

Abstract
Methodologies that facilitate high-throughput proteomic analysis are a key step toward moving proteome investigations into clinical translation. Data independent acquisition (DIA) has potential as a high-throughput analytical method due to the reduced time needed for sample analysis, as well as its highly quantitative accuracy. However, a limiting feature of DIA methods is the sensitivity of detection of low abundant proteins and depth of coverage, which other mass spectrometry approaches address by two-dimensional fractionation (2D) to reduce sample complexity during data acquisition. In this study, we developed a 2D-DIA method intended for rapid- and deeper-proteome analysis compared to conventional 1D-DIA analysis. First, we characterized 96 individual fractions obtained from the protein standard, NCI-7, using a data-dependent approach (DDA), identifying a total of 151,366 unique peptides from 11,273 protein groups. We observed that the majority of the proteins can be identified from just a few selected fractions. By performing an optimization analysis, we identified six fractions with high peptide number and uniqueness that can account for 80% of the proteins identified in the entire experiment. These selected fractions were combined into a single sample which was then subjected to DIA (referred to as 2D-DIA) quantitative analysis. Furthermore, improved DIA quantification was achieved using a hybrid spectral library, obtained by combining peptides identified from DDA data with peptides identified directly from the DIA runs with the help of DIA-Umpire. The optimized 2D-DIA method allowed for improved identification and quantification of low abundant proteins compared to conventional unfractionated DIA analysis (1D-DIA). We then applied the 2D-DIA method to profile the proteomes of two breast cancer patient-derived xenograft (PDX) models, quantifying 6,217 and 6,167 unique proteins in basal- and luminal-tumors, respectively. Overall, this study demonstrates the potential of high-throughput quantitative proteomics using a novel 2D-DIA method.
Graphical Abstract
The ultimate goal of human proteomics research is to be able to analyze and understand all of the proteins expressed in certain cell or tissue type including post-translational modifications essential for cellular function for biological or pathological investigations.1,2 Due to the high complexity of the human proteome, the development of sample processing methods, mass spectrometry technologies, and data analytics, in combination and independently, have been leveraged to identify and quantify almost all genes at the protein level.3−5 Liquid chromatography followed by tandem mass spectrometry (LC−MS/MS) using data-dependent acquisition (DDA) is the conventional approach used for proteomic analysis due to its simplicity and relative high-throughput. 6,7 Typically, DDA methods incorporate a MS1 scan for all peptide peaks to obtain precursor m/z information and then perform a fragmenting ion scan (MS2 scan) of selected peaks dependent on the MS1-scan data. Obtaining information on one peptide per MS/MS fragment scan has the advantage of obtaining a high-purity MS2 scan, but peptide features not selected for fragmentation are lost opportunities due to the limitation of instrument cycle time.8−10 This intrinsic feature of DDA limits its application for complex samples, such as mammalian tissues, due to reduced peak capacity on chromatography columns and rate-limiting cycle time of the mass spectrometer.11 To circumvent this drawback of DDA, recent studies have combined advanced mass spectrometry instrumentation and optimized MS scan parameters to increase protein identifications from single-shot sample analysis.12
Data independent acquisition (DIA) is one strategy to overcome the described limitations of DDA. In the DIA method, the MS2 scan is performed through a predefined MS1 m/z isolation window, fragmenting all precursor ions within the selected window range, repeating until MS2 spectra for all the MS1 windows are acquired.13,14 In addition, DIA has improved quantitative accuracy and reproducibility for protein quantification, using a similar principle of traditional selected reaction monitoring (SRM) methods, wherein transitions (MS2 peptide fragmentation spectra) of all peptides are recorded.15−18 Because of this inherent advantage, DIA has great potential for quantitative proteomic analysis.19,20
To reduce the peak complexity and increase sensitivity, additional dimensions of ion separation such as ion mobility have been developed to allow for the identification of more proteins in a single shot through additional ion separation.21−23 For example, high-field asymmetric waveform ion mobility spectrometer (FAIMS) coupled with Orbitrap mass spectrometer results in quantifying 7,818 of human proteins in 4 h.22 In addition, multidimensional fractionation strategies are widely used to achieve comprehensive proteomic analysis by decreasing the sample complexity and increasing the peak capacity of the LC separation. In particular, fractionation using basic reverse phase liquid chromatography (bRPLC) has shown remarkable potential as a prior process of onlineLC.24,25 Optimized fractionation coupled with a LC−MS/MS workflow has allowed for the identification of 14,200 protein isoforms (from 12,200 protein coding-genes) and 584,000 unique peptides in a single experiment.26 However, the described method requires extensive sample fractionation and instrument acquisition time, severely reducing sample throughput.
In this study, we describe a novel single-shot proteomic approach, integrating the positive characteristics of peptide fractionation (reducing sample complexity) and DIA (protein quantitation accuracy), which we refer to as single-shot 2DDIA. We demonstrated that our 2D-DIA approach provided reproducible, quantitative performance, as well as improved sensitivity for the detection of low abundance peptides/proteins compared to existing single-shot DDA and DIA methods.
MATERIALS AND METHODS
Enzymatic Digestion and Basic-pH Reverse Phase Fractionation.
Four replicates of NCI-7 cells,27 which were developed as clinical proteomic reference material based on seven NCI-60 cell lines that expressed 92% of NCI-60 genome or 88% of human genome, and three replicates from each of the patient-derived xenograft (PDX) models (basal breast cancer and luminal breast cancer), were prepared and digested with trypsin (Pierce Trypsin Protease, MS grade) as described in a previous study.27,28 Briefly, equal amounts of proteins (0.3 mg) were dissolved in 8 M urea lysis buffer (8 M urea, 75 mM NaCl, 50 mM Tris-HCl, pH 8.0), followed by reduction and alkylation with 5 mM dithiothreitol for 1 h at 37 °C and 10 mM iodoacetamide for 45 min in darkness. Samples were diluted to 2 M urea with 50 mM Tris, and trypsin was added at a ratio 1:50 = enzyme: protein. The mixture was incubated for 16 h at RT. Digested peptides were desalted using C18 cartridge (Waters, Sep-Pak Vac 1 cm3 100 mg) after acidifying to adjust to pH 2.0 and dried by Speed Vac. The peptide concentration was determined by nanodrop (Nanodrop lite, Thermo Scientific), and 200 μg of peptides were used for the DIA-MS analysis.
The 3% of peptides (6 ug) were saved as “unfractionated peptides”, and the remaining 97% (194 ug) of peptides were fractionated using bRPLC (1220 Infinity series, Agilent) with reverse phase column (Agilent Zorbax 300 Extend-C18; 4.6 × 250 mm) under mid-pH mobile phase (solvent A: 2% ACN in 5 mM ammonium formate, pH 8.0, B: 90% ACN in 5 mM ammonium formate, pH 8.0). Peptide samples were loaded through a 1 mL sample loop to the column and separated using the following gradient: time (min)/B% ∼ 7/0 (isocratic), 7−13/0−16 (linear), 13−73/16−40 (linear), 73−77/40−44 (linear), 77−82/44−60 (linear), and 82−96/60 (isocratic) with a 1 mL/min flow rate. The fraction collector (Agilent, G1364C Analyt-FC) collected 96 fractions (A1 ∼ H12) from 1 to 97 min with a 1 min time slice, and then each fraction was dried and stored at −80 °C until MS analysis.
LC−MS/MS Setting up for DDA/DIA.
All DDA and DIA analyses were performed by Orbitrap Fusion Lumos Tribrid mass spectrometer with Thermo Scientific EASY-nLC 1200 system. Before injecting the samples, iRT peptides (Biognosys, Ki-3002) were added to each sample (1 μg) respectively and the mixed samples were separated on the analytical column (house-made column, 0.75 μm I.D. × 26.5 cm length packed with ReproSil-Pur 120 C18-AQ, 1.9 μm) using the following LC gradient [min/B%: 0−1 min/4% B (isocratic), 1−61 min/ 4−30% B (linear gradient), 61−65 min/30−60% B (linear gradient), 65−66 min/60−90% B (linear), 66−70 min/90% B (isocratic), 70−71 min/90−50% B (linear), and 71−80/50% B (isocratic)]. The temperature of the column was maintained at 50 °C, and the flow rate was at 200 nL/min during the analysis. For DDA setting, the precursor ions were acquired with 120 K resolution at 200 m/z for 350−1650 m/z range and the AGC value was set as 4E105. The top 20 highest intensity precursor ions were fragmented respectively by HCD using 34% normalized collision energy (NCE), and fragment ions were acquired with 15K resolution with 5E104 of AGC value for 50 ms of injection time. For DIA setting, the DIA segments, MS1, and MS2 resolutions were set as in the following Table S1. Isolated ions from the corresponding MS1 window were fragmented by HCD with 34% NCE, and all of fragmented ions were acquired with 3E106 of the AGC value for 120 ms of maximum injection time.
Exploratory DDA Data Analysis.
The initial exploratory analysis of DDA data was performed as follows. The MS/MS spectra were searched with SEQUEST search engine of Proteome Discoverer software against the human protein sequence database (UniProt/SwissProt 2017–04 release) appended with the Biognosys iRT peptide sequences. The precursor and fragment mass tolerances were set to 10 and 20 ppm, respectively. The oxidation of methionine and N-terminal protein acetylation were set as variable modifications, while carbamidomethylation of cysteine was set as fixed modification. Peptide to spectrum matches (PSMs) were processed using the Percolator and then filtered to 1% protein and PSM-level false discovery rate (FDR). The log2iBAQ protein intensity (an intensity-based measure of absolute protein abundance) was calculated based on the following equation: log2 (∑intensity/#theoretical peptides).29,30
Construction of Spectral Libraries Using LibMatic.
Spectral libraries were generated from DDA data and DIA data acquired in the current study using a LibMatic pipeline, which implements our previously outlined strategy for DIA quantification using combined (hybrid) DDA and DIA-derived libraries.31−33 First, all DDA (NCI7 cell line data) and DIA (NCI7 and PDX data) raw files were converted into the mzXML file format using msconvert.exe with centroid spectra option. The NCI7 DDA mzXML data were searched with three different database search algorithms (X! Tandem, MSGF+, and Comet)34−36 against a human protein sequence database (UniProt/SwissProt; downloaded 2018–03-22), appended with an equal number of decoy sequences as well as Biognosys iRT peptide sequences. Precursor tolerance was set to 10 ppm, and only tryptic peptides with up to two missed cleavages were allowed. The oxidation of methionine was set as a variable modification, while carbamidomethylation of cysteine was set as a fixed modification (note that X! Tandem, by default, considers several additional common modifications, including N-terminal protein acetylation). Database search results from each search engine were individually processed (via the Philosopher toolkit; https://github.com/Nesvilab/philosopher) using PeptideProphet37 and then combined using iProphet,38 resulting in one iproph.pep.xml file for each DDA run.
Direct identification of peptides from the DIA data was performed with the help of DIA-Umpire.32 The DIA-Umpire signal extraction (SE) module was used to process each DIA mzXML file to generate the so-called pseudo MS/MS spectra (assembled separately into three output files based on the quality of the corresponding precursor peptide signal in MS1 data, Q1, Q2, and Q3). After conversion to the mzXML format, DIA pseudo-MS/MS spectra were searched using the MSFragger39 search engine against the same human sequence data set as for DDA data (NCI7 DIA data) or against a combined mouse plus human UniProt/Swiss database (PDX DIA data). For each run, MSFragger search results from the three input mzXML files (Q1, Q2, and Q3) were individually processed using PeptideProphet and then combined using iProphet into a single iproph.pep.xml file.
With the use of the DDA and DIA-derived peptide identification results described above, spectral libraries were generated for each of the three analyses described in the manuscript (Table S2): (1) NCI7 DDA (all 96 fractions) combined with NCI7 DIA; (2) NCI7 DDA (6 fractions selected for 2D DIA analysis only) combined with NCI7 DIA; and (3) NCI7 DDA (6 fractions selected for 2D DIA analysis only) combined with PDX DIA. For each analysis, all iproph.pep.xml (both DDA and DIA) were processed together with ProteinProphet40 to perform joint protein inference, resulting in one combined.prot.xml file encompassing both DDA and DIA-identified peptides. All peptide identifications (DDA and DIA) were filtered using this combined protein inference file using Philosopher (filter command), generating a list of proteins filtered to 1% protein-level FDR. In addition, as part of that step, all peptides shared between multiple proteins were assigned as razor peptides to only one protein in the combined list which had the most peptide evidence overall (using “razor” option of the Philosopher filter command). Second, retention times of peptides identified in each DDA or DIA run were aligned, using nonlinear alignment, against one of the DIA run selected as a reference run (which showed the best average correlation coefficient against all other DIA runs in that experiment). The original retention times in the mzXML and iproph.pep.xml files were then replaced with the aligned retention times. Third, the aligned iprophet.pep.xml and mzXML files were used to build the spectral libraries, separately for DDA and DIA data, using a SpectraST and msproteomicstools toolbox (https://github.com/msproteomicstools/msproteomicstools) essentially as previously described.31 In short, SpectraST41 was run to build the spectral library using MS/MS spectra identified with the PeptideProphet probability score passing the 1% peptide ion FDR level (the threshold reported by the Philosopher filter command). Retention times of the library peptides were rescaled to the iRT scale using the spectrast2spectrast_irt.py program. The consensus spectral library (.splib file) was converted into a transition list format using a spectrast2tsv.py command with the following options: −g −17.03, −18.01 −1250, 2000 −s b,y −x 1,2 −o 3 −n 6 −p 0.05 −d −e −k. The resulting DDA and DIA-derived libraries were converted to Spectronaut-compatible format and further processed to correct peptide to protein mappings to match those determined by the joint protein inference analysis described above (i.e., peptide−protein mappings reported in the Philosopher-generated reports). Finally, the DDA and DIA libraries were combined into a single library. In doing so, if a peptide ion was present in both the DDA and DIA library, the entry from the DIA library was selected for the combined spectral library. More information regarding the pipeline for building hybrid spectral libraries from DDA and DIA data (LibMatic pipeline), including associated scripts, can be found at https://github.com/Nesvilab/LibMatic.
Peptide Quantification Using Spectronaut.
The spectral libraries, built as described above for each analysis (Table S2), were loaded into Spectronaut (versions 12.4, Biognosys, Schlieren, Switzerland) and analyzed using default settings. In short, the MS1 and MS2 tolerance for extracted ion chromatography (XIC) were set as “dynamic”. The indexed retention time value of peptides were calculated by linear regression of empirical retention time of iRT peptides and XIC retention time window set as “dynamic” with 1 correction factor. The decoy method was set as “mutated”, and 1% FDR was applied at the precursor and protein levels (Q value ≤ 0.01).42 After removing interfering signals, selected 3 peptide ions were used for protein quantification. The protein quantity was normalized based on local regression described by Callister et al.43 The protein inference was set as “from search engine”, i.e., based on the peptide-protein grouping as provided by the spectral library.
RESULTS AND DISCUSSION
Spectral Library Built Using 96 Fraction DDA.
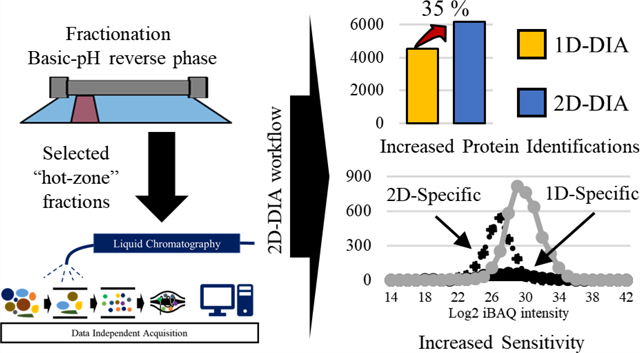
Spectral libraries were used for DIA-based proteomes. Library building consists of DDA mass spectrometry analysis of either the same sample or similar sample composition. In addition, sample fractionation can reduce sample complexity by separating peptides based on their unique amino acid composition; however, this does not result in uniform distribution of peptides derived from individual proteins. To fully optimize our 2D-DIA approach, and identify a “hot zone” of peptide distribution for maximum protein identification, we performed extensive fractionation of a reference standard, NCI-7,27 followed by single-shot DDA analysis of the individual fractions.27 After identifying the respective fractions that would result in the highest number of identifications at the protein-level, select fractions were pooled and subjected to DIA analysis. The optimized 2D-DIA method was then utilized to investigate the proteome of two patient-derived breast cancer xenograft models. Our overall experimental approach is illustrated in Figure 1.
Figure 1.

Overall experimental design for single shot “2D-DIA”.
Comprehensive Proteome Profile Using bRPLC Fractions.
Using a modified basic reversed-phase liquid chromatography (bRPLC) separation protocol (Materials and Methods section), we fractionated the NCI-7 reference standard, obtaining 96 individual samples that were subsequently analyzed via ESI-LC-MS/MS using a DDA approach. This method allowed us to identify a total of 151,366 of peptides from 11,273 of human proteins. Our search parameter required two peptides in order to identify a protein. Individual fractions corresponded to 1 min of the bRPLC gradient time. On the basis of our mass spectrometry results, most peptides eluted between 10 and 60 min (corresponding to fractions A10-E12) (Figure 2A). Due to uneven separation of peptides via fractionation, fewer peptides were identified in early fractions (A01−A09) and later fractions (F01−H12), respectively. We observed a high density of peptide identification in fractions B02 to C10, identifying 7,000 peptides from 3,000 proteins (Figure 2A). Since fractions were based on bRPLC gradient time and not peptide peak width, peptides and consequently proteins were identified across sequential fractions. The combination of peptide redundancy across fractions and limited number of peptides required for protein identification resulted in only a small number of fractions needed to obtain 80% of total numbers of peptides and proteins identified from all 96 fractions (Figure 2B,C). When selecting various subsets of fractions for the analysis, we observed that peptide uniqueness (defined here as the proportion of all peptides identified in that fraction that are fraction-specific) in each fraction was an important metric affecting the total number of proteins identified. Out of all 96 fractions, 27 fractions were considered to have a high peptide uniqueness, resulting in 122,789 peptides identified (Figure 2B). At the protein level, the total number of proteins identified showed rapid saturation after inclusion of just a few fractions with high peptide uniqueness (Figure 2C). Although the identification of more unique peptides generally results in more protein identifications, an important caveat to consider is the number of unique peptide sequences contributed by each protein (with larger proteins contributing more unique peptides). Examining this relationship of unique peptide identification compared to unique protein identifications, we found that six fractions (B04 to B09) displayed the highest degree of peptide uniqueness, while the six best fractions for protein coverage were B05, B06, B09, B10, C4, and C10. We identified 8,730 proteins in these six fractions, representing ∼80% of the proteins identified from 96 fractions Also, due to redundancy of peptide identification across fractions, we found an inverse relationship of increasing analysis time (i.e., analyzing additional fractions) and the number of identified peptides and proteins (Figure S1).
Figure 2.

Protein and peptide characterization of NCI-7 cell through 96 DDA runs. (A) The number of PSMs, peptides, and proteins in each fraction, respectively. The density bar (bottom of graph) indicates the peptide uniqueness (no. unique peptide/no. total peptides) of each fraction. (B) The number of peptides identified with an increasing number of fractions. (C) Same as (B), at the protein level.
Optimization of 2D-DIA Analysis Parameters.
In addition to determining which bRPLC fractions would be optimal for protein identification, we examined several DIA mass spectrometry data acquisition parameters for the optimal peptide identification and peptide/protein quantification, including MS2 resolution and the m/z range for DIA windows. As shown in Table 1, additional metrics such as scan cycle times and the number of data points per peak were influenced by the selected MS2 parameters and impacted the total number of proteins identified. We optimized the data acquisition parameters, with 15K MS2 resolution and 24 DIA windows resulting in the highest number of proteins identified while providing a sufficient number of data points per peak for accurate quantification (>5 points, on average, per fragment peak). Furthermore, we were able to improve the number of detected and quantified peptides and proteins by using hybrid (i.e., combined DDA-based and direct DIA-based) spectral libraries derived using a LibMatic pipeline (see the Materials and Methods section) compared to using spectral libraries created using DDA data alone (Table S3).
Table 1.
Optimization of DIA Parameter According to MS2 Resolution and Number of DIA Window
| index | MS1 resolution | MS2 resolution | no. DIA window | cycle time (sec) | data points per peak | no. protein ID |
|---|---|---|---|---|---|---|
| 1 | 120 K | 15 K | 44 | 5 | <3 | 4000 |
| 2 | 15 K | 30 | 3.04 | 4 | 4342 | |
| 3 | 15 K | 24 | 2.25 | 6 | 4431 | |
| 4 | 30 K | 14 | 1.67 | 7 | 3800 |
We also hypothesized that by incorporating a second dimension (2D fractionation) in our DIA analysis, we could reduce sample complexity and increase the dynamic range of peptides detected, which could increase the number of identified peptides from low abundance proteins relative to conventional 1D (i.e., single-shot analysis of unfractionated samples) DIA strategies. We chose the six fractions (B04−B09) derived from NCI-7 with the highest degree of peptide uniqueness to maximize the protein coverage and pooled the peptides from those fractions. We evaluated the reproducibility of quantification by analyzing three replicates of pooled B04−B09 samples (obtained from three independent fractionation experiments). As a comparison, NCI-7 peptides were also analyzed with 1D-DIA. In 1D-DIA analysis, the identified peptides had a median CV (across three replicates) of 6% for peptide quantification and 5% for protein quantification. In 2D-DIA analysis, a median CV of 11% and 10% were obtained for peptide and protein quantification, respectively (Figure 3A). Even though the 2D-DIA method had slightly higher median CVs than the 1D-DIA method for both peptide- and protein-level quantification, we observed that the 2D-DIA method significantly increased the number of quantified proteins compared to 1D-DIA or 1D-DDA methods (Figure 3B). A total of 6,219 proteins were identified in 2D-DIA, almost doubling the number of protein identifications compared to 1D-DDA (3,185) and providing a 35% increase comparing with the number of protein identifications from 1DDIA (4,586). Filtering for quantitative confidence (<20% CV), we still observed a higher number of proteins identified using our 2D-DIA method, identifying 4,540 with 2D-DIA compared to 3,733 using 1D-DIA. Although the median CV% of 2D-DIA was slightly higher than 1D-DIA, the number of reliably quantified proteins was higher in the 2D-DIA data sets due to the significant increase in the total number of identified and quantified proteins. Even when leveraging the fast scan speed of the state-of-the-art Orbitrap Fusion Lumos Tribrid mass spectrometer used in this work, the number of peptides quantified in 1D-DDA was lower than that in both 1D-DIA and 2D-DIA methods, potentially due to the limited number of peptide features that can be detected and measured within one hour gradient time. At the peptide level, a total of 27,730 peptides was identified using 1D-DDA, compared to the 37,664 and 35,038 peptides identified using 1D-DIA and 2DDIA methods, respectively. Interestingly, we detected a higher number of peptides identified in our 1D-DIA data set relative to the 2D-DIA data set (Figure 3B). When we investigated the average number of peptides per protein, we found the 1D-DIA method had a higher peptide/protein ratio than the 2D-DIA (8.2 vs 5.6). Although the DIA method acquires all fragment ions in the selected m/z window, intensities of fragment ions are affected by individual peptide dynamics due to the limited ion counts in the c-trap and the complexity of fragment ions from several peptides. Thus, the peptide species derived from highly abundant proteins have an increased likelihood of detection relative to peptide species from low abundance proteins. As a result, 2D-DIA had less peptide coverage than 1D-DIA. However, the reduction in sample complexity due to fractionation enabled us to identify more low abundance proteins. Comparison of the two data sets showed that 2,118 proteins were identified only in 2D-DIA, while 485 proteins were identified in the 1D-DIA analysis only (Figure 3C). Using log2iBAQ intensities (see the Materials and Methods section) as measures of protein abundance, we observed a wider range of protein abundance in the 2D-DIA data set (22−35) relative to the 1D-DIA data set (26−35). Further investigation revealed that proteins uniquely identified using the 1D-DIA method displayed a log2iBAQ range of 25−34, which was similar to the range of commonly identified proteins between the two data sets. In contrast, proteins uniquely identified in the 2D-DIA data set had a lower log2iBAQ range (22−30), an exclusive feature to 2D-DIA relative to those identified in 1D-DIA or shared between the two data sets (Figure 3C).
Figure 3.

Comparison of 2D-DIA with 1D-DIA. (A) Coefficient variation (CV) was calculated from 3 replicates at the peptide (line) and protein (dot) levels, respectively. Asterisk (*) indicates the median CV (%). (B) The number of peptides and protein identified in 1D-DDA, 1D-DIA, and 2D-DIA. Asterisk (**) indicates the average number of peptides per protein. (C) Distribution of intensity-based absolute abundances of proteins (iBAQ) identified using 1D-DDA, 1D-DIA, and 2D-DIA methods.
Quantitative Proteome Analysis of Two Breast PDX Tumors through 2D-DIA.
The optimized 2D-DIA workflow was further applied to investigate the proteomes of two PDX tumors models, comprising of breast basal and luminal tumors (Figure S2).44 Three sample preparations were generated and analyzed using the 2D-DIA approach. Resulting 2D-DIA data were searched using a combined spectral library that included the mouse proteome (released by Biognosys, 11,505 precursor entries) and human plus mouse proteome (NCI-7 DDA + PDX DIA library, 69,113 precursor entries, Table S2) from xenograft material containing both mouse and human proteins.28,45 From three replicate DIA runs, totally 6,217 and 6,167 proteins were identified from basal and luminal PDX models, respectively, including 4,419 and 4,408 proteins from the human proteome that did not share peptides with the mouse proteome. Note that due to the relatively small size of the mouse subset of the spectral library, the proteome coverage for mouse proteins was lower than expected. Using a specific spectral library for the PDX model should result in a higher mouse proteome coverage. Applying a filter of CV < 20%, 5,016 (3,555 human proteins) and 5,114 (3,663 human proteins) from basal and luminal were quantified. To further evaluate the quantitative performance of the 2D-DIA method, we correlated reported protein abundances from replicate analyses of the same tumor sample, observing high correlation and reproducibility. Pearson correlation (r) among intratumor analyses was over 0.95, while intertumor protein abundance correlation was lower (r = 0.65) as expected (Figure S3A). The median CVs of the quantified proteins were 7.5% and 5.6% for basal and luminal tumors, respectively, with protein CVs lower than peptide CVs (Figure S3B). Hierarchical clustering clearly separated the two molecular subtypes of breast cancer (Figure 4C). Next, we explored the differential expression of proteins between the basal and luminal subtypes by filtering proteins based on 2-fold change and p-value < 0.001 (Figure 4B). In total, 477 proteins were identified with increased levels in the basal subtype and 889 proteins with increased levels in the luminal subtype. Among them, several known triple-negative breast cancer markers, such as WDHD1, GSTP1 and SMARCA5, were identified with increased expression in the basal subtype.46,47 Similarly, several known markers of the luminal subtype, PARP1, FOXA1, GATA3, and SLC9A3R1 showed increased expression in the luminal samples (Figure 4C).48−50 Gene ontology analysis showed that overexpressed proteins in the basal subtype were significantly enriched (p-value <0.01) for biological processes including cell proliferation, cell division, G1/S transition of mitotic cell cycle, extracellular matrix organization, and cholesterol biosynthetic process, whereas down-regulated proteins were enriched in oxidation reduction, fatty acid beta-oxidation, type I interferon signaling pathway, mitochondrial electron transport ubiquinol to cytochrome c, and tricarboxylic acid cycle (Figure 4D). The high proliferation and mitotic rates are known as a characteristic of the basal breast cancer subtype because of low expression of the RB1 and CCND1 genes, as well as high expression of E2F3 and CCNE genes.51,52 Overall, our 2D-DIA data recapitulated previous findings observed in triple negative breast cancer experiments, proving additional confirmation of the overall good quality of the quantitative data generated using our 2D-DIA method.
Figure 4.

Quantitative proteomic analysis of PDX models (basal and luminal subtypes) using 2D-DIA. (A) The total number of identified protein groups that were human-only (green), from either human or mouse (yellow) or mouse-only (orange). The number of proteins with a quantification CV < 20% (dashed bars). (B) Volcano plot showing the Fold Change and the p-values (t test) between the basal and luminal subtypes. The known breast cancer markers were highlighted in red. (C) Hierarchical clustering analysis for protein expression between the basal and luminal subtypes. (D) Gene ontology analysis of up- and down-regulated proteins in the basal vs luminal subtype.
CONCLUSIONS
Recent improvements in the accuracy, sensitivity, and resolution of mass spectrometers have enabled high throughput, in-depth proteome characterization. However, spatial or temporal limits, such as loading capacity on columns or c-trap, and the instrument cycle time, make it difficult to identify low abundance proteins in highly complex biological/clinical samples. The conventional 1D-DIA method has been shown to overcome some of these constraints; however, many low abundance proteins remain unidentified in a typical analysis due to the high dynamic range of protein abundances in complex samples. In this study, we developed an improved 2D-DIA method by reducing the sample complexity of the entire proteome via sample fractionation. Characterization of the peptides derived from NCI-7 cell in each fraction from 2D fractionation showed that a combination of several fractions can achieve sufficient protein identification, and those fractions were evaluated with DIA. The optimized 2D-DIA method demonstrated that analyzing a pool of just a few fractions from a complex sample could improve the number of protein identifications, as well as the ability to identify relatively low abundance proteins, as compared to conventional 1D-DIA analysis. In addition, despite a slight decrease in the reproducibility due to variability of the 2D fractionation processes, it was still possible to achieve a median CV values of around 10%, demonstrating the potential of this method as a quantitative assay. Given the emergence of the field of personalized medicine, the demand for personalized proteomics in the clinical field is increasing. The maintenance of the instruments and the cost of the analysis per sample are a major hurdle for researchers who want to analyze hundreds of samples. Our results show that 2D-DIA allows identification of over 6,000 proteins within 1 h gradient time in cell lines and PDX samples. We expect that the 2D-DIA method will become a useful approach for quantitative proteomic analysis that is able to achieve excellent results while minimizing the overall time and costs of the analysis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by grants from the National Institutes of Health, National Cancer Institute, the Clinical Proteomic Tumor Analysis Consortium (CPTAC, U24CA210985, and U24CA210967), NCI U01CA152813, and NIGMS R01GM094231.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b04418.
Conditions for optimization of DIA method (Table S1), information of spectral libraries that used in this experiment (Table S2), comparison DIA results by both spectral libraries (DDA only and DDA/DIA combining, Table S3), and link to raw data: https://cptac-data-portal.georgetown.edu/cptac/s/S052 (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.9b04418
The authors declare no competing financial interest.
Contributor Information
Kyung-Cho Cho, Department of Pathology, Johns Hopkins, University School of Medicine, Baltimore, Maryland 21231, United States.
David J. Clark, Department of Pathology, Johns Hopkins, University School of Medicine, Baltimore, Maryland 21231, United States.
Michael Schnaubelt, Department of Pathology, Johns Hopkins, University School of Medicine, Baltimore, Maryland 21231, United States.
Guo Ci Teo, Department of Pathology, University of Michigan, Ann Arbor, Michigan 48109, United States.
Felipe da Veiga Leprevost, Department of Pathology, University of Michigan, Ann Arbor, Michigan 48109, United, States.
William Bocik, Antibody Characterization Laboratory, Cancer Research Technology Program, Frederick National Laboratory for Cancer Research, Frederick, Maryland 21701, United States.
Emily S. Boja, Office of Cancer Clinical Proteomics Research, National Cancer Institute, Bethesda, Maryland 20892, United, States
Tara Hiltke, Office of Cancer Clinical Proteomics Research, National Cancer Institute, Bethesda, Maryland 20892, United States.
Alexey I. Nesvizhskii, Department of Pathology and Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan 48109, United, States
Hui Zhang, Department of Pathology, Johns Hopkins, University School of Medicine, Baltimore, Maryland 21231, United States.
REFERENCES
- (1).Hao JJ; Zhi X; Wang Y; Zhang Z; Hao Z; Ye R; Tang Z; Qian F; Wang Q; Zhu J Sci. Rep. 2017, 7, 42436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Huang Z; Ma L; Huang C; Li Q; Nice EC Proteomics 2017, 17 (6), 1600240. [DOI] [PubMed] [Google Scholar]
- (3).Kelstrup CD; Bekker-Jensen DB; Arrey TN; Hogrebe A; Harder A; Olsen JV J. Proteome Res. 2018, 17 (1), 727–738. [DOI] [PubMed] [Google Scholar]
- (4).Kim MS; Pinto SM; Getnet D; Nirujogi RS; Manda SS; Chaerkady R; Madugundu AK; Kelkar DS; Isserlin R; Jain S; Thomas JK; Muthusamy B; Leal-Rojas P; Kumar P; Sahasrabuddhe NA; Balakrishnan L; Advani J; George B; Renuse S; Selvan LD; Patil AH; Nanjappa V; Radhakrishnan A; Prasad S; Subbannayya T; Raju R; Kumar M; Sreenivasamurthy SK; Marimuthu A; Sathe GJ; Chavan S; Datta KK; Subbannayya Y; Sahu A; Yelamanchi SD; Jayaram S; Rajagopalan P; Sharma J; Murthy KR; Syed N; Goel R; Khan AA; Ahmad S; Dey G; Mudgal K; Chatterjee A; Huang TC; Zhong J; Wu X; Shaw PG; Freed D; Zahari MS; Mukherjee KK; Shankar S; Mahadevan A; Lam H; Mitchell CJ; Shankar SK; Satishchandra P; Schroeder JT; Sirdeshmukh R; Maitra A; Leach SD; Drake CG; Halushka MK; Prasad TS; Hruban RH; Kerr CL; Bader GD; Iacobuzio-Donahue CA; Gowda H; Pandey A Nature 2014, 509 (7502), 575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Omenn GS; Lane L; Lundberg EK; Overall CM; Deutsch EW J. Proteome Res. 2017, 16 (12), 4281–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gillet LC; Leitner A; Aebersold R Annu. Rev. Anal. Chem. 2016, 9 (1), 449–72. [DOI] [PubMed] [Google Scholar]
- (7).Aebersold R; Mann M Nature 2003, 422 (6928), 198–207. [DOI] [PubMed] [Google Scholar]
- (8).Tabb DL; Vega-Montoto L; Rudnick PA; Variyath AM; Ham AJ; Bunk DM; Kilpatrick LE; Billheimer DD; Blackman RK; Cardasis HL; Carr SA; Clauser KR; Jaffe JD; Kowalski KA; Neubert TA; Regnier FE; Schilling B; Tegeler TJ; Wang M; Wang P; Whiteaker JR; Zimmerman LJ; Fisher SJ; Gibson BW; Kinsinger CR; Mesri M; Rodriguez H; Stein SE; Tempst P; Paulovich AG; Liebler DC; Spiegelman CJ Proteome Res. 2010, 9 (2), 761–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Michalski A; Cox J; Mann MJ Proteome Res. 2011, 10 (4), 1785–93. [DOI] [PubMed] [Google Scholar]
- (10).Geromanos SJ; Vissers JP; Silva JC; Dorschel CA; Li GZ; Gorenstein MV; Bateman RH; Langridge JI Proteomics 2009, 9 (6), 1683–95. [DOI] [PubMed] [Google Scholar]
- (11).Shishkova E; Hebert AS; Coon JJ Cell Syst 2016, 3 (4), 321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Meier F; Geyer PE; Virreira Winter S; Cox J; Mann M Nat. Methods 2018, 15 (6), 440–448. [DOI] [PubMed] [Google Scholar]
- (13).Gillet LC; Navarro P; Tate S; Rost H; Selevsek N; Reiter L; Bonner R; Aebersold R Mol. Cell. Proteomics 2012, 11 (6), O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rost HL; Rosenberger G; Navarro P; Gillet L; Miladinovic SM; Schubert OT; Wolski W; Collins BC; Malmstrom J; Malmstrom L; Aebersold R Nat. Biotechnol. 2014, 32 (3), 219–23. [DOI] [PubMed] [Google Scholar]
- (15).Muntel J; Xuan Y; Berger ST; Reiter L; Bachur R; Kentsis A; Steen HJ Proteome Res. 2015, 14 (11), 4752–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Collins BC; Hunter CL; Liu Y; Schilling B; Rosenberger G; Bader SL; Chan DW; Gibson BW; Gingras AC; Held JM; Hirayama-Kurogi M; Hou G; Krisp C; Larsen B; Lin L; Liu S; Molloy MP; Moritz RL; Ohtsuki S; Schlapbach R; Selevsek N; Thomas SN; Tzeng SC; Zhang H; Aebersold R Nat. Commun. 2017, 8 (1), 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rosenberger G; Koh CC; Guo T; Rost HL; Kouvonen P; Collins BC; Heusel M; Liu Y; Caron E; Vichalkovski A; Faini M; Schubert OT; Faridi P; Ebhardt HA; Matondo M; Lam H; Bader SL; Campbell DS; Deutsch EW; Moritz RL; Tate S; Aebersold R Sci. Data 2014, 1, 140031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Selevsek N; Chang CY; Gillet LC; Navarro P; Bernhardt OM; Reiter L; Cheng LY; Vitek O; Aebersold R Mol. Cell. Proteomics 2015, 14 (3), 739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jayabalan N; Lai A; Nair S; Guanzon D; Scholz-Romero K; Palma C; McIntyre HD; Lappas M; Salomon C Proteomics 2018, 1800164. [DOI] [PubMed] [Google Scholar]
- (20).Kim YJ; Sweet SM; Egertson JD; Sedgewick AJ; Woo S; Liao WL; Merrihew GE; Searle BC; Vaske C; Heaton R; MacCoss MJ; Hembrough TJ Proteome Res. 2018, DOI: 10.1021/acs.jproteome.8b00699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Meier F; Brunner AD; Koch S; Koch H; Lubeck M; Krause M; Goedecke N; Decker J; Kosinski T; Park MA; Bache N; Hoerning O; Cox J; Rather O; Mann M Mol. Cell. Proteomics 2018, 17 (12), 2534–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hebert AS; Prasad S; Belford MW; Bailey DJ; McAlister GC; Abbatiello SE; Huguet R; Wouters ER; Dunyach JJ; Brademan DR; Westphall MS; Coon JJ Anal. Chem. 2018, 90 (15), 9529–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Swearingen KE; Moritz RL Expert Rev. Proteomics 2012, 9 (5), 505–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang Y; Yang F; Gritsenko MA; Wang Y; Clauss T; Liu T; Shen Y; Monroe ME; Lopez-Ferrer D; Reno T; Moore RJ; Klemke RL; Camp DG, 2nd; Smith RD Proteomics 2011, 11 (10), 2019–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhang H; Liu T; Zhang Z; Payne SH; Zhang B; McDermott JE; Zhou JY; Petyuk VA; Chen L; Ray D; Sun S; Yang F; Chen L; Wang J; Shah P; Cha SW; Aiyetan P; Woo S; Tian Y; Gritsenko MA; Clauss TR; Choi C; Monroe ME; Thomas S; Nie S; Wu C; Moore RJ; Yu KH; Tabb DL; Fenyo D; Bafna V; Wang Y; Rodriguez H; Boja ES; Hiltke T; Rivers RC; Sokoll L; Zhu H; Shih IM; Cope L; Pandey A; Zhang B; Snyder MP; Levine DA; Smith RD; Chan DW; Rodland KD; Investigators C. Cell 2016, 166 (3), 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bekker-Jensen DB; Kelstrup CD; Batth TS; Larsen SC; Haldrup C; Bramsen JB; Sorensen KD; Hoyer S; Orntoft TF; Andersen CL; Nielsen ML; Olsen JV Cell Syst 2017, 4 (6), 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Clark DJ; Hu Y; Bocik W; Chen L; Schnaubelt M; Roberts R; Shah P; Whiteley G; Zhang HJ Proteome Res. 2018, 17 (6), 2205–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhou JY; Chen L; Zhang B; Tian Y; Liu T; Thomas SN; Chen L; Schnaubelt M; Boja E; Hiltke T; Kinsinger CR; Rodriguez H; Davies SR; Li S; Snider JE; Erdmann-Gilmore P; Tabb DL; Townsend RR; Ellis MJ; Rodland KD; Smith RD; Carr SA; Zhang Z; Chan DW; Zhang HJ Proteome Res. 2017, 16 (12), 4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Schwanhausser B; Busse D; Li N; Dittmar G; Schuchhardt J; Wolf J; Chen W; Selbach M Nature 2011, 473 (7347), 337–42. [DOI] [PubMed] [Google Scholar]
- (30).Nagaraj N; Wisniewski JR; Geiger T; Cox J; Kircher M; Kelso J; Paabo S; Mann M Mol. Syst. Biol. 2011, 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Navarro P; Kuharev J; Gillet LC; Bernhardt OM; MacLean B; Rost HL; Tate SA; Tsou CC; Reiter L; Distler U; Rosenberger G; Perez-Riverol Y; Nesvizhskii AI; Aebersold R; Tenzer S Nat. Biotechnol. 2016, 34 (11), 1130–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tsou CC; Avtonomov D; Larsen B; Tucholska M; Choi H; Gingras AC; Nesvizhskii AI Nat. Methods 2015, 12 (3), 258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tsou CC; Tsai CF; Teo GC; Chen YJ; Nesvizhskii AI Proteomics 2016, 16 (15−16), 2257–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bjornson RD; Carriero NJ; Colangelo C; Shifman M; Cheung KH; Miller PL; Williams KJ Proteome Res. 2008, 7 (1), 293–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kim S; Pevzner PA Nat. Commun. 2014, 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Eng JK; Jahan TA; Hoopmann MR Proteomics 2013, 13 (1), 22–4. [DOI] [PubMed] [Google Scholar]
- (37).Keller A; Nesvizhskii AI; Kolker E; Aebersold R Anal. Chem. 2002, 74 (20), 5383–92. [DOI] [PubMed] [Google Scholar]
- (38).Shteynberg D; Deutsch EW; Lam H; Eng JK; Sun Z; Tasman N; Mendoza L; Moritz RL; Aebersold R; Nesvizhskii AI Mol. Cell. Proteomics 2011, 10 (12), M111 007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI Nat. Methods 2017, 14 (5), 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Nesvizhskii AI; Keller A; Kolker E; Aebersold R Anal. Chem. 2003, 75 (17), 4646–58. [DOI] [PubMed] [Google Scholar]
- (41).Lam H; Deutsch EW; Eddes JS; Eng JK; Stein SE; Aebersold R Nat. Methods 2008, 5 (10), 873–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Reiter L; Rinner O; Picotti P; Huttenhain R; Beck M; Brusniak MY; Hengartner MO; Aebersold R Nat. Methods 2011, 8 (5), 430–5. [DOI] [PubMed] [Google Scholar]
- (43).Callister SJ; Barry RC; Adkins JN; Johnson ET; Qian WJ; Webb-Robertson BJ; Smith RD; Lipton MS J. Proteome Res. 2006, 5 (2), 277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Huang KL; Li S; Mertins P; Cao S; Gunawardena HP; Ruggles KV; Mani DR; Clauser KR; Tanioka M; Usary J; Kavuri SM; Xie L; Yoon C; Qiao JW; Wrobel J; Wyczalkowski MA; Erdmann-Gilmore P; Snider JE; Hoog J; Singh P; Niu B; Guo Z; Sun SQ; Sanati S; Kawaler E; Wang X; Scott A; Ye K; McLellan MD; Wendl MC; Malovannaya A; Held JM; Gillette MA; Fenyo D; Kinsinger CR; Mesri M; Rodriguez H; Davies SR; Perou CM; Ma C; Reid Townsend R; Chen X; Carr SA; Ellis MJ; Ding L Nat. Commun. 2017, 8, 14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Mertins P; Tang LC; Krug K; Clark DJ; Gritsenko MA; Chen L; Clauser KR; Clauss TR; Shah P; Gillette MA; Petyuk VA; Thomas SN; Mani DR; Mundt F; Moore RJ; Hu Y; Zhao R; Schnaubelt M; Keshishian H; Monroe ME; Zhang Z; Udeshi ND; Mani D; Davies SR; Townsend RR; Chan DW; Smith RD; Zhang H; Liu T; Carr SA Nat. Protoc. 2018, 13 (7), 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jin Q; Mao X; Li B; Guan S; Yao F; Jin F Tumor Biol. 2015, 36 (3), 1895–902. [DOI] [PubMed] [Google Scholar]
- (47).Louie SM; Grossman EA; Crawford LA; Ding L; Camarda R; Huffman TR; Miyamoto DK; Goga A; Weerapana E; Nomura DK Cell Chem. Biol. 2016, 23 (5), 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Karn T; Ruckhaberle E; Hanker L; Muller V; Schmidt M; Solbach C; Gatje R; Gehrmann M; Holtrich U; Kaufmann M; Rody A Breast Cancer Res. Treat. 2011, 130 (2), 409–20. [DOI] [PubMed] [Google Scholar]
- (49).Ademuyiwa FO; Thorat MA; Jain RK; Nakshatri H; Badve S Mod. Pathol. 2010, 23 (2), 270–5. [DOI] [PubMed] [Google Scholar]
- (50).Mangia A; Scarpi E; Partipilo G; Schirosi L; Opinto G; Giotta F; Simone G Oncotarget 2017, 8 (39), 65730–65742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Livasy CA; Karaca G; Nanda R; Tretiakova MS; Olopade OI; Moore DT; Perou CM Mod. Pathol. 2006, 19 (2), 264–71. [DOI] [PubMed] [Google Scholar]
- (52).Gauthier ML; Berman HK; Miller C; Kozakeiwicz K; Chew K; Moore D; Rabban J; Chen YY; Kerlikowske K; Tlsty TD Cancer Cell 2007, 12 (5), 479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.