

Abstract
During DNA replication, the genetic information of a cell is copied. Subsequently, identical genetic information is segregated reliably to the two daughter cells through cell division. Meanwhile, DNA replication is intrinsically linked to the process of chromatin duplication, which is required for regulating gene expression and establishing cell identities. Understanding how chromatin is established, maintained or changed during DNA replication represents a fundamental question in biology. Recently, we developed a method to directly visualize chromatin components at individual replication forks undergoing DNA replication. This method builds upon the existing chromatin fiber technique, and combines it with cell-type-specific chromatin labeling and superresolution microscopy. In this method, a short pulse of nucleoside analog labels replicative regions in the cells of interest. Chromatin fibers are subsequently isolated and attached to a glass slide, after which a laminar flow of lysis buffer extends the lysed chromatin fibers parallel with the direction of the flow. Fibers are then immunostained for different chromatin-associated proteins and mounted for visualization using superresolution microscopy. Replication foci or “bubbles” are identified by the presence of the incorporated nucleoside analog. For researchers experienced in molecular biology and superresolution microscopy, this protocol typically takes two to three days from sample preparation to data acquisition, with an additional day for data processing and quantification.
Keywords: DNA replication, chromatin fiber, sister chromatids, histones, histone modifications, superresolution microscopy, leading strand, lagging strand
Introduction
In all organisms, DNA is bound and organized by DNA-binding proteins to create chromatin structure. By regulating the accessibility of DNA, chromatin structure regulates gene expression, thereby defining cellular identities and dictating their functions1. The process of DNA replication represents a significant challenge to maintain the integrity of chromatin structure2. In order to maintain proper chromatin structure during the process of DNA duplication3–5, DNA-associated proteins must also increase their levels and re-distribute6–9.
For eukaryotes, the predominant feature responsible for defining the chromatin landscape is the nucleosome structure, composed of 147-base pairs of DNA wound around the histone octamer, consisting of two copies each of the histone proteins H2A, H2B, H3 and H410,11. During DNA replication, old octamers ahead of the fork must be disassembled to allow for replication fork passage. Following disassembly, retained old histones could be re-deposited to newly synthesized DNA in a process known as histone recycling12. As old histones are being recycled, new histones should be recruited and incorporated onto newly synthesized DNA to restore nucleosome density13. Together, the two processes of old histone recycling and new histone incorporation are collectively referred to as the process of RCNA (Replication-Coupled Nucleosome Assembly)14.
Different models have been proposed to explain how intrinsic differences in leading versus lagging strand synthesis may impact the process of RCNA. As the continuously synthesized leading strand has a slight temporal advantage over the lagging strand, it has been postulated that old histones could be preferentially recycled by the leading strand. Electron microscopy studies have demonstrated that the leading strand does tend to incorporate histones slightly before the lagging strand, as evidenced by the fact that nucleosomes are re-established ~225bp behind the advancing fork at the leading strand, compared to ~285bp on the lagging strand15. Despite these differences shown by electron microscopy, other studies have previously demonstrated a variety of histone incorporation patterns at the replication fork, ranging from asymmetric to symmetric. Some studies have shown that old histones are equally distributed between sister chromatids16, whereas others have shown that old histone recycling has a strand preference during the process of RCNA. However, preferential recycling of old histones has been observed on the leading strand17–19 as well as on the lagging strand20, raising questions as to whether a consensus pattern for RCNA exists.
In recent years, several sequencing-based techniques have been developed to investigate histone inheritance patterns during DNA replication. Yu et al. utilized a method called eSPAN (enrichment and Sequencing of Protein-Associated Nascent DNA) to map old versus new histone distribution at newly replicated DNA in yeast Saccharomyces cerevisiae21. This study showed a slight preference of old histones towards the lagging strand. In another study, Petryk et al. used a similar method termed SCAR-seq (Sister Chromatids After Replication by DNA sequencing) to investigate histone inheritance patterns in cultured mESCs (mouse Embryonic Stem Cells) and found a slight bias of old histones towards the leading strand22. Our recent studies profiling RCNA in early-stage male germ cells in Drosophila melanogaster revealed that old histones tend to be incorporated towards the leading strand, though lagging strand bias can be detected in a small population of replication forks23. Taken together, these studies demonstrate a surprising plasticity in RCNA, and suggest that factors such as developmental stage and genomic context may regulate histone incorporation patterns at the replication fork. To facilitate this study using small populations of cells from developing tissues or specific cells from living organisms, we have developed a method termed SRCF (SuperResolution of Chromatin Fibers) to directly visualize histone distribution patterns on replicative DNA.
Advantages of the SRCF technique:
Sequencing-based techniques have proven to be powerful for studying the underlying molecular mechanisms of RCNA. However, these methods typically require large number of cells, making it challenging to employ these techniques to study cells from tissues in living organisms. In contrast, SRCF is compatible with small cell numbers, which greatly facilitates the study of histone inheritance patterns in cells from tissues in vivo. In our experiments studying RCNA in the Drosophila testes, we were able to generate high quality chromatin fiber data with as few as ~1,000 cells. SRCF is also compatible with higher cell numbers, as experiments with cultured Drosophila Kc cells were performed with ~15,000–80,000 cells. Additionally, using cell-type- and/or differentiation stage-specific drivers to express tagged histones, we can selectively label and profile histone incorporation patterns in specific cells from a heterogeneous tissue, further improving the spatial and temporal precision.
Furthermore, SRCF can be used to simultaneously study the distribution of multiple proteins at the same replication fork. For example, we have utilized this technique to label lagging strand-enriched proteins, such as PCNA (Proliferating Cell Nuclear Antigen) and RPA (Replication Protein-A), as well as particular post-translational modifications associated with either old or new histones. By selectively labeling the lagging strand and old histone-enriched modifications, we have been able to study strand bias during histone inheritance without having the genomic sequence information of replication origins. This is very useful for studies in higher order multicellular organisms, as most origins remain poorly characterized in these systems. Additionally, SRCF is a single-molecule technique that allows for the detection of RCNA events at individual forks, which may be lost using techniques with bulk populations of cells and data analyses to achieve an average assessment. Thus, the SRCF technique could achieve an unprecedented opportunity to investigate the processes of RCNA with single-molecule resolution, cell type specificity, and temporal precision during development of multicellular organisms.
Limitations of the SRCF technique:
While SRCF offers many advantages, this technique also has several disadvantages at its current stage. First, further development is needed to retrieve the sequence information of chromatin fibers. For now, we do not know the genomic regions where different degrees of asymmetry in histone inheritance pattern occur, for example, histone asymmetry could occur mostly at the developmentally relevant genes, such as “stemness” genes and differentiation genes in specific lineages. Further improvement to overcome some of these limitations will be the next step, such as combining SRCF with DNA FISH (fluorescence in situ hybridization) to selectively label regions of interest. Another limitation of the SRCF technique is that it is low-throughput, making it challenging to generate large datasets in a timely fashion, due to several reasons. First, not all sample preparation will produce extended chromatin fibers that adhered to the glass surface well. In our hands, we found that Fisherbrand™ Superfrost™ Plus Microscope Slides can increase fiber retention compared to standard glass slides. While other slides with great adhesive properties, such as poly-Lysine coated glass slides, may further increase fiber retention, they might hinder effective fiber extension and spreading, which is also needed to produce high-quality fibers for imaging and data analysis. Notably, great care must also be taken during the washing and immunostaining steps to avoid dislodging adhered fibers. Furthermore, as fibers are not always deposited and located in a predictable manner at the glass slide, it could be challenging to find fibers during imaging. Ample time (detailed in the protocol) must be allowed to scan slides in order to ensure that scarcely distributed chromatin fibers can be found and imaged. Third, even though chromatin fibers can be found and successfully superresolved, they may not be suitable for data analysis, either. This could be due to loss of the proteins of interest during the experiment, or the separation between sister chromatids is insufficient to spatially resolve sister chromatids, even with the superresolution microscopy. Together, these factors should be considered and evaluated when using the SRCF technique.
Experimental Design:
Overview:
The SRCF technique builds upon the previously established chromatin fiber technique24–28 to isolate and visualize replicating chromatin. This technique was utilized to study histone inheritance patterns in early-stage germ cells from the Drosophila testes, as described in Wooten et. al.23. In this protocol, we utilize manual dissection to isolate adult fly testes, followed by removing the basal parts of the testes containing later-stage germ cells (Step 1 in Figure 1). We then digest the testis tips to generate a suspension of cells, which are subsequently deposited onto a glass slide. Cells are then lysed to release chromatin from the confines of the nuclei. Following lysis, a laminar flow of the lysis buffer containing chromatin is introduced to extend and linearize chromatin fibers over the surface of a glass slide. Chromatin fibers are then fixed and immunostained against proteins of interest. Finally, chromatin fibers are imaged with superresolution microscopes. Images are then analyzed and quantified to determine the relative abundance of key chromatin-associated components between sister chromatids. To verify the calling of replicating regions on chromatin fibers, control experiments are performed on post-replicative tissues that do not contain any cell actively undergoing S-phase. This control is important to ensure EdU labeling of chromatin fibers is specific to replicative cells. We also recommend that at least three independent biological replicates are performed on each experimental condition to produce statically meaningful results. We will now describe the key steps in each stage of SRCF in details.
Figure 1:
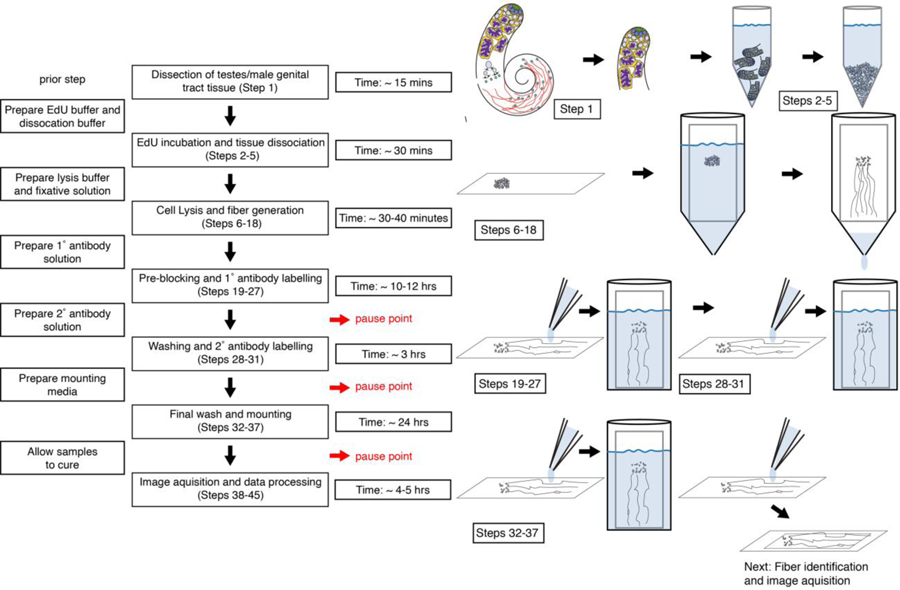
A flow chart of chromatin fiber protocol with estimated time for each key steps and potential pause points.
Fiber preparation and staining:
The first step of this protocol involves physically isolating the tissue of interest. For our experiments, we dissect whole testes from the abdomen of adult males of Drosophila melanogaster (Figure 1). Following removing the basal part, testis tips enriched with the cells of interest (i.e. germline stem cells) are incubated with the thymidine analogue 5-Ethynyl-2′-deoxyuridine (EdU) to label replicating genomic regions and allow for identification of replicative sister chromatids.
Next, we lyse the cells: To facilitate cell lysis, tissues are treated with a mixture of collagenase/dispase to digest them into a suspension of cells, which are then spun down onto a glass slide. This centrifugation is done by spinning cells down onto a slide at 900 RPM for 4 min using a Thermo Scientific Shandon Cytospin3, which produces a 5mm diameter circle with cells at the center of a slide. It is important to note here that for SRCF experiments involving cultured cells, steps involving isolation and dissociation of the tissues are not needed. Cultured cells can be directly incubated in the culture medium containing EdU for thirty minutes, after which cells are spun down onto a glass slide, as described previously. Once deposited cells onto the slide, the slide is placed in a 50ml conical tube filled with a hypotonic lysis buffer adapted from McKnight and Miller et al.28. This buffer is to lyse the cells as gentle as possible, in order to maximize protein retention on chromatin fibers. Testis samples are incubated in the lysis buffer for 10 minutes until most cells are lysed. The lysing time is an important variable, which should be adjusted empirically. If chromatin fibers fail to show strong signals of associated proteins, it is likely that the lysis time is too long, resulting in stripping off most proteins. Conversely, if the majority of cells are not fully lysed or chromatin fibers are not fully extended on the slide, lysing time may need to be increased.
Following cell lysis, lysis buffer is slowly drained from a 50ml conical tube with a hole at the bottom at a rate of approximately 10ml/min. In order to generate long and continuous fibers, it is important that lysis buffer is drained in a slow and steady manner. A steady flow rate of lysis buffer will generate a more even hydrodynamic drag force, which generates longer and straighter fibers desirable for imaging and analysis. To ensure steady flow rate, we modify a 50ml conical tube with a small hole at the bottom (hole size can be adjusted to alter flow rate). This hole is drilled with a 25-guage hypodermic needle to produce a hole with ~.5mm diameter (Picture S1). During the 10-minute lysis incubation, the cap of the conical is screwed tightly closed to prevent premature buffer flow (Pictures S2, S3). At the end of the incubation period, the cap is screwed slightly open to allow the lysis buffer to slowly flow out of the conical tube. This flow rate can be adjusted by the hole size and the tightness of the cap of the conical tube. Chromatin fibers attached to the slide are then fixed and immunostained to label proteins of interest. Click-chemistry reaction is performed to conjugate fluorophores to EdU at the replicating regions.
Identification of replicative regions at chromatin fibers:
To identify replicative fibers, slides are visually scanned through the microscope eyepiece at the DAPI channel to identify the place where chromatin fibers are deposited (Figure 2a). This visual scanning should be done with as low lamp power as possible to minimize photobleaching (note: refer to superresolution microscope User Manual to determine how to adjust scanning lamp power). Once chromatin fibers are identified by DAPI, fibers are scanned for the presence of EdU to pinpoint replicative fibers (Figure 2b). Replicative regions or “bubbles” are distinguished as EdU-positive regions showing a transition from a single fiber structure to a double fiber structure (Figure 2b, 2c). Moreover, EdU-positive regions along the chromatin fiber are frequently associated with an increase in DNA dye intensity, termed DAPI-bright regions showing greater than a 2-fold increase in DAPI-intensity relative to the surrounding DAPI-dim regions, from the same chromatin fiber (Figure 3a). This greater than 2-fold increase in DAPI intensity likely reflects the two-fold increase in DNA content associated with replication and the superhelical torsion associated with the advancing replication fork. Previous studies have demonstrated that negative supercoiling is present behind the replication fork, whereas positive supercoiling is ahead of the advancing replication fork29–31. The slightly unwound and open nature of the negatively supercoiled DNA favors the intercalation of many small molecules, including DNA dyes such as DAPI and Yoyo332,33. Following identification of replicative regions at the chromatin fibers, the signals and distribution of key proteins or post-translational histone modifications, such as PCNA, RPA, H3K27me3 and H3K14Ac, will be further imaged. Superresolution microscopy will increase the probability to distinguish replicative sister chromatids (Figure 2a–d, Figure 3).
Figure 2: A flow chart of chromatin fiber image acquisition and data analysis procedure.
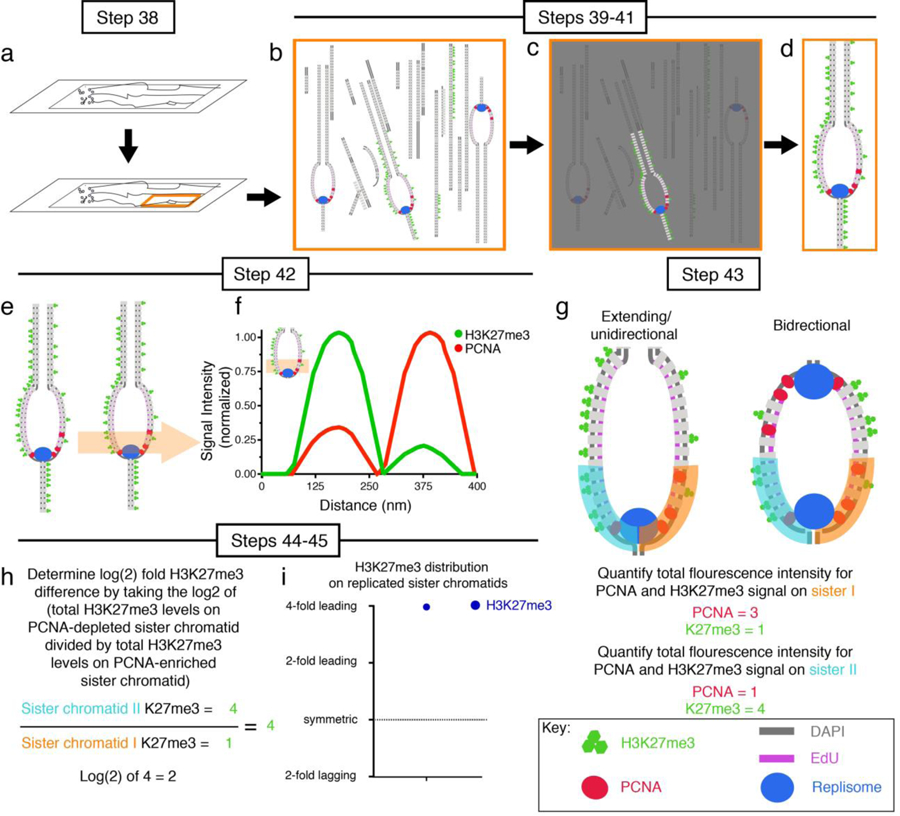
(a) Identify DAPI-positive regions of the slide containing chromatin fibers. (b) Scan chromatin fibers to identify EdU-positive ones. (c) Identify chromatin fibers labeled with all proteins of interest (H3K27me3 and PCNA are shown as examples). (d) Imaging chromatin fiber of interest. (e) Drawn line plot across replicative region at the chromatin fiber. (f) Graph line plot to qualitatively display distribution of histone modification (e.g. H3K27me3) and chromatin-associated protein (e.g. PCNA) at sister chromatids. (g) Draw 2μm long line segments along sister chromatid I and quantify fluorescence (orange line). Repeat for sister chromatid II (cyan line). Figure shows both quantification line plots (sister I and sister II) drawn on extending/unidirectional replicon as well as bidirectional replication bubbles. Total fluorescence levels can by calculated by summing all fluorescence signal in line plot region. (h) Determine log2-fold difference in protein(s) of interest (e.g. H3K27me3) between sister chromatids. (i) Presentation of data in a scatter plot with log2 ratios on Y-axis.
Figure 3: Superresolution imaging of chromatin fibers resolves sister chromatids.
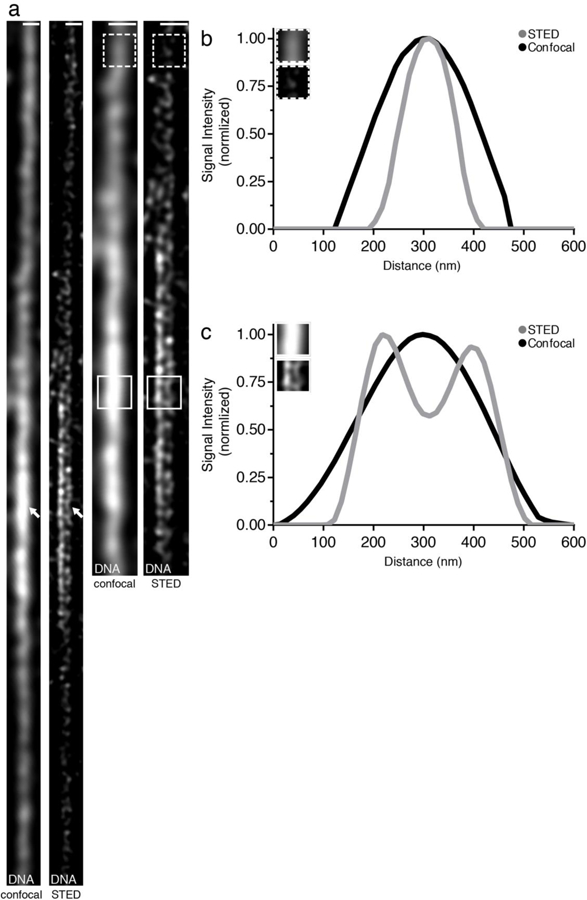
(a) Confocal and STED images of chromatin fiber isolated from Drosophila testis tips stained with DNA dye Yoyo3. (b) Line plot of Yoyo3 signal at a Yoyo3-dim region shows a single fiber structure using both confocal and STED (dashed box). (c) Line plot of Yoyo3 signal at a Yoyo3-bright region shows double fiber structure using STED but not with confocal (solid box). Scale bar in (a): 500 nm.
Superresolution imaging:
For our experiments, STED (Stimulated Emission Depletion) microscopy34 superresolution images are acquired using a Leica TCS SP8 STED microscope with a 1.4 NA 100X STED white objective. For 3-channel STED imaging, we find that the optimal conditions are with the 592 nm continuous wave (CW) and 775 nm pulsed depletion lasers. In our experimental setup, the Alexa 488 channel is depleted with the STED 592nm laser, whereas the Alexa 568 channel and the Alexa 647 channel are both depleted with the STED 775nm laser. Images are acquired as a single z-plane since there is no need for z-stacks on chromatin fibers. To recognize drift during imaging, specimens are mounted with 100 nm TetraSpeck microsphere beads as fiducial markers. Following image acquisition, we perform deconvolution using the Scientific Volume Imaging (SVI) Huygens Professional software package, in order to improve image resolution as well as to correct instrument aberration and blurring. This software package is ideally suited for post-processing images, as it is completely integrated with the Leica LAS-X software and hardware to achieve improved calculated/theoretical PSF (point-spread function) for improved resolution and image quality.
Airyscan high spatial resolution images35 are acquired using a Zeiss LSM 800 microscope with a plan-APOCHROMAT 63x oil objective and Airyscan detector. Airyscan images are processed with Zeiss Airyscan processing software. Other superresolution techniques, such as SIM (Structured Illumination Microscopy36), should be compatible with our methods. Optical reconstruction techniques, such as PALM (Photoactivated Localization Microscopy37) and STORM (Stochastic Optical Reconstruction Microscopy38), should also be compatible, though it may be more technically challenging due to image acquisition conditions required for these techniques39.
Quantification of protein distribution difference on sister chromatids:
Data analyses are conducted using the Java image processing program FIJI. To capture distribution difference of histones, histone modifications and other proteins on sister chromatid fibers, images are imported into FIJI and line plots are drawn across sister chromatids to measure average fluorescence intensity at replicative regions (Figure 2e–f). To measure histone distribution differences between sister chromatids, we divide replication regions longer than 2μm into 2μm-long line segments along the length of the analyzed chromatin fiber region (Figure 2g). The 2μm-length is chosen as this is the average size of individual replicons with 30-minute EdU pulses in our experiments23. Given the estimated average synthesizing rate of DNA polymerase40, the 2μm length at chromatin fiber should represent approximately 15–60 kb of genomic DNA, depending on the stretching and/or relative chromatin compaction at the corresponding chromatin fiber region. For replicative regions shorter than 2μm, the entire length of the region containing resolvable sister chromatids is used for analyses.
To compare histone distribution patterns among multiple biological replicates, we normalize datasets using the following strategy: First, we quantify fluorescence levels for the protein of interest (e.g. different post-translational histone modification immunofluorescence signals, such as H3K27me3 or H3K14Ac) at each segment of chromatin fibers harboring resolvable sister chromatids. We then divide the fluorescence intensity from the brighter sister chromatid fiber side by the fluorescence intensity from the comparatively dimmer sister chromatid fiber side, to generate a ≥ 1 ratio of the relative ratio between sister chromatids. We utilized this strategy to quantify data with no strandness information (i.e. leading strand versus lagging strand).
To obtain strandness information in SRCF experiments, we use lagging strand-enriched proteins, such as RPA70 or PCNA23,41. To quantify strand-specific distribution of post-translational histone modifications on replicating or newly replicated sister chromatids, signal intensity at the leading strand (RPA70-depleted or PCNA-depleted strand) is divided by the signal intensity at the lagging strand (RPA70-enriched or PCNA-enriched):
The raw ratios are then converted into a log2 scale, such that the leading strand enrichment would appear as a positive value and the lagging strand enrichment would appear as a negative value, in an equally distributed manner from the symmetric point (1:1 ratio, Figure 2h–i). To obtain the average fold enrichment as an unconverted ratio, the following calculation is done:
Application of the SRCF technique to study RCNA in cultured Drosophila Kc cells
As we have already used the SRCF technique to profiles histone inheritance patterns in cells from tissue such as Drosophila testes23, we next used SRCF to investigate histone inheritance patterns in a symmetrically expanding population of cultured Drosophila Kc cells. This experiment make it possible to compare SRCF-generated results with previous sequencing-based data using yeast21 and cultured mESCs22, since all three experimental designs utilize symmetrically dividing cells.
In Kc cells, we use DAPI to label DNA, EdU to label replicative regions, PCNA to label the lagging strand, H3 lysine 27 trimethylation (H3K27me3) as a proxy for old histones and H3 lysine 14 acetylation (H3K14Ac) as a proxy for new histones, according to previous biochemical assays on their enrichment on old histone and new histone, respectively42,43. Using SRCF on Kc cells, we found that old histones, marked by H3K27me3, tend to have a bias towards the leading strand whereas new histones, marked by H3K14ac, tend to have a bias towards the lagging strand (Figure 4). Quantification of these data reveals that old histones are 1.51-fold more enriched towards the leading strand, whereas new histones are 1.34-fold enriched towards the lagging strand (Figure 4c). Furthermore, using the criteria to classify fibers as leading strand-enriched, symmetric, or lagging strand-enriched23, we find that for old histone-enriched H3K27me3, 45% of fibers show leading strand bias, 45% are symmetric and only 10% of fibers show lagging strand enrichment in Kc cells (Figure 4f). For new histone-enriched H3K14ac, 55% show a lagging strand bias, 33% are symmetric and only 11% show bias towards the leading strand (Figure 4f). Notably, asymmetries of the old histone distribution in Kc cells are more subtle than the asymmetries observed in the Drosophila testis samples, where SRCF studies reveal a 2.03-fold H3K27me3 enrichment towards the leading strand and 68% leading strand biased fibers23. Differences in old histone strand bias between these two systems (Kc cells versus testis samples enriched with early-stage germline) suggest that developmental programs play an important role in regulating histone distribution patterns during RCNA.
Figure 4: Distributions of old histone-enriched H3K27me3 and new histone-enriched H3K14Ac on Kc cell-derived chromatin fibers.
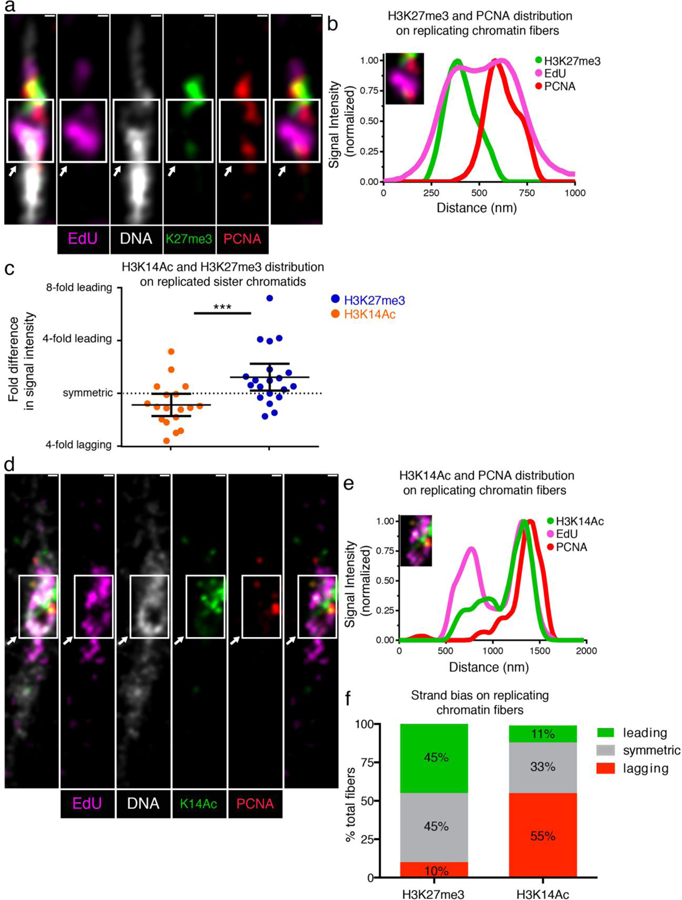
(a) Airyscan image of chromatin fiber labeled with EdU showing H3K27me3 and PCNA distributions at a replicative region on Kc cell-derived chromatin fiber. The transition from single to double fibers colocalizes with the EdU incorporation site (arrow). (b) Line plot shows PCNA, H3K27me3 and EdU distribution across the replicative region (solid box). (c) Quantification of the log2 ratios of H3K27me3 fluorescence intensity at: PCNA-depleted sister chromatid/PCNA-enriched sister chromatid (Average fold enrichment at the PCNA-depleted leading strand = 1.52; n = 20); and log2 ratios of H3K14Ac fluorescence intensity at: PCNA-depleted sister chromatid/PCNA-enriched sister chromatid (Average fold enrichment at the PCNA-enriched lagging strand = 1.34; n = 18). Individual data points and mean values are shown. Y-axis is with log2 scale. Error bars represent 95% confidence intervals. *** P < 10−3, Mann-Whitney U two-sided test (P = 0.0008), Mann-Whitney U score 69.0. (d) Airyscan image of chromatin fiber labeled with EdU showing H3K14Ac and PCNA distribution. The arrow indicates the replication region. (e) Upper line plot shows PCNA, H3K14Ac and EdU distribution across the replicating region (solid box). (f) Classification of PCNA-labeled fibers into leading strand-enriched (inter-sister ratio >1.4-fold), lagging strand-enriched (inter-sister ratio < −1.4) and symmetric (−1.4 < inter-sister ratio < 1.4) for H3K27me3 and H3K14ac. Scale bar in (a) and (d): 250 nm.
Next, to compare our dataset using SRCF to histone inheritance patterns obtained using sequencing-based methods in other systems, we compared our data to published results in yeast21 (see footnote1). To facilitate a direct comparison between our results and results obtained from sequencing based techniques, we converted all data to log2-fold difference of old histone distribution between the leading strand and the lagging strand (Figure 5): A two-fold bias of old histone towards the leading strand would appear as a value of +1. Conversely, a two-fold bias of old histone towards the lagging strand would appear as a value of −1. Overall, SRCF studies in Drosophila Kc cells and the Drosophila testes revealed old histone bias towards the leading strand (Figure 5b). More pronounced asymmetry in old histone distribution was observed in the Drosophila testes, compared to Kc cells. The budding yeast Saccharomyces cerevisiae showed an even more subtle asymmetry, as differences in old histone distribution differed by only a log2-fold differences of −0.33, or approximately a 1.25-fold bias toward the lagging strand (Figure 5b). It is also important to note that despite the overall bias of old histone towards the leading strand, SRCF studies in both Kc cells (Figure 4) and testes23 showed a low percentage of biased old histone towards the lagging strand. These results suggest that in certain contexts, either at the specific genomic regions or in particular cell types, old histones can be preferentially incorporated towards the lagging strand. More studies in the future will be needed to understand these context-dependent histone incorporation patterns.
Figure 5: Comparison of histone distribution patterns in different systems using distinct methods.
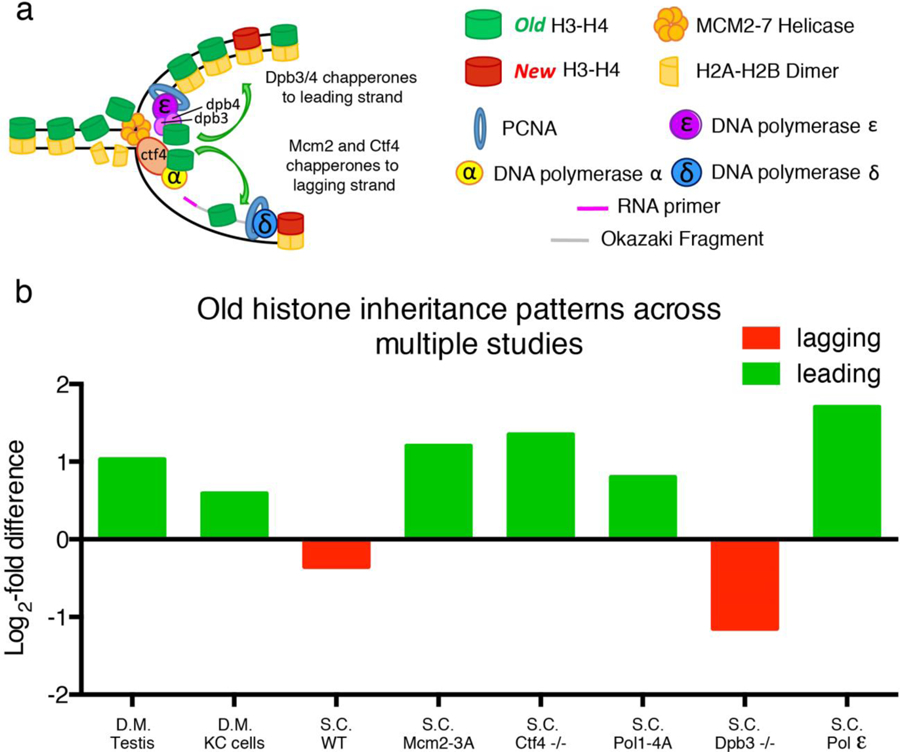
(a) A cartoon shows molecular players responsible for histone incorporation at the replication fork. (b) Bar graphs compare log2-fold enrichment of old histone on leading versus lagging strand in different systems under different experimental conditions. S.C. = Saccharomyces cerevisiae; D.M. = Drosophila melanogaster; WT: wild-type; −/−: homozygous loss-of-function mutant. MCM2–3A indicates a mutant MCM2 protein unable to properly chaperone histones. POL1–4A indicates a mutant Pol α unable to properly tether Ctf4. S.C. Pol ε indicates distribution of the leading strand Polymerase epsilon on sister chromatids. Green bars indicate biases towards the leading strand and red bars indicate biases towards the lagging strand.
Recent studies have revealed several key molecular chaperones involved in depositing histones to the leading strand or the lagging strand during DNA replication. For example, mutations of key residues in the replicative helicase MCM2 (MCM2–3A) result in a loss of its histone chaperoning role during RCNA44. Studies in mESCs demonstrate that MCM2–3A mutant cells have an exaggerated bias in histone inheritance pattern, such that the majority of old histones are recycled by the leading strand22. These studies demonstrate that wild-type MCM2 plays a crucial role in facilitating old histone incorporation towards the lagging strand during DNA replication in mESCs. Similarly, loss of MCM2’s chaperoning ability in yeast results in a significant shift in old histone inheritance towards the leading strand (1.20 log2-fold or 2.30-fold old histone bias towards leading strand)45. For comparison, similar eSPAN studies looking at the distribution of the leading strand polylerase ε (Pol ε) on replicating DNA identified a 1.7 log2-fold or 3.25-fold bias towards the leading strand41,46. As Pol ε is thought to be exclusively localized towards the leading strand during DNA replication47, this 3.25-fold biased distribution probably represent the maximum bias that can be detected using the eSPAN method. Therefore, the histone asymmetry observed under MCM2 mutant conditions is quite extreme, as the asymmetry is comparable to the strand preferences observed for Pol ε.
In addition to MCM2, other key replication proteins have been shown to play important roles in chaperoning histones during DNA replication. For example, Ctf4 appears to facilitate old histone deposition onto the lagging strand, as loss of Ctf4 results in a significant shift in old histone incorporation towards the leading strand (1.35 log2-fold or 2.55-fold bias towards the leading strand, Figure 5b)45. Tethering of Ctf4 to Pol α is critical for the role of Ctf4 as a histone chaperone. Loss of the association between Ctf4 and Pol α by expressing a mutant Pol α (Pol1–4A) also results in a shift of old histone incorporation towards the leading strand (0.8 log2-fold or 1.74-fold bias towards the leading strand, Figure 5b).
Key replication proteins have also been found to facilitate old histone deposition onto the leading strand. For example, Pol ε contains two non-essential subunits Dpb3 and Dpb4, which have important roles in chaperoning histones during RCNA. Loss-of-function of either subunit results in an increased bias in old histone incorporation towards the lagging strand, from a 0.33 log2-fold (or 1.26-fold) lagging strand bias in wild-type yeast to a 1.15 log2-fold (or 2.22-fold) lagging strand bias in Dpb3/Dpb4 double mutant yeast21 (Figure 5b). Together, these studies demonstrate that replication proteins play important and unappreciated roles in regulating the incorporation of old histones during the process of RCNA.
Interestingly, histone inheritance patterns characterized using SRCF in the Drosophila testes23 show an asymmetric old histone distribution more similar to yeast cells mutant for MCM2, Ctf4 and Pol α than to wild-type yeast cells. These observations suggest that chaperones such as MCM2 and/or Ctf4, may be differentially regulated in certain developmental contexts of multicellular organisms to yield a more pronounced asymmetric histone incorporation pattern. Our SRCF-based results using Kc cells support this hypothesis, as Kc cells show a more modest asymmetry of old histone distribution at the leading strand when compared to Drosophila testis sample (0.59 log2-fold or 1.51-fold bias for Kc cells versus 1.02 log2-fold or 2.03-fold bias for testes).
In summary, although it remains elusive what molecular factors are responsible for regulating different histone incorporation patterns, recent results generated from imaging-based technique (e.g. SRCF) as well as sequencing-based techniques (e.g. SCAR-seq and eSPAN) suggest that the process of RCNA is a dynamic and highly regulated process capable of producing distinct outcomes in different developmental contexts. Importantly, the SRCF technique provides a complementary strategy to study the process of RCNA using a small number of cells, which will greatly assist in characterizing the process of RCNA in diverse developmental contexts where limit cell number may preclude the use of sequencing-based techniques. Future work will lead to a better understanding of the range and diversity of RCNA patterns across a wide range of cell types and organisms. Given the important roles that RCNA has in regulating chromatin states, it is highly likely that changes in RCNA may help determine cell fates and pattern tissues during development, homeostasis, and regeneration of multicellular organisms, including mammals.
Materials:
Equipment:
Dissecting forceps (Dumont tweezer #55 World Precision Instruments© order number 500235)
Dissecting dish (Micro test/staining dish, ProSciTech © Product Code: H422)
ThermoScientific® Shandon Cytospin 3
Modified 50ml conical (MilliporeSigma® Eppendorf® Conical Tubes, catalogue number EP0030122267 – modified as shown in Picture S1. Hole made at the base of 50ml conical with a 2 gauge needle (Picture S1).
Humid chamber – made from an old P1000 tip box as shown in Picture S4–S5.
Coplin Jar (United Scientific Supplies Coplin Staining Jar, Fisher Scientific®, catalog number S17495A)
Superresolution microscope (STED, Zeiss Airyscan) with deconvolution/post-processing software (Huygens Professional software package for STED, Zeiss Airyscan processing software for Airyscan)
Fiji: A Java image processing program
Denville 260D Brushless Microcentrifuge (Denville Inc.©, catalog number: C0260–24)
Reagents:
Schneider’s Drosophila medium (Gibco™, Catalog# 21720001)
Invitrogen™ Click-iT™ Plus EdU Cell Proliferation Kit for Imaging, Alexa Fluor™ 647 dye (Catalog number: C10340)
Dulbecco’s Phosphate Buffered Saline (Gibco™ DPBS, calcium, magnesium. catalog number: 14040117)
Collagenase/dispase (MilliporeSigma®, SKU-pack size: 10269638001)
Clean glass slides (Fisherbrand™ Superfrost™ Plus Microscope Slides, ThermoFisher Scientific® catalog number: 12–550-15)
Glass coverslips (MilliporeSigma®, Corning® cover glasses, SKU number CLS2980225)
1.5ml Eppendorf tubes (MilliporeSigma® TubeEppendorf® Safe-Lock microcentrifuge tubes, SKU T9661)
Bovine Serum Albumin (MilliporeSigma®, catalog number: A2153)
Normal Goat Serum (ThermoFisher Scientific® Invitrogen™ catalogue number: 31872)
ProLong™ Diamond Antifade Mountant with DAPI (ThermoFisher Scientific®, Catalog number: P36966) Caution! Dangerous if touched. Always wear gloves when working with substances containing DNA intercalating agents such as DAPI.
TetraSpeck microsphere beads as fiducial markers (ThermoFisher Scientific® Thermo Fisher Catalog number: T7279)
Joy Non-ultra Dishwashing Liquid, Lemon Scent (Proctor and Gamble®)
Sodium Chloride (NaCl, MilliporeSigma® catalogue number: S9888)
Tris Base (NaCl, MilliporeSigma®, product number: TRIS-RO)
Formaldehyde (MilliporeSigma® catalogue number: F8775) Caution! Dangerous if touched or inhaled. Always wear gloves when working with formaldehyde and perform work in a well-ventilated hood when working with formaldehyde.
Sucrose (MilliporeSigma®, catalog number: S7903)
25 gauge hypodermic needle (MilliporeSigma®, catalog number: Z192406)
Phosphate-Buffered Saline (PBS) (ThermoFisher Scientific® catalog number: 10010023)
Triton detergent (ThermoFisher Scientific® Thermo Scientific™Triton™ X-100 Surfact-Amps™ Detergent Solution, Catalog number 85111)
PCNA antibody (Santa Cruz Biotechnology Inc. Catalog number: sc-56, RRID:CVCL_Z834, http://antibodyregistry.org/AB_628110)
H3K27me3 antibody (MilliporeSigma®, H3K27me3 Millipore Cat# 07–449, RRID:AB_310624, http://antibodyregistry.org/AB_310624)
H3K14Ac antibody (Abcam Cat# ab52946, RRID:AB_880442, http://antibodyregistry.org/AB_880442)
Reagents Setup:
Dissolve all components to prepare for the lysis buffer (100 mM NaCl, 25 mM Tris-base, 0.2% Joy detergent, pH 10) in Milli-Q water. Lysis Buffer can be made ahead of time and stored for up to six months at room temperature.
Dissolve all components of Sucrose/Formalin Mixture (1M sucrose 10% formaldehyde) in Milli-Q water. Sucrose/Formalin solution should be made fresh on the day of fiber experiment.
Fixative Solutions: All formaldehyde-based solutions should be diluted in 1xPBS from formaldehyde stock on the day of experiment.
Bovine serum albumin stock solution (2% BSA). Dissolve BSA in 1xPBS. Solution can be stored at 4° for no more than two months.
Normal goat serum (NGS) stock solution (2.5% NGS). Dissolve NGS in 1xPBS. Solution can be stored at 4° for no more than two months.
Biological reagents:
Kc 167 cells DGRC Cat# 1, RRID:CVCL_Z834 (https://web.expasy.org/cellosaurus/CVCL_Z834)
Procedure:
Drosophila testis dissection and fiber preparation: Time 2–3hrs
[All steps hereafter are performed at room temperature (RT) unless otherwise specified.]
1| For chromatin fiber preparation, we typically dissected 10–15 male flies. Testes were dissected in Schneider’s Drosophila medium. For dissections, it is best to dissect testes out from all flies and store them in a separate well of a dissecting dish, which contains Schneider’s medium. A more detailed description on testis dissection can be found in48. In brief, we first place an anesthetized male in a dissection well containing ~100μl of Schneider’s medium. Using dissecting forceps, exert slight pressure at the top of the abdomen. Pressure should be sufficient to hold the fly steady during dissection, but not too much to crush the abdomen, as this could damage the testes. Once being held steady by one dissecting forceps, use the other dissecting forceps to grasp the fly by the external genitalia and slowly pull out the testes and associated genital tract into the dissection well containing Schneider’s medium. If the testes do not come out cleanly, use the forceps as a needle to tease the testes out of the abdomen. Do not try to grab the testes near the apical tip with the forceps, as this will cause damage to the early-stage germline cells which we will study. Separate testes from genital tract and transfer them into a fresh drop of Schneider’s medium.
2| All dissected testes can then be transferred en masse into an eppendorf tube containing Schneider’s medium with 10μM EdU analogue, followed by rotating on a nutator for 15 minutes to ensure robust EdU incorporation.
3| Take the eppendorf tube off nutator and allow testes to settle. This takes approximately 30 seconds.
4| Aspirate Schneider’s medium using P1000 pipette.
5| Resuspend testes in new media containing dissociation buffer (Dulbecco’s PBS with Mg2+ and Ca2+ with collagenase/dispase added to a final concentration of 2mg/ml) with 10μM EdU analogue. This solution is to break apart the whole testis into single cells while incorporating EdU analogue at the active replication regions. Following resuspension, transfer testes to a 37°C water bath for optimal collagenase/dispase activity and incubated for 15 minutes (total EdU incubation time: 30 minutes). Note: To achieve thorough tissue disruption, eppendorf tubes should be shaken lightly every 5 minutes during incubation to assist tissue dissociation. Additional incubation time can be used as needed to ensure complete breakdown of tissue into single-cell suspension. However, any extension of lysis time should be carefully noted, as this also extends EdU pulse duration.
6| Following tissue dissociation, pellet cell suspension at 900 rpm (revolutions per minute) for four minutes using a Shandon Cytospin3 centrifuge. This step will concentrate and deposit cells onto a small, ~5mm diameter circle at Fisherbrand Superfrost Plus Microscope slides. Note: While cells are being spun down, a 50ml conical tube containing 25ml of lysis buffer should be prepared (Pictures S2–S3). This conical tube is modified with a small hole (~0.5 mm) at the bottom to allow lysis buffer to flow out (Picture S1). It is important to move cells quickly from spinning-down step to the subsequent lysis step to prevent desiccation and potential inhibition of proper cell lysis.
7| Place glass slides into the 50ml conical tube containing 25ml lysis buffer placed at a 35° angle (Pictures S2–S3) and incubate for 10 minutes. (Note: Cap should be tightly screwed on at this time point to prevent lysis buffer from flowing out.)
8| After a 10-minute incubation, slightly and carefully unscrew cap from the conical tube and allow lysis buffer to flow out at a slow, steady rate. Flow rate should be approximately 10ml/min, for a total flow time of 2.5 minutes.
9| Once the lysis buffer has completely flowed out of the conical tube, remove slides from tube and add 10μl of sucrose/formalin solution (1M sucrose; 10% formaldehyde) to the surface of the slide. When adding sucrose/formalin mixture, be sure not to add directly onto newly made chromatin fibers. Rather, add mixture slowly to the edge of the slide (away from fibers) and allow liquid to slowly flow overtop of the chromatin fibers. Then place a glass coverslip on top and incubate for two minutes.
10| Transfer slide into liquid nitrogen and incubate for two minutes.
11| Remove cover slip with razor blade and rapidly transfer the slide to cold (−20°C) 95% ethanol for ten minutes.
12| Remove slide from ethanol and place at 45° angle for ~2 minutes (or until almost all ethanol has evaporated from slide but not completely dry).
13| Add 1ml of fixative solution [0.5% formaldehyde in 1xPBST (1xPBS with 0.1% Triton)] to the slide and incubate for two minutes.
Caution! Formaldehyde can be dangerous if being touched or inhaled. Always wear gloves and work in a well-ventilated hood when working with formaldehyde.
14| Drain fixative solution and transfer the slide to a coplin jar containing 50ml 1xPBS.
15| Remove slides from the coplin jar and place at 45° to allow 1xPBS to efficiently drain off the surface of the slide but not completely dry out.
16| Discard 1xPBS from the coplin jar and refill with 50mls fresh 1xPBS.
17| Place slides back in the coplin jar with fresh 1xPBS to wash slides.
18| Repeat steps 16–17 twice.
Procedure
Pre-blocking and antibody labeling: Time 12–16hrs
19| Remove slides from the coplin jar after the final wash and place at a 45° angle for one minute to allow the residual PBS to drain. Note: PBS should be largely gone but slides should not be totally dry. After a minute has passed, place slides in a humid chamber and cover with 500μl of blocking solution (2% BSA in 1xPBST) for 30 minutes. Humid chamber is made from empty P1000 tip boxes with the top covered with tin foil (Pictures S4–S5). 5mls of water was put at the bottom of the humid chamber to soak a few pieces of paper towel and the top is placed on to prevent desiccation of the slide during this blocking step.
20| Drain blocking buffer and proceed to antibody labeling protocol.
21| Dilute primary antibodies to appropriate dilution in blocking solution (2% BSA in 1xPBST).
22| Place slides at 45° angle for one minute to drain residual blocking buffer from pre-blocking step 19.
23| Add 200μls of diluted primary antibody solution to each slide.
24| Cut a small piece of parafilm (2cm x 4cm) and place it on top of the drop of primary antibody solution.
25| Place slides into the humidity chamber and incubate overnight at 4°C.
Pause point: Slides can be incubated for 6–30 hours.
26| Remove slides from humidity chamber and drain excess antibody solution.
(Note: antibody solution could be saved and reused for subsequent fiber experiments.)
27| Repeat steps 15–18 to wash slides.
28| Place the slide in humid chamber and cover with 500 l secondary blocking buffer (2.5% NGS in 0.1% PBST). Incubate for 30 minutes.
29| Remove the slide from humidity chamber and place slides at 45° angle for 1 minute to drain residue secondary blocking buffer.
30| Dilute secondary antibodies in the blocking buffer to create secondary solution.
31| Place slides in humid chamber and cover with 300μl secondary solution. Cut a small piece of parafilm (2cm x 4cm) and place on top of the antibody solution. Incubate for 2 hrs RT.
Pause point: Slides can be incubated for 4 to 8 hours at 4°C.
Procedure
Final wash and mounting: Time 24hrs
32| Remove the slide from humidity chamber and drain excess secondary antibody solution.
33| Repeat steps 15–18 to wash slides.
34| Place slides at 45° angle for two minutes to drain excess 1xPBS solution.
35| Lay slides flat and cover with 15μl 100 nm TetraSpeck microsphere beads (Thermo Fisher Catalog# T7279), diluted 1:100 from stock solution. Wait for almost all liquid to evaporate (no more than five minutes). Slides should be almost dry but not completely devoid of moisture before adding the mounting media.
36| Lay slides flat and cover with 15μl ProLong® Diamond mounting media containing DAPI. Caution! DAPI can be harmful if it comes in direct contact with skin. Always wear gloves when working with DAPI.
37| Let the slide sit foe one day at room temperature in a dark place prior to imaging.
Pause point: Slides can be stored for up to two weeks at 4°C prior to imaging. Note: slides stored at 4°C should be warmed to RT prior to imaging.
Procedure
Observation and Image Acquisition: Time 4–5hrs (add 2–3hrs for post-imaging deconvolution processing)
38| Scan slides thoroughly for DAPI signals to identify regions of the slide with chromatin fibers. When scanning, lamp power should be kept as low as possible to minimize photobleaching of fluorophores, but high enough so that chromatin fibers can be readily detected when viewing through microscope eyepiece. Instructions on adjusting lamp power should be available in the User Manual of the superresolution microscope.
39| Scan fiber-positive regions for EdU signal to identify chromatin fibers that are actively undergoing DNA replication. Identify replication bubbles by finding EdU-positive regions where fibers show a clear transition from a single fiber structure to a double fiber structure, or directly proximal to the site of EdU incorporation. It is important to note that superresolution microscopy (Step 41) is often necessary to resolve the transition from single fiber to double fiber structure. Therefore, identification of replication bubble structure often occurs during data analysis and after superresolution images have been acquired and processed. It is also important to note that EdU-positive regions were also associated with a significant increase in DAPI signal. DAPI-bright regions are identified as regions showing greater than a 2-fold increase in DAPI-intensity relative to the neighboring DAPI-dim regions from the same chromatin fiber. As a timesaving measure, DAPI-bright regions can be used to assist in finding replicating regions.
40| Scan other fluorescent channels where other proteins of interest are labeled (e.g. H3K27me3 or PCNA) to verify that the identified replicative chromatin fibers from Step 39 have retained key proteins of interest.
41| Once replicative regions have been identified, superresolution images should be acquired to assist in visualization of replicating sister chromatids. For STED superresolution imaging, images in our datasets were acquired using a Leica TCS SP8 STED microscope with a 1.4 NA 100X STED white objective. STED depletion for three-color imaging was performed using the 592 nm continuous wave (CW) and 775 nm pulsed depletion lasers. Fluorophores labeled with Alexa 488 were superresolved (depleted) with STED 592 nm, whereas fluorophores labeled with Alexa 568 and 647 were superresolved (depleted) with STED 775 nm depletion laser. Laser power for both excitation and depletion can vary from experiment to experiment. Therefore, when beginning a superresolution experiment, it is important to take the time to set the laser power for the 488, 568 and 647 excitation beams to maximize fluorophore brightness (without saturating the detector) while minimizing background noise. For setting laser power with the depletion lasers, it is important to start with a low laser power (to minimize photobleaching) and increase power until fluorophores begin to be depleted to produce superresolved images. It is important to note that higher laser power for the depletion laser produces a greater theoretical resolution. Therefore, depletion laser power should be increased as much as possible (without photobleaching) to achieve maximum resolution. As a reference, typical depletion laser power for our experiments is as follows: 1.4380W for 592 nm; 1.5010W for 775 nm. However, due to inherent variability in fluorophore strength and efficiency of antibody labeling, depletion laser power must be empirically determined prior to image acquisition in order to maximize resolving power and image quality. In order to minimize drift between channels, it is important to acquire images of isolated fibers as a single z-plane. Following image acquisition, instrument aberration and blurring should be corrected with deconvolution using the Scientific Volume Imagine (SVI) Huygens Professional software package. For all pictures, it is important to take careful notes of the following: slide name, date, antibody conditions, genetic background and EdU incubation time. Other superresolution strategies can be employed to increase frequency of spatially resolvable sister chromatids. For example, Zeiss Airyscan imaging is compatible with this technique. Other superresolution methods, such as SIM, STORM and PALM should also be compatible with this technique. However, STORM and PALM may be technically more challenging.
Procedure
Data analysis: Time ~10–15 minutes per replicative chromatin fiber.
42| All data analyses can be performed using the Java image processing program FIJI. Once images have been imported to FIJI, draw a line plot across replication bubble to qualitatively show distribution difference between the analyzed sister chromatids. Line plot should be drawn approximately orthogonal to the replication bubble and should be of sufficient width to encompass all signals at the edge of the replication fork. Note: Line-plots are merely meant to qualitatively show the data and should not be taken as a precise quantification. To more precisely quantify differences between sister chromatids proceed to Steps 43–45.
43| To quantify protein level differences between sister chromatids, first identify the edge of the replication bubble by finding the junction where a single fiber structure transitions to a double fiber structure (at or directly proximal to the EdU incorporation site). For extending replication bubbles (i.e. fork progresses toward one direction at a single replicon and PCNA is localized at one side of the replication bubble) and bidirectional bubbles (i.e. fork progresses toward both directions and PCNA is localized at both sides of the replication bubble) longer than 2μm, draw a 2μm line in length extending from the edge of the replication bubble along one sister chromatid to be designated as sister chromatid I (Figure 2g). Two microns was chosen as this represents the average size of 30-minute EdU incorporation in chromatin fiber experiments23. Line should be drawn with sufficient width to capture all signal associated with the individual sister chromatid. Once drawn, total fluorescence signal for all relevant proteins (PCNA, H3K27me3 or H3K14Ac) should be calculated and recorded. Using the drawing tools provided by the FIJI imaging processing program, a mark should be made on the chromatin fiber to indicate where the 2μm line plot ends. This mark should be used as the starting point for the next line plot to be drawn. After recording data for sister chromatid I, a second line will be drawn to acquire data for sister chromatid II. A line is drawn from the edge of the replication bubble along sister chromatid II. It is important that this line is of identical width and length to allow for unbiased comparison of protein levels between sister chromatid I and sister chromatid II. Once drawn, calculate the total fluorescence signal for all relevant proteins (PCNA, H3K27me3 or H3K14Ac). For bidirectional replication bubbles (PCNA localized at both sides of the replication bubble) shorter than 2μm, line plots should be drawn with appropriate length to capture each PCNA-positive region located at the edge of the replication bubble separately. For extending replication bubbles shorter than 2μm in length, lines should be drawn throughout the length where both sister chromatids can be clearly visualized.
44| Determine the log2-fold H3K27me3 difference between leading strand and lagging strand by taking the log2 (total H3K27me3 levels on PCNA-depleted sister chromatid/total H3K27me3 levels on PCNA-enriched sister chromatid).
45| Plot log2-fold difference for proteins of interest. By dividing the values on the PCNA-depleted sister chromatid by the PCNA-enriched sister chromatid, proteins with a leading strand (PCNA-depleted) bias should appear as positive values, whereas proteins with lagging strand (PCNA-enriched) bias should appear as negative values.
Timing:
Step 1: Tissue dissection: ~15 minutes (time may vary depending on tissue; estimate given for Drosophila testis)
Steps 2–5: EdU incubation and tissue dissociation: 30–40 minutes
Steps 6–18: Cell lysis and fiber generation: 30–40 minutes
Steps 19–27: Pre-blocking and 1° antibody labeling: 10–12 hours
Steps 28–31: Wash and 2° antibody labeling: 3 hours
Steps 32–37: Final wash and mounting: 24 hours
Steps 38–41: Image acquisition: 4–5 hours
Steps 42–45: Data analysis: 15 minutes (per replicating chromatin fiber)
Anticipated Results:
Superresolved chromatin fiber (SRCF) experiments will generate glass slides coated with replicative chromatin fibers. With the aid of superresolution microscopy, these replicative chromatin fibers can be spatially resolved into newly synthesized sister chromatids. These fibers can be stained for key proteins of interest, which allows for investigating the distribution of multiple proteins simultaneously at the replication fork. Figures 4a and 4d show examples of replicative (EdU-positive) chromatin fibers showing distinct distributions of post-translational histone modifications at the replication fork. The distribution of proteins at the replication fork can be visualized and quantified using the protocol described above. It is important to note that many of the chromatin fibers identified at the “observation and identification” stage will not be suitable for imaging as they may not contain all the markers required for proper data analysis. For example, in case where our experimental goal was to explore H3K27me3 distribution patterns at the replication fork, many fibers could not be used for data analysis, as they did not retain any H3K27me3 signal at the replication bubble. Furthermore, if fibers have finished replication before the process of fiber preparation, it is possible that PCNA may be unloaded, preventing identification of the lagging strand. Without this information, it is impossible to assign strandness to the replicating/recently replicated chromatin fibers. Many of the fibers identified could be from unreplicative cells, or from G2-phase or G1-phase cells, which are unsuitable for further analysis. For these reasons, it is often necessary to search each prepared slide extensively to find chromatin fibers suitable for superresolution imaging and data analyses.
Troubleshooting:
| Problems | Steps* | Possible Reasons | Possible Solutions |
|---|---|---|---|
| Cells deposited on slides are not lysed/poorly lysed. | Step 6 and Step 7 | Step 6: Too much time taken between spinning down cells and proceeding to cell lysis. Step 7: Insufficient lysis time. |
Step 6: Transfer slides more quickly from the spinning stage (Step 6) to the lysis stage (Step 7) to prevent desiccation. Step 7: Extend Step 7 to allow for more complete lysis. |
| Slide contains no chromatin fibers | Steps 8–9, Steps 16–18, 27, 33 | Steps 8–9: Chromatin fibers did not adhere to the slide strongly enough to remain attached throughout the subsequent immunostaining procedures. Steps 16–18, 27, 33: Washing steps dislodge chromatin fibers. |
Steps 8–9: Allow slides to briefly (<10 seconds) desiccate prior to addition of sucrose/formalin mixture. This has been found to increase fiber retention rates, though at the cost of increased background noise at the subsequent imaging steps. Steps 16–18, 27, 33: Show extreme care when transferring slides during washing steps. Try not to disrupt the surface of the slide with mechanical forces at these steps. |
| Protein labeling on chromatin fibers is poor or nonexistent | Steps 7,8 | Step 7: Lysis buffer salt concentration is too high, and proteins have been strip off DNA. Step 8: Lysis step is too strong and proteins have been strip off DNA. |
Step 7: Certain proteins (particularly histone H2A/H2B dimers) show lower retention rates with 100μM NaCl. Lowering salt concentration to 80–90μM can improve protein retention in certain cases. Step 8: Lysis time may be decreased to increase protein retention. Step 8: Use an alternative lysis method described hereafter. |
| Sister chromatids cannot be resolved | Step 41 | Step 41: Microscope resolution is insufficient to resolved sister chromatids. | Step 41: Utilize microscopy technique with greater spatial resolution. Step 8: Use an alternative lysis method described hereafter. |
Steps refer to those of the protocol when the described problems likely occur. However, problems would not be detected until Steps 38–40.
Alternative Steps 6–8:
6| Following tissue dissociation, pellet cell suspension at 1000 rpm for 6 minutes using a Denville 260D Brushless Microcentrifuge.
7| Aspirate supernatant and resuspend cells in 20μl lysis buffer. Transfer cells in lysis buffer to surface of glass slide.
8| Incubate cells for 10 minutes, covered, to prevent desiccation. Proceed to Step 9 of the original protocol.
This lysis method does not generate chromatin fibers as long, or as uniformly oriented as does the method described in the main protocol. However, it can be tried as an alternative method to increase the efficiency of protein retention and sister chromatid visualization if experimenter encounters problems with the method described in the main protocol.
Other data that support the findings of this study are available from the corresponding author upon request.
No custom code was used in this manuscript.
Supplementary Material
Picture S1: A modified 50ml conical tube with a small hole made at the bottom with a 25-gauge hypodermic needle.
Picture S2: Modified 50ml conical containing 25ml lysis buffer with cap screwed on, angled at 35° in preparation for lysis and fiber generation steps.
Picture S3: A zoom-in picture of S2.
Picture S4: Humid chamber made with a P1000 tip box. The lid is wrapped with tin foil to minimize light exposure to slides placed inside. Lid should be placed on when slides are inside.
Picture S5: Humid chamber with lid off.
Acknowledgements:
We thank Shelby Blythe and Eric Wieschaus for the RPA70-EGFP and PCNA-EGFP fly lines. We thank Barbara Mellone and Sharon Pavanacherry for suggestions on the chromatin fiber technique. We thank Johns Hopkins Integrated Imaging Center for confocal and Airyscan imaging; Carnegie Institute Imaging Center for STED microscopy work. Supported by NIH 5T32GM007231 and F31GM115149–01A1 (M.W.), NIH T32GM007231 (J.S.), NIH R01GM33397 (J.G.), NIH R35GM127075, the Howard Hughes Medical Institute, and Johns Hopkins University startup funds (X.C.)
Footnotes
Competing interests statement: The authors declare no competing interests.
Due to differences in techniques and data analyses, a direct comparison between the SRCF data and SCAR-seq using mESCs is not feasible at the current stage (personal communications with Dr. Groth).
References:
- 1.Kornberg RD & Lorch Y Chromatin structure and transcription. Annu Rev Cell Biol 8, 563–87 (1992). [DOI] [PubMed] [Google Scholar]
- 2.Snedeker J, Wooten M & Chen X The Inherent Asymmetry of DNA Replication. Annu Rev Cell Dev Biol 33, 291–318 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burgers PMJ & Kunkel TA Eukaryotic DNA Replication Fork. Annu Rev Biochem 86, 417–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell SP & Dutta A DNA replication in eukaryotic cells. Annu Rev Biochem 71, 333–74 (2002). [DOI] [PubMed] [Google Scholar]
- 5.DePamphilis ML Review: nuclear structure and DNA replication. J Struct Biol 129, 186–97 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Alabert C, Jasencakova Z & Groth A Chromatin Replication and Histone Dynamics. Adv Exp Med Biol 1042, 311–333 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Ramachandran S, Ahmad K & Henikoff S Capitalizing on disaster: Establishing chromatin specificity behind the replication fork. Bioessays 39(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller TC & Costa A The architecture and function of the chromatin replication machinery. Curr Opin Struct Biol 47, 9–16 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Ehrenhofer-Murray AE, Kamakaka RT & Rine J A role for the replication proteins PCNA, RF-C, polymerase epsilon and Cdc45 in transcriptional silencing in Saccharomyces cerevisiae. Genetics 153, 1171–82 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luger K, Mader AW, Richmond RK, Sargent DF & Richmond TJ Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–60 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Arents G, Burlingame RW, Wang BC, Love WE & Moudrianakis EN The nucleosomal core histone octamer at 3.1 A resolution: a tripartite protein assembly and a left-handed superhelix. Proc Natl Acad Sci U S A 88, 10148–52 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Annunziato AT Assembling chromatin: the long and winding road. Biochim Biophys Acta 1819, 196–210 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Hammond CM, Stromme CB, Huang H, Patel DJ & Groth A Histone chaperone networks shaping chromatin function. Nat Rev Mol Cell Biol 18, 141–158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Worcel A, Han S & Wong ML Assembly of newly replicated chromatin. Cell 15, 969–77 (1978). [DOI] [PubMed] [Google Scholar]
- 15.Sogo JM, Stahl H, Koller T & Knippers R Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. J Mol Biol 189, 189–204 (1986). [DOI] [PubMed] [Google Scholar]
- 16.Jackson V & Chalkley R A new method for the isolation of replicative chromatin: selective deposition of histone on both new and old DNA. Cell 23, 121–34 (1981). [DOI] [PubMed] [Google Scholar]
- 17.Leffak IM, Grainger R & Weintraub H Conservative assembly and segregation of nucleosomal histones. Cell 12, 837–45 (1977). [DOI] [PubMed] [Google Scholar]
- 18.Seidman MM, Levine AJ & Weintraub H The asymmetric segregation of parental nucleosomes during chrosome replication. Cell 18, 439–49 (1979). [DOI] [PubMed] [Google Scholar]
- 19.Weintraub H Cooperative alignment of nu bodies during chromosome replication in the presence of cycloheximide. Cell 9, 419–22 (1976). [DOI] [PubMed] [Google Scholar]
- 20.Roufa DJ & Marchionni MA Nucleosome segregation at a defined mammalian chromosomal site. Proc Natl Acad Sci U S A 79, 1810–4 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu C et al. A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science 361, 1386–1389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petryk N et al. MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 361, 1389–1392 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Wooten M et al. Asymmetric histone inheritance via strand-specific incorporation and biased replication fork movement. Nat Struct Mol Biol 26, 732–743 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen SM, Chastain PD 2nd, Cordeiro-Stone M & Kaufman DG. DNA replication and the GINS complex: localization on extended chromatin fibers. Epigenetics Chromatin 2, 6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmad K & Henikoff S Histone H3 variants specify modes of chromatin assembly. Proc Natl Acad Sci U S A 99 Suppl 4, 16477–84 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blower MD, Sullivan BA & Karpen GH Conserved Organization of Centromeric Chromatin in Flies and Humans. Developmental Cell 2, 319–330 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang CH et al. Islands of retroelements are major components of Drosophila centromeres. PLoS Biol 17, e3000241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKnight SL & Miller OL Jr. Electron microscopic analysis of chromatin replication in the cellular blastoderm Drosophila melanogaster embryo. Cell 12, 795–804 (1977). [DOI] [PubMed] [Google Scholar]
- 29.Kuzminov A When DNA Topology Turns Deadly - RNA Polymerases Dig in Their R-Loops to Stand Their Ground: New Positive and Negative (Super)Twists in the Replication-Transcription Conflict. Trends Genet 34, 111–120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koster DA, Crut A, Shuman S, Bjornsti MA & Dekker NH Cellular strategies for regulating DNA supercoiling: a single-molecule perspective. Cell 142, 519–30 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang JC Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 3, 430–40 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Ljungman M & Hanawalt PC Localized torsional tension in the DNA of human cells. Proc Natl Acad Sci U S A 89, 6055–9 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LaMarr WA, Yu L, Nicolaou KC & Dedon PC Supercoiling affects the accessibility of glutathione to DNA-bound molecules: positive supercoiling inhibits calicheamicin-induced DNA damage. Proc Natl Acad Sci U S A 95, 102–7 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hell SW & Wichmann J Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett 19, 780–2 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Sivaguru M et al. Comparative performance of airyscan and structured illumination superresolution microscopy in the study of the surface texture and 3D shape of pollen. Microsc Res Tech 81, 101–114 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Gustafsson MG Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc 198, 82–7 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Betzig E et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–5 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Rust MJ, Bates M & Zhuang X Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3, 793–5 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glushonkov O, Réal E, Boutant E, Mély Y & Didier P Optimized protocol for combined PALM-dSTORM imaging. Sci Rep 8, 8749 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Techer H et al. Replication dynamics: biases and robustness of DNA fiber analysis. J Mol Biol 425, 4845–55 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Yu C et al. Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol Cell 56, 551–63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alabert C et al. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev 29, 585–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin S, Yuan ZF, Han Y, Marchione DM & Garcia BA Preferential Phosphorylation on Old Histones during Early Mitosis in Human Cells. J Biol Chem 291, 15342–57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang H et al. A unique binding mode enables MCM2 to chaperone histones H3-H4 at replication forks. Nat Struct Mol Biol 22, 618–26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gan H et al. The Mcm2-Ctf4-Polalpha Axis Facilitates Parental Histone H3-H4 Transfer to Lagging Strands. Mol Cell 72, 140–151 e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gan H et al. Checkpoint Kinase Rad53 Couples Leading- and Lagging-Strand DNA Synthesis under Replication Stress. Mol Cell 68, 446–455 e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou JC et al. CMG-Pol epsilon dynamics suggests a mechanism for the establishment of leading-strand synthesis in the eukaryotic replisome. Proc Natl Acad Sci U S A 114, 4141–4146 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White-Cooper H Spermatogenesis: analysis of meiosis and morphogenesis. Methods Mol Biol 247, 45–75 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Picture S1: A modified 50ml conical tube with a small hole made at the bottom with a 25-gauge hypodermic needle.
Picture S2: Modified 50ml conical containing 25ml lysis buffer with cap screwed on, angled at 35° in preparation for lysis and fiber generation steps.
Picture S3: A zoom-in picture of S2.
Picture S4: Humid chamber made with a P1000 tip box. The lid is wrapped with tin foil to minimize light exposure to slides placed inside. Lid should be placed on when slides are inside.
Picture S5: Humid chamber with lid off.