

SUMMARY
The tumor microenvironment (TME) is critical for tumor progression. However, the establishment and function of TME remain obscure due to its complex cellular composition. Using a mouse genetic system called Mosaic Analysis with Double Markers (MADM), we delineated TME evolution at single-cell resolution in SHH-activated medulloblastoma that originate from unipotent granule neuron progenitors in the brain. First, we found that astrocytes within the TME (TuAstrocytes) were trans-differentiated from tumor GNPs, which normally never differentiate into astrocytes. Second, we identified that TME-derived IGF1 promotes tumor progression. Third, we uncovered that IGF1 is produced by tumor-associated microglia, in response to IL4 stimulation. Finally, we found that IL4 is secreted by TuAstrocytes. Collectively, our studies reveal an evolutionary process that produces a multi-lateral network within the TME of medulloblastoma: a fraction of tumor cells trans-differentiate into TuAstrocytes, which in turn produce IL4 that stimulates microglia to produce IGF1 to promote tumor progression.
INTRODUCTION
A tumor is a living organ. Cancer cells actively interact with the tumor microenvironment (TME) at multiple levels of complexity for their survival (Balkwill et al., 2012; Hanahan and Coussens, 2012; Quail and Joyce, 2013). First, TME is consisted of multiple cell types, including blood vessels (Chung et al., 2010; Folkman, 1971), fibroblasts (Gascard and Tlsty, 2016; Kalluri, 2016; Shiga et al., 2015), immune cells (Binnewies et al., 2018; de Visser et al., 2006; Gajewski et al., 2013), and other tissue-resident cells (Tabuso et al., 2017). Second, TME cells crosstalk among themselves to form a supportive environment for tumor cells (DeNardo et al., 2009; Gabrilovich et al., 2012; Kumar et al., 2018; Stockmann et al., 2014). Finally, TME dynamically co-evolves with tumor cells from initiation, to progression, and to full malignancy (Polyak et al., 2009; Weinberg, 2008). TME can even be reprogrammed during cancer therapy and contribute to therapeutic resistance (Ahmed and Haass, 2018; Shee et al., 2018; Straussman et al., 2012; Su et al., 2018; Sun, 2016). Therefore, it is imperative to deconstruct the complexity of TME: how a heterogeneous TME is established, and how the crosstalk between TME cells supports tumor progression.
While histopathological analyses of patient samples can reveal TME composition and correlation with prognosis, animal models are needed to delineate the origin of each TME components and to provide mechanistic insights into TME functions (Arina et al., 2016; Ozdemir et al., 2014; Wyckoff et al., 2004). To maximize the temporospatial resolution of TME analysis, our lab developed a mouse genetic system termed Mosaic Analysis with Double Markers (MADM) (Zong et al., 2005). From a heterozygous mouse, MADM relies on Cre/loxP guided inter-chromosomal mitotic recombination to generate sparse, GFP-labeled cells homozygous-null for a given tumor suppressor gene (TSG) and RFP-labeled wildtype sibling cells in somatic tissues. The rarity of TSG-null cells, resulting from low-efficiency of recombination between two homologous chromosomes, not only mimics the clonal origin of cancer in human patients but also provides unprecedented resolution for phenotypic analysis; and the immediate labeling of TSG-mutant cells enables one to track them throughout the entire tumorigenic process (Liu et al., 2011). Careful choice of a Cre transgene that specifically expresses in tumor but not TME cells would allow one to investigate the recruitment, activation, and organization of non-labeled TME cells in relation to GFP-labeled tumor cells from tumor initiation to full malignancy.
Medulloblastoma is the most common malignant pediatric brain tumor arising in the developing cerebellum (Gilbertson and Ellison, 2008), and the SHH-activated subtype contains multiple TME cell types (Bailey and Cushing, 1925; Wright, 1910), including neurons, endothelial cells, microglia/macrophages, and astrocytes (Supplemental Fig. 1A,B). Intriguingly, while medulloblastoma cells proliferate uncontrollably in vivo, propagating them in vitro has been very difficult, requiring serial transplantation in the brains of immunodeficient mice to expand human medulloblastoma cells (Shu et al., 2008). A potential explanation for this apparent paradox is that medulloblastoma cells rely on supportive signals from TME cells that are absent in vitro. In fact, the presence of an astrocytic component in medulloblastoma has been linked to poor prognosis even though its origin was unclear (Rickert and Paulus, 2005).
In addition to the clinical importance, studying the TME in SHH-subtype of medulloblastoma, which has an ostensibly simple tumor organization, could provide critical proof-of-principle groundwork. Years of research on this tumor type (Goodrich et al., 1997; Schuller et al., 2008; Yang et al., 2008) provide a vast amount of knowledge as well as well-characterized tools and techniques for this cancer type. First, the cell of origin for medulloblastoma, granule neuron precursor (GNP), is a unipotent progenitor that does not give rise to any other cell types but granule neurons (Fig. 1A–C) (Zhang and Goldman, 1996), allowing one to use GNP-specific Math1-Cre (Matei et al., 2005) to mutate and label tumor but not TME cells in a mouse model. Second, tumor GNPs can be specifically labeled by the Math1-GFP reporter transgene (Rose et al., 2009) to facilitate in vivo analysis, and can be highly purified through percoll gradient centrifugation for in vitro analysis (Oliver et al., 2005). Third, genetic mutations contributing to this tumor type have been well studied: while heterozygous Ptch1 mutation leads to upregulation of SHH signaling and significant tumor risks (Goodrich et al., 1997; Mullor et al., 2002; Pietsch et al., 1997), p53 loss results in precipitous survival rate in human patients (Tabori et al., 2010) and shortened latency with full penetrance of tumor formation in mouse models (Wetmore et al., 2001). Finally, the brain is an ‘immune-privileged’ organ in which microglia are the elementary tissue-resident macrophages, while brain-specific astrocytes serve as non-professional immune cells (Burda and Sofroniew, 2014; Colombo and Farina, 2016; Prinz et al., 2017), providing a relatively simple system to decipher the interactions among cell types. As tumor progresses, if blood-derived immune cells infiltrate into the tumor mass as in neurological diseases or brain infections (Harris et al., 2012; Hoglund and Maghazachi, 2014), they can be readily identified for further functional studies. Here we created a MADM-based model for medulloblastoma, with which we discovered an intricate TME network formed through trans-differentiation and multilateral paracrine signaling.
Figure 1. MADM-based medulloblastoma model reveals that some tumor GNPs trans-differentiate into astrocytes.
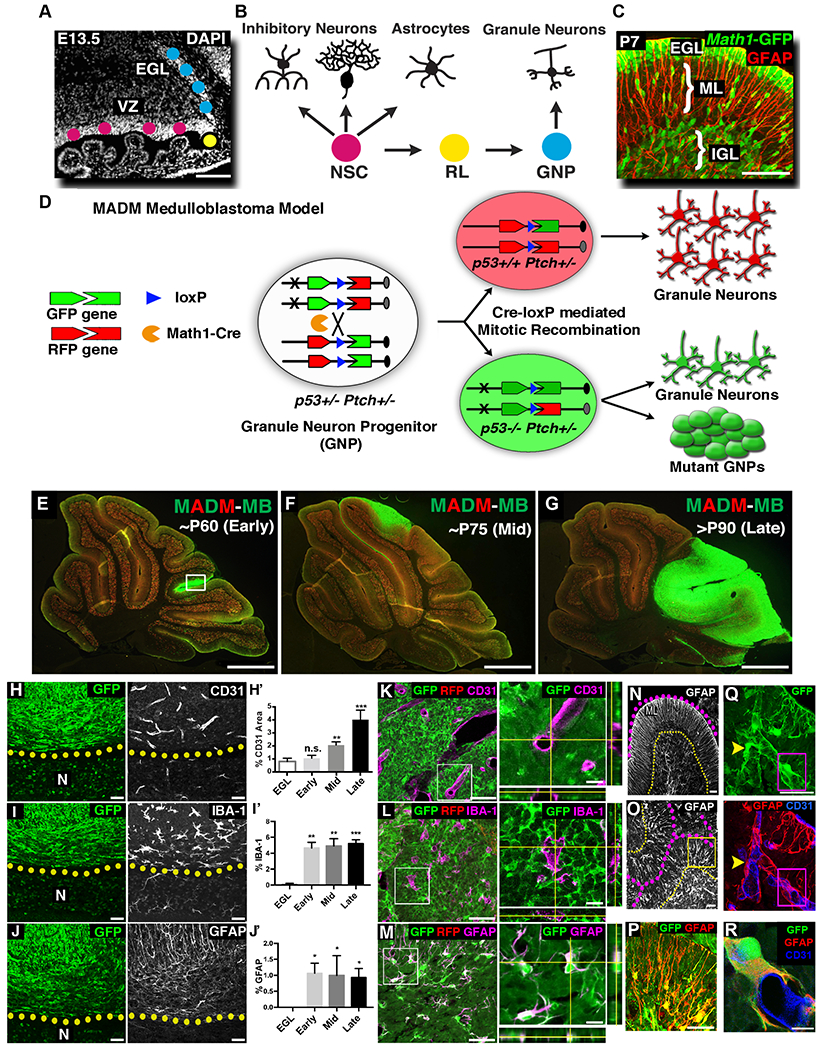
A-B, The cerebellum develops from two physically separated progenitor pools: multipotent NSCs in the ventricular zone (VZ) and unipotent GNPs in the external granule layer (EGL). C, At neonatal age, GNPs (Math1-GFP+) proliferate exponentially in the EGL, then migrate along radial processes of Bergmann glia (GFAP+) in the molecular layer (ML) into the inner granule layer (IGL) and terminally differentiated into mature granule neurons. D, MADM-based medulloblastoma model that generates RFP+, p53+/+ and GFP+, p53−/− GNPs in Ptch1+/− mouse. E-G, MADM-based medulloblastoma model had relatively consistent tumor progression kinetics. H,H’, Tumor-associated vessels had larger lumen sizes than adjacent normal tissue (N). Compared to normal EGL, blood vessel coverage gradually increased as tumors progressed (n=3, 4, 4 and 3, respectively). I,I’, Microglia are few in the EGL, but significantly increase during tumor progression (n=3 each). J,J’ Astrocyte cell bodies are absent in EGL but are significantly present in the tumor mass (n=3 each). K-M, Blood vessels (K) and microglia (L) did not express GFP. In stark contrast, all GFAP+ cells within the tumor mass (M) were GFP+ (see also Supplemental video). N, In the normal cerebellum, GFAP+ Bergmann glia extend radial processes through ML (outlined with dotted lines). O, Similar radial organization of GFAP+ processes could be found in focal areas in the tumor mass (outlined with dotted lines). P, Higher magnification of the boxed region in O. Q,R, Tumor-derived astrocytes (GFP/GFAP+) intimately interacted with blood vessels (CD31+), often wrapping an entire vessel (R, higher magnification of the boxed region in Q).
Scale bars: A=100μm, C= 50μm, E-G= 500μm, H-J, K-M (left panels), N-P= 50μm, K-M (right panels) and Q, R= 10μm.
Data are Mean±SD, one-way ANOVA; n.s. not significant, *p<0.05; **p<0.01; ***p<0.001.
See also Figure S1
RESULTS
Establishment of MADM-based and other genetic models for medulloblastoma
To pinpoint the contributions of diverse TME cells to SHH-activated subtype of medulloblastoma (hereinafter referred to as medulloblastoma), we established a MADM-based genetic model, in which sporadic Ptch1-heterozygous, p53-null, GFP+ GNPs were generated by Math1-Cre (Matei et al., 2005) in an otherwise Ptch1-heterozygous, p53-heterozygous mouse (Fig. 1D). We chose Math1-Cre as it faithfully labels GNPs (Yang et al., 2008) (Supplemental Fig. 1C) but not potential TME cell types (Supplemental Fig. 1D), enabling us to investigate the establishment and evolution of TME throughout tumorigenesis (Fig. 1E–G, illustrated in Supplemental Fig. 1E).
In addition to the MADM-based model (model A, Supplemental Table 1), three slightly modified mouse models were used in this study to address specific questions. In model B, Math1-Cre and a Cre reporter (Madisen et al., 2010) were used to specifically inactivate genes in GNPs and keep track of tumor lineage even after they differentiate into other cell types; in model C, Math1-GFP (Rose et al., 2009) was used as tumor cell marker to distinguish them from TME cells; and in model D, a Cre-free medulloblastoma model allowed us to designate Cre activity at desired time or in desired TME cell types for functional analysis. All of these are genetic models with the intact immune system, and are free of injury-related complications common for tumor-grafting models.
Progressive accumulation of TME cells as tumor progresses, and the discovery of trans-differentiation of astrocyte-like cells from tumor cells
Immunofluorescence-based histological analysis of tumor sections at various stages from the MADM model revealed that all types of TME cells observed in human medulloblastoma are abundantly present (Fig. 1H–J). Interestingly, distinct TME cells progressed at different pace: while the coverage of CD31+ endothelial cells gradually increased as tumors progressed (Fig. 1H’), both IBA-1+ tumor-associated microglia/macrophages (TAMs) and GFAP+ astrocytes were drastically elevated at tumor onset and then remained at a constant composition throughout (Fig. 1I’,J’). Since microglia and astrocytes are minimally present in the normal external granule layer (EGL) from where tumor initiates (Fig. 1I’,J’, first bar), their sudden and abundant presence in even the smallest tumors implies their critical importance for malignancy. Finally, compared to normal brain regions (N in Fig. 1H–J), the enlarged vessel lumen (Fig. 1H), reduced ramification of IBA-1+ cells (Fig. 1I), and elevated GFAP+ expression (Fig. 1J) in tumor regions collectively imply that these TME cells co-evolve with tumor cells.
To investigate the tumor-TME interactions, we closely examined the distribution of TME cells among GFP+ tumor GNPs (referred to as TuGNPs hereinafter)(Fig. 1K–M). We observed intermixed patterns of a minor population of TME cells intimately associating with tumor cells. While endothelial cells and TAMs lacked GFP labeling (Fig. 1K,L), unexpectedly all astrocytes (GFAP+) in the tumor mass were GFP-positive in every tumor examined (Fig. 1M, Supplemental Video 1). Because we confirmed that Math1-Cre faithfully labels GNPs but not astrocytes in normal brains (Supplemental Fig. 1D), it suggested that the GFAP+ GFP+ cells in medulloblastoma are likely TuGNP-derived rather than normal astrocytes.
In addition to GFAP, another marker for astrocytes BLBP also showed overlapped staining with GFP (Supplemental Fig. 1F,F’). Importantly, BLBP+ GFP+ cells were confined to the tumor region but never found in adjacent normal regions (Supplemental Fig. 1F’ versus F”), suggesting a tumor-specific phenomenon. To further examine the nature of these cells, we established a medulloblastoma model in which the GNP lineage is labeled with tdTomato (model B, Supplemental Table 1) and astrocytes are labeled by an astrocyte-specific Aldh1L1-GFP BAC transgene (Gong et al., 2003). The observed co-expression of GFP and tdTomato (Supplemental Fig. 1G) lent further support for the astrocytic nature of these TuGNP-derived cells. Finally, we also noted a remarkable morphological resemblance of these GFAP+ cells to normal astrocytes. Phenocopying Bergmann glia in normal cerebellum (Fig. 1N), a radial arrangement of GFP+ GFAP fibers was highly evident at the edge of early- to mid-stage tumors (Fig. 1O,P). Additionally, GFP-labeled astrocyte-like cells intimately associated with blood vessels throughout tumor regions (Fig. 1Q,R), reminiscent of another salient feature of normal astrocytes (Abbott et al., 2006; Iadecola and Nedergaard, 2007). In summary, the cellular resolution provided by MADM not only revealed the co-evolution process of TME cells during tumor progression, but also led to an unexpected finding of potential trans-differentiation of TuGNPs to astrocytes.
Validation of astrocyte identity of cells trans-differentiated from TuGNPs and the human relevance
While intrigued, we are acutely aware of alternative interpretations of the observed trans-differentiation. One possibility is promoter leakage: while Math1-Cre faithfully labels GNPs in the normal brain, the Math1 promoter could be mis-activated in astrocytes after they are recruited into the tumor mass. While this possibility cannot be ruled out by conventional Cre reporters, it was readily excluded by the dual-color design of MADM because promoter leakage would result in yellow astrocytes (illustrated in Fig. 2A), which were never observed in all tumors (Fig. 2B,B’). The possibility of cell fusion between astrocytes and GFP-labeled TuGNPs was ruled out because we never observed binucleated cells after examining more than 1,000 BLBP+GFP+ cells from 10 individual tumors (Supplemental Fig. 2A). Just in case nuclei fuse after cell fusion resulting in single-nucleus fused cells, we established a medulloblastoma model in which all GNPs are labeled with GFP (model D, Supplemental Table 1) while all astrocytes are labeled with tdTomato (Supplemental Fig. 2B) to capture such events based on the generation of yellow cells (illustrated in Supplemental Fig. 2C). The fact that yellow TME astrocytes were never observed after examining >100 tdTomato+ cells in 4 tumor-bearing mice (Supplemental Fig. 2D), definitively ruled out the possibility of cell fusion. Finally, we ruled out the possibility of GFP protein transfer from TuGNPs to astrocytes because in model C (Supplemental Table 1) all astrocytes are GFP-negative even though all tumor cells are labeled by Math1-GFP (data not shown).
Figure 2. Validation of tumor-to-astrocyte trans-differentiation.
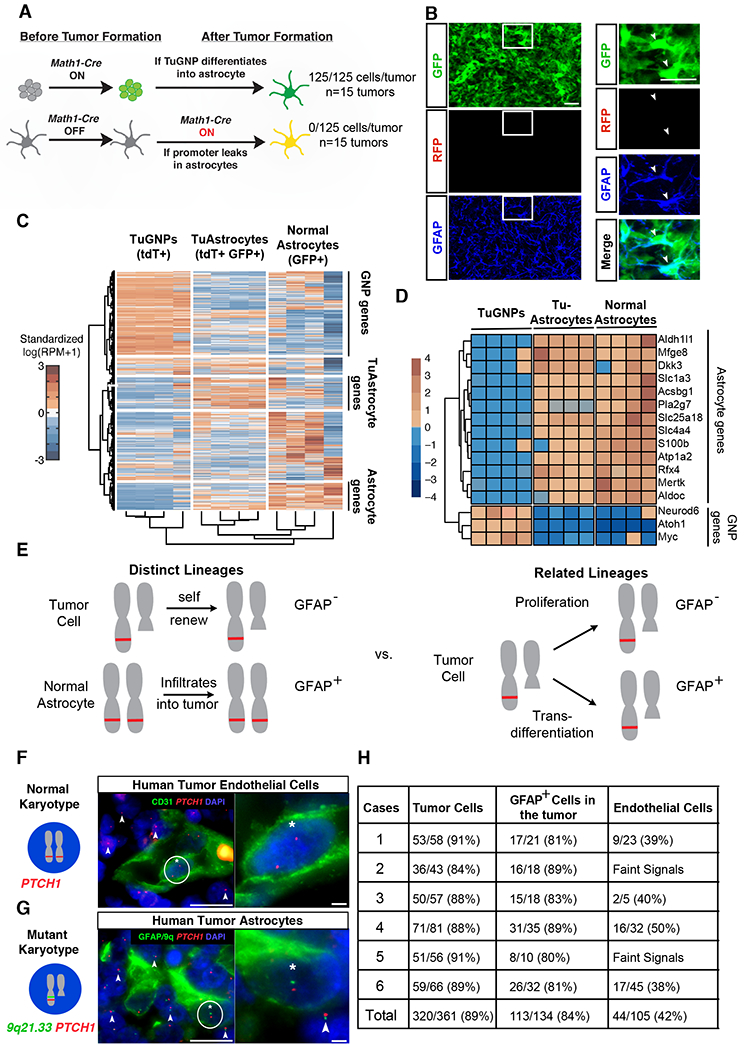
A, Schematic explanation on how the MADM system could definitively rule out the “Cre leakage” interpretation. B-B’ Representative images from the MADM model show that GFAP+ cells are not yellow. C, Hierarchical clustering of RNA sequencing data for TuGNPs, normal astrocytes, and TuAstrocytes. D, Many astrocytic markers are elevated in TuAstrocyte, while signature TuGNP genes are decreased in TuAstrocytes. E, Schematic illustration on how karyotypic analysis of astrocytes in human medulloblastoma could reveal their lineage relationship with tumor cells. F, Endothelial cells (CD31+) have a normal karyotype (two red FISH signals, asterisk-marked cell has zoom-in on the right), while surrounding tumor cells have one red signal (arrowheads), indicating the loss of one PTCH1-containing chromosomal fragment. G, The loss of one allele from the 9q21.33 region to the PTCH1 locus was seen in GFAP+ cells (asterisk-marked cell has zoom-in on the right), same as surrounding tumor cells (arrowheads). H, Quantification showed that the majority of TuGNP and TuAstrocytes in all six patient samples share the same karyotypic aberration.
Scale bars: B and B’=20μm, F and G (left panel) =25μm, F and G (right panel) =10μm
See also Figure S2
To pinpoint the identity of these “astrocyte-like” cells more definitively, we profiled their transcriptome using laser-capture microdissection (LCM) followed by RNA-sequencing. To visualize target cells in the tumor mass without immunostaining, we used a mouse model in which TuGNPs are red, tumor-derived astrocyte-like cells are yellow, and normal astrocytes are green (Supplemental Fig. 2E, model B). After extensive optimization of the tissue-processing procedure to avoid diffusive loss of fluorescent proteins in unfixed tissue (Singh et al., 2019; Wang and Janes, 2013)(Supplemental Fig. 2F and see “Methods”), we were able to visualize both GFP and tdTomato and collected ~250 astrocyte-like cells from each tumor sample. After extracting RNA, amplifying cDNA (Janes et al., 2010; Wang and Janes, 2013), and confirming the purity of LCM-collected astrocyte-like cells, TuGNPs, and normal cerebellar astrocytes (Supplemental Fig. 2G,H), we performed RNA-sequencing, followed by unsupervised clustering of transcriptomic profiles (n=4 each). We found that astrocyte-like tumor cells clustered more closely to normal astrocytes than to TuGNPs (Fig. 2C). Notably, almost all of the TuGNP-specific transcripts were downregulated in astrocyte-like cells (Fig. 2C top, 2D bottom), while many astrocyte-specific transcripts were upregulated (Fig. 2C bottom, 2D top), suggesting extensive trans-differentiation. Since the astrocyte-like cells closely resemble but still differ from normal astrocytes in ~2500 uniquely expressed transcripts (Fig. 2C, middle), we herein refer to them as “TuAstrocytes”.
Finally, we investigated whether the astrocytic component long recognized in human medulloblastomas (Burger et al., 1987; Mannoji et al., 1981) (Supplemental Fig. 1A,B) was descended from TuGNPs with two approaches. First, we assessed whether patient-derived medulloblastoma cells could trans-differentiate into astrocytes in xenografts. To avoid culture-induced artifacts, we used a primary human SHH-subgroup medulloblastoma that has been serially xenografted into the brains of NOD/SCID mice and authenticated by gene expression profiling along the passages (Shu et al., 2008; Zhao et al., 2012). We consistently observed human GFAP-specific immunoreactivity within all xenografted tumors examined (Supplemental Fig. 2I,J). Since these tumor cells had been grafted through multiple passages to eliminate non-tumor cells, these human GFAP+ cells most likely were trans-differentiated from TuGNPs. For our second approach, we exploited the fact that the PTCH1 locus on chromosome 9q in the human genome is recurrently lost in human desmoplastic medulloblastoma (Schofield et al., 1995). We predicted that, if astrocytes in the tumor were not lineage-related to tumor cells, they would have a normal karyotype (Fig. 2E, left); while if astrocytes are lineage-related to tumor cells, then astrocytes would have the identical chromosomal loss as surrounding tumor cells (Fig. 2E, right). Among 21 cases of SHH-activated subtype human medulloblastoma, six were identified with 9q loss that warranted further study. We assessed the chromosomal integrity in each astrocyte within the tumor mass by two-color fluorescent in situ hybridization (FISH) for PTCH1 (9q22.33) and an adjacent locus (9q21.33) along with immunofluorescent staining of GFAP. To ensure that these patients are not germline carriers for this karyotypic aberration that would confound data interpretation, we verified that many endothelial cells in the tumor mass had a normal karyotype (Fig. 2F). The fact that GFAP-positive cells had a significant enrichment for monoallelic PTCH1-9q21.33 loss to the same level of tumor cells (Fig. 2G) in six out of six cases (summarized in Fig. 2H) suggests that these astrocytes share a common lineage with tumor cells harboring the same genetic lesion.
After exhaustively examining all conceivable alternative interpretations, we conclude that TuAstrocytes in the TME are derived from TuGNPs in both mouse models and human medulloblastoma patients.
TuAstrocytes appear to support tumor progression
To determine the function of TuAstrocytes, we first sought to determine whether TuAstrocytes display cancer stem cell characteristics since GFAP is a marker gene for not only astrocytes but also neural stem cells (Doetsch et al., 1999). Because the purification of viable TuAstrocytes was not feasible (data not shown), we established a mouse medulloblastoma model in which TuAstrocytes are labeled with RFP upon tamoxifen injection (Fig. 3A, Model D), with an expectation to find RFP+ TuGNPs after an extended period if TuAstrocytes function as cancer stem cells. However, no red TuGNPs were observed two weeks after Tamoxifen injection (Fig. 3B). In case more time was needed for TuAstrocytes to give rise to TuGNPs, we transplanted unlabeled primary tumors from this model into NOD/SCID mice, administered tamoxifen after tumors started growing, and waited for two months. The fact that we still did not observe any RFP+ TuGNPs (Fig. 3C) does not support a cancer stem cell role of TuAstrocytes.
Figure 3. TuAstrocytes seem not behaving as cancer stem cells.
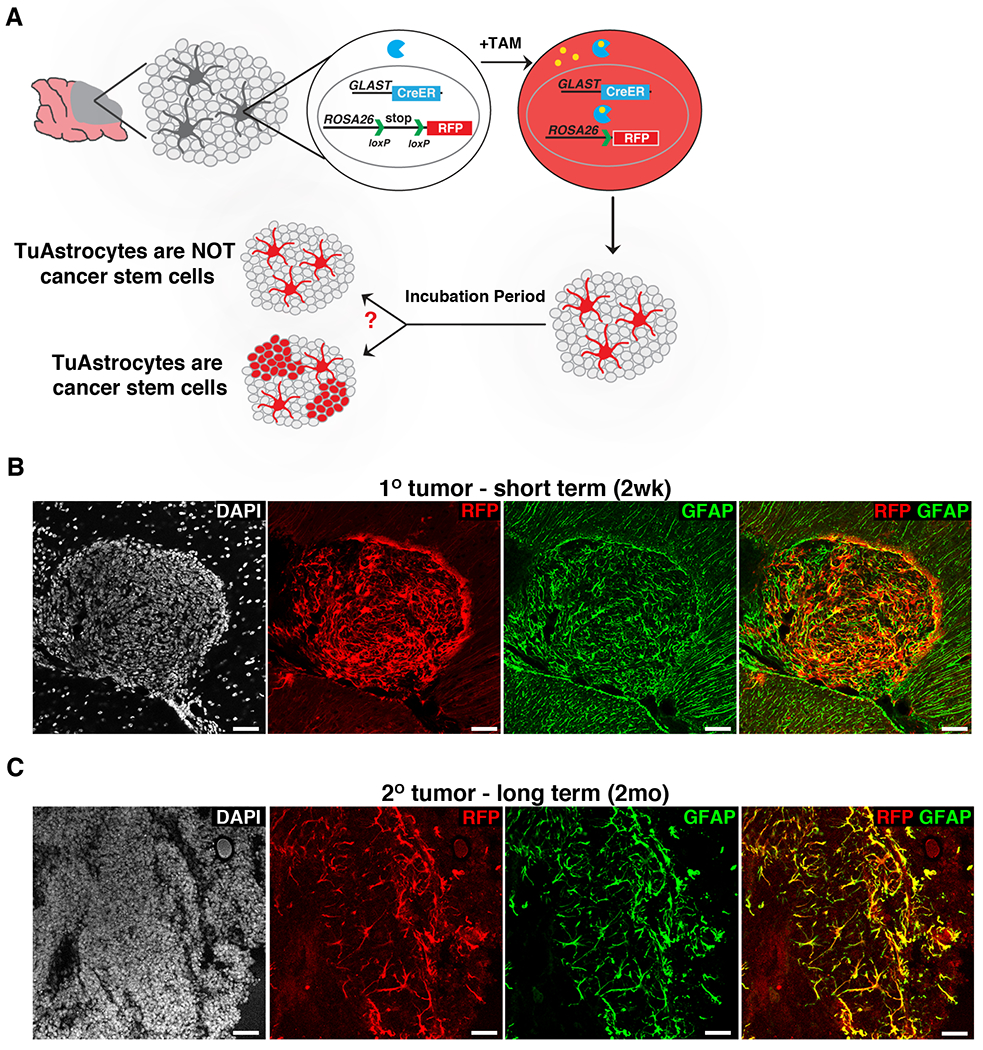
A, Schematic illustration of the mouse model and expected outcomes. B, TuAstrocytes did not give rise to TuGNPs two weeks after tamoxifen injection. C, TuAstrocytes did not give rise to TuGNPs even after two months.
Scale bars: B-C=50μm.
See also Figure S3
Next, we probed into tumor-supporting roles of TuAstrocytes with two experiments. First, we examined the correlation between the presence of TuAstrocytes and the progression of pre-neoplastic lesions (PNLs) (Oliver et al., 2005), among which only some progress to full malignancy. After identifying PNLs on the surface of the cerebellum in Ptch1+/− mice at P35, we assessed correlation between overall content of astrocytes and the proliferative status of GNPs (Supplemental Fig. 3A,B). Across more than 30 PNLs, we found that the presence of astrocytes strongly correlated with GNP proliferation and vice versa (p < 10−5, Fisher exact test; Supplemental Fig. 3C), suggesting that astrocytes might play important roles in PNL progression. Second, when purified TuGNPs were cultured in vitro, we observed that, while dispersed cells stopped proliferating quickly, those aggregating cells showed robust proliferation (Supplemental Fig. 3D). To investigate whether TuAstrocytes were involved, we prepared RFP+ TuGNPs from the Aldh1L1-GFP model used for LCM experiment (model B, Supplemental Fig. 2E). While all cells were red initially, GFP-positive TuAstrocytes started to appear in aggregates as early as day-3 in culture (Supplemental Fig. 3E,G) and increased substantially by day-6 (Supplemental Fig. 3F,G), indicating the concurrence between spontaneous TuGNP-to-TuAstrocytes trans-differentiation and sphere expansion in the culture. Taken together, we conclude that TuAstrocytes are not cancer stem cells, rather likely play a supportive role toward TuGNPs.
IGF1 is a tumor-supporting TME factor
Next we asked how TuAstrocytes might promote tumor progression by examining a panel of eight growth factors known to be important for brain development and tumor progression. First, we performed qRT-PCR to compare their expression levels between tumors and normal brains, hypothesizing that candidate factors should consistently express at higher levels in tumors. We identified three candidate factors, IGF1, HGF, and PDGF-A, which were consistently ≥2-fold higher in tumors (Fig. 4A). To distinguish TME-derived from tumor-intrinsic factors, we performed paired qRT-PCR of these three factors between the tumor mass and purified TuGNPs, and found that IGF1 and HGF but not PDGF-A were TME-derived factors (Fig. 4B). Next, we found that IGF1 but not HGF promoted the proliferation of TuGNPs in cell culture in a dose-dependent manner (Fig. 4C,D). Finally, the importance of IGF1 to human medulloblastoma was supported by the IGF1-dependent proliferation of a human SHH medulloblastoma line (Shu et al., 2008; Zhao et al., 2012) (Fig. 4E).
Figure 4. IGF1 is a TME factor that is necessary for tumor progression.
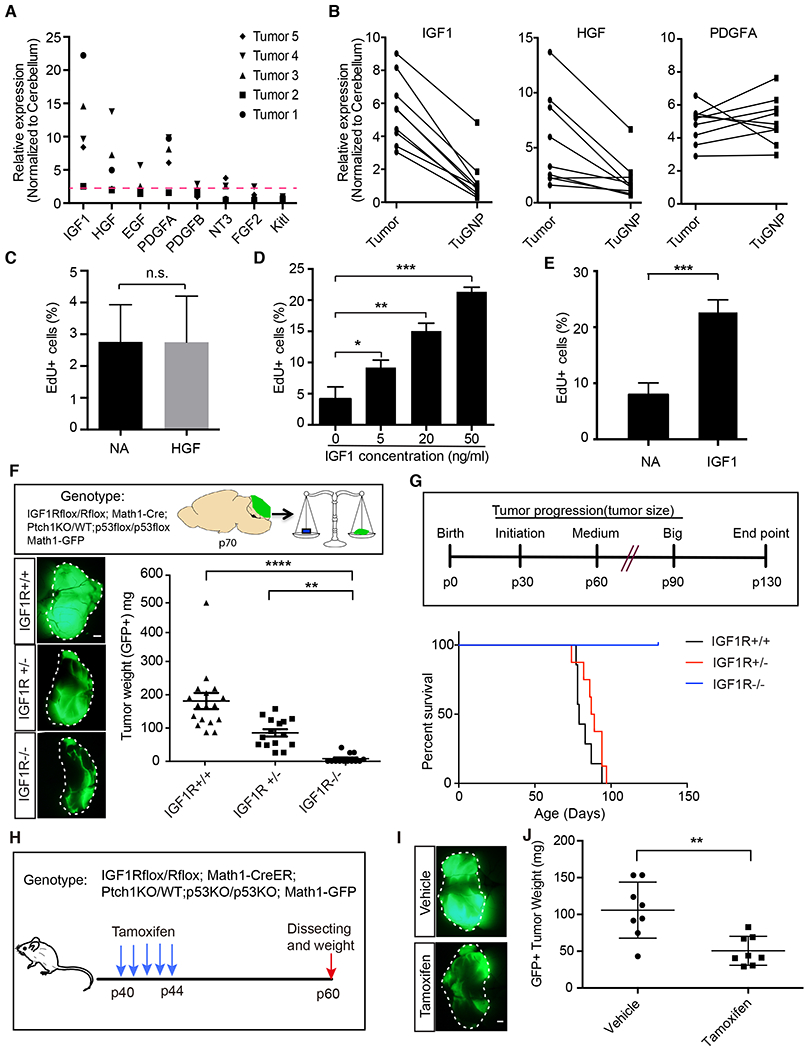
A, Expression of growth factors in tumors in comparison to normal cerebelli. B, Expression levels of IGF1 and HGF, but not PDGFA, were much higher in the tumor mass than in purified tumor GNPs (n=8). C, HGF did not promote tumor cell proliferation. D,E, IGF1 promoted cell proliferation of both mouse (D) and human tumor cells (E). F, Inactivation of IGF1R in GNPs delayed tumor progression (n≥12 for each genotype). G, Inactivation of IGF1R in GNPs prolonged mouse survival. While all IGF1R+/+ (n=7) and IGF1R−/+ (n=8) tumor mice died before P90, all IGF1R-null mice (n=14) survived till the experimental endpoint (P130). H, The medulloblastoma model in which IGF1R can be inactivated after tumor onset. I, Tamoxifen (150 mg/kg) induced inactivation of IGF1R in tumor GNPs suppressed tumor progression (n=8 for each group).
Scale bar: F and I=1mm. Data are Means±SD, Student’s t-test (C, D and E) or one-way ANOVA (F and I); n.s: not significant,**p<0.01; ***p<0.001; ****p<0.0001.
See also Figure S4
To determine whether IGF1 signaling is critical for medulloblastoma progression, which is well known for its reliance on Shh signaling (Goodrich et al., 1997; Mullor et al., 2002; Pietsch et al., 1997), we established a mouse model in which IGF1R is genetically deleted specifically in GNPs (model C plus IGF1R-flox alleles, herein referred to as IGF1R-CKO, Supplemental Fig. 4A). We found that IGF1R is critical for both tumor initiation (Supplemental Fig. 4B,C) and progression (Fig. 4F) as tumor sizes were much smaller in IGF1R-CKO mice compared to the control group at both stages. IGF1R-CKO mice survived much longer than IGF1R-WT and IGF1R-het mice (Fig. 4G). Histological examination of brains at the study endpoint (P130) revealed few tumor cells in IGF1R-CKO brains, indicating the indispensable role of IGF1R signaling for medulloblastoma. It should be noted that since Math1-Cre is expressed throughout GNP development, the lack of tumor formation in this model could be caused by developmental defects of GNPs upon IGF1R inactivation. To rule out this possibility, we established another mouse model in which IGF1R deletion is mediated by Tamoxifen-dependent Math1-CreER (Fig. 4H, model D). After confirming that Tamoxifen had no impact on tumor progression (Supplemental Fig. 4D), we injected Tamoxifen to inactivate IGF1R in TuGNPs after tumor formation (P40). The reduction of tumor size in Tamoxifen-treated mice in comparison to vehicle controls (Fig. 4I,J) suggests that acute inactivation of IGF1 signaling in TuGNPs halted tumor progression. To probe into the cause of tumor size reduction, we further examined apoptotic index and cell cycle exit index of TuGNPs (illustrated in Supplemental Fig. 4E), and found that IGF1R-null TuGNPs are much more likely to die and to exit the cell cycle than IGF1R-WT TuGNPs (Supplemental Fig. 4F–I).
In summary, these data suggested that TME-derived IGF1 signaling is critical not only for the initiation but also for the sustained progression of medulloblastoma.
IGF1 is secreted by TAMs rather than by TuAstrocytes
Because TuAstrocytes are trans-differentiated from TuGNPs, we established a complementary mouse model to inactivate IGF1 with Math1-Cre (model C plus IGF1-flox alleles), expecting it to phenocopy the IGF1R knockout model. Surprisingly, the IGF1-KO model failed to slow tumor progression, leading to our suspicion that TuAstrocytes might not be the cellular source of IGF1. Using RNA in situ hybridization to detect IGF1 and immunofluorescent staining to identify cell type, we found that IGF1 is specifically produced by IBA-1+ tumor-associated myeloid cells (TAMs) but not TuAstrocytes (Fig. 5A, B). Interestingly, while ~70% of TAMs were IGF1+, no detectable IGF1 was seen in microglia in the normal brain regions (normal versus tumor region in Fig. 5A middle panel, quantified in 5C). After affinity-based purification of TAMs and microglia, we further verified by qRT-PCR that the expression level of IGF1 is more than an order of magnitude greater in TAMs than in normal microglia (Fig. 5D). Co-culturing purified TAMs and TuGNPs showed that TAMs promoted TuGNP proliferation in a density-dependent manner (Fig. 5E,F), which is IGF1-dependent because IGF1-sequestering protein IGFBP3 (Ranke, 2015) completely abrogated the tumor-supporting activity of TAMs (column 3 versus 2 in Fig. 5G). To investigate the role of TAM-secreted IGF1 in vivo, we established another medulloblastoma model in which IGF1 is inactivated by myeloid-specific CSF1R-Cre (Deng et al., 2010)(Fig. 5H, model D). We found that TAM-specific deletion of IGF1 resulted in much smaller tumors at P35 compared to tumors in the IGF1-WT conditions (Fig. 5I,J, n>10), suggesting the supportive role of IGF1 from TAMs for tumor initiation.
Figure 5. IGF1 produced by TAMs is critical for tumor progression.
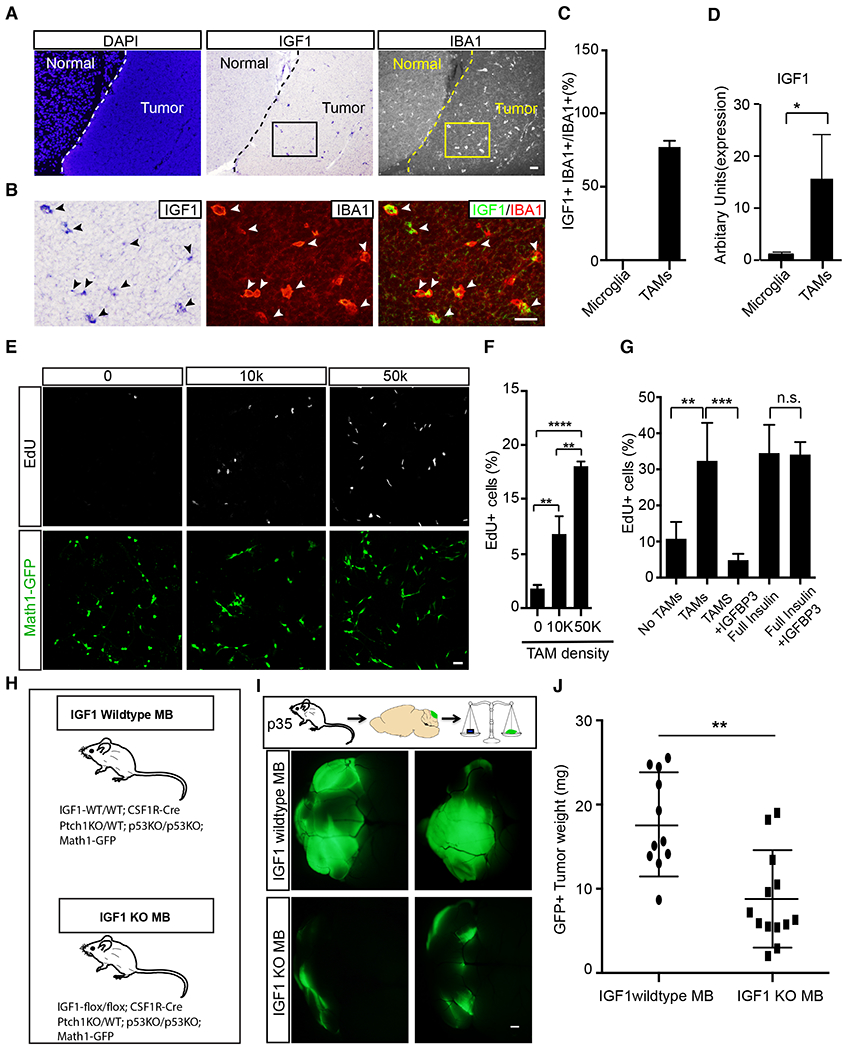
A,B, In situ hybridization showed that IGF1 was produced by IBA1+ tumor associated myeloid cells (TAMs) but not by normal microglia. C, The proportion of IGF 1-producing IBA1+ TAMs (n=4). D, qRT-PCR comparison of IGF 1 expression level between acute purified microglia and TAMs. E-F, Proliferation of tumor GNPs was enhanced by acutely purified TAMs in a density-dependent fashion. G, TAM-promoted proliferation of tumor GNPs was abrogated by IGF 1-blocking IGFBP3. General toxicity of IGFBP3 was ruled out by the fact that it couldn’t block the proliferation of tumor GNPs induced by insulin, which activates IGF1R but cannot be blocked by IGFBP3. H, Medulloblastoma mouse model with IGF1 knockout in TAMs. I,J Representative images (I) and tumor weight (J) in IGF1−/− tumor mice (n=13) and IGF1-WT controls (n=11) at P35.
Scale bar: A=50μm, B and E=20μm, I=1mm. Data are Means±SD, one-way ANOVA (F and G) or Student’s t-test (C, D and J) *p<0.05; **p<0.01; ***p<0.001
See also Figure S5
TAMs are locally activated microglia and not circulating monocyte-derived macrophages
Why do most TAMs but few normal microglia express IGF1? One possible explanation could be their distinct origins. Derived from embryonic yolk sac and shaped by the brain environment, microglia are intrinsically different from macrophages derived from circulating monocytes (Ginhoux et al., 2010; Goldmann et al., 2016; Gomez Perdiguero et al., 2015; Gosselin et al., 2014). Therefore, the elevated IGF1 expression in TAMs could mean that TAMs originated from circulating-monocyte derived macrophages, especially because we noted that TAMs had highly distinct morphologies from resident microglia (Supplemental Fig. 5A–F). Our initial attempt to pinpoint the origin of TAMs based on their marker expression was unsuccessful due to its puzzling pattern: while they stained positively for microglia marker P2RY12 (Butovsky et al., 2014), they also expressed monocyte-derived macrophage marker CCR2 (Kurihara et al., 1997; Siebert et al., 2000) (Supplemental Fig. 5G,H). Even more puzzling was the lack of expression of TMEM119 (Supplemental Fig. 5I), which has been previously shown to remain stable in microglia even in the presence of brain inflammation and injuries (Bennett et al., 2016). While these efforts turned out to be inconclusive, it nevertheless revealed a strong influence of TME factors on TAM states.
Next we turned to a lineage tracing method to distinguish microglia from monocyte-derived macrophages. Using this approach, initially labeled monocytes would be replaced by unlabeled monocytes derived from stem cells due to their fast turnover but microglia would remain labeled due to slow and local replenishment (Fig. 6A) (Goldmann et al., 2013; Yona et al., 2013). We carefully re-validated the system to rule out possible interferences by genetic mutations in the medulloblastoma model (Supplemental Fig. 6). To avoid the slim possibility that monocytes labeled in early tumor lesions lodging in the tumor mass might fail to turnover, we injected Tamoxifen at P7, long before tumor initiation. When we examined brain sections at P40 when tumors were still small and at P60 when tumors fully developed, we found that all TAMs in the tumor mass were labeled by tdTomato, suggesting that they originated from the microglia (Fig. 6B–E). Complementary to the lineage tracing experiment, RNA-seq analysis of purified TAMs followed by Hierarchical Cluster Analysis also indicated that TAMs shared more transcriptomic similarity to microglia than to circulating monocyte-derived macrophages (Fig. 6F,G). Notably, TAMs displayed significant alterations of gene expression when compared to normal microglia, and demonstrated even greater deviation from normal microglia than LPS-treated microglia (Fig. 6G), suggesting that they were heavily shaped by local TME factors. Finally, in addition to microglia, one recent paper reported that meningeal macrophages also turn over slowly (Goldmann et al., 2016), which could also be a source for labeled TAMs. Dextran dye labeling of meningeal macrophages (Fig. 6H,I) followed by 3- and 7-days monitoring did not detect infiltration (Fig. 6J,K) suggests that meningeal macrophages are unlikely to be a significant source of TAMs. Taken together, these data suggest that, TAMs originate from microglia, and undergo significant morphological and functional changes that are likely induced by TME factors.
Figure 6. TAMs originate from brain-resident microglia.
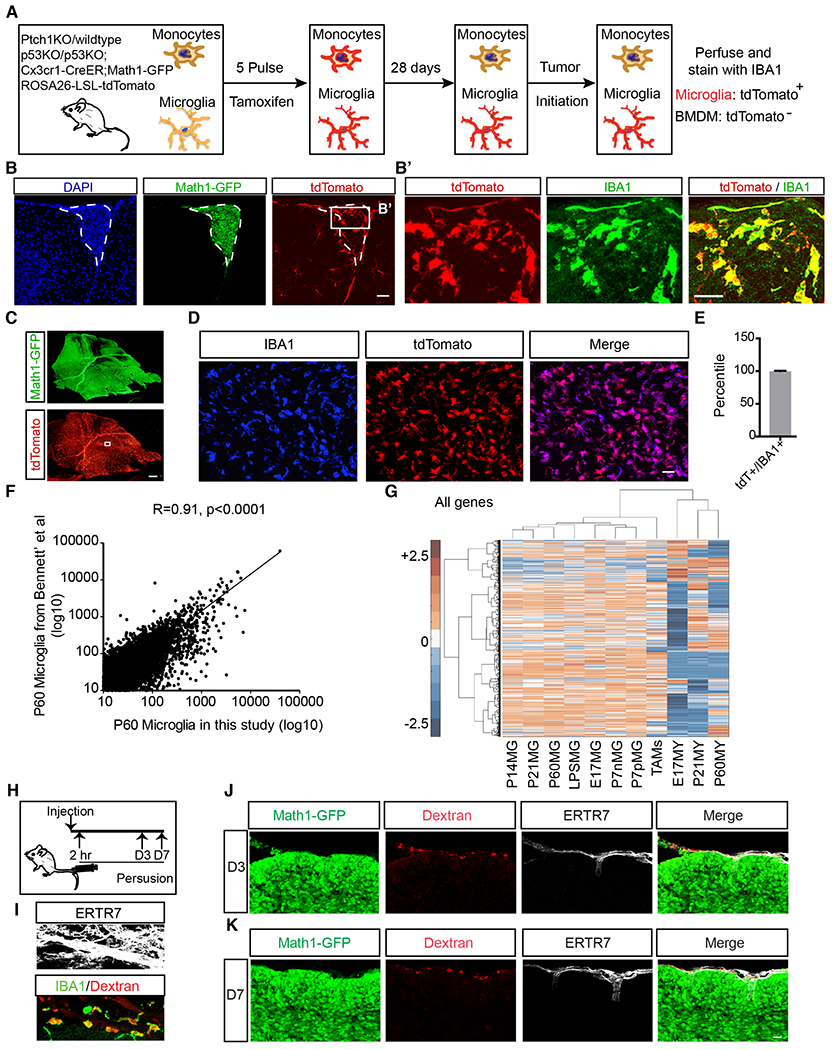
A, Schematic of the lineage tracing experiment. B,B’ TdTomato+ TAMs were present in small tumors (outlined with dotted line, n=3). C-E, tdTomato+ TAMs overwhelmed the Math1-GFP+ tumor mass in full-blown tumors. n=5. F, Pearson correlation of transcriptomic data of P60 microglia between this study and published dataset by Bennett et al (Bennett et al., 2016) indicates that these two sets of data are highly correlated, allowing us to perform Hierarchical Cluster Analysis of pooled data in panel G. G, Hierarchical Cluster Analysis of transcriptomes showed that gene expression profile of TAMs clusters with microglia rather than circulating myeloid cells. [P: postnatal; E: embryonic; numbers indicate embryonic or postnatal ages. MG: microglia; MY: circulating myeloid cells; P7nMG: TMEM119-microglia at P7; P7pMG: TMEM119+ microglia at P7] H, Schematic illustration of dextran-labeling of meningeal macrophages. I, The majority of meningeal macrophages were labeled by dextran via tail vein injection (75.79+7.77%, IBA1+Dextran+/IBA1+). J-K, Macrophages within meninges (ERTR7+) did not infiltrate into Math1-GFP+ tumor mass 3-day (J) and 7-day (K) post dextran injection (n=3).
Scale bar, B, B’and D =30μm, C= 200μm; J and K=40μm. n=5 mice for each group. Data are Means±SD, Student’s t-test. n.s.: not significant; ****p<0.0001.
See also Figure S6
IL4 signaling is responsible for stimulating increased IGF1 expression by TAMs
Because TAMs originate from microglia that normally do not express high level of IGF1, we pursued the hypothesis that IGF1 expression in TAMs is caused by the alteration of their immune state induced by TME factors. We first performed comprehensive bioinformatic analysis of RNA-seq data of TAMs in comparison to normal microglia with a focus on tolerogenic signature genes (Chanmee et al., 2014) previously demonstrated to promote tumorigenesis (Noy and Pollard, 2014). Among 4898 upregulated and 4521 downregulated genes, we found an overall tolerogenic signature, including a bias toward IL-10 production, negative regulation of adaptive immunity, negative regulation of inflammatory responses, factors associated with Th2 immunity, and IL-4 mediated signaling event (Supplemental Fig. 7A).
Next, we examined two pathways that are best known for IGF 1-induction in macrophages/microglia, namely IL4 and TNFα (Fournier et al., 1995; Lake et al., 1994; Spadaro et al., 2017; Wynes and Riches, 2003). We found that genes in the IL-4 pathway are globally elevated in TAMs, especially those elicited in a STAT6-dependent manner (Fig. 7A). In support of this finding, we observed a significant increase of differentially expressed genes regulated by IL-4 related kinases in macrophages, including JAK2 and ERK1/2 (Supplemental Fig. 7B). On the contrary, genes known to be elicited in macrophages stimulated with TNFα were generally downregulated, including genes dependent on TNFa signaling viaNFkB and API (Fig. 7B, blue bars), whereas genes that are thought to be suppressed by TNFα were upregulated (Fig. 7B, red bar). These findings are in accordance with our Luminex profiling data that indicated an increase of IL-4 but not TNFα in the tumor mass (Fig. 7C, Supplemental Table 2). To investigate the functional relevance, we purified TAMs from the tumor mass and demonstrated significant increase of IGF 1 expression upon IL4 stimulation (Fig. 7D). In a complementary experiment, we established a mouse medulloblastoma model with an IL4-null background (model C, Supplemental Fig. 7C). While IL4-KO did not alter the number of TAMs in the tumor mass (Fig. 7E, Supplemental Fig. 7D), it led to a significant decrease of IGF 1 expression in the tumor mass (Fig. 7F). Therefore, this set of experiments indicated that IL4 is sufficient and necessary to promote IGF1 expression in TAMs.
Figure 7. IL4 produced by TuAstrocytes promotes IGF1 expression in TAMs.
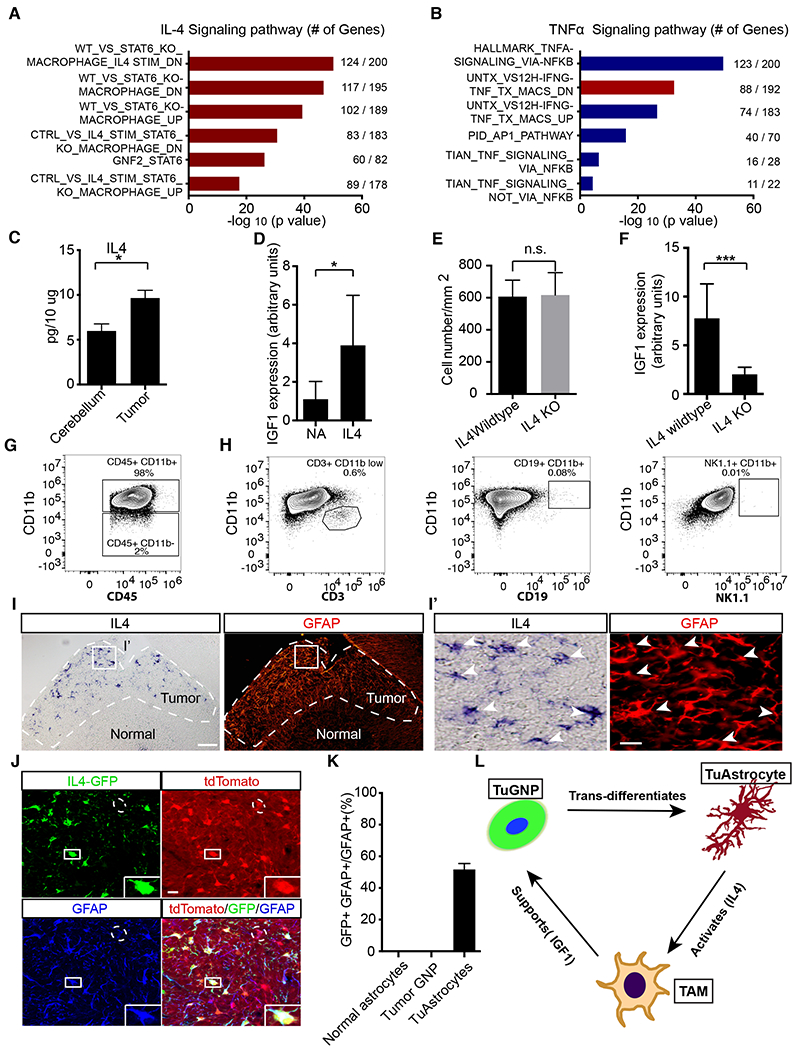
A,B, Pathway analysis of differentially expressed genes in TAMs revealed significantly up-regulated signatures associated with IL4 but not TNFa signaling. C, Luminex profiling showed that IL4 level was elevated in the tumor in comparison to normal cerebellum (n=3). D, IL4 stimulation of acutely purified TAMs led to elevated IGF1 level in culture (n=5). E, Quantification of the cell density of TAMs in IL4 wildtype and IL4 KO tumors. F, IL4 knockout led to reduced IGF1 expression level in the tumor mass (n=8 for each genotype). G, Flow cytometry analysis demonstrated that CD11b+ myeloid cells were the dominant population of immune cells in the tumor (n=10). H, Negligible numbers of T, B and NK cells were present (n=4). I-I’, In situ hybridization showed that IL4 was produced by GFAP+ cells in the tumor mass. J, All IL4-GFP+ cells were tdTomato+ and GFAP+, suggesting that IL4 was produced by TuAstrocytes (n=3). K, Quantitative analysis showed that IL4 was expressed by −50% TuAstrocytes but not by normal astrocytes or TuGNPs. L, Working model of an intricately organized TME network in medulloblastoma: a small fraction of tumor GNPs trans-differentiates into TuAstrocytes, which produce IL4 to activate tumor-associated microglia (TAMs), which in turn secrete IGF1 to promote tumor progression.
Scale bar: I =50μm, I′ and J=20μm. Data are Means±SD, Studen t’s t-test; *p<0.05; **p<0.01; ***p<0.001.
IL4 is produced by TuAstrocytes
Finally, we set out to determine the cellular source of IL4. Since it is known that Th2 lymphocytes are the major source of IL4 (Bradding et al., 1992; Le Gros et al., 1990; Swain et al., 1990), we first comprehensively catalogue the immune cell types in the TME, after carefully removing immune cells in blood vessels with intracardiac perfusion with PBS. Our data indicated that >98% of CD45+ immune cells in medulloblastoma were positive for the myeloid marker CD1 lb (Fig. 7G). Considering the possibility of CD11b expression in activated T and B lymphocytes (Christensen et al., 2001; Ghosn et al., 2008; Kawai et al., 2005; Wagner et al., 2001) and mature NK cells (Omi et al., 2014), we performed further flow cytometry analysis for T cells (CD3), B cells (CD19), and NK cells (NK1.1). This analysis, (Fig. 7H) along with the lack of immuno-staining of the T cell marker CD3 in the tumor mass (Supplemental Fig. 7E,F) indicated that only negligible numbers of T, B, and NK cells were present within the tumor mass. This finding corroborates well with reports in the literature that the predominant immune component in human medulloblastoma is CD11b+ myeloid cells (Margol et al., 2015; Rossi et al., 1991). Taken together, the lack of infiltrated T cells led us to postulate that, rather than Th2 lymphocytes, other TME cells must be the source of IL4.
We performed in situ analysis for IL4 together with immuno-staining for cell type-specific markers to identify IL-4 producing cells. To our surprise, we identified GFAP+ astrocytes as the major source of IL4 (Fig. 7I,I’). To investigate whether IL4 is produced by normal or tumor-derived astrocytes, we established a mouse model that incorporates both tumor-lineage marker (tdTomato) and an IL4-GFP reporter transgene (Mohrs et al., 2001)(model B, illustrated in Supplemental Fig. 7G), and found that all IL4-GFP+ cells expressed both GFAP and tdTomato, suggesting that IL4 is specifically produced by TuAstrocytes rather than normal astrocytes (Fig. 7J,K, Supplemental Fig. 7H).
DISCUSSION
Through comprehensive lineage tracing, molecular profiling, and functional studies, we have demonstrated that TME components in the ostensibly simple medulloblastoma form an intricate network to support tumor progression (Fig. 7L). Our work clearly illustrates the importance of studying co-evolution of TME and tumor cells at high spatiotemporal resolution using immuno-competent, injury-free genetic models, and exemplifies how tumor plasticity contribute to cellular heterogeneity in the tumor mass, and how multiple TME cells form a local community to promote tumor progression.
The MADM system provides temporospatial resolution to deconvolute the complexity of TME
The importance of TME for tumor progression has been well supported by a wealth of reports (Balkwill et al., 2012; Hanahan and Coussens, 2012; Quail and Joyce, 2013). While tumor angiogenesis was the initial focus (Folkman, 1971), other TME cells including fibroblasts, immune cells, and other tissue-resident cells quickly took the stage (Binnewies et al., 2018; Gascard and Tlsty, 2016; Kalluri, 2016; Shiga et al., 2015; Tabuso et al., 2017). To develp novel cancer treatment strategies that target TME, one must thoroughly study the establishment and functions of TME, with greater attention to time, space, cell lineage, as well as homotypic and heterotypic cell-cell interactions. Therefore, it is imperative to use high resolution in vivo tools to delineate hierarchical organizations, reprogramming potentials, as well as clonal behaviors within TME. The Confetti mouse is an example of such a tool that allows lineage tracing of multiple clones in vivo, which revealed not only the neutral competition mechanism of intestinal crypt homeostasis (Snippert et al., 2010), but also how cancer stem cells maintain tumor architecture to sustain intestinal adenomas (Schepers et al., 2012). On the other hand, the MADM system is more suitable for dual lineage analysis of the fate of one mutant and one wildtype cell from the same progenitor cell (Zong et al., 2005). While previous application of MADM was focused on cell-autonomous gene functions in cancer (Gonzalez et al., 2018; Liu et al., 2011; Muzumdar et al., 2007), this study took advantage of its lineage tracing capability to uncover tumor-TME relationships. The fact that such complexity of interactions could be unveiled from an ostensibly “simple” cancer further emphasizes the importance of untangling the inner works of TME in more complex tumors.
The trans-differentiation of TuGNPs to TuAstrocytes: community-building behaviors in the tumor mass
Relying on the lineage tracing capability of MADM, we unexpectedly discovered the phenomenon of tumor-to-TME trans-differentiation. Fully aware of potential technical caveats, we painstakingly and exhaustively pursued all imaginable alternative explanations to exclude the possibility of cell fusion, protein transfer, ectopic Cre expression, and mis-expression of cell markers. Most importantly, karyotyping-based analysis of human medulloblastoma samples also demonstrated a lineage relationship between tumor GNPs and the astrocyte-like component in the tumor mass. The complementary nature between mouse genetics and human sample analysis greatly strengthens our confidence of this finding.
It should be noted that, while the differentiation of tumor cells into other cell types have been reported before, the novelty of our findings lies within two aspects. First, the trans-differentiation from GNPs to astrocytes has to overcome significant barriers within the developmental program since the GNP pool and astrocyte-producing neural stem cell pool are segregated temporally (from E9.5 and on during mouse embryonic development) and spatially (GNPs reside in EGL on the surface of cerebellar primordium, while NSCs reside in ventricular zone, the inner most layer of the neural tube at the fourth ventricle). Second, while the functional contribution of trans-differentiated TuAstrocytes was unveiled in this study, it remains to be seen if transdifferentiated cells in other tumor types also play such a significant functional role.
Still there are many unanswered questions. First, what is the mechanism of trans-differentiation? Second, how is the ~1% composition of TuAstrocytes maintained throughout tumor progression? Finally, while we observed tantalizing hints of direct support of TuAstrocytes to tumor progression (Supplemental Fig. 3D–G), our study took an unexpected turn toward the discovery of TuAstrocyte-TAM-tumor cell network. In the future, it will be important to investigate the direct supportive factors from TuAstrocytes toward tumor cells, as well as to explore their role in BBB maintenance since SHH-subtype medulloblastoma displayed little BBB disruption in stark contrast to the Wnt-activated subtype (Phoenix et al., 2016).
Harness the uniqueness of neuro-immunological network for brain tumor treatment
While our study discovered an anti-inflammatory immune network formed by TuAstrocytes and microglia in medulloblastoma, in other situations microglia and astrocytes mutually activate each other to establish a pro-inflammatory environment (Liddelow et al., 2017; Vainchtein et al., 2018). It is conceivable that one could re-polarize TuAstrocytes and TAMs if the proper conditions can be indentified. Of course, such efforts must be grounded in the appreciation that microglia have a distinct developmental origin from macrophages (Ginhoux et al., 2010; Gomez Perdiguero et al., 2015) and tend to only mount subdued immune responses when aggravated (Bennett et al., 2018; Bennett et al., 2016; Gosselin et al., 2014; Gosselin et al., 2017), and that astrocytes are not ‘classical’ immune cells and often play injury-repairing roles in the brain (Anderson et al., 2016; Burda and Sofroniew, 2014; Farina et al., 2007; Groves et al., 2018).
In addition to activating the innate immune system in the brain, attracting adaptive immune cells into the brain parenchyma as found in neurological diseases could facilitate brain tumor treatment (Baruch et al., 2013; Filiano et al., 2015; Hemmer et al., 2015). While many checkpoint blockade-based immunotherapy clinical trials for brain tumors are currently underway (clinicaltrials.gov), limited successes (Filley et al., 2017; Xue et al., 2017) emphasize the need to carefully consider the unique immunological environment of the brain. While the infiltration of T cells was previously reported in a glioma model (Quail et al., 2016), few T, B, or NK cells were found in our medulloblastoma model, which is in concordance with previous findings in human medulloblastoma samples (Margol et al., 2015; Rossi et al., 1991). While this difference could be attributed either to different tumor types or to grafting-induced injuries in the previous model, the possible absence of T cells nevertheless raises the concern on the effectiveness of T cell-based immunotherapy for brain tumors. Even if T cells reach the tumor mass, critical cytokines for T cells such as IL2 and IL15 were too low in our medulloblastoma model (Supplemental Table 2) to support their survival. This finding corroborates well with recent clinical research that demonstrated the presence of cytotoxic T cells had no correlation with overall survival of medulloblastoma patients (Vermeulen et al., 2018), and that T-cell exhaustion is extremely severe in glioma (Woroniecka et al., 2018). Last but not least, one must take into consideration of the fact that the brain is extremely sensitive to chemical and electrical imbalances and could be severely harmed by cytokine storm or other overt immunological responses. Therefore, the efficacy of T cell based immunotherapy in brain tumors would only manifest after we develop a firm grasp of neuro-immunology (Binnewies et al., 2018).
Understand the intrinsic stability of tumor built upon multilateral paracrine circuitry to devise effective treatment strategies
While detailed molecular mechanisms warrant further exploration, our study clearly revealed an under-appreciated complexity of TME organization in medulloblastoma. Rather than a one-on-one TME-tumor crosstalk, our data uncovered an intricate TME community in which trans-differentiation and a multilateral paracrine network support the robust growth of tumor cells. It should be noted that, while signaling plasticity could partially explain the problem of therapeutic resistance in cancer, cellular plasticity and multi-cell network could also contribute to tumor robustness. For example, an elegant modeling work demonstrated that a two-cell circuitry with reciprocally supportive paracrine factors could form a stable system to maintain tissue homeostasis that can withstand perturbations (Zhou et al., 2018). Conceivably, the three-cell system in medulloblastoma opens up even more channels of paracrine communications and would likely withstand severe perturbations during cancer therapy. While this study was focused on the SHH-subtype of medulloblastoma, it showcases a powerful technical platform that can be applied to deconstruct TME network within other cancer types (Binnewies et al., 2018; Palucka and Coussens, 2016). The deepened understanding of TME-tumor cell relationships carries great promise toward paradigm-shifting therapeutic strategies to cut off multiple paracrine crosstalks to fundamentally undermine the robustness of cancer.
STAR* METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
All Plasmid, Cell lines, tumor mouse model generated in this study will be made available upon request to the Lead Contact. Further information and requests for resources may be directed to, and will be fulfilled by the Lead Contact, Hui Zong (hz9s@virginia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice.
C57BL6 was obtained from Charles River. MADM-ML pair TG11ML, GT11ML JAX# 030578 (Henner et al., 2013), IGF1R-flox (Dietrich et al., 2000), IGF1-flox (Liu et al., 1998), CSF1R-iCre (Deng et al., 2010), CX3CR1-CreERT2 (Yona et al., 2013), Ptch1-KO JAX# 003081 (Goodrich et al., 1997), p53-KO JAX# 002101 (Jacks et al., 1994), p53-flox JAX# 008462 (Marino et al., 2000), Math1-Cre JAX# 011104 (Matei et al., 2005), Math1-CreERT2 (Machold and Fishell, 2005), Math1-GFP JAX# 013593 (Rose et al., 2009), IL4-GFP reporter (Mohrs et al., 2001), IL4-KO (Kuhn et al., 1991), and Rosa26-LSL-tdTomato JAX# 007908 (Madisen et al., 2010) were obtained from Jackson Laboratory or colleagues. Aldh1L1-GFP was ordered from Genesat (MMRRC# 011015-UCD) (Heintz, 2004). For all in vivo experiments, the ages of mice were indicated in figures or figure legends. Mice were separated into experimental groups on the basis of genotype and age. Female and male mice were used for all experiments to exclude gender effects. All animal procedures, including housing conditions and husbandry routines were performed according to the protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Virginia. Statistical tests were used to predetermine sample size of tumor mice. All mice are healthy, immune-competent except for genetic mutatations introduced for experimental purposes. Prior to experiments, they were not involved in previous procedures and remained durg- and test-naïve.
To test distinct hypotheses, in total we used ~10 mouse models in this study, which were derived from 4 basic models depicted in Supplemental Table 1.
METHOD DETAILS
Tamoxifen Administration
For adult mice, tamoxifen citrate tablets (20 mg/tablet; Mylan) were ground and dissolved in saline at 20 mg/ml and delivered via oral gavage.
Tumor dissection and tumor weight measurement.
To obtain the tumor weight, tumor-bearing mice were sacked at corresponding age. Tumor tissues (GFP+) was carefully dissected out under fluorescence microscopy and weighed by analytical balances.
Luminex.
Tumor or normal cerebellum tissues were dissected out and flash-frozen in liquid nitrogen. Frozen tissue pieces were then ground up and lysed in Tissue Extraction Reagent I (Life Technologies) supplemented with protease inhibitors and phosphatase inhibitors. The protein concentration of the lysate was determined by Pierce™ BCA protein Assay Kit (Thermo Scientific). All samples were adjusted to lmg/ml with Tissue Extraction Reagent I and submitted to UVA Flow Cytometry Core Facility to run a 32-plex cytokine/chemokine/growth factor panel on Luminex MAGPIX system. This 32-plex panel includes IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12, IL-13, IL-15, IL-17, Eotaxin, CXCL-10, KC/GRO, MCP-1, ΜΠΜ, MIP-2, MIG, LIF, Rantes, LIX, IFN, TNF, GM-CSF, M-CSF, G-CSF and VEGF. 25 μg total proteins extracted from tumor tissue and adjacent normal cerebellum were used for Luminex analysis. The concentration of each cytokine/chemokine/growth factor was normalized to total protein amount.
Quantitative PCR (qPCR).
We performed real-time PCR as described previously (Liu et al., 2011). Briefly, Total RNA was isolated from cells/tissues using TRIzol reagent (Invitrogen) followed by phenol/chloroform extraction and ethanol precipitation according to the method previously described (Miller et al., 2009). To extract total RNA from small quantity of cells/tissues or to obtain high quality RNA, RNAqueous-Micro kit (Ambion) or RNeasy micro kit (Qiagen) was used. cDNA was generated by using iScript cDNA synthesis kit (Bio-Rad). qPCR was performed based on SYBR green method using SyBR green/Rox PCR Master Mix (Kapa Biosystems). In some cases, qPCR was performed with Taqman method using gene-specific probes (ABI Applied Biosystems). All qPCR reactions were carried out according to manufacturer’s suggestions on ABI StepOnePlus real-time PCR system. Relative levels of cDNA for the interest genes were normalized with the level of those of Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) or M-CSF receptor (CSF1R) (when myeloid cells are concerned) in the same cDNA templates. The detailed primer sequences (for SyBR qPCR) and Taqman probes can be found in the resource table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-GFAP antibody | Dako | Cat# Z0334, RRID:AB_10013382 |
| rabbit anti-GFAP antibody | Abcam | Cat# ab7260, RRID:AB_30580 |
| rabbit anti-BLBP antibody | Millipore | Cat# AB9558, RRID:AB_2314014 |
| chicken anti-GFP antibody | Aves Labs | Cat# GFP-1020 RRID:AB_10000240 |
| goat anti-c-Myc Antibody | Novus Biochemicals | Cat# NB600-338 RRID:AB_10001879 |
| rat anti-BrdU antibody | Accurate Chemical & Scientific Corporation | Cat# OBT0030 RRID:AB_2313756 |
| sheep Anti-Digoxigenin Fab fragments Antibody, AP Conjugated | Roche | Cat# 11093274910, RRID:AB_514497 |
| Armenian Hamster anti-CD3 | eBioscience | Cat# 14-0031-82 RRID:AB_467049 |
| rabbit anti-NeuN | Abcam | Cat# ab177487, RRID:AB_2532109 |
| rabbit anti-P2ry12 | AnaSpec | Cat# 55043A, RRID:AB_2298886 |
| rabbit anti-ER-TR7 | Abcam | Cat# ab51824; RRID:AB_881651 |
| goat anti-PDGFRalpha | R&D | Cat# AF1062; RRID:AB_2236897 |
| rabbit anti-TMEM119 | Abcam | Cat# ab185333; RRID:AB_2687894 |
| rabbit anti-Iba 1 | Wako | Cat# 019-19741, RRID:AB_839504 |
| rabbit anti-cleaved caspase 3 | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| rabbit anti-CD31 | Thermo Fisher Scientific | Cat# MA5-16337, RRID:AB_2537856 |
| mouse anti-hNA | millipore | Cat# MAB1281, RRID:AB_94090 |
| mouse anti-hGFAP | Covance | Cat# SMI-21R-100, RRID:AB_509978 |
| mouse anti-Ki67 | BD Biosciences | Cat# 556003, RRID:AB_396287 |
| rabbit anti-CD68 | Bio-Rad | Cat# MCA1957GA, RRID:AB_324217 |
| Alexa Flour 647-CD192(CCR2) | eBioscience | Cat# 150612, RRID:AB_2616984 |
| Anti-mouse CD16/32 | eBioscience | Cat# 14-0161-82, RRID:AB_467133 |
| PE/Cy7-CD45 | BioLegend | Cat# 103223, RRID:AB_313006 |
| BV421-CD11b | BD Bioscience | Cat# 562605, RRID:AB_11152949 |
| PE-CD3 | eBioscience | Cat# 14-0031-82, RRID:AB_467049 |
| PE/Cy7-CD19 | eBioscience | Cat#25-0193-82, clone1D3 ,RRID:AB_925735) |
| APC-CD45 | BD Bioscience | Cat# 559864 clone 30-F11, RRID:AB_398672) |
| Biotin-NK1.1 | BioLegend | Cat#108703, clone PK136 RRID:AB_313390 |
| SA-BV605 | BD Bioscience | Cat#563260 RRID:AB_2738383 |
| R-phycoerythrin goat anti-rabbit IgG | Life Technologies | Cat#P2771MP N/A |
| Alexa-488 Donkey Anti-Mouse IgG (H+L) | Thermo Fisher Scientific | Cat# A21202, RRID: Ab_141607 |
| Alexa-555 Donkey Anti-Mouse IgG (H+L) | Thermo Fisher Scientific | at# A31570, RRID: B_2536180 |
| Alexa-647 Donkey Anti-Mouse IgG (H+L) | Thermo Fisher Scientific | Cat# A31571, RRID: AB_162542 |
| Alexa-488 Donkey Anti-Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# 711-545-152, RRID:AB_2313584 |
| Alexa-555 Donkey Anti-Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# A31572, RRID: AB_162543 |
| Alexa-647 Donkey Anti- Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# 711-605-152, RRID:AB_2492288 |
| Alexa-555 Donkey Anti-Rat IgG (H+L) | Thermo Fisher Scientific | Cat# 712-165-150, RRID:AB_2340666 |
| Alexa-647 Donkey Anti- Armenian Hamster IgG (H+L) | Thermo Fisher Scientific | Cat# 127-495-160, RRID:AB_2338995 |
| Alexa-647 Donkey Anti-Rat IgG (H+L) | Jackson Immuno Research Labs | Cat# 712-605-153, RRID:AB_2340694 |
| Bacterial and virus strains | ||
| NEB® 5-alpha Competent E. coli (High Efficiency) | NEB | C2987 |
| Biological Samples | ||
| Mouse tumor, cortex, spinal cord | This study | N/A |
| Human medulloblastoma | John Hopkins Hospital pathology core | N/A |
| Chemicals | ||
| Dextran −10 KD, Tetramethylrhodamine | Thermo Fisher Scientific | D1817 |
| Tamoxifen | Sigma | T5648 |
| Tamoxifen tablet | Actavis | NDC 0591-2473-30 |
| sunflower oil | Sigma | S5007 |
| MinElute PCR Purification Kit | Qiagen | ID:28004 |
| iScript™ cDNA Synthesis Kit, 100 x 20 μl rxns | Bio-Rad | #1708891 |
| AMPure XP bead | Bechman | REF A63881 |
| Qubit assay | Life Technology | REF Q32850 |
| Superscript III | Invitrogen | 18080-044 |
| RNAqueous-Micro kit | Ambion | AM1931 |
| LCM caps | Applied Biosystems | LCM0214 |
| Proteinase K | Sigma | P2308 |
| RNAse Η | Amersham | E70054Z |
| 5x terminal transferase buffer | Invitrogen | 16314-015 |
| dNTP | Roche | 11277049001 |
| High Fidelity PCR system | Roche | 11732650001 |
| PMSF | Sigma | P7626 |
| Terminal transferase | Roche | 3333575001 |
| 10x ThermoPol buffer | New England Biolabs | B9005S |
| AmpliTaq polymerase | Applied Biosystems | N8080156 |
| 0.5ml thin-walled PCR tube | Applied Biosystems | N8010611 |
| 0.2ml thin-walled PCR tube | Applied Biosystems | N8010612 |
| SP6 RNA polymerase | Roche | Cat# 11 487 671 001 |
| T7 RNA polymerase | Roche | Cat# 11 881 775 001 |
| Protector RNase Inhibitor | Roche | Cat# 03 335 399 001 |
| BCIP®/NBT Alkaline Phosphatase Substrate | Roche | B5655 |
| DNase 1 recombinant | Roche | Cat# 04 716 728 001 |
| TRIzol reagent | ThermoFisher Scientific | Cat#15596018 |
| pGEM®-T Easy Vector Systems | Promega | A1360 |
| Peptides and Recombinant Proteins | ||
| Mouse IL4 | R&D | Cat# 404-ML/CF |
| Insulin from bovine pancreas | Sigma-Aid rich | I6634 |
| Human HGF | Peprotech | Cat# 100-39 |
| Mouse HGF | Peprotech | Cat# 315-23 |
| Mouse IGF1 | Peprotech | Cat# 250-19 |
| Human IGF1 | Peprotech | Cat# 100-11 |
| IGFBP3 | Sigma | Cat# SRP3067-25UG |
| Critical commercial Assays | ||
| CD11b MACS beads | Miltenyi Biotec | Cat# 130-093-634 |
| Luminex Panel | Millipore | Cat# MCYTOMAG-70K |
| EdU Click assay | Invitrogen | Cat# A10277 |
| DIG RNA Labeling Mix | Roche | Cat# 11 277 073 910 |
| Deposited Data | ||
| LCM Tumor Astrocyte RNAseq | This paper | GEO: GSE111734 |
| Tumor associate microglia RNAseq | This paper | GEO: GSE109750 |
| microglia RNAseq | Bennett et al., 2016 | NCBI BioProject: PRJNA307271 |
| Experimental Models: Cell Lines | ||
| Tumor cell primary culture | This study | N/A |
| Tumor Associate microglia primary culture | This study | N/A |
| Tumor sphere primary culture | This study | N/A |
| Human Medulloblastoma Cell | Shu et al., 2008 | N/A |
| Experimental Models: Organisms/Strains | ||
| TG11ML, GT11ML | Henner et al., 2013 | JAX#030578 |
| IGF1 Rflox/flox | Dietrich et al., 2000 | JAX#012251 |
| IGF1flox/flox | Liu et al., 1998 | JAX#016831 |
| CSFIR-iCre | Deng et al., 2010 | JAX#021024 |
| CX3CR1-CreERT2 | Yona et al., 2013 | N/A |
| Ptch1-KO | Goodrich et al., 1997 | JAX#003081 |
| p53-KO | Jacks et al., 1994 | JAX#002101 |
| p53-flox | Marino et al., 2000 | JAX#008462 |
| Math1-Cre | Matei et al., 2005 | JAX#011104 |
| Math1-CreERT2 | Machold et al., 2005 | JAX#007684 |
| Math1-GFP | Rose et al., 2009 | JAX#013593 |
| IL4-GFP reporter | Mohrs et al., 2001 | JAX#004190 |
| IL4-KO | Kuhn et al., 1991 | JAX#002253 |
| Rosa26-LSL-tdT omato | Madisen et al., 2010 | JAX#007908 |
| Aldh1l1-GFP | Heintz et al., 2004 | GENESAT (MMRRC#011015-UCD) |
| Oligonucleotides | ||
| See Supplemental Table 3 | This study | N/A |
| Software and Algorithms | ||
| GSEA | Broad Institute | http://www.broadinstitute.org/gsea/index.jsp |
| GraphPad Prism 6.00 | GraphPad | www.graphpad.com |
| R language | R Core Team (2016) The R Project for Statistical Computing | http://www.r-project.org |
| Matlab | The MathWorks | https://www.mathworks.com/products/matlab.html |
Immunofluorescence staining and image processing.
In brief (Liu et al., 2011), mice were anaesthetized (60mg/kg ketamine; 12 mg/kg xylazine) and perfused through the right cardiac ventricle with PBS, followed by 4% paraformaldehyde (PFA). Brains were then post-fixed overnight at 4°C, cryoprotected in 30% sucrose (two days at 4°C) and embedded into optimal cutting temperature (O.C.T.) prior to cryo-sectioning on a cryostat. 12-20 μm sections were captured onto plus slides and dried at room temperature for 1 hour. Tissue slices were then rehydrated with PBS, blocked in 10% serum in PBST (PBS + 0.3-0.5% Triton X) for 1 hour at room temperature and incubated with primary antibodies (diluted in 1% serum PBST) overnight at 4°C. After several washes, Alexa dye conjugated secondary antibodies (Life Technologies) were applied at 1:500 for 2 hours at room temperature. Following several washes with PBS and counterstaining with DAPI, tissue slices were mounted in 70% glycerol with coverslips. Primary antibodies used were listed below: GFP (Chicken, Aves Labs), IBA1 (Rabbit, DAKO or Goat, Abeam), P2ryl2 (Rabbit, Abeam), TMEM119 (Rabbit, Abeam), GFAP (Rabbit, DAKO), PDGFRa (Goat, R&D), NeuN (Rabbit, Abeam), ER-TR7 (Rat, Abeam). To perform CD3e (Armenian Hamster, eBioscience) staining, spleen and tumor tissues were dissected out without perfusion and fixed with 4% PFA at room temperature for one hour. We used BrdU incorporation method to identify proliferating cells (Wojtowicz and Kee, 2006). Mice were first intraperitoneally injected with BrdU (50 mg/kg) prior to perfusion. To visualize incorporated BrdU in cells/tissues, slices were pre-treated with 2M HCL to denature the DNAs and expose BrdU incorporated regions for BrdU antibody (Rat, Bio-rad) to bind.
Images were acquired on confocal system (Zeiss LSM700 or Zeiss LSM880) and processed using ImageJ or Adobe Photoshop CS 6.0 (San Jose, CA). Morphology of Microglia and TAMs was traced by Neurolucida software (Nikon).
Quantification of TME cells throughout tumor development
Tissue sections from ≥3 mice at 3 distinct stages (early, mid, late) of tumor development were immunostained for GFP/RFP fluorescent proteins, CD31 to visualize blood vessels, IBA-1 to visualize microglia/macrophages, BLBP to visualize astrocyte cell bodies, and DAPI for all nuclei. Since medulloblastoma originates from granular neuron precursors (GNPs) in the external germinal layer (EGL), we chose EGL rather than adult cerebellum as the control for comparison to tumor regions. We dissected cerebellum from postnatal day 6 (P6) pups, performed the immunofluorescence staining with the corresponding marker proteins as in tumors. Three images were taken per tumor/EGL at 200X magnification throughout the tumor/ P6 cerebellum. Due to ambiguity in determining individual tumor GNPs, given the dense cellularity of these tumors, DAPI was used as a proxy for total tumor cell number and in the EGL. Some TME cells will also be included in this number, but their presence is so low, compared to GFP+ tumor cells, that inclusion of these cells in total tumor cell number will not significantly affect interpretation. Blood vessel density was determined by quantifying the % area occupied by CD31+ signal. Microglia/macrophage numbers were determined by counting the number of IBA-1+ cells per field of view. Astrocyte numbers were determined by counting the number of BLBP+ cells per field of view. The percentage of TME cell population numbers was calculated by dividing the # of marker-positive cells by the # of total DAPI+ cells in the same field of view and multiplied by 100 to output a percentage value. ImageJ was used for image processing and counting. Excel and Graph Pad Prism were used to perform statistical analysis and a one-way ANOVA was applied to determine significance and error bars represent standard deviation.
RNA In situ hybridization.
Please refer all probes (IGF1 and IL4) for in situ hybridization to Allen Brain atlas. In brief (Zhao et al., 2009), DNA fragment amplified by PCR were ligated into pGEMT-easy vector. The plasmids containing the relevant DNA fragment were purified and then linearized by restriction enzyme. The linearized plasmids were used as template to generate labeled RNA probes with Digoxin by transcription in vitro (Roche). RNA probes (1μg/ml) were hybridized with cryosectioned slices (12 μm) at 64°C overnight. BCIP/NBT substrates were used to visualize the hybridized cells/tissues. Immunofluorescence staining was carried out after hybridization if we need to pinpoint the particular cell types with positive RNA hybridization signals. In that case, the hybridized slides were further fixed with 4% paraformaldehyde (PFA) for fifteen minutes at room temperature, blocked with 5% donkey serum for one hour at room temperature, followed by primary (IBA, Ki67 and GFAP) and secondary antibody incubation at 4°C overnight. The nuclei were counterstained with DAPI (4′, 6-diamidino-2-phenylindole). At least three tumors were analyzed. Images were acquired from Olympus fluorescence microscope and processed by imageJ.
Fluorescent In Situ Hybridization of chromosome loci with immunofluorescence
Tissue microarray (TMA) paraffin sections were mounted on silanized slides, baked for 5 minutes at 90 °C, and then de-paraffinized in xylene. Slides were briefly dehydrated in 100% ethanol followed by 35 minutes in hot 1 mM EDTA. Sections were rinsed in dH2O and incubated with GFAP (Abcam) diluted in CAS Block (Invitrogen Corporation). Slides were washed in 1X PBS and then incubated with FITC conjugated fluorescent secondary antibody (Abcam) diluted in CAS Block. There is an additional wash in 1XPBS followed by fixation in 4% paraformaldehyde (USB Corporation) for 20 minutes. Slides were dehydrated in an ethanol series and air dried. 20 μl of PTCH1/CEP9 (with GFAP) probe working solution (Empire genomics) was applied to the hybridization area, a 24x50 mm coverslip is placed over the top, and the edges of the coverslip were sealed with a continuous bead of rubber cement. The slide and probe were co-denatured at 95°C for 4 minutes and hybridized 24 hours or more at 37°C in a humidified chamber. Slides were then washed in 2X saline sodium citrate buffer (SSC)/0.1%NP40 at 70°C for 1 minute and DAPI counterstain (Vector Laboratories) was applied as well as a glass coverslip. Visualization of the dual FISH/immunoflourescence signals was accomplished by use of a fluorescent microscope with standard filters.
Laser Capture Microdissection
We chose GFP-guided single cell Laser Capture Microdissection over tissue dissociation considering that non-physiological noises at transcription level could occur during tissue dissociation. For example, an immune signature would be hard to interpret because it is known that glia cells tend to get into an “activated” state after going through a dissociation process (Bennett et al., 2016).
The laser capture microdissection (LCM) experiment was designed and modified according to previous studies (Janes et al., 2010; Wang and Janes, 2013). Briefly, tumor brains were removed and slowly lowered into liquid nitrogen-cooled methyl-butane. The brains were kept fully submerged for 1 minute then store at −80°C. Tumor brain was cut at 8 μm thickness on a cryostat and sections were collected on positively-charged slides. Slides were fixed in 100% ethanol for 5 minutes at room temperature and then transferred into 100% xylene for 5 minutes at room temperature. Slide was then air-dried for 5 minutes. Laser capture microdissection of individual cells was performed on an ArcturusXT LCM system (Applied Biosystems) with an infrared laser using CapSure HS LCM caps after optimization of laser power and duration.
Since paraformaldehyde fixation would negatively affect the RNA quality and the subsequent sequencing, all samples were snap frozen without fixation. We found that GFP molecules quickly diffuse away from labeled cells once tissue sections melt or encounter moisture. After extensive troubleshooting we found that pre-chilling slides in the cryostat could prevent melting once tissue sections touch the slide. This trick gave us a couple of precious seconds to dip the slide with still-frozen sections into ethanol to not only fix the RNA but also remove water from sections to prevent GFP loss.
We sectioned the tissue at 8μm to minimize contamination from the z-axis. Initially we tried to use UV laser and found that single cell cutting with UV laser completely destroyed RNAs in the cell, most likely due to the high energy level of UV laser. When we switched to lower-energy IR laser that melts a small point of wax onto each target cell to pull it off, we found that the strength of attachment of sections to slides needs to be optimized every time for precise cell collection. If they attached too loosely, the IR laser would pull off too many surrounding tissue with the target cells. On the other hand, if tissue sections attached too tightly, the IR laser would fail to pick up individual target cells into the collection cap. We controled the attachment strength by warming up pre-chilled slides for ~1 second (touch finger tip under the slide) then immediately proceeded to LCM. If we encountered problems, the time could be lengthened slightly to increase attachment strength, or shortened slightly to reduce it.
RNA extraction, cDNA amplification, library construction and RNA sequencing of LCM micro-dissected tissues/cells
RNA extraction and cDNA amplification procedures were performed as described under optimized conditions for 50 microdissected cells. For each sample, 250 cells were microdissected per LCM cap and split into five 50-cell technical replicates (as a control, one replicate did not undergo reverse transcription) after elution from the cap. Reamplified cDNA (~500 bp 3’ ends) was purified away from primer concatemers by two rounds of purification with 0.7× volume of AMPure XP beads (Beckman) according to the manufacturer’s recommendations, and purified cDNA was quantified by Qubit assay (Life Technologies). For each sample, 1 ng was tagmented with the Nextera XT kit (Illumina) and sequenced as 75 bp paired-end reads on aNextSeq instrument using v2 reagents (Illumina). 14–22 million reads per sample were filtered for signal to noise (chastity filtered), assessed for overall quality with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and then mapped with STAR (Dobin et al., 2013) against the mouse genome build mm 10. Data were quantified as reads per million (RPM), and log(RPM+l) values were clustered hierarchically in MATLAB after row standardization with a Euclidean distance metric and Ward’s linkage.
Primary tumor GNP purification and in vitro culture
Primary tumor GNPs were purified with percoll gradient method (Lee et al., 2009). In brief, tumor mice were anaesthetized (60mg/kg ketamine; 12 mg/kg xylazine) and tumor tissues were dissected out and digested with Papain (Worthington Biochemical) for 45 minutes at room temperature. Digested tumor pieces were then triturated into single cell dissociates and cleared by passing through 70um cell strainer. The cleared dissociates were applied to percoll gradient (35%/65%) followed by centrifugation at 1300g for 30 minutes. The cells located at the interface of these two gradient layers are mainly tumor GNPs. These cells were carefully collected and transferred into a new polystyrene tube, washed and spun down at 1000g for 5 minutes. To evaluate the effects of growth factors (such as IGF1 and HGF), 1x105 cells were seeded onto PDL pre-coated coverslips in Neurobasal media supplemented with B27 minus insulin. 4 hours post seeding, IGF1 or HGF was added at varying concentrations. After 2 days in culture, EdU was added at 0.1mM for 3 hours prior to cell harvesting. The proliferation of tumor GNPs was evaluated by EdU incorporation. All experiments were repeated at least three times. Images were acquired on Olympus fluorescence microscope and processed by ImageJ program.
Isolation of primary Tumor Associated Microglia/Macrophages (TAMs).
This purification method was adapted from adult microglia purification method (Nikodemova and Watters, 2012). All steps were performed on ice whenever possible. To get rid of circulating immune cells from brain/tumor tissues, mice underwent trans-cardiac perfusion with sterile ice-cold PBS before tumors were dissected out. Dissected out tumor pieces were digested with Papain (Worthington Biochemical) at 37°C for 45 minutes and dissociated by trituration. Tissue debris was removed by passing tumor dissociates through a 70um cell strainer. Cleared tumor dissociates were then applied to Percoll gradient (18.6%/62.5%) centrifugation at 1300g for 30 minutes to get rid of myelin and cell debris. The cells that were collected from interface between 18.6% and 62.5% Percoll layers were then captured by anti-CD11b magnetic beads (Meltinyi Biotec, Germany) to enrich tumor associated CD11b+ myeloid populations (TAMs). Finally TAMs were separated out in a magnetic field using MS columns (Meltinyi Biotec, Germany) or through autoMACS Pro Separator.
Co-culture of TAMs with Tumor GNPs.
TAMs were isolated as described above. To ensure the sterility during culture, after trans-cardiac perfusion, tumor mouse was decapitated and soaked completely in betadine before the tumor pieces were dissected out. Also in the final step of TAMs purification, TAMs were manually separated out through MS columns in a laminar hood. The viability of TAMs were evaluated by Trypan blue staining and cell counting under the microscope based on dye exclusion. We consistently reached more than 90% viability. The purity was assessed by counting the contaminated Math1-GFP+ tumor cells and by Iba1 staining. Usually we have less than 10% tumor cell contaminated and Iba1+ cells are approximately 90%. To set up co-culture with tumor cells, acutely prepared TAMs were seeded up to 10K onto coverslips and cultured in Neurobasal media supplemented with B27 (Life Technologies) and 10ng/ml M-CSF (Peprotech) overnight to recover. Next day, TAMs were washed twice with D-PBS. GFP+ tumor GNPs were seeded on top of TAMs at 100K in Neurobasal media supplemented with B27 minus insulin and 5ng/ml M-CSF. After 2 days in culture, EdU was added at 0.1mM for 3 hours prior to cell harvesting. The proliferation of GFP+ tumor GNPs was evaluated by EdU incorporation.
Flow cytometry.
Immune cells were isolated from tumors as described above. After percoll gradient centrifugation, myelin-free cell suspension was incubated with anti-CD45 magnetic beads (Meltinyi Biotec, Germany) at 4°C for 15 minutes to capture all immune cells in the tumors. All the tumor associated immune cells (CD45+) were then separated out through autoMACS Pro Separator. To pin down the identities of these immune cells, surface marker staining followed by flow cytometry was performed with these immune cells. All the staining procedures were carried out at 4°C and in PBS with 0.5% BSA+ 2mM EDTA to maximize the cell viability. To reduce the non-specific immunofluorescence staining, the cells were pre-incubated with anti-mouse CD16/32, a Fc receptor blocker (eBioscience) for 10 minutes. Then the cells were incubated in in an antibody cocktail consisting BV421 anti-CD11b, APC anti-CD45 (BD Bioscience), PE-Cy7 anti-CD19, PE anti-CD3e (eBioscience), biotinylated anti-NK1.1, Zombie Aqua Live/dead dye (BioLegend) for 30 minutes. Cells were then washed with PBS/0.5%BSA/2mM EDTA, and centrifuged at 350g for 10 minutes at 4°C. Pellets were re-suspended and incubated with BV605 conjugated streptavidin for 30 minutes, followed by centrifugation at 350g for 10 minutes at 4°C. Cell pellets were re-suspended in PBS and proceed for flow analysis. In a separate analysis of myeloid cell surface markers, the cells were pre-incubated with anti-mouse CD16/32, a Fc receptor blocker (eBioscience) for 10 minutes. Then anti-P2ry12 (AnaSpec, rabbit polyclonal) was added at 1:400 and incubated for 30 minutes. Cells were washed with PBS/0.5%BSA/2mM EDTA, and centrifuged at 350g for 10 minutes at 4°C. Pellets were re-suspended in an antibody cocktail consisting BV421 anti-CD11b (BD Bioscience), 647 anti-CD192 (CCR2), BV605 anti-CD11c, PE-Cy7 anti-CD45, Zombie Aqua Live/dead dye (BioLegend), PE goat anti-rabbit secondary antibody (ThermoFisher Scientific) and incubated for 30 minutes, followed by centrifugation at 350g for 10 minutes at 4°C. Cell pellets were re-suspended in PBS and proceed for flow analysis with BD LSRFortessa cytometer. Data were analyzed with FlowJo v10 software.
RNAseq and data analysis of TAMs
Total RNAs of TAMs and normal microglia from age matched mice were extracted using the RNeasy micro kit (QIAGEN) according to manufacturer’s protocol. RNA Quality was assessed by Bioanalyzer (Agilent Technologies) and samples with RNA integrity >8 were used for library construction. First and second-strand cDNA synthesis, RNAseq library construction and sequencing were carried out by BGI Americas. High-quality libraries were sequenced by the Illumina® Hiseq2500 sequencer to obtain 75bp paired-end reads. Reads alignment to mmlO mouse reference genome, data processing and TPM (Total read per million) calculation were done by using package in the R statistical computing environment (Anders and Huber, 2010; Patro et al., 2014; Soneson et al., 2015). After estimating transcript abundances with Sailfish (Patro et ak, 2014), we converted the abundance estimates to counts of reads mapping to GENCODE genes using the tximport package in the R statistical computing environment (Soneson et ak, 2015). We then used the DESeq2 Bioconductor package (Love et ak, 2014) to normalize count data, estimate dispersion, and fit a negative binomial model for each gene. GENCODE/Ensembl gene IDs mapping to predicted genes were excluded and the Benjamini-Hochberg False Discovery Rate procedure was used to re-estimate the adjusted p-values for GENCODE/Ensembl gene IDs mapping to known genes. Genes were filtered for low abundance by selecting on genes with a TPM > 20 in at least one group. Pathway analyses were performed using a compilation of previously defined pathways from consortiums such as GO via the Broad Institute’s MSigDB resource (Subramanian source). Fisher’s exact tests were performed to determine significant pathway enrichment. All codes used for analyses are available upon request.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was determined using GraphPad Prism 6 using unpaired Student’s two-tailed t-test, one-way ANOVA or two-way ANOVA, according to test requirements. No inclusion/exclusion criteria were pre-established. Grubbs’ Outlier Test was used to determine outliers, which were excluded from final analysis. A P value of <0.05 (indicated by one asterisk), <0.01 (indicated by two asterisks), <0.001 (indicated by three asterisks), <0.0001 (indicated by four asterisks) or were considered significant.
DATA AND CODE AVAILABILITY
RNA-sequencing data of TuAstrocyte were deposited in GEO (Accession number is GSE111734). RNA-sequencing data of TAMs were deposited in GEO (Accession number is GSE109750). This study did not generate any unique code..
Supplementary Material
SUPPLEMENTAL VIDEO CAPTION. Related to Figure 1.
3D rendering of the image in Figure 1M.
Supplemental Table 1. Related to STAR Methods section “EXPERIMENTAL MODEL AND SUBJECT DETAILS, Mice”, and to the first section in “RESULTS, Establishment of MADM-based and other genetic models for medulloblastoma”. Medulloblastoma models used in this paper.
Supplemental Table 2. Related to Figure 7. Luminex profiling of cytokines and chemokines. Tumor tissues were dissected out based on Math1-GFP expression and lysed in Tissue Extraction Reagent I (Invitrogen) supplemented with protease inhibitors. Normal cerebelli from age-matched mice were processed in the same fashion as control samples. Protein concentration of the lysates was determined by BCA protein quantification method. All lysates were carefully adjusted to 1.5mg/ml with lysis buffer, and 25μl each was applied to mouse 32-plex cytokine/chemokine luminex panel. The absolute quantity of cytokine/chemokine present in cell lysate was expressed as picogram per 10ug of tissue lysate. Four tumors and three normal cerebella were used for this study.
Supplemental Table 3. Related to “Key Resources Table, Oligonucleotides section”. Oligonucleotides used in this paper. This is a list of oligonucleotides that were used for mouse genotyping, qRT-PCR, and probes for in situ hybridization.
Supplemental Figure 1. Related to Figure 1. TME cells in medulloblastoma and the characterization of Math1-Cre. A, Representative images and the scoring scheme for TME cell composition in human SHH-subtype medulloblastoma samples. B, Data summary of TME cell composition from A. C, In adult MADM-WT mice, only NeuN+ granule neurons in IGL were labeled, demonstrating the faithful expression of Math1-Cre in GNPs. D, There was no MADM labeling of potential TME cells, including blood vessels (CD31), microglia (IBA-1), and astrocytes (BLBP, GFAP). E, In the MADM model, tumor GNPs are GFP+ while TME cells are expected to be unlabelled, allowing cellular resolution analyses of tumor-TME interactions from tumor initiation throughout progression. F, GFP+ astrocytes are present only in tumors (F’), but never in tumor-free brain regions (F”). G, A mouse medulloblastoma model in which all cells from GNP lineage is tdTomato+ while astrocytes are labeled by Aldh1L1-GFP. If tumor-associated astrocytes were derived from tumor cells, they should be yellow (Aldh1L1-GFP+ and tdTomato+).
[WM= White matter]
Scale bars: A, D and F=50μm, F’ and F”=10μm, G=25μm
Supplemental Figure 2. Related to Figure 2. Validation of tumor-to-astrocyte trans-differentiation with multiple approaches.
A, BLBP+ tumor-associated astrocytes had only one nucleus (representative image of >100 cells/tumor in 10 tumors), suggesting that GFP-labeling of astrocyte-like cells did not result from cell fusion. B-D, Mouse model and tamoxifen dosing scheme (B), experimental outcome predictions (C), and a representative image (D) of a mouse medulloblastoma model, in which astrocytes were labeled with tdTomato following the excision of the “floxed stop cassette” by GLAST-CreER upon tamoxifen injection, while TuGNPs were labeled by Math1-GFP. The fact that none of the tdTomato-labeled cells expressed GFP suggests that cell fusion did not occur. E, Tumor-associated astrocytes were labeled with Aldh1L1-GFP in a medulloblastoma model in which tumor lineage is labeled with tdTomato. This enables distinction of tumor-associated astrocytes (yellow) from normal astrocytes (green) and tumor GNPs (red). F, Circled region indicated one micro-dissected tumor-associated astrocyte with minimal contamination of neighboring cells. The gamma scaling in the GFP channel was adjusted to 0.5 to highlight GFP fluorescence clearly above tissue autofluorescence. G-H, Purity verification of LCM-dissected tumor-associated astrocytes vs. TuGNPs before RNA-sequencing. qRT-PCR showed minimal Math1 expression (Math1: 72.92±16.01 TuGNPs vs. 5.252±1.13 tumor-derived astrocytes, p=0.0002 t-test, n=4) and strong enrichment of Aldh1L1 expression (Aldh1L1: 0.078±0.053 TuGNPs vs. 20.15±4.892 TuAstrocytes, p=0.0002 t-test, n=4) in the tumor-derived astrocytes pool. I, Anti-human GFAP antibody specifically labels human astrocytes (arrowhead) in mouse brain grafted with human medulloblastoma cells. J, Serially xenografted SHH-subtype human medulloblastoma cells (passage 12 shown here) gave rise to sporadic cells with astrocytic morphology that stained positive for human GFAP protein throughout the tumor mass, suggesting trans-differentiating activity of human medulloblastoma cells could be maintained in the long-term.
[GLAST= glutamate transporter],
Scale bar: A=10μm, F =20μm, D, I and J= 50μm. Data are Means±SD. Student’s t-test. n.s.: not significant; ***p<0.001.
Supplemental Figure 3. Related to Figure 3. Supportive role for TuAstrocytes in medulloblastoma progression. A-C, In PNLs, the proliferation of TuGNPs strongly correlated with the presence of TuAstrocytes. D, TuGNPs (Math1-GFP+) proliferated actively (EdU+) in spheres (arrows) but not as dispersed cells (circled). E-G, TuAstrocytes (Aldh1L1-GFP+) gradually appeared in tumor spheres.
Scale bars: A, B, D, E and F= 50μm;
Supplemental Figure 4. Related to Figure 4. Inactivation of IGF1 receptor (IGF1R) in GNPs delays tumor progression. A, Medulloblastoma mouse model with IGF1R knockout in GNPs. B, Mice with different genotypes (IGF1R−/−; IGF1R−/+; IGF1R+/+) were dissected at postnatal day 40 and tumor tissues (Math1-GFP+) were collected and weighted. Dotted line indicates cerebellum area. C, Graph of tumor weight from tumor mice of different genotypes: IGF1R−/− (n=14), IGF1R+/− (n=20) and IGF1R+/+ (n=11). D, Tamoxifen (150 mg/kg) did not affect tumor progression in comparison to vehicle control. n=10 for each group. E, Schematic illustration on IGF1R inactivation in tumor cells. After 5 pulse Tamoxifen gavage, the tumor was harvested and apoptotic index and cell cycle exit index were quantified in tissue sections. F-G, Elevated apoptotic cells in Tamoxifen-treated group in comparison to vehicle group. H-I, There were more cells that exited cell cycle (BrdU+ Ki67−) in Tamoxifen-treated group in comparison to vehicle group.
Scale bar: B=1mm, F and H =20μm. Data are Means±SD, one-way ANOVA (C) or Student’s t-test (D G and I). n.s.: not significant, *p<0.05; **p<0.01; ***p<0.001.
Supplemental Figure 5. Related to Figure 5. TAMs display activated morphology and express cell markers of both microglia and monocyte-derived macrophages. A-B, Morphologies of TAMs were traced by neurolucida program in big tumors (B), in comparison to microglia in normal cerebellum (A). C, Schematic diagram of branching pattern analysis. D-F, Morphological analysis of microglia vs TAMs showed that TAMs had enlarged cell bodies (D) and shortened processes (E,F) in comparison to normal microglia. n=213 normal microglia and 288 TAMs from 5 tumor mice. G. Flow cytometry analysis showed that TAMs expressed both microglia marker (p2ry12) and macrophage marker (CCR2). H, Immunofluorescent staining of P2ry12 (red, microglia specific marker) and IBA1 (green, pan-microglia/macrophage marker) showed that TAMs expressed P2ry12. I, Immunofluorescent staining of TMEM119 (red, microglia specific marker) and IBA1 (green, pan-microglia/macrophage marker) showed that TAMs were TMEM119− while normal microglia were TMEM119+.
Scale bar: H and I=30μm. Data are Means±SD, one-way ANOVA. n.s.: not significant, *p<0.05; **p<0.01; ***p<0.001.
Supplemental Figure 6. Related to Figure 6. Validation of the lineage tracing genetic tool to differentiate microglia from circulating monocytes in the medulloblastoma model. A, Schematic diagram of the tool validation: In brief, pups were subcutaneously injected with Tamoxifen (50 mg/kg) at postnatal day four to day eight, long before tumor initiation. One day (acute) or Twenty-eight days (4 weeks) after injection, peripheral blood cells were collected for flow analysis and brains were collected for immunofluorescence staining. B, Histogram of flow analysis of tdTomato+ cells in the peripheral blood showed that tdTomato-labeled cells at acute phase were turned over by 4 weeks (still long before tumor formation). C, Quantification of flow analysis of tdTomato+ cells in the peripheral blood. D, Immunofluorescent staining showed that all microglia (IBA1+, green) were tdTomato+ (red) in both acute and 4 weeks group. E, Quantification from immunofluorescent staining showed that microglia had rare turnover in the cerebellum.
Scale bar: D=30μm, n=5 mice for each group. Data are Means±SD, Student’s t-test. n.s.: not significant; ****p<0.0001.
Supplemental Figure 7. Related to Figure 7. IL4 produced by TuAstrocyte activate TAMs. A. TAMs exhibited robust tolerogenic gene program expression. Pathway analysis of the significantly up- and down-regulated transcripts in tumor-associated microglia (focusing on immune signatures) revealed robust differential expression of genes associated with IL-4 signaling, IL-10 production, regulation of adaptive immunity, and macrophage/microglia activation. Shown in red were the significantly up-regulated genes in tumor associated microglia relative to normal microglia from age-matched mice, whereas blue denoted down-regulated genes. Immune signature genes were pulled from the MSigDB database. B, We observed significantly differentially regulated gene signatures associated with additional IL-4/STAT6 signaling components, JAK2 and ERK1/2. Shown is the level of significance of a given pathway (the negative log of the p value) as well as the number of significantly differentially expressed genes in a given pathway over the total number of genes reported in that pathway. P values were determined using Fisher’s exact tests. n=5 tumor mice for RNAseq . C, In situ hybridization showed that IL4 mRNA could not be detected in IL4 KO tumor mice, validating the effectiveness of gene KO. D, Immunofluorescent staining of IBA1 showed that the cell density of TAMs was similar between IL4 wildtype and IL4 KO tumors. E-F, Immunofluorescent staining of CD3 shows that there were no detectable T cells in the tumor mass. G, Schematic diagram of how to use a medulloblastoma model containing both IL4-GFP reporter and tdTomato tumor lineage marker to investigate the identity of IL4-producing cells. H, IL4-GFP overlapped with GFAP and tdTomato staining in the tumor mass, suggesting that IL4 was produced by tumor-derived astrocytes. n=3 tumor mice (at least) for each panel.
Scale bar: C=200μm, D, E F and H=20μm. Data are Means±SEM, Student’s t-test. *p<0.05. n.s.: not significant, *p<0.01.**p<0.001.
ACKNOWLEDGEMENTS
We thank Tajie H. Harris, Chris Doe, David Rowitch, Praveen Raju, Melanie Rutkowski, and Jonathan Kipnis for critical discussions and insightful feedback for our manuscript; Ryan A. Llewellyn, Noel C. Derecki, James C. Cronk, and UVa Flow Cytometry Core Facility for help with flow analysis; Fujun Qin for helping with GSEA analysis; UVa Advanced Microscopy Facility for imaging analysis; and Steffen Jung for providing CX3CR1-CreER mouse line. This project was partially supported by grants to H.Z. (DoD W81XWH-11-1-0557, NIH R01NS097271) K.A.J (R01CA194470 and U01CA215794), T.P.B (R21HL143025), and C.G.E. (R01NS055089). P.B.V. was supported by NIH/NINDS pre-doctoral fellowship (F31-NS076313). J.S.A.P. is supported by Mark Foundation Fellowship from the Cancer Research Institute, a NCI 1K99CA237728-01, and the Burroughs Wellcome PDEP award. H.Z. and K.A.J. were Pew Scholars in Biomedical Sciences, supported by the Pew Charitable Trusts.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Abbott NJ, Ronnback L, and Hansson E (2006). Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7, 41–53. [DOI] [PubMed] [Google Scholar]
- Ahmed F, and Haass NK (2018). Microenvironment-Driven Dynamic Heterogeneity and Phenotypic Plasticity as a Mechanism of Melanoma Therapy Resistance. Frontiers in oncology 8, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, and Sofroniew MV (2016). Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arina A, Idel C, Hyjek EM, Alegre ML, Wang Y, Bindokas VP, Weichselbaum RR, and Schreiber H (2016). Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc Natl Acad Sci U S A 113, 7551–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, and Cushing H (1925). Medulloblastoma Cerebelli: a Common Type of Midcerebellar Glioma of Childhood. Arch Neurol Psychiatry 2, 192. [Google Scholar]
- Balkwill FR, Capasso M, and Hagemann T (2012). The tumor microenvironment at a glance. Journal of cell science 125, 5591–5596. [DOI] [PubMed] [Google Scholar]
- Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, Mirlas-Neisberg N, Cardon M, Vaknin I, Cahalon L, et al. (2013). CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc Natl Acad Sci U S A 110, 2264–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, Plowey ED, and Barres BA (2018). A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 98, 1170–1183 e1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, et al. (2016). New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 113, E1738–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradding P, Feather IH, Howarth PH, Mueller R, Roberts JA, Britten K, Bews JP, Hunt TC, Okayama Y, Heusser CH, et al. (1992). Interleukin 4 is localized to and released by human mast cells. The Journal of experimental medicine 176, 1381–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, and Sofroniew MV (2014). Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger PC, Grahmann FC, Bliestle A, and Kleihues P (1987). Differentiation in the medulloblastoma. A histological and immunohistochemical study. Acta Neuropathol 73, 115–123. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, et al. (2014). Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanmee T, Ontong P, Konno K, and Itano N (2014). Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 6, 1670–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JE, Andreasen SO, Christensen JP, and Thomsen AR (2001). CD11b expression as a marker to distinguish between recently activated effector CD8(+) T cells and memory cells. International immunology 13, 593–600. [DOI] [PubMed] [Google Scholar]
- Chung AS, Lee J, and Ferrara N (2010). Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer 10, 505–514. [DOI] [PubMed] [Google Scholar]
- Colombo E, and Farina C (2016). Astrocytes: Key Regulators of Neuroinflammation. Trends in immunology 37, 608–620. [DOI] [PubMed] [Google Scholar]
- de Visser KE, Eichten A, and Coussens LM (2006). Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 6, 24–37. [DOI] [PubMed] [Google Scholar]
- DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, and Coussens LM (2009). CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Zhou JF, Sellers RS, Li JF, Nguyen AV, Wang Y, Orlofsky A, Liu Q, Hume DA, Pollard JW, et al. (2010). A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am J Pathol 176, 952–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich P, Dragatsis I, Xuan S, Zeitlin S, and Efstratiadis A (2000). Conditional mutagenesis in mice with heat shock promoter-driven cre transgenes. Mamm Genome 11, 196–205. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, and Alvarez-Buylla A (1999). Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97, 703–716. [DOI] [PubMed] [Google Scholar]
- Farina C, Aloisi F, and Meinl E (2007). Astrocytes are active players in cerebral innate immunity. Trends in immunology 28, 138–145. [DOI] [PubMed] [Google Scholar]
- Filiano AJ, Gadani SP, and Kipnis J (2015). Interactions of innate and adaptive immunity in brain development and function. Brain research 1617, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filley AC, Henriquez M, and Dey M (2017). Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget 8, 91779–91794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J (1971). Tumor angiogenesis: therapeutic implications. N Engl J Med 285, 1182–1186. [DOI] [PubMed] [Google Scholar]
- Fournier T, Riches DW, Winston BW, Rose DM, Young SK, Noble PW, Lake FR, and Henson PM (1995). Divergence in macrophage insulin-like growth factor-I (IGF-I) synthesis induced by TNF-alpha and prostaglandin E2. J Immunol 155, 2123–2133. [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, and Bronte V (2012). Coordinated regulation of myeloid cells by tumours. Nature reviews Immunology 12, 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, and Fu YX (2013). Innate and adaptive immune cells in the tumor microenvironment. Nature immunology 14, 1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascard P, and Tlsty TD (2016). Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev 30, 1002–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosn EE, Yang Y, Tung J, Herzenberg LA, and Herzenberg LA (2008). CD11b expression distinguishes sequential stages of peritoneal B-1 development. Proc Natl Acad Sci U S A 105, 5195–5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson RJ, and Ellison DW (2008). The origins of medulloblastoma subtypes. Annu Rev Pathol 3, 341–365. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M, et al. (2016). Origin, fate and dynamics of macrophages at central nervous system interfaces. Nature immunology 17, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Muller PF, Wolf Y, Varol D, Yona S, Brendecke SM, Kierdorf K, Staszewski O, Datta M, et al. (2013). A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci 16, 1618–1626. [DOI] [PubMed] [Google Scholar]
- Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, et al. (2015). Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, et al. (2003). A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925. [DOI] [PubMed] [Google Scholar]
- Gonzalez PP, Kim J, Galvao RP, Cruickshanks N, Abounader R, and Zong H (2018). p53 and NF 1 loss plays distinct but complementary roles in glioma initiation and progression. Glia 66, 999–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, and Scott MP (1997). Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, et al. (2014). Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves A, Kihara Y, Jonnalagadda D, Rivera R, Kennedy G, Mayford M, and Chun J (2018). A Functionally Defined In Vivo Astrocyte Population Identified by c-Fos Activation in a Mouse Model of Multiple Sclerosis Modulated by S1P Signaling: Immediate-Early Astrocytes (ieAstrocytes). eNeuro 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Coussens LM (2012). Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322. [DOI] [PubMed] [Google Scholar]
- Harris TH, Banigan EJ, Christian DA, Konradt C, Tait Wojno ED, Norose K, Wilson EH, John B, Weninger W, Luster AD, et al. (2012). Generalized Levy walks and the role of chemokines in migration of effector CD8+ T cells. Nature 486, 545–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz N (2004). Gene expression nervous system atlas (GENSAT). Nat Neurosci 7, 483. [DOI] [PubMed] [Google Scholar]
- Hemmer B, Kerschensteiner M, and Korn T (2015). Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol 14, 406–419. [DOI] [PubMed] [Google Scholar]
- Henner A, Ventura PB, Jiang Y, and Zong H (2013). MADM-ML, a Mouse Genetic Mosaic System with Increased Clonal Efficiency. PLoS One 8, e77672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglund RA, and Maghazachi AA (2014). Multiple sclerosis and the role of immune cells. World journal of experimental medicine 4, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, and Nedergaard M (2007). Glial regulation of the cerebral microvasculature. Nat Neurosci 10, 1369–1376. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, and Weinberg RA (1994). Tumor spectrum analysis in p53-mutant mice. Current biology : CB 4, 1–7. [DOI] [PubMed] [Google Scholar]
- Janes KA, Wang CC, Holmberg KJ, Cabral K, and Brugge JS (2010). Identifying single-cell molecular programs by stochastic profiling. Nat Methods 7, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R (2016). The biology and function of fibroblasts in cancer. Nat Rev Cancer 16, 582–598. [DOI] [PubMed] [Google Scholar]
- Kawai K, Tsuno NH, Matsuhashi M, Kitayama J, Osada T, Yamada J, Tsuchiya T, Yoneyama S, Watanabe T, Takahashi K, et al. (2005). CD11b-mediated migratory property of peripheral blood B cells. The Journal of allergy and clinical immunology 116, 192–197. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Rajewsky K, and Muller W (1991). Generation and analysis of interleukin-4 deficient mice. Science 254, 707–710. [DOI] [PubMed] [Google Scholar]
- Kumar MP, Du J, Lagoudas G, Jiao Y, Sawyer A, Drummond DC, Lauffenburger DA, and Raue A (2018). Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell reports 25, 1458–1468 e1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara T, Warr G, Loy J, and Bravo R (1997). Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. The Journal of experimental medicine 186, 1757–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake FR, Noble PW, Henson PM, and Riches DW (1994). Functional switching of macrophage responses to tumor necrosis factor-alpha (TNF alpha) by interferons. Implications for the pleiotropic activities of TNF alpha. The Journal of clinical investigation 93, 1661–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gros G, Ben-Sasson SZ, Seder R, Finkelman FD, and Paul WE (1990). Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. The Journal of experimental medicine 172, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Greene LA, Mason CA, and Manzini MC (2009). Isolation and culture of postnatal mouse cerebellar granule neuron progenitor cells and neurons. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L, et al. (2011). Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JL, Grinberg A, Westphal H, Sauer B, Accili D, Karas M, and LeRoith D (1998). Insulin-like growth factor-I affects perinatal lethality and postnatal development in a gene dosage-dependent manner: manipulation using the Cre/loxP system in transgenic mice. Molecular endocrinology 12, 1452–1462. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machold R, and Fishell G (2005). Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron 48, 17–24. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannoji H, Takeshita I, Fukui M, Ohta M, and Kitamura K (1981). Glial fibrillary acidic protein in medulloblastoma. Acta Neuropathol 55, 63–69. [DOI] [PubMed] [Google Scholar]
- Margol AS, Robison NJ, Gnanachandran J, Hung LT, Kennedy RJ, Vali M, Dhall G, Finlay JL, Erdreich-Epstein A, Krieger MD, et al. (2015). Tumor-associated macrophages in SHH subgroup of medulloblastomas. Clin Cancer Res 21, 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, and Berns A (2000). Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14, 994–1004. [PMC free article] [PubMed] [Google Scholar]
- Matei V, Pauley S, Kaing S, Rowitch D, Beisel KW, Morris K, Feng F, Jones K, Lee J, and Fritzsch B (2005). Smaller inner ear sensory epithelia in Neurog 1 null mice are related to earlier hair cell cycle exit. Dev Dyn 234, 633–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MR, Robinson KJ, Cleary MD, and Doe CQ (2009). TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat Methods 6, 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrs M, Shinkai K, Mohrs K, and Locksley RM (2001). Analysis of type 2 immunity in vivo with a bicistronic IL-4 reporter. Immunity 15, 303–311. [DOI] [PubMed] [Google Scholar]
- Mullor JL, Sanchez P, and Ruiz i Altaba A (2002). Pathways and consequences: Hedgehog signaling in human disease. Trends Cell Biol 12, 562–569. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Luo L, and Zong H (2007). Modeling sporadic loss of heterozygosity in mice by using mosaic analysis with double markers (MADM). Proc Natl Acad Sci U S A 104, 4495–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikodemova M, and Watters JJ (2012). Efficient isolation of live microglia with preserved phenotypes from adult mouse brain. J Neuroinflammation 9, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy R, and Pollard JW (2014). Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TT, Lin SM, and Wechsler-Reya RJ (2005). Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development 132, 2425–2439. [DOI] [PubMed] [Google Scholar]
- Omi A, Enomoto Y, Kiniwa T, Miyata N, and Miyajima A (2014). Mature resting Ly6C(high) natural killer cells can be reactivated by IL-15. European journal of immunology 44, 2638–2647. [DOI] [PubMed] [Google Scholar]
- Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palucka AK, and Coussens LM (2016). The Basis of Oncoimmunology. Cell 164, 1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Mount SM, and Kingsford C (2014). Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotechnol 32, 462–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoenix TN, Patmore DM, Boop S, Boulos N, Jacus MO, Patel YT, Roussel MF, Finkelstein D, Goumnerova L, Perreault S, et al. (2016). Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 29, 508–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, Sorensen N, Berthold F, Henk B, Schmandt N, et al. (1997). Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res 57, 2085–2088. [PubMed] [Google Scholar]
- Polyak K, Haviv I, and Campbell IG (2009). Co-evolution of tumor cells and their microenvironment. Trends in genetics : TIG 25, 30–38. [DOI] [PubMed] [Google Scholar]
- Prinz M, Erny D, and Hagemeyer N (2017). Ontogeny and homeostasis of CNS myeloid cells. Nature immunology 18, 385–392. [DOI] [PubMed] [Google Scholar]
- Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, Holland EC, Sutton JC, and Joyce JA (2016). The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352, aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, and Joyce JA (2013). Microenvironmental regulation of tumor progression and metastasis. Nat Med 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranke MB (2015). Insulin-like growth factor binding-protein-3 (IGFBP-3). Best practice & research Clinical endocrinology & metabolism 29, 701–711. [DOI] [PubMed] [Google Scholar]
- Rickert CH, and Paulus W (2005). Prognosis-related histomorphological and immunohistochemical markers in central nervous system tumors of childhood and adolescence. Acta Neuropathol 109, 69–92. [DOI] [PubMed] [Google Scholar]
- Rose MF, Ren J, Ahmad KA, Chao HT, Klisch TJ, Flora A, Greer JJ, and Zoghbi HY (2009). Math1 is essential for the development of hindbrain neurons critical for perinatal breathing. Neuron 64, 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi ML, Buller JR, Heath SA, Carey MP, Carboni P Jr., Koutsoubelis G, and Coakham HB (1991). The monocyte/macrophage infiltrate in 35 medulloblastomas: a paraffin-wax study. Tumori 77, 36–40. [DOI] [PubMed] [Google Scholar]
- Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, and Clevers H (2012). Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 337, 730–735. [DOI] [PubMed] [Google Scholar]
- Schofield D, West DC, Anthony DC, Marshal R, and Sklar J (1995). Correlation of loss of heterozygosity at chromosome 9q with histological subtype in medulloblastomas. Am J Pathol 146, 472–480. [PMC free article] [PubMed] [Google Scholar]
- Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, et al. (2008). Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 14, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shee K, Yang W, Hinds JW, Hampsch RA, Varn FS, Traphagen NA, Patel K, Cheng C, Jenkins NP, Kettenbach AN, et al. (2018). Therapeutically targeting tumor microenvironment-mediated drug resistance in estrogen receptor-positive breast cancer. The Journal of experimental medicine 215, 895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, and Takeyama H (2015). Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 7, 2443–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Q, Wong KK, Su JM, Adesina AM, Yu LT, Tsang YT, Antalffy BC, Baxter P, Perlaky L, Yang J, et al. (2008). Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells 26, 1414–1424. [DOI] [PubMed] [Google Scholar]
- Siebert H, Sachse A, Kuziel WA, Maeda N, and Bruck W (2000). The chemokine receptor CCR2 is involved in macrophage recruitment to the injured peripheral nervous system. J Neuroimmunol 110, 177–185. [DOI] [PubMed] [Google Scholar]
- Singh S, Wang L, Schaff DL, Sutcliffe MD, Koeppel AF, Kim J, Onengut-Gumuscu S, Park KS, Zong H, and Janes KA (2019). In situ 10-cell RNA sequencing in tissue and tumor biopsy samples. Scientific reports 9, 4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, et al. (2010). Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143, 134–144. [DOI] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadaro O, Camell CD, Bosurgi L, Nguyen KY, Youm YH, Rothlin CV, and Dixit VD (2017). IGF1 Shapes Macrophage Activation in Response to Immunometabolic Challenge. Cell reports 19, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockmann C, Schadendorf D, Klose R, and Helfrich I (2014). The impact of the immune system on tumor: angiogenesis and vascular remodeling. Frontiers in oncology 4, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. (2012). Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487, 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S, Chen J, Yao H, Liu J, Yu S, Lao L, Wang M, Luo M, Xing Y, Chen F, et al. (2018). CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 172, 841–856 e816. [DOI] [PubMed] [Google Scholar]
- Sun Y (2016). Tumor microenvironment and cancer therapy resistance. Cancer letters 380, 205–215. [DOI] [PubMed] [Google Scholar]
- Swain SL, Weinberg AD, English M, and Huston G (1990). IL-4 directs the development of Th2-like helper effectors. J Immunol 145, 3796–3806. [PubMed] [Google Scholar]
- Tabori U, Baskin B, Shago M, Alon N, Taylor MD, Ray PN, Bouffet E, Malkin D, and Hawkins C (2010). Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol 28, 1345–1350. [DOI] [PubMed] [Google Scholar]
- Tabuso M, Homer-Vanniasinkam S, Adya R, and Arasaradnam RP (2017). Role of tissue microenvironment resident adipocytes in colon cancer. World journal of gastroenterology 23, 5829–5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao-Inoue H, Dorman LC, et al. (2018). Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359, 1269–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen JF, Van Hecke W, Adriaansen EJM, Jansen MK, Bouma RG, Villacorta Hidalgo J, Fisch P, Broekhuizen R, Spliet WGM, Kool M, et al. (2018). Prognostic relevance of tumor-infiltrating lymphocytes and immune checkpoints in pediatric medulloblastoma. Oncoimmunology 7, e1398877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner C, Hansch GM, Stegmaier S, Denefleh B, Hug F, and Schoels M (2001). The complement receptor 3, CR3 (CD11b/CD18), on T lymphocytes: activation-dependent up-regulation and regulatory function. European journal of immunology 31, 1173–1180. [DOI] [PubMed] [Google Scholar]
- Wang L, and Janes KA (2013). Stochastic profiling of transcriptional regulatory heterogeneities in tissues, tumors and cultured cells. Nature protocols 8, 282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA (2008). Coevolution in the tumor microenvironment. Nat Genet 40, 494–495. [DOI] [PubMed] [Google Scholar]
- Wetmore C, Eberhart DE, and Curran T (2001). Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res 61, 513–516. [PubMed] [Google Scholar]
- Wojtowicz JM, and Kee N (2006). BrdU assay for neurogenesis in rodents. Nat Protoc 1, 1399–1405. [DOI] [PubMed] [Google Scholar]
- Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, Elsamadicy AA, Cui X, Koyama S, Jackson C, et al. (2018). T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res 24, 4175–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JH (1910). Neurocytoma or Neuroblastoma, a Kind of Tumor Not Generally Recognized. The Journal of experimental medicine 12, 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, and Condeelis J (2004). A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res 64, 7022–7029. [DOI] [PubMed] [Google Scholar]
- Wynes MW, and Riches DW (2003). Induction of macrophage insulin-like growth factor-I expression by the Th2 cytokines IL-4 and IL-13. J Immunol 171, 3550–3559. [DOI] [PubMed] [Google Scholar]
- Xin M, Yue T, Ma Z, Wu FF, Gow A, and Lu QR (2005). Myelinogenesis and axonal recognition by oligodendrocytes in brain are uncoupled in Olig1-null mice. J Neurosci 25, 1354–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S, Hu M, Iyer V, and Yu J (2017). Blocking the PD-1/PD-L1 pathway in glioma: a potential new treatment strategy. Journal of hematology & oncology 10, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schuller U, Machold R, Fishell G, Rowitch DH, et al. (2008). Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 14, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, and Goldman JE (1996). Developmental fates and migratory pathways of dividing progenitors in the postnatal rat cerebellum. J Comp Neurol 370, 536–550. [DOI] [PubMed] [Google Scholar]
- Zhao CT, Li K, Li JT, Zheng W, Liang XJ, Geng AQ, Li N, and Yuan XB (2009). PKCdelta regulates cortical radial migration by stabilizing the Cdk5 activator p35. Proc Natl Acad Sci U S A 106, 21353–21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Liu Z, Yu L, Zhang Y, Baxter P, Voicu H, Gurusiddappa S, Luan J, Su JM, Leung HC, et al. (2012). Global gene expression profiling confirms the molecular fidelity of primary tumor-based orthotopic xenograft mouse models of medulloblastoma. Neuro Oncol 14, 574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Franklin RA, Adler M, Jacox JB, Bailis W, Shyer JA, Flavell RA, Mayo A, Alon U, and Medzhitov R (2018). Circuit Design Features of a Stable Two-Cell System. Cell 172, 744–757 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Espinosa JS, Su HH, Muzumdar MD, and Luo L (2005). Mosaic analysis with double markers in mice. Cell 121, 479–492 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTAL VIDEO CAPTION. Related to Figure 1.
3D rendering of the image in Figure 1M.
Supplemental Table 1. Related to STAR Methods section “EXPERIMENTAL MODEL AND SUBJECT DETAILS, Mice”, and to the first section in “RESULTS, Establishment of MADM-based and other genetic models for medulloblastoma”. Medulloblastoma models used in this paper.
Supplemental Table 2. Related to Figure 7. Luminex profiling of cytokines and chemokines. Tumor tissues were dissected out based on Math1-GFP expression and lysed in Tissue Extraction Reagent I (Invitrogen) supplemented with protease inhibitors. Normal cerebelli from age-matched mice were processed in the same fashion as control samples. Protein concentration of the lysates was determined by BCA protein quantification method. All lysates were carefully adjusted to 1.5mg/ml with lysis buffer, and 25μl each was applied to mouse 32-plex cytokine/chemokine luminex panel. The absolute quantity of cytokine/chemokine present in cell lysate was expressed as picogram per 10ug of tissue lysate. Four tumors and three normal cerebella were used for this study.
Supplemental Table 3. Related to “Key Resources Table, Oligonucleotides section”. Oligonucleotides used in this paper. This is a list of oligonucleotides that were used for mouse genotyping, qRT-PCR, and probes for in situ hybridization.
Supplemental Figure 1. Related to Figure 1. TME cells in medulloblastoma and the characterization of Math1-Cre. A, Representative images and the scoring scheme for TME cell composition in human SHH-subtype medulloblastoma samples. B, Data summary of TME cell composition from A. C, In adult MADM-WT mice, only NeuN+ granule neurons in IGL were labeled, demonstrating the faithful expression of Math1-Cre in GNPs. D, There was no MADM labeling of potential TME cells, including blood vessels (CD31), microglia (IBA-1), and astrocytes (BLBP, GFAP). E, In the MADM model, tumor GNPs are GFP+ while TME cells are expected to be unlabelled, allowing cellular resolution analyses of tumor-TME interactions from tumor initiation throughout progression. F, GFP+ astrocytes are present only in tumors (F’), but never in tumor-free brain regions (F”). G, A mouse medulloblastoma model in which all cells from GNP lineage is tdTomato+ while astrocytes are labeled by Aldh1L1-GFP. If tumor-associated astrocytes were derived from tumor cells, they should be yellow (Aldh1L1-GFP+ and tdTomato+).
[WM= White matter]
Scale bars: A, D and F=50μm, F’ and F”=10μm, G=25μm
Supplemental Figure 2. Related to Figure 2. Validation of tumor-to-astrocyte trans-differentiation with multiple approaches.
A, BLBP+ tumor-associated astrocytes had only one nucleus (representative image of >100 cells/tumor in 10 tumors), suggesting that GFP-labeling of astrocyte-like cells did not result from cell fusion. B-D, Mouse model and tamoxifen dosing scheme (B), experimental outcome predictions (C), and a representative image (D) of a mouse medulloblastoma model, in which astrocytes were labeled with tdTomato following the excision of the “floxed stop cassette” by GLAST-CreER upon tamoxifen injection, while TuGNPs were labeled by Math1-GFP. The fact that none of the tdTomato-labeled cells expressed GFP suggests that cell fusion did not occur. E, Tumor-associated astrocytes were labeled with Aldh1L1-GFP in a medulloblastoma model in which tumor lineage is labeled with tdTomato. This enables distinction of tumor-associated astrocytes (yellow) from normal astrocytes (green) and tumor GNPs (red). F, Circled region indicated one micro-dissected tumor-associated astrocyte with minimal contamination of neighboring cells. The gamma scaling in the GFP channel was adjusted to 0.5 to highlight GFP fluorescence clearly above tissue autofluorescence. G-H, Purity verification of LCM-dissected tumor-associated astrocytes vs. TuGNPs before RNA-sequencing. qRT-PCR showed minimal Math1 expression (Math1: 72.92±16.01 TuGNPs vs. 5.252±1.13 tumor-derived astrocytes, p=0.0002 t-test, n=4) and strong enrichment of Aldh1L1 expression (Aldh1L1: 0.078±0.053 TuGNPs vs. 20.15±4.892 TuAstrocytes, p=0.0002 t-test, n=4) in the tumor-derived astrocytes pool. I, Anti-human GFAP antibody specifically labels human astrocytes (arrowhead) in mouse brain grafted with human medulloblastoma cells. J, Serially xenografted SHH-subtype human medulloblastoma cells (passage 12 shown here) gave rise to sporadic cells with astrocytic morphology that stained positive for human GFAP protein throughout the tumor mass, suggesting trans-differentiating activity of human medulloblastoma cells could be maintained in the long-term.
[GLAST= glutamate transporter],
Scale bar: A=10μm, F =20μm, D, I and J= 50μm. Data are Means±SD. Student’s t-test. n.s.: not significant; ***p<0.001.
Supplemental Figure 3. Related to Figure 3. Supportive role for TuAstrocytes in medulloblastoma progression. A-C, In PNLs, the proliferation of TuGNPs strongly correlated with the presence of TuAstrocytes. D, TuGNPs (Math1-GFP+) proliferated actively (EdU+) in spheres (arrows) but not as dispersed cells (circled). E-G, TuAstrocytes (Aldh1L1-GFP+) gradually appeared in tumor spheres.
Scale bars: A, B, D, E and F= 50μm;
Supplemental Figure 4. Related to Figure 4. Inactivation of IGF1 receptor (IGF1R) in GNPs delays tumor progression. A, Medulloblastoma mouse model with IGF1R knockout in GNPs. B, Mice with different genotypes (IGF1R−/−; IGF1R−/+; IGF1R+/+) were dissected at postnatal day 40 and tumor tissues (Math1-GFP+) were collected and weighted. Dotted line indicates cerebellum area. C, Graph of tumor weight from tumor mice of different genotypes: IGF1R−/− (n=14), IGF1R+/− (n=20) and IGF1R+/+ (n=11). D, Tamoxifen (150 mg/kg) did not affect tumor progression in comparison to vehicle control. n=10 for each group. E, Schematic illustration on IGF1R inactivation in tumor cells. After 5 pulse Tamoxifen gavage, the tumor was harvested and apoptotic index and cell cycle exit index were quantified in tissue sections. F-G, Elevated apoptotic cells in Tamoxifen-treated group in comparison to vehicle group. H-I, There were more cells that exited cell cycle (BrdU+ Ki67−) in Tamoxifen-treated group in comparison to vehicle group.
Scale bar: B=1mm, F and H =20μm. Data are Means±SD, one-way ANOVA (C) or Student’s t-test (D G and I). n.s.: not significant, *p<0.05; **p<0.01; ***p<0.001.
Supplemental Figure 5. Related to Figure 5. TAMs display activated morphology and express cell markers of both microglia and monocyte-derived macrophages. A-B, Morphologies of TAMs were traced by neurolucida program in big tumors (B), in comparison to microglia in normal cerebellum (A). C, Schematic diagram of branching pattern analysis. D-F, Morphological analysis of microglia vs TAMs showed that TAMs had enlarged cell bodies (D) and shortened processes (E,F) in comparison to normal microglia. n=213 normal microglia and 288 TAMs from 5 tumor mice. G. Flow cytometry analysis showed that TAMs expressed both microglia marker (p2ry12) and macrophage marker (CCR2). H, Immunofluorescent staining of P2ry12 (red, microglia specific marker) and IBA1 (green, pan-microglia/macrophage marker) showed that TAMs expressed P2ry12. I, Immunofluorescent staining of TMEM119 (red, microglia specific marker) and IBA1 (green, pan-microglia/macrophage marker) showed that TAMs were TMEM119− while normal microglia were TMEM119+.
Scale bar: H and I=30μm. Data are Means±SD, one-way ANOVA. n.s.: not significant, *p<0.05; **p<0.01; ***p<0.001.
Supplemental Figure 6. Related to Figure 6. Validation of the lineage tracing genetic tool to differentiate microglia from circulating monocytes in the medulloblastoma model. A, Schematic diagram of the tool validation: In brief, pups were subcutaneously injected with Tamoxifen (50 mg/kg) at postnatal day four to day eight, long before tumor initiation. One day (acute) or Twenty-eight days (4 weeks) after injection, peripheral blood cells were collected for flow analysis and brains were collected for immunofluorescence staining. B, Histogram of flow analysis of tdTomato+ cells in the peripheral blood showed that tdTomato-labeled cells at acute phase were turned over by 4 weeks (still long before tumor formation). C, Quantification of flow analysis of tdTomato+ cells in the peripheral blood. D, Immunofluorescent staining showed that all microglia (IBA1+, green) were tdTomato+ (red) in both acute and 4 weeks group. E, Quantification from immunofluorescent staining showed that microglia had rare turnover in the cerebellum.
Scale bar: D=30μm, n=5 mice for each group. Data are Means±SD, Student’s t-test. n.s.: not significant; ****p<0.0001.
Supplemental Figure 7. Related to Figure 7. IL4 produced by TuAstrocyte activate TAMs. A. TAMs exhibited robust tolerogenic gene program expression. Pathway analysis of the significantly up- and down-regulated transcripts in tumor-associated microglia (focusing on immune signatures) revealed robust differential expression of genes associated with IL-4 signaling, IL-10 production, regulation of adaptive immunity, and macrophage/microglia activation. Shown in red were the significantly up-regulated genes in tumor associated microglia relative to normal microglia from age-matched mice, whereas blue denoted down-regulated genes. Immune signature genes were pulled from the MSigDB database. B, We observed significantly differentially regulated gene signatures associated with additional IL-4/STAT6 signaling components, JAK2 and ERK1/2. Shown is the level of significance of a given pathway (the negative log of the p value) as well as the number of significantly differentially expressed genes in a given pathway over the total number of genes reported in that pathway. P values were determined using Fisher’s exact tests. n=5 tumor mice for RNAseq . C, In situ hybridization showed that IL4 mRNA could not be detected in IL4 KO tumor mice, validating the effectiveness of gene KO. D, Immunofluorescent staining of IBA1 showed that the cell density of TAMs was similar between IL4 wildtype and IL4 KO tumors. E-F, Immunofluorescent staining of CD3 shows that there were no detectable T cells in the tumor mass. G, Schematic diagram of how to use a medulloblastoma model containing both IL4-GFP reporter and tdTomato tumor lineage marker to investigate the identity of IL4-producing cells. H, IL4-GFP overlapped with GFAP and tdTomato staining in the tumor mass, suggesting that IL4 was produced by tumor-derived astrocytes. n=3 tumor mice (at least) for each panel.
Scale bar: C=200μm, D, E F and H=20μm. Data are Means±SEM, Student’s t-test. *p<0.05. n.s.: not significant, *p<0.01.**p<0.001.
Data Availability Statement
RNA-sequencing data of TuAstrocyte were deposited in GEO (Accession number is GSE111734). RNA-sequencing data of TAMs were deposited in GEO (Accession number is GSE109750). This study did not generate any unique code..