

Abstract
Munc13‐1 is crucial for neurotransmitter release and, together with Munc18‐1, orchestrates assembly of the neuronal SNARE complex formed by syntaxin‐1, SNAP‐25, and synaptobrevin. Assembly starts with syntaxin‐1 folded into a self‐inhibited closed conformation that binds to Munc18‐1. Munc13‐1 is believed to catalyze the opening of syntaxin‐1 to facilitate SNARE complex formation. However, different types of Munc13‐1‐syntaxin‐1 interactions have been reported to underlie this activity, and the critical nature of Munc13‐1 for release may arise because of its key role in bridging the vesicle and plasma membranes. To shed light into the mechanism of action of Munc13‐1, we have used NMR spectroscopy, SNARE complex assembly experiments, and liposome fusion assays. We show that point mutations in a linker region of syntaxin‐1 that forms intrinsic part of the closed conformation strongly impair stimulation of SNARE complex assembly and liposome fusion mediated by Munc13‐1 fragments, even though binding of this linker region to Munc13‐1 is barely detectable. Conversely, the syntaxin‐1 SNARE motif clearly binds to Munc13‐1, but a mutation that disrupts this interaction does not affect SNARE complex assembly or liposome fusion. We also show that Munc13‐1 cannot be replaced by an artificial tethering factor to mediate liposome fusion. Overall, these results emphasize how very weak interactions can play fundamental roles in promoting conformational transitions and strongly support a model whereby the critical nature of Munc13‐1 for neurotransmitter release arises not only from its ability to bridge two membranes but also from an active role in opening syntaxin‐1 via interactions with the linker.
Keywords: conformational transition, membrane fusion, Munc13, Munc18, neurotransmitter release, SNAREs, syntaxin‐1
Significance Statement.
A large protein called Munc13‐1 is crucial for neurotransmitter release, a process that is key for communication between neurons and hence for brain function. The study presented here sheds light into the mechanism of action of Munc13‐1, showing that very weak interactions of Munc13‐1 with syntaxin‐1, one of the SNARE proteins involved in the release, mediates a conformational change in syntaxin‐1 that is critical for neurotransmitter release.
1. INTRODUCTION
Neurotransmitter release by Ca2+‐evoked synaptic vesicle exocytosis is an exquisitely regulated process that is crucial for communication between neurons. Release involves tethering of synaptic vesicles to specialized areas of the presynaptic membrane called active zones, priming of the vesicles to a release‐ready state and fast (<1 ms) fusion of the vesicle and plasma membranes when an action potential causes Ca2+ influx into the presynaptic terminal.1 Extensive characterization of the machinery that controls these steps has provided key insights into the mechanism of neurotransmitter release2, 3, 4, 5 and allowed reconstitution of some of the basic steps that lead to synaptic exocytosis with the central components of this machinery,6, 7, 8, 9 leading to defined models for their functions. Central for membrane fusion are the neuronal SNAP receptors (SNAREs) syntaxin‐1, SNAP‐25, and synaptobrevin, which form a tight SNARE complex that consists of a four‐helix bundle and brings the two membranes together.10, 11, 12, 13 N‐ethylmaleimide sensitive factor (NSF) and soluble NSF adaptor proteins (SNAPs) disassemble SNARE complexes to recycle the SNAREs,10, 14 whereas Munc18‐1 and Munc13s coordinate SNARE complex assembly in an NSF‐SNAP‐resistant manner,6, 15 and synaptotagmin‐1 acts as the Ca2+ sensor that triggers fast, synchronous release through interactions with phospholipids16 and the SNAREs,17, 18, 19 in a tight interplay with complexins.20, 21, 22
The critical involvement of Munc18‐1 and Munc13s in SNARE complex assembly explains, at least in part, the total abrogation of neurotransmitter release observed in the absence of these proteins.23, 24, 25 Assembly is initiated by binding of Munc18‐1 to a self‐inhibited “closed” conformation of syntaxin‐1 that involves the intramolecular binding of its N‐terminal Habc domain26 to the SNARE motif27, 28 (see domain diagram in Figure 1a and model on the left of Figure 1b). The transition from the Munc18‐1‐syntaxin‐1 complex to the SNARE complex requires Munc13‐1, which bridges the vesicle and plasma membranes7, 29 and is believed to help opening syntaxin‐130, 31, 32, 33 while Munc18‐1 binds also to the synaptobrevin SNARE motif, placing it near the syntaxin‐1 SNARE motif and thus providing a template for SNARE complex assembly.34, 35, 36, 37 Synaptobrevin binding is hindered by the furled conformation of a Munc18‐1 loop,36 and a gain‐of‐function mutation in Munc18‐1 that facilitates SNARE complex formation or a so‐called LE mutation in syntaxin‐1 that opens its conformation27 can partially rescue the severe phenotypes observed in the absence of Unc13,30, 38 the invertebrate homolog of Munc13s. Note also that the syntaxin‐1 LE mutation increases the vesicular release probability in mice.39 These findings suggest that the energy barriers to SNARE complex assembly provided by the Munc18‐1 furled loop and the syntaxin‐1 closed conformation are critical to render neurotransmitter release strictly dependent on Munc13/Unc13 and enable a wide variety of regulatory presynaptic plasticity processes that depend on Munc13s40 and underlie multiple forms of information processing in the brain.41 Recent reconstitution data suggest that release is restricted to this highly regulated pathway of SNARE complex assembly because αSNAP inhibits membrane fusion caused by alternative, constitutive pathways of SNARE complex assembly by multiple mechanisms and the αSNAP block can only be bypassed when Munc18‐1 binds to the syntaxin‐1 closed conformation.42
Figure 1.
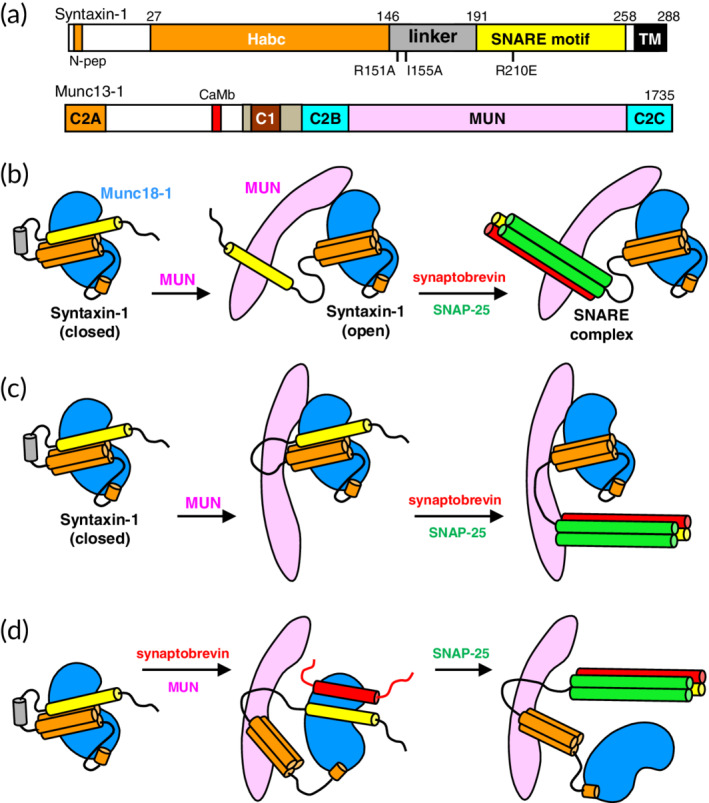
Models proposed for how the Munc13‐1 MUN domain facilitates the opening of syntaxin‐1 and SNARE complex formation. (a) Domain diagrams of syntaxin‐1 and Munc13‐1. SNARE indicates the SNARE motif and N‐pep indicates the N‐peptide of syntaxin‐1, which contributes to Munc18‐1 binding.2 Mutations used in this study are indicated below the syntaxin‐1 diagram. A calmodulin‐binding sequence of Munc13‐1 is labeled CaMb. Numbers on the right above the diagrams indicate the length of the proteins. (b–d) The diagram on the left of all models illustrates how Munc18‐1 (blue) binds to the N‐peptide and the closed conformation of syntaxin‐1, which involves intramolecular interactions between the Habc domain (orange) and the SNARE motif (yellow), as well as a specific structure in the linker (gray). An initial model postulated that the Munc13‐1 MUN domain (pink) helps to open syntaxin‐1 by binding to its SNARE motif and then templates SNARE complex assembly (synaptobrevin, red; SNAP‐25, green)31 (b). A revised model proposed that the MUN domain binds to the linker region and changes its conformation to open syntaxin‐1 and then template SNARE complex assembly33 (c). The model that we favor also postulates that the MUN domain binds to the syntaxin‐1 linker to open syntaxin‐1 but synaptobrevin is recruited to Munc18‐1, which templates SNARE complex assembly (d)
These results have established the functional importance of the syntaxin‐1 closed conformation, but the mechanism by which Munc13‐1 facilitates the opening of syntaxin‐1 has been controversial, in part because of the diversity of Munc13‐1‐syntaxin‐1 interactions that have been described. Initial yeast‐two‐hybrid assays revealed binding of a fragment spanning part of the syntaxin‐1 Habc domain to a C‐terminal fragment containing the Munc13‐1 C2C domain, whereas pulldown assays implicated sequences upstream of the C2C domain,43 which are part of what is now known as the MUN domain44 (see domain structure in Figure 1a). Subsequent analyses by NMR spectroscopy showed that the MUN domain accelerates the transition from the syntaxin‐1‐Munc18‐1 complex to the SNARE complex31 and that the MUN domain indeed binds weekly to syntaxin‐1, but the interaction involves primarily the N‐terminal half of the SNARE motif; the syntaxin‐1 N‐terminal region including the Habc domain or the Munc13‐1 C2C domain did not appear to contribute significantly to binding.29, 31 These results led to a model whereby the MUN domain “pulls away” the SNARE motif to open syntaxin‐1 and facilitate SNARE complex assembly (Figure 1b). Conversely, another study suggested that the activity of the MUN in stimulating SNARE complex formation involves interactions with the linker region joining the Habc domain and the SNARE motif of syntaxin‐1, rather than with the SNARE motif, leading to a model whereby the MUN domain destabilizes the closed conformation and templates SNARE complex assembly33 (Figure 1c). This model was supported by the observation that mutations in the linker region (R151A, I155A; Figure 1a) impair MUN domain‐catalyzed SNARE complex formation, liposome fusion in a reconstitution assay, and neurotransmitter release in neurons. This study also suggested that an R210E mutation in the syntaxin‐1 SNARE motif that was used to support the functional importance of its interaction with the MUN domain31 did not affect MUN domain‐catalyzed SNARE complex formation, but the effects of this mutation on liposome fusion and neurotransmitter release were not tested.33
It is also important to note that, while the initial experiments in C. elegans revealed robust rescue of Unc13 null phenotypes by the open syntaxin‐1 LE mutant,30 subsequent studies observed much weaker rescue,45 and the syntaxin‐1 LE mutant yielded very limited or no rescue of phenotypes in Munc13‐1 KO or Munc13‐1/2 double KO mice.8, 39 Moreover, the syntaxin‐1 LE mutant also rescues phenotypes of C. elegans lacking Unc10/RIM,46 and the demonstration of the physiological relevance of the membrane–membrane bridging activity of Munc13‐129 provided a natural explanation for the critical nature of this protein for neurotransmitter release that is independent of its role in opening syntaxin‐1. These findings raise two key questions: Is the syntaxin‐1 opening activity of Munc13‐1 as important as previously thought? What is the relative functional importance of this activity compared to that of membrane–membrane bridging by Munc13‐1?
To address these questions and clarify the mechanism underlying the role of Munc13‐1 in facilitating SNARE complex assembly, we have employed a combination of NMR spectroscopy and other biophysical experiments with fusion assays using reconstituted proteoliposomes. Our data show that, although the Munc13‐1 MUN domain clearly binds to the syntaxin‐1 SNARE motif, the R210E mutation does not affect liposome fusion, while the individual R151A and I155A mutations strongly impair fusion. Moreover, we find that Munc13‐1 cannot be replaced by an artificial membrane–membrane tether in reconstitution assays incorporating Munc18‐1, NSF, and αSNAP. These results strongly support the notion that the fundamental role of Munc13‐1 in synaptic vesicle fusion involves not only its membrane–membrane bridging activity but also its ability to facilitate SNARE complex assembly through interactions with the syntaxin‐1 linker region.
2. RESULTS
2.1. NMR analysis of SNARE‐Munc13‐1 MUN domain interactions
Previous NMR studies analyzed interactions between Munc13‐1 and syntaxin‐1 using 1H‐15N heteronuclear single quantum coherence (HSQC) spectra of uniformly 15N labeled samples of syntaxin‐1 fragments spanning different regions in the presence or absence of the Munc13‐1 MUN domain or a fragment spanning the MUN and C2C domains.29, 31 These spectra provide power tools to detect and analyze protein–protein interactions,47 and the observation that these large Munc13‐1 fragments (70 and 93 kDa, respectively) caused almost no perturbation of the 1H‐15N HSQC spectra of fragments spanning the cytoplasmic region of syntaxin‐1 [syntaxin‐1(2–253)] or the syntaxin‐1 N‐terminal region including the Habc domain and linker sequence [syntaxin‐1(2–180)]29, 31 showed that these syntaxin‐1 and Munc13‐1 fragments do not bind to each other or interact very weakly. In contrast, the Munc13‐1 MUN domain broadened beyond detection of the cross‐peaks corresponding to residues 200–226 of a fragment spanning the syntaxin‐1 SNARE motif [syntaxin‐1(191–253)] at similar concentrations, showing that the MUN domain binds to the N‐terminal half of the SNARE motif.31 The binding was of moderate affinity (K D = 46 μM), but the interaction was clearly stronger than any potential interaction of MUN with syntaxin‐1 N‐terminal sequences.
Since the linker region of syntaxin‐1 (residues 147–190) was proposed to be the bona fide binding target of the MUN domain,33 we tested whether we could observe binding using 15N‐labeled fragments spanning part of the linker (residues 138–180) and the entire linker (residues 138–200). Both fragments had a tendency to aggregate but we were able to obtain 1H‐15N HSQC spectra of reasonable quality at 15–20 μM concentrations (Figure 2a,b; black contours). The addition of equimolar amounts of the Munc13‐1 MUN domain caused only small perturbations of a few cross‐peaks of syntaxin‐1(138–180) and syntaxin‐1(138–200) (Figure 2a,b; red contours), showing that, if there is any interaction between these protein fragments, it is very weak. To explore whether there might be some cooperativity between the linker region and the SNARE motif for binding to the MUN domain, we also performed experiments with a 15N‐labeled fragment corresponding to residues 138–232 of syntaxin‐1. In this case, we did observe broadening beyond the detection of several cross‐peaks (Figure 2c), but they most likely correspond to the SNARE motif and not to the linker region, given the limited number of cross‐peaks that broadened. A control experiment with a fragment spanning the N‐terminal half of the SNARE motif [syntaxin‐1(191–236)] showed broadening beyond the detection of many of the cross‐peaks upon addition of the MUN domain (Figure 2d), which is consistent with our previous results obtained with syntaxin‐1(191–253).31 These results show that MUN interacts more strongly with the SNARE motif than with the linker region of syntaxin‐1.
Figure 2.
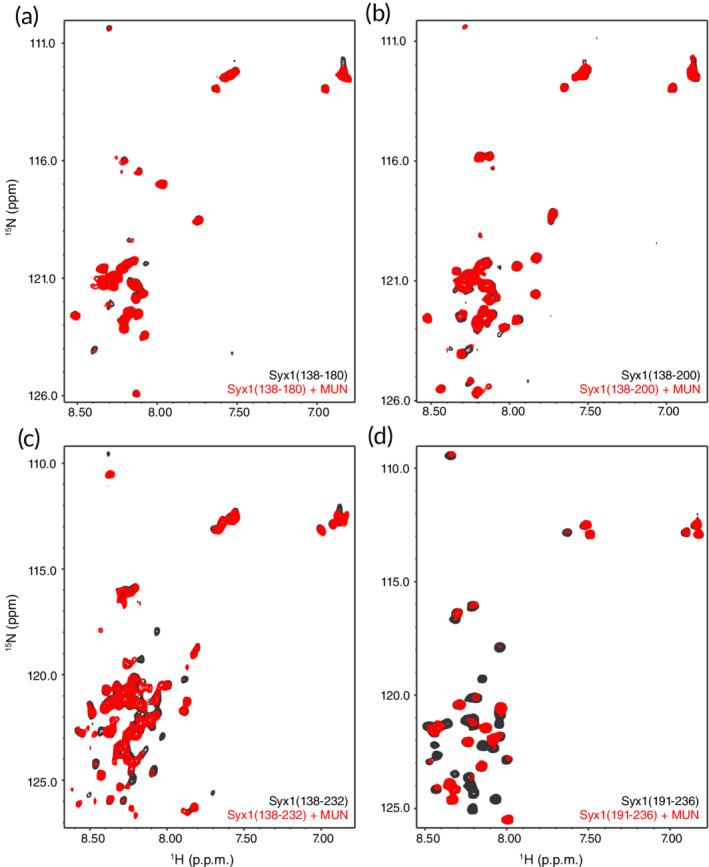
Analysis of interactions between the Munc13‐1 MUN domain and syntaxin‐1 by NMR spectroscopy. (a–d) 1H‐15N HSQC spectra of syntaxin‐1(138–180) (a), syntaxin‐1(138–200) (b), syntaxin‐1(138–232) (c) or syntaxin‐1(191–236) (d), in the absence (black contours) or presence (red contours) of equimolar amounts of MUN domain. The protein concentrations ranged from 15 to 25 μM
Interactions of the Munc13‐1 MUN domain with the other SNAREs may also be important for its mechanism of action, as Munc13‐1 was shown to prevent the formation of complexes with antiparallel SNARE motifs.8 Previously, we had shown that the Munc13‐1 MUN domain binds to the juxtamembrane region of synaptobrevin36 but this interaction is likely non‐specific, as this sequence also binds to Munc18‐148 and to membranes,49 and its promiscuity can be attributed to the presence of three aromatic and multiple basic residues in its sequence. Since interactions between the Munc13‐1 MUN domain and SNAP‐25 have not been studied, we prepared 15N‐labeled SNAP‐25 and analyzed how its 1H‐15N HSQC spectrum is affected by the MUN domain. The 1H‐15N HSQC spectrum of SNAP‐25 was rather well resolved considering its relatively large size (206 residues) and the lack of tertiary structure (Figure 3, black contours). The addition of the MUN domain caused almost no perturbation of the SNAP‐25 cross‐peaks (Figure 3, red contours), showing that, if there is any binding between MUN and SNAP‐25, the affinity is very weak.
Figure 3.
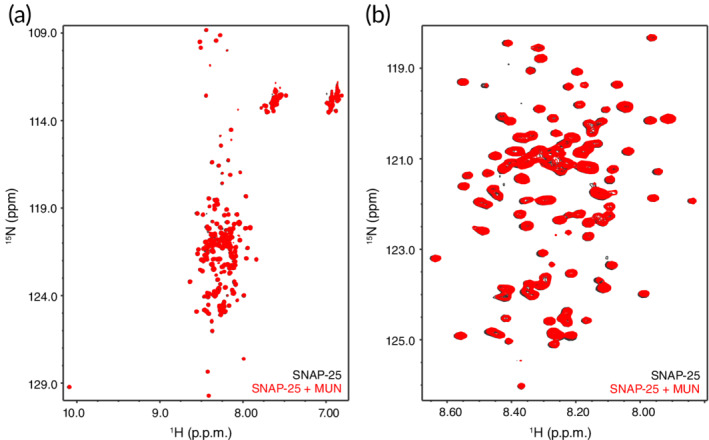
Analysis of interactions between the Munc13‐1 MUN domain and SNAP‐25 by NMR spectroscopy. (a) 1H‐15N HSQC spectra of 19 μM 15N‐labeled SNAP‐25 in the absence (black contours) or presence (red contours) of 19 μM MUN domain. (b) Expansion of the spectra of panel (a) showing the crowded region in the center of the spectra
2.2. Interactions involving the syntaxin‐1 linker are crucial for liposome fusion
The NMR data favor the proposal that the Munc13‐1 MUN domain acts via interactions with the syntaxin‐1 SNARE motif, which was also supported by previous FRET assays showing that the MUN domain accelerates the transition from the wild type (WT) syntaxin‐1‐Munc18‐1 complex to the SNARE complex but not when syntaxin‐1 contains the R210E mutation in the SNARE motif.31 These assays were performed with a donor FRET probe attached to residue 61 of the synaptobrevin SNARE motif and an acceptor probe attached to residue 249 on the SNARE motif of syntaxin‐1. Subsequently, Cong Ma's laboratory performed analogous assays but placing the acceptor probe on residue 197 of the C‐terminal SNAP‐25 SNARE motif and found that SNARE complex assembly was impaired by the R151A and I155A mutations, but not by the R210E mutation, leading to the proposal that the MUN domain acts through interactions with the linker region of syntaxin‐1 rather than its SNARE motif.33 The reasons for the different results obtained with the R210E mutant and for changing the location of the acceptor probe were unclear.
To clarify this controversy, we performed similar FRET assays but with two modifications. First, instead of the separate SNAP‐25 SNARE motifs that were used in the previous studies, we used full‐length SNAP‐25 because the covalent linkage of the SNARE motifs is expected to influence the kinetics of SNARE complex assembly. Second, since very weak but potentially relevant interactions of the MUN domain or Munc18‐1 with the SNARE motifs might be perturbed by the fluorescent probes, we placed the fluorescent probes in residues preceding the SNARE motifs. Thus, we labeled residue 26 of a fragment spanning the cytoplasmic region of synaptobrevin (residues 1–96) with a BODIPY‐FL donor probe and residue 136 of SNAP‐25 with an acceptor tetramethylrhodamine (TMR) probe. This design is unlikely to affect the mechanism of SNARE complex assembly, as we have previously shown that the efficiency of liposome fusion in reconstitution experiments was not affected by attachment of fluorescent probes on residue 26 of synaptobrevin and residue 186 of syntaxin‐1 (preceding its SNARE motif similarly to R136 with respect to the C‐terminal SNARE motif of SNAP‐25).42 Here, we labeled SNAP‐25 rather than syntaxin‐1 because we wanted to compare the behavior of WT syntaxin‐1 and the three syntaxin‐1 mutants. Using these probes, we monitored the decrease in the donor fluorescence emission intensity due to FRET with the acceptor as the SNARE complex forms.
We found that, as expected, WT, R210E, R151A, and I155A syntaxin‐1 assembled into SNARE complexes with synaptobrevin(1–96) and SNAP‐25 with relatively fast speed, and that preincubation of the corresponding syntaxin‐1 version with an excess of Munc18‐1 strongly inhibited the reaction (black and red curves, respectively, in the insets of Figure 4a–d). Moreover, we observed that the Munc13‐1 MUN domain stimulated SNARE complex assembly in the presence of Munc18‐1 in reactions performed with WT and R210E syntaxin‐1, but not in those carried out with R151A and I155A syntaxin‐1 (Figure 4a–d, blue curves). These conclusions were supported by quantification of the relative donor fluorescence observed at particular time points in repeat experiments (e.g., at 1,300 s, Figure 4e). We note that there was some variability in these assays, perhaps because relatively high concentrations of MUN domain (e.g., 30 μM) are required to observe stimulation, and both the MUN domain and Munc18‐1 (used at 10 μM) have limited solubility, which may lead to inconsistent activity. In addition, the decrease in the donor fluorescence intensity observed for the reaction with the R210E syntaxin‐1 mutant in the absence of Munc18‐1 and MUN domain was considerably slower than that observed for WT syntaxin‐1 and the R151A and I155A mutants but plateaued at lower relative fluorescence intensity. The reasons underlying these differences are unclear, but they may arise from aggregation in the reactions performed with the R210E mutant. Although the results of these kinetic assays need to be interpreted with caution because of these caveats, they clearly correlate with those reported by the Ma laboratory.33
Figure 4.
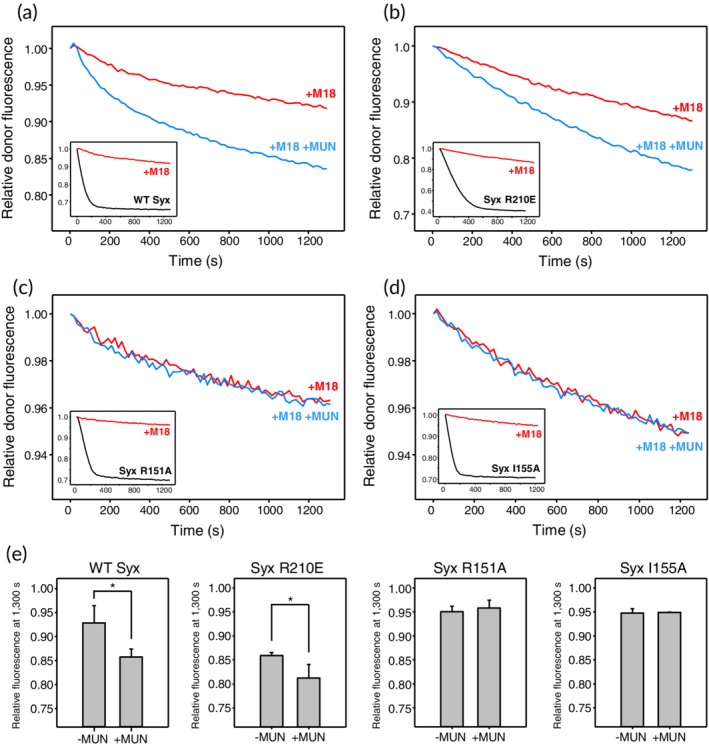
The R151A and I155A mutations in the syntaxin‐1 linker region but not the R210E mutation in its SNARE motif disrupt stimulation of SNARE complex formation by the Munc13‐1 MUN domain. (a–d) SNARE complex assembly assays monitored from the decrease in the fluorescence emission intensity of a donor BODIPY‐FL label attached to residue 26 of synaptobrevin(1–96) due to FRET with a TMR acceptor attached to residue 136 of SNAP‐25 as a function of time. Syntaxin‐1(2–253) had a WT sequence (a) or bore the R210E (b), R151A (c) or I155A (d) mutation. The insets show the results obtained upon mixing BODIPY‐FL‐synaptobrevin(1–96) with syntaxin‐1(2–253) and TMR‐SNAP‐25 without Munc18‐1 (black curves) or pre‐incubating syntaxin‐1(2–253) with an excess of Munc18‐1 (red curves). The main plots compare the latter (red curves) with analogous experiments performed in the presence of the MUN domain (blue curves) to illustrate whether the MUN domain stimulates SNARE complex assembly. All data were normalized to the first point of each kinetic trace. (e) Quantification of the relative donor fluorescence emission intensities observed after 1,300 s in SNARE complex assembly assays that were started with syntaxin‐1(2–253) pre‐incubated with Munc18‐1 and were performed in the absence and presence of the MUN domain. All experiments were performed in triplicate. Values indicate means ±SDs. Statistical significance and p values were determined by one‐way analysis of variance (ANOVA) with the Holm‐Sidak test (*p < .05)
We turned to our reconstitution assays of liposome fusion, which provide a more reliable means to recapitulate the events that lead to SNARE complex formation and membrane fusion. The standard assays that we use monitor lipid and content mixing50 between liposomes containing synaptobrevin (V‐liposomes) and liposomes containing syntaxin‐1 and SNAP‐25 (T‐liposomes) in the presence of NSF, αSNAP, Munc18‐1, and a Munc13‐1 fragment spanning the C1, C2B, MUN, and C2C domains (C1C2BMUNC2C).7 In our hands, these assays yield more reproducible results than those that were employed to test the effects of a double R151A, I155A mutation, which instead used syntaxin‐1‐Munc18‐1 liposomes and a Munc13‐1 fragment spanning only the C1, C2B, and MUN domains (C1C2BMUN), and did not include NSF and αSNAP.33 The presence of NSF and αSNAP helps to remove off‐pathway intermediates (e.g., syntaxin‐1 aggregates or syntaxin‐1‐SNAP‐25 heterodimers) and generates the syntaxin‐1‐Munc18‐1 freshly in situ, while the C1C2BMUNC2C fragment is much more active than C1C2BMUN because of the key role of the C2C domain in membrane–membrane bridging,7, 29 allowing highly efficient Ca2+‐dependent fusion at C1C2BMUNC2C concentrations on the order of 100–500 nM.29, 51
In experiments performed with T‐liposomes containing WT syntaxin‐1, we observed fast Ca2+‐dependent liposome fusion with V‐liposomes when NSF, αSNAP, Munc18‐1, and Munc13‐1 C1C2BMUNC2C were included, but no fusion was observed in control experiments where Munc18‐1 and/or C1C2BMUNC2C were omitted (Figure 5a,b), as expected. Highly efficient Ca2+‐dependent fusion was also observed when syntaxin‐1 bore the R210E mutation, but the I155A mutation and particularly the R151A mutation strongly impaired fusion (Figure 5c–f). These effects were not due to poor incorporation of proteins into the liposomes, as the different liposome preparations contained comparable amounts of syntaxin‐1 and SNAP‐25 (Figure 5g). Moreover, the R151 and I155 side chains are exposed on the surface of the syntaxin‐1 closed conformation and do not contact Munc18‐1 in the binary syntaxin‐1‐Munc18‐1 complex. Hence, it is unlikely that the effects of the R151A and I155A mutations arise because they destabilize the closed conformation or impair Munc18‐1 binding. Nevertheless, we analyzed the binding of R151A and I155A syntaxin‐1(2–253) mutants to Munc18‐1 by isothermal titration calorimetry (ITC) to rule out these possibilities. We found that both syntaxin‐1(2–253) mutants bound to Munc18‐1 with low nM affinities that are comparable to that measured with WT syntaxin‐1(2–253) (Figure 6; K D = 2.3, 4.6, and 4.8 nM for WT, R151A, and I155A, respectively). Note that the differences between these K D values are not significant because affinities below 10 nM cannot be distinguished under the conditions of these experiments, and for the same reason we cannot rule out that the mutations indeed affect Munc18‐1‐syntaxin‐1(2–253) binding to some degree. However, it is clear that the R151A and I155A syntaxin‐1(2–253) mutants retain very strong binding to Munc18‐1 and hence it seems unlikely that perturbation of syntaxin‐1‐Munc18‐1 interactions underlies the dramatic impairment of liposome fusion caused by these mutations.
Figure 5.
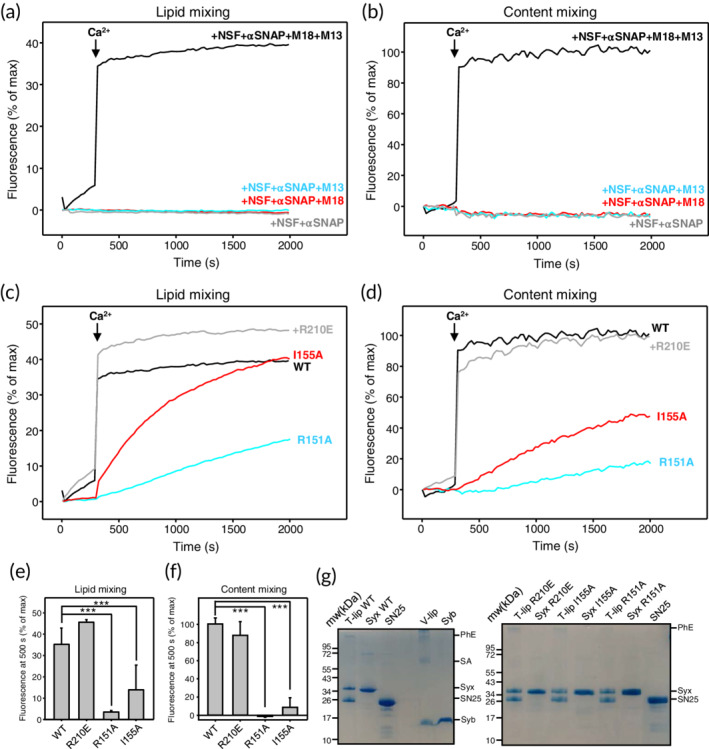
The R151A and I155A mutations in the syntaxin‐1 linker region but not the R210E mutation in its SNARE motif impair liposome fusion dependent on Munc18‐1 and Munc13‐1 C1C2BMUNC2C. (a–d) Lipid mixing (a, c) between V‐ and T‐liposomes was monitored from the fluorescence de‐quenching of Marina Blue lipids and content mixing (b, d) was monitored from the increase in the fluorescence signal of Cy5‐streptavidin trapped in the V‐liposomes caused by FRET with PhycoE‐biotin trapped in the T‐liposomes upon liposome fusion. In (a, b), the assays were performed with T‐liposomes that contained SNAP‐25 and WT syntaxin‐1 in the presence of NSF and αSNAP with or without Munc18‐1 (M18), and/or Munc13‐1 C1C2BMUNC2C (M13) as indicated. In (c, d), assays were performed in the presence of NSF, αSNAP, Munc18‐1, and C1C2BMUNC2C with T‐liposomes containing WT (black curves), R210E (gray curves), R151A (blue curves), or I155A (red curves) syntaxin‐1. All experiments were started in the presence of 100 μM EGTA and 5 μM streptavidin, and Ca2+ (600 μM) was added at 300 s. (e, f) Quantification of the amounts of lipid (e) or content (f) mixing observed after 500 s in experiments analogous to those shown in panels (c, d). All experiments were performed in triplicate. Values indicate means ±SDs. Statistical significance and p values were determined by one‐way analysis of variance (ANOVA) with the Holm‐Sidak test (***p < .001). (g) SDS‐PAGE gels stained with Coomassie blue of the V‐liposomes and the different T‐liposome preparations used for the liposome fusion assays. Control samples containing isolated WT and mutant syntaxin‐1 (Syx) proteins, as well as SNAP‐25 and synaptobrevin (Syb) are also shown. The positions of molecular weight markers are shown on the left of both gels
Figure 6.
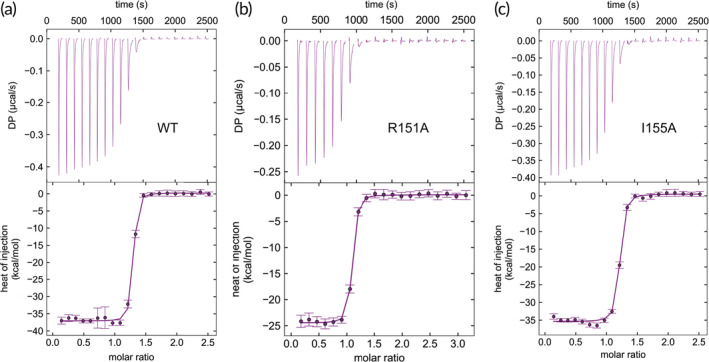
The R151A and I155A mutations in syntaxin‐1 do not impair Munc18‐1 binding. (a–c) Titrations of WT (a), R151A (b) or I155A (c) syntaxin‐1(2–253) onto Munc18‐1. The upper panels show the baseline‐ and singular‐value‐decomposition‐corrected thermograms for the respective experiments. The circles in the lower panels are the integrated heats of injection, with the error bars representing estimated errors for these values. The lines in these panels represent the respective fits of the data to a single binding site “A + B <‐> AB” model. The K D values derived from the titrations, with 68.3% confidence intervals indicated in parenthesis, are WT 2.3 nM (1.2–3.7 nM), R151A 4.6 nM (3.5–5.9 nM); I155A 4.8 nM (2.8–7.3 nM)
It also seems unlikely that mutations in the linker region alter interactions with SNAP‐25 and synaptobrevin, thus affecting SNARE complex assembly because the linker region does not participate in the formation of the four‐helix bundle. Nevertheless, to rule out this possibility we performed another set of experiments where we analyzed fusion of V‐liposomes with T‐liposomes bearing WT, R151A, or I155A syntaxin‐1 in the presence of Munc13‐1 C1C2BMUNC2C. We previously showed that C1C2BMUNC2C stimulates Ca2+‐independent fusion between V‐liposomes and T‐liposomes containing WT syntaxin‐1 in the absence of NSF, αSNAP, and Munc18‐1, which can be attributed to facilitation of SNARE complex formation between synaptobrevin on the V‐liposomes and syntaxin‐1‐SNAP‐25 heterodimers on the T‐liposomes by C1C2BMUNC2C, at least in part due to its membrane‐bridging activity7, 42 (see further discussion below). Because syntaxin‐1 needs to be open to form heterodimers with SNAP‐25, this pathway of membrane fusion is not expected to be affected by the mutations in the syntaxin‐1 linker. Indeed, we obtained comparable results in these assays with WT, R151A, and I155A syntaxin‐1: fusion between T‐ and V‐liposomes alone was highly inefficient, as expected, and addition of C1C2BMUNC2C stimulated fusion to a similar extent for WT syntaxin‐1 and the linker mutants (Figure 7). We note that the amount of fusion observed under these conditions for WT syntaxin‐1 is somewhat variable because of variability in the amount of SNAP‐25 incorporated into the T‐liposomes42 but, even with this variability, it was clear that the R151A and I155A mutations did not cause drastic impairments of liposome fusion such as those observed in the experiments performed in the presence of Munc18‐1, C1C2BMUNC2C, NSF, and αSNAP.
Figure 7.
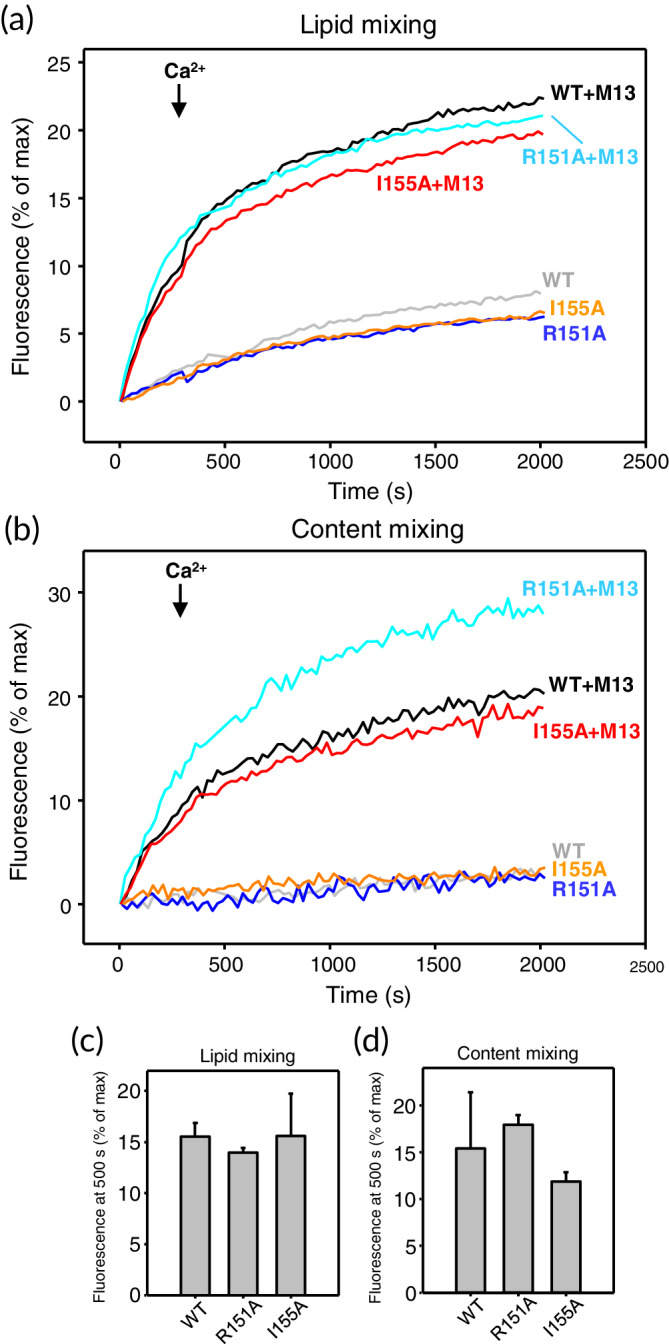
The R151A and I155A mutations do not impair fusion involving syntaxin‐1‐SNAP‐25 heterodimers. (a, b) Lipid mixing (a) between V‐ and T‐liposomes was monitored from the fluorescence de‐quenching of Marina Blue lipids and content mixing (b) was monitored from the increase in the fluorescence signal of Cy5‐streptavidin trapped in the V‐liposomes caused by FRET with PhycoE‐biotin trapped in the T‐liposomes upon liposome fusion. In (a, b), assays were performed in the absence or presence of Munc13‐1 C1C2BMUNC2C with T‐liposomes containing WT (gray and black curves), R151A (blue and cyan curves), or I155A (orange and red curves) syntaxin‐1. All experiments were started in the presence of 100 μM EGTA and 5 μM streptavidin, and Ca2+ (600 μM) was added at 300 s. (c, d) Quantification of the amounts of lipid (c) or content (d) mixing observed after 500 s in experiments analogous to those shown in panels (a–d). All experiments were performed in triplicate. Values indicate means ±SDs
2.3. Evidence for a direct MUN‐syntaxin‐1 linker interaction
The dramatic effects of the R151A and I155A single point mutations in the reconstitution assays performed in the presence of Munc18‐1, Munc13‐1 C1C2BMUNC2C, NSF, and αSNAP, but not in the presence of C1C2BMUNC2C alone, are remarkable and suggest that interactions involving the mutated residues are crucial for the events that lead to opening of syntaxin‐1 and subsequent membrane fusion. These results contrast with the NMR data presented here (Figure 2) and in previous studies,29, 31 which showed very little or no cross‐peak perturbations in analyses of interactions between the MUN domain and syntaxin‐1 fragments including the syntaxin‐1 linker by 1H‐15N HSQC or 1H‐13C heteronuclear multiple quantum coherence (HMQC) spectra. These results suggest that interactions between the MUN domain and the linker are very weak but are sufficient to lower the energy barrier for SNARE complex formation, and they can occur more efficiently in experiments performed with membranes (e.g., the liposome fusion assays) that in those performed in solution (e.g., the FRET assays of SNARE complex assembly) because the interactions of the Munc13‐1 C1, C2B, and C2C domains with the membranes dramatically increase the local concentration of the MUN domain near syntaxin‐1. Since analyses by 1H‐15N HSQC or 1H‐13C HMQC spectra in solution at high concentrations are precluded by the limited solubility of the MUN domain, we sought to obtain evidence for a direct interaction between the syntaxin‐1 linker and the MUN domain using a very highly sensitive method based on the observation of Gd3+‐induced paramagnetic relaxation effects (PREs). Because Gd3+ induces very strong PREs on nuclei in its proximity and the relaxation can be transferred between free and bound forms of a protein, a Gd3+ tag attached to a protein may induce substantial broadening of cross‐peaks from another protein even if the two proteins bind for only a small fraction of time.
We prepared two mutant versions of syntaxin‐1(2–253) where we attached a DOTA‐Gd3+ tag on residue 159 or 186 (referred to as Syx‐159‐Gd and Syx‐186‐Gd, respectively). The side chains of these residues are near residues 151 and 155 but point in different directions (see inset of Figure 8a) such that the attached Gd3+ tag may induce PREs on the MUN domain if it binds at residues 151 and 155 without disrupting the interaction. Note that a fluorescent tag on residue 186 did not hinder liposome fusion in our reconstitution assays.42 We first examined whether Syx‐159‐Gd induced PREs in the 1H‐13C HMQC spectra of samples of the Munc13‐1 MUN domain that were specifically 1H‐13C‐labeled at the methyl groups of Ile, Leu, Met, and Val residues on an otherwise perdeuterated background (2H,13CH3‐ILMV‐MUN), taking advantage of the high sensitivity offered by these spectra even for large proteins with this labeling scheme.52 Note also that syntaxin‐1(2–253) is known to adopt the closed conformation under the conditions of these experiments.27, 53 Syx‐159‐Gd caused only limited perturbations in the 1H‐13C HMQC spectrum of the MUN domain (Figure 8a), but did cause a clear broadening of specific cross‐peaks (more readily observable in the expansions of Figure 8b,c). Interestingly, Syx‐186‐Gd caused less or no broadening on these cross‐peaks (Figure 8d,e), showing that the broadening caused by Syx‐159‐Gd is specifically caused by the tag at this position. Although more detailed studies using this approach will be necessary to fully interpret these data, the results do support the notion that the linker of closed syntaxin‐1 binds directly to the MUN domain.
Figure 8.
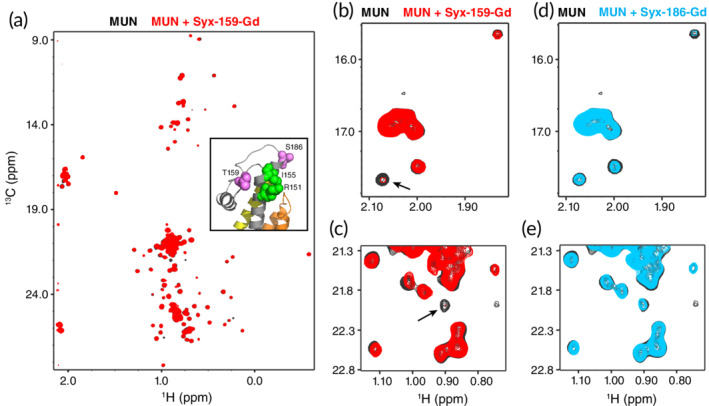
Analysis of Munc13‐1 MUN domain‐syntaxin‐1 interactions using Gd3+‐induced PREs. (a) 1H‐13C HMQC spectra of 8 μM 2H,13CH3‐ILMV‐MUN domain in the absence (black contours) or presence (red contours) of 24 μM Syx‐159‐Gd. The inset shows a ribbon diagram of the region containing the linker in the structure of the syntaxin‐1‐Munc18‐1 complex66 (PDB accession code http://bioinformatics.org/firstglance/fgij//fg.htm?mol=3C98), where the Habc domain is colored in orange, the linker in gray and the SNARE motif in yellow, residues R151 and I155 are shown as green spheres, and residues T159 and S186 (where the Gd3+ tags were placed) are shown as violet spheres. (b, c) Expansions of the spectra shown in panel (a) but plotted at different levels to illustrate the broadening caused on specific cross‐peaks by Syx‐159‐Gd (arrows). (d, e) Expansions of the same regions of 1H‐13C HMQC spectra of 8 μM 2H,13CH3‐ILMV‐MUN domain in the absence (black contours) or presence (blue contours) of 24 μM Syx‐186‐Gd
2.4. Munc13‐1 cannot be replaced by an artificial linker in Munc18‐1‐dependent liposome fusion
Overall, our data support the notion that a key function of Munc13‐1, in addition to bridging the two membranes, is to open syntaxin‐1. This model predicts that Munc13‐1 cannot be replaced by a factor that merely bridges membranes and cannot interact specifically with syntaxin‐1. To test this prediction, we took advantage of the recent observation that a dimeric fusion protein of GST with a PX domain that binds tightly to PI3P catalyzes fusion between liposomes bearing the yeast vacuolar homolog of synaptobrevin and liposomes bearing the cognate syntaxin‐1‐SNAP‐25 homologs when both liposome populations contained PI3P.54 Thus, we prepared syntaxin‐1‐SNAP‐25 liposomes and synaptobrevin liposomes containing 1% PI3P (referred to as T*‐liposomes and V*‐liposomes, respectively), and investigated whether Munc13‐1 C1C2BMUNC2C can be functionally replaced by the GST‐PX fusion protein.
In these experiments, we focused on measuring content mixing, which provides a reliable means to assess membrane fusion. We observed efficient Ca2+‐independent liposome fusion in the presence of NSF, αSNAP, Munc18‐1, and Munc13‐1 C1C2BMUNC2C that is not observed normally with liposomes lacking PI3P (Figure 5b) and was further accelerated by the addition of Ca2+ (Figure 9a). No content mixing was observed in analogous experiments lacking C1C2BMUNC2C, and inclusion of GST‐PX instead of C1C2BMUNC2C did not restore fusion (Figure 9a). Thus, GST‐PX was completely unable to stimulate fusion that is initiated with the closed syntaxin‐1‐Munc18‐1 complex.
Figure 9.
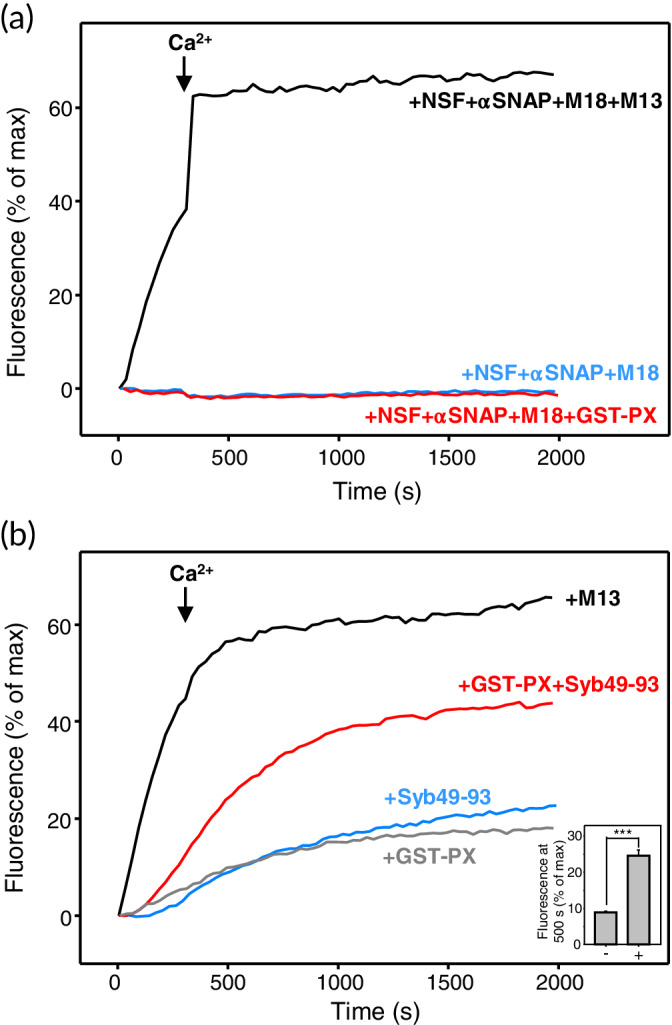
Munc13‐1 C1C2BMUNC2C cannot be replaced with an artificial tether to stimulate liposome fusion. (a) Content mixing between V*‐liposomes and T*‐liposomes (i.e., V‐liposomes and T‐liposomes containing 1% PI3P) was monitored from the increase in the fluorescence signal of Cy5‐streptavidin trapped in the V*‐liposomes caused by FRET with PhycoE‐biotin trapped in the T*‐liposomes upon liposome fusion. The assays were performed in the presence of NSF, αSNAP, and Munc18‐1 (M18) in the absence (blue curve) or presence of Munc13‐1 C1C2BMUNC2C (M13; black curve) or GST‐PX (red curve). (b) Content mixing between V*‐liposomes and T*‐liposomes was monitored as in panel (a) but in the absence of NSF, αSNAP, and Munc18‐1, and the presence of Munc13‐1 C1C2BMUNC2C (black curve), GST‐PX (gray curve), Syb49–93 (blue curve), or GST‐PX and Syb49–93 (red curve). All experiments were started in the presence of 100 μM EGTA and 5‐μM streptavidin, and Ca2+ (600 μM) was added at 300 s. The inset shows quantification of the amounts of content mixing observed after 500 s in experiments performed in the presence of Syb49–93 without (−) or with (+) GST‐PX. All experiments were performed in triplicate. Values indicate means ± SDs. Statistical significance and the p‐value were determined by one‐way analysis of variance (ANOVA) with the Holm‐Sidak test (***p < .001)
We also tested whether GST‐PX could replace Munc13‐1 C1C2BMUNC2C in fusion reactions between V*‐liposomes and T*‐liposomes in the absence of Munc18‐1, NSF, and αSNAP. We observed only a small amount of fusion in the presence of GST‐PX, which was much less efficient than that observed in the presence of C1C2BMUNC2C (Figure 9b). In these experiments, SNARE complex formation is expected to be hindered by the formation of syntaxin‐1‐SNAP‐25 heterodimers with a 2:1 stoichiometry where the second syntaxin‐1 molecule replaces synaptobrevin in the four‐helix bundle (reviewed in Ref. 40). Such inhibition can be overcome by inclusion of a peptide spanning the C‐terminal half of the synaptobrevin SNARE motif [residues 49–93; Syb(49–93)], which displaces the second syntaxin‐1 molecule and thus generates an activated complex that readily binds to synaptobrevin.55 Hence, we examined whether GST‐PX could stimulate fusion when syntaxin‐1‐SNA‐25 complexes were activated by Syb49–93. We observed only a small amount of fusion between T*‐liposomes and V*‐liposomes in the presence of Syb49–93, which was comparable to the fusion observed in the presence of GST‐PX alone, but considerably more efficient fusion occurred when GST‐PX and Syb49–93 were added together (Figure 9b). These results suggest a synergy whereby Syb49–93 activates syntaxin‐1‐SNAP‐25 complexes and GST‐PX tethers the liposome to stimulate fusion. It is noteworthy however that, even with this synergy, fusion was less efficient than that induced by Munc13‐1 C1C2BMUNC2C in the absence of Syb49–93. These results suggest that in these experiments Munc13‐1 not only acts as a membrane–membrane tether but in addition also has an activating or templating function that facilitates SNARE complex assembly.
3. DISCUSSION
Great advances have been made to understand the mechanism of neurotransmitter release, establishing that Munc18‐1 and Munc13‐1 play a crucial role in orchestrating assembly of the neuronal SNARE complex that mediates synaptic vesicle fusion and enabling a wide variety of presynaptic plasticity processes that are critical for brain function. However, key aspects of the molecular events that lead to SNARE complex assembly remain poorly understood. The critical nature of Munc13‐1 for neurotransmitter release was believed to arise from its ability to mediate opening of the conformation of syntaxin‐1 and thus facilitate the transition from the closed syntaxin‐1‐Munc18‐1 complex to the SNARE complex.30, 31, 32, 33 However, different types of interactions between Munc13‐1 and syntaxin‐1 were proposed to underlie this activity, and the finding that the ability of Munc13‐1 to bridge two membranes is crucial for neurotransmitter release7, 29 suggested an alternative mechanism underlying the crucial role of Munc13‐1 in release. The results presented here strongly support the notion that the ability to facilitate syntaxin‐1 opening indeed constitutes a fundamental function of Munc13‐1 and that binding of the Munc13‐1 MUN domain to the linker region of syntaxin‐1 is critical for this ability. Thus, the available data suggest that Munc13‐1 performs at least two critical roles, membrane bridging, and syntaxin‐1 opening. Both activities likely synergize with each other and with the templating function of Munc18‐1 to overcome the energy barriers that hinder SNARE complex assembly starting with the obligatory Munc18‐1‐closed syntaxin‐1 complex.
Our results and those of previous studies emphasize the difficulty of elucidating molecular mechanisms involving dynamic interactions and transitions between states that require large conformational changes such as the conversion from the binary complex of closed syntaxin‐1 with Munc18‐1 to the SNARE complex containing open syntaxin‐1. As we pointed out previously,31 such transitions can be mediated by weak interactions that lower energy barriers, as a 10‐fold increase in the rate of a reaction requires only 1.4 kcal/mol of energy, which corresponds to a very weak binding affinity. The problem that arises is how to detect these weak interactions and how to distinguish them from non‐specific interactions that in fact may be stronger. In the context of the present study, binding of the MUN domain to the syntaxin‐1 SNARE motif is relatively weak (K D = 46 μM31) but still stronger than binding to the syntaxin‐1 linker, which is barely detectable even at 15–20 μM concentrations (Figure 2). Nevertheless, the syntaxin‐1 SNARE motif‐MUN interaction does not appear to be relevant for syntaxin‐1 opening, as this interaction is disrupted by the R210E mutation31 but this mutation does not affect liposome fusion in our reconstitution assays (Figure 5c–f). It is most likely that this interaction arises because of the high promiscuity of the syntaxin‐1 SNARE motif, which binds to many different proteins.40, 56
Conversely, binding of the Munc13‐1 MUN domain to the syntaxin‐1 linker region is very weak but our data and previous results33 suggest that this interaction is crucial for the mechanism of neurotransmitter release. This conclusion is supported by our data showing that the R151A and I55A mutations prevent the stimulation of SNARE complex assembly caused by the MUN domain (Figure 4) and strongly impair liposome fusion in our reconstitution assays (Figure 5c–f), as well as by data obtained previously with similar assays and by the impairment caused by these mutations on the ability of syntaxin‐1A to rescue neurotransmitter release in syntaxin‐1A/B knockdown neurons.33 These mutations involve side chains that are exposed on the surface of the closed conformation of syntaxin‐1 and do not participate in Munc18‐1 binding. Hence, it is very unlikely that these mutations substantially affect the stability of the closed conformation or the interaction with Munc18‐1, which was supported by our ITC data (Figure 6). Moreover, R151 and I155 are far from the SNARE motif and hence do not participate in binding to synaptobrevin and SNAP‐25, and the notion that the syntaxin‐1 linker interacts directly with the MUN domain is supported by our PRE data (Figure 8). Hence, it is most likely that the effects of the R151A and I155A mutations on SNARE complex assembly, liposome fusion, and neurotransmitter arise because they disrupt binding of the MUN domain to the syntaxin‐1 linker. The functional importance of MUN domain‐syntaxin‐1 linker interactions is also supported by the finding that binding of a peptide spanning part of the linker to a fragment spanning the two central subdomains of the MUN domain was impaired by mutation of N1128 and F1131 (referred to NF),33 a mutation that impairs rescue of neurotransmitter release by Unc13 in unc13 C. elegans nulls.32
The low affinity of the MUN‐linker interaction explains the fact that high concentrations of the MUN domain are required to stimulate the transition from the closed syntaxin‐1‐Munc18‐1 complex to the SNARE complex.31 However, such stimulation can be performed much more efficiently by the Munc13‐1 C1C2BMUNC2C fragment in the context of the liposome fusion assays even at much lower concentrations because binding of the C1‐C2B region to the liposomes containing the syntaxin‐1‐Munc18‐1 complex dramatically increases the local concentration of the MUN domain. Indeed, the model of Figure 10 shows that such binding could readily place the region of the MUN domain containing the NF sequence that was implicated in catalyzing syntaxin‐1 opening32 near residues R151 and I155 of the syntaxin‐1 linker. It is also plausible that membrane bridging by Munc13‐1 causes allosteric changes that enhance the affinity of the MUN domain for syntaxin‐1. Note also that, in addition to syntaxin‐1 opening, SNARE complex formation requires the recruitment of synaptobrevin, which is now generally believed to occur via the interaction of the synaptobrevin SNARE motif with Munc18‐134, 35, 36, 37 and leads to the model proposed in Figure 1d. Such recruitment is expected to be facilitated by binding of the Munc13‐1 C2C domain to the vesicle membrane29 and requires unfurling of the Munc18‐1 loop that covers the synaptobrevin binding site.36 Since the Munc13‐1 MUN domain binds weakly to Munc18‐1,31 and also binds to the template complex formed by Munc18‐1, syntaxin‐1, and synaptobrevin,57 an intriguing possibility is that the MUN domain helps to unfurl the Munc18‐1 loop. Once the template complex is formed, SNAP‐25 needs to be incorporated and the interactions of the synaptobrevin and syntaxin‐1 SNARE motifs with Munc18‐1 need to be released to form the SNARE four‐helix bundle.
Figure 10.
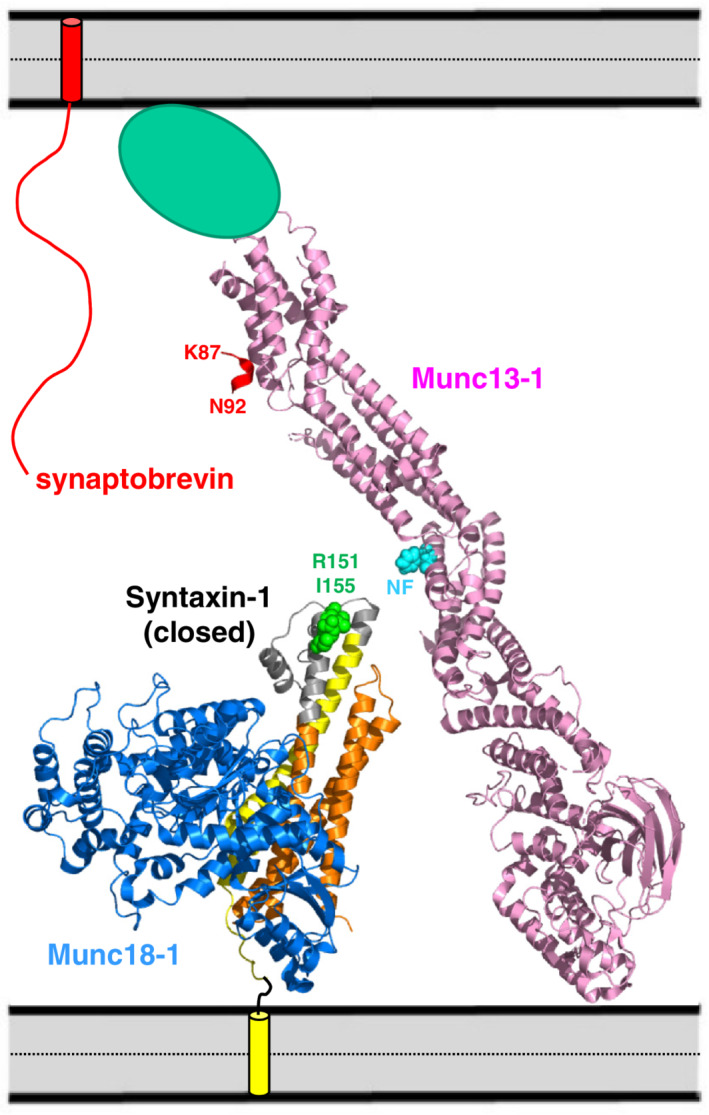
Speculative model of how Munc13‐1 C1C2BMUNC2C, the closed syntaxin‐1‐Munc18‐1 complex and synaptobrevin may be arranged between the synaptic vesicle and plasma membranes before initiation of SNARE complex assembly. The crystal structures of Munc13‐1 C1C2BMUN51 and the closed syntaxin‐1‐Munc18‐1 complex66 (PDB accession codes http://bioinformatics.org/firstglance/fgij//fg.htm?mol=5UE8 and http://bioinformatics.org/firstglance/fgij//fg.htm?mol=3C98, respectively) are represented by ribbon diagrams. Munc18‐1 is in blue, syntaxin‐1 in orange (Habc domain), gray (linker) and yellow (SNARE motif) and Munc13‐1 C1C2BMUN in pink. Residues R151 and I155 of syntaxin‐1 are shown as green spheres and the NF sequence of Munc13‐1 (N1128, F1131) is shown as blue spheres. The C2C domain, which was not part of the structure of C1C2BMUN, is shown as a green ellipsis. The synaptobrevin SNARE motif is shown as a wiggly curve to illustrate its unstructured nature before interacting with other proteins.49 The TM regions of syntaxin‐1 and synaptobrevin are represented by cylinders inserted into the apposed membranes. The diagram illustrates how Munc13‐1 C1C2BMUNC2C can bridge the two membranes through interactions of the C1 and C2B domains with one membrane (bottom) and the C2C domain with the other membrane (top),7, 29 while the region containing the NF sequence in the center of the MUN domain can readily come into proximity to the linker region of syntaxin‐1 containing R151 and I155 in its complex with Munc18‐1. A more slanted orientation of Munc13‐1 with respect to the membranes is expected to bring them closer and facilitate binding of synaptobrevin to Munc18‐1.51 A short red ribbon near the top represents the juxtamembrane region of synaptobrevin as observed in the crystal structure of its complex with the MUN domain58 (PDB accession code http://bioinformatics.org/firstglance/fgij//fg.htm?mol=6A30). The location of residues K87 and N92 are labeled to show that this binding mode orients the C‐terminus away from the membrane and hence it is highly unlikely that this interaction can occur while the TM region of synaptobrevin is inserted into the membrane
These observations indicate than an intricate set of conformational rearrangements, the release of existing interactions, and the formation of new ones is required for the transition from the syntaxin‐1‐Munc18‐1 complex to the SNARE complex, and that there is a delicate balance between the interactions that hinder or favor this transition. Hence, it is not surprising that even very weak interactions that are difficult or impossible to detect by most available biochemical methods can play a critical role in tilting this balance. Note also that multiple SNARE complexes likely mediate membrane fusion and therefore there may be strong cooperativity between the events that lead to fusion. Such cooperativity likely underlies the remarkably dramatic impairment of liposome fusion and neurotransmitter caused by the R151A and I155S mutations33 (Figure 5c–f), or by single point mutations in the Munc13‐1 C2C domain that disrupt the ability of Munc13‐1 to bridge two membranes,29 as well as the strong gains of function caused by point mutations in Munc18‐1 that unfurl the loop involved in synaptobrevin binding and/or facilitate SNARE complex formation.36, 38 The finding that one of these Munc18‐1 mutations can partially rescue the strong phenotypes observed in unc13 nulls in C. elegans 38 emphasizes the close interrelationship between the various molecular events that lead to syntaxin‐1 opening and SNARE complex formation.
The highly efficient fusion observed in our reconstitution assays that included NSF, αSNAP, Munc18‐1 and Munc13‐1 C1C2BMUNC2C, and the complete absence of fusion when C1C2BMUNC2C was replaced with the artificial tether GST‐PX (Figure 9a), show that Munc13‐1 C1C2BMUNC2C does not act simply as a membrane–membrane tether and further illustrate the critical importance of the syntaxin‐1 opening function of Munc13‐1. Munc13‐1 C1C2BMUNC2C was known to induce efficient Ca2+‐independent fusion between T‐liposomes and V‐liposomes in the absence of NSF, C1C2BMUNC2C, and Munc18‐1,7, 42 but it is interesting that fusion between T*‐liposomes and V*‐liposomes is more efficient than that caused by GST‐PX with the help of the Syb49–93 activating peptide (Figure 9b). Since syntaxin‐1 bound to SNAP‐25 is already open, these findings suggest that Munc13‐1 C1C2BMUNC2C also has a function beyond its membrane–membrane tethering role in these experiments and that such function involves interactions with the SNAREs. This notion is supported by single‐molecule fluorescence experiments showing that the Munc13‐1 MUN domain hinders the formation of antiparallel SNARE complexes and favors the proper parallel configuration.8
In this context, a recent report suggested that interactions of the Munc13‐1 MUN domain with the juxtamembrane region of synaptobrevin contribute to its activity in stimulating SNARE complex assembly and described a crystal structure of a peptide corresponding to this region bound to the MUN domain.58 The functional importance of this interaction was supported by mutagenesis,58 and the synaptobrevin juxtamembrane region was reported to contribute to the binding of the MUN domain to the syntaxin‐1‐Munc18‐1‐synaptobrevin template.57 However, as explained above, this region of synaptobrevin is known to bind also to Munc18‐148 and to membranes,49 and its high promiscuity likely arises because of the presence of three aromatic and multiple basic residues in its sequence. Moreover, a model of C1C2BMUNC2C with C2C domain bound to the membrane containing synaptobrevin and with the juxtamembrane peptide bound to the MUN domain, based on the available crystal structures,51, 58 shows that the peptide is in the wrong orientation, with the C‐terminus pointing away from the membrane (Figure 10). Although the overall orientation of C1C2BMUNC2C is likely more slanted to facilitate approximation of the two membranes and binding of synaptobrevin to Munc18‐1, it is difficult to envision how residue N92 of synaptobrevin in this location can be linked to the transmembrane region, which starts at residue 95. It seems more likely that, when synaptobrevin is membrane‐anchored, the basic/hydrophobic juxtamembrane region binds to the membrane rather than to the MUN domain. Nevertheless, it is plausible that there is some arrangement that allows simultaneous interactions of MUN with the synaptobrevin juxtamembrane region and of the C2C domain with the membrane. Structural studies in the presence of membranes will be necessary to distinguish these possibilities.
Although our NMR experiments did not detect any interactions of the Munc13‐1 MUN domain with SNAP‐25 (Figure 3) or with the synaptobrevin SNARE motif, it is plausible that such interactions exist but cannot be detected in our experiments and may be favored at the high local concentrations existing when all these proteins are localized on the membranes. Such interactions could underlie the stimulation of Ca2+‐independent fusion between T‐liposomes and V‐liposomes caused by Munc13‐1 C1C2BMUNC2C. The mechanism of SNARE complex formation under these conditions does not require Munc18‐1 and is expected to be very different from that occurring in the presence of NSF, αSNAP, and Munc18‐1, which starts with the closed syntaxin‐1‐Munc18‐1 complex after NSF and αSNAP disassemble the syntaxin‐1‐SNAP‐25 complexes. However, the activity of Munc13‐1 C1C2BMUNC2C in both pathways may be facilitated similarly by the potential interactions of Munc13‐1 with synaptobrevin and/or SNAP‐25. Note in this context that the HOPS tethering complex that controls yeast vacuolar fusion, performing functions similar to those of Munc18‐1 and Munc13‐1,59 catalyzes SNARE complex formation by interacting with each of the SNARE motifs of the four yeast vacuolar SNAREs.60 It is also possible that the Munc13‐1 C1, C2B, and/or C2C domains also participate in interactions that facilitate SNARE complex assembly. These possibilities will need to be studied on a membrane environment or ideally between two membranes. Clearly, much remains to be learned about the molecular mechanisms underlying assembly of the SNARE complex, even with all the advances that have already been made.
4. EXPERIMENTAL PROCEDURES
4.1. Recombinant proteins
The following proteins were expressed in E. coli BL21 (DE3) cells and purified as previously described6, 15, 27, 29, 61, 62: full‐length rat syntaxin‐1A, rat syntaxin‐1A (2–253), full‐length rat SNAP‐25A (C84S, C85S, C90S, C92S), full‐length rat synaptobrevin 2, rat synaptobrevin 2 49–93, full‐length Chinese hamster NSF (a kind gift from Minglei Zhao), full‐length Bos Taurus αSNAP, full‐length rat Munc18‐1, and a rat Munc13‐1 fragment fragments spanning the MUN domain (residues 859–1531 Δ1408–1452) or the C1, C2B, MUN, and C2C domains (C1C2BMUNC2C; residues 529–1735 Δ1408–1452). Constructs to express rat syntaxin‐1A fragments spanning residues 138–180, 138–200, 138–232, and 191–236 were prepared starting from DNA encoding the rat syntaxin‐1A(2–253) fragment using standard recombinant DNA techniques and custom‐designed primers. The proteins were expressed and purified as described previously for syntaxin‐1A (2–253).53 The following mutants were obtained using QuickChange site‐directed mutagenesis (Stratagene) on the R210E, R151A, and I155A mutations in full‐length syntaxin‐1A, and in syntaxin‐1A 2–253, the T159C and S186C mutant syntaxin‐1(2–253) (also containing a C145S mutation), the R136C mutation in SNAP‐25A (also bearing the C84S, C85S, C90S, C92S mutations). The synaptobrevin(1–96) L26C mutant was prepared by inserting a STOP codon in a previously described construct encoding full‐length rat synaptobrevin 2 bearing the L26C mutation.15 All mutant proteins were purified as the wild type proteins. Purified fusion protein of GST with the PX domain of Vam754 was a kind gift from William Wickner. To obtain uniformly 15N‐labeled proteins, we used M9 minimal expression media with 15NH4Cl as the sole nitrogen source (1 g/L). 2H,13CH3‐ILMV‐labeled Munc13‐1 MUN domain was produced using M9 expression media in D2O (Sigma) with 2H‐glucose (Sigma) as the sole carbon source (3 g/L), and adding [3,3‐2H2] 13C‐methyl alpha‐ketobutyric acid (80 mg/L), [3‐2H] 13C‐dimethyl alpha‐ketoisovaleric acid (80 mg/L), and 13C‐methyl methionine (250 mg/L) (Cambridge Isotope Laboratories) to the cell cultures 30 min prior to induction with 0.4 μM Isopropyl β‐d‐1‐thiogalactopyranoside overnight at 16°C.
4.1.1. Protein labeling
Single cysteine mutant proteins were labeled with BODIPY‐FL [for synaptobrevin(1–96) L26C] or TMR (for SNAP‐25 R136C, C84S, C85S, C90S, C92S) using maleimide reactions (Thermo Fisher) as described.15 To attach the Gd3+ tag on syntaxin‐1(2–253) C145S,T159C or syntaxin‐1(2–253) C145S,S186C, 1,4,7,10‐tetraazacyclododecane‐1,4,7‐tris‐acetic acid‐10‐maleimidoethylacetamide (DOTA) was dissolved in 50 mM MOPS, 150 mM NaCl and the pH was adjusted to 7.0 and an equimolar amount of GdCl3 was added. A 10‐fold excess of the resulting Gd3+‐DOTA complex was added to syntaxin‐1(2–253) C145S,T159C in the presence of TCEP, the pH was adjusted to 7.2, and the reaction was allowed to proceed for 3 hr at room temperature. The unreacted tag was removed by gel filtration chromatography on a Superdex 75 in buffer containing 20 mM HEPES, 125 mM KCl, 1 mM TCEP, pH 7.4.
4.2. NMR spectroscopy
NMR spectra were acquired on Agilent DD2 spectrometers operating at 800 or 600 MHz. 1H‐15N HSQC spectra63 were performed at 20°C with samples dissolved in 20 mM HEPES pH 7.4, 125 mM KCl, 1 mM TCEP, containing 7% D2O. 1H‐13C HMQC spectra52 were acquired at 25°C in the same buffer. Protein concentrations are described in the figure legends. All data were processed with NMRpipe64 and analyzed with NMRView.65
4.2.1. FRET assays to monitor SNARE complex assembly
FRET experiments were performed at 28°C on a PTI Quantamaster 400 spectrofluorometer (T‐format) equipped with a rapid Peltier temperature controlled four‐position sample holder. SNARE complex formation was monitored from the decrease in the fluorescence emission intensity of BODIPY‐FL attached to residue 26 of synaptobrevin(1–96) due to FRET with TMR attached to residue 136 of SNAP‐25 as a function of time. The excitation wavelength was 485 nm and the fluorescence emission intensity was measured at 513 nm. Assays were performed in a buffer containing 25 mM HEPES pH 7.4, 150 mM KCl, 10% glycerol, 1 mM TCEP. For experiments performed in the absence of Munc18‐1 or Munc13‐1 MUN domain, SNARE complex formation was initiated by mixing 0.5 μM BODIPY‐FL‐synaptobrevin(1–96) with 8 μM TMR‐SNAP‐25 and 8 μM WT, R210E, R151A, or I151A syntaxin‐1(2–253). Additional experiments were performed with samples of 8 μM WT, R210E, R151A or I151A syntaxin‐1(2–253) that were pre‐incubated at room temperature for 1 hr with 10 μM Munc18‐1 and then mixed with 0.5 μM BODIPY‐FL‐synaptobrevin(1–96) and 8 μM TMR‐SNAP‐25 in the absence or presence of 30‐μM Munc13‐1 MUN domain. Data points were collected every 17 s (1 s acquisition). Only a small amount of photobleaching of the donor was observed under these conditions in control experiments with donors alone.
4.2.2. Simultaneous lipid mixing and content mixing assays
Assays to simultaneously monitor lipid and content mixing between V‐liposomes and T‐liposomes containing WT or mutant syntaxin‐1A (Figure 5) were performed as described previously in detail61 except for a few modifications. Briefly, V‐liposomes with full‐length synaptobrevin contained 39% POPC, 19% DOPS, 19% POPE, 20% cholesterol, 1.5% NBD‐PE, and 1.5% Marina Blue DHPE. T‐liposomes with full‐length WT, R210E, R151A or I155S syntaxin‐1A and full‐length SNAP25 contained 38% POPC, 18% DOPS, 20% POPE, 20% cholesterol, 2% PIP2, and 2% DAG. Dried lipid mixtures were resuspended in 25 mM HEPES pH 7.4, 150 KCl, 1 mM TCEP, 10% glycerol, 2% β‐OG. Purified SNARE proteins and fluorescently labeled content mixing molecules were added to the lipid mixtures to make the syntaxin‐1:SNAP25:lipid ratio 1:5:800 and Phycoerythrin‐Biotin (4 μM) for T‐liposomes, and the synaptobrevin:lipid ratio 1:500 and Cy5‐Streptavidin (8 μM) for V‐liposomes. The mixtures were incubated at room temperature and dialyzed against the reaction buffer (25 mM HEPES pH 7.4, 150 mM KCl, 1 mM TCEP, 10% glycerol) with 2 g/L Amberlite XAD‐2 beads (Sigma) three times at 4°C. Proteoliposomes were purified by floatation on a three‐layer histodenz gradient (35%, 25%, and 0%) and harvested from the topmost interface. To simultaneously measure lipid mixing from de‐quenching of Marina Blue lipids and content mixing from the development of FRET between Phycoerythrin‐Biotin trapped in T‐liposomes and Cy5‐streptavidin trapped in V‐liposomes, T‐liposomes (0.25 mM lipid) were mixed with V‐liposomes (0.125 mM lipid) in a total volume of 200 μl. Acceptor T‐liposomes were first incubated with 0.4 μM NSF, 2 μM αSNAP, 2.5 mM MgCl2, 2 mM ATP, 0.1 mM EGTA, and 1 μM Munc18‐1 at 37°C for 25 min. They were then mixed with donor V‐liposomes, 0.5 μM Munc13‐1 C1C2BMUNC2C, and 1 μM excess SNAP25. Control experiments without Munc18‐1 and/or C1C2BMUNC2C were also performed. All experiments were performed at 30°C and 0.6 mM Ca2+ was added at 300 s. The fluorescence signal from Marina Blue (excitation at 370 nm, emission at 465 nm) and Cy5 (excitation at 565 nm, emission at 670 nm) were recorded to monitor lipid and content mixing, respectively. At the end of the reaction, 1% β‐OG was added to solubilize the liposomes and the lipid mixing data were normalized to the maximum fluorescence signal. Most experiments were performed in the presence of 5‐μM streptavidin, and control experiments without streptavidin were performed to measure the maximum Cy5 fluorescence after detergent addition for normalization of the content mixing data. Experiments were performed in triplicates and the results were verified with additional experiments performed with different liposome preparations. Analogous procedures were followed for the experiments of Figure 9, except that only T‐liposomes containing WT syntaxin‐1A were used, and both V‐ and T‐liposomes contained 1% PI3P and 1% less DOPS. In this set of experiments, we performed assays where the T‐liposomes were first incubated with 0.4 μM NSF, 2 μM αSNAP, 2.5 mM MgCl2, 2 mM ATP, 0.1 mM EGTA, and 1 μM Munc18‐1 at 37°C for 25 min, and then mixed with V‐liposomes in the presence of 1 μM excess SNAP25 with or without 0.2 μM Munc13‐1 C1C2BMUNC2C or 0.5 μM GST‐PX. Additional experiments were performed by directly mixing V‐ and T‐liposomes in the presence of 0.2 μM Munc13‐1 C1C2BMUNC2C, 0.5 μM GST‐PX, 10 μM Syb49–93 or 0.5 μM GST‐PX and 10 μM Syb49–93.
4.3. Isothermal titration calorimetry
ITC experiments were performed using a Microcal ITC200 system (Malvern) at 25°C. WT, R210E, R151A or I155A syntaxin‐1(2–253) (60 μM) was titrated into the cell containing Munc18‐1 (5 μM) in phosphate‐buffered saline. All proteins were dialyzed in this buffer before the experiments. The data were processed and analyzed as described.36
ACKNOWLEDGMENTS
We thank Shih‐Chia Tso for assistance in acquiring the ITC data and William Wickner for kindly providing purified GST‐PX protein. The Agilent DD2 console of the 800 MHz spectrometer used for the research presented here was purchased with a shared instrumentation grant from the NIH (S10OD018027 to JR). Bradley Quade was supported by NIH Training Grant T32 GM008297. This work was supported by grant I‐1304 from the Welch Foundation (to JR) and by NIH Research Project Award R35 NS097333 (to JR).
Magdziarek M, Bolembach AA, Stepien KP, Quade B, Liu X, Rizo J. Re‐examining how Munc13‐1 facilitates opening of syntaxin‐1. Protein Science. 2020;29:1440–1458. 10.1002/pro.3844
Magdalena Magdziarek and Agnieszka A. Bolembach are co‐first authors.
Magdalena Magdziarek is on leave from the Department of Crystallography, Faculty of Chemistry, Adam Mickiewicz University, Poznan, Poland.
Agnieszka A. Bolembach is on leave from the Department of Chemical Biology, Faculty of Biotechnology, University of Wrocław, Wrocław, Poland.
Funding information NIH, Grant/Award Numbers: R35 NS097333, S10OD018027, T32 GM008297; Welch Foundation, Grant/Award Number: I‐1304
REFERENCES
- 1. Sudhof TC. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rizo J. Mechanism of neurotransmitter release coming into focus. Protein Sci. 2018;27:1364–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brunger AT, Choi UB, Lai Y, Leitz J, Zhou Q. Molecular mechanisms of fast neurotransmitter release. Annu Rev Biophys. 2018;47:469–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sudhof TC, Rothman JE. Membrane fusion: Grappling with SNARE and SM proteins. Science. 2009;323:474–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma C, Su L, Seven AB, Xu Y, Rizo J. Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science. 2013;339:421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu X, Seven AB, Camacho M, et al. Functional synergy between the Munc13 C‐terminal C1 and C2 domains. Elife. 2016;5:e13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lai Y, Choi UB, Leitz J, et al. Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron. 2017;95:591–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim J, Zhu Y, Shin YK. Preincubation of t‐SNAREs with complexin I increases content‐mixing efficiency. Biochemistry. 2016;55:3667–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly‐disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. [DOI] [PubMed] [Google Scholar]
- 11. Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick‐freeze/deep‐etch electron microscopy. Cell. 1997;90:523–535. [DOI] [PubMed] [Google Scholar]
- 12. Poirier MA, Xiao W, Macosko JC, Chan C, Shin YK, Bennett MK. The synaptic SNARE complex is a parallel four‐stranded helical bundle. Nat Struct Biol. 1998;5:765–769. [DOI] [PubMed] [Google Scholar]
- 13. Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 a resolution. Nature. 1998;395:347–353. [DOI] [PubMed] [Google Scholar]
- 14. Mayer A, Wickner W, Haas A. Sec18p (NSF)‐driven release of Sec17p (alpha‐SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. [DOI] [PubMed] [Google Scholar]
- 15. Prinslow EA, Stepien KP, Pan YZ, Xu J, Rizo J. Multiple factors maintain assembled trans‐SNARE complexes in the presence of NSF and alphaSNAP. Elife. 2019;8:e38880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fernandez‐Chacon R, Konigstorfer A, Gerber SH, et al. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. [DOI] [PubMed] [Google Scholar]
- 17. Brewer KD, Bacaj T, Cavalli A, et al. Dynamic binding mode of a Synaptotagmin‐1‐SNARE complex in solution. Nat Struct Mol Biol. 2015;22:555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou Q, Lai Y, Bacaj T, et al. Architecture of the synaptotagmin‐SNARE machinery for neuronal exocytosis. Nature. 2015;525:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou Q, Zhou P, Wang AL, et al. The primed SNARE‐complexin‐synaptotagmin complex for neuronal exocytosis. Nature. 2017;548:420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tang J, Maximov A, Shin OH, Dai H, Rizo J, Sudhof TC. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. [DOI] [PubMed] [Google Scholar]
- 21. Giraudo CG, Eng WS, Melia TJ, Rothman JE. A clamping mechanism involved in SNARE‐dependent exocytosis. Science. 2006;313:676–680. [DOI] [PubMed] [Google Scholar]
- 22. Schaub JR, Lu X, Doneske B, Shin YK, McNew JA. Hemifusion arrest by complexin is relieved by Ca2+−synaptotagmin I. Nat Struct Mol Biol. 2006;13:748–750. [DOI] [PubMed] [Google Scholar]
- 23. Verhage M, Maia AS, Plomp JJ, et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. [DOI] [PubMed] [Google Scholar]
- 24. Richmond JE, Davis WS, Jorgensen EM. UNC‐13 is required for synaptic vesicle fusion in C. elegans . Nat Neurosci. 1999;2:959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Varoqueaux F, Sigler A, Rhee JS, et al. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13‐mediated vesicle priming. Proc Natl Acad Sci U S A. 2002;99:9037–9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fernandez I, Ubach J, Dulubova I, Zhang X, Sudhof TC, Rizo J. Three‐dimensional structure of an evolutionarily conserved N‐terminal domain of syntaxin 1A. Cell. 1998;94:841–849. [DOI] [PubMed] [Google Scholar]
- 27. Dulubova I, Sugita S, Hill S, et al. Conformational switch in syntaxin during exocytosis: Role of munc18. EMBO J. 1999;18:4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Misura KM, Scheller RH, Weis WI. Three‐dimensional structure of the neuronal‐Sec1‐syntaxin 1a complex. Nature. 2000;404:355–362. [DOI] [PubMed] [Google Scholar]
- 29. Quade B, Camacho M, Zhao X, et al. Membrane bridging by Munc13‐1 is crucial for neurotransmitter release. Elife. 2019;8:e42806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Richmond JE, Weimer RM, Jorgensen EM. An open form of syntaxin bypasses the requirement for UNC‐13 in vesicle priming. Nature. 2001;412:338–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma C, Li W, Xu Y, Rizo J. Munc13 mediates the transition from the closed syntaxin‐Munc18 complex to the SNARE complex. Nat Struct Mol Biol. 2011;18:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang X, Wang S, Sheng Y, et al. Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic‐vesicle priming. Nat Struct Mol Biol. 2015;22:547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang S, Choi UB, Gong J, et al. Conformational change of syntaxin linker region induced by Munc13s initiates SNARE complex formation in synaptic exocytosis. EMBO J. 2017;36:816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parisotto D, Pfau M, Scheutzow A, et al. An extended helical conformation in domain 3a of Munc18‐1 provides a template for SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor) complex assembly. J Biol Chem. 2014;289:9639–9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM. A direct role for the Sec1/Munc18‐family protein Vps33 as a template for SNARE assembly. Science. 2015;349:1111–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sitarska E, Xu J, Park S, et al. Autoinhibition of Munc18‐1 modulates synaptobrevin binding and helps to enable Munc13‐dependent regulation of membrane fusion. Elife. 2017;6:e24278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiao J, He M, Port SA, et al. Munc18‐1 catalyzes neuronal SNARE assembly by templating SNARE association. Elife. 2018;7:e41771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park S, Bin NR, Yu B, et al. UNC‐18 and tomosyn antagonistically control synaptic vesicle priming downstream of UNC‐13 in Caenorhabditis elegans . J Neurosci. 2017;37:8797–8815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gerber SH, Rah JC, Min SW, et al. Conformational switch of syntaxin‐1 controls synaptic vesicle fusion. Science. 2008;321:1507–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rizo J, Sudhof TC. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices‐guilty as charged? Annu Rev Cell Dev Biol. 2012;28:279–308. [DOI] [PubMed] [Google Scholar]
- 41. Regehr WG. Short‐term presynaptic plasticity. Cold Spring Harb Perspect Biol. 2012;4:a005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stepien KP, Prinslow EA, Rizo J. Munc18‐1 is crucial to overcome the inhibition of synaptic vesicle fusion by alphaSNAP. Nat Commun. 2019;10:4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Betz A, Okamoto M, Benseler F, Brose N. Direct interaction of the rat unc‐13 homologue Munc13‐1 with the N terminus of syntaxin. J Biol Chem. 1997;272:2520–2526. [DOI] [PubMed] [Google Scholar]
- 44. Basu J, Shen N, Dulubova I, et al. A minimal domain responsible for Munc13 activity. Nat Struct Mol Biol. 2005;12:1017–1018. [DOI] [PubMed] [Google Scholar]
- 45. McEwen JM, Madison JM, Dybbs M, Kaplan JM. Antagonistic regulation of synaptic vesicle priming by Tomosyn and UNC‐13. Neuron. 2006;51:303–315. [DOI] [PubMed] [Google Scholar]
- 46. Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML. A post‐docking role for active zone protein rim. Nat Neurosci. 2001;4:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rizo J, Rosen MK, Gardner KH. Enlightening molecular mechanisms through study of protein interactions. J Mol Cell Biol. 2012;4:270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu Y, Su L, Rizo J. Binding of Munc18‐1 to synaptobrevin and to the SNARE four‐helix bundle. Biochemistry. 2010;49:1568–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brewer KD, Li W, Horne BE, Rizo J. Reluctance to membrane binding enables accessibility of the synaptobrevin SNARE motif for SNARE complex formation. Proc Natl Acad Sci U S A. 2011;108:12723–12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zucchi PC, Zick M. Membrane fusion catalyzed by a Rab, SNAREs, and SNARE chaperones is accompanied by enhanced permeability to small molecules and by lysis. Mol Biol Cell. 2011;22:4635–4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu J, Camacho M, Xu Y, et al. Mechanistic insights into neurotransmitter release and presynaptic plasticity from the crystal structure of Munc13‐1 C1C2BMUN. Elife. 2017;6:e22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ruschak AM, Kay LE. Methyl groups as probes of supra‐molecular structure, dynamics and function. J Biomol NMR. 2010;46:75–87. [DOI] [PubMed] [Google Scholar]
- 53. Chen X, Lu J, Dulubova I, Rizo J. NMR analysis of the closed conformation of syntaxin‐1. J Biomol NMR. 2008;41:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Song H, Wickner W. Tethering guides fusion‐competent trans‐SNARE assembly. Proc Natl Acad Sci U S A. 2019;116:13952–13957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pobbati AV, Stein A, Fasshauer D. N‐ to C‐terminal SNARE complex assembly promotes rapid membrane fusion. Science. 2006;313:673–676. [DOI] [PubMed] [Google Scholar]
- 56. Jahn R, Scheller RH. SNAREs—engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. [DOI] [PubMed] [Google Scholar]
- 57. Shu T, Jin H, Rothman JE, Zhang Y. Munc13‐1 MUN domain and Munc18‐1 cooperatively chaperone SNARE assembly through a tetrameric complex. Proc Natl Acad Sci U S A. 2020;117:1036–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang S, Li Y, Gong J, et al. Munc18 and Munc13 serve as a functional template to orchestrate neuronal SNARE complex assembly. Nat Commun. 2019;10:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wickner W, Rizo J. A cascade of multiple proteins and lipids catalyzes membrane fusion. Mol Biol Cell. 2017;28:707–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Song H, Orr AS, Lee M, Harner ME, Wickner WT. HOPS recognizes each SNARE, assembling ternary trans‐complexes for rapid fusion upon engagement with the 4th SNARE. Elife. 2020;9:e53559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu X, Seven AB, Xu J, et al. Simultaneous lipid and content mixing assays for in vitro reconstitution studies of synaptic vesicle fusion. Nat Protoc. 2017;12:2014–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen X, Arac D, Wang TM, Gilpin CJ, Zimmerberg J, Rizo J. SNARE‐mediated lipid mixing depends on the physical state of the vesicles. Biophys J. 2006;90:2062–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang O, Kay LE, Olivier JP, Forman‐Kay JD. Backbone 1H and 15N resonance assignments of the N‐terminal SH3 domain of drk in folded and unfolded states using enhanced‐sensitivity pulsed field gradient NMR techniques. J Biomol NMR. 1994;4:845–858. [DOI] [PubMed] [Google Scholar]
- 64. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRpipe ‐ A multidimensional spectral processing system based on Unix pipes. J Biomol NMR. 1995;6:277–293. [DOI] [PubMed] [Google Scholar]
- 65. Johnson BA, Blevins RA. NMR View ‐ A computer‐program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. [DOI] [PubMed] [Google Scholar]
- 66. Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D. Munc18a controls SNARE assembly through its interaction with the syntaxin N‐peptide. EMBO J. 2008;27:923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]