

Summary
Many metabolic diseases disrupt endoplasmic reticulum (ER) homeostasis, but little is known about how metabolic activity is communicated to the ER. Here, we show in hepatocytes and other metabolically active cells that decreasing the availability of substrate for the tricarboxylic acid (TCA) cycle diminished NADPH production, elevated glutathione oxidation, led to altered oxidative maturation of ER client proteins, and attenuated ER stress. This attenuation was prevented when glutathione oxidation was disfavored. ER stress was also alleviated by inhibiting either TCA-dependent NADPH production or Glutathione Reductase. Conversely, stimulating TCA activity increased NADPH production, glutathione reduction, and ER stress. Validating these findings, deletion of the Mitochondrial Pyruvate Carrier—which is known to decrease TCA cycle activity and protect the liver from steatohepatitis—also diminished NADPH, elevated glutathione oxidation, and alleviated ER stress. Together, our results demonstrate a novel pathway by which mitochondrial metabolic activity is communicated to the ER through the relay of redox metabolites.
Subject Areas: biological sciences, cell biology, functional aspects of cell biology
Graphical Abstract
Highlights
-
•
Inhibiting nutrient catabolism alleviates ER stress in metabolically active cells
-
•
NADPH production and glutathione redox link TCA activity to ER homeostasis
-
•
ER client protein oxidation, maturation, and ERAD respond to metabolic activity
-
•
Cells lacking the Mitochondrial Pyruvate Carrier are resistant to ER stress
biological sciences; cell biology; functional aspects of cell biology
Introduction
As the gateway to the secretory pathway, the endoplasmic reticulum (ER) must properly synthesize and fold nascent secretory and membrane proteins. Many disease states, including obesity and its comorbidities, disrupt this process and cause ER stress (Mohan et al., 2019). Thus, it is important to understand how metabolic activity is communicated to the ER.
The ER and mitochondrial networks, although not connected to each other by secretory pathway traffic, are intertwined both physically and functionally. The ER makes close physical contacts with mitochondria to facilitate the exchange of metabolites (Raffaello et al., 2016, Vance, 2014, Yoboue et al., 2018). The ER also communicates with mitochondria via signaling from the unfolded protein response (UPR), which is activated by ER stress and signals through the three ER-resident stress sensors IRE1, PERK, and ATF6 (Walter and Ron, 2011). The UPR regulates mitochondrial activity at several levels, including enhancing mitochondrial protein quality control, augmenting ER-mitochondrial interactions and calcium signaling, and contributing to mitochondrial depolarization and initiation of apoptosis (Fan and Simmen, 2019, Gutierrez and Simmen, 2018, Rainbolt et al., 2014). By these pathways, the mitochondria can respond to alterations in ER homeostasis. However, the converse—how alterations in mitochondrial activity are transmitted to the ER—is less understood.
The impact of metabolic activity on ER homeostasis is most evident from the association of ER stress with obesity—particularly in highly metabolically active tissues such as the liver, pancreas, and adipose (Cnop et al., 2012). Lipotoxicity (i.e., damage caused by the inappropriate accumulation of lipids in non-adipose tissue), inflammation, and oxidative stress have all been shown to contribute to obesity-associated ER stress (Fu et al., 2012, Salvado et al., 2015). Yet, independent of diet content, feeding after a fast is sufficient to elicit ER stress in the liver (Gomez and Rutkowski, 2016, Oyadomari et al., 2008, Pfaffenbach et al., 2010) and to alter the extent of physical contacts between the ER and mitochondria (Theurey et al., 2016). These findings suggest that ER homeostasis is acutely and intrinsically connected with metabolism even apart from the problems brought on by obesity.
The tricarboxylic acid (TCA) cycle is the central hub of metabolism, participating in both catabolism and anabolism. Acetyl-CoA enters the cycle after either oxidative breakdown of fatty acyl-CoAs within the mitochondria or after conversion from pyruvate—generated from glycolysis in the cytosol—by the mitochondrial pyruvate dehydrogenase complex. Canonically, TCA activity yields NADH and FADH2 for the electron transport chain. However, the TCA cycle also provides precursors for biosynthetic pathways (Owen et al., 2002). In addition, the cycle can produce NADPH through the activity of isozymes that reside either in the mitochondria (isocitrate dehydrogenase [IDH] 2) or in the cytosol (IDH1 and malic enzyme/ME1) (Rydstrom, 2006). Therefore, activity of the TCA cycle is likely to influence cellular processes by mechanisms beyond the production of ATP from the electron transport chain.
NADPH is used as a cofactor by Glutathione Reductase to reduce oxidized glutathione (GSSG→2GSH), and likewise by thioredoxin reductase to reduce oxidized thioredoxin. Both these molecules contribute to defense against oxidative stress (Sies et al., 2017), and both have connections to ER protein biogenesis. Thioredoxin has been shown to be a source of electrons for reduction and isomerization of disulfide bonds of ER client proteins (Poet et al., 2017). The oxidized form of glutathione (GSSG) was formerly thought to reoxidize protein disulfide isomerase (PDI) in the ER lumen to promote ER disulfide bond formation. However, since the discovery of alternative pathways for PDI reoxidation (Frand and Kaiser, 1999, Tu et al., 2000, Zito et al., 2010b), the role of glutathione in ER homeostasis is now much less clear (Delaunay-Moisan et al., 2017, Tsunoda et al., 2014). Whether elevated GSSG might be beneficial to ER function under some cellular conditions but detrimental in others is also unknown.
Despite the centrality of the TCA cycle to cellular function and hints that its activity might be tied to ER stress (Mogilenko et al., 2019, Xin et al., 2018), a contribution to ER homeostasis has not been directly investigated. Here, we used primary hepatocytes and other metabolically active cell types to investigate the relationship between mitochondrial metabolic activity and ER stress. We show that TCA cycle activity links lipid and carbohydrate catabolism to ER homeostasis through production of NADPH and redox regulation of glutathione. Our findings delineate a novel pathway by which mitochondrial metabolic activity is sensed by the ER and suggest a role for GSSG in the process.
Results
Inhibition of β-Oxidation Alleviates ER Stress in Metabolically Active Cell Types
We have previously shown in the liver and in hepatoma cells in vitro that inhibiting β-oxidation diminishes ER stress signaling (Tyra et al., 2012). To identify the pathway by which β-oxidation and ER homeostasis are linked, we first asked whether etomoxir (ET), which blocks β-oxidation by inhibiting the CPT1-dependent transport of fatty acyl-CoAs into the mitochondria for oxidation (Weis et al., 1994, Yao et al., 2018), could diminish ER stress signaling in primary hepatocytes in vitro as it does in the liver in vivo (see Transparent Methods). Treatment of primary hepatocytes with the ER stressor tunicamycin (TM) upregulated UPR-responsive mRNAs, and ET cotreatment suppressed this effect (Figure 1A). A similar attenuation of UPR signaling was seen after knockdown of Cpt1a (Figure S1A). Dampened ER stress signaling was also evident from diminishment of the splicing of the IRE1α nuclease target Xbp1 (Figure 1B) and of the upregulation of the stress-regulated factor CHOP (Figure 1C). Because these events are differentially regulated by the three limbs of the UPR, our data suggest that ET attenuates signaling from all three UPR pathways.
Figure 1.

Inhibiting β-Oxidation Attenuates UPR Activity in Metabolically Active Cells and Protects against Different ER Stressors
(A–C) Mouse primary hepatocytes were treated with vehicle or 250 ng/mL tunicamycin (TM) in the presence or absence of 25 μg/mL ET (ET) for 8 h. Activation of the UPR was assessed by qRT-PCR of a sampling of UPR target genes (A), splicing of Xbp1 by conventional RT-PCR (B), and CHOP protein expression by immunoblot (C). In (B) and (C), and elsewhere, each lane represents an independently treated well. Loading control for immunoblot was calnexin, which does not change in response to TM or ET.
(D and E) Same as (A) except using C2C12 mouse myoblasts (D) or mouse primary brown adipocytes (E).
(F) mRNA expression of UPR markers was assessed by qRT-PCR in primary hepatocytes treated with 200 μM palmitate (PA) or oleate (OA) and 25 μg/mL ET for 8 h. For this and subsequent figures error bars represent means ± S.D.M. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by two-tailed t test and Benjamini-Hochberg adjustment for multiple comparisons unless indicated otherwise. p values between 0.05 and 0.1 are given as values. NS, p > 0.1.
See also Figure S1.
We next determined if the apparent protective effects of ET were limited to one cell type or to one ER stressor. Using the selection of UPR-regulated mRNAs in Figure 1A as an index of ER stress signaling, we found that ET also diminished UPR activation in C2C12 myoblasts (Figure 1D) and immortalized (data not shown) and primary (Figure 1E) brown adipocytes. Hepatocytes, myocytes, and brown adipocytes are characterized by particularly high metabolic activity (Frayn et al., 2006), as adipose, skeletal muscle, and particularly liver are among the top contributors to whole-body basal metabolic activity (Wang et al., 2012). In contrast, ET had no significant effect on the expression of UPR target genes in primary mouse embryonic fibroblasts (Figure S1B). In primary hepatocytes subjected to ER stress by palmitate loading, ET likewise diminished ER stress signaling (Figure 1F). (As expected, the unsaturated fatty acid oleate did not cause ER stress.) Palmitate is thought to elicit ER stress not by disrupting protein folding per se but by altering ER membrane fluidity, activating the UPR stress sensors through their transmembrane domains (Volmer et al., 2013). Thus, ET diminishes ER stress signaling induced by stressors that act by distinct mechanisms.
The effects of ET on ER stress signaling could be due to improvement of ER homeostasis in some way, or to simple inhibition of UPR signaling. The first model predicts that ER homeostasis will be preserved by ET, whereas the second predicts that it will be compromised, because impairment of the UPR is known to aggravate ER stress (Walter and Ron, 2011). There are relatively few assays that are capable of examining ER homeostasis apart from UPR signaling, but one such assay is the disruption of ER ultrastructure, which is caused by ER stress and exacerbated when the UPR is compromised (e.g. Harding et al., 2001, Heazlewood et al., 2008, Lee et al., 2005). Consistent with previous reports (Finnie, 2001, Rutkowski et al., 2006), TM elicited marked ER dilation accompanied by a loss of lamellar structure and overall disorganization, although some areas of grossly normal ER were present and were predominantly juxtaposed near mitochondria. In contrast, these disruptions were largely prevented by cotreatment with ET (Figure 2A) as confirmed by two independent, blinded scorers (Figure 2B). Furthermore, although ET diminished ER stress signaling, it did not block it entirely. Higher doses of TM elicited UPR activation in ET-treated cells to an extent comparable to that observed with lower doses in non-ET-treated cells (Figure 2C). Therefore, the UPR in ET-treated cells remained competent for signaling. Together, these results suggest that inhibiting β-oxidation alleviates ER stress. ET did not inhibit protein synthesis (Figure S2), suggesting that ET does not alleviate ER stress simply by reducing the nascent ER client protein load.
Figure 2.

Inhibiting β-Oxidation Improves ER Ultrastructure
(A) Primary hepatocytes were treated with vehicle, 250 ng/mL TM, 25 μg/mL ET, or TM and ET for 8 h and fixed in 2.5% glutaraldehyde. Fixed cells were imaged by transmission electron microscopy. Inset panels are indicated and shown on the right. Blue arrowheads represent areas of structurally normal ER, whereas orange arrowheads represent dysmorphic ER. Scale bars, 2 μm.
(B) Images were scored blindly based on an approximation of the percentage of “dilated” ER in each image. Normal (blue): <25% of ER in an image dilated, moderate (gray): 25%–75% dilated, and severe (orange): >75% dilated. Thus, the graph represents the percentage of images falling into each category. n = 9–10 cell images per group.
(C) Primary hepatocytes were treated with 25 μg/mL ET in the presence of 0.5, 2, or 10 μg/ml TM for 8 h, and expression of Bip and Chop was detected by qRT-PCR. Note that the relative magnitude of UPR activation upon TM treatment varied from experiment to experiment, which is why induction of CHOP was less robust at even the highest dose of TM in this experiment than in response to 0.25 μg/mL TM in Figure 1A.
See also Figure S2.
Inhibiting β-Oxidation Protects ER Function through Glutathione Redox
We and others have previously shown that ET raises the cellular ratio of oxidized (GSSG) to reduced (GSH) glutathione (Merrill et al., 2002, Pike et al., 2010, Tyra et al., 2012). To determine if glutathione redox plays a role in the protective effects of ET, we used 2-Acetylamino-3-[4-(2-acetylamino-2-carboxy-ethylsulfanylthiocarbonylamino)phenylthiocarbamoylsulfanyl]propionic acid (2-AAPA) to inhibit Glutathione Reductase (Seefeldt et al., 2009, Zhao et al., 2009), an enzyme with both mitochondrial and cytosolic activities that couples NADPH oxidation to GSSG reduction. We predicted that this treatment would phenocopy the protective effects of ET. As expected, treatment with 2-AAPA diminished cellular GSH levels and increased GSSG, thus significantly elevating the GSSG/GSH ratio, similar to the effects of ET (Figure 3A). Also, similarly to ET, 2-AAPA treatment alleviated ER stress, as determined by attenuated Xbp1 splicing (Figure 3B) and diminished upregulation of UPR target genes (Figure 3C).
Figure 3.

Stimulating Glutathione Oxidation Phenocopies Inhibition of β-Oxidation
(A) Primary hepatocytes were treated with 25 μg/mL ET or 25 μM 2-AAPA for 8 h. Levels of total (GSxtot), reduced (GSH), and oxidized (GSSG) glutathione were measured fluorimetrically, and the ratio of GSSG to GSH was calculated. Values are expressed relative to untreated cells.
(B and C) (B) Splicing of Xbp1 mRNA was measured by conventional RT-PCR, and (C) mRNA expression of UPR markers was measured by qRT-PCR in cells treated for 8 h with TM and/or 2-AAPA.
(D) Levels of total, reduced, and oxidized glutathione were measured in primary hepatocytes treated with 10 μM auranofin (AuF) for 8 h.
(E) mRNA expression of UPR markers after treatment with AuF for 8 h was quantified.
We next tested the converse prediction: that promoting glutathione reduction would cause ER stress. We treated hepatocytes with auranofin, which inhibits redox-active selenoproteins, including thioredoxin reductases and, at the dose used here, glutathione peroxidases 1, 2, 3, 4, and 6 (Chaudiere and Tappel, 1984, Roberts and Shaw, 1998, Scarbrough et al., 2012). Glutathione peroxidases generate GSSG through the reduction of H2O2. In the liver, glutathione peroxidase 1 (GPX1) is two orders of magnitude more abundant than any thioredoxin reductase or other GPX (Lai et al., 2008). Auranofin treatment elevated cellular GSH levels and diminished GSSG, leading to a decrease in the GSSG/GSH ratio (Figure 3D). Auranofin treatment alone was sufficient to induce a modest level of ER stress, seen in upregulation of UPR target genes in the absence of any other ER stress stimulus (Figure 3E).
Similar to 2-AAPA treatment, knockdown of Glutathione Reductase elevated the GSSG/GSH ratio (Figure S3A) and diminished ER stress signaling (Figure 4A). 2-AAPA did not have any additional protective effect when Glutathione Reductase was knocked down (Figure S3B), implying that the drug's effects on ER stress were specific to Glutathione Reductase. 2-AAPA accentuated the protective effects of ET (Figure S3C). This effect might be attributable to the more direct and rapid effects of 2-AAPA on the GSSG/GSH ratio compared with ET (Figure S3D). Even though 2-AAPA did not elevate the GSSG/GSH ratio above the effects of ET at the 8 h time point (see Figure 4D), it is likely that the combination of 2-AAPA and ET would elevate the GSSG/GSH ratio at earlier time points compared with ET alone, thus diminishing the early transcriptional induction of UPR targets, which would still be evident when mRNAs were collected at 8 h. Alternatively, we cannot exclude that either ET or 2-AAPA (or both) might mitigate ER stress partially through separate pathways.
Figure 4.

Glutathione Oxidation Is Required for Inhibition of β-Oxidation to Alleviate ER Stress
(A) mRNA expression of Glutathione Reductase (Gsr) and UPR markers Bip and Chop was quantified in primary hepatocytes transfected with a non-targeting control dsiRNA or either of two dsiRNAs targeting Gsr and treated for 8 h with TM.
(B) mRNA expression of UPR markers was assessed in primary hepatocytes transfected with a non-targeting control dsiRNA or dsiRNAs targeting Glutathione Peroxidase 1 (Gpx1).
(C) Hepatocytes were treated for 8 h with TM, ET, and/or AuF as indicated. mRNA expression of UPR markers was measured by qRT-PCR.
(D) The effect of AuF or AAPA on the GSSG/GSH ratio in cells treated with TM and ET was measured and is expressed relative to the ratio in untreated cells. For (D), significance was calculated by one-way ANOVA with Tukey honestly signifiant differene (HSD) post-hoc test for comparisons.
See also Figure S3.
As with auranofin, knockdown of GPX1 diminished the GSSG/GSH ratio (Figure S3A) and was sufficient to cause ER stress (Figure 4B). Therefore, genetic manipulation of glutathione redox phenocopied the effects of pharmacological manipulation on ER stress. Finally, auranofin blocked the ability of ET to alleviate ER stress (Figure 4C) and also blocked the ET-induced elevation of the GSSG/GSH ratio (Figure 4D). Together, these results show that ER homeostasis is sensitive to modulation of the GSSG/GSH ratio. As yet, whether the ER senses these changes to glutathione directly or indirectly—for example, through the activity of other codependent pathways such as the thioredoxin pathway—is not clear.
Inhibiting β-Oxidation Alters the Redox Maturation of ER Client Proteins
Given the association of GSSG with the oxidative protein folding environment in the ER, we examined whether ET altered the oxidative state of client proteins in the ER lumen. This was done by monitoring the oxidation of albumin, an endogenous hepatocyte protein that forms 17 disulfide bonds (Peters and Davidson, 1982). We tested the resistance of albumin to reduction by treating cells with DTT, lysing them, and then modifying free sulfhydryls with polyethylene glycol (PEG) maleimide, which retards migration on SDS-PAGE (Winther and Thorpe, 2014). Albumin could not be modified by PEG maleimide in the absence of DTT, because nascent albumin is rapidly and quantitatively oxidized, so unoxidized or partially oxidized albumin is undetectable among the steady-state pool (Peters and Davidson, 1982). Treatment with DTT led to almost complete reduction of albumin in otherwise untreated cells. However, ET caused a significant increase in the proportion of albumin that remained oxidized (Figure 5A). These findings were mirrored by treating cells with 2-AAPA, which also rendered albumin somewhat resistant to reduction, and contrasted by auranofin, which promoted reduction (Figure 5B). These results suggest that ET makes the ER lumen relatively resistant to the reducing effects of DTT.
Figure 5.

Enhancing Glutathione Oxidation Influences ER Oxidative Protein Maturation
(A) Primary hepatocytes were treated with 25 μg/mL ET for 4 h followed by addition of 10 mM DTT for 0, 30, or 60 min. Protein lysates were treated with 4 μM MM(PEG)24 (PEG-mal) before SDS-PAGE and immunoblotting to detect endogenous albumin. The percentage of oxidized albumin is given below each group, with statistical comparison by t test between ET-treated and non-treated cells.
(B) Hepatocytes were treated with 2-AAPA or AuF for 4 h before DTT and (PEG-mal) as in (A). Statistical comparisons were by one-way ANOVA with Tukey post-hoc test.
(C) Hepatocytes were treated with vehicle or 25 μg/mL ET for 4 h, pulse labeled with 35S Met/Cys for 15 min, and chased in complete hepatocyte medium with 5 mM unlabeled Met/Cys and containing vehicle or ET (and 5 μM MG-132 as indicated). At lysis, N-ethyl maleimide (NEM) was added to block free thiols, and endogenous α1-antitrpysin was immunoprecipitated from cell lysates. Samples were separated by non-reducing SDS-PAGE. The immature form (“ER”), complex glycosylated form (“post-ER”), and disulfide-bonded dimer (“S-S”) are indicated.
(D) Hepatocytes were pretreated for 3 h 15 min with 25 μg/mL ET and/or 500 ng/mL TM and pulse labeled for 30 min with 35S Met/Cys. Lysates were collected after the pulse, and media were collected after 3 or 8 h of chase with unlabeled Met/Cys media containing TM and/or ET as indicated. For (C) and (D), each condition was performed in two independently treated wells.
As a more direct measure of ER oxidation, we examined the processing of the endogenous ER client protein α1-antitrypsin (α1AT) by pulse-chase and non-reducing electrophoresis. α1AT has a single unpaired cysteine residue. Treatment of cells with ET yielded an ~100-kDa species of α1AT (Figure 5C) that disappeared upon reducing SDS-PAGE (data not shown), meaning that it is very likely an intermolecular α1AT disulfide-linked dimer. Together, these results suggest that ET alters the oxidative modification of ER client proteins, even though Glutathione Reductase is localized outside the ER.
The formation of intermolecular disulfide bonds has been associated with disease-causing misfolded alleles of human α1AT (Ronzoni et al., 2016, Ushioda et al., 2008), raising the possibility that ET might compromise α1AT protein folding. Seemingly in support of this idea, our pulse chase revealed that a greater percentage of α1AT chains in ET-treated cells were in the immature ER-localized form (confirmed by EndoH-sensitivity; data not shown) at early times in the chase period (Figure 5C). However, by later chase points essentially all α1AT chains had matured to the post-ER complex glycosylated form in the ET-treated cells, such that by 5 h, comparable amounts of α1AT had matured in both groups (Figure 5C). Furthermore, cotreatment of cells with the proteasome inhibitor MG-132 did not stabilize the immature ER-localized form of α1AT or the disulfide-bonded dimer, suggesting that neither of these forms was terminally misfolded and degraded by proteasome-dependent ER-associated degradation (ERAD). Rather, it appeared that ET delayed, but did not block, the anterograde maturation of α1AT through the secretory pathway. This finding was supported by examining global protein secretion, again using a pulse chase. At early chase points, ET diminished the appearance of radiolabeled proteins in the media both in cells treated with vehicle and in cells treated with TM. However, by later chase points, this difference was lost (Figure 5D), again suggesting that ET retarded, but did not block, secretory pathway protein processing.
We considered the possibility that diminished ER stress, improved ER ultrastructure, and delayed protein maturation and secretion might together be explained by more stringent ER quality control. We therefore examined the ability of ET to reverse ER stress associated with overexpression of the null Hong Kong (NHK) protein. NHK is encoded by a frameshift mutation of α1AT that results in a truncated and terminally misfolded product that is retained in the ER and undergoes ERAD; its expression causes ER stress in diverse cell types (Nagasawa et al., 2007, Ordonez et al., 2013, Sifers et al., 1988, Wu et al., 2007). We transduced primary hepatocytes with a recombinant adenovirus expressing NHK in a doxycycline (Dox)-inducible manner (Ad-TetOn-NHK). As a control, cells were transduced with adenovirus constitutively overexpressing GFP instead (Ad-GFP). No ER stress was observed in Ad-TetOn-NHK-transduced cells in the absence of Dox (not shown). As expected, Dox treatment elicited ER stress in cells transduced with Ad-TetOn-NHK, but not in cells expressing Ad-GFP. The stress induced by NHK was markedly diminished by ET (Figure 6A). Overexpression of NHK disrupted ER structure, resulting in the appearance of amorphous ER puncta that were diminished by ET treatment (Figures 6B and S4). This finding was substantiated by blinded analysis of electron microscopic images (Figure 6C). Furthermore, we observed an apparent enhancement of the clearance of NHK by ERAD. Eight hours after induction (but not at 4 h), steady-state expression of NHK was lower in ET-treated cells than in vehicle-treated cells, and this distinction was lost when degradation of NHK was blocked by MG-132 (Figure 6D). Although the mechanisms by which ET enhances ERAD remain under investigation, our results in Figures 5 and 6 point to effects of inhibiting β-oxidation—and by extension promoting glutathione oxidation—on ER protein oxidative maturation and quality control.
Figure 6.

Inhibiting β-Oxidation Attenuates ER Stress Induced by Overexpression of a Constitutively Misfolded Protein
(A) Primary hepatocytes were infected with Ad-CMV-eGFP or Ad-TetOn-NHK at an MOI of 1. NHK expression was induced with 500 ng/mL doxycycline (Dox) in the presence or absence of 25 μg/mL ET. mRNA expression of UPR markers was measured by qRT-PCR after 8 h of treatment.
(B) Primary hepatocytes infected with Ad-TetOn-NHK and treated with Dox and/or ET for 8 h were fixed in 2.5% glutaraldehyde for TEM. Orange arrowheads in insets denote dysmorphic ER. Images from which these insets were taken, along with scale bars, are in Figure S4.
(C) Images were scored as described in Figure 2; n = 10 cell images per group.
(D) Primary hepatocytes infected with Ad-TetOn-NHK were treated with Dox and ET as in (A) for 4 or 8 h in the presence or absence of 5 μM MG-132. NHK expression was analyzed by immunoblot. Loading control was calnexin.
See also Figure S4.
TCA Cycle Activity Links Oxidative Metabolism to ER Homeostasis
How is β-oxidation linked to glutathione redox? Glutathione Reductase requires NADPH to reduce glutathione, but β-oxidation yields NADH and FADH2 rather than NADPH. However, the end product of β-oxidation is acetyl-CoA, which enters the TCA cycle by condensation with oxaloacetate. We therefore speculated that ET might diminish production of NADPH from the TCA cycle, thereby resulting in elevated GSSG. In this model, it is not β-oxidation per se that is linked to ER homeostasis, but rather TCA cycle activity and the resultant production of NADPH. This model first predicted that ET would diminish levels of both acetyl-CoA and NADPH—predictions that we then confirmed (Figures 7A and 7B). The model also predicted that impairing TCA-dependent NADPH production would elevate the GSSG/GSH ratio and alleviate ER stress, similarly to ET treatment. Three TCA cycle isozymes generate NADPH in the liver: mitochondrial IDH2, cytosolic IDH1, and cytosolic ME1. Using dicer-substrate small interfering RNA (dsiRNAs), we specifically knocked down mRNA expression of each of these (Figure S5A), resulting in all cases in a surprisingly robust diminishment of the NADPH/NADP+ ratio (Figure S5B). Knockdown of each gene also diminished GSH levels, and knockdown of Idh2 and Me1 increased GSSG levels, thus elevating the GSSG/GSH ratio (Figure 7C). Consistent with these results, each knockdown also alleviated ER stress (Figure 7D).
Figure 7.

ER Homeostasis Responds to TCA-Dependent NADPH Production
(A and B) (A) Acetyl-CoA levels or (B) the NADPH/NADP+ ratio was measured in primary hepatocytes treated with ET for 8 h. The NADPH/NADP+ ratio is expressed relative to untreated cells.
(C) Glutathione was quantified in cells with dsiRNA-mediated knockdown of Idh1, Idh2, or Me1 (first oligo pair from Figure 7D in each case). A non-targeting dsiRNA was used as a control.
(D) Idh1, Idh2, or Me1 was knocked down as in (C) with either of two separate dsiRNAs before treatment with TM for 8 h. Relative mRNA expression of Bip, Chop, and Derl3 was measured by qRT-PCR.
(E–G) Primary hepatocytes were treated with 4 μM dichloroacetate (DCA) or TM for 8 h. The NADPH/NADP+ ratio (E), Xbp1 mRNA splicing (F), and UPR target gene expression (G) were analyzed as in previous figures.
See also Figure S5.
The Pyruvate Dehydrogenase complex oxidizes pyruvate produced by glycolysis to acetyl-CoA and is a major control point of TCA cycle activity for carbohydrate metabolism (Gray et al., 2014). Pyruvate Dehydrogenase Kinases (PDKs) phosphorylate and inactivate this enzyme complex. To determine the effects of stimulating TCA cycle activity, we treated hepatocytes with dichloroacetate (DCA), which stimulates substrate entry into the TCA cycle by inhibiting PDKs (Constantin-Teodosiu et al., 1999, Wu et al., 2018). Dichloroacetate treatment substantially elevated the NADPH/NADP+ ratio (Figure 7E). In addition, DCA treatment alone caused ER stress to an extent that was nearly as robust as the bona fide ER stressor TM, as seen by splicing of Xbp1 (Figure 7F) and upregulation of UPR target genes (Figure 7G). Therefore, these data provide direct evidence that mitochondrial oxidative catabolic activity causes ER stress, and that inhibiting production of NADPH by the TCA cycle alleviates ER stress.
Ablation of the Mitochondrial Pyruvate Carrier Alleviates ER Stress
Our results are consistent with a model whereby the production of NADPH by the TCA cycle links activity of the cycle to ER oxidative protein maturation through glutathione. If this model is correct, then genetic manipulations that diminish TCA flux should confer resistance to ER stress. We tested this idea in cells lacking the Mitochondrial Pyruvate Carrier (MPC). The MPC is composed of two subunits, MPC1 and MPC2, and mediates mitochondrial import of pyruvate (Bricker et al., 2012, Herzig et al., 2012). Loss of either subunit eliminates MPC activity, and liver-specific ablation of the MPC diminishes liver damage and inflammation in mice on obesogenic diets (Gray et al., 2015, McCommis et al., 2015, McCommis et al., 2017). In the absence of MPC activity, import of pyruvate into the mitochondria is greatly diminished, meaning that less acetyl-CoA can be produced by Pyruvate Dehydrogenase and that TCA cycle flux is diminished (Gray et al., 2015, Rauckhorst et al., 2017). We first examined the effects of deleting MPC1 by CRISPR in C2C12 myocytes (Oonthonpan et al., 2019). We found that loss of MPC1 diminished the NADPH/NADP+ ratio and elevated the GSSG/GSH ratio in these cells (Figures 8A and 8B). Diminished UPR activation at both the protein (Figure 8C) and mRNA (Figure 8D) levels indicated that MPC1-deficient cells were remarkably resistant to ER stress induced by TM or by the ER calcium-disrupting agent thapsigargin. These findings were confirmed in primary hepatocytes taken from mice lacking MPC2 in the liver (Mpc2LKO), when compared with cells from mice with intact alleles (Mpc2fl/fl). In cells lacking MPC2, both the upregulation of UPR target genes (Figure 8E) and Xbp1 mRNA splicing (Figures 8F and 8G) were attenuated. These findings provide direct genetic evidence that ER homeostasis is responsive to the availability of TCA cycle substrates.
Figure 8.

Deletion of the Mitochondrial Pyruvate Carrier Alleviates ER stress
(A and B) The NADPH/NADP+ ratio (A) and the GSSG/GSH ratio (B) were measured as mentioned earlier in parental wild-type and Mpc1−/− C2C12 myoblasts.
(C) Expression of UPR markers BiP, CHOP, phosphorylated eIF2α, and MPC1 was analyzed by immunoblot in wild-type and Mpc1−/− C2C12 myocytes treated with 100 ng/mL TM, 1 μg/mL TM, or 100 nM thapsigargin (TG). Loading control was actin. Hairlines are for visual aid only.
(D) Relative mRNA expression of UPR markers from cells treated similarly to (C) was quantified by qRT-PCR.
(E) Primary hepatocytes from Mpc2fl/fl and Mpc2LKO mice were treated with 250 ng/mL TM, and gene expression was measured by qRT-PCR.
(F) Splicing of Xbp1 in TM-treated hepatocytes from Mpc2fl/fl or Mpc2LKO mice was assessed by conventional RT-PCR. The ordering of the second and third groups was rearranged by cut/paste to remain consistent with the rest of the data in the article, but the image was otherwise unmodified.
(G) Xbp1 splicing was quantified from 8 independent samples (4 from cells taken from a male mouse, 4 from cells taken from a female mouse).
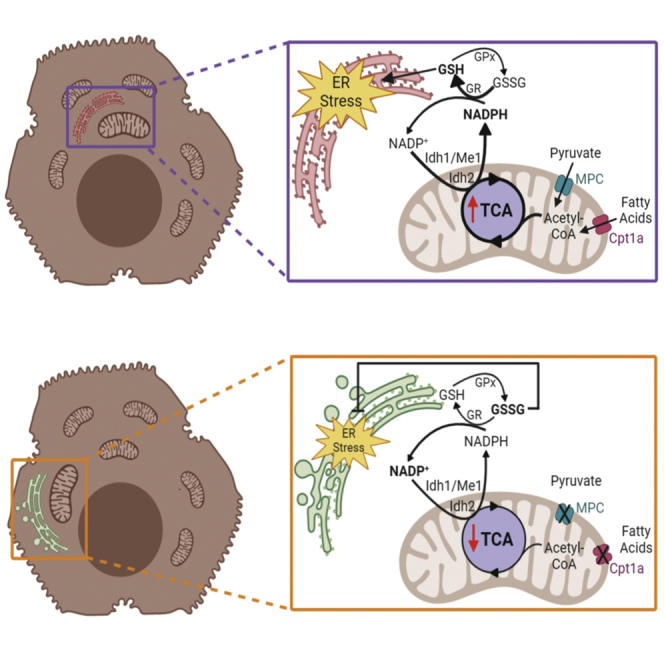
(H) Model for how TCA cycle activity is linked to ER homeostasis.
Discussion
Our results elucidate a pathway by which the ER senses mitochondrial metabolic activity (Figure 8H). We propose that NADPH and GSSG convey TCA cycle status: decreasing the availability of acetyl-CoA—either from lipid or carbohydrate catabolism—dampens NADPH production and disfavors glutathione reduction, with the increased GSSG ultimately—either directly or indirectly—altering ER oxidative protein maturation and protecting ER homeostasis. Conversely, enhancing acetyl-CoA availability ultimately favors GSH over GSSG and causes ER stress. That these relationships were seen in hepatocytes, myocytes, and brown adipocytes, but were conspicuously absent in fibroblasts, hints that oxidative metabolism might represent an intrinsic challenge to ER functionality and might explain why feeding itself appears to be an ER stressor. Notably, proliferating fibroblasts exhibit high glycolytic activity and a high GSSG/GSH ratio, and very low TCA activity, with cycle truncation before IDH activity (Lemons et al., 2010). Therefore, it is plausible that inhibiting FAox when TCA activity is already low and GSSG already high has no further effect, potentially explaining why fibroblasts are refractory to protection by ET.
That ER stress accompanies increased TCA cycle activity does not necessarily signify cellular dysfunction. It is possible that ER stress induced by enhanced metabolic activity might “prime” the ER by eliciting prophylactic ER stress. Brief exposures to stress are known to precondition cells against subsequent stresses (Rutkowski and Kaufman, 2007). Metabolic fluxes are, at least outside the context of overnutrition and obesity, transient. Insulin action on the liver during feeding is known to promote both oxidative catabolism through PDH dephosphorylation (Moule and Denton, 1997, Wieland et al., 1972) and protein biogenesis through mammalian target of rapamycin activation (Howell and Manning, 2011). UPR activation induced by TCA flux could enhance the functionality of the ER in anticipation of the increase in protein synthesis—including synthesis of ER client proteins—that follows in the post-absorptive state.
Although our data suggest that production of NADPH by the TCA cycle is a key event in our proposed pathway, it is not yet clear how NADPH production corresponds to actual activity of the cycle—given that metabolites can enter and exit the cycle at multiple points depending on nutritional conditions—or how the cellular compartmentalization of NADPH production contributes to the transmission of the NADPH status to the glutathione system. The NADPH/NADP+ ratio is regulated by nutritional state, exercise, diet, and circadian rhythms (Bradshaw, 2019, Ying, 2008). Only a portion of this NADPH is generated by the TCA cycle; other contributors include the pentose phosphate pathway of glucose metabolism and one-carbon folate metabolism, both of which are cytosolic. The fractional contribution of these pathways to total NADPH pools varies by cell type (Fan et al., 2014). We found here that inhibition of either the mitochondrial (IDH2) or cytosolic (IDH1 and ME1) NADPH-producing enzymes diminished ER stress, and we have found previously that inhibition of the pentose phosphate pathway does likewise (Tyra et al., 2012). These findings suggest that inhibiting NADPH production from any source protects ER function. Moreover, the fact that knockdown of any single TCA-dependent NADPH-producing enzyme has such robust effects on the NADPH/NADP+ ratio (Figure S5B) suggests that these individual pathways are interconnected in a way that is not yet clear. Furthermore, the fact that altering NADPH production either in the mitochondria or in the cytosol has the same effects on the GSSG/GSH ratio and on ER stress points to mechanisms for transmission of NADP(H) and/or glutathione status across membranes. Whether this exchange involves trans-mitochondrial transport of NADP(H) itself, or of glutathione, or of some other metabolite is not yet clear.
A related question is how elevated GSSG in the mitochondria and/or cytosol is transmitted to the ER. Does a change in the total cellular GSSG/GSH ratio ultimately change that ratio in the ER, or does GSSG exert its effects on the ER environment indirectly, perhaps through the thioredoxin pathway, which also relies on NADPH and which is functionally linked to glutathione redox (Miller and Schmidt, 2019, Toledano et al., 2013)? To date, no mechanism for import of GSSG into the ER has been described. One possibility is that diminishing the cytosolic level of GSH suppresses its import into the ER, thereby elevating the ER GSSG/GSH ratio as well. However, given that the ratio of GSH to GSSG in the cytosol is approximately 100:1, it seems unlikely that cytosolic GSH is limiting in this way. Alternatively, perhaps sites of ER-mitochondrial contact facilitate spatially restricted exchange of glutathione as they do for other metabolites such as calcium and reactive oxygen species (Joseph et al., 2019). Ultimately, determining how elevated GSSG outside the ER alters ER oxidative protein maturation will require compartment-specific monitoring and manipulation of the glutathione redox state.
Our results are surprising also in their implication that oxidized glutathione has a beneficial role for ER function. It is well established that reducing agents elicit robust ER stress and activate the UPR, which speaks to the importance of the ER oxidative environment in the protein folding process. However, reducing equivalents are needed as well, to activate the ER oxidase ERO1 (Kim et al., 2012) and to reduce disulfide isomerases so that they can catalyze reduction of improper disulfide bonds (Schwaller et al., 2003). Disruption of this capacity impairs the secretory pathway transport of model proteins with non-sequential disulfide bonds that must undergo such isomerization to avoid being trapped in non-native conformations (Poet et al., 2017). This need for reducing power might account for the delayed maturation of secretory client proteins that we observed in ET-treated cells (Figures 5C and 5D), although it appears to still allow for eventual secretion. How oxidation and reduction are balanced within the ER lumen is not well understood, nor is it understood the extent to which a cell recognizes a hyperoxidizing ER lumen as a stress. It is notable that treatments that favor ER oxidation such as diamide or overexpression of constitutively active ERO1 elicit only modest ER stress (Hansen et al., 2012) (data not shown). The understanding of ER redox is further confounded by the observations that neither ablation of both ERO1 isoforms (Zito et al., 2010a) nor apparent depletion of ER glutathione (Tsunoda et al., 2014) appreciably impacts ER protein oxidation capacity or stress sensitivity except for specialized substrates. Furthermore, because glutathione redox homeostasis is tied to cysteine and glutamate levels, and glutamate feeds into the TCA cycle as α-ketoglutarate, TCA activity, ER homeostasis, and glutathione redox can also be connected through amino acid sufficiency. Perhaps exemplifying this point, inhibition of the cystine/glutamate exchanger results in activation of an integrated stress response (but probably not a bona fide ER stress response) in cancer cells (Dixon et al., 2014), and this response is activated by, among other stimuli, amino acid depletion (Donnelly et al., 2013). We have observed that protein synthesis rates in ET-treated cells are particularly sensitive to amino acid deprivation (data not shown), suggesting a link between ET treatment and amino acid pools that might involve cysteine, glutamate, and glutathione. The relative importance of each of these pathways to ER homeostasis might also vary by cell type.
Our results also raise the question of what aspect(s) of ER functional capacity are altered by TCA cycle activity and glutathione redox. Activation of the UPR generally serves as a readout for ER stress, but it is not clear what diminished UPR signaling implies in terms of actual accumulation of unfolded proteins, particularly because some stressors, such as palmitate loading, activate the UPR apparently independent of the protein folding process (Volmer et al., 2013). There are relatively few approaches for disentangling truly dysfunctional protein processing from whatever criteria the UPR uses to perceive ER stress, which are still not well understood. For example, one might expect a dysfunctional ER to secrete proteins more slowly. On the other hand, perhaps enhanced ER retention time and chaperone association of client proteins facilitates proper protein folding under stressful conditions, which would be consistent with the delayed secretion but diminished ER stress by ET that we observed. One intriguing—but speculative—possibility is that GSSG might paradoxically protect the ER by exacerbating protein misfolding, if doing so renders misfolded client proteins more readily recognized and either refolded or cleared rather than futilely engaged with the ER quality control machinery. It will thus be interesting to test whether the protective effects of GSSG are negated by ablation of specific ER chaperones, trafficking factors, or ERAD components that facilitate protein clearance. The redox status and activity of BiP will be of particular interest, given its roles in protein folding, ERAD, protein translocation, and UPR signaling (Pobre et al., 2019), and the fact that oxidative modifications can alter the nature of its associations with nascent proteins (Wang et al., 2014) and with the translocation channel (Ponsero et al., 2017). Whatever the case, we infer that inhibiting TCA activity actually protects ER homeostasis in part because of its effects on ER ultrastructure. In principle, a hyperoxidizing ER might simply blunt the UPR rather than improve ER function, because activation of at least the UPR sensors ATF6 and IRE1 can be inhibited by oxidation (Nadanaka et al., 2007, Wang et al., 2018). However, there is no evidence that enforcing reduction of these sensors is sufficient for their activation, meaning there would then be no reason to expect stimuli, such as DCA or GPX impairment, that elevate GSH to activate the UPR on their own.
Although our findings extend to other cell types beyond hepatocytes, they seem particularly germane to liver disease. Obesity is the leading cause of non-alcoholic fatty liver disease (NAFLD) which, along with its downstream consequences—steatohepatitis, cirrhosis, and liver cancer—is the most common liver disease in the world (Araujo et al., 2018). ER stress and dysregulation of the UPR are associated with NAFLD/non-alcoholic steatohepatosis (NASH) in humans (Gonzalez-Rodriguez et al., 2014, Lake et al., 2014, Lebeaupin et al., 2015, Lebeaupin et al., 2018), and in mice on NASH-promoting diets (Charlton et al., 2011, Rahman et al., 2007). ER stress might promote NASH by aggravating diet-induced steatosis (Lee et al., 2008, Oyadomari et al., 2008, Rutkowski et al., 2008), and ER stress has also been shown to directly activate inflammatory signaling cascades (Lebeaupin et al., 2018, Özcan et al., 2004, Willy et al., 2015) and to promote hepatocyte cell death (Iracheta-Vellve et al., 2016, Olivares and Henkel, 2015). NAFLD is associated with increased TCA flux in the liver (Rauckhorst et al., 2017, Satapati et al., 2012, Sunny et al., 2011), raising the question of whether NAFLD progression is accelerated by ER stress arising from this increased flux, and whether diminished hepatic TCA flux in mice lacking MPC activity protects the liver at least in part by alleviating or preventing ER stress. On one hand, oxidative stress is recognized to contribute to NAFLD progression, and glutathione is known to protect against NAFLD progression (Liu et al., 2015), although most of those studies have examined glutathione synthesis rather than glutathione redox. On the other hand, our observation of elevated GSSG in cells lacking MPC1 (Figure 8B) suggests that elevated GSSG is not incompatible with protection from NAFLD. Whether TCA-dependent NADPH production contributes to NAFLD is not known. IDH1 and IDH2 function in vivo has mostly been examined in the context of transforming mutations in gliomas that change IDH activity. Both IDH1 and IDH2 are widely expressed, and to our knowledge no liver-specific deletion of either has been created. Thus, the role of the axis identified here in NAFLD has not been directly tested.
In conclusion, we have identified a novel NADPH- and glutathione-dependent pathway through which TCA cycle activity impacts ER homeostasis. We expect that this pathway will be relevant to the physiology and pathophysiology of liver, muscle, adipose, and other highly metabolically active cell types.
Limitations of the Study
This work was performed predominantly in mouse primary hepatocytes, with certain experiments also performed in immortalized myoblasts or in primary brown adipocytes. How our findings relate to other cell types and to in vivo versus in vitro contexts is not yet clear. It is also likely that the effects of TCA cycle manipulation on NADPH production will affect other NADPH-dependent pathways that have been implicated in ER homeostasis—particularly the thioredoxin pathway—in unforeseen ways. Last, although we demonstrate that diminishing TCA cycle activity improves ER structure, diminishes UPR signaling, and alters secretory protein maturation, neither how protein folding and processing is altered nor how changes to cytosolic glutathione redox are transmitted to the ER is clear yet. These are topics for future investigation.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, D. Thomas Rutkowski (thomas-rutkowski@uiowa.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact upon request.
Data and Code Availability
This study did not generate large datasets. The published article contains all individual data points generated during the study.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors would like to thank the University of Iowa Viral Vector Core and Central Microscopy Research Facility—especially Jian Shao—for technical assistance. Funding sources were as follows: D.T.R.: R01GM115424 (National Institutes of Health, U.S.A. (NIH)); E.R.G.: T32GM067795 (NIH); M.J.P.: R01DK106104 (NIH); B.N.F.: R01DK104735 (NIH); E.B.T.: R01DK104998 (NIH); and Central Microscopy Research Facility: S10RR018998 (NIH) (for JEOL JEM-1230 Transmission Electron Microscope).
Author Contributions
Conceptualization: E.R.G. and D.T.R.; Investigation: E.R.G., K.S.M., M.M., A.Q.K.-M., and D.T.R.; Writing – Original Draft: E.R.G. and D.T.R.; Writing – Review and Editing: all authors; Supervision: M.J.P., B.N.F., E.B.T., and D.T.R.; Funding Acquisition: E.R.G., M.J.P., B.N.F., E.B.T., and D.T.R.
Declaration of Interests
B.N.F. is a shareholder and member of the Scientific Advisory Board for Cirius Therapeutics. K.S.M. received research support from Cirius Therapeutics between 2017 and 2019. E.B.T. receives research grant funding for MPC-related research administered through the University of Iowa from Cirius Therapeutics and Poxel SA.
Published: May 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101116.
Supplemental Information
References
- Araujo A.R., Rosso N., Bedogni G., Tiribelli C., Bellentani S. Global epidemiology of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: what we need in the future. Liver Int. 2018;38(Suppl 1):47–51. doi: 10.1111/liv.13643. [DOI] [PubMed] [Google Scholar]
- Bradshaw P.C. Cytoplasmic and mitochondrial NADPH-coupled redox systems in the regulation of aging. Nutrients. 2019;11:e504. doi: 10.3390/nu11030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker D.K., Taylor E.B., Schell J.C., Orsak T., Boutron A., Chen Y.C., Cox J.E., Cardon C.M., Van Vranken J.G., Dephoure N. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton M., Krishnan A., Viker K., Sanderson S., Cazanave S., McConico A., Masuoko H., Gores G. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2011;301:G825–G834. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudiere J., Tappel A.L. Interaction of gold(I) with the active site of selenium-glutathione peroxidase. J. Inorg. Biochem. 1984;20:313–325. doi: 10.1016/0162-0134(84)85030-8. [DOI] [PubMed] [Google Scholar]
- Cnop M., Foufelle F., Velloso L.A. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 2012;18:59–68. doi: 10.1016/j.molmed.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D., Simpson E.J., Greenhaff P.L. The importance of pyruvate availability to PDC activation and anaplerosis in human skeletal muscle. Am. J. Physiol. 1999;276:E472–E478. doi: 10.1152/ajpendo.1999.276.3.E472. [DOI] [PubMed] [Google Scholar]
- Delaunay-Moisan A., Ponsero A., Toledano M.B. Reexamining the function of glutathione in oxidative protein folding and secretion. Antioxid. Redox Signal. 2017;27:1178–1199. doi: 10.1089/ars.2017.7148. [DOI] [PubMed] [Google Scholar]
- Dixon S.J., Patel D.N., Welsch M., Skouta R., Lee E.D., Hayano M., Thomas A.G., Gleason C.E., Tatonetti N.P., Slusher B.S. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly N., Gorman A.M., Gupta S., Samali A. The eIF2alpha kinases: their structures and functions. Cell. Mol. Life Sci. 2013;70:3493–3511. doi: 10.1007/s00018-012-1252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Ye J., Kamphorst J.J., Shlomi T., Thompson C.B., Rabinowitz J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y., Simmen T. Mechanistic connections between endoplasmic reticulum (ER) redox control and mitochondrial metabolism. Cells. 2019;8:e1071. doi: 10.3390/cells8091071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnie J.W. Effect of tunicamycin on hepatocytes in vitro. J. Comp. Pathol. 2001;125:318–321. doi: 10.1053/jcpa.2001.0510. [DOI] [PubMed] [Google Scholar]
- Frand A.R., Kaiser C.A. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol. Cell. 1999;4:469–477. doi: 10.1016/s1097-2765(00)80198-7. [DOI] [PubMed] [Google Scholar]
- Frayn K.N., Arner P., Yki-Jarvinen H. Fatty acid metabolism in adipose tissue, muscle and liver in health and disease. Essays Biochem. 2006;42:89–103. doi: 10.1042/bse0420089. [DOI] [PubMed] [Google Scholar]
- Fu S., Watkins S.M., Hotamisligil G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012;15:623–634. doi: 10.1016/j.cmet.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Gomez J.A., Rutkowski D.T. Experimental reconstitution of chronic ER stress in the liver reveals feedback suppression of BiP mRNA expression. Elife. 2016;5:e20390. doi: 10.7554/eLife.20390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rodriguez A., Mayoral R., Agra N., Valdecantos M.P., Pardo V., Miquilena-Colina M.E., Vargas-Castrillon J., Lo Iacono O., Corazzari M., Fimia G.M. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5:e1179. doi: 10.1038/cddis.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray L.R., Sultana M.R., Rauckhorst A.J., Oonthonpan L., Tompkins S.C., Sharma A., Fu X., Miao R., Pewa A.D., Brown K.S. Hepatic mitochondrial pyruvate carrier 1 is required for efficient regulation of gluconeogenesis and whole-body glucose homeostasis. Cell Metab. 2015;22:669–681. doi: 10.1016/j.cmet.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. 2014;71:2577–2604. doi: 10.1007/s00018-013-1539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez T., Simmen T. Endoplasmic reticulum chaperones tweak the mitochondrial calcium rheostat to control metabolism and cell death. Cell Calcium. 2018;70:64–75. doi: 10.1016/j.ceca.2017.05.015. [DOI] [PubMed] [Google Scholar]
- Hansen H.G., Schmidt J.D., Soltoft C.L., Ramming T., Geertz-Hansen H.M., Christensen B., Sorensen E.S., Juncker A.S., Appenzeller-Herzog C., Ellgaard L. Hyperactivity of the Ero1alpha oxidase elicits endoplasmic reticulum stress but no broad antioxidant response. J. Biol. Chem. 2012;287:39513–39523. doi: 10.1074/jbc.M112.405050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H.P., Zeng H., Zhang Y., Jungries R., Chung P., Plesken H., Sabatini D.D., Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Heazlewood C.K., Cook M.C., Eri R., Price G.R., Tauro S.B., Taupin D., Thornton D.J., Png C.W., Crockford T.L., Cornall R.J. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008;5:e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S., Raemy E., Montessuit S., Veuthey J.L., Zamboni N., Westermann B., Kunji E.R., Martinou J.C. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337:93–96. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- Howell J.J., Manning B.D. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol. Metab. 2011;22:94–102. doi: 10.1016/j.tem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iracheta-Vellve A., Petrasek J., Gyongyosi B., Satishchandran A., Lowe P., Kodys K., Catalano D., Calenda C.D., Kurt-Jones E.A., Fitzgerald K.A. Endoplasmic reticulum stress-induced hepatocellular death pathways mediate liver injury and fibrosis via stimulator of interferon genes. J. Biol. Chem. 2016;291:26794–26805. doi: 10.1074/jbc.M116.736991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph S.K., Booth D.M., Young M.P., Hajnoczky G. Redox regulation of ER and mitochondrial Ca(2+) signaling in cell survival and death. Cell Calcium. 2019;79:89–97. doi: 10.1016/j.ceca.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Sideris D.P., Sevier C.S., Kaiser C.A. Balanced Ero1 activation and inactivation establishes ER redox homeostasis. J. Cell Biol. 2012;196:713–725. doi: 10.1083/jcb.201110090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K.K., Kolippakkam D., Beretta L. Comprehensive and quantitative proteome profiling of the mouse liver and plasma. Hepatology. 2008;47:1043–1051. doi: 10.1002/hep.22123. [DOI] [PubMed] [Google Scholar]
- Lake A.D., Novak P., Hardwick R.N., Flores-Keown B., Zhao F., Klimecki W.T., Cherrington N.J. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol. Sci. 2014;137:26–35. doi: 10.1093/toxsci/kft230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeaupin C., Proics E., de Bieville C.H., Rousseau D., Bonnafous S., Patouraux S., Adam G., Lavallard V.J., Rovere C., Le Thuc O. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015;6:e1879. doi: 10.1038/cddis.2015.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeaupin C., Vallee D., Rousseau D., Patouraux S., Bonnafous S., Adam G., Luciano F., Luci C., Anty R., Iannelli A. Bax inhibitor-1 protects from nonalcoholic steatohepatitis by limiting inositol-requiring enzyme 1 alpha signaling in mice. Hepatology. 2018;68:515–532. doi: 10.1002/hep.29847. [DOI] [PubMed] [Google Scholar]
- Lee A.H., Chu G.C., Iwakoshi N.N., Glimcher L.H. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.H., Scapa E.F., Cohen D.E., Glimcher L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemons J.M., Feng X.J., Bennett B.D., Legesse-Miller A., Johnson E.L., Raitman I., Pollina E.A., Rabitz H.A., Rabinowitz J.D., Coller H.A. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 2010;8:e1000514. doi: 10.1371/journal.pbio.1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Baker S.S., Baker R.D., Zhu L. Antioxidant mechanisms in nonalcoholic fatty liver disease. Curr. Drug Targets. 2015;16:1301–1314. doi: 10.2174/1389450116666150427155342. [DOI] [PubMed] [Google Scholar]
- McCommis K.S., Chen Z., Fu X., McDonald W.G., Colca J.R., Kletzien R.F., Burgess S.C., Finck B.N. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metab. 2015;22:682–694. doi: 10.1016/j.cmet.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCommis K.S., Hodges W.T., Brunt E.M., Nalbantoglu I., McDonald W.G., Holley C., Fujiwara H., Schaffer J.E., Colca J.R., Finck B.N. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology. 2017;65:1543–1556. doi: 10.1002/hep.29025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill C.L., Ni H., Yoon L.W., Tirmenstein M.A., Narayanan P., Benavides G.R., Easton M.J., Creech D.R., Hu C.X., McFarland D.C. Etomoxir-induced oxidative stress in HepG2 cells detected by differential gene expression is confirmed biochemically. Toxicol. Sci. 2002;68:93–101. doi: 10.1093/toxsci/68.1.93. [DOI] [PubMed] [Google Scholar]
- Miller C.G., Schmidt E.E. Disulfide reductase systems in liver. Br. J. Pharmacol. 2019;176:532–543. doi: 10.1111/bph.14498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilenko D.A., Haas J.T., L'Homme L., Fleury S., Quemener S., Levavasseur M., Becquart C., Wartelle J., Bogomolova A., Pineau L. Metabolic and innate immune cues merge into a specific inflammatory response via the UPR. Cell. 2019;177:1201–1216.e19. doi: 10.1016/j.cell.2019.03.018. [DOI] [PubMed] [Google Scholar]
- Mohan S., R P.R.M., Brown L., Ayyappan P., G R.K. Endoplasmic reticulum stress: a master regulator of metabolic syndrome. Eur. J. Pharmacol. 2019;860:172553. doi: 10.1016/j.ejphar.2019.172553. [DOI] [PubMed] [Google Scholar]
- Moule S.K., Denton R.M. Multiple signaling pathways involved in the metabolic effects of insulin. Am. J. Cardiol. 1997;80:41A–49A. doi: 10.1016/s0002-9149(97)00457-8. [DOI] [PubMed] [Google Scholar]
- Nadanaka S., Okada T., Yoshida H., Mori K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell. Biol. 2007;27:1027–1043. doi: 10.1128/MCB.00408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa K., Higashi T., Hosokawa N., Kaufman R.J., Nagata K. Simultaneous induction of the four subunits of the TRAP complex by ER stress accelerates ER degradation. EMBO Rep. 2007;8:483–489. doi: 10.1038/sj.embor.7400933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares S., Henkel A.S. Hepatic Xbp1 gene deletion promotes endoplasmic reticulum stress-induced liver injury and apoptosis. J. Biol. Chem. 2015;290:30142–30151. doi: 10.1074/jbc.M115.676239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oonthonpan L., Rauckhorst A.J., Gray L.R., Boutron A.C., Taylor E.B. Two human patient mitochondrial pyruvate carrier mutations reveal distinct molecular mechanisms of dysfunction. JCI Insight. 2019;4:e126132. doi: 10.1172/jci.insight.126132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordonez A., Snapp E.L., Tan L., Miranda E., Marciniak S.J., Lomas D.A. Endoplasmic reticulum polymers impair luminal protein mobility and sensitize to cellular stress in alpha1-antitrypsin deficiency. Hepatology. 2013;57:2049–2060. doi: 10.1002/hep.26173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen O.E., Kalhan S.C., Hanson R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002;277:30409–30412. doi: 10.1074/jbc.R200006200. [DOI] [PubMed] [Google Scholar]
- Oyadomari S., Harding H.P., Zhang Y., Oyadomari M., Ron D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7:520–532. doi: 10.1016/j.cmet.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özcan U., Cao Q., Yilmaz E., Lee A.H., Iwakoshi N.N., Ozdelen E., Tuncman G., Gorgun C., Glimcher L.H., Hotamisligil G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Peters T., Jr., Davidson L.K. The biosynthesis of rat serum albumin. In vivo studies on the formation of the disulfide bonds. J. Biol. Chem. 1982;257:8847–8853. [PubMed] [Google Scholar]
- Pfaffenbach K.T., Nivala A.M., Reese L., Ellis F., Wang D., Wei Y., Pagliassotti M.J. Rapamycin inhibits postprandial-mediated X-box-binding protein-1 splicing in rat liver. J. Nutr. 2010;140:879–884. doi: 10.3945/jn.109.119883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike L.S., Smift A.L., Croteau N.J., Ferrick D.A., Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta. 2010;1807:726–734. doi: 10.1016/j.bbabio.2010.10.022. [DOI] [PubMed] [Google Scholar]
- Pobre K.F.R., Poet G.J., Hendershot L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: getting by with a little help from ERdj friends. J. Biol. Chem. 2019;294:2098–2108. doi: 10.1074/jbc.REV118.002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poet G.J., Oka O.B., van Lith M., Cao Z., Robinson P.J., Pringle M.A., Arner E.S., Bulleid N.J. Cytosolic thioredoxin reductase 1 is required for correct disulfide formation in the ER. EMBO J. 2017;36:693–702. doi: 10.15252/embj.201695336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponsero A.J., Igbaria A., Darch M.A., Miled S., Outten C.E., Winther J.R., Palais G., D'Autreaux B., Delaunay-Moisan A., Toledano M.B. Endoplasmic reticulum transport of glutathione by Sec61 is regulated by Ero1 and Bip. Mol. Cell. 2017;67:962–973.e5. doi: 10.1016/j.molcel.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A., Mammucari C., Gherardi G., Rizzuto R. Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 2016;41:1035–1049. doi: 10.1016/j.tibs.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S.M., Schroeder-Gloeckler J.M., Janssen R.C., Jiang H., Qadri I., Maclean K.N., Friedman J.E. CCAAT/enhancing binding protein beta deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology. 2007;45:1108–1117. doi: 10.1002/hep.21614. [DOI] [PubMed] [Google Scholar]
- Rainbolt T.K., Saunders J.M., Wiseman R.L. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 2014;25:528–537. doi: 10.1016/j.tem.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Rauckhorst A.J., Gray L.R., Sheldon R.D., Fu X., Pewa A.D., Feddersen C.R., Dupuy A.J., Gibson-Corley K.N., Cox J.E., Burgess S.C. The mitochondrial pyruvate carrier mediates high fat diet-induced increases in hepatic TCA cycle capacity. Mol. Metab. 2017;6:1468–1479. doi: 10.1016/j.molmet.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts J.R., Shaw C.F., 3rd Inhibition of erythrocyte selenium-glutathione peroxidase by auranofin analogues and metabolites. Biochem. Pharmacol. 1998;55:1291–1299. doi: 10.1016/s0006-2952(97)00634-5. [DOI] [PubMed] [Google Scholar]
- Ronzoni R., Berardelli R., Medicina D., Sitia R., Gooptu B., Fra A.M. Aberrant disulphide bonding contributes to the ER retention of alpha1-antitrypsin deficiency variants. Hum. Mol. Genet. 2016;25:642–650. doi: 10.1093/hmg/ddv501. [DOI] [PubMed] [Google Scholar]
- Rutkowski D.T., Arnold S.M., Miller C.N., Wu J., Li J., Gunnison K.M., Mori K., Sadighi Akha A.A., Raden D., Kaufman R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski D.T., Kaufman R.J. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem. Sci. 2007;32:469–476. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Rutkowski D.T., Wu J., Back S.H., Callaghan M.U., Ferris S.P., Iqbal J., Clark R., Miao H., Hassler J.R., Fornek J. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell. 2008;15:829–840. doi: 10.1016/j.devcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydstrom J. Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta. 2006;1757:721–726. doi: 10.1016/j.bbabio.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Salvado L., Palomer X., Barroso E., Vazquez-Carrera M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol. Metab. 2015;26:438–448. doi: 10.1016/j.tem.2015.05.007. [DOI] [PubMed] [Google Scholar]
- Satapati S., Sunny N.E., Kucejova B., Fu X., He T.T., Mendez-Lucas A., Shelton J.M., Perales J.C., Browning J.D., Burgess S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012;53:1080–1092. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarbrough P.M., Mapuskar K.A., Mattson D.M., Gius D., Watson W.H., Spitz D.R. Simultaneous inhibition of glutathione- and thioredoxin-dependent metabolism is necessary to potentiate 17AAG-induced cancer cell killing via oxidative stress. Free Radic. Biol. Med. 2012;52:436–443. doi: 10.1016/j.freeradbiomed.2011.10.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller M., Wilkinson B., Gilbert H.F. Reduction-reoxidation cycles contribute to catalysis of disulfide isomerization by protein-disulfide isomerase. J. Biol. Chem. 2003;278:7154–7159. doi: 10.1074/jbc.M211036200. [DOI] [PubMed] [Google Scholar]
- Seefeldt T., Zhao Y., Chen W., Raza A.S., Carlson L., Herman J., Stoebner A., Hanson S., Foll R., Guan X. Characterization of a novel dithiocarbamate glutathione reductase inhibitor and its use as a tool to modulate intracellular glutathione. J. Biol. Chem. 2009;284:2729–2737. doi: 10.1074/jbc.M802683200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H., Berndt C., Jones D.P. Oxidative stress. Annu. Rev. Biochem. 2017;86:715–748. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- Sifers R.N., Brashears-Macatee S., Kidd V.J., Muensch H., Woo S.L. A frameshift mutation results in a truncated alpha 1-antitrypsin that is retained within the rough endoplasmic reticulum. J. Biol. Chem. 1988;263:7330–7335. [PubMed] [Google Scholar]
- Sunny N.E., Parks E.J., Browning J.D., Burgess S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–810. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurey P., Tubbs E., Vial G., Jacquemetton J., Bendridi N., Chauvin M.A., Alam M.R., Le Romancer M., Vidal H., Rieusset J. Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 2016;8:129–143. doi: 10.1093/jmcb/mjw004. [DOI] [PubMed] [Google Scholar]
- Toledano M.B., Delaunay-Moisan A., Outten C.E., Igbaria A. Functions and cellular compartmentation of the thioredoxin and glutathione pathways in yeast. Antioxid. Redox Signal. 2013;18:1699–1711. doi: 10.1089/ars.2012.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda S., Avezov E., Zyryanova A., Konno T., Mendes-Silva L., Pinho Melo E., Harding H.P., Ron D. Intact protein folding in the glutathione-depleted endoplasmic reticulum implicates alternative protein thiol reductants. Elife. 2014;3:e03421. doi: 10.7554/eLife.03421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu B.P., Ho-Schleyer S.C., Travers K.J., Weissman J.S. Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science. 2000;290:1571–1574. doi: 10.1126/science.290.5496.1571. [DOI] [PubMed] [Google Scholar]
- Tyra H.M., Spitz D.R., Rutkowski D.T. Inhibition of fatty acid oxidation enhances oxidative protein folding and protects hepatocytes from endoplasmic reticulum stress. Mol. Biol. Cell. 2012;23:811–819. doi: 10.1091/mbc.E11-12-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushioda R., Hoseki J., Araki K., Jansen G., Thomas D.Y., Nagata K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–572. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- Vance J.E. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta. 2014;1841:595–609. doi: 10.1016/j.bbalip.2013.11.014. [DOI] [PubMed] [Google Scholar]
- Volmer R., van der Ploeg K., Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. U S A. 2013;110:4628–4633. doi: 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P., Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang J., Pareja K.A., Kaiser C.A., Sevier C.S. Redox signaling via the molecular chaperone BiP protects cells against endoplasmic reticulum-derived oxidative stress. Elife. 2014;3:e03496. doi: 10.7554/eLife.03496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.M., Qiu Y., Yang Z., Kim H., Qian Q., Sun Q., Zhang C., Yin L., Fang D., Back S.H. IRE1alpha prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci. Signal. 2018;11:eaao4617. doi: 10.1126/scisignal.aao4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Ying Z., Bosy-Westphal A., Zhang J., Heller M., Later W., Heymsfield S.B., Muller M.J. Evaluation of specific metabolic rates of major organs and tissues: comparison between nonobese and obese women. Obesity (Silver Spring) 2012;20:95–100. doi: 10.1038/oby.2011.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis B.C., Cowan A.T., Brown N., Foster D.W., McGarry J.D. Use of a selective inhibitor of liver carnitine palmitoyltransferase I (CPT I) allows quantification of its contribution to total CPT I activity in rat heart. Evidence that the dominant cardiac CPT I isoform is identical to the skeletal muscle enzyme. J. Biol. Chem. 1994;269:26443–26448. [PubMed] [Google Scholar]
- Wieland O.H., Patzelt C., Loffler G. Active and inactive forms of pyruvate dehydrogenase in rat liver. Effect of starvation and refeeding and of insulin treatment on pyruvate-dehydrogenase interconversion. Eur. J. Biochem. 1972;26:426–433. doi: 10.1111/j.1432-1033.1972.tb01783.x. [DOI] [PubMed] [Google Scholar]
- Willy J.A., Young S.K., Stevens J.L., Masuoka H.C., Wek R.C. CHOP links endoplasmic reticulum stress to NF-kappaB activation in the pathogenesis of nonalcoholic steatohepatitis. Mol. Biol. Cell. 2015;26:2190–2204. doi: 10.1091/mbc.E15-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winther J.R., Thorpe C. Quantification of thiols and disulfides. Biochim. Biophys. Acta. 2014;1840:838–846. doi: 10.1016/j.bbagen.2013.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.Y., Satapati S., Gui W., Wynn R.M., Sharma G., Lou M., Qi X., Burgess S.C., Malloy C., Khemtong C. A novel inhibitor of pyruvate dehydrogenase kinase stimulates myocardial carbohydrate oxidation in diet-induced obesity. J. Biol. Chem. 2018;293:9604–9613. doi: 10.1074/jbc.RA118.002838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Rutkowski D.T., Dubois M., Swathirajan J., Saunders T., Wang J., Song B., Yau G.D., Kaufman R.J. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell. 2007;13:351–364. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Xin Y., Dominguez Gutierrez G., Okamoto H., Kim J., Lee A.H., Adler C., Ni M., Yancopoulos G.D., Murphy A.J., Gromada J. Pseudotime ordering of single human beta-cells reveals states of insulin production and unfolded protein response. Diabetes. 2018;67:1783–1794. doi: 10.2337/db18-0365. [DOI] [PubMed] [Google Scholar]
- Yao C.H., Liu G.Y., Wang R., Moon S.H., Gross R.W., Patti G.J. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of beta-oxidation. PLoS Biol. 2018;16:e2003782. doi: 10.1371/journal.pbio.2003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- Yoboue E.D., Sitia R., Simmen T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018;9:331. doi: 10.1038/s41419-017-0033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Seefeldt T., Chen W., Wang X., Matthees D., Hu Y., Guan X. Effects of glutathione reductase inhibition on cellular thiol redox state and related systems. Arch. Biochem. Biophys. 2009;485:56–62. doi: 10.1016/j.abb.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito E., Chin K.T., Blais J., Harding H.P., Ron D. ERO1-beta, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J. Cell Biol. 2010;188:821–832. doi: 10.1083/jcb.200911086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito E., Melo E.P., Yang Y., Wahlander A., Neubert T.A., Ron D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell. 2010;40:787–797. doi: 10.1016/j.molcel.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate large datasets. The published article contains all individual data points generated during the study.