

Abstract
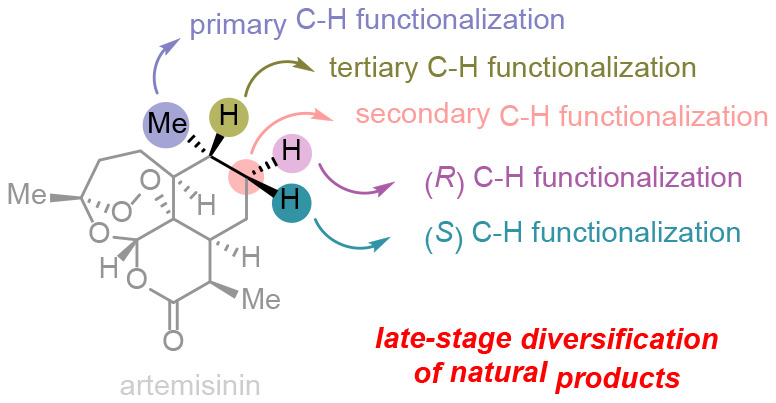
Late-stage diversification of natural products is an efficient way to generate natural product derivatives for drug discovery and chemical biology. Benefiting from the development of site-selective synthetic methodologies, late-stage diversification of natural products has achieved notable success. This outlook will outline selected examples of novel methodologies for site-selective transformations of reactive functional groups and inert C–H bonds that enable late-stage diversification of complex natural products. Accordingly, late-stage diversification provides an opportunity to rapidly access various derivatives for modifying lead compounds, identifying cellular targets, probing protein–protein interactions, and elucidating natural product biosynthetic relationships.
Short abstract
Selected examples of late-stage diversification of natural products are discussed to show recent development of synthetic methods and how late-stage diversification benefits chemical biology studies and drug discovery.
Introduction
Natural products play an important role in chemistry and drug discovery.1,2 Over the years, chemists have sought to develop novel methods and strategies for their effective chemical synthesis3 in order to elucidate their biological functions for novel therapeutic agents4,5 and determine their biosynthesis for synthetic biology.6 These studies have been motivated by the historic involvement of natural products such as penicillin (1), taxol (2), artemisinin (3), and vinblastine (4) in drug discovery (Figure 1). Over the last three decades, nearly 50% of newly approved small molecule drugs have been natural products or derivatives thereof.5
Figure 1.
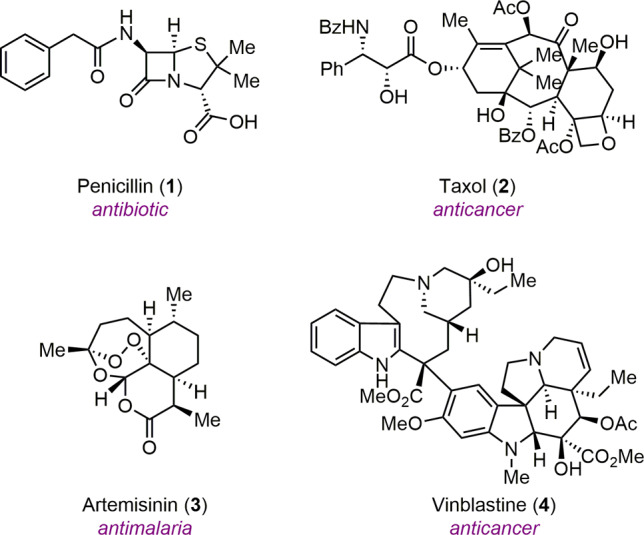
Natural products in human medicine.
Natural product diversification is essential in drug discovery, particularly in the optimization of pharmacological properties and investigation of structure–activity relationships (SARs). Driven by the discovery of biologically relevant natural product-like derivatives, several strategies have been employed to prepare these molecules, including diverted total synthesis (DTS).7,8 Since traditional synthesis of natural product derivatives from simple starting materials can be laborious and ineffective, an alternative approach is to derivatize natural products directly via selective reactions, which may shorten synthetic routes and provide a more effective means of producing these compounds. However, late-stage diversification of natural products has been underexplored owing to the synthetic challenge of performing selective functionalizations in the presence of the diverse functional groups often found in natural products.
Recently, a series of remarkable advances in synthetic organic methodologies, including developments in site-selective catalysts,9,10 state-of-the-art C–H functionalization,11,12 photochemistry,13,14 electrochemistry,15,16 and biocatalysis,17−19 have been achieved, which have led to the emergence of many approaches to directly functionalize complex molecules. These selective transformations have provided a great opportunity for the rapid late-stage modification of various natural products.
Over the past decade, the late-stage diversification of natural products has undergone explosive growth.20−24 This approach has enabled efficient access to lead compounds and natural product-based probes. Herein, we report selected examples of late-stage diversification of complex natural products and the impacts of this approach on organic synthesis as well as chemical biology and drug discovery. The examples provided herein will focus on the site-selective functionalization of natural products or their protected derivatives. The use of natural products as starting materials to synthesize diverse molecules is not covered, as this topic has been reviewed elsewhere.20 The aim of this outlook is not to provide a comprehensive review of achievements in this area but, rather, to showcase some key ideas of this expanding field. Accordingly, we sincerely apologize for the inevitable omissions in our coverage of the literature.
Synthetic Methodology Development
Many complex natural products contain the same reactive functional groups in high frequency, such as polyols, polyenes, and polyarenes. Selective functionalization of one specific reactive group in the presence of others without the use of protecting groups represents a powerful tool for synthesizing diverse analogues. Therefore, many new reagents, catalysts, and reactions have been developed for site- and chemoselective transformations.9 The Miller group has developed various site-selective modification reactions for complex natural products based on peptide catalysts.10 In 2012, the group reported the site-selective diversification of the complex natural product vancomycin (5).25,26 Vancomycin (5) is a glycopeptide antibiotic that inhibits bacterial cell wall biosynthesis through binding to the acyl-d-alanyl-d-alanine (dAla-dAla) moiety of bacterial peptidoglycan. Vancomycin-induced drug resistance emerged in 1988.27 One mechanism of vancomycin resistance in pathogens involves the formation of bacterial peptidoglycan with other residues, such as d-alanyl-d-lactate (dAla-dLac), instead of dAla-dAla.28 As bacterial resistance to vancomycin has become a worldwide problem, the synthesis of novel derivatives has become essential.29,30 In order to synthesize novel vancomycin derivatives, Miller and co-workers first employed their peptide catalysts for site-selective thiocarbonylation/deoxygenation of alcohols of minimally protected vancomycin derivative 6 (Scheme 1A).25 In the presence of achiral catalyst N-methylimidazole (NMI), Z6-OH was acylated preferentially over G6-OH (7:8 = 1:5). This unexpected selectivity was presumably due to the steric hindrance of the primary hydroxy group G6-OH. After a library of peptide catalysts was screened, 21 was observed to greatly enhance the selectivity of Z6-thionocarbonate (7: 8 = 1:21) on a 500 mg scale. In order to achieve selectivity for G6-OH, new catalysts were designed based on X-ray crystallographic data of a dAla–dAla-containing peptide bound to vancomycin. The authors speculated that replacement of the second dAla residue in the catalyst with histidine may put the reactive imidazole closer to G6-OH. This hypothesis led to the discovery of catalyst 22, which enabled highly selective thiocarbonylation of G6-OH (7: 8 = 24:1) on a 500 mg scale. After deoxygenation and global deprotection, novel deoxy-vancomycin derivatives 9 and 10 were obtained.
Scheme 1. (A) Late-Stage Site-Selective Diversification of Vancomycin. (B) MIC Values (μg/mL) for Vancomycin Derivatives. (C) Selected Natural Products for Late-Stage Diversification via Site-Selective Transformation of Reactive Functional Groups.

Inspired by the success of the selective thiocarbonylation reactions, the Miller group further explored the potential utility of their peptide catalysts for the site-selective lipidation of vancomycin (Scheme 1A).26 Both catalysts 21 and 22 retained the same selectivity as before at Z6 (11:12:13 = 0:1:17) and G6 (11:12:13 = 6:1:0), respectively. After a library of peptide catalysts was screened, 23 was observed to enable selective lipidation at the G4 position (11:12:13 = 1:5:0). Various acyl chains with different lengths were installed at G4 using catalyst 23. After global deprotection, novel lipidated vancomycin derivatives 14–20 were obtained. The authors proposed that the major contributing factor leading to the observed selectivity was the specific hydrogen-bonding interactions between 6 and each catalyst, which would put the reactive site of the substrate near the imidazole moiety. The cocrystal structure of a catalyst–substrate complex that was determined in a subsequent mechanistic study was consistent with this hypothesis.31
With these new vancomycin derivatives in hand, the antibiotic activities were measured (Scheme 1B). Compounds 14, 15, and 16 showed increased activity against both vancomycin-sensitive and -resistant bacteria. Among them, G4-lipidated vancomycin 15 was found to be 64 times more reactive against the VanB pathogen than vancomycin. The derivatives with shorter lipid chains at the G4 position (17–20) were less potent than vancomycin, which was consistent with the previous report that vancomycin derivatives with increased lipophilicity had better activity against both sensitive and resistant bacterial strains.32 Although deoxy-vancomycin derivatives showed comparable (9) or lower (10) activity relative to vancomycin, careful analysis revealed that Z6-deoxy-vancomycin 10 existed in two interconvertible conformations at room temperature, which affected its biological activity and led to reduced antibiotic activity compared with vancomycin. This study revealed that Z6-OH may form a hydrogen bond with the C6-amide carbonyl group to maintain the particular conformation of vancomycin, which is essential for its bioactivity.
In addition to the site-selective modification of vancomycin,33 peptide catalysts have also been applied toward the site-selective modification of other complex natural products (Scheme 1C),10,22 including acylation34,35 and deoxygenation36 of erythromycin A, acylation of apoptolidin A,37 and phosphorylation38 and bromination39 of teicoplanin A2-A (see the Supporting Information).
C–H bonds are the most common and inert functional groups in complex natural products. The explosive growth of C–H bond activation chemistry has allowed for the selective functionalization of C–H bonds in the presence of various other functional groups.11,12,23 Because of its abundance and the presence of sterically and electronically diverse C–H bonds, the sesquiterpene (+)-sclareolide (27) has become a commonly used testing platform to validate the application of a wide range of C–H functionalization reactions (Scheme 2).
Scheme 2. (A) Late-Stage Diversification of (+)-Sclareolide (27) via Selective C–H Functionalization. (B) Chemical Environment of (+)-Sclareolide.

The addition of oxygen or nitrogen atoms to bioactive small molecules not only changes their physicochemical properties (e.g., solubility),40 but it also enhances their hydrogen bonding interactions with target proteins.41 Notable advances in selective C–H oxidation have been achieved during the past decade.42,43 For example, when 27 was subjected to oxidation using strong oxidant methyl(trifluoromethyl)dioxirane (TFDO), the C3 oxidized product 28 was preferentially formed along with the C2 oxidized product 29 in a 3.5:1 ratio.44 C3 oxidation was favored presumably due to electronic effects since the C3 position is located further away from the electron-withdrawing lactone group, making the C–H bonds at this position more electron-rich.45 In 2010, the White group developed an iron catalyst, Fe(PDP) 30, that could catalyze highly selective sp3 C–H oxidation reactions.46 When this catalyst was applied toward the oxidation of 27, the ratio of C2:C3 oxidized product was improved to 1.4:1 in 78% overall yield. In 2017, Baran and co-workers reported a C2-selective oxidation of 27 via electrochemical oxidation,44 where reticulated vitreous carbon (RVC) and nickel were used as the anode and cathode, respectively. The oxidized sclareolide 29 was afforded in 47% yield (63% brsm) and with high regioselectivity (C2/C3 = 5.6:1) on a 50 g scale. The applicability of this reaction was further demonstrated by converting 29 to meroterpenoid analogue (+)-oxo-yahazunone (31) in six steps. Furthermore, enzymes have been shown to selectively install a β-hydroxy group at the C3 position of (+)-sclareolide (27).47−49 In 2011, the Fasan group reported that P450BM3 FL#62 II-H8 selectively catalyzes C3 β-hydroxylation to afford 32 in 83% yield (50 mg scale).47 Inspired by this work, Renata and co-workers achieved C3 β-hydroxylation in a scalable manner using variant P450BM3 MeRO1 (1857 V328A).49 The ability to procure 32 in gram quantities enabled the synthesis of five 3-OH α-pyrone meroterpenoids in 6–7 steps. In addition to the introduction of oxygen functionality to (+)-sclareolide (27), the Baran and Du Bois groups independently reported the selective amination of 27 via rhodium-catalyzed C–H insertion of a nitrene.50,51 When a combination of Du Bois’ catalyst Rh2(esp)2 (34) and sulfonamide was utilized, nitrene insertion occurred specifically at the C2 equatorial position to afford 35 in 60% yield.
Selective halogenation of natural products has also emerged as a popular area of research due to the important pharmacological properties of organo-halide compounds, including numerous halogen-containing drugs.52−54 For example, Groves and co-workers reported a manganese-porphyrin-mediated aliphatic C–H chlorination using sodium hypochlorite as the chlorine source.55 Mn(TMP)Cl (36) catalyzed β-chlorination of (+)-sclareolide (27) preferentially at the C2 position (C2/C3 = 7:1) to afford 37 in 42% yield. This strategy was further applied to fluorination (38 and 39)56 and azidation (40)57 of 27 with similar selectivity toward the C2 and C3 positions.58,59 Highly chemo- and stereoselective halogenation of (+)-sclareolide (27) was further addressed by Alexanian and co-workers via an N-centered radical-mediated process. The functionalization of 27 using N-bromoamide 41 under visible light irradiation afforded the C2 β-bromination product 42 as a single regio- and stereoisomer in 67% isolated yield.60 Then, the Alexanian group collaborated with the Vanderwal group to test the chlorination of 27 using the same strategy to synthesize (+)-chlorolissoclimide (44), an antitumor natural product, which contains a β-chloride at the C2 position. Using N-chloroamide 43 under visible light irradiation, (+)-sclareolide (27) was converted to the desired C2 β-chlorination product 37 in 82% yield with significant selectivity and efficiency on a gram scale.61 Thus, with ample amounts of 37 in hand, the first synthesis of (+)-chlorolissoclimide (44) was realized in nine steps starting from 27. Success in selective halogenation further inspired the Alexanian group to apply the same protocol to C–H xanthylation.62 The xanthylation of 27 occurred with the same regio- and stereoselectivity to give 46 in 55% yield. The latter was further converted to various derivatives via one-step transformations, including deuteration, thiolation, and trifluoromethylthiolation. The selective functionalization of (+)-sclareolide (27) at C2 can be attributed to both electronic and steric effects. The C–H bonds in ring A are more electron-rich than those in rings B and C because they are furthest away from the electron-withdrawing γ-lactone of ring C. The methyl groups at C4 and C6 make the C1 and C3 positions more sterically hindered than C2. The 1,3-diaxial interaction between the C2 axial β hydrogen and the C4 axial methyl group led to the activation of the C2 equatorial α C–H bond by strain release in the transition state (Scheme 2B).50 In contrast, P450 variants are able to selectively achieve C3 β-hydroxylation via specific substrate–enzyme interactions that place the C3 position close to the reactive site in the enzyme.63
As demonstrated by the studies described above that employed vancomycin (5) and (+)-sclareolide (27) as model substrates, many recently developed reactions show great potential for the late-stage regio- and stereoselective functionalization of reactive groups or inert C–H bonds in natural products. In turn, late-stage diversification of abundant natural products can provide novel chiral building blocks for the efficient synthesis of complex, bioactive molecules.
Lead Modification
In order to develop new therapeutic agents, the generation of drug or drug candidate analogues is essential for addressing problems associated with suboptimal bioactivity and pharmacological properties of existing small-molecule therapies. The newly developed strategies for site-selective functionalization of inert C–H bonds open the door to access diverse drug derivatives that may be inaccessible by traditional methods or de novo synthesis.
Artemisinin (3) was first isolated in 1971 from a traditional Chinese medicine.64,65 Artemisinin (3) and its derivatives (47, 48, 49) are recommended by the World Health Organization (WHO) as the most effective drugs for the treatment of malaria (Scheme 3A).66 Because of the short half-life of these compounds in vivo as well as emerging drug resistance,67 artemisinins are often used in combination with one or two long-acting drugs with different modes of action, a treatment paradigm referred to as artemisinin-based combination therapy (ACT).68 Therefore, new drugs are needed to address the aforementioned pharmacological limitations. Synthetic efforts toward development of improved bioactive molecules have mainly focused on the lactone ring (C9 and C10), the modification of which has delivered successful drugs such as dihydroartemisinin (47), artemether (48), and artesunate (49).66 The White group achieved the C6 oxidized product 50 in 54% yield by employing Fe(PDP) 30 without disrupting the sensitive endoperoxide moiety (Scheme 3B).69 The observed selectivity is controlled by the innate electronic properties of artemisinin, where the tertiary C–H bond at C6 is remote from the electron-withdrawing lactone, acetal, and endoperoxide. The bulkier catalyst 51 can alter the inherent substrate selectivity to favor oxidation at the electron-rich and less sterically hindered C7 position to afford the C7 ketone 52 in 52% yield.70 In 2012, Fasan and co-workers reported P450BM3 FL#62 variants that could catalyze C7 and C6a hydroxylation.71 These two positions had remained inaccessible with respect to hydroxylation using iron-based catalysts69,70 or via microbial transformation.72,73 Following site-saturation mutagenesis of the P450BM3 FL#62 active site, three efficient mutants (X-E12, II-H10, and IV-H4) were identified that catalyzed hydroxylation of C6a (53), C7(R) (54), and C7(S) (55) with 94%, 100%, and 100% selectivity, respectively, with over 90% isolated yield on a 100 mg scale (Scheme 3B). 55 was further converted into 7(R)-fluoroartemether (56) and 7(R)-fluoroartesunate (57) in three steps.
Scheme 3. (A) (+)-Artemisinin Based Antimalaria Drugs. (B) Modification of (+)-Artemisinin (3) via Late-Stage Diversification. (C) Phase I and Phase II Metabolism of Artemisinins. (D) Selected Examples of Artemisinin Analogues with Improved Metabolic Stability.
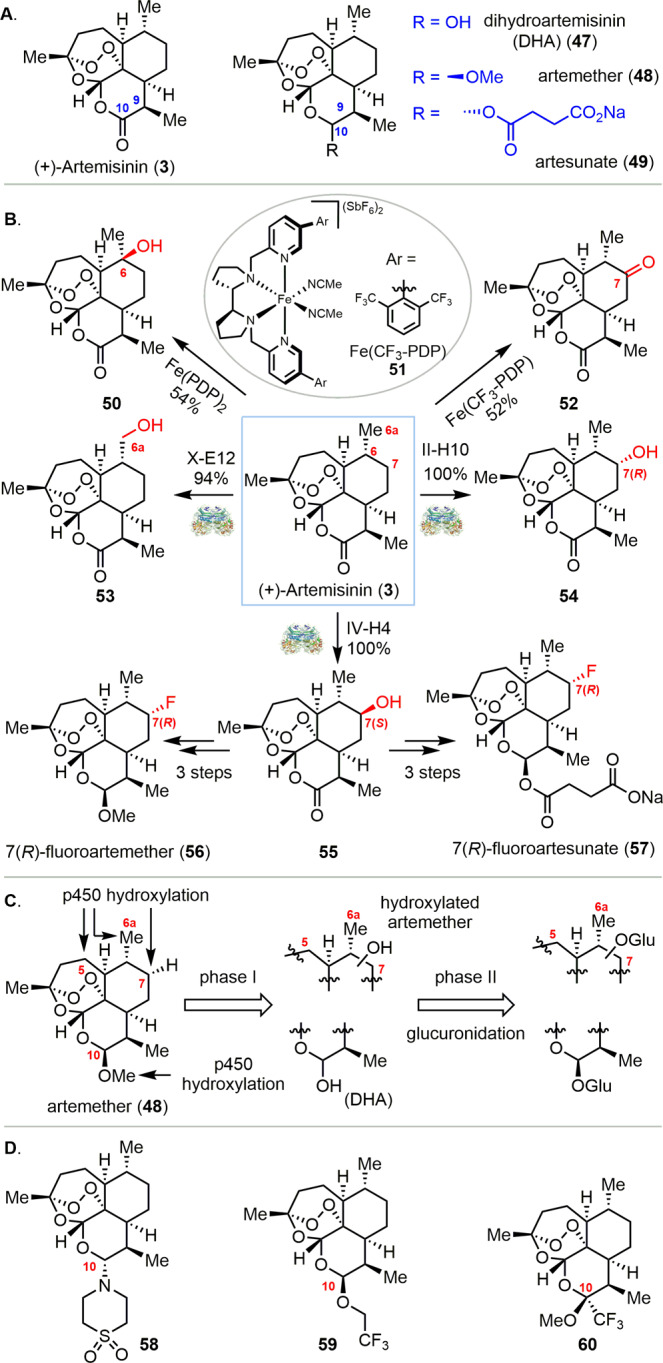
Phase I metabolism of artemisinin-based drugs (artemisinins) mainly targets the C10 methoxy group and the C5, C6a, and C7 carbons through cytochrome P450-mediated hydroxylation to afford dihydroartemisinin (DHA) and hydroxylated artemisinins (Scheme 3C).74,75 These oxidations are followed by rapid glucuronidation and excretion, resulting in a short half-life for artemisinins in vivo. Significant efforts have been aimed toward developing new artemisinin analogues to improve their metabolic stability, with synthetic strategies having primarily focused on generating C10-substituted analogues. These efforts have afforded a C10-alkylamino-artemisinin, artemisone 58, with improved antimalarial activity and metabolic stability achieved by blocking phase I metabolism at the C10 position (Scheme 3D).76 Fluorine substitution at metabolically labile sites can prevent unwanted oxidation, leading to increased bioavailability due to the high chemical stability of the C–F bond. Fluorination can also slow down or block metabolism at adjacent sites due to inductive effects.53 Several such fluorinated artemisinin derivatives have exhibited increased bioactivity in vivo(77) (e.g., 59(78) and 60;79 (Scheme 3D)). The 7(R) C–F bonds in 56 and 57 may also prevent phase I metabolic transformation at the C7 β hydrogen, thereby extending the half-life of these potential drug molecules.75 Meanwhile, generation of 53 and 54 on preparative scales could allow for access to new compounds in which bioactivity is maintained, but metabolic processing is minimized. These artemisinin analogues could also be used to study the properties of hydroxylated artemisinin metabolites.80
In another case, the Herzon group applied hydroxyl-directed iridium-catalyzed C–H silylation toward the late-stage diversification of (+)-pleuromutilin (61) (Scheme 4A).81 (+)-Pleuromutilin (61) acts as an effective antibiotic for the treatment of Gram-positive bacterial infections via binding to the peptidyl transferase center of bacterial ribosomes, inhibiting protein synthesis. This unique mode of action has curtailed the emergence of pleuromutilin-resistant bacterial strains.82 Surprisingly, 12-epi-mutilin derivatives (e.g., 65) show significant broad-spectrum activity against Gram-negative and drug resistant bacteria.83 To overcome antibiotic resistance and develop new anti-Gram-negative agents, novel analogues of (+)-pleuromutilin are required for further evaluation. A number of pleuromutilin analogous were prepared by direct functionalization of the C22 hydroxyl group by sulfonylation followed by standard SN2 substitution (Scheme 4A).83,84 These analogues included the clinically used drug retapamulin (62), lefamulin (63), which is in phase III clinical trials, and the veterinary agent tiamulin (64).
Scheme 4. (A) Pleuromutilin-Based Antibiotics. (B) Modification of (+)-Pleuromutilin (61) via Late-Stage Diversification.

Recently, Hartwig and co-workers reported a strategy for aliphatic C–H hydroxylation using a free alcohol as a directing group.85−87 Silylation of alcohol 66 generated silyl ether 67, with this step followed by iridium- or rhodium-catalyzed intramolecular silylation of alkyl C–H bonds to afford oxasilolane intermediate 68. The latter then underwent Tamao-Fleming oxidation to yield 1,3- or 1,4-diol 69. Except for the C11 or C14 hydroxyl groups, the absence of reactive functional groups in the core structure of pleuromutilin led to only limited success in directly functionalizing its tricyclic skeleton, including C7 oxidation,46,88 C7,51 C13,89 and C1690 amination, and C20 deuteration91 (see the Supporting Information). Using Hartwig’s iridium-catalyzed C–H silylation/oxidation reaction directed by the native C11 or C14 hydroxyl group, the Herzon group was able to achieve C16- (70, 71), C17- (72, 73), and C18- (74) monooxidized derivatives, C15/C16- (75) and C17/C18- (76) dioxidized products, and the C6-normethyl product (77). Moreover, four amine derivatives (78–81) were prepared from the C17-oxidized product (73) (Scheme 4B). This late-stage modification strategy enabled efficient access to these analogues for assessment of structure–activity relationships that had not been extensively investigated before.46,51,88−91
As exemplified by the natural product drugs artemisinin and pleuromutilin, late-stage diversification using state-of-the-art methods can enable rapid preparation of derivatives that would remain inaccessible using conventional chemical synthesis.92−95 Thus, this methodology offers a new opportunity to investigate novel bioactive lead compounds to address pharmacological limitations for further drug development.
Target Identification
During the development of drug candidates, the target identification process is essential for determining the binding target(s) and mode(s) of action of bioactive molecules. The discovery process could unveil new target proteins and molecular pathways, which might further promote the advancement of first-in-class drug discovery.96,97 The target identification process often requires natural product-derived chemical probes, which must retain activities comparable to those of the parent compounds. Conventional functionalization of natural products mostly depends on the pre-existing reactive sites present in the native structure, which are often required for biological activity. Recent developments in the direct functionalization of inert C–H bonds in natural products provide the opportunity to access bioactive chemical probes to explore the cellular targets of the parent compounds.
An elegant example of using late-stage C–H functionalization for the synthesis of bioactive probes to facilitate target identification was reported by the Romo group.98 Eupalmerin acetate (EuPA) (82), a cembranolide diterpene, displays promising anticancer activity against several cancer cell lines (Scheme 5A). In order to identify the functional target protein(s) of EuPA to explore its potential as a therapeutic agent, an effective natural product-based chemical probe is required. Synthesis of the corresponding probe via conventional chemical transformations is very challenging due to the limited number of reactive functional group handles within the EuPA skeleton. Romo and co-workers applied rhodium-catalyzed selective C–H amination to EuPA to obtain probe EuPAyne (84) in one step as a 1:1 diastereomeric mixture at the macrocyclic allylic position without affecting the bioactive enone double bond, which may form a covalent bond with nucleophilic residues of the target protein(s). The antiproliferative activity of EuPAyne (84) was only slightly lower than that of EuPA (82) in HL 60 cells (IC50 = 3.0 μM for EuPA versus 14 μM for EuPAyne), presumably due to the installation of the alkyne handle at a remote position relative to the functional enone moiety. The fact that probe EuPAyne (84) retained most of its activity suggested that it could be used to identify the cellular target(s) of natural product EuPA (82). An initial gel proteomic profiling study and competitive ABPPSILAC enabled quantitative proteome analysis to identify three high-affinity targets: cancer cell proliferation related protein Derlin-1 (DERL1), cancer cell overexpressed cytochrome b5 type B (CYB5B), and thromboxane A synthase (TBXAS1) (Scheme 5B). This result suggested that EuPA (82) may have multiple modes of action against cancer cells. Competition experiments in the overexpressed 293T cells showed that these target proteins interacted with EuPAyne (84) selectively, which indicated that EuPAyne (84) may serve as an effective probe to further investigate the biological roles of these proteins in cancer cell proliferation.
Scheme 5. (A) Chemical Probe Synthesis of Eupalmerin Acetate (EuPA) (82). (B) Target Identification of Anticancer Natural Product Eupalmerin Acetate (EuPA) (82).

This study demonstrates how late-stage diversification of natural products via selective C–H functionalization provides a rapid method for the synthesis of natural product-based probes to enable the identification of potential cellular targets. More and more selective transformations,99 especially selective C–H functionalization reactions, that continue to emerge are anticipated to enable the efficient synthesis of bioactive probes, even for natural products that lack reactive functional group handles.
Biosynthetic Implications
Key chemical transformations that occur in natural product biosynthetic pathways are widely mimicked by synthetic chemists to construct complex molecules.100,101 Moreover, such biomimetic syntheses provide insight into how molecules are formed in nature, which can help to confirm the originally proposed biogenetic hypothesis.102,103
Isodon diterpenoids, a large family of terpenoid natural products,104,105 are biosynthetically derived from geranylgeranyl-diphosphate (GGPP) via series terpenoid cyclase-catalyzed carbocationic rearrangements.106,107 According to the initial biosynthetic hypothesis, jungermannenone diterpenoids (86) are derived from ent-kaurane diterpenoids (85) via two possible carbocationic rearrangement pathways (Scheme 6A).108,109 During the synthesis of complex Isodon diterpenoids by Lei and co-workers,110,111 initial attempts to convert ent-kaurane-type skeletons into jungermannenone-type skeletons via carbocationic rearrangement on a model substrate were unsuccessful. Interconversion between ent-kauranes and jungermannenones was ultimately achieved via late-stage photochemical rearrangement of the bicyclo[3.2.1]octene moiety (Scheme 6B). Upon 254 nm UV light irradiation, ent-kaurane-type analogue 89 was directly converted to (−)-jungermannenone C (90) in 58% yield along with 28% recovered 89. When (−)-jungermannenone C (90) was irradiated with 365 nm UV light, 89 was generated in 21% yield along with 71% recovered (−)-90. This photoinduced skeletal rearrangement may have proceeded via photochemical 1,3-acyl migration of the β,γ-unsaturated ketone112,113 involving a caged radical pair (91) or a biradical intermediate (92).114−118 The interconversion between 89 and (−)-jungermannenone C (90) as well as between (+)-12-hydroxy-1α,6α-diacetoxy-ent-kaura-9(11),16-dien-15-one (93) and (−)-1α,6α-diacetoxyjungermannenone C (94) also occurred smoothly under sunlight, which indicated that this photoinduced rearrangement is likely involved in the biosynthesis of ent-kauranes and jungermannenones. Lei and co-worker further applied this late-stage photochemical rearrangement to other ent-kaurane- and jungermannenone-type diterpenoids (95–100) as well as complex sesquiterpenoids (101–106), with the results showing that all of these substrates could smoothly undergo photochemical rearrangement. These studies showed that this late-stage photoradical process is very likely involved in natural product biosynthetic pathways, which may further inspire organic chemists to design new strategies for complex natural product synthesis and late-stage functionalization.
Scheme 6. (A) Biogenetic Hypothesis of Jungermannenone-Type Diterpenoids. (B) Late-Stage Photoinduced Skeleton Rearrangements of Terpenoids.

Protein–Protein Interactions
The important role of natural products in regulating protein–protein interactions has long been recognized.119 One prominent example is FK506 (tacrolimus, 107), an immunosuppressive macrolide that specifically recruits immunophilin FKBP and calcineurin to form a ternary complex.120,121 Elucidation of its mechanism-of-action not only represented a landmark discovery in the field of chemical biology,122 but it also led to the development of chemical inducers of dimerization (CIDs).123,124 Specifically, the presence of a terminal olefin in FK506 (107) (C37–C38 alkene) has enabled the facile late-stage derivatization of this natural product (Scheme 7). A number of synthetic FK1012s (e.g., 108) have been prepared, eliminating the inhibitory activity of the parent compound toward calcineurin while maintaining its high affinity for FKBP12. Consequently, dimerization of fusion proteins equipped with the FKBP12 domain can be artificially regulated with precise temporal resolution in a dose-dependent, small-molecule controlled manner, which has proved useful in the investigation of various signaling pathways. FK506 heterodimers have also been reported.125 Most recently, the Shokat group developed bifunctional molecules (e.g., 109) that recruit the oncogenic mutant K-Ras protein to intracellularly abundant FKBP12, demonstrating a promising strategy for interfering with Ras activity and blocking its downstream signaling pathway.126
Scheme 7. Modulating Protein–Protein Interactions by Late-Stage Derivatization of FK506.

Summary and Outlook
Although direct functionalization of natural products at a late stage is challenging due to their inherent structural complexity and diversity, many new synthetic methodologies have been developed for late-stage diversification via selective functionalization of reactive functional groups and C–H bonds. Recent development of site-selective reactions has enabled late-stage modification of natural product-based drugs to access various analogues for facile assessment of structure–activity relationships. Direct and selective C–H functionalization makes the synthesis of bioactive chemical probes more straightforward and efficient than traditional methods. In addition, the information acquired from studies that employ late-stage diversification strategies may also provide insight into natural product biosynthetic relationships.
However, the broader application of late-stage diversification of natural products is still limited by currently available synthetic methods. Accordingly, additional efforts should be directed toward enriching the current chemical reaction toolbox that enables effective diversification of highly functionalized molecules. Current site-selective transformations, such as site-selective C–H bond functionalization, mainly depend on the intrinsic reactivity properties of the natural product itself, which are dictated by the sterics and electronics of particular sites on the molecule. The development of nondirected,127 catalyst-controlled strategies rather than substrate-controlled site-selective synthetic methodologies is critical for overriding the innate reactivity of the substrate in order to access diverse sites on natural products. With respect to new catalyst development, enzymes have a great potential to achieve diverse site selectivity. Because nature has evolved various enzymes, such as cytochromes P450, to selectively functionalize particular sites on natural product scaffolds via specific substrate–enzyme interactions,128 discovery of new enzymes and their variants63 offers a new possibility in natural product diversification. Because of the limitations of synthetic methods, late-stage diversification of natural products in drug discovery has not been widely investigated. With the enrichment of new catalysts and reactions, efficient late-stage diversification of natural products will afford sufficient analogues and bioactive probes to facilitate chemical biological studies aimed toward addressing existing biological problems. Indeed, it is urgent to deepen our understanding of the interactions between natural products and their target proteins. The flexible late-stage modification could potentially leverage this knowledge to achieve the powerful chemically induced proximity,129 including the development of proteolysis-targeting chimeras (PROTACs).130 Another promising application would be in the “bump-and-hole” approach, which aims to elucidate the function of a specific gene product.131 The development of versatile substrate analogues is essential for the successful reengineering of the protein–small molecule interface.
To fully explore the potential utility of late-stage diversification of natural products, collaborations among diverse research fields, such as synthetic chemistry, medicinal chemistry, and chemical biology, are needed. With the emergence of new methods, new opportunities and applications will appear. We anticipate that late-stage diversification of natural products will continue to stimulate the emergence of new applications of nature products in drug discovery and biomedical research.
Acknowledgments
We thank Dr. Houhua Li and Dr. Hiuchun Lam for helpful discussions and Dr. Matthew D. Demars for proofreading the manuscript. Financial support from the National Natural Science Foundation of China Grant (21625201, 21961142010, 21661140001, 91853202, and 21521003), the National Key Research and Development Program of China (2017YFA0505200), and the Beijing Outstanding Young Scientist Program (BJJWZYJH01201910001001) is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.9b00916.
Scheme S1. Late-stage selective bromination of vancimycin. Scheme S2. (A) Late-stage selective acylation of erythromycin A. (B) Late-stage selective deoxygenation of erythromycin A. Scheme S3. Late-stage selective acylation of apoptolidin A. Scheme S4. Late-stage selective acylation of teicoplanin A2-2. Scheme S5. Late-stage diversification of thiostrepton via Rh-catalyzed site- and stereoselective arylation. Scheme S6. Late-stage diversification of thiostrepton via site-selective C–H amidation. Scheme S7. Late-stage diversification of betulin and betulinic acid via C–H oxidation. Scheme S8. Late-stage diversification of pleuromutilin skeleton. (A) Microbial oxidation. (B) C7 oxidation and C16 amination. (C) C7 amination. (D) C13 amination. (E) C20 deuteration. Scheme S9. Late-stage diversification of pleuromutilin skeleton via ring system distortion (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Nicolaou K. C.; Montagnon T.. Molecules That Changed the World; Wiley-VCH, 2008. [Google Scholar]
- Corey E. J.; Kürti L.; Czako B.. Molecules and Medicine; Wiley-VCH, 2007. [Google Scholar]
- Newhouse T.; Baran P. S.; Hoffmann R. W. The economies of synthesis. Chem. Soc. Rev. 2009, 38, 3010–3021. 10.1039/b821200g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehn F. E.; Carter G. T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discovery 2005, 4, 206–220. 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Paddon C. J.; Keasling J. D. Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 2014, 12, 355–367. 10.1038/nrmicro3240. [DOI] [PubMed] [Google Scholar]
- Wilson R. M.; Danishefsky S. J. Small Molecule Natural Products in the Discovery of Therapeutic Agents: The Synthesis Connection. J. Org. Chem. 2006, 71, 8329–8351. 10.1021/jo0610053. [DOI] [PubMed] [Google Scholar]
- Danishefsky S. J. On the potential of natural products in the discovery of pharma leads: A case for reassessment. Nat. Prod. Rep. 2010, 27, 1114–1116. 10.1039/c003211p. [DOI] [PubMed] [Google Scholar]
- Mahatthananchai J.; Dumas A. M.; Bode J. W. Catalytic Selective Synthesis. Angew. Chem., Int. Ed. 2012, 51, 10954–10990. 10.1002/anie.201201787. [DOI] [PubMed] [Google Scholar]
- Giuliano M. W.; Miller S. J. Site-Selective Reactions with Peptide Based Catalysts. Top. Curr. Chem. 2015, 372, 157–202. 10.1007/128_2015_653. [DOI] [PubMed] [Google Scholar]
- Godula K.; Sames D. C-H Bond Functionalization in Complex Organic Synthesis. Science 2006, 312, 67–72. 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F. Evolution of C-H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc. 2016, 138, 2–24. 10.1021/jacs.5b08707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skubi K. L.; Blum T. R.; Yoon T. P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida J.; Kataoka K.; Horcajada R.; Nagaki A. Modern strategies in electroorganic synthesis. Chem. Rev. 2008, 108, 2265–2299. 10.1021/cr0680843. [DOI] [PubMed] [Google Scholar]
- Horn E. J.; Rosen B. R.; Baran P. S. Synthetic Organic Electrochemistry: An Enabling and Innately Sustainable Method. ACS Cent. Sci. 2016, 2, 302–308. 10.1021/acscentsci.6b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkovic S. J.; Hammes-Schiffer S. A Perspective on Enzyme Catalysis. Science 2003, 301, 1196–1202. 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- Lewis J. C.; Coelho P. S.; Arnold F. H. Enzymatic Functionalization of Carbon-Hydrogen Bonds. Chem. Soc. Rev. 2011, 40, 2003–2021. 10.1039/C0CS00067A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwizer F.; Okamoto Y.; Heinisch T.; Gu Y.; Pellizzoni M. M.; Lebrun V.; Reuter R.; Köhler V.; Lewis J. C.; Ward T. R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. 10.1021/acs.chemrev.7b00014. [DOI] [PubMed] [Google Scholar]
- Morrison K. C.; Hergenrother P. J. Natural products as starting points for the synthesis of complex and diverse compounds. Nat. Prod. Rep. 2014, 31, 6–14. 10.1039/C3NP70063A. [DOI] [PubMed] [Google Scholar]
- Robles O.; Romo D. Chemo- and site-selective derivatizations of natural products enabling biological studies. Nat. Prod. Rep. 2014, 31, 318–334. 10.1039/C3NP70087A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugrue C. R.; Miller S. J. Applications of Nonenzymatic Catalysts to the Alteration of Natural Products. Chem. Rev. 2017, 117, 11894–11951. 10.1021/acs.chemrev.7b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimov O. R. R.; Hartwig J. F. Transition-Metal-Catalyzed Selective Functionalization of C(sp3)-H Bonds in Natural Products. Angew. Chem., Int. Ed. 2018, 57, 4234–4241. 10.1002/anie.201710330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessner N. D. P450 Monooxygenases Enable Rapid Late-Stage Diversification of Natural Products via C-H Bond Activation. ChemCatChem 2019, 11, 2226–2242. 10.1002/cctc.201801829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler B. S.; Laemmerhold K. M.; Miller S. J. Catalytic Site-Selective Thiocarbonylations and Deoxygenations of Vancomycin Reveal Hydroxyl-Dependent Conformational Effects. J. Am. Chem. Soc. 2012, 134, 9755–9761. 10.1021/ja302692j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoganathan S.; Miller S. J. Structure Diversification of Vancomycin through Peptide-Catalyzed, Site-Selective Lipidation: A Catalysis-Based Approach To Combat Glycopeptide-Resistant Pathogens. J. Med. Chem. 2015, 58, 2367–2377. 10.1021/jm501872s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclercq R.; Derlot E.; Duval J.; Courvalin P. Plasmid-Mediated Resistance to Vancomycin and Teicoplanin in Enterococcus Faecium. N. Engl. J. Med. 1988, 319, 157–161. 10.1056/NEJM198807213190307. [DOI] [PubMed] [Google Scholar]
- Pootoolal J.; Neu J.; Wright G. D. Glycopeptide antibiotic resistance. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 381–408. 10.1146/annurev.pharmtox.42.091601.142813. [DOI] [PubMed] [Google Scholar]
- Ashford P.-A.; Bew S. P. Recent advances in the synthesis of new glycopeptide antibiotics. Chem. Soc. Rev. 2012, 41, 957–978. 10.1039/C1CS15125H. [DOI] [PubMed] [Google Scholar]
- Okano A.; Isley N. A.; Boger D. L. Total Syntheses of Vancomycin-Related Glycopeptide Antibiotics and Key Analogues. Chem. Rev. 2017, 117, 11952–11993. 10.1021/acs.chemrev.6b00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.; Le B. V.; Hajare H. S.; Baxter R. H. G.; Miller S. J. X-Ray Crystal Structure of Teicoplanin A2-2 Bound to a Catalytic Peptide Sequence via the Carrier Protein Strategy. J. Org. Chem. 2014, 79, 8550–8556. 10.1021/jo501625f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerns R.; Dong S. D.; Fukuzawa S.; Carbeck J.; Kohler J.; Silver L.; Kahne D. The role of hydrophobic substituents in the biological activity of glycopeptide antibiotics. J. Am. Chem. Soc. 2000, 122, 12608–12609. 10.1021/ja0027665. [DOI] [Google Scholar]
- Pathak T. P.; Miller S. J. Site-Selective Bromination of Vancomycin. J. Am. Chem. Soc. 2012, 134, 6120–6123. 10.1021/ja301566t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis C. A.; Miller S. J. Site-Selective Derivatization and Remodeling of Erythromycin A by Using Simple Peptide-Based Chiral Catalysts. Angew. Chem., Int. Ed. 2006, 45, 5616–5619. 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- Lewis C. A.; Merkel J.; Miller S. J. Catalytic Site-Selective Synthesis and Evaluation of a Series of Erythromycin Analogs. Bioorg. Med. Chem. Lett. 2008, 18, 6007–6011. 10.1016/j.bmcl.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan P. A.; Miller S. J. An Approach to the Site-Selective Deoxygenation of Hydroxy Groups Based on Catalytic Phosphoramidite Transfer. Angew. Chem., Int. Ed. 2012, 51, 2907–2911. 10.1002/anie.201109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis C. A.; Longcore K. E.; Miller S. J.; Wender P. A. An Approach to the Site-Selective Diversification of Apoptolidin A with Peptide-Based Catalysts. J. Nat. Prod. 2009, 72, 1864–1869. 10.1021/np9004932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.; Miller S. J. Asymmetric Catalysis at a Distance: Catalytic, Site-Selective Phosphorylation of Teicoplanin. J. Am. Chem. Soc. 2013, 135, 12414–12421. 10.1021/ja406067v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak T. P.; Miller S. J. Chemical Tailoring of Teicoplanin with Site-Selective Reactions. J. Am. Chem. Soc. 2013, 135, 8415–8422. 10.1021/ja4038998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an example of the hydroxyl group increased solubility of bioactive compounds, see:Leu Y.-L.; Chen C.-S.; Wu Y.-J.; Chern J.-W. Benzyl Ether-Linked Glucuronide Derivative of 10-Hydroxycamptothecin Designed for Selective Camptothecin-Based Anticancer Therapy. J. Med. Chem. 2008, 51, 1740–1746. 10.1021/jm701151c. [DOI] [PubMed] [Google Scholar]
- For an example of the hydroxyl group increased anti-HIV activity via hydrogen bonding interaction, see:Xu L.; Liu H.; Hong A.; Vivian R.; Murray B. P.; Callebaut C.; Choi Y. C.; Lee M. S.; Chau J.; Tsai L. K.; Stray K. M.; Strickley R. G.; Wang J.; Tong L.; Swaminathan S.; Rhodes G. R.; Desai M. C. Structure–activity relationships of diamine inhibitors of cytochrome P450 (CYP) 3A as novel pharmacoenhancers. Part II: P2/P3 region and discovery of cobicistat (GS-9350. Bioorg. Med. Chem. Lett. 2014, 24, 995–999. 10.1016/j.bmcl.2013.12.057. [DOI] [PubMed] [Google Scholar]
- Newhouse T.; Baran P. S. If C-H Bonds Could Talk: Selective C-H Bond Oxidation. Angew. Chem., Int. Ed. 2011, 50, 3362–3374. 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M. C.; Zhao J. Aliphatic C-H Oxidations for Late-Stage Functionalization. J. Am. Chem. Soc. 2018, 140, 13988–14009. 10.1021/jacs.8b05195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata Y.; Yan M.; Liu Z.; Bao D. H.; Chen J.; Starr J. T.; Baran P. S. Scalable, Electrochemical Oxidation of Unactivated C-H Bonds. J. Am. Chem. Soc. 2017, 139, 7448–7451. 10.1021/jacs.7b03539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L.; Paton R. S.; Eschenmoser A.; Newhouse T. R.; Baran P. S.; Houk K. N. Enhanced Reactivity in Dioxirane C–H Oxidations via Strain Release: A Computational and Experimental Study. J. Org. Chem. 2013, 78, 4037–4048. 10.1021/jo400350v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. S.; White M. C. Combined Effects on Selectivity in Fe Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- Zhang K.; El Damaty S.; Fasan R. P450 Fingerprinting Method for Rapid Discovery of Terpene Hydroxylating P450 Catalysts with Diversified Regioselectivity. J. Am. Chem. Soc. 2011, 133, 3242–3245. 10.1021/ja109590h. [DOI] [PubMed] [Google Scholar]
- Hall E. A.; Sarkar M. R.; Lee J. H. Z.; Munday S. D.; Bell S. G. Improving the Monooxygenase Activity and the Regio- and Stereoselectivity of Terpenoid Hydroxylation Using Ester Directing Groups. ACS Catal. 2016, 6, 6306–6317. 10.1021/acscatal.6b01882. [DOI] [Google Scholar]
- Li J.; Li F.; King-Smith E.; Renata H. Merging chemoenzymatic and radical-based retrosynthetic logic for rapid and modular synthesis of oxidized meroterpenoids. Nat. Chem. 2020, 12, 173–179. 10.1038/s41557-019-0407-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.; Eschenmoser A.; Baran P. S. Strain Release in C-H Bond Activation?. Angew. Chem., Int. Ed. 2009, 48, 9705–9708. 10.1002/anie.200904474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappini N. D.; Mack J. B. C.; Du Bois J. Intermolecular C(sp3)-H Amination of Complex Molecules. Angew. Chem., Int. Ed. 2018, 57, 4956–4959. 10.1002/anie.201713225. [DOI] [PubMed] [Google Scholar]
- Müller K.; Faeh C.; Diederich F. Fluorine in Pharmaceuticals: Looking beyond Intuition. Science 2007, 317, 1881–1886. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Gál B.; Bucher C.; Burns N. Z. Chiral Alkyl Halides: Underexplored Motifs in Medicine. Mar. Drugs 2016, 14, 206. 10.3390/md14110206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Groves J. T. Manganese Porphyrins Catalyze Selective C-H Bond Halogenations. J. Am. Chem. Soc. 2010, 132, 12847–12849. 10.1021/ja105548x. [DOI] [PubMed] [Google Scholar]
- Liu W.; Huang X.; Cheng M.-J.; Nielsen R. J.; Goddard W. A. III; Groves J. T. Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Poyrphyrin. Science 2012, 337, 1322–1325. 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]
- Huang X.; Bergsten T. M.; Groves J. T. Manganese-Catalyzed Laste-Stage Aliphatic C-H Aziation. J. Am. Chem. Soc. 2015, 137, 5300–5303. 10.1021/jacs.5b01983. [DOI] [PubMed] [Google Scholar]
- For a Photocatalytic C3-Fluorination of (+)-sclareolide, see:Halperin S. D.; Fan H.; Chang S.; Martin R. E.; Britton R. A Convenient Photocatalytic Fluorination of Unactivated C-H Bonds. Angew. Chem., Int. Ed. 2014, 53, 4690–4693. 10.1002/anie.201400420. [DOI] [PubMed] [Google Scholar]
- For Fe(OAc)2 catalyzed late-stage azidation of natural products, see:Karimov R. R.; Sharma A.; Hartwig J. F. Late Stage Azidation of Complex Molecules. ACS Cent. Sci. 2016, 2, 715–724. 10.1021/acscentsci.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt V. A.; Quinn R. K.; Brusoe A. T.; Alexanian E. J. Site Selective Aliphatic C–H Bromination Using N-Bromoamides and Visible Light. J. Am. Chem. Soc. 2014, 136, 14389–14392. 10.1021/ja508469u. [DOI] [PubMed] [Google Scholar]
- Quinn R. K.; Könst Z. A.; Michalak S. E.; Schmidt Y.; Szklarski A. R.; Flores A. R.; Nam S.; Horne D. A.; Vanderwal C. D.; Alexanian E. J. Site-Selective Aliphatic C-H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc. 2016, 138, 696–702. 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaplyski W. L.; Na C. G.; Alexanian E. J. C-H Xanthylation: A Synthetic Platform for Alkane Functionalization. J. Am. Chem. Soc. 2016, 138, 13854–13857. 10.1021/jacs.6b09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasan R. Tuning P450 Enzymes as Oxidation Catalysts. ACS Catal. 2012, 2, 647–666. 10.1021/cs300001x. [DOI] [Google Scholar]
- A new sesquiterpene lactone-qinghaosu [in Chinese]. Kexue Tongbao 1977, 3, 142. [Google Scholar]
- Tu Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220. 10.1038/nm.2471. [DOI] [PubMed] [Google Scholar]
- Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization, April 2015. [PubMed]
- Noedl H.; Se Y.; Schaecher K.; Smith B. L.; Socheat D.; Fukuda M. M. Evidence of Artemisinin-Resistant Malaria in Western Cambodia. N. Engl. J. Med. 2008, 359, 2619–2620. 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- Artemisinin Derivatives: Summary of Nonclinical Safety Data Introductory Remarks; World Health Organization, January 24, 2016.
- Chen M. S.; White M. C. A Predictably Selective Aliphatic C-H Hydroxylation Reaction. Science 2007, 318, 783–787. 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- Gormisky P. E.; White M. C. Catalyst-Controlled Aliphatic C– H Oxidations with a Predictive Model for Site-Selectivity. J. Am. Chem. Soc. 2013, 135, 14052–14055. 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Shafer B. M.; Demars M. D. II; Stern H. A.; Fasan R. Controlled Oxidation of Remote sp3 C–H Bonds in Artemisinin via P450 Catalysts with Fine-Tuned Regio- and Stereoselectivity. J. Am. Chem. Soc. 2012, 134, 18695–18704. 10.1021/ja3073462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan J. X.; Zhang Y. X.; Guo H. Z.; Han J.; Ning L. L.; Guo D. A. J. J. Nat. Prod. 2002, 65, 1693–1695. 10.1021/np020113r. [DOI] [PubMed] [Google Scholar]
- Liu J. H.; Chen Y. G.; Yu B. Y.; Chen Y. J. Bioorg. Med. Chem. Lett. 2006, 16, 1909–1912. 10.1016/j.bmcl.2005.12.076. [DOI] [PubMed] [Google Scholar]
- Svensson U. S. H.; Ashton M. Identification of the human cytochrome P450 enzymes involved in the in vitro metabolism of artemisinin. Br. J. Clin. Pharmacol. 1999, 48, 528–535. 10.1046/j.1365-2125.1999.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill P. M.; Posner G. H. J. Med. Chem. 2004, 47, 2945–2964. 10.1021/jm030571c. [DOI] [PubMed] [Google Scholar]
- Haynes R. K.; Fugmann B.; Stetter J.; Rieckmann K.; Heilmann H. D.; Chan H. W.; Cheung M. K.; Lam W. L.; Wong H. N.; Croft S. L.; Vivas L.; Rattray L.; Stewart L.; Peters W.; Robinson B. L.; Edstein M. D.; Kotecka B.; Kyle D. E.; Beckermann B.; Gerisch M.; Radtke M.; Schmuck G.; Steinke W.; Wollborn U.; Schmeer K.; Romer A. Angew. Chem., Int. Ed. 2006, 45, 2082–2088. 10.1002/anie.200503071. [DOI] [PubMed] [Google Scholar]
- Bégué J. P.; Bonnet-Delpon D. Fluoroartemisinins: Metabolically More Stable Antimalarial Artemisinin Derivatives. ChemMedChem 2007, 2, 608–624. 10.1002/cmdc.200600156. [DOI] [PubMed] [Google Scholar]
- Nga T. T. T.; Ménage C.; Bégué J. P.; Bonnet-Delpon D.; Gantier J. C.; Pradines B.; Doury J. C.; Thac T. D. Synthesis and Antimalarial Activities of Fluoroalkyl Derivatives of Dihydroartemisinin. J. Med. Chem. 1998, 41, 4101–4108. 10.1021/jm9810147. [DOI] [PubMed] [Google Scholar]
- Magueur G.; Crousse B.; Charneau S.; Grellier P.; Bégué J. P.; Bonnet-Delpon D. Fluoroartemisinin: Trifluoromethyl Analogues of Artemether and Artesunate. J. Med. Chem. 2004, 47, 2694–2699. 10.1021/jm0310333. [DOI] [PubMed] [Google Scholar]
- Kumar Ramu K.; Baker J. K. Synthesis, Characterization, and Antimalarial Activity of the Glucuronides of the Hydroxylated Metabolites of Arteether. J. Med. Chem. 1995, 38, 1911–1921. 10.1021/jm00011a011. [DOI] [PubMed] [Google Scholar]
- Ma X.; Kucera R.; Goethe O. F.; Murphy K. S.; Herzon S. B. Directed C–H Bond Oxidation of (+)-Pleuromutilin. J. Org. Chem. 2018, 83, 6843–6892. 10.1021/acs.joc.8b00462. [DOI] [PubMed] [Google Scholar]
- Paukner S.; Riedl R. Pleuromutilins: Potent Drugs for Resistant Bugs-Mode of Action and Resistance. Cold Spring Harbor Perspect. Med. 2017, 7, a027110 10.1101/cshperspect.a027110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goethe O.; Heuer A.; Ma X.; Wang Z.; Herzon S. B. Antibacterial properties and clinical potential of pleuromutilins. Nat. Prod. Rep. 2019, 36, 220–247. 10.1039/C8NP00042E. [DOI] [PubMed] [Google Scholar]
- Fazakerley N. J.; Procter D. J. Synthesis and synthetic chemistry of pleuromutilin. Tetrahedron 2014, 70, 6911–6930. 10.1016/j.tet.2014.05.092. [DOI] [Google Scholar]
- Simmons E. M.; Hartwig J. F. Catalytic Functionalization of Unactivated Primary C-H Bonds Directed by an Alcohol. Nature 2012, 483, 70–73. 10.1038/nature10785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Driess M.; Hartwig J. F. Iridium-Catalyzed Regioselective Silylation of Secondary Alkyl C–H Bonds for the Synthesis of 1,3-Diols. J. Am. Chem. Soc. 2014, 136, 6586–6589. 10.1021/ja5026479. [DOI] [PubMed] [Google Scholar]
- Karmel C.; Li B.; Hartwig J. F. Rhodium-Catalyzed Regioselective Silylation of Alkyl C–H Bonds for the Synthesis of 1,4- Diols. J. Am. Chem. Soc. 2018, 140, 1460–1470. 10.1021/jacs.7b11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson R. L.; Matson J. A.; Brzozowski D. B.; LaPorte T. L.; Springer D. M.; Patel R. N. Hydroxylation of Mutilin by Streptomyces griseus and Cunninghamella echinulata. Org. Process Res. Dev. 2002, 6, 482–487. 10.1021/op020028q. [DOI] [Google Scholar]
- Uccello D. P.; Miller S. M.; Dieterich N. A.; Stepan A. F.; Chung S.; Farley K. A.; Samas B.; Chen J.; Montgomery J. I. The Synthesis of C-13 Functionalized Pleuromutilins via C-H Amidation and Subsequent Novel Rearrangement Product. Tetrahedron Lett. 2011, 52, 4247–4251. 10.1016/j.tetlet.2011.06.028. [DOI] [Google Scholar]
- Paradine S. M.; Griffin J. R.; Zhao J.; Petronico A. L.; Miller S. M.; White M. C. A Manganese Catalyst for Highly Reactive yet Chemoselective Intramolecular C(sp3)-H Amination. Nat. Chem. 2015, 7, 987–994. 10.1038/nchem.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Hartwig J. F. Iridium-Catalyzed H/D Exchange at Vinyl Groups without Olefin Isomerization. Angew. Chem., Int. Ed. 2008, 47, 5783–5787. 10.1002/anie.200801992. [DOI] [PubMed] [Google Scholar]
- For a recent example of diversification of pleuromutilin using ring system distortion strategy, See:Llabani E.; Hicklin R. W.; Lee H. Y.; Motika S. E.; Crawford L. A.; Weerapana E.; Hergenrother P. J. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat. Chem. 2019, 11, 521–531. See the Supporting Information 10.1038/s41557-019-0261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For late-stage diversification of thiostrepton via Rh-catalyzed site- and stereoselective arylation, see:Key H. M.; Miller S. J. Site- and Stereoselective Chemical Editing of Thiostrepton by Rh-Catalyzed Conjugate Arylation: New Analogues and Collateral Enantioselective Synthesis of Amino Acids. J. Am. Chem. Soc. 2017, 139, 15460–15466. See the Supporting Information 10.1021/jacs.7b08775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For Late-stage diversification of thiostrepton via site-selective C–H Amidation, see:Scamp R. J.; de Ramon E.; Paulson E. K.; Miller S. C.; Ellman J. A. Co (III)-Catalyzed C–H Amidation of Dehydroalanine for the Site-Selective Structural Diversification of Thiostrepton. Angew. Chem., Int. Ed. 2020, 59, 890–895. See the Supporting Information 10.1002/anie.201911886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For Late-stage diversification of betulin and betulinic acid via C–H oxidation, see:Michaudel Q.; Journot G.; Regueiro-Ren A.; Goswami A.; Guo Z.; Tully T. P.; Zou L.; Ramabhadran R. O.; Houk K. N.; Baran P. S. Improving Physical Properties via C-H Oxidation: Chemical and Enzymatic Approaches. Angew. Chem., Int. Ed. 2014, 53, 12091–12096. See the Supporting Information 10.1002/anie.201407016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenone M.; Dancik V.; Wagner B. K.; Clemons P. A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013, 9, 232–240. 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson E. E. Natural products as chemical probes. ACS Chem. Biol. 2010, 5, 639–653. 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Cisar J. S.; Zhou C.-Y.; Vera B.; Williams H.; Rodríguez A. D.; Cravatt B. F.; Romo D. Simultaneous Structure-Activity Studies and Arming of Natural Products by C–H Amination Reveal Cellular Targets of Eupalmerin Acetate. Nat. Chem. 2013, 5, 510–517. 10.1038/nchem.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the target identification of fumagillol via O-H insertion, see:Chamni S.; He Q. L.; Dang Y.; Bhat S.; Liu J. O.; Romo D. ACS Chem. Biol. 2011, 6, 1175–1181. 10.1021/cb2002686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poupon E.; Nay B.. Biomimetic Organic Synthesis; Wiley-VCH, 2011. [Google Scholar]
- Razzak M.; De Brabander J. K. Lessons and revelations from biomimetic syntheses. Nat. Chem. Biol. 2011, 7, 865–875. 10.1038/nchembio.709. [DOI] [PubMed] [Google Scholar]
- Heathcock C. H. Nature knows best: An amazing reaction cascade is uncovered by design and discovery. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 14323–14327. 10.1073/pnas.93.25.14323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R. M. Natural Products Synthesis: Enabling Tools To Penetrate Nature’s Secrets of Biogenesis and Biomechanism. J. Org. Chem. 2011, 76, 4221–4259. 10.1021/jo2003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.-D.; Huang S.-X.; Han Q.-B. Diterpenoids from Isodon species and their biological activities. Nat. Prod. Rep. 2006, 23, 673–698. 10.1039/b604174d. [DOI] [PubMed] [Google Scholar]
- Liu M.; Wang W.-G.; Sun H.-D.; Pu J.-X. Diterpenoids from Isodon species: an update. Nat. Prod. Rep. 2017, 34, 1090–1140. 10.1039/C7NP00027H. [DOI] [PubMed] [Google Scholar]
- Hong Y. J.; Tantillo D. J. Formation of beyerene, kaurene, trachylobane, and atiserene diterpenes by rearrangements that avoid secondary carbocations. J. Am. Chem. Soc. 2010, 132, 5375–5386. 10.1021/ja9084786. [DOI] [PubMed] [Google Scholar]
- Tantillo D. J. The carbocation continuum in terpene biosynthesis-where are the secondary cations?. Chem. Soc. Rev. 2010, 39, 2847–2854. 10.1039/b917107j. [DOI] [PubMed] [Google Scholar]
- Nagashima F.; Kondoh M.; Fujii M.; Takaoka S.; Watanabe Y.; Asakawa Y. Novel cytotoxic kaurane-type diterpenoids from the New Zealand liverwort Jungermannia species. Tetrahedron 2005, 61, 4531–4544. 10.1016/j.tet.2005.03.010. [DOI] [Google Scholar]
- Li L.-M.; Li G.-Y.; Xiao W.-L.; Zhou Y.; Li S.-H.; Huang S.-X.; Han Q.-B.; Ding L.-S.; Lou L.-G.; Sun H.-D. A new rearranged and a new seco-ent-kaurane diterpenoids from Isodon parvifolius. Tetrahedron Lett. 2006, 47, 5187–5190. 10.1016/j.tetlet.2006.05.025. [DOI] [Google Scholar]
- Hong B.; Liu W.; Wang J.; Wu J.; Kadonaga Y.; Cai P.-J.; Lou H.-X.; Yu Z.-X.; Li H.; Lei X. Photoinduced Skeletal Rearrangements Reveal Radical-Mediated Synthesis of Terpenoids. Chem 2019, 5, 1671–1681. 10.1016/j.chempr.2019.04.023. [DOI] [Google Scholar]
- Wu J.; Kadonaga Y.; Hong B.; Wang J.; Lei X. Enantioselective Total Synthesis of (+)-Jungermatrobrunin A. Angew. Chem., Int. Ed. 2019, 58, 10879–10883. 10.1002/anie.201903682. [DOI] [PubMed] [Google Scholar]
- Paquette L. A.; Meehan G. V. Photorearrangement of β, γ-unsaturated ketones. application to the synthesis of bridged bicyclic ketones. J. Org. Chem. 1969, 34, 450–454. 10.1021/jo01254a041. [DOI] [Google Scholar]
- Houk K. N. The photochemistry and spectroscopy of β, γ-unsaturated carbonyl compounds. Chem. Rev. 1976, 76, 1–74. 10.1021/cr60299a001. [DOI] [Google Scholar]
- Givens R. S.; Chae W. K. Photorearrangements of bicyclo[3.2.l]oct-2-en-7-ones. a substituent effect study. mechanistic studies in photochemistry. 19. J. Am. Chem. Soc. 1978, 100, 6278–6280. 10.1021/ja00487a078. [DOI] [Google Scholar]
- Givens R. S.; Chae W. K.; Matuszewski B. Singlet-triplet reactivity of α, β, γ-unsaturated ketone: mechanistic studies in photochemistry. J. Am. Chem. Soc. 1982, 104, 2456–2466. 10.1021/ja00373a021. [DOI] [Google Scholar]
- Reimann B.; Sadler D. E.; Schaffner K. Gas-phase photochemistry of α, β, γ-unsaturated ketone. Concerted and radical mechanisms of the 1,3-acetyl shift in 1,2-dimethylcyclopent-2-enyl methyl ketone. J. Am. Chem. Soc. 1986, 108, 5527–5530. 10.1021/ja00278a026. [DOI] [Google Scholar]
- Wilsey S.; Bearpark M. J.; Bernardi F.; Olivucci M.; Robb M. A. Mechanism of the oxadi-π-methane and [1,3]-acyl sigmatropic rearrangements of β, γ-enones: a theoretical study. J. Am. Chem. Soc. 1996, 118, 176–184. 10.1021/ja9522802. [DOI] [Google Scholar]
- Xu G.; Elkin M.; Tantillo D. J.; Newhouse T. R.; Maimone T. J. Traversing Biosynthetic Carbocation Landscapes in the Total Synthesis of Andrastin and Terretonin Meroterpenes. Angew. Chem., Int. Ed. 2017, 56, 12498–12502. 10.1002/anie.201705654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milroy L.-G.; Grossmann T. N.; Hennig S.; Brunsveld L.; Ottmann C. N.; Hennig, s.; Brunsveld, l.; Ottmann, C. Modulators of Protein–Protein Interactions. Chem. Rev. 2014, 114, 4695–4748. 10.1021/cr400698c. [DOI] [PubMed] [Google Scholar]
- Liu J.; Farmer J. D.; Lane W. S.; Friedman J.; Weissman I.; Schreiber S. L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. 10.1016/0092-8674(91)90124-H. [DOI] [PubMed] [Google Scholar]
- Griffith J. P.; Kim J. L.; Kim E. E.; Sintchak M. D.; Thomson J. A.; Fitzgibbon M. J.; Fleming M. A.; Caron P. R.; Hsiao K.; Navia M. A. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 1995, 82, 507–522. 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]
- Bierer B. E.; Somers P. K.; Wandless T. J.; Burakoff S. J.; Schreiber S. L. Probing immunosuppressant action with a nonnatural immunophilin ligand. Science 1990, 250, 556–559. 10.1126/science.1700475. [DOI] [PubMed] [Google Scholar]
- Spencer D. M.; Wandless T. J.; Schreiber S. L.; Crabtree G. R. Controlling signal transduction with synthetic ligands. Science 1993, 262, 1019–1024. 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- Fegan A.; White B.; Carlson J. C. T.; Wagner C. R. Chemically Controlled Protein Assembly: Techniques and Applications. Chem. Rev. 2010, 110, 3315–3336. 10.1021/cr8002888. [DOI] [PubMed] [Google Scholar]
- Belshaw P.; Ho S. N.; Crabtree G. R.; Schreiber S. L. Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 4604–4607. 10.1073/pnas.93.10.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Shokat K. M. Bifunctional Small-Molecule Ligands of K-Ras Induce Its Association with Immunophilin Proteins. Angew. Chem., Int. Ed. 2019, 58, 16314–16319. 10.1002/anie.201910124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig J. F.; Larsen M. A. Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities. ACS Cent. Sci. 2016, 2, 281–292. 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro A. W.; Girvan H. M.; McLean K. J. Variations on a (T)heme - Novel Mechanisms, Redox Partners and Catalytic Functions in the Cytochrome P450 Superfamily. Nat. Prod. Rep. 2007, 24, 585–609. 10.1039/B604190F. [DOI] [PubMed] [Google Scholar]
- Stanton B. Z.; Chory E. J.; Crabtree G. R. Chemically induced proximity in biology and medicine. Science 2018, 359, eaao5902 10.1126/science.aao5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira M.; Calabrese M. F.; Bullock A. N.; Crews C. M. Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discovery 2019, 18, 949–963. 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- Islam K. The Bump-and-Hole Tactic: Expanding the Scope of Chemical Genetics. Cell Chem. Biol. 2018, 25, 1171–1184. 10.1016/j.chembiol.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.