

Abstract
Most genetic susceptibility to cutaneous melanoma remains to be discovered. Meta-analysis genome-wide association study (GWAS) of 36,760 melanoma cases (67% newly-genotyped) and 375,188 controls identified 54 significant loci with 68 independent SNPs. Analysis of risk estimates across geographical regions and host factors suggests the acral melanoma subtype is uniquely unrelated to pigmentation. Combining this meta-analysis with nevus count and hair color GWAS, and transcriptome association approaches, uncovered 31 potential secondary loci, for a total of 85 cutaneous melanoma susceptibility loci. These findings provide substantial insights into cutaneous melanoma genetic architecture, reinforcing the importance of nevogenesis, pigmentation, and telomere maintenance together with identifying potential new pathways for cutaneous melanoma pathogenesis.
Cutaneous melanoma is a deadly malignancy with increasing incidence and burden in fair-skinned populations worldwide1. Increased risk for cutaneous melanoma is caused by high exposure to ultraviolet radiation2, as well as host factors including family history3,4, pigmentary phenotypes5, number of melanocytic nevi6,7, longer telomeres8,9, and immunosuppression10.
Identified melanoma genetic risk variants include rare, highly penetrant mutations in genes such as CDKN2A11,12 and POT113,14, as well as more common variants (e.g., lower-penetrance variants in MC1R)15,16. Genome-wide association studies (GWAS) of cutaneous melanoma susceptibility in populations of European ancestry have identified 21 genetic loci reaching genome-wide significance (P < 5 × 10−8)17-24. Additional approaches, including family-based analyses of cutaneous melanoma25,26, combining cutaneous melanoma and nevus count GWAS27 and transcriptome-wide association studies (TWAS)28 have identified further loci that, despite not containing SNPs reaching P < 5 × 10−8 in a cutaneous melanoma-only GWAS, most likely influence melanoma risk.
This meta-analysis of cutaneous melanoma susceptibility is more than three times the effective sample size of previous cutaneous melanoma GWAS, providing unprecedented power to identify cutaneous melanoma susceptibility variants and enhanced distinction of independent variants in known cutaneous melanoma susceptibility regions. We report here 68 independent cutaneous melanoma associated variants across 54 loci that confirm the importance of key functional pathways and highlight previously unknown cutaneous melanoma etiologic routes (Tables 1-2). Stratified analyses showed a lack of involvement of the pigmentation pathway for acral melanoma, in line with observational data29. The combined analysis of cutaneous melanoma, nevus and hair color GWAS data, and use of expression data through TWAS, identified 31 secondary, potential loci.
Results
Study overview
We performed a GWAS meta-analysis of cutaneous melanoma susceptibility with 30,134 clinically-confirmed cutaneous melanoma cases (Online Methods), 6,626 self-reported cutaneous melanoma cases and 375,188 cutaneous melanoma -free controls from the United Kingdom, United States, Australia, Northern and Western Europe as well as the Mediterranean – a highly sun exposed population often under-represented in cutaneous melanoma studies (Supplementary Table 1). Of these, 24,756 cases (67%) and 358,734 controls (96%) had not been included in any previous melanoma GWAS.
Separately, we performed total (clinically confirmed cases + self-reported cases from 23andMe, Inc. and a subset of UK Biobank cases with only self-reported cutaneous melanoma status) and confirmed-only cutaneous melanoma meta-analyses to determine the power gained by including self-reported cutaneous melanoma cases. Risk loci were deemed genome-wide significant when variants had fixed effects meta-analysis P-values < 5 × 10−8 (Pmeta); where variants exhibited notable heterogeneity (I2 > 31%)30 random effects P-values (Pmeta_r) were also required to be < 5 × 10−8 (Online Methods). Q-Q plots (Supplementary Figure 1) and linkage disequilibrium score regression31 (LDSC; Online Methods) intercepts showed minimal inflation for individual studies (mostly < 1.04; Supplementary Table 1), indicating adequate control of population stratification.
Before including the self-report GWAS data, we used LDSC31 to verify their genetic correlation (Rg) with the confirmed-only GWAS meta-analysis (Supplementary Note; Supplementary Table 2). Based on the high Rg and similarity in h2 estimates for self-report and clinically confirmed cutaneous melanoma cases (Supplementary Note), we performed an overall total cutaneous melanoma meta-analysis (h2total= 0.11, 95% CI = 0.08–0.15). The lambda and LDSC intercept for the total cutaneous melanoma meta-analysis indicated that the majority of inflation is due to polygenic signal (λ = 1.165, intercept = 1.054, ratio = 0.17; Supplementary Table 2). A similar h2total (12%) was estimated using genetic effect-size distribution inference from summary level data (GENESIS; Online Methods)32.
Conditional and joint analysis of the total cutaneous melanoma meta-analysis summary statistics using GCTA33 identified a total of 54 loci meeting our requirements for genome-wide significance (Online Methods; Figure 1, Extended Data Figure 1-2). Results for loci previously reported by cutaneous melanoma GWAS reaching significance in the total meta-analysis are presented in Table 1. Results for loci not previously reported by a cutaneous melanoma GWAS are summarized in Table 2. In addition to the 54 lead variants, 14 independent variants with linkage disequilibrium (LD) r2EUR < 0.05 with lead variants at or near 6 loci (TERT, AGR3, CDKN2A, OCA2, MC1R, and TP53) were identified (Supplementary Table 3). Individual regional association plots for the association signals have been provided as Supplementary Data 1. Conditional and joint analysis of summary data identified a further 9 variants at or near SLC45A2, IRF4, AGR3, CCND1, GPRC5A, FTO, and MC1R; however, these additional variants were not carried forward, having either Pmeta > 5 × 10−8 in the single variant analysis or excess heterogeneity (I2 > 31%) and Pmeta_r < 5 × 10−8 (Supplementary Table 4). In addition, we used GENESIS (Online Methods), which enables a reformulation of the variance explained by associated SNPs to estimate a theoretical optimal area under the curve (AUC), rather than formally testing this using a training and prediction set32 to estimate the potential AUC. The estimated AUC was 66.8%, compared to ~64% in the 2015 cutaneous melanoma meta-analysis23. This estimate does not include any host factors and would require benchmarking in a prospective study for validation.
Figure 1. Manhattan plot for the total cutaneous melanoma meta-analysis.
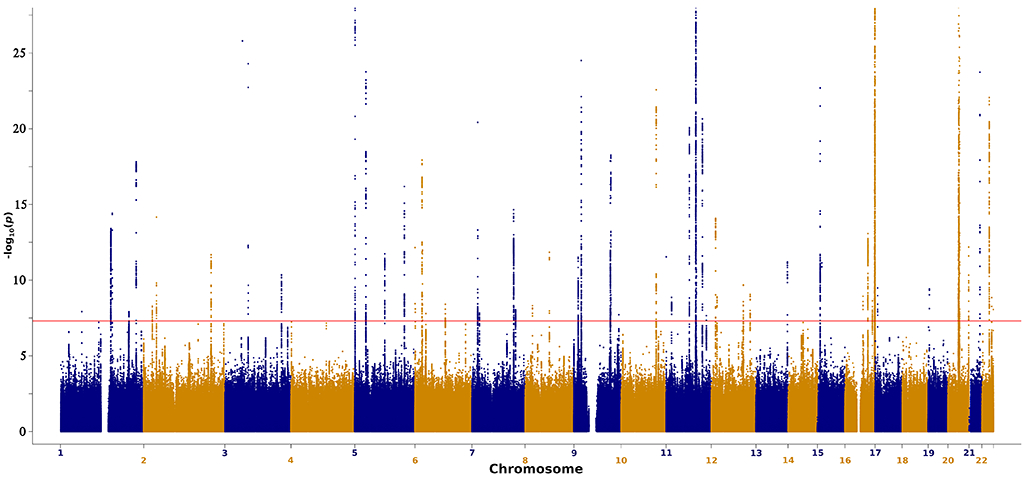
−log10 of two-sided P-values for SNPs derived from a fixed-effects inverse variance weighted meta-analysis of logistic regression GWAS (Y-axis) plotted against SNP chromosome positions for the total meta-analysis (36,760 melanoma cases and 375,188 controls; for full details of analysis and covariates included see the Online Methods). The y-axis is limited to −log10(1×10−25) to truncate strong signals at loci such as MC1R and ASIP. The full plot is displayed in Extended Data Figure 2. To account for multiple testing, SNPs with a P-value less than 5×10−8 are deemed significant.
Previous cutaneous melanoma GWAS have identified 21 genome-wide significant loci17-24. Family-based methods or the combination of cutaneous melanoma with nevus count have identified a further 12 loci including IRF4, MITF, HDAC4, and GPRC5A25-27. The lead SNPs from many of these loci are associated with pigmentation, tanning response, nevus count, and telomere maintenance (Supplementary Table 5). Other SNPs are proximal to DNA repair genes. Some loci are associated with more than one trait (Tables 1-2). Our analysis confirms 19 of the 21 loci previously reaching genome-wide significance (Table 1; Supplementary Note). The total cutaneous melanoma meta-analysis also confirms the previously reported IRF4 and MITF associations25-27,34,35, as well as 6 regions previously identified only by combining nevus count and cutaneous melanoma GWAS data27 (Table 2; Supplementary Note). These results demonstrate the ability of cross-trait GWAS to identify disease loci. The remaining 27 loci have not previously been reported as cutaneous melanoma susceptibility loci (Table 2; Supplementary Table 3). The results for the pathologically confirmed-only cutaneous melanoma cases (N = 30,134; Supplementary Table 1) are reported in the Supplementary Note, Extended Data Figure 2-3, Supplementary Table 6. Our full meta-analysis identified 11 loci not found in the confirmed-only GWAS meta-analysis, demonstrating the advantage of including the 6,626 self-reported cutaneous melanoma cases and over 290,000 controls (Supplementary Table 1). Results for SNPs with a fixed or random P < 5 × 10−7, from the total meta-analysis are reported in Supplementary Table 7.
Melanoma associations by sex, age at diagnosis and subtype
We performed separate GWAS by sex, age at cutaneous melanoma diagnosis (≤40, 40–60, ≥60 years) and major cutaneous melanoma subtypes (superficial spreading (SS), lentigo maligna (LM), nodular melanoma (NM), and acral lentiginous (AL)) to identify variants associated with select subgroups (Supplementary Table 8). Our analysis identified no additional variants after adjustment for multiple testing (5 × 10−8/9), suggesting that if such variants exist they are undetectable at our current sample size.
We also performed polygenic risk score (PRS) analyses based on the lead independent genome-wide significant SNPs for nevus count (10 variants; Online Methods) and hair color (276 variants; Online Methods) to explore further whether either trait’s association with cutaneous melanoma differs across phenotypic subtypes (significance threshold = 0.05/28; Online Methods). We observed no significant differences in the distribution of the tested PRSs by sex or age at cutaneous melanoma diagnosis. We did, however, detect differences in the distribution of the hair color PRS for the acral lentiginous subtype compared to all non-acral subtypes (P = 2.1 × 10−4). Our analyses indicated that genetically-predicted pigmentation in acral lentiginous cases was no different to controls (P = 0.65, Extended Data Figure 4) and darker than in superficial spreading, lentigo maligna and nodular melanoma cases (P = 5.3 × 10−5, 0.01, 4.8 × 10−4, respectively). These findings provide strong genetic evidence that the pigmentation pathway is far less important for risk of acral lentiginous melanoma than for other subtypes of cutaneous melanoma. No significant differences were observed by subtype for the nevus count PRS.
Variant annotation with cutaneous melanoma risk phenotypes
To investigate possible biological pathways underlying cutaneous melanoma signals, variants independently associated with cutaneous melanoma in the total meta-analysis were evaluated in GWAS of telomere length, tanning response, pigmentation and nevus count (Online Methods, Table 1 and 2, Supplementary Tables 5,7-8). Using a Bonferroni-corrected threshold of phenotype P-value < 0.00074 (0.05/68 independent SNPs), 18 of the 35 novel loci are associated with tanning response or pigmentation (Table 2, Supplementary Table 5), further indicating the importance of pigmentation pathways in cutaneous melanoma susceptibility. Several new loci, including rs12473635 near DTNB and rs78378222 near TP53, are associated with nevus count, reinforcing the role of nevi in cutaneous melanoma susceptibility. Furthermore, four novel loci have previously been associated with telomere length (rs3950296/TERC, rs4731207/POT1, rs2967383/MPHOSPH6, and rs143190905/RTEL136) (Table 2, Supplementary Table 5) providing additional support for the role of telomere maintenance in cutaneous melanoma susceptibility following earlier findings that genetic determinants of telomere length are generally associated with melanoma risk13,14,37. Other newly-discovered lead variants are not associated with these phenotypes, suggesting novel pathways.
Additional approaches to identify melanoma risk loci
To identify further loci influencing cutaneous melanoma risk and provide a more nuanced annotation of discovered cutaneous melanoma risk loci, we used a range of secondary approaches with correction for multiple testing (Online Methods). To explore the overlap between cutaneous melanoma loci and established risk factor phenotypes, we combined our total cutaneous melanoma GWAS meta-analysis with a nevus count GWAS meta-analysis (N = 65,777; Online Methods) and separately with a UKBB hair color GWAS (N = 352,662; Online Methods). For the total cutaneous melanoma GWAS meta-analysis and nevus count the Rg is 0.57 (SE = 0.11, P-value = 2.39 × 10−7), and for hair color scored from light hair to dark (Online Methods) the Rg is 0.290 (SE = 0.096, P-value = 0.0025). Pairwise GWAS (GWAS-PW)38 was used to determine whether loci were associated with only one trait or pleiotropic with both cutaneous melanoma and either nevus count or hair color (Online Methods). Loci previously-reported through the combination of cutaneous melanoma and nevus GWAS27 are now confirmed by our larger cutaneous melanoma GWAS meta-analysis (Table 2). Together these analyses identified secondary potential loci not associated at genome-wide significance levels in the total cutaneous melanoma GWAS meta-analysis. At the Bonferroni-corrected threshold of 1.25 × 10−8 (Online Methods), they included 8 loci jointly significant for cutaneous melanoma and nevus count, 17 for cutaneous melanoma and hair color, and 4 with cutaneous melanoma, nevus count and hair color (Table 3, Supplementary Table 9- 10).
In parallel, we examined data from a recently-established cell-type specific melanocyte cis-expression quantitative trait loci (eQTL) dataset28 as well as tissue-based cis-eQTL datasets available through Genotype-Tissue Expression (GTEx)39 resource to identify additional susceptibility loci using a transcriptome prediction mapping strategy (or transcriptome-wide association study; TWAS)40,41. TWAS utilizing these expression datasets enabled gene-based testing for significant cis genetic correlations between imputed gene expression and cutaneous melanoma risk, aiding identification of additional susceptibility loci (Online Methods). While identification of significant genes by TWAS does not establish causation, it can indicate plausible gene candidates to be utilized in pathway analyses and investigated in future functional studies. This analysis built on a previous melanocyte TWAS that analyzed data from a prior cutaneous melanoma GWAS meta-analysis28 and identified significant novel associations between cutaneous melanoma and imputed gene expression of five genes at four loci. Importantly, the CBWD1 locus on chromosome 9 was later identified as a genome-wide significant cutaneous melanoma+nevus count pleiotropic locus27 (Table 3, Supplementary Table 9), and the other three loci (ZFP90 on chromosome 16, HEBP1 on chromosome 12, and MSC/RP11–383H13.1 on chromosome 8) are now at genome-wide significance with cutaneous melanoma in this larger GWAS meta-analysis (Table 2). This confirmation supports the TWAS approach for both identifying new loci and nominating potentially functional genes at GWAS-discovered loci (Tables 1-2).
To empirically identify the target tissues for cutaneous melanoma risk variants, we used partitioned LD score regression42 to determine the proportion of total cutaneous melanoma GWAS meta-analysis heritability that could be captured by genes expressed in melanocytes and in 50 GTEx tissue types. We found that partitioned cutaneous melanoma heritability was most enriched in genes specifically expressed in melanocytes (2.76-fold, P = 3.12 × 10−6 for top 4,000 genes; Extended Data Figure 5), followed by three other skin-related tissues (GTEx sun-exposed and non-sun-exposed skin, transformed skin fibroblasts). This enrichment was much stronger than the one based on the previously published melanoma GWAS23. We then focused on these four tissues for discovery of new loci, applying Bonferroni correction for multiple comparisons based on the number of genes tested within each tissue set (Online Methods). TWAS using the melanocyte dataset (Supplementary Table 11, Supplementary Table 3) identified a total of 40 significant genes. Combining genes within 1 Mb of each other into discrete loci, 32 genes were located within 13 formally genome-wide significant cutaneous melanoma GWAS loci, and eight genes were identified within six novel loci. Considering the other skin-related tissues collectively (Supplementary Table 12, Supplementary Table 3), TWAS identified a single significant gene at one additional novel locus, as well as genes within 15 GWAS-significant loci. The TWAS using all GTEx tissues is reported in Supplementary Table 13.
In aggregate, these complementary approaches identified a total of 85 discrete loci (Figure 2; Supplementary Table 14): 54 formally significant at P < 5 × 10−8 in the total cutaneous melanoma meta-analysis (Table 1-2, Supplementary Table 3), and the remainder supported by one or more of the secondary analyses (Table 3-4, Supplementary Tables 7-10,14) and likely representing additional cutaneous melanoma risk loci, but requiring a larger sample size to reach genome-wide significance. In order to annotate cutaneous melanoma GWAS loci for candidate susceptibility genes for pathway analyses as well as future functional studies, we turned to eQTL colocalization analyses. These approaches identified multiple pathways that may play a role in developing melanoma and are described in the Supplementary Note.
Figure 2. Overlap of loci identified by primary and secondary analyses.
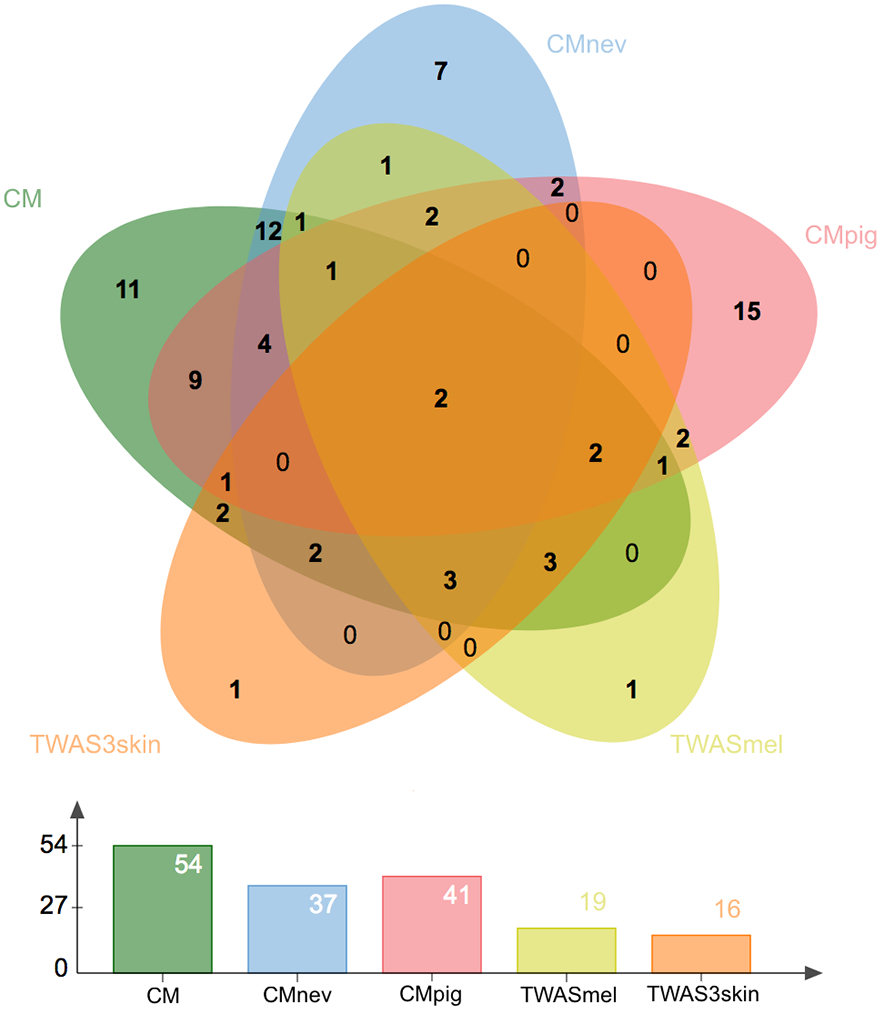
Loci identified in the total cutaneous melanoma meta-analysis (CM, green, Supplementary Table 3), the pleiotropic analysis with nevus count (CMnev, blue, Supplementary Table 9) and hair color (CMpig, red, Supplementary Table 10), melanocyte TWAS (TWASmel, yellow, Supplementary Table 10), and TWAS using the expression of three skin tissues (TWAS3skin, orange, Supplementary Table 12).
Discussion
We report the largest cutaneous melanoma GWAS meta-analysis to date with over three times the effective sample size of prior analyses (Supplementary Table 1). We identified 68 independent cutaneous melanoma -associated variants across 54 loci. TWAS analysis, eQTL colocalization and multi-marker genomic annotations, identified promising gene candidates at many of these risk loci. Joint pairwise GWAS with the cutaneous melanoma-related traits of nevus count and hair color, and TWAS identified a further 31 independent loci that, while not formally reaching genome-wide significance for cutaneous melanoma alone, represent potential additional risk loci. Our cutaneous melanoma meta-analysis also confirmed several loci previously identified only by TWAS28, supporting the value of TWAS in identifying additional genes associated with cutaneous melanoma (Table 4). In total, our integrative analysis identified 85 loci associated with cutaneous melanoma susceptibility (Tables 1-4, Figure 2), constituting a substantial increase from the 21 loci previously identified by cutaneous melanoma susceptibility GWAS alone (Table 1), in addition to those found by family-based approaches or in combination with nevus GWAS data (Table 2).
Our analyses showed strong genetic correlation between self-reported and clinically-confirmed cases (Supplementary Table 2; Supplementary Note), and inclusion of self-reported cases enabled the identification of 11 additional cutaneous melanoma susceptibility loci (Supplementary Tables 3,6; Supplementary Note), indicating that self-reported cutaneous melanoma cases are a valuable and reliable resource for genomic cutaneous melanoma studies. Furthermore, we assessed cutaneous melanoma genetic susceptibility across several geographic regions, including the often-underrepresented Mediterranean population. Interestingly, we found little evidence for difference in cutaneous melanoma locus effect estimates by contributing GWAS (Supplementary Figure 2) or differences in effect size and allele frequency by geographic regions (Supplementary Figure 3), beyond minor variation in pigmentation genes (e.g., rs6059655 near ASIP and rs1805007 near MC1R). The stratified analysis based on cutaneous melanoma histological subtypes identified acral lentiginous melanomas as being uniquely unassociated with pigmentation loci, in line with observational data29. In contrast, the stratified analyses based on age at diagnosis and gender found no evidence for differences in the distribution of nevus-related or pigmentation-related loci.
The discovery of new loci and genes augments our understanding of cutaneous melanoma risk and provides many new insights into cutaneous melanoma etiology. Many of the loci previously associated with nevus count27 or pigmentation57 are also associated with cutaneous melanoma (Table 2) confirming the close relationship between these traits. Specifically, of 10 loci previously significantly-associated in a joint analysis of cutaneous melanoma and Nevus, but not associated with cutaneous melanoma alone27, 6 are now associated with cutaneous melanoma alone (Table 2), demonstrating the benefits of conducting joint analyses. The remaining 4 loci reach P < 5 × 10−8 in the joint cutaneous melanoma+Nevus analysis (Supplementary Table 9); 3 of which are significant at the Bonferroni corrected threshold of 1.25 × 10−8 (Table 3). In turn, we conducted further pleiotropic analyses and identified secondary loci associated with a combination of both these traits and cutaneous melanoma, but not significantly associated with cutaneous melanoma alone (Table 3). Loci found in such joint analyses are of value as they would likely be associated with cutaneous melanoma alone in a sufficiently large cutaneous melanoma GWAS meta-analysis. These joint analyses provide a direct biological interpretation that several GWAS risk loci may act through nevus development, in line with clinical evidence. Interestingly, following these expanded pleiotropic analyses, many loci were associated with neither nevus count or hair color, indicating that many risk variants act outside of these classic cutaneous melanoma risk phenotypes (Tables 1-2).
The discovery of many new loci, when added to the existing catalog of melanoma risk loci, augments our understanding of the genetic architecture of cutaneous melanoma, as discussed in the Supplementary Note. It is important to note that confirmation of the genes we have identified are causal for cutaneous melanoma, and the biological understanding of how variants at these loci influence cutaneous melanoma, remains to be functionally established. For example, melanocyte eQTL and TWAS analyses indicated PARP1 expression was associated with cutaneous melanoma risk SNPs at 1q4228,58. While PARP1 is an established DNA repair gene, extensive functional characterization of the cutaneous melanoma risk locus over PARP1 demonstrated that its role in cutaneous melanoma appears to be through regulation of melanocyte proliferation, senescence, and transcriptional regulation of the key melanoma oncogene MITF 58. Despite the need for follow-up functional studies, a preliminary, complex model of pathways potentially important for the development of melanoma is emerging through the candidate genes suggested by this and prior work, including pathways mediating protection against UV-induced DNA damage and DNA repair, telomere maintenance, immunity, melanocyte differentiation, and cell adhesion.
For example, we identified an association between multiple independent variants at the TP53 locus, rs78378222 and rs1641548, and cutaneous melanoma further reinforcing the potential importance of DNA repair and genome integrity for cutaneous melanoma susceptibility (see Supplementary Note). Rare germline mutations in TP53 lead to Li-Fraumeni syndrome59 which is associated with early onset of cancer, including cutaneous melanoma60. Notably, one of the common sequence variants we found to be associated with cutaneous melanoma has previously been shown to alter TP53 mRNA levels by disruption of TP53 polyadenylation. TP53 responds to cellular stresses to regulate target gene expression resulting in DNA repair, cell cycle arrest, apoptosis, and cellular senescence61,62; variation resulting in loss of normal TP53 function could result in clonal expansion of cells that carry accumulated mutations, which may explain the association with both cutaneous melanoma and nevus count.
This study also adds to a growing body of evidence supporting a key role for telomere maintenance in cutaneous melanoma susceptibility8,9,13,14,37,51,63, with cutaneous melanoma risk loci associated with telomere length or located near prominent telomere maintenance genes or loci, including POT1, TERC, RTEL1, MPHOSPH6, and OBFC1. Additional previously-identified GWAS loci are located near CCND1 (rs4354713), ATM (rs1801516), and PARP1 (rs2695237), all genes with established roles in telomere maintenance, DNA repair, and regulation of senescence64,65.
The well-established role of immunity in melanoma biology has fueled a search for an association between variation within the HLA region and melanoma risk66-68. While several studies have investigated associations between HLA alleles and cutaneous melanoma, these studies have largely been conducted on small, underpowered datasets and have not been consistently replicated69-79. Here, we report identification of a genome-wide significant association between cutaneous melanoma susceptibility and rs28986343 at the HLA locus (see Supplementary Note). This additional evidence for a role for immunity adds to previous28 and current TWAS and colocalization analyses suggesting association between rs408825 and expression of the innate immunity gene MX2. Additionally, many risk alleles for the autoimmune melanocyte-related disorder vitiligo48,80 are protective for cutaneous melanoma with the lead SNPs either identical (rs1126809/TYR; rs6059655/ASIP), or in strong LD with cutaneous melanoma lead SNPs (rs251464 near PPARGC1B for vitiligo, rs32578 for melanoma, LD r2EUR = 0.73; rs72928038 near BACH2 for vitiligo, rs6908626 for melanoma, r2EUR = 0.95; rs1129038 near OCA2 for vitiligo, rs12913832 for melanoma, r2EUR = 0.99). While the vitiligo and cutaneous melanoma associations share many similar loci, suggesting a role for immunity, we cannot rule out their action on cutaneous melanoma risk being through pigmentation or protection against UV damage. Taken as a whole, these data suggest further investigation into these potentially immune-related associations, and more broadly the role of immunity in melanoma risk.
New loci emerging from these analyses suggest a role of genes or networks regulating the development and differentiation of the melanocytic lineage. The cutaneous melanoma meta-analysis identified a locus near FOXD3, while the pleiotropic cutaneous melanoma+Nevus analysis and TWAS locus identified a novel locus significantly associated with allelic expression of NOTCH2 in melanocytes (Supplementary Note). FOXD3 participates as a part of a larger gene regulatory network governing the development of melanocytes from the neural crest, at least in part through transcriptional repression of one of the earliest markers of melanoblast development (and melanoma predisposition gene), MITF81,82. NOTCH2, as well as NOTCH1, appear to play roles in both development of the melanocyte lineage as well as maintenance of melanocyte stem cells53,83, and NOTCH signaling has been shown to lead to de-differentiation of melanocytes to multipotent neural crest stem-like cells84. These two new candidate susceptibility genes join previously-identified loci also harboring genes involved in melanocyte fate. Whole-genome and targeted sequencing studies of melanoma-prone families led to the identification of a functional intermediate-penetrance missense mutation of MITF associated with both melanoma and nevus count (MITF p.E318K)25,26, a variant that was rediscovered by this population-based meta-analysis (rs149617956, P = 5.17 × 10−25, OR = 0.38). Additionally, a previously-identified melanoma and nevus risk locus85 is located ~200 kb from SOX10, another key regulator of melanocyte development and differentiation and direct transcriptional activator of MITF. These genes, and others in this gene regulatory network, have likewise been variously implicated in the progression of melanoma86-90.
The identification of a cutaneous melanoma risk locus for which risk genotype strongly correlates with higher melanocyte-specific expression of CDH1, encoding E-cadherin, suggests a potential role for cell-cell adhesion in melanoma risk (see Supplementary Note). E-cadherin plays a crucial role in cell-cell adhesion, epithelial-mesenchymal transition (EMT) and carcinoma progression. Germline mutations in this gene are associated with a variety of tumors including gastric91, breast92, and potentially colorectal cancer93. In human skin, E-cadherin is typically expressed on the cell surface of both melanocytes and keratinocytes and is considered the major adhesion molecule between these two cell types54,55. During melanoma progression, expression of E-cadherin is typically lost, with a concurrent switch to expression of N-cadherin, facilitating preferential association with fibroblasts and vascular endothelial cells55. In contrast to loss of E-cadherin expression with melanoma progression, we find the cutaneous melanoma risk allele at this locus to be associated with higher expression of CDH1. Interestingly, melanocytes in non-lesional skin of vitiligo patients have been found to have loss of or discontinuously distributed E-cadherin expression. This loss of E-cadherin induces reduced adhesiveness to the basal layer under oxidative and mechanical stress, leading melanocytes to migrate passively to the exterior of the skin, and die by apoptosis94. Thus, germline variation leading to higher melanocyte CDH1 could act as a protective mechanism, allowing cells damaged by oxidative stress to remain in the skin and survive without dying. A similar mechanism has been recently identified in breast cancer metastasis, where E-cadherin acts as a survival factor by limiting reactive oxygen-mediated apoptosis95.
In summary, our large, international genetic meta-analysis showcases the utility of including self-reported cutaneous melanoma cases, complementary analytical approaches, and data from multiple sources to expand our understanding of cutaneous melanoma risk. While the biological mechanisms underlying many of the existing and novel cutaneous melanoma risk loci remain to be confirmed or discovered by post-GWAS functional studies and even larger GWAS, these data suggest potential pathways novel to melanoma susceptibility, and highlight nevus formation, pigmentation and telomere maintenance, the three pathways that appear to dominate the landscape of melanoma susceptibility.
Table 1.
Loci previously identified in cutaneous melanoma susceptibility GWAS. CHR, BP: hg19 positional information. rsID: dbSNP142 rs number. Publications. We also summarize Supplementary Table 3; Gene prioritizes the functional target if known, followed by melanocyte or skin tissue TWAS data, or finally the closest protein coding gene; ‘Multiple’ indicates three or more genes. GWS: We indicate with yes (Y) or no (N) whether this locus is genome-wide significant (P < 5 × 10−8) in the total meta-analysis. The effect allele (EA) and non-effect allele (NEA) are listed, as are the effect allele Frequency in the HRC reference panel107; total fixed-effects inverse-variance weighted meta-analysis of logistic regression two-sided P-value (Pmeta) and Odds Ratio (OR) are from an additive model and are reported per-allele with respect to the EA. Reported results are for the total meta-analysis (36,760 melanoma cases and 375,188 controls; for full details of analysis and covariates included see Online Methods). We also indicate whether this locus is associated with other traits: Nevi: Pleiotropically associated with cutaneous melanoma and nevus count (Online Methods; Supplementary Table 9); Hair: Pleiotropically associated with cutaneous melanoma and hair color (Online Methods; Supplementary Table 10). Tanning response (Tan) and Telomere length (Telo) indicates the lead SNP is associated with these traits when corrected for multiple testing (Online Methods, Supplementary Table 5).
| CHR:BP | rsID | Pub | Gene | EA/ NEA |
Freq | Pmeta | OR | Nevi | Hair | Tan | Telo |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1:150,938,571 | rs8444 | 108 | Multiple | G/A | 0.645 | 3.89 × 10−14 | 1.08 | - | - | Y | - |
| 1:226,603,635 | rs2695237 | 27,108,109 | PARP1 | T/C | 0.628 | 1.53 × 10−18 | 1.10 | Y | - | - | - |
| 2:38,298,139 | rs1800440 | 23,27 | CYP1B1 | T/C | 0.824 | 6.97 × 10−15 | 1.10 | Y | - | Y | - |
| 2:202,143,928 | rs10931936a | 20 | CASP8 | T/C | 0.281 | 2.17 × 10−8 | 1.08 | - | - | - | - |
| 5:1,323,212 | rs13178866a | 20,110,111 | TERT | C/T | 0.554 | 2.59 × 10−18 | 0.87 | - | Y | - | Y |
| 5:33,951,693 | rs16891982a | 20,34,111 | SLC45A2 | C/G | 0.122 | 1.96 × 10−28 | 0.51 | - | Y | Y | - |
| 6:21,163,919 | rs6914598 | 23 | CDKAL1 | T/C | 0.683 | 1.18 × 10−18 | 0.91 | - | - | Y | - |
| 7:17,134,708 | rs117132860b | 23,57 | AGR3 | G/A | 0.981 | 3.83 × 10−21 | 0.71 | Y | - | Y | - |
| 9:21,803,880 | rs871024a | 18,27 | MTAP, CDKN2A | C/A | 0.477 | 2.72 × 10−23 | 1.18 | Y | Y | - | - |
| 9:109,054,417 | rs10739220 | 23,27 | TMEM38B | C/T | 0.260 | 1.34 × 10−18 | 1.10 | Y | Y | - | - |
| 10:105,694,301 | rs7902587 | 23,27 | OBFC1 | C/T | 0.904 | 2.68 × 10−23 | 0.86 | Y | - | - | Y |
| 11:69x,380,898 | rs4354713 | 20,23 | CCND1 | A/G | 0.356 | 8.50 × 10−21 | 1.10 | - | Y | - | - |
| 11:89,017,961 | rs1126809a | 18 | TYR | G/A | 0.757 | 4.78 × 10−37 | 0.83 | - | Y | Y | - |
| 11:108,175,462 | rs1801516 | 20 | ATM | G/A | 0.856 | 2.22 × 10−21 | 1.14 | Y | - | - | - |
| 15:28,365,618 | rs12913832a | 19,23 | OCA2 | A/G | 0.335 | 4.85 × 10−12 | 0.88 | - | Y | Y | - |
| 16:89,986,117 | rs1805007a | 18 | MC1R | C/T | 0.937 | 5.86 × 10−52 | 0.57 | Y | Y | Y | - |
| 20:32,665,748 | rs6059655a | 17,18 | ASIP | A/G | 0.061 | 2.52 × 10−42 | 1.45 | - | Y | Y | - |
| 21:42,743,496 | rs408825 | 20 | MX2 | C/T | 0.413 | 1.03 × 10−32 | 0.89 | - | - | Y | - |
| 22:38,545,942 | rs132941 | 18,27,35 | MAFF | T/C | 0.549 | 8.80 × 10−23 | 1.10 | Y | - | Y | - |
Variant meta-analysis results are heterogeneous (I2 > 31%) and random effects estimates are presented.
While this locus overlaps the previously reported IRF4 or AGR3 locus, the lead variants are independent.
Table 2.
Novel loci not previously identified in cutaneous melanoma GWAS. CHR, BP: hg19 position. rsID: dbSNP142 rs number. Gene prioritizes the functional target if known, followed by melanocyte or skin tissue TWAS data, or finally the closest protein coding gene; multiple indicates three or more genes (Supplementary Table 3). The effect allele (EA) and non-effect allele (NEA) are listed, as are the effect allele Frequency in the HRC reference panel107; total fixed-effects inverse-variance weighted meta-analysis of logistic regression two-sided P-values and Odds Ratio (OR) are with respect to the EA. Reported results are for the total meta-analysis (36,760 melanoma cases and 375,188 controls; for full details of analysis and covariates included see Online Methods). Nevi: Associated with cutaneous melanoma+nevus count (Online Methods; Supplementary Table 9); Hair: Associated with cutaneous melanoma+hair color (Online Methods; Supplementary Table 10). Tanning response (Tan) and Telomere length (Telo) indicate lead SNP is associated with these traits when corrected for multiple testing (Online Methods, Supplementary Table 5).
| CHR:BP | rsID | Gene | EA/ NEA |
Freq | Pmeta | OR | Nevi | Hair | Tan | Telo |
|---|---|---|---|---|---|---|---|---|---|---|
| 1:63,727,542 | rs670318 | FOXD3 | T/C | 0.047 | 1.21 × 10−8 | 0.86 | - | - | Y | - |
| 1:154,994,978 | rs76798800 | ZBTB7B, ADAM15, GBA | G/T | 0.753 | 3.86 × 10−15 | 0.92 | Y | - | Y | - |
| 1:205,181,062 | rs2369633 | DSTYK | T/C | 0.083 | 1.24 × 10−8 | 1.10 | - | -e- | Y | - |
| 2:25,778,637 | rs12473635 | DTNB | T/C | 0.776 | 5.17 × 10−9 | 0.93 | Y | - | - | - |
| 3:70,014,091 | rs149617956a | MITF | G/A | 0.998 | 9.00 × 10−14 | 0.39 | - | Y | Y | - |
| 3:169,493,283 | rs3950296b | TERC | C/G | 0.747 | 4.47 × 10−11 | 1.08 | Y | - | - | Y |
| 5:90,262,612 | rs12523094c | GPR98 | T/C | 0.567 | 1.74 × 10−6c | 1.07 | - | Y | Y | - |
| 5:149,211,868 | rs32578b,d | PPARGC1B | G/A | 0.658 | 6.58 × 10−17 | 1.09 | Y | - | Y | - |
| 6:1,145,265 | rs12215602a | IRF4 | G/A | 0.721 | 7.91 × 10−9 | 0.94 | Y | - | Y | - |
| 6:22,719,379 | rs72834823 | HDGFL1 | T/A | 0.819 | 1.04 × 10−12 | 1.10 | Y | - | Y | - |
| 6:32,748,953 | rs28986343 | HLA-DQB2 | C/T | 0.952 | 1.61 × 10−8 | 1.15 | - | - | - | - |
| 6:91,005,743 | rs6908626 | BACH2 | G/T | 0.844 | 3.92 × 10−9 | 1.09 | - | - | - | - |
| 7:22,115,454 | rs12539524 | RAPGEF5 | C/T | 0.846 | 1.65 × 10−8 | 0.93 | - | - | - | - |
| 7:124,396,645 | rs4731207 | POT1 | G/A | 0.540 | 2.22 × 10−15 | 0.93 | Y | - | - | Y |
| 7:130,738,666 | rs7778378 | MKLN1 | C/T | 0.248 | 8.93 × 10−9 | 0.93 | Y | Y | - | - |
| 8:21,951,009 | rs6994183 | FAM160B2 | A/T | 0.866 | 4.84 × 10−9 | 0.92 | - | - | - | - |
| 8:72,864,240 | rs13263376c | RP11-383H13.1, MSC | G/A | 0.364 | 2.28 × 10−8c | 0.93 | Y | - | Y | - |
| 9:12,587,153 | rs10960710 | TYRP1 | G/T | 0.393 | 3.08 × 10−12 | 0.93 | - | Y | Y | - |
| 9:110,711,586 | rs1339759b | KLF4 | C/G | 0.666 | 5.61 × 10−19 | 1.10 | Y | - | - | - |
| 9:134,457,580 | rs3780269 | RAPGEF1 | G/A | 0.691 | 1.92 × 10−8 | 0.94 | Y | - | - | - |
| 11:16,041,305 | rs7941496 | SOX6 | G/T | 0.516 | 1.40 × 10−9 | 1.06 | Y | - | Y | - |
| 11:120,195,702 | rs12290699 | TMEM136 | T/C | 0.745 | 2.20 × 10−8 | 0.94 | - | - | - | - |
| 12:13,070,752 | rs1056927b,c | Multiple | A/G | 0.561 | 2.74 × 10−9b | 0.93 | Y | - | - | - |
| 12:17,275,460 | rs4237963 | LMO3 | T/A | 0.207 | 1.27 × 10−9 | 0.93 | - | - | - | - |
| 12:96,378,807 | rs10859996 | HAL, RP11-256L6.3 | C/T | 0.635 | 2.09 × 10−10 | 1.07 | - | - | - | - |
| 12:116,580,291 | rs113469387 | MED13L | G/A | 0.907 | 8.76 × 10−10 | 0.91 | - | Y | Y | - |
| 13:113,535,949 | rs1278768 | MCF2L | G/C | 0.488 | 6.33 × 10−12 | 0.94 | - | - | Y | - |
| 15:33,277,710 | rs117648907b | FNM1 | C/T | 0.983 | 7.29 × 10−12 | 0.80 | Y | - | - | - |
| 16:68,822,971 | rs4420522 | Multiple, CDH1 | A/G | 0.690 | 8.34 × 10−14 | 0.93 | Y | Y | - | - |
| 16:82,217,153 | rs2967383 | MPHOSPH6 | G/T | 0.267 | 2.24 × 10−9 | 1.06 | - | - | - | Y |
| 17:7,571,752 | rs78378222 | TP53 | T/G | 0.989 | 3.33 × 10−10 | 0.76 | Y | - | - | - |
| 19:3,540,539 | rs12984831b | MFSD12 | G/C | 0.984 | 3.86 × 10−10 | 0.65 | Y | - | Y | - |
| 20:62,291,767 | rs143190905 | RETL1 | G/T | 0.907 | 6.54 × 10−13 | 1.15 | - | - | - | Y |
| 22:45,622,684 | rs5766565 | KIAA0930 | A/G | 0.647 | 1.44 × 10−9 | 1.06 | Y | Y | Y | - |
| 22:50,722,408 | rs79966207 | PLXNB2 | T/C | 0.849 | 8.68 × 10−9 | 0.92 | - | Y | - | - |
Previously associated pleiotropically with cutaneous melanoma and nevus count27.
Variant meta-analysis results are heterogeneous (I2>31%) and random effects estimates are presented. For rs12523094/GPR98 while the lead SNP selected in conditional mapping is heterogenous, other SNPs in LD pass this requirement (e.g., rs12173258, r2EUR = 0.9, Pmeta = 1.09 × 10−11, I2 = 29.6).
Previously associated with tanning response57.
Joint cutaneous melanoma+hair color P-value is greater than multiple testing corrected threshold of 1.25 × 10−8 (Supplementary Table 10).
Table 3.
Novel pleiotropic associations with cutaneous melanoma and nevus count or hair color. Reported cutaneous melanoma P-values are from the total fixed-effects inverse-variance weighted meta-analysis of logistic regression two-sided P-values from GWAS representing a total of 36,760 melanoma cases and 375,188 controls (Online Methods). Results for the lead variants from pleiotropic loci (lead SNP reaching P < 5 × 10−8 following a Stouffers sample size weighted meta-analysis of cutaneous melanoma P-values and either Nevus GWAS meta-analysis (N = 65,777) or Hair Color GWAS (N=352,662) and GWAS-PW Model 3 prior probability of association (PPA) > 0.5, Online Methods) distinct to those in the total cutaneous melanoma meta-analysis (Table 1, 2). CHR, BP: hg19 positional information. rsID: dbSNP142 rs number. Gene prioritizes genes that the variant is an eQTL for in GTEx skin datasets or otherwise is the closest protein coding gene; multiple indicates three or more genes. We report the total cutaneous melanoma meta-analysis P (CM P), and the CM+nevus or CM+hair color Stouffer’s meta-analysis fixed effect P-value. Full results can be found in Supplementary Tables 7 and 10.
| CHR:BP | rsID | Gene | CM P | CM + Nevus P | CM + Hair P |
|---|---|---|---|---|---|
| 1:24787947 | rs195720 | NIPAL3 | 7.97 × 10−6 | - | 2.24 × 10−12 |
| 1:78450517 | rs34517439 | DNAJB4 | 2.23 × 10−4 | - | 2.17 × 10−12 |
| 1:214673271 | rs7533482 | PTPN14 | 2.79 × 10−5 | - | 2.45 × 10−13 |
| 2:135430709 | rs6745983 | TMEM163 | 1.69 × 10−3 | - | 7.00 × 10−13 |
| 2:214065880 | rs16849932 | IKZF2 | 1.46 × 10−3 | - | 1.18 × 10−10 |
| 2:240065356 | rs11677464a | HDAC4 | 4.00 × 10−5 | 1.10 × 10−9 | - |
| 4:37470753 | rs11730662 | KIAA1239 | 1.82 × 10−3 | 1.19 × 10−8 | - |
| 5:56011357 | rs7714232 | MAP3K1 | 6.99 × 10−4 | - | 3.32 × 10−22 |
| 6:7189567 | rs75818295 | RREB1 | 1.87 × 10−3 | - | 8.27 × 10−10 |
| 6:11637483 | rs548304 | ADTRP | 2.67 × 10−5 | - | 1.46 × 10−10 |
| 6:15503696 | rs10949304 | DTNBP1 | 1.7 × 10−3 | 4.96 × 10−9 | - |
| 6:50790642 | rs2857482 | TFAP2B | 3.59 × 10−5 | 3.44 × 10−10 | - |
| 6:151577739, 6:151577830 | rs10434895, rs10434896b | AKAP12 | 8.17 × 10−8, 7.88 × 10−8 |
7.71 × 10−10 | 2.07 × 10−42 |
| 8:131138979 | rs111595456 | ASAP1 | 3.86 × 10−4 | 2.83 × 10−10 | - |
| 9:211762, 9:235201 | rs520015, rs593179a,c | CBWD1 | 8.95 × 10−7, 3.78 × 10−6 |
4.13 × 10−12 | 1.10 × 10−43 |
| 10:5767177 | rs76154345a | GDI2 | 4.43 × 10−6 | 7.80 × 10−11 | - |
| 10:111889779 | rs11194997 | MXI1 | 3.45 × 10−6 | - | 2.70 × 10−11 |
| 11:7543519 | rs11041426 | PPFIBP2 | 2.73 × 10−4 | - | 1.66 × 10−33 |
| 11:62203865 | rs10897275 | AHNAK | 6.47 × 10−5 | - | 2.47 × 10−33 |
| 11:91616691 | rs12225068 | FAT3 | 3.80 × 10−5 | - | 6.48 × 10−10 |
| 13:76351286 | rs474240 | LMO7 | 2.53 × 10−4 | - | 9.28 × 10−9 |
| 13:114744546 | rs75414584 | RASA3 | 6.31 × 10−3 | - | 4.62 × 10−12 |
| 14:64390030 | rs10873172a,d | SYNE2 | 6.29 × 10−8 | 5.95 × 10−13 | 6.47 × 10−27 |
| 14:69226931 | rs11625064d | ZFP36L1 | 3.33 × 10−6 | 2.09 × 10−10 | 1.83 × 10−19 |
| 14:92795912 | rs4904871 | SLC24A4 | 2.06 × 10−4 | - | 2.15 × 10−278 |
| 14:103923475 | rs2273699 | MARK3 | 5.27 × 10−5 | - | 1.21 × 10−16 |
| 15:48400199 | rs2675345 | SLC24A5 | 4.92 × 10−3 | - | 1.09 × 10−9 |
| 16:54118132, 16:54131939 | rs62034121, rs62034139a,e | FTO | 1.16 × 10−9, 4.56 × 10−9 | 4.69 × 10−14 | - |
| 16:55322732 | rs12930459a | IRX6 | 1.82 × 10−5 | 4.89 × 10−9 | - |
Locus previously reported as pleiotropically associated with cutaneous melanoma and nevus count, but not significant for cutaneous melanoma alone here.
Lead SNP for Pigment (rs10434895) and nevus (rs10434895) are in LD r2EUR = 1.0.
Lead SNP for Pigment (rs520015) and nevus (rs593179) are in LD r2EUR = 0.63.
Same lead SNP.
Lead SNP for Pigment (rs62034121) and nevus (rs62034139) are in LD r2EUR = 0.88.
Table 4.
Genes identified by TWAS outside of regions identified in the total cutaneous melanoma GWAS meta-analysis. For each gene with a Bonferroni-corrected P-value cutoff in melanocytes (PTWAS < 3.22 × 10−6), or skin-related tissue types (PTWAS < 5.28 × 10−7) that does not overlap with an existing cutaneous melanoma region we report the local peak cutaneous melanoma variant from the total confirmed plus self-report GWAS meta-analysis, and TWAS Z score. Full results for all genes with a PTWAS < 1.48 × 10−5 can be found in Supplementary Tables 10,12. CBWD1 and C9orf66 are within 1 Mb of each other and are merged into a single locus. * RP11-676J12.7 was identified using sun-exposed skin expression data from GTEx (Supplementary Table 12), while all other genes were identified using melanocyte gene expression.
| TWAS | Locus Peak CM Variant | ||||
|---|---|---|---|---|---|
| Gene | Z | P | rsID | CHR:BP | CM P |
| NIPAL3 | 4.84 | 1.28 × 10−6 | rs2294524 | 1:24,770,594 | 2.74 × 10−7 |
| RCAN3 | 4.83 | 1.33 × 10−6 | rs2294524 | 1:24,770,594 | 2.74 × 10−7 |
| NOTCH2 | 4.81 | 1.50 × 10−6 | rs2793830 | 1:120,466,108 | 3.80 × 10−7 |
| PTPN14 | −4.84 | 1.30 × 10−6 | rs6693492 | 1:214,685,978 | 2.68 × 10−5 |
| CBWD1 | −4.81 | 1.51 × 10−6 | rs478882 | 9:205,964 | 1.64 × 10−6 |
| C9orf66 | 5.05 | 4.48 × 10−7 | rs478882 | 9:205,964 | 1.64 × 10−6 |
| SYNE2 | 5.19 | 2.06 × 10−7 | rs12881652 | 14:64,400,120 | 2.12 × 10−7 |
| IRX6 | −4.80 | 1.62 × 10−6 | rs12919110 | 16:55,319,789 | 1.27 × 10−6 |
| RP11-676J12.7* | −5.55 | 2.79 × 10−8 | rs1703824 | 17:813,324 | 1.59 × 10−5 |
Online Methods
Quality control metrics, imputation and association analysis
Data cleaning was performed using Illumina GenomeStudio/BeadStudio (v2.0.4 San Diego, CA, USA) and PLINK (v1.90b5.4) 96,97. Full details of the sample collections and genotyping arrays used for each GWAS are reported in the Supplementary Note. Prior to imputation any SNP with either minor allele frequency (MAF) < 0.01, Hardy-Weinberg Equilibrium (HWE) P-value < 5 x 10−4 in controls or < 5 x 10−10 in cases was removed. Similarly, any individual was removed who was missing > 3% of variants, had heterozygosity values either > 0.05 or < −0.05 or 3 sd from the mean, whose genetically-predicted sex did not match their recorded sex, or who was determined to be non-European based on principal component analysis (PCA). In addition, one of any pair of individuals estimated to be related with identity by descent (IBD) pihat > 0.15 was removed.
The Harvard, BNMS, and 23andMe GWAS were imputed to 1000 Genomes Project phase 1 v3; for all other sets (Supplementary Table 1) imputation was conducted using the Michigan Imputation Server with the Haplotype Reference Consortium panel (HRC version 1) and run using Minimac3 98. Following imputation, any imputed variant with imputation quality score r2 < 0.5 or MAF < 0.0001 was rejected. As rare SNPs where one allele is missing in the case or control group can lead to very large (or infinite) OR estimates, variants with an OR < 1 × 10−4 (the minimum reported by PLINK) or > 1 × 106 were also filtered. To handle variants with the same name (e.g., triplicate SNPs), variant IDs were converted to the format CHR:BP:A1A2 prior to meta-analysis.
Logistic regression under an additive model with ORs calculated on a per-allele basis was then conducted using PLINK (v1.90b5.4) 96,97 with either geographic region (in GenoMEL Phase 1 and 2 data) or principal components as covariates to account for potential population stratification. Individual studies were checked for evidence of inflation by producing QQ plots (Supplementary Figure 1) and calculating the corresponding inflation factor λ and LDSC intercept (Supplementary Table 1).
Where individual studies have deviated from this protocol, details are included in the study description in the Supplementary Note. All reported tests are two-sided.
Meta-analysis and conditional-and-joint-analysis to identify independent loci
Meta-analyses of the GWAS were conducted in one stage using both inverse-variance weighted fixed effects and random effects meta-analysis 99 as implemented in PLINK v1.90b5.4 96,97. Meta-analyses were conducted for confirmed only cases, and in the total set including self-report sets (23andMe, Inc. and a portion of UK Biobank).
Conditional and joint analysis of summary GWAS meta-analysis data was performed using Genome-wide Complex Trait Analysis (GCTA, v1.26.0) to identify independently associated variants 33. To ensure we were only detecting completely independent SNPs the collinearity threshold (--cojo-collinear) was set to R2 = 0.05. The threshold for genome-wide significance 5 × 10−8 and fixed effect meta-analysis p-values and log(OR) effect sizes were analysed.
Linkage-disequilibrium (LD) between SNPs for the conditional and joint analysis of summary data in GCTA (v1.26.0) reported in the manuscript was calculated using a reference population of 5,000 individuals selected randomly from the portion of the UK Biobank population determined to be European by PCA (LDEUR). Variants were converted to best guess genotype (threshold 0.3). Best guess data were cleaned for missingness > 3%, HWE P < 1 × 10−6, MAF < 0.001
To limit the chance of false positive claims of novel SNP/loci, we further filtered the list of 77 conditionally independent variants (Supplementary Table 4) to those (i) genome-wide significant (P < 5 × 10−8) in single SNP and joint conditional analysis, and (ii) as recommended30 where there was evidence of heterogeneity between studies (I2 > 31%) the random effect P-value also needed to be < 5 × 10−8. Passing variants were further checked to ensure that MAFs and effect sizes were consistent across studies and that the result was not driven by a single study (Supplementary Figure 2-3). The 68 retained variants were combined into 54 loci using a concatenating 1 Mb window (Supplementary Table 3). Regional association plots for all 54 loci were interactively plotted by LDassoc (https://ldlink.nci.nih.gov/)100 and included as Supplementary Data 1.
Multiple testing corrections
The primary aim of our study was to perform a GWAS meta-analysis of cutaneous melanoma risk. For this primary analysis our significance threshold was set at p < 5 × 10−8. Following this primary analysis, we conducted two classes of secondary analyses: 1) joint analysis of melanoma with a risk phenotype (Nevus or Pigmentation) and 2) TWAS.
To ensure robust adjustment for multiple testing, within the joint cutaneous melanoma-nevus and cutaneous melanoma-pigmentation GWAS analyses we Bonferroni-corrected for each of the two risk factor phenotypes (pigmentation and nevus count), as well as accounting for the two classes of secondary analysis (joint GWAS and TWAS). The resulting significance threshold was (5 × 10−8)/(2 × 2) = 1.25 × 10−8. Loci reaching this corrected threshold are indicated in bold in Supplementary Tables 7 and 10.
TWAS was performed on expression data from melanocytes, and then separately on the three skin tissues within GTEx (sun-exposed, not-sun-exposed, and fibroblasts) as these were the most enriched tissues in terms of enrichment for cutaneous melanoma heritability after melanocytes (Extended Data Figure 5) and are likely to be involved in cutaneous melanoma development.
For the melanocyte TWAS analysis, we Bonferroni corrected the significance threshold by the number of tested genes in melanocytes multiplied by the 2 classes of secondary tests and further for the 2 tissue sets; 0.05/(3878 genes × 2 classes × 2 tissue sets) = 3.22 × 10−6.
For the GTEx skin TWAS analysis we Bonferroni corrected for the total number of tested genes across the tissues multiplied by two classes of secondary tests and further for the 2 tissue sets; 0.05/( ( 8879 + 7458 + 7353 genes) × 2 classes × 2 tissue sets = 5.28 × 10−7.
The accuracy of p-value calculation for rare SNPs where case/control numbers are imbalanced
The non-normality of the test statistics may cause severely inflated P-values due to violation of asymptotic approximations, particularly for imbalanced case-control ratios. While we addressed this for extreme cases by filtering very rare SNPs (Online Methods), we also investigated whether this could be inflating the P-value of rare SNPs included in the meta-analysis by performing 5 × 108 simulations. For each simulation, we first generated genotype data for 21 studies with the same sample size as in our meta-analysis (Supplementary Table 1) assuming Hardy Weinberg equilibrium for variants with MAF = 0.01.
We then performed association testing for each study and calculated the test statistics to derive an empirical P-value of 6.4 × 10−8 when using an asymptotic P-value of 5 × 10−8 as the threshold. While imbalanced case-control ratios had minimal impact on the calculation of asymptotic p-values for SNPs with MAF = 0.01, as the empirical P-value was slightly larger than genome-wide significance we further explored the results of our meta-analysis. Three of our 68 reported variants have a MAF less than 0.01: rs149617956 with MAF = 0.002, rs79356439 with MAF = 0.008 and rs3212371 with MAF = 0.003. All three variants had asymptotic p-values < 5 × 10−12. We performed 5 × 108 simulations for each of the variants using their MAF, and found no simulations had a nominal P-value < 5 × 10−12. These simulations indicate that the actual p-values for these three SNPs are less than 1/(5 × 108) = 2 ×10−9, and have reached genome-wide significance.
Joint analyses of cutaneous melanoma and nevus count and pigmentation
Nevus GWAS meta-analysis
Using beta meta-analysis weighted by SE as implemented in PLINK 1.90b5.4, we combined the recently published nevus meta-analysis (N = 52,506) 27 which excluded samples with melanoma but may include a small portion of overlap with the controls used for some melanoma GWAS datasets; participants of the QSkin study with nevus count that are non-overlapping and unrelated (IBD pihat < 0.15) to the QSkin melanoma case control set (N = 12,930) and the final set of participants not previously included from the Brisbane Twin Nevus Morphology study (N = 341) 27. The total sample size was 65,777.
Pigmentation GWAS
A GWAS for hair color was performed on 352,662 UK Biobank samples not included in the melanoma GWAS who self-reported having either blonde, light brown, dark brown or black hair (coded as 1, 2, 3 and 4). Hair color was then treated as a continuous variable and regressed on imputed genotype adjusting for principal comments using the same approach as for the melanoma GWAS.
Joint analyses
The melanoma results were then jointly analyzed first with nevus count and then with hair color. Two approaches were taken. Firstly the total confirmed plus self-report cutaneous melanoma GWAS meta-analysis results were combined with the separate nevus and pigmentation GWAS data using Stouffer’s method (P-value weighted by per SNP sample N) as implemented in METAL (version 2011-03-25)101. LD calculations were performed in PLINK using a reference panel of 10,000 white British UK Biobank individuals as implemented in the FUMA platform (v1.3.5)102 was used to identify independent SNPs with P < 5 × 10−8; independent SNPs within 1 Mb were considered to be single loci. Secondly, the melanoma and pigmentation/nevus GWAS results were analyzed using GWAS-PW (v0.21)38, which estimates the posterior probability of four possible models for each genetic region: (i) association with cutaneous melanoma only, (ii) association with the second trait only, (iii) association with both traits (pleiotropic), (iv) association with both traits, but co-located and independent (v) no association with either trait. Given that nevus count and pigmentation are believed to act directly on melanoma risk, model (iv) seemed unrealistic so we only considered models (i), (ii), (iii) and (v). For nevus count, SNPs were assigned to blocks using the recommended boundaries for GWAS-PW (https://bitbucket.org/nygcresearch/ldetect-data). For cutaneous melanoma and hair color, 50 SNP windows were used for blocks as the default LD blocks contained multiple independent hair color loci. Following the approach taken by27, any locus with a lead SNP reaching P < 1.25 × 10−8 for the combined cutaneous melanoma and nevus/hair color analysis and with a posterior probability > 0.5 that the locus is associated with both traits (model 3) to ensure that the association is not driven by a single trait was declared to be pleiotropically associated with both traits.
Analysis of pigmentation and nevi polygenic risk score across melanoma subtypes
For each subject in our study, we calculated two polygenic scores (PRS), using 276 genetic variants associated with pigmentation and 10 genetic variants associated with nevus count. Nevus count SNPs were derived from the same nevus GWAS meta-analysis used for the pleiotropic analysis (N = 65,597), with independent lead SNPs with P < 5 × 10−8 identified using LD calculations performed in PLINK using a reference panel of 10,000 white British UK Biobank individuals as implemented in the FUMA platform (v1.3.5)102, with the LD r2 cut off for independence < 0.05. Pigmentation PRS SNPs were selected from the hair color GWAS used for the pleiotropic analysis (N= 352,662), with independent lead SNPs with P < 5 × 10−8 and LD calculations performed in PLINK using a reference panel of 10,000 white British UK Biobank individuals as implemented in the FUMA platform, with the LD r2 cut off for independence < 0.025. PRS were calculated for each subject by applying the regression coefficient (from the GWAS of pigmentation or nevus count) to the genotype dosages. We then tested whether PRS distribution differed between males and females, across age groups, and histology subtypes. In total, we performed 27 comparisons and thus any comparison with p-value less than 0.05/27 (=0.00186) was declared as statistically significant.
GENESIS estimation of heritability and polygenic risk
We used GENESIS (https://github.com/yandorazhang/GENESIS)32 (Version 2019-06-01) to estimate the genetic architecture (number of causal SNPs and their effect size distribution) using the summary level statistics from the GWAS meta-analysis. Quantile-quantile plot comparing the p-values generated from this fitted distribution against the observed p-values suggested a three component Gaussian mixture model for the effect size distribution. Based on this estimated genetic architecture, we calculated the heritability at the observational scale and the number of SNPs reaching genome-wide significance for a given GWAS with known sample size. Similarly, GENESIS calculated the AUC for an additive polygenic risk prediction model built based on a discovery GWAS of known sample size.
UK Biobank melanoma risk phenotype GWAS
Four pigmentary GWAS were performed on UK Biobank participants not included in the melanoma GWAS (1) Ease of tanning with 367,229 UK Biobank samples who self-reported their ability to tan as either ‘Get very tanned’, ‘Get moderately tanned’, ‘Get mildly or occasionally tanned’ or ‘Never tan, only burn’ (coded as 1, 2, 3 and 4). Ease of tanning was treated as a continuous variable and regressed on imputed genotypes adjusting for principal components using the same approach as for the melanoma GWAS of UK Biobank data. (2) Skin color with 370,260 UK Biobank samples who self-reported having either ‘Very fair’, ‘Fair’, ‘Light olive’, ‘Dark olive’, ‘Brown’, or ‘Black’ skin color (coded as 1, 2, 3 and 4). Skin color was treated as a continuous variable and regressed on imputed genotype adjusting for principal components using the same approach as for the melanoma GWAS of UK Biobank data. (3) Number of childhood sunburns with 320,345 UK Biobank samples who self-reported their sunburn incidents pre-sixteen years old. The data were dichotomized into none and at least one pre-sixteen sunburn incident categories (coded as 1, 2). Number of childhood sunburns was treated as a binary variable and regressed using a logistic model on imputed genotype adjusting for principal comments using the same approach as for the melanoma GWAS of UK Biobank data. (4) Red hair with 120,925 UK Biobank samples who self-reported having either ‘red hair’ or other (coded as 1 or 2). Red hair was treated as a binary variable and regressed using a logistic model on imputed genotype adjusting for principal comments using the same approach as for the melanoma GWAS of UK Biobank data.
Linkage disequilibrium (LD) score regression
As LD score regression (LDSC) is sensitive to the quality of input SNPs, GWAS or meta-analysis variants were filtered to the list of high quality HapMap SNPs provided103. Using LD Score regression v1.0.0 genomic inflation (Lambda), intercept and SNP-heritability (h2) were estimated. h2 estimates were converted to the liability scale using the lifetime population prevalence for cutaneous melanoma in Australia (0.0588)104.
LD score regression of tissue-specific genes
Cutaneous melanoma heritability enrichment for SNPs around tissue-specific genes was assessed by stratified LD score regression as described previously 28,42 and implemented in the LDSC program v1.0.0 (https://github.com/bulik/ldsc). Briefly, RNA-seq data for all 50 GTEx (v7) tissue types and primary melanocyte were quantified as RPKM using RNA-SeQC (v1.18) 105 and quantile normalized to reduce batch effect. Tissue-specific genes were defined by calculating the t-statistic of each gene for a given tissue, excluding all samples from the same tissue category. Tissue category assignment for GTEx tissue types was based on the previous publications 28,106, and melanocytes were defined as “skin” category together with two types of skin and transformed skin fibroblasts from the GTEx. We selected the top 1,000, 2,000, and 4,000 tissue-specific genes from the t-statistic analysis, and added 100 Kb each to the transcription start site and transcription end site to define tissue-specific genes annotation. Stratified LD score regression was then applied on a joint SNP annotation to estimate the heritability enrichment against the total cutaneous melanoma GWAS data from the current study.
Colocalization of cutaneous melanoma GWAS and eQTLs
We performed colocalization analyses of cutaneous melanoma GWAS signals with eQTL signals from our melanocyte and 48 GTEx (v7) tissue eQTL datasets (note that 2 tissue types that were included for LDSC using expression data were not included here as well as in TWAS analyses due to lack of eQTL data from GTEx), using eQTL and GWAS CAusal Variants Identification in Associated Regions (eCAVIAR, v2.0, http://genetics.cs.ucla.edu/caviar/, https://github.com/fhormoz/caviar)43. Consistent with the previous study, we used 50 SNPs upstream and downstream of each cutaneous melanoma GWAS lead SNP to extract both GWAS and eQTL summary statistics to be used as the input for eCAVIAR analysis. The LD matrix was calculated using the unphased 1000 Genomes reference set. For the CLPP score calculation, we allowed a maximum number of two causal SNPs in each locus. For a given cutaneous melanoma GWAS locus, an eGene with a CLPP score above 1% (0.01) was considered to display a positive co-localization. To avoid reporting spurious effects, we applied a conservative criterion and only reported variants displaying LD r2 > 0.9 with the cutaneous melanoma GWAS lead SNP and eQTL P-value below a Bonferroni-corrected cutoff of each dataset (0.05/number of eGenes tested for each tissue dataset).
TWAS
We performed transcriptome-wide association studies (TWAS) for the cutaneous melanoma GWAS meta-analysis data using TWAS/FUSION (http://gusevlab.org/projects/fusion/) as previously described28,41. TWAS was performed in three separate groups, using eQTL datasets from 1) melanocytes, 2) three skin tissues (sun-exposed, not-sun-exposed, and fibroblasts) within GTEx (V7), and 3) the rest of GTEx tissue types (a total of 45) by imputing the gene expression phenotypes for the total cutaneous melanoma GWAS meta-analysis data. The analysis parameters were set to allow for multiple prediction models, independent reference LD, additional feature statistics and cross-validation results41. The total cutaneous melanoma GWAS meta-analysis summary statistics were included with no significance thresholding. For GTEx data, we downloaded the precomputed expression reference weights for GTEx gene expression (v7) RNA-seq across 48 tissue types from the TWAS/FUSION website (http://gusevlab.org/projects/fusion/). We computed functional weights from the primary melanocyte RNA-seq data28 one gene at a time. Genes that failed quality control during the heritability check (using minimum heritability P-value 0.01) were excluded from further analyses. We restricted the cis-locus to 500 Kb on either side of the gene boundary.
Data Availability
Genome-wide summary statistics for the confirmed meta-analysis have been made publicly available at dbGaP (phs001868.v1.p1), with the exclusion of genomic data from 23andMe. Results for SNPs with a fixed or random P < 5 × 10−7, from the total meta-analysis are reported in Supplementary Table 7. The total meta-analysis includes self-report cutaneous melanoma GWAS data from the UK Biobank and 23andMe. The raw genetic and phenotypic UK Biobank data used in this study, which were used under license, are available from: http://www.ukbiobank.ac.uk/. The genome-wide summary statistics from 23andMe, Inc. data were obtained under a data transfer agreement. Further information about obtaining access to the 23and Me, Inc. summary statistics are available from: https://research.23andme.com/collaborate/.
Extended Data
Extended Data Fig. 1. Quantile-Quantile plot of total CM meta-analysis.
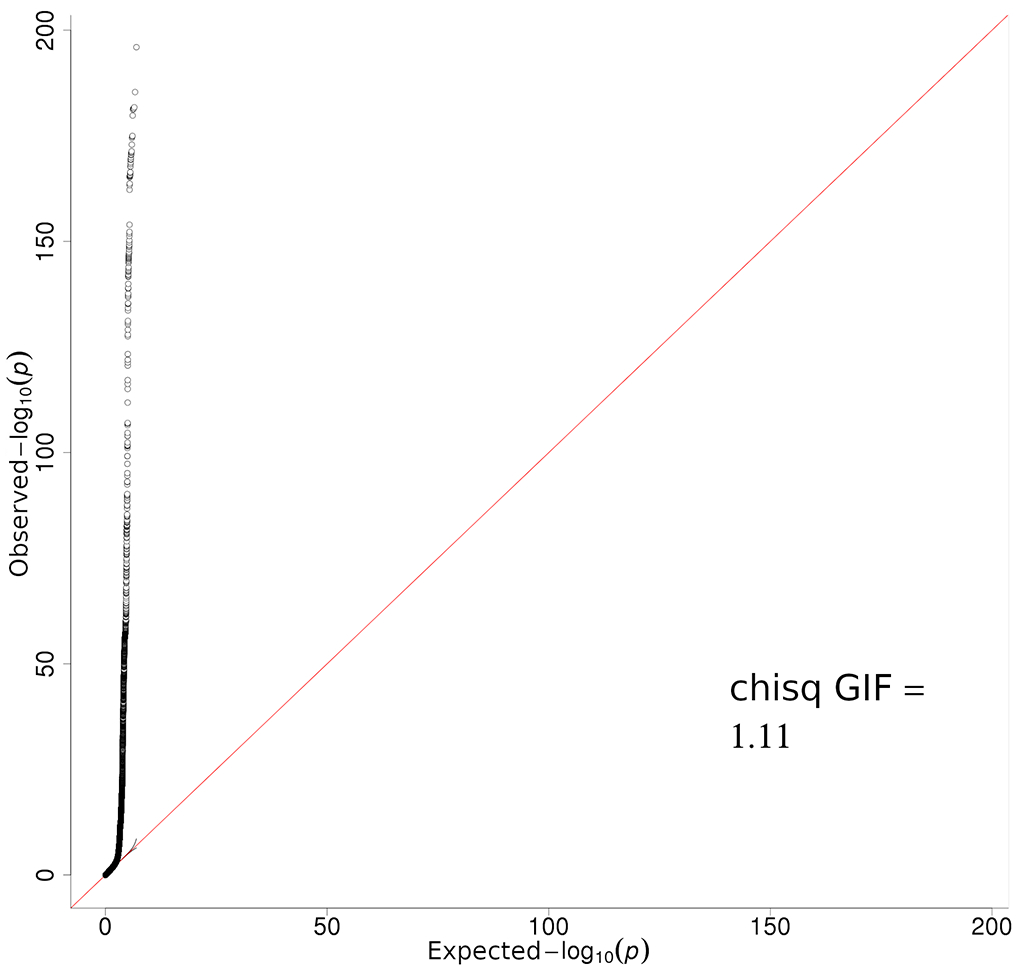
Quantile-quantile plots of negative log10 two sided P-value derived from a fixed-effects inverse-variance weighted meta-analysis of log(OR) effect-sizes derived from the logistic regression GWAS listed in Supplementary Table 1. All confirmed and self-report cases are included, with a total sample size of 36,760 melanoma cases and 375,188 controls.
Extended Data Fig. 2. Manhattan plots of melanoma risk loci from total and confirmed-only GWAS-meta-analyses.
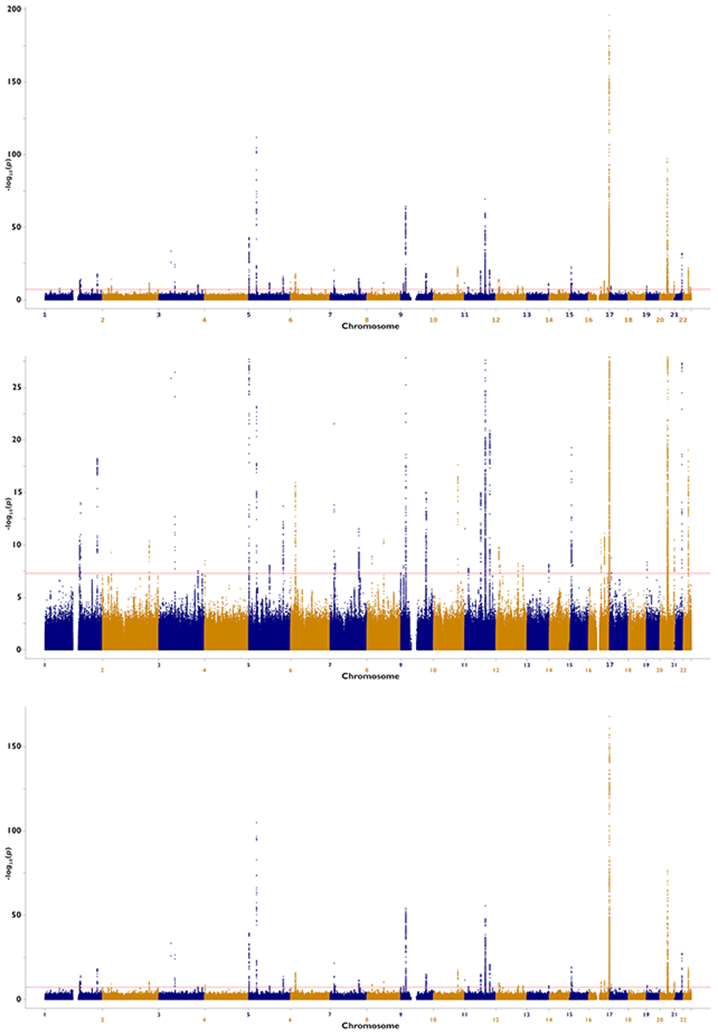
Negative log10 two sided P-value derived from a fixed-effects inverse-variance weighted meta-analysis of log(OR) effect-sizes derived from the logistic regression GWAS (y-axis) are plotted by their chromosome position. The confirmed-only analysis included 30,134 cases with histopathologically confirmed CM, and 81,415 controls. The total CM meta-analysis includes all confirmed and self-report cases, with a total sample size of 36,760 CM cases and 375,188 controls. Multiple-testing corrected genome-wide significance threshold was P<5×10−8. We display in order the total CM meta-analysis without limiting the y-axis; the pathologically confirmed CM cases only meta-analysis with the y-axis limited to 1×10−25 and without a limit to more clearly display loci other than MC1R.
Extended Data Fig. 3. Quantile-Quantile plot of confirmed-only CM meta-analysis.
Quantile-quantile plots of negative log10 two sided P-value derived from a fixed-effects inverse-variance weighted meta-analysis of log(OR) effect-sizes derived from the logistic regression GWAS listed in Supplementary Table 1. Only cases with histopathologically confirmed CM are included, with a total sample size of 30,134 melanoma cases and 81,415 controls.
Extended Data Fig. 4. Distribution of pigmentation polygenic risk scores across melanoma histological subtypes.
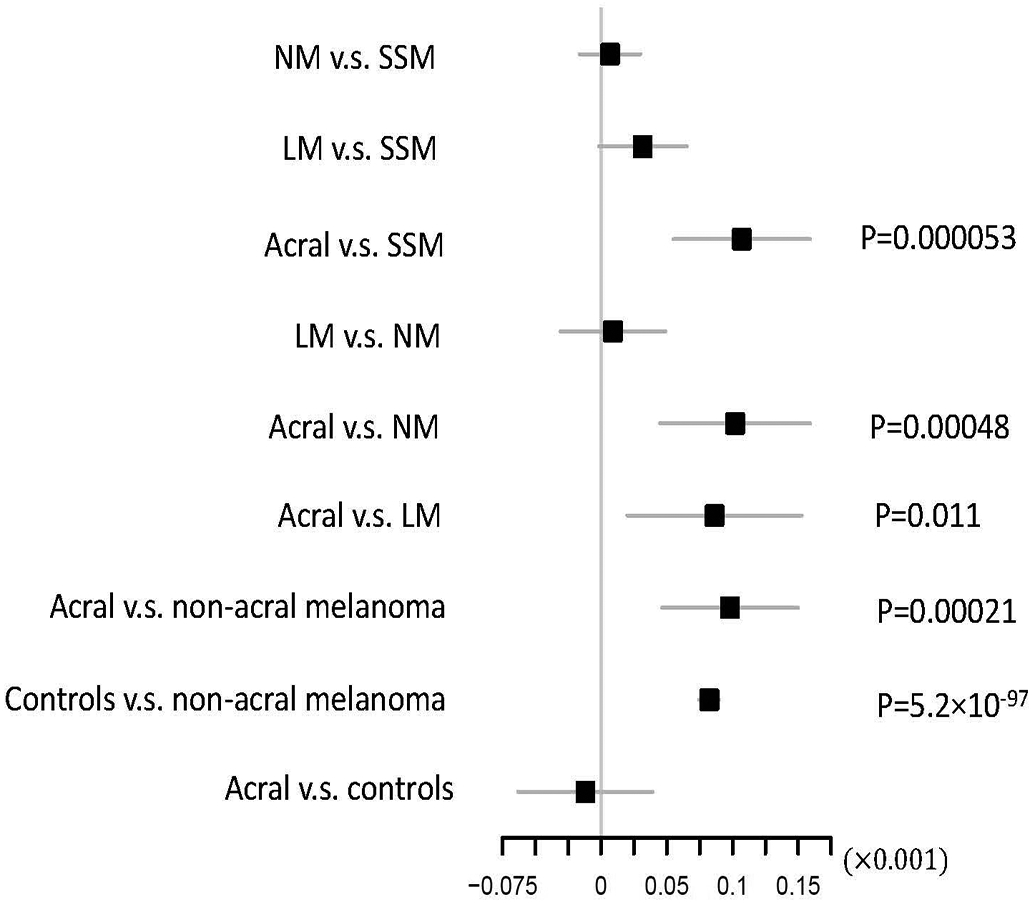
The figure shows whether PRS defined based on SNPs associated with hair colour differ across CM histological types (Online Methods; SSM: superficial spreading melanoma; NM: nodular melanoma; LM: lentigo melanoma; Acral: acral lentiginous melanoma). The higher the PRS the lighter the hair colour. When comparing subtype 1 vs. subtype 2, we report the effect size for the linear regression of PRS on subtype 1, including study and principal components as covariates to control for population stratification. The regression coefficient, 95% confidence interval, and statistical significance are shown. The positive beta indicates the PRS is higher in subtype 2 (e.g., non-acral melanomas). This analysis included 9828 SSM, 2137 NM, 900 LM, 353 acral melanoma cases and 44676 controls. Two-sided t-statistic was used for testing significance. P-values reported were not adjusted for multiple comparison.
Extended Data Fig. 5. LD score regression plots.
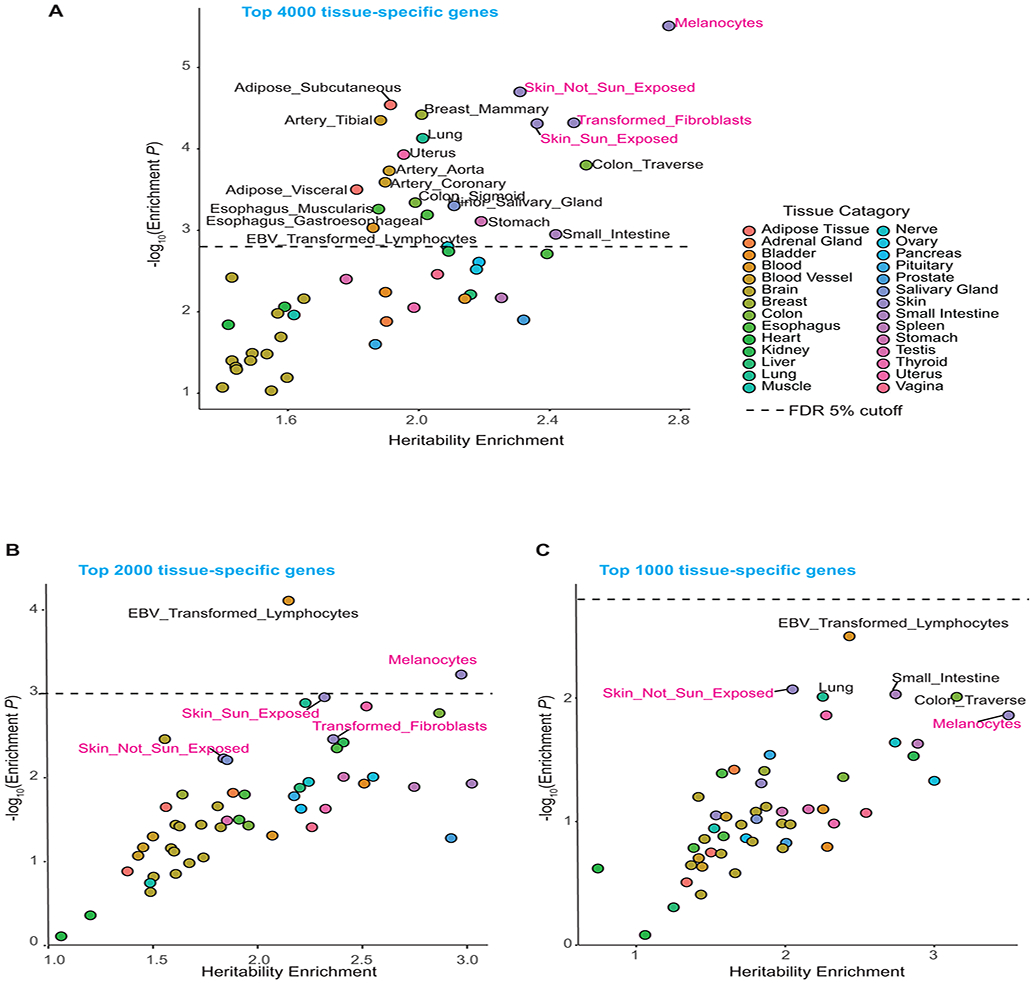
LD score regression was performed for the top 4000 (A) 2000 (B) and 1000 (C) tissue-specific genes from melanocyte and GTEx tissue types (v7 datasets), to assess the enrichment of melanoma heritability in these genomic regions using summary statistics from Total CM GWAS meta-analysis. The level of enrichment and P-values are shown, with an FDR = 0.05 cutoff marked as a dashed horizontal line (See Online Methods for statistical test). Tissue categories are color-coded, and a subset of top individual tissue types are shown on the plot. Tissue types from “Skin” category including melanocytes are highlighted in magenta.
Extended Data Fig. 6. Effect sizes for confirmed-only meta-analysis versus UKBB self-report set.
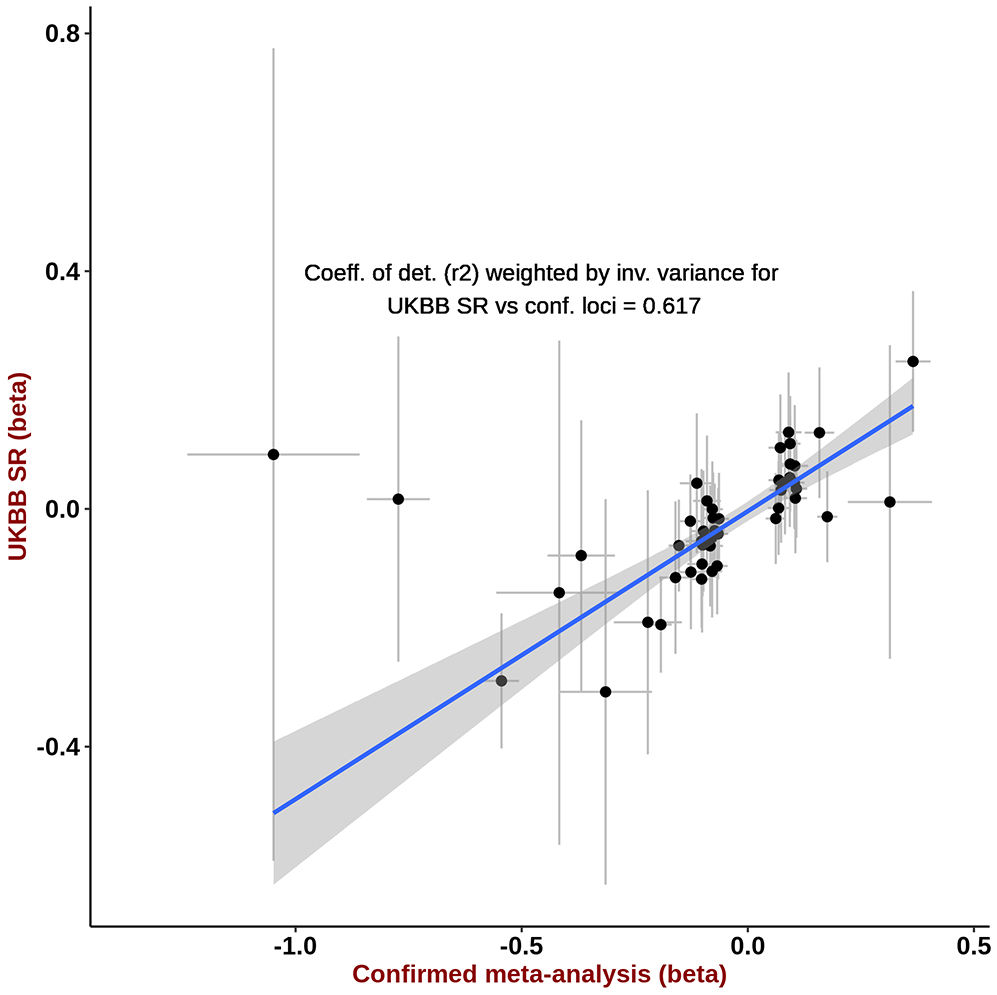
For each independent genome-wide significant (P<5×10−8) lead SNP from the confirmed only meta-analysis (30,134 melanoma cases and 81,415 controls), we plot on the Y-axis UK Biobank self-report GWAS (UKBB SR) log(OR) and standard error from a logistic regression GWAS (1,802 self-report CM cases and 7,208 controls) and on the X-axis we plot the log(OR) and standard error from a fixed-effects inverse-variance weighted meta-analysis of log(OR) effect-sizes derived from the logistic regression GWAS for confirmed melanoma cases listed in Supplementary Table 1. We also report the r2 correlation from the linear regression of UKBB SR log(OR) on the confirmed met-analysis estimates, weighted by their standard error.
Supplementary Material
Acknowledgments
NCI
This study was supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Department of Health and Human Services.
AOCS/OCAC/SEARCH
AOCS/OCAC/SEARCH is accessible via European Genome-Phenome Archive. We acknowledge their support and data, and the contribution of the study nurses, research assistants and all clinical and scientific collaborators in generation of these data. We also acknowledge their funding sources; OCAC (NIH U19CA148112), SEARCH team (Cancer Research UK C490/A16561), AOCS (U.S. Army Medical Research and Materiel Command under DAMD17‐01‐1‐0729, The Cancer Council Victoria, Queensland Cancer Fund, The Cancer Council New South Wales, The Cancer Council South Australia, The Cancer Foundation of Western Australia, The Cancer Council Tasmania and the NHMRC (ID400413 and ID400281, as well as support from S. Boldeman, the Agar family, Ovarian Cancer Action (UK), Ovarian Cancer Australia and the Peter MacCallum Foundation).
MelaNostrum Consortium
We thank the participants of the MelaNostrum Consortium from Italy (Genoa, L’Aquila, Rome, Padua, Milan, Florence, Bergamo), Spain (Valencia, Barcelona), Greece (Athens), and Cyprus (Nicosia) who provided data and biospecimens for this study. The Consortium is partially supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, NCI, NIH, DHHS. Funding for the University of Genoa and Genetics of Rare Cancers, Ospedale Policlinico San Martino: Italian Ministry of Health 5×1000 per la Ricerca Corrente to Ospedale Policlinico San Martino and AIRC IG 15460. The research at the Melanoma Unit in Barcelona: The Spanish Fondo de Investigaciones Sanitarias grants PI15/00716 and PI15/00956 co-financed by FEDER “Una manera de hacer Europa”; CIBER de Enfermedades Raras of the Instituto de Salud Carlos III, Spain, co-financed by European Development Regional Fund “A way to achieve Europe” ERDF; AGAUR 2014_SGR_603 of the Catalan Government, Spain; European Commission, Contract No. LSHC-CT-2006–018702 (GenoMEL) and by the European Commission under the 7th Framework Programme, Diagnostics; “Fundació La Marató de TV3” 201331–30, Catalonia, Spain; “Fundación Científica de la Asociación Española Contra el Cáncer” GCB15152978SOEN, Spain, and CERCA Programme/Generalitat de Catalunya. Melanoma research at the Department of Dermatology, University of L’Aquila, Italy: Italian Ministry of the University and Scientific Research (PRIN-2012 grant 2012JJX494).
Q-MEGA/QTWIN
The Q-MEGA/QTWIN study was supported by the Melanoma Research Alliance, the NIH NCI (CA88363, CA83115, CA122838, CA87969, CA055075, CA100264, CA133996 and CA49449), the National Health and Medical Research Council of Australia (NHMRC) (200071, 241944, 339462, 380385, 389927,389875, 389891, 389892,389938, 443036, 442915, 442981, 496610, 496675, 496739, 552485, 552498, APP1049894), the Cancer Councils New South Wales, Victoria and Queensland, the Cancer Institute New South Wales, the Cooperative Research Centre for Discovery of Genes for Common Human Diseases (CRC), Cerylid Biosciences (Melbourne), the Australian Cancer Research Foundation, The Wellcome Trust (WT084766/Z/08/Z) and donations from Neville and Shirley Hawkins. Stuart MacGregor acknowledges fellowship support from the Australian National Health and Medical Research Council and from the Australian Research Council.
Please see the Supplementary Note for additional acknowledgments.
References
- 1.Karimkhani C et al. The global burden of melanoma: results from the Global Burden of Disease Study 2015. British Journal of Dermatology 177, 134–140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Secretan B et al. WHO International Agency for Research on Cancer Monograph Working Group A review of human carcinogens—Part E: tobacco, areca nut, alcohol, coal smoke, and salted fish. Lancet Oncol. 10, 1033–1034 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Ford D et al. Risk of cutaneous melanoma associated with a family history of the disease. Int. J. Cancer 62, 377–381 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Olsen CM, Carroll HJ & Whiteman DC Familial melanoma: a meta-analysis and estimates of attributable fraction. Cancer Epidemiol. Biomarkers Prev 19, 65–73 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Olsen CM, Carroll HJ & Whiteman DC Estimating the attributable fraction for melanoma: a meta-analysis of pigmentary characteristics and freckling. Int. J. Cancer 127, 2430–2445 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Chang YM et al. A pooled analysis of Melanocytic nevus phenotype and the risk of cutaneous melanoma at different latitudes. International Journal of Cancer (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olsen CM, Carroll HJ & Whiteman DC Estimating the attributable fraction for cancer: A meta-analysis of nevi and melanoma. Cancer Prev. Res 3, 233–245 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Bataille V et al. Nevus size and number are associated with telomere length and represent potential markers of a decreased senescence in vivo. Cancer Epidemiol. Biomarkers Prev. (2007). [DOI] [PubMed]
- 9.Han J et al. A prospective study of telomere length and the risk of skin cancer. J. Invest. Dermatol (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green AC & Olsen CM Increased risk of melanoma in organ transplant recipients: systematic review and meta-analysis of cohort studies. Acta Derm. Venereol 95, 923–927 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Kamb A et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat. Genet 8, 23–26 (1994). [DOI] [PubMed] [Google Scholar]
- 12.Berwick M et al. The prevalence of CDKN2A germ-line mutations and relative risk for cutaneous malignant melanoma: an international population-based study. Cancer Epidemiol. Biomarkers Prev. 15, 1520–1525 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Robles-Espinoza CD et al. POT1 loss-of-function variants predispose to familial melanoma. Nat. Genet (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi J et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat. Genet 46, 482–486 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palmer JS et al. Melanocortin-1 receptor polymorphisms and risk of melanoma: is the association explained solely by pigmentation phenotype? Am. J. Hum. Genet 66, 176–186 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landi MT et al. MC1R, ASIP, and DNA repair in sporadic and familial melanoma in a mediterranean population. J. Natl. Cancer Inst (2005). [DOI] [PubMed] [Google Scholar]
- 17.Brown KM et al. Common sequence variants on 20q11.22 confer melanoma susceptibility. Nat. Genet 40, 838–840 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bishop DT et al. Genome-wide association study identifies three loci associated with melanoma risk. Nat. Genet (2009). [DOI] [PMC free article] [PubMed]
- 19.Amos CI et al. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum. Mol. Genet 20, 5012–5023 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrett JH et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat. Genet (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macgregor S et al. Genome-wide association study identifies a new melanoma susceptibility locus at 1q21.3. Nat. Genet 43, 1114–1118 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iles MM et al. A variant in FTO shows association with melanoma risk not due to BMI. Nat. Genet. 45, 428–32, 432e1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Law MH et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat. Genet (2015). [DOI] [PMC free article] [PubMed]
- 24.Ransohoff KJ et al. Two-stage genome-wide association study identifies a novel susceptibility locus associated with melanoma. Oncotarget 8, 17586–17592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yokoyama S et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. (2011). [DOI] [PMC free article] [PubMed]
- 26.Bertolotto C et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 480, 94–98 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Duffy DL et al. Novel pleiotropic risk loci for melanoma and nevus density implicate multiple biological pathways. Nat. Commun 9, 4774 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang T et al. Cell-type-specific eQTL of primary melanocytes facilitates identification of melanoma susceptibility genes. Genome Res. 28, 1621–1635 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elder DE, Massi D, Willemze R & Scolyer R WHO Classification of Skin Tumours, 500 (International Agency for Research on Cancer, 2018). [Google Scholar]
- 30.Higgins JPT & Thompson SG Quantifying heterogeneity in a meta-analysis. Stat. Med (2002). [DOI] [PubMed]
- 31.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Qi G, Park J-H & Chatterjee N Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat. Genet 50, 1318–1326 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Yang J et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duffy DL et al. Multiple Pigmentation Gene Polymorphisms Account for a Substantial Proportion of Risk of Cutaneous Malignant Melanoma. J. Invest. Dermatol 130, 520–528 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duffy DL et al. IRF4 variants have age-specific effects on nevus count and predispose to melanoma. Am. J. Hum. Genet (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delgado DA et al. Genome-wide association study of telomere length among South Asians identifies a second RTEL1 association signal. J. Med. Genet 55, 64–71 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iles MM et al. The effect on melanoma risk of genes previously associated with telomere length. J. Natl. Cancer Inst 106(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pickrell JK et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet 48, 709–717 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Consortium, G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gamazon ER et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet 47, 1091–1098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gusev A et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet 48, 245–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hormozdiari F et al. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet 99, 1245–1260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stacey SN et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat. Genet 43, 1098–1103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chahal HS et al. Genome-wide association study identifies 14 novel risk alleles associated with basal cell carcinoma. Nat. Commun 7, 12510 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ostrom QT et al. Sex-specific glioma genome-wide association study identifies new risk locus at 3p21.31 in females, and finds sex-differences in risk at 8q24.21. Sci. Rep 8, 7352 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melin BS et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat. Genet 49, 789–794 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin Y et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat. Genet (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Y, Wu H, Zhao M, Chang C & Lu Q The Bach Family of Transcription Factors: A Comprehensive Review. Clin. Rev. Allergy Immunol 50, 345–356 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Milovic-Holm K, Krieghoff E, Jensen K, Will H & Hofmann TG FLASH links the CD95 signaling pathway to the cell nucleus and nuclear bodies. EMBO J. 26, 391–401 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aoude LG et al. Nonsense mutations in the shelterin complex genes ACD and TERF2IP in familial melanoma. J. Natl. Cancer Inst 107(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rafnar T et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat. Genet 41, 221–227 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumano K et al. Both Notch1 and Notch2 contribute to the regulation of melanocyte homeostasis. Pigment Cell Melanoma Res. 21, 70–78 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Tang A et al. E-cadherin is the major mediator of human melanocyte adhesion to keratinocytes in vitro. J. Cell Sci 107 (Pt 4), 983–992 (1994). [DOI] [PubMed] [Google Scholar]
- 55.Hsu MY, Wheelock MJ, Johnson KR & Herlyn M Shifts in cadherin profiles between human normal melanocytes and melanomas. J. Investig. Dermatol. Symp. Proc 1, 188–194 (1996). [PubMed] [Google Scholar]
- 56.Study C et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat. Genet 40, 1426–1435 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Visconti A et al. Genome-wide association study in 176,678 Europeans reveals genetic loci for tanning response to sun exposure. Nat. Commun 9, 1684 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi J et al. A common intronic variant of PARP1 confers melanoma risk and mediates melanocyte growth via regulation of MITF. Nat. Genet 49, 1326–1335 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Li FP & Fraumeni JF Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann. Intern. Med 71, 747–752 (1969). [DOI] [PubMed] [Google Scholar]
- 60.Curiel-Lewandrowski C, Speetzen LS, Cranmer L, Warneke JA & Loescher LJ Multiple primary cutaneous melanomas in Li-Fraumeni syndrome. Arch. Dermatol 147, 248–250 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Beausejour CM Reversal of human cellular senescence: roles of the p53 and p16 pathways. The EMBO Journal 22, 4212–4222 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuilman T, Michaloglou C, Mooi WJ & Peeper DS The essence of senescence. Genes Dev. 24, 2463–2479 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rachakonda S et al. Telomere length, telomerase reverse transcriptase promoter mutations, and melanoma risk. Genes Chromosomes Cancer (2018). [DOI] [PubMed]
- 64.Choi J et al. A common intronic variant of PARP1 confers melanoma risk and mediates melanocyte growth via regulation of MITF. Nat. Genet 49(2017). [DOI] [PubMed] [Google Scholar]
- 65.Derheimer FA & Kastan MB Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 584, 3675–3681 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Demenais F et al. A linkage study between HLA and cutaneous malignant melanoma or precursor lesions or both. J. Med. Genet 21, 429–435 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bale SJ et al. Hereditary malignant melanoma is not linked to the HLA complex on chromosome 6. Int. J. Cancer 36, 439–443 (1985). [DOI] [PubMed] [Google Scholar]
- 68.Holland EA, Beaton SC, Kefford RF & Mann GJ Linkage analysis of familial melanoma and chromosome 6 in 14 Australian kindreds. Genes Chromosomes Cancer 19, 241–249 (1997). [DOI] [PubMed] [Google Scholar]
- 69.Barger BO, Acton RT, Soong SJ, Roseman J & Balch C Increase of HLA-DR4 in melanoma patients from Alabama. Cancer Res. 42, 4276–4279 (1982). [PubMed] [Google Scholar]
- 70.Rovini D, Sacchini V, Codazzi V, Vaglini M & Illeni MT HLA antigen frequencies in malignant melanoma patients. A second study. Tumori 70, 29–33 (1984). [DOI] [PubMed] [Google Scholar]
- 71.Hors J et al. HLA and Familial Malignant Melanoma. Histocompatibility Testing 1984, 407–410 (1984). [Google Scholar]
- 72.Lee JE, Reveille JD, Ross MI & Platsoucas CD HLA-DQB1* 0301 association with increased cutaneous melanoma risk. International journal of cancer 59, 510–513 (1994). [DOI] [PubMed] [Google Scholar]
- 73.Muto M et al. HLA class I polymorphism and the susceptibility to malignant melanoma. Tissue Antigens 47, 447–449 (1996). [PubMed] [Google Scholar]
- 74.Kageshita T et al. Molecular genetic analysis of HLA class II alleles in Japanese patients with melanoma. Tissue Antigens 49, 466–470 (1997). [DOI] [PubMed] [Google Scholar]
- 75.Bateman AC, Turner SJ, Theaker JM & Howell WM HLA-DQB1*0303 and *0301 alleles influence susceptibility to and prognosis in cutaneous malignant melanoma in the British Caucasian population. Tissue Antigens 52, 67–73 (1998). [DOI] [PubMed] [Google Scholar]
- 76.Lombardi ML et al. Molecular analysis of HLA DRB1 and DQB1 polymorphism in Italian melanoma patients. J. Immunother 21, 435–439 (1998). [DOI] [PubMed] [Google Scholar]
- 77.Luongo V et al. HLA allele frequency and clinical outcome in Italian patients with cutaneous melanoma. Tissue Antigens 64, 84–87 (2004). [DOI] [PubMed] [Google Scholar]
- 78.Campillo JA et al. HLA class I and class II frequencies in patients with cutaneous malignant melanoma from southeastern Spain: the role of HLA-C in disease prognosis. Immunogenetics 57, 926–933 (2006). [DOI] [PubMed] [Google Scholar]
- 79.Planelles D et al. HLA class II polymorphisms in Spanish melanoma patients: homozygosity for HLA-DQA1 locus can be a potential melanoma risk factor. Br. J. Dermatol 154, 261–266 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Jin Y et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet 44, 676–680 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Curran K et al. Interplay between Foxd3 and Mitf regulates cell fate plasticity in the zebrafish neural crest. Dev. Biol 344, 107–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thomas AJ & Erickson CA FOXD3 regulates the lineage switch between neural crest-derived glial cells and pigment cells by repressing MITF through a non-canonical mechanism. Development 136, 1849–1858 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schouwey K, Larue L, Radtke F, Delmas V & Beermann F Transgenic expression of Notch in melanocytes demonstrates RBP-Jkappa-dependent signaling. Pigment Cell Melanoma Res. 23, 134–136 (2010). [DOI] [PubMed] [Google Scholar]
- 84.Zabierowski SE et al. Direct reprogramming of melanocytes to neural crest stem-like cells by one defined factor. Stem Cells 29, 1752–1762 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Falchi M et al. Genome-wide association study identifies variants at 9p21 and 22q13 associated with development of cutaneous nevi. Nat. Genet 41, 915–919 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Garraway LA et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature (2005). [DOI] [PubMed]
- 87.Abel EV & Aplin AE FOXD3 is a mutant B-RAF-regulated inhibitor of G(1)-S progression in melanoma cells. Cancer Res. 70, 2891–2900 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weiss MB, Abel EV, Dadpey N & Aplin AE FOXD3 modulates migration through direct transcriptional repression of TWIST1 in melanoma. Mol. Cancer Res 12, 1314–1323 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Golan T et al. Interactions of Melanoma Cells with Distal Keratinocytes Trigger Metastasis via Notch Signaling Inhibition of MITF. Mol. Cell (2015). [DOI] [PubMed]
- 90.Cronin JC et al. SOX10 ablation arrests cell cycle, induces senescence, and suppresses melanomagenesis. Cancer Res. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guilford P et al. E-cadherin germline mutations in familial gastric cancer. Nature 392, 402–405 (1998). [DOI] [PubMed] [Google Scholar]
- 92.Hansford S et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol 1, 23–32 (2015). [DOI] [PubMed] [Google Scholar]
- 93.Kim HC et al. The E-cadherin gene (CDH1) variants T340A and L599V in gastric and colorectal cancer patients in Korea. Gut 47, 262–267 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wagner RY et al. Altered E-Cadherin Levels and Distribution in Melanocytes Precede Clinical Manifestations of Vitiligo. J. Invest. Dermatol 135, 1810–1819 (2015). [DOI] [PubMed] [Google Scholar]
- 95.Padmanaban V et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573, 439–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 96.Purcell S et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Das S et al. Next-generation genotype imputation service and methods. Nat. Genet 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.DerSimonian R & Laird N Meta-analysis in clinical trials. Control. Clin. Trials 7, 177–188 (1986). [DOI] [PubMed] [Google Scholar]
- 100.Machiela MJ & Chanock SJ LDassoc: an online tool for interactively exploring genome-wide association study results and prioritizing variants for functional investigation. Bioinformatics 34, 887–889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Willer CJ, Li Y & Abecasis GR METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics (2010). [DOI] [PMC free article] [PubMed]
- 102.Watanabe K, Taskesen E, Van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat. Commun (2017). [DOI] [PMC free article] [PubMed]
- 103.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Australian Institute of, H. & Welfare. Cancer in Australia: Actual incidence data from 1982 to 2013 and mortality data from 1982 to 2014 with projections to 2017. Asia Pac. J. Clin. Oncol 14, 5–15 (2018). [DOI] [PubMed] [Google Scholar]
- 105.DeLuca DS et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28, 1530–1532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McCarthy S et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet 48, 1279–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.MacGregor S et al. Two novel loci for melanoma susceptibility on chromosome bands 1q42.12 and 1q21.3. Nat. Genet (2011). [Google Scholar]
- 109.Peña-Chilet M et al. Genetic variants in PARP1 (rs3219090) and IRF4 (rs12203592) genes associated with melanoma susceptibility in a Spanish population. BMC Cancer 13(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Law MH et al. Meta-analysis combining new and existing data sets confirms that the TERT-CLPTM1L locus influences melanoma risk. J. Invest. Dermatol 132, 485–487 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Antonopoulou K et al. Updated field synopsis and systematic meta-analyses of genetic association studies in cutaneous melanoma: the MelGene database. J. Invest. Dermatol 135, 1074–1079 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genome-wide summary statistics for the confirmed meta-analysis have been made publicly available at dbGaP (phs001868.v1.p1), with the exclusion of genomic data from 23andMe. Results for SNPs with a fixed or random P < 5 × 10−7, from the total meta-analysis are reported in Supplementary Table 7. The total meta-analysis includes self-report cutaneous melanoma GWAS data from the UK Biobank and 23andMe. The raw genetic and phenotypic UK Biobank data used in this study, which were used under license, are available from: http://www.ukbiobank.ac.uk/. The genome-wide summary statistics from 23andMe, Inc. data were obtained under a data transfer agreement. Further information about obtaining access to the 23and Me, Inc. summary statistics are available from: https://research.23andme.com/collaborate/.