportin-1 can still abrogate FUS phase separation in the absence of the PY-NLS under conditions of high concentrations (Yoshizawa et al., 2018). The majority of ALS-causing mutations resides in the NLS of FUS and impairs its recognition by transportin-1. Phase separation of FUS with ALS-causing mutations in the PY-NLS was also blocked by transportin-1, although the effect was less pronounced compared to wild-type FUS. Interestingly, transportin-1 does not solely bind to the PY-NLS of FUS, but the high-affinity interaction with the NLS enables the formation of weak and dynamic interactions between transportin-1 and other regions of FUS, including its N-terminal low-complexity domain, the RGG/RG regions, as well as the RRM and the ZnF domains (Yoshizawa et al., 2018). These additional binding sites may explain the effect of Transportin-1 on mutant FUS, despite the disruption of its canonical NLS.
Previous studies have shown that both the N-terminal and C-terminal regions of FUS are required for it to phase-separate and aggregate (Boeynaems et al., 2017; Kato et al., 2012; Patel et al., 2015; Schwartz et al., 2013; Sun et al., 2011). Tethering of both regions is necessary for the LLPS of FUS, through the formation of cation-π interactions between tyrosines in the N-terminal domain and arginines in the C-terminal RGG motifs (Lin et al., 2017; Qamar et al., 2018; Yoshizawa et al., 2018). Increasing the number of these arginines or inhibiting their methylation can promote phase separation of FUS. Of interest, hypomethylation of FUS has been described in FUS-FTD (Dormann et al., 2012), although how FUS methylation is lost in these patients is currently unknown. This might be due to neuron-specific differences in methylase and/or demethylase activity, although no mutations in arginine methyltransferases have been described to date. Loss of arginine methylation promoted phase separation of FUS in vitro, while dimethylated FUS underwent LLPS but at a very high concentration compared to unmethylated FUS (Hofweber et al., 2018). Therefore, hypomethylation of FUS can contribute to its aggregation in disease (Gittings et al., 2019). On the other hand, hypomethylated FUS shows a higher binding affinity to transportin-1 compared to dimethylated FUS (Dormann et al., 2012; Suárez-Calvet et al., 2016). This could explain why transportin-1 strongly co-aggregates with hypomethylated FUS in FUS-FTD cases but not with methylated FUS in familial FUS-ALS (Brelstaff et al., 2011; Neumann et al., 2012; Takeuchi et al., 2013). Importantly, transportin-1 efficiently suppressed phase separation of hypomethylated FUS in vitro and in SH-SY5Y cells (Qamar et al., 2018). Thus, the question of why transportin-1 is unable to suppress aggregation of hypomethylated FUS in FTD-FUS remains to be answered.
In yeast and human cells, transportin-1 overexpression reduced the number of cytoplasmic FUS foci and restored FUS localization to the nucleus (Guo et al., 2018). In addition, it reduced the partitioning of FUS into SGs, without affecting SG biogenesis nor by directly increasing transportin-1-dependent FUS nuclear import (Guo et al., 2018; Hofweber et al., 2018). On the other hand, drug-induced inhibition of endogenous transportin-1 increased the number of cells with FUS-positive SGs (Hofweber et al., 2018). Elevating transportin-1 levels rescued mutant FUSR521H-induced toxicity in HEK293T cells and restored the expression of FUS mRNA targets in mutant FUSR521H ALS-patient fibroblasts. This protective effect was also confirmed in fly models of FUS proteinopathy, where silencing transportin-1 enhanced the rough eye degeneration phenotype, whereas increasing transportin-1 expression in motor neurons rescued survival defects in vivo (Guo et al., 2018). Of interest, transportin-1 also was identified as one of the strongest suppressors of toxicity in yeast and Drosophila models of poly(PR) pathology (Boeynaems et al., 2016; Jovičić et al., 2015), as well as in other G4C2 fly models (Freibaum et al., 2015; Lee et al., 2016), suggesting it might play a key role in the pathogenesis of both FUS-ALS and C9-ALS/FTD cases. Poly(PR) can interact with transportin-1 and impair its nuclear import activity, thus causing the cytoplasmic accumulation of its cargo (Boeynaems et al., 2016). Knock-down of importins TNPO1, IPO11 and KPNA3 enhanced rough eye degeneration in poly(PR)25-expressing flies, whereas overexpression of the same importins rescued yeast toxicity (Boeynaems et al., 2016; Jovičić et al., 2015). Overexpression of importins in yeast did not change the levels or distribution of poly(PR) aggregates, suggesting that importins rescued toxicity by restoring normal NCT (Jovičić et al., 2015). In addition, upregulation of TNPO3, IPO9 and XPO5 also abrogated poly(PR) toxicity in yeast (Jovičić et al., 2015), and KPNB1 knock-down in poly(PR) and poly(GR) flies enhanced eye degeneration (Boeynaems et al., 2016; Lee et al., 2016).
Aside from transportin-1, the importin-α/β complex was shown in vitro to strongly reduce fibrillization of TDP-43 by binding to its cNLS. However, the import complex had neither an effect on TDP-43 fibrils lacking a cNLS, nor on FUS which harbors a PY-NLS (Guo et al., 2018). Thus, the NLS appears to act as a specific key initiation signal for nuclear import, chaperoning and dis-aggregation of RBPs with prion-like domains. On the other hand, importin-β1 expression was able to reduce the aggregation of TDP-CTF which lacks the N-terminal cNLS of TDP-43 in neuroblastoma cells (Chou et al., 2018). This would suggest that importin-β1 does not reduce TDP-CTF aggregation through binding the cNLS via importin-α, or that other factors not present in in vitro experiments are required for this process to occur. Since transportin-1 can still reduce the LLPS of FUS proteins lacking a PY-NLS when added at high concentrations, multivalent interactions at different regions may also be able to facilitate this process (Yoshizawa et al., 2018). It will be of interest to see what the substrate-specificity and mode of action for karyopherin family proteins is, and how they may modulate LLPS of other RBPs under normal physiological and disease conditions.
8. Discussion
The discovery of NCT defects as a common denominator of ALS/FTD can be surely considered a major milestone in the research on these invariably fatal and yet untreatable diseases. Although many questions still remain unanswered, this discovery impacts our understanding of disease mechanisms and their connection to aging, as well as the development of innovative therapeutic approaches focused on NCT modifiers.
Mutations in several genes have been identified in ALS, collectively implicating four main pathways, including 1) RNA processing, 2) protein quality control and degradation, 3) cytoskeletal integrity and trafficking, and 4) mitochondrial function and transport (Cook and Petrucelli, 2019). Despite the wide range of genes and cellular functions involved, all familial and sporadic ALS patients present with almost indistinguishable clinical symptoms, regardless of the primary cause of the disease. This suggests that all these different pathways may converge onto a common mechanism of disease, which eventually leads to motor neuron dysfunction and death. The collective evidence gathered by several labs using a variety of cellular, animal and human models summarized in this review support the hypothesis that such a unifying mechanism may be the disruption of the NCT pathway.
As we have discussed, experimental evidence directly links cellular stress, RNA(e.g. HRE) and protein aggregation (e.g. TDP-43, DPRs), and cytoskeletal disruption (e.g. PFN, tau) to nuclear transport defects, possibly caused by the sequestration and/or mislocalization of nucleoporins and NTRs. This in turn leads to the cytoplasmic accumulation of shuttling proteins such as TDP-43 and FUS, favoring their aggregation via LLPS/LSPT, a hallmark of ALS. The formation of these aggregates further sequesters transport factors and nucleoporins, leading to a vicious cycle that culminates in neuronal cell death (Figure 3). Changes to RNA export, splicing and post-transcriptional regulation as a consequence of NCT defects have been reported (Freibaum et al., 2015; Giampetruzzi et al., 2019; Woerner et al., 2016), potentially affecting many downstream ALS-relevant pathways. Stress and disruption of metabolic homeostasis, common phenomena in ALS, have also been shown to induce the rearrangement of the cytoskeleton, further feeding this toxic loop (Figure 4). Despite this compelling hypothesis, the exact mechanisms and initial steps that trigger the loss or mislocalization of NPC proteins and NCT defects in the familial and sporadic forms of ALS, as well as the consequences of such defects that lead to cell death, still remain unclear. This relationship is difficult to disentangle, since many of the observed defects, such as TDP-43 mislocalization and RNA dysregulation, may be both the cause and consequence of NCT defects. In addition, some of the basic aspects of NPC function, maintenance and repair in postmitotic cells are not well understood. A common defect linked to NCT dysfunction is the mislocalization and aggregation of nucleoporins and transport proteins. The NCT transport machinery is part of the endomembrane system, which provides internal compartmentalization in eukaryotic cells. NPCs and NTRs, clathrin, COPI, and COPII vesicle coats are involved in membrane-associated cargo trafficking and share a common evolutionary origin and conserved structural similarity (Onischenko and Weis, 2011), containing many β-propellers and α-solenoid–like domains (Rout and Field, 2017). It will be interesting to see if these related intracellular transport pathways, such as ER–Golgi transport, are affected in a similar manner to NCT (Burk and Pasterkamp, 2019; Schreij et al., 2016). Numerous studies discussed in this review article have also observed morphological defects in the nuclear envelope and lamina, such as deep invaginations into the nucleoplasm, but their frequency, contribution to NCT defects, and relevance to the neurodegenerative disease phenotypes is currently unclear.
Figure 3: Cellular disease mechanisms associated with NCT defects in ALS/FTD.
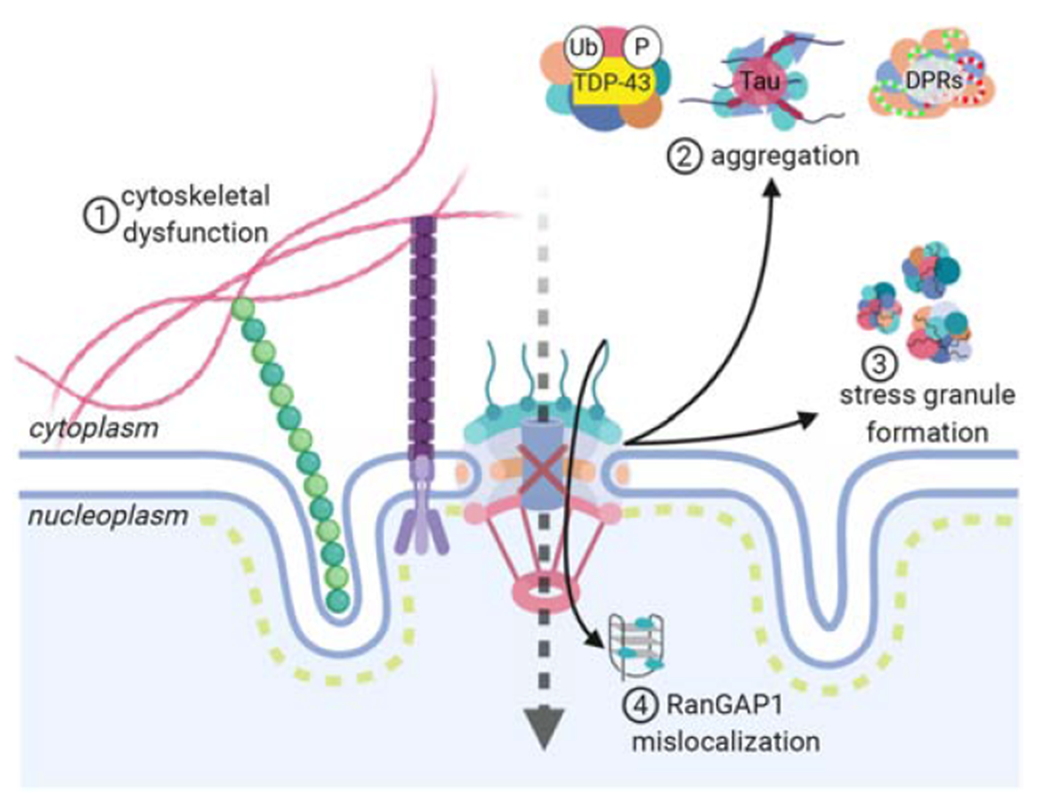
Cytoskeletal dysfunction can lead to an increase in the force exerted on the nucleus, causing the formation of nuclear invaginations and disrupting the integrity of the nuclear lamina (1). Abnormal protein aggregation (2) and stress granule formation (3) sequester nucleoporins and transport proteins in the cytoplasm, affecting the integrity of the nuclear pore and impairing nuclear import. C9-ALS-associated HREs bind and sequester RanGAP1 in the nucleus, thereby disrupting the nuclear gradient of Ran and leading to NCT defects (4).
Figure 4: All roads lead to NCT defects in ALS/FTD.
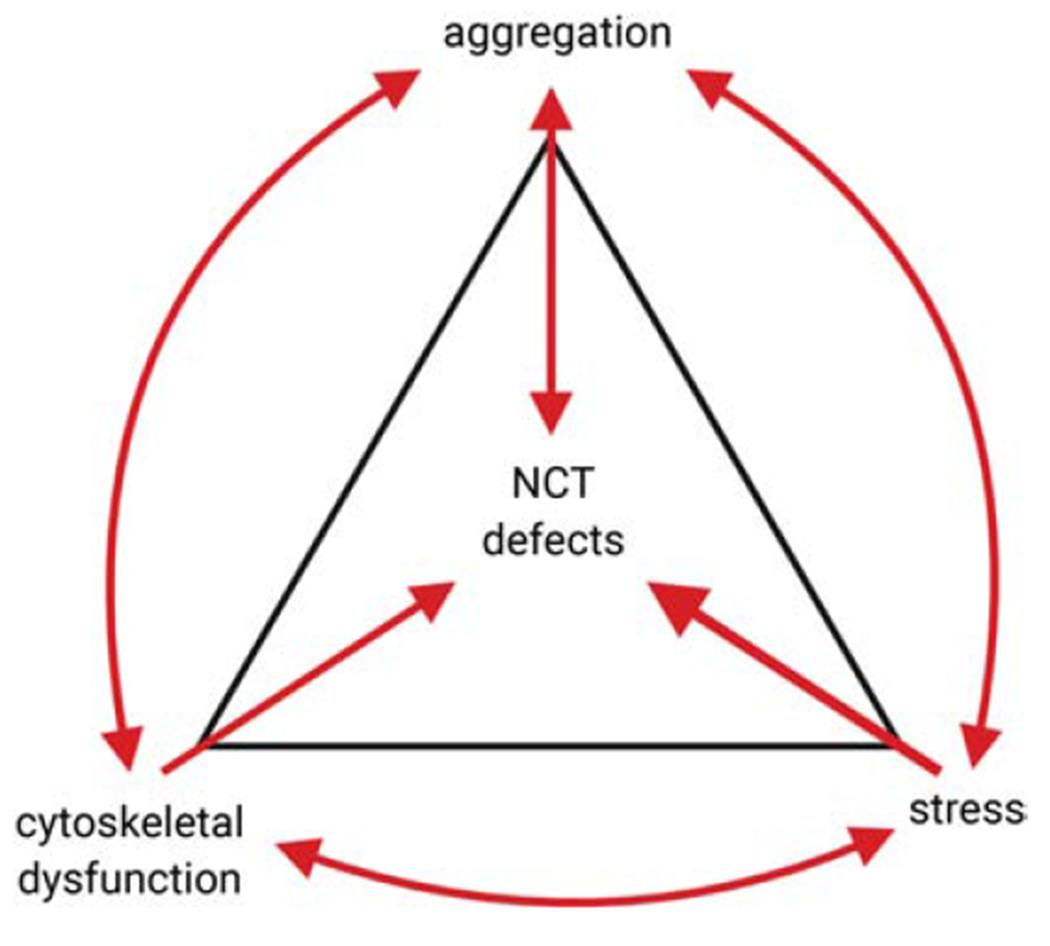
Cellular stress, protein aggregation, and cytoskeletal dysfunction initiated by disease-causing mutations or by environmental factors such as ageing, all have been independently shown to initiate a cascade of events that culminate in the disruption of normal NCT of proteins and RNAs. However, these pathways are not independent of each other, and each one can induce additional damage by further promoting protein aggregation, cytoskeletal disruption, and cellular stress. We hypothesize that an insult in any of these pathways can result in triggering this vicious cycle, resulting in NCT defects and neuronal death in ALS/FTD.
The discovery of NCT defects in neurodegenerative diseases has generated much interest in both the academic and biotech world because of the potential for new drug discovery and development. Several studies have found a protective effect of selective inhibitors of exportin-1 in cellular and animal models of ALS (Chou et al., 2018; Giampetruzzi et al., 2019; Zhang et al., 2015). This class of selective inhibitors of nuclear transport (SINE) compounds have originally been developed as potential therapeutics for cancer, where exportin-1 upregulation is commonly found and associated with poor prognosis (Jans et al., 2019; Wang and Liu, 2019). While it has been speculated that in the context of neurodegenerative disease models these compounds may prevent nuclear export of TDP-43, further studies have shown that TDP-43 may not be actively exported via exportin-1 (Archbold et al., 2018; Ederle et al., 2018; Pinarbasi et al., 2018). Different hypotheses have been put forward to explain the efficacy of SINE compounds in preclinical ALS models. These inhibitors might act indirectly, by inhibiting export of other nuclear proteins. They may also prevent formation of SGs, reduce neuroinflammation, or counteract defects in protein import and re-establish a balance between these processes. Regardless of the exact mechanism, preclinical trials testing the efficacy of these compounds are ongoing, and more strategies are being developed as new promising therapies for the treatment of ALS/FTD and related disorders.
Highlights.
Nucleocytoplasmic transport (NCT) defects are a common feature of sporadic and familiar forms of ALS/FTD
Protein aggregates sequester nucleoporins and transport factors, driving cytoplasmic mislocalization and aggregation in a positive feedback loop.
Protein sequestration, cytoskeletal dysfunction, and stress response are major drivers of NCT pathology.
NCT modulators appear as promising targets for therapeutic development.
Acknowledgments
Funding
This work was supported by the ALS Association (18-IIA-418 to C.F.) and National Institutes of Health/NINDS (R01 NS91749 to W.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
None.
Bibliography
- Aizawa H, Yamashita T, Kato H, Kimura T, Kwak S, 2019. Impaired Nucleoporins Are Present in Sporadic Amyotrophic Lateral Sclerosis Motor Neurons that Exhibit Mislocalization of the 43-kDa TAR DNA-Binding Protein. J. Clin. Neurol 15, 62–67. doi: 10.3988/jcn.2019.15.1.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramburu IV, Lemke EA, 2017. Floppy but not sloppy: Interaction mechanism of FG-nucleoporins and nuclear transport receptors. Semin. Cell Dev. Biol 68, 34–41. doi: 10.1016/j.semcdb.2017.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archbold HC, Jackson KL, Arora A, Weskamp K, Tank EM-H, Li X, Miguez R, Dayton RD, Tamir S, Klein RL, Barmada SJ, 2018. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Rep 8, 4606. doi: 10.1038/s41598-018-22858-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskaran P, Shaw C, Guthrie S, 2018. TDP-43 causes neurotoxicity and cytoskeletal dysfunction in primary cortical neurons. PLoS One 13, e0196528. doi: 10.1371/journal.pone.0196528 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bellouze S, Baillat G, Buttigieg D, de la Grange P, Rabouille C, Haase G, 2016. Stathmin 1/2-triggered microtubule loss mediates Golgi fragmentation in mutant SOD1 motor neurons. Mol. Neurodegener 11, 43. doi: 10.1186/s13024-016-0111-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokhuis AM, Groen EJN, Koppers M, van den Berg LH, Pasterkamp RJ, 2013. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794. doi: 10.1007/s00401-013-1125-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Kovacs D, Konijnenberg A, Timmerman E, Volkov A, Guharoy M, De Decker M, Jaspers T, Ryan VH, Janke AM, Baatsen P, Vercruysse T, Kolaitis R-M, Daelemans D, Taylor JP, Kedersha N, Anderson P, Impens F, Sobott F, Schymkowitz J, Rousseau F, Fawzi NL, Robberecht W, Van Damme P, Tompa P, Van Den Bosch L, 2017. Phase separation of c9orf72 dipeptide repeats perturbs stress granule dynamics. Mol. Cell 65, 1044–1055.e5. doi: 10.1016/j.molcel.2017.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, De Baets G, Scheveneels W, Steyaert J, Cuijt I, Verstrepen KJ, Callaerts P, Rousseau F, Schymkowitz J, Cruts M, Van Broeckhoven C, Van Damme P, Gitler AD, Robberecht W, Van Den Bosch L, 2016. Drosophila screen connects nuclear transport genes to DPR pathology in C9ALS/FTD. Sci. Rep 6, 20877. doi: 10.1038/srep20877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi B, Faure AJ, Seuma M, Schmiedel JM, Tartaglia GG, Lehner B, 2019. The mutational landscape of a prion-like domain. Nat. Commun 10, 4162. doi: 10.1038/s41467-019-12101-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Sapp P, McKenna-Yasek D, Brown RH, Hayward LJ, 2010. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet 19, 4160–4175. doi: 10.1093/hmg/ddq335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzo F, Mirra A, Carrì MT, 2017. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neurosci. Lett 636, 3–8. doi: 10.1016/j.neulet.2016.04.065 [DOI] [PubMed] [Google Scholar]
- Breitsprecher D, Goode BL, 2013. Formins at a glance. J. Cell Sci 126, 1–7. doi: 10.1242/jcs.107250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelstaff J, Lashley T, Holton JL, Lees AJ, Rossor MN, Bandopadhyay R, Revesz T, 2011. Transportin1: a marker of FTLD-FUS. Acta Neuropathol. 122, 591–600. doi: 10.1007/s00401-011-0863-6 [DOI] [PubMed] [Google Scholar]
- Burk K, Pasterkamp RJ, 2019. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 137, 859–877. doi: 10.1007/s00401-019-01964-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke B, 2019. Chain reaction: LINC complexes and nuclear positioning. [version 1; peer review: 3 approved]. F1000Res. 8. doi: 10.12688/f1000research.16877.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai N, Gitler AD, 2018. Yeast screen for modifiers of C9orf72 poly(glycine-arginine) dipeptide repeat toxicity. FEMS Yeast Res 18. doi: 10.1093/femsyr/foy024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W-L, Tarn W-Y, 2009. A role for transports in deposition of TTP to cytoplasmic RNA granules and mRNA decay. Nucleic Acids Res. 37, 6600–6612. doi: 10.1093/nar/gkp717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H, Blackbourn LW, Huang C-L, Errigo A, Yin Y, Lu J, Ayala M, Zhang S-C, 2014. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 14, 796–809. doi: 10.1016/j.stem.2014.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew J, Cook C, Gendron TF, Jansen-West K, Del Rosso G, Daughrity LM, Castanedes-Casey M, Kurti A, Stankowski JN, Disney MD, Rothstein JD, Dickson DW, Fryer JD, Zhang Y-J, Petrucelli L, 2019. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol. Neurodegener 14, 9. doi: 10.1186/s13024-019-0310-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C-C, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin-Asp PG, Chen YH, Duong DM, Seyfried NT, Powers MA, Kukar T, Hales CM, Gearing M, Cairns NJ, Boylan KB, Dickson DW, Rademakers R, Zhang Y-J, Petrucelli L, Sattler R, Zarnescu DC, Glass JD, Rossoll W, 2018. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci 21, 228–239. doi: 10.1038/s41593-017-0047-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Petrucelli L, 2019. Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron 101, 1057–1069. doi: 10.1016/j.neuron.2019.02.032 [DOI] [PubMed] [Google Scholar]
- Cornelison GL, Levy SA, Jenson T, Frost B, 2019. Tau-induced nuclear envelope invagination causes a toxic accumulation of mRNA in Drosophila. Aging Cell 18, e12847. doi: 10.1111/acel.12847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crampton N, Kodiha M, Shrivastava S, Umar R, Stochaj U, 2009. Oxidative stress inhibits nuclear protein export by multiple mechanisms that target FG nucleoporins and Crm1. Mol. Biol. Cell 20, 5106–5116. doi: 10.1091/mbe.E09-05-0397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo MA, Raices M, Panowski SH, Hetzer MW, 2009. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 136, 284–295. doi: 10.1016/j.cell.2008.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw R, Gruenbaum Y, Medalia O, 2018. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 28, 34–45. doi: 10.1016/j.tcb.2017.08.004 [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung G-YR, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R, 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang P-W, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD, 2013. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. doi: 10.1016/j.neuron.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, Kremmer E, Ansorge O, Mackenzie IRA, Neumann M, Haass C, 2012. Arginine methylation next to the PY-NLS modulates Transports binding and nuclear import of FUS. EMBO J. 31, 4258–4275. doi: 10.1038/emboj.2012.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ederle H, Funk C, Abou-Ajram C, Hutten S, Funk EBE, Kehlenbach RH, Bailer SM, Dormann D, 2018. Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1. Sci. Rep 8, 7084. doi: 10.1038/s41598-018-25007-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ, Dujardin S, Amaral AS, Grima JC, Bennett RE, Tepper K, DeTure M, Vanderburg CR, Corjuc BT, DeVos SL, Gonzalez JA, Chew J, Vidensky S, Gage FH, Mertens J, Troncoso J, Mandelkow E, Salvatella X, Lim RYH, Petrucelli L, Wegmann S, Rothstein JD, Hyman BT, 2018. Tau protein disrupts nucleocytoplasmic transport in alzheimer’s disease. Neuron 99, 925–940. e7. doi: 10.1016/j.neuron.2018.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rüb U, Auburger G, Trojanowski JQ, Lee VM-Y, Van Deerlin VM, Bonini NM, Gitler AD, 2010. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. doi: 10.1038/nature09320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, Rossoll W, 2012. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet 21, 3703–3718. doi: 10.1093/hmg/dds205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanara P, Banerjee J, Hueck RV, Harper MR, Awada M, Turner H, Husted KH, Brandt R, Hellerstein MK, 2007. Stabilization of hyperdynamic microtubules is neuroprotective in amyotrophic lateral sclerosis. J. Biol. Chem 282, 23465–23472. doi: 10.1074/jbc.M703434200 [DOI] [PubMed] [Google Scholar]
- Fiserova J, Goldberg MW, 2010. Relationships at the nuclear envelope: lam ins and nuclear pore complexes in animals and plants. Biochem. Soc. Trans 38, 829–831. doi: 10.1042/BST0380829 [DOI] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee K-H, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao F-B, Taylor JP, 2015. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. doi: 10.1038/nature14974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Bardai FH, Feany MB, 2016. Lamin dysfunction mediates neurodegeneration in tauopathies. Curr. Biol 26, 129–136. doi: 10.1016/j.cub.2015.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura K, Suzuki T, Yasuda Y, Murata M, Katahira J, Yoneda Y, 2010. Identification of importin alphal as a novel constituent of RNA stress granules. Biochim. Biophys. Acta 1803, 865–871. doi: 10.1016/j.bbamcr.2010.03.020 [DOI] [PubMed] [Google Scholar]
- Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, Shorter J, Da Cruz S, Cleveland DW, 2019Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 102, 339–357. e7. doi: 10.1016/j.neuron.2019.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Petrucelli L, 2018. Disease mechanisms of C9ORF72 repeat expansions. Cold Spring Harb. Perspect. Med 8. doi: 10.1101/cshperspect.a024224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampetruzzi A, Danielson EW, Gumina V, Jeon M, Boopathy S, Brown RH, Ratti A, Landers JE, Fallini C, 2019. Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat. Commun 10, 3827. doi: 10.1038/s41467-019-11837-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpel P, Lee YL, Sobota RM, Calvi A, Koullourou V, Patel R, Mamchaoui K, Nédélec F, Shackleton S, Schmoranzer J, Burke B, Cadot B, Gomes ER, 2017. Nesprin-1α-Dependent Microtubule Nucleation from the Nuclear Envelope via Akap450 Is Necessary for Nuclear Positioning in Muscle Cells. Curr. Biol 27, 2999–3009. e9. doi: 10.1016/j.cub.2017.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittings LM, Foti SC, Benson BC, Gami-Patel P, Isaacs AM, Lashley T, 2019. Heterogeneous nuclear ribonucleoproteins R and Q accumulate in pathological inclusions in FTLD-FUS. Acta Neuropathol. Commun 7, 18. doi: 10.1186/s40478-019-0673-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MW, 2017. Nuclear pore complex tethers to the cytoskeleton. Semin. Cell Dev. Biol 68, 52–58. doi: 10.1016/j.semcdb.2017.06.017 [DOI] [PubMed] [Google Scholar]
- Gros-Louis F, Dupré N, Dion P, Fox MA, Laurent S, Verreault S, Sanes JR, Bouchard J-P, Rouleau GA, 2007. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat. Genet 39, 80–85. doi: 10.1038/ng1927 [DOI] [PubMed] [Google Scholar]
- Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O’Donovan K, Fare CM, Diaz Z, Singh N, Zhang ZC, Coughlin M, Sweeny EA, DeSantis ME, Jackrel ME, Rodell CB, Burdick JA, King OD, Gitler AD, Lagier-Tourenne C, Pandey UB, Chook YM, Taylor JP, Shorter J, 2018. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 173, 677–692. e20. doi: 10.1016/j.cell.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampoelz B, Andres-Pons A, Kastritis P, Beck M, 2019. Structure and assembly of the nuclear pore complex. Annu. Rev. Biophys 48, 515–536. doi: 10.1146/annurev-biophys-052118-115308 [DOI] [PubMed] [Google Scholar]
- Hans F, Glasebach H, Kahle PJ, 2019. Multiple distinct pathways lead to hyperubiquitylated insoluble TDP-43 protein independent of its translocation into stress granules. J. Biol. Chem doi: 10.1074/jbc.RA119.010617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayama R, Rout MP, Fernandez-Martinez J, 2017. The nuclear pore complex core scaffold and permeability barrier: variations of a common theme. Curr. Opin. Cell Biol 46, 110–118. doi: 10.1016/j.ceb.2017.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensel N, Claus P, 2018. The actin cytoskeleton in SMA and ALS: how does it contribute to motoneuron degeneration? Neuroscientist 24, 54–72. doi: 10.1177/1073858417705059 [DOI] [PubMed] [Google Scholar]
- Hergesheimer RC, Chami AA, de Assis DR, Vourc’h P, Andres CR, Corcia P, Lanznaster D, Blasco H, 2019. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: a resolution in sight? Brain 142, 1176–1194. doi: 10.1093/brain/awz078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzer MW, 2010. The role of the nuclear pore complex in aging of post-mitotic cells. Aging (Albany, NY) 2, 74–75. doi: 10.18632/aging.100125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieda M, 2017. Implications for diverse functions of the LINC complexes based on the structure. Cells 6. doi: 10.3390/cells6010003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofweber M, Hutten S, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, Ruepp M-D, Simons M, Niessing D, Madl T, Dormann D, 2018. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 173, 706–719. e13. doi: 10.1016/j.cell.2018.03.004 [DOI] [PubMed] [Google Scholar]
- Hülsmann BB, Labokha AA, Görlich D, 2012. The permeability of reconstituted nuclear pores provides direct evidence for the selective phase model. Cell 150, 738–751. doi: 10.1016/j.cell.2012.07.019 [DOI] [PubMed] [Google Scholar]
- Hutten S, Dormann D, 2019. Nucleocytoplasmic transport defects in neurodegeneration - Cause or consequence? Semin. Cell Dev. Biol doi: 10.1016/j.semcdb.2019.05.020 [DOI] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, McCluskey LF, Trojanowski JQ, Lee VM-Y, 2008. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol 173, 182–194. doi: 10.2353/ajpath.2008.080003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong C-X, 2016. Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol 12, 15–27. doi: 10.1038/nrneurol.2015.225 [DOI] [PubMed] [Google Scholar]
- Ito D, Hatano M, Suzuki N, 2017. RNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration. Sci. Transl. Med 9. doi: 10.1126/scitranslmed.aah5436 [DOI] [PubMed] [Google Scholar]
- Jahed Z, Soheilypour M, Peyro M, Mofrad MRK, 2016. The LINC and NPC relationship - it’s complicated! J. Cell Sci 129, 3219–3229. doi: 10.1242/jcs.184184 [DOI] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, Parker R, 2016. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 164, 487–498. doi: 10.1016/j.cell.2015.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäkel S, Mingot J-M, Schwarzmaier P, Hartmann E, Görlich D, 2002. Importins fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed basic domains. EMBO J. 21, 377–386. doi: 10.1093/emboj/21.3.377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamali T, Jamali Y, Mehrbod M, Mofrad MRK, 2011. Nuclear pore complex: biochemistry and biophysics of nucleocytoplasmic transport in health and disease. Int. Rev. Cell Mol. Biol 287, 233–286. doi: 10.1016/B978-0-12-386043-9.00006-2 [DOI] [PubMed] [Google Scholar]
- Jans DA, Martin AJ, Wagstaff KM, 2019. Inhibitors of nuclear transport. Curr. Opin. Cell Biol 58, 50–60. doi: 10.1016/j.ceb.2019.01.001 [DOI] [PubMed] [Google Scholar]
- Jovičić A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD, 2015. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci 18, 1226–1229. doi: 10.1038/nn.4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneb HM, Folkmann AW, Belzil VV, Jao L-E, Leblond CS, Girard SL, Daoud H, Noreau A, Rochefort D, Hince P, Szuto A, Levert A, Vidal S, André-Guimont C, Camu W, Bouchard J-P, Dupré N, Rouleau GA, Wente SR, Dion PA, 2015. Deleterious mutations in the essential m RNA metabolism factor, hGlel, in amyotrophic lateral sclerosis. Hum. Mol. Genet 24, 1363–1373. doi: 10.1093/hmg/ddu545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellos G, Zhou J, Patel H, Ridgway RA, Huels D, Gurniak CB, Sandilands E, Carragher NO, Sansom OJ, Witke W, Brunton VG, Frame MC, 2015. ADF and cofilin1 control actin stress fibers, nuclear integrity, and cell survival. Cell Rep. 13, 1949–1964. doi: 10.1016/j.celrep.2015.10.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL, 2012. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767. doi: 10.1016/j.cell.2012.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalfallah Y, Kuta R, Grasmuck C, Prat A, Durham HD, Vande Velde C, 2018. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci. Rep 8, 7551. doi: 10.1038/S41598-018-25767-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi B, Hartmann H, May S, Möhl C, Ederle H, Michaelsen M, Schludi MH, Dormann D, Edbauer D, 2017. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum. Mol. Genet 26, 790–800. doi: 10.1093/hmg/ddw432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Taylor JP, 2017. Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 96, 285–297. doi: 10.1016/j.neuron.2017.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Zhan L, Hanson KA, Tibbetts RS, 2012. High-content RNA screening identifies the Type 1 inositol triphosphate receptor as a modifier of TDP-43 localization and neurotoxicity. Hum. Mol. Genet 21, 4845–4856. doi: 10.1093/hmg/dds321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita Y, Ito H, Hirano A, Fujita K, Wate R, Nakamura M, Kaneko S, Nakano S, Kusaka H, 2009. Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol 68, 1184–1192. doi: 10.1097/NEN.0b013e3181bc3bec [DOI] [PubMed] [Google Scholar]
- Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, Moccia R, Cassel SH, Chen K, Wainger BJ, Woolf CJ, Eggan K, 2019. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci 22, 167–179. doi: 10.1038/s41593-018-0300-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodiha M, Chu A, Matusiewicz N, Stochaj U, 2004. Multiple mechanisms promote the inhibition of classical nuclear import upon exposure to severe oxidative stress. Cell Death Differ. 11, 862–874. doi: 10.1038/sj.cdd.4401432 [DOI] [PubMed] [Google Scholar]
- Kodiha M, Tran D, Qian C, Morogan A, Presley JF, Brown CM, Stochaj U, 2008. Oxidative stress mislocalizes and retains transport factor importin-alpha and nucleoporins Nup153 and Nup88 in nuclei where they generate high molecular mass complexes. Biochim. Biophys. Acta 1783, 405–418. doi: 10.1016/j.bbamcr.2007.10.022 [DOI] [PubMed] [Google Scholar]
- Kounakis K, Tavernarakis N, 2019. The cytoskeleton as a modulator of aging and neurodegeneration. Adv. Exp. Med. Biol 1178, 227–245. doi: 10.1007/978-3-030-25650-0_12 [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW, 2009. Rethinking ALS: the FUS about TDP-43. Cell 136, 1001–1004. doi: 10.1016/j.cell.2009.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattante S, Ciura S, Rouleau GA, Kabashi E, 2015. Defining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD). Trends Genet. 31, 263–273. doi: 10.1016/j.tig.2015.03.005 [DOI] [PubMed] [Google Scholar]
- Lee K-H, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A, Maxwell BA, Kim NC, Temirov J, Moore J, Kolaitis R-M, Shaw TI, Bai B, Peng J, Kriwacki RW, Taylor JP, 2016. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 167, 774–788. e17. doi: 10.1016/j.cell.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke EA, 2016. The multiple faces of disordered nucleoporins. J. Mol. Biol 428, 2011–2024. doi: 10.1016/j.jmb.2016.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Goryaynov A, Yang W, 2016. The selective permeability barrier in the nuclear pore complex. Nucleus 7, 430–446. doi: 10.1080/19491034.2016.1238997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Currie SL, Rosen MK, 2017. Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J. Biol. Chem 292, 19110–19120. doi: 10.1074/jbc.M117.800466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S-C, Polymenidou M, Cleveland DW, 2013. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, Matchett BJ, Mittag T, Temirov J, Hsiung G-YR, Krieger C, Murray ME, Kato M, Fryer JD, Petrucelli L, Zinman L, Weintraub S, Mesulam M, Keith J, Zivkovic SA, Hirsch-Reinshagen V, Roos RP, Züchner S, Graff-Radford NR, Petersen RC, Caselli RJ, Wszolek ZK, Finger E, Lippa C, Lacomis D, Stewart H, Dickson DW, Kim HJ, Rogaeva E, Bigio E, Boylan KB, Taylor JP, Rademakers R, 2017. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95, 808–816. e9. doi: 10.1016/j.neuron.2017.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima K, lino H, Hihara S, Funakoshi T, Watanabe A, Nishimura M, Nakatomi R, Yahata K, Imamoto F, Hashikawa T, Yokota H, Imamoto N, 2010. Nuclear pore formation but not nuclear growth is governed by cyclin-dependent kinases (Cdks) during interphase. Nat. Struct. Mol. Biol 17, 1065–1071. doi: 10.1038/nsmb.1878 [DOI] [PubMed] [Google Scholar]
- Mahboubi H, Seganathy E, Kong D, Stochaj U, 2013. Identification of novel stress granule components that are involved in nuclear transport. PLoS One 8, e68356. doi: 10.1371/journal.pone.0068356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrioli J, Mediani L, Alberti S, Carra S, 2019. ALS and FTD: Where RNA metabolism meets protein quality control. Semin. Cell Dev. Biol doi: 10.1016/j.semcdb.2019.06.003 [DOI] [PubMed] [Google Scholar]
- Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis-Marks MR, Needham PG, Copley KE, Hurtle B, Portz B, Pyles NJ, Guo L, Calder CB, Wills ZP, Pandey UB, Kofler JK, Brodsky JL, Thathiah A, Shorter J, Donnelly CJ, 2019. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 102, 321–338. e8. doi: 10.1016/j.neuron.2019.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo E-C, Krach F, Yang D, Sen A, Fulzele A, Wozniak JM, Gonzalez DJ, Kankel MW, Gao F-B, Bennett EJ, Lécuyer E, Yeo GW, 2018. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 172, 590–604. e13. doi: 10.1016/j.cell.2017.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FJ, Pratt GA, Van Nostrand EL, Batra R, Huelga SC, Kapeli K, Freese P, Chun SJ, Ling K , Gelboin-Burkhart C, Fijany L, Wang HC, Nussbacher JK, Broski SM, Kim HJ, Lardelli R, Sundararaman B, Donohue JP, Javaherian A, Lykke-Andersen J, Finkbeiner S, Bennett CF, Ares M, Burge CB, Taylor JP, Rigo F, Yeo GW, 2016. Protein-RNA Networks Regulated by Normal and ALS-Associated Mutant HNRNPA2B1 in the Nervous System. Neuron 92, 780–795. doi: 10.1016/j.neuron.2016.09.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura Y, 2016. Mechanistic Insights from Structural Analyses of Ran-GTPase-Driven Nuclear Export of Proteins and RNAs. J. Mol. Biol 428, 2025–2039. doi: 10.1016/j.jmb.2015.09.025 [DOI] [PubMed] [Google Scholar]
- Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, Ohkubo T, Rodriguez M, Lin N, Wu D, Bennett CF, Rigo F, Da Cruz S, Ravits J, Lagier-Tourenne C, Cleveland DW, 2019. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci 22, 180–190. doi: 10.1038/s41593-018-0293-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J, Paquola ACM, Ku M, Hatch E, Böhnke L, Ladjevardi S, McGrath S, Campbell B, Lee H, Herdy JR, Gonçalves JT, Toda T, Kim Y, Winkler J, Yao J, Hetzer MW, Gage FH, 2015. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 17, 705–718. doi: 10.1016/j.stem.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaleva S, Lemke EA, 2018. Beyond the Transport Function of Import Receptors: What’s All the FUS about? Cell 173, 549–553. doi: 10.1016/j.cell.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milles S, Huy Bui K, Koehler C, Eltsov M, Beck M, Lemke EA, 2013. Facilitated aggregation of FG nucleoporins under molecular crowding conditions. EMBO Rep. 14, 178–183. doi: 10.1038/embor.2012.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y, Saiwaki T, Yamashita J, Yasuda Y, Kotera I, Shibata S, Shigeta M, Hiraoka Y, Haraguchi T, Yoneda Y, 2004. Cellular stresses induce the nuclear accumulation of importin alpha and cause a conventional nuclear import block. J. Cell Biol 165, 617–623. doi: 10.1083/jcb.200312008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Qamar S, Lin JQ, Schierle GSK, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FTS, Michel CH, Kronenberg-Versteeg D, Li Y, Yang S-P, Wakutani Y, Meadows W, Ferry RR, Dong L, Tartaglia GG, Favrin G, Lin W-L, Dickson DW, Zhen M, Ron D, Schmitt-Ulms G, Fraser PE, Shneider NA, Holt C, Vendruscolo M, Kaminski CF, St George-Hyslop P, 2015. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 88, 678–690. doi: 10.1016/j.neuron.2015.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagara Y, Tateishi T, Yamasaki R, Hayashi S, Kawamura M, Kikuchi H, linuma KM, Tanaka M, Iwaki T, Matsushita T, Ohyagi Y, Kira J, 2013. Impaired cytoplasmic-nuclear transport of hypoxia-inducible factor-1α in amyotrophic lateral sclerosis. Brain Pathol. 23, 534–546. doi: 10.1111/bpa.12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM-Y, 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- Neumann M, Valori CF, Ansorge O, Kretzschmar HA, Munoz DG, Kusaka H, Yokota O, Ishihara K, Ang L-C, Bilbao JM, Mackenzie IRA, 2012. Transports 1 accumulates specifically with FET proteins but no other transports cargos in FTLD-FUS and is absent in FUS inclusions in ALS with FUS mutations. Acta Neuropathol. 124, 705–716. doi: 10.1007/s00401-012-1020-6 [DOI] [PubMed] [Google Scholar]
- Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, et al. , 2018. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283. e6. doi: 10.1016/j.neuron.2018.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, Gallo J-M, Hortobágyi T, Shaw CE, Rogelj B, 2010. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain 133, 1763–1771. doi: 10.1093/brain/awq111 [DOI] [PubMed] [Google Scholar]
- Nousiainen HO, Kestilä M, Pakkasjärvi N, Honkala H, Kuure S, Tallila J, Vuopala K, Ignatius J, Herva R, Peltonen L, 2008. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat. Genet 40, 155–157. doi: 10.1038/ng.2007.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny K, Jung T, Grune T, Höhn A, 2014. Accumulation of modified proteins and aggregate formation in aging. Exp. Gerontol 57, 122–131. doi: 10.1016/j.exger.2014.05.016 [DOI] [PubMed] [Google Scholar]
- Oberstadt M, Claßen J, Arendt T, Holzer M, 2018. TDP-43 and Cytoskeletal Proteins in ALS. Mol. Neurobiol 55, 3143–3151. doi: 10.1007/s12035-017-0543-1 [DOI] [PubMed] [Google Scholar]
- Onischenko E, Weis K, 2011. Nuclear pore complex-a coat specifically tailored for the nuclear envelope. Curr. Opin. Cell Biol 23, 293–301. doi: 10.1016/j.ceb.2011.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski LA, Hall AC, Mekhail K, 2017. Ataxin-2: From RNA Control to Human Health and Disease. Genes (Basel) 8. doi: 10.3390/genes8060157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padavannil A, Sarkar P, Kim SJ, Cagatay T, Jiou J, Brautigam CA, Tomchick DR, Sali A, D’Arcy S, Chook YM, 2019. Importin-9 wraps around the H2A-H2B core to act as nuclear importer and histone chaperone. Elite 8. doi: 10.7554/eLife.43630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paonessa F, Evans LD, Solanki R, Larrieu D, Wray S, Hardy J, Jackson SP, Livesey FJ, 2019. Microtubules Deform the Nuclear Membrane and Disrupt Nucleocytoplasmic Transport in Tau-Mediated Frontotemporal Dementia. Cell Rep. 26, 582–593. e5. doi: 10.1016/j.celrep.2018.12.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, Maghelli N, Royer LA, Weigert M, Myers EW, Grill S, Drechsel D, Hyman AA, Alberti S, 2015. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 162, 1066–1077. doi: 10.1016/j.cell.2015.07.047 [DOI] [PubMed] [Google Scholar]
- Pinarbasi ES, Cağatay T, Fung HYJ, Li YC, Chook YM, Thomas PJ, 2018. Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci. Rep 8, 7083. doi: 10.1038/S41598-018-25008-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol G, Söderqvist H, Radu A, 2002. Age-associated reduction of nuclear protein import in human fibroblasts. Biochem. Biophys. Res. Commun 294, 354–358. doi: 10.1016/S0006-291X(02)00492-8 [DOI] [PubMed] [Google Scholar]
- Qamar S, Wang G, Randle SJ, Ruggeri FS, Varela JA, Lin JQ, Phillips EC, Miyashita A, Williams D, Ströhl F, Meadows W, Ferry R, Dardov VJ, Tartaglia GG, Farrer LA, Kaminski Schierle GS, Kaminski CF, Holt CE, Fraser PE, Schmitt-Ulms G, Klenerman D, Knowles T, Vendruscolo M, St George-Hyslop P, 2018. FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-π Interactions. Cell 173, 720–734. e15. doi: 10.1016/j.cell.2018.03.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh N, Pandey UB, 2017. Autophagy dysregulation in ALS: when protein aggregates get out of hand. Front. Mol. Neurosci 10, 263. doi: 10.3389/fnmol.2017.00263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, et al. , 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez CM, Todd PK, 2019. New pathologic mechanisms in nucleotide repeat expansion disorders. Neurobiol. Dis 130, 104515. doi: 10.1016/j.nbd.2019.104515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, Field MC, 2017. The evolution of organellar coat complexes and organization of the eukaryotic cell. Annu. Rev. Biochem 86, 637–657. doi: 10.1146/annurev-biochem-061516-044643 [DOI] [PubMed] [Google Scholar]
- Sama RRK, Fallini C, Gatto R, McKeon JE, Song Y, Rotunno MS, Penaranda S, Abdurakhmanov I, Landers JE, Morfini G, Brady ST, Bosco DA, 2017. ALS-linked FUS exerts a gain of toxic function involving aberrant p38 MAPK activation. Sci. Rep 7, 115. doi: 10.1038/s41598-017-00091-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas JN, Toyama BH, Xu T, Yates JR, Hetzer MW, 2012. Extremely long-lived nuclear pore proteins in the rat brain. Science 335, 942. doi: 10.1126/science.1217421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HB, Görlich D, 2016. Transport selectivity of nuclear pores, phase separation, and membraneless organelles. Trends Biochem. Sci 41, 46–61. doi: 10.1016/j.tibs.2015.11.001 [DOI] [PubMed] [Google Scholar]
- Schreij AMA, Fon EA, McPherson PS, 2016. Endocytic membrane trafficking and neurodegenerative disease. Cell Mol. Life Sci 73, 1529–1545. doi: 10.1007/s00018-015-2105-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Wang X, Podell ER, Cech TR, 2013. RNA seeds higher-order assembly of FUS protein. Cell Rep. 5, 918–925. doi: 10.1016/j.celrep.2013.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Travesa A, Martell SW, Forbes DJ, 2015. Analysis of the initiation of nuclear pore assembly by ectopically targeting nucleoporins to chromatin. Nucleus 6, 40–54. doi: 10.1080/19491034.2015.1004260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM, Roth FP, Herz J, Peng J, Moore MJ, Yu G, 2011. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem 286, 1204–1215. doi: 10.1074/jbc.M110.190884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J, Yamashita T, Nakano Y, Morihara R, Li X, Feng T, Liu X, Huang Y, Fukui Y, Hishikawa N, Ohta Y, Abe K, 2017. Aberrant distributions of nuclear pore complex proteins in ALS mice and ALS patients. Neuroscience 350, 158–168. doi: 10.1016/j.neuroscience.2017.03.024 [DOI] [PubMed] [Google Scholar]
- Shang Y, Huang EJ, 2016. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 1647, 65–78. doi: 10.1016/j.brainres.2016.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi KY, Mori E, Nizami ZF, Lin Y, Kato M, Xiang S, Wu LC, Ding M, Yu Y, Gall JG, McKnight SL, 2017. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. USA 114, E1111–E1117. doi: 10.1073/pnas.1620293114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Kukreti R, Saso L, Kukreti S, 2019. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 24. doi: 10.3390/molecules24081583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivadasan R, Hornburg D, Drepper C, Frank N, Jablonka S, Hansel A, Lojewski X, Sterneckert J, Hermann A, Shaw PJ, Ince PG, Mann M, Meissner F, Sendtner M, 2016. C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat. Neurosci 19, 1610–1618. doi: 10.1038/nn.4407 [DOI] [PubMed] [Google Scholar]
- Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, et al. , 2014. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. doi: 10.1016/j.neuron.2014.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DA, Stepto A, Au WH, Adachi Y, Diaper DC, Hall R, Rekhi A, Boudi A, Tziortzouda P, Lee Y-B, Smith B, Bridi JC, Spinelli G, Dearlove J, Humphrey DM, Gallo J-M, Troakes C, Fanto M, Soller M, Rogelj B, Parsons RB, Shaw CE, Hortobágyi T, Hirth F, 2018. A feedback loop between dipeptide-repeat protein, TDP-43and karyopherin-α mediates C9orf72-related neurodegeneration. Brain 141, 2908–2924. doi: 10.1093/brain/awy241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soniat M, Chook YM, 2015. Nuclear localization signals for four distinct karyopherin-β nuclear import systems. Biochem. J 468, 353–362. doi: 10.1042/BJ20150368 [DOI] [PubMed] [Google Scholar]
- Steggerda SM, Paschal BM, 2002. Regulation of nuclear import and export by the GTPase Ran. Int Rev Cytol 217, 41–91. doi: 10.1016/S0074-7696(02)17012-4 [DOI] [PubMed] [Google Scholar]
- Steyaert J, Scheveneels W, Vanneste J, Van Damme P, Robberecht W, Callaerts P, Bogaert E, Van Den Bosch L, 2018. FUS-induced neurotoxicity in Drosophila is prevented by downregulating nucleocytoplasmic transport proteins. Hum. Mol. Genet 27, 4103–4116. doi: 10.1093/hmg/ddy303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stochaj U, Rassadi R, Chiu J, 2000. Stress-mediated inhibition of the classical nuclear protein import pathway and nuclear accumulation of the small GTPase Gsplp. FASEB J. 14, 2130–2132. doi: 10.1096/fj.99-0751fje [DOI] [PubMed] [Google Scholar]
- Stoffler D, Feja B, Fahrenkrog B, Walz J, Typke D, Aebi U, 2003. Cryo-electron tomography provides novel insights into nuclear pore architecture: implications for nucleocytoplasmic transport. J. Mol. Biol 328, 119–130. doi: 10.1016/s0022-2836(03)00266-3 [DOI] [PubMed] [Google Scholar]
- Suárez-Calvet M, Neumann M, Arzberger T, Abou-Ajram C, Funk E, Hartmann H, Edbauer D, Kremmer E, Göbi C, Resch M, Bourgeois B, Madl T, Reber S, Jutzi D, Ruepp M-D, Mackenzie IRA, Ansorge O, Dormann D, Haass C, 2016. Monomethylated and unmethylated FUS exhibit increased binding to Transportin and distinguish FTLD-FUS from ALS-FUS. Acta Neuropathol. 131, 587–604. doi: 10.1007/s00401-016-1544-2 [DOI] [PubMed] [Google Scholar]
- Sugiura T, Matsuda S, Kurosaka S, Nakai N, Fukumoto K, Takahashi T, Maruyama H, Imaizumi K, Matsumoto M, Takumi T, 2016. Translocated in liposarcoma regulates the distribution and function of mammalian enabled, a modulator of actin dynamics. FEBS J. 283, 1475–1487. doi: 10.1111/febs.13685 [DOI] [PubMed] [Google Scholar]
- Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, Gitler AD, 2011. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 9, e1000614. doi: 10.1371/journal.pbio.1000614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi R, Toyoshima Y, Tada M, Shiga A, Tanaka H, Shimohata M, Kimura K, Morita T, Kakita A, Nishizawa M, Takahashi H, 2013. Transportin 1 accumulates in FUS inclusions in adult-onset ALS without FUS mutation. Neuropathol Appl Neurobiol 39, 580–584. doi: 10.1111/nan.12022 [DOI] [PubMed] [Google Scholar]
- Talbot K, Ansorge O, 2006. Recent advances in the genetics of amyotrophic lateral sclerosis and frontotemporal dementia: common pathways in neurodegenerative disease. Hum. Mol. Genet 15 Spec No 2, R182–7. doi: 10.1093/hmg/ddl202 [DOI] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR, Hetzer MW, 2013. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154, 971–982. doi: 10.1016/j.cell.2013.07.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanneste J, Vercruysse T, Boeynaems S, Sicart A, Van Damme P, Daelemans D, Van Den Bosch L, 2019. C9orf72-generated poly-GR and poly-PR do not directly interfere with nucleocytoplasmic transport. Sci. Rep 9, 15728. doi: 10.1038/S41598-019-52035-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallar BJ, Alberts AS., 2003. The formins: active scaffolds that remodel the cytoskeleton. Trends Cell Biol. 13, 435–446. doi: 10.1016/S0962-8924(03)00153-3 [DOI] [PubMed] [Google Scholar]
- Wang AY, Liu H, 2019. The past, present, and future of CRM1/XP01 inhibitors. Stem Cell Investig. 6, 6. doi: 10.21037/sci.2019.02.03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J-Y, Yu I-S, Huang C-C, Chen C-Y, Wang W-P, Lin S-W, Jeang K-T, Chi Y-H, 2015. Sun1 deficiency leads to cerebellar ataxia in mice. Dis. Model. Mech 8, 957–967. doi: 10.1242/dmm.019240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weberruss M, Antonin W, 2016. Perforating the nuclear boundary - how nuclear pore complexes assemble. J. Cell Sci 129, 4439–4447. doi: 10.1242/jcs.194753 [DOI] [PubMed] [Google Scholar]
- Weishaupt JH, Hyman T, Dikic I, 2016. Common molecular pathways in amyotrophic lateral sclerosis and frontotemporal dementia. Trends Mol. Med 22, 769–783. doi: 10.1016/j.molmed.2016.07.005 [DOI] [PubMed] [Google Scholar]
- Wiggan O, Schroder B, Krapf D, Bamburg JR, DeLuca JG, 2017. Cofilin Regulates Nuclear Architecture through a Myosin-II Dependent Mechanotransduction Module. Sci. Rep 7, 40953. doi: 10.1038/srep40953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, Hipp MS, 2016. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA Science 351, 173–176. doi: 10.1126/science.aad2033 [DOI] [PubMed] [Google Scholar]
- Wu C, Jain MR, Li Q, Oka SI, Li W, Kong A-NT, Nagarajan N, Sadoshima J, Simmons WJ, Li H, 2014. Identification of novel nuclear targets of human thioredoxin 1. Mol. Cell Proteomics 13, 3507–3518. doi: 10.1074/mcp.M114.040931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-H, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM, Kost JE, Gonzalez-Perez P, Fox AD, Adams J, Taroni F, Tiloca C, Leclerc AL, Chafe SC, Mangroo D, Moore MJ, Zitzewitz JA, Xu Z-S, van den Berg LH, Glass JD, Siciliano G, Cirulli ET, Goldstein DB, Salachas F, Meininger V, Rossoll W, Ratti A, Gellera C, Bosco DA, Bassell GJ, Silani V, Drory VE, Brown RH, Landers JE, 2012. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. doi: 10.1038/nature11280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, Keith J, Zinman L, Rogaeva E, Robertson J, 2015. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann. Neurol 78, 568–583. doi: 10.1002/ana.24469 [DOI] [PubMed] [Google Scholar]
- Yamashita T, Akamatsu M, Kwak S, 2017. Altered Intracellular Milieu of ADAR2-Deficient Motor Neurons in Amyotrophic Lateral Sclerosis. Genes (Basel) 8. doi: 10.3390/genes8020060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Shen Y, Garre E, Hao X, Krumlinde D, Cvijović M, Arens C, Nyström T, Liu B, Sunnerhagen P, 2014. Stress granule-defective mutants deregulate stress responsive transcripts. PLoS Genet. 10, e1004763. doi: 10.1371/journal.pgen.1004763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Yiting, Qu R, Fan T, Zhu X, Feng Y, Yang Yuchao, Deng T, Peng Y, Huang W, Ouyang J, Dai J, 2018. Cross-talk between microtubules and the linker of nucleoskeleton complex plays a critical doi: 10.1186/s13287-018-0836-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Clatterbuck-Soper SF, Jackrel ME, Shorter J, Mili S, 2017. FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J. Cell Biol 216, 1015–1034. doi: 10.1083/jcb.201608022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa T, Ali R, Jiou J, Fung HYJ, Burke KA, Kim SJ, Lin Y, Peeples WB, Saltzberg D, Soniat M, Baumhardt JM, Oldenbourg R, Sali A, Fawzi NL, Rosen MK, Chook YM, 2018. Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell 173, 693–705. e22. doi: 10.1016/j.cell.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Daigle JG, Cunningham KM, Coyne AN, Ruan K, Grima JC, Bowen KE, Wadhwa H, Yang P, Rigo F, Taylor JP, Gitler AD, Rothstein JD, Lloyd TE, 2018. Stress granule assembly disrupts nucleocytoplasmic transport. Cell 173, 958–971. e17. doi: 10.1016/j.cell.2018.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu S-L, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD, 2015. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. doi: 10.1038/nature14973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Grima JC, Rothstein JD, Lloyd TE, 2016. Nucleocytoplasmic transport in C9orf72-mediated ALS/FTD. Nucleus 7, 132–137. doi: 10.1080/19491034.2016.1172152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y-J, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu Y-F, Katzman RB, Gass J, Murray ME, Shinohara M, Lin W-L, Garrett A, Stankowski JN, Daughrity L, Tong J, Perkerson EA, Yue M, Chew J, Castanedes-Casey M, Kurti A, Wang ZS, Liesinger AM, Baker JD, Jiang J, Lagier-Tourenne C, Edbauer D, Cleveland DW, Rademakers R, Boylan KB, Bu G, Link CD, Dickey CA, Rothstein JD, Dickson DW, Fryer JD, Petrucelli L, 2016. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci 19, 668–677. doi: 10.1038/nn.4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Beck MV, Griffith JD, Deshmukh M, Dokholyan NV, 2018. Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 115, 4661–4665. doi: 10.1073/pnas.1800187115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilman A, 2018. Aggregation, phase separation and spatial morphologies of the assemblies of FG nucleoporins. J. Mol. Biol 430, 4730–4740. doi: 10.1016/j.jmb.2018.07.011 [DOI] [PubMed] [Google Scholar]