

High-throughput large-droplet sorting is demonstrated by an array of sequentially addressable dielectrophoretic sources.
Abstract
Droplet microfluidics has become a powerful tool in precision medicine, green biotechnology, and cell therapy for single-cell analysis and selection by virtue of its ability to effectively confine cells. However, there remains a fundamental trade-off between droplet volume and sorting throughput, limiting the advantages of droplet microfluidics to small droplets (<10 pl) that are incompatible with long-term maintenance and growth of most cells. We present a sequentially addressable dielectrophoretic array (SADA) sorter to overcome this problem. The SADA sorter uses an on-chip array of electrodes activated and deactivated in a sequence synchronized to the speed and position of a passing target droplet to deliver an accumulated dielectrophoretic force and gently pull it in the direction of sorting in a high-speed flow. We use it to demonstrate large-droplet sorting with ~20-fold higher throughputs than conventional techniques and apply it to long-term single-cell analysis of Saccharomyces cerevisiae based on their growth rate.
INTRODUCTION
Droplet microfluidics has become an established tool in biomedical research for a diverse range of applications, such as chemical assays (1, 2), biological assays (3–5), synthetic biology (6), digital polymerase chain reaction (7), drug screening (8), and clinical diagnostics (9–11). It exploits immiscible phases of liquids to produce emulsions of monodisperse droplets that can serve as isolated reaction and transport vessels, which can be manipulated, mixed, analyzed, and sorted by various techniques (12). Unlike continuous flows in microfluidic devices, isolated drops allow independent monitoring of local conditions in the liquid volume of each droplet. This capability is useful for single-cell analysis because protein secretion, enzyme activity, and proliferation of individual cells or a clonal colony initiated from a single cell can be studied in an isolated environment (13–15) and potentially selected on the basis of desirable biological and biotechnological properties.
Despite these advantages, conventional high-throughput droplet sorting approaches have been limited to small droplets (<10 pl in volume or <27 μm in diameter), while large droplets (>100 pl in volume or >57 μm in diameter) offer other numerous advantages over small droplets, including higher cellular viability (16), growth rates (17), and secretion rates (16) for encapsulated cells, as well as increased reverse transcription efficiency (18) following cell lysis. Furthermore, some larger cells or cell clusters (e.g., algae, tumor spheroids, and embryoid bodies) do not physically fit into smaller droplets. The main challenge that prohibits high-throughput large-droplet sorting is, at the most fundamental level, the increased inertia compared to the surface-free energy of these droplets, resulting in a trade-off relation between sorting throughput and droplet volume. At high sorting rates, larger droplets break into smaller pieces more easily than smaller droplets due to the external force required to redirect the droplet to a separate outlet (19). To avoid this problem during sorting of large droplets, the sorting rate must be reduced by orders of magnitude (20) to ensure the structural integrity of the droplets and thus the reliability of the results and reproducibility of an experiment. To slow down sorting to such a degree is to forego the advantages of droplet microfluidics over standard microwell plate assays.
In this article, we present a sequentially addressable dielectrophoretic array (SADA) chip that overcomes the aforementioned trade-off, enabling high-throughput sorting of large droplets without any damage to the droplets that reduces sort accuracy and the ability to perform downstream assays. The SADA chip is an on-chip array of electrodes, which are activated and deactivated in a sequence synchronized to the speed and position of a target droplet passing nearby. As the droplet passes each electrode in the array, the electrode delivers a small dielectrophoretic force (FDEP), the sum of which gently pulls the target droplet in the direction of sorting (Fig. 1A and figs. S1 and S2; Materials and Methods). In addition to the gentle manipulation, the functionality of sequentially activating and deactivating the electrodes enables the SADA chip to sort out a single target droplet without affecting nontarget droplets in the electrode-array region (Fig. 1A and figs. S1 and S2; Materials and Methods). To demonstrate the ability of our device, we performed SADA-based on-chip fluorescence-activated sorting of large single cells or cell clusters in large droplets (~100 pl) at a record throughput of 4400 droplets/s. Furthermore, we demonstrated substantially improved cellular function (viability and secretion rate) for various cell types when using large droplets and sorting them with our SADA sorter. Last, we used the SADA sorter to demonstrate sorting of budding yeast cells in larger droplets (~1 nl) based on their growth rate differences. Our method extends high-throughput droplet microfluidics to volumes compatible with cell cultivation, thus opening up single-cell analysis and selection based on new critical cellular processes.
Fig. 1. Sequentially addressable dielectrophoretic array.
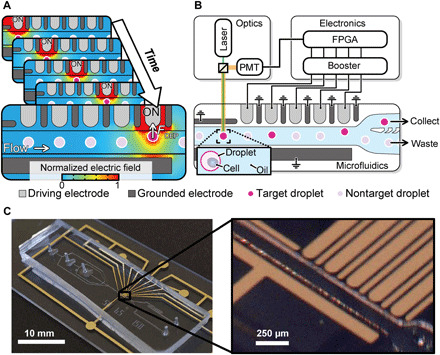
(A) Concept and simulation of the SADA. (B) Schematic of the SADA-based fluorescence-activated droplet sorter (SADA sorter in short). An animated movie of the functionality of the SADA sorter is available in movie S1. (C) Pictures of the constructed SADA sorter. Photo credit: Akihiro Isozaki, University of Tokyo.
RESULTS
Schematic, functionality, and evaluation of the SADA
As shown in Fig. 1B, the SADA chip consists of an upper array of five driving electrodes and six grounded electrodes (alternating side by side), a large grounded electrode in the lower region, a microchannel between the upper and lower electrodes, and a continuous phase in the microchannel (see Materials and Methods and fig. S3 for fabrication details and physical dimensions of the SADA chip). When a stream of interspersed target and nontarget droplets flows into the SADA, weak dielectrophoretic forces are applied to a target droplet flowing downstream by sequentially activating the driving electrodes. AC voltage is applied to the driving electrodes with timings matched to the position and speed of the target droplet. Specifically, when the target droplet is traveling by the first electrode, the first electrode is activated and pulls the droplet. Then, as the droplet continues to travel by the second electrode, the second electrode is activated, while the first electrode is deactivated. The same process occurs for the rest of the electrode array, accumulating overall in a sufficient displacement of the target droplet (Fig. 1A and movie S1). Because the applied electric field is localized, nontarget droplets are unaffected by the manipulation of the adjacent target droplet. The device featured in the figure has five driving electrodes, but this number can be increased to suit the needs of the application (for example, to facilitate a larger total displacement or, for a set displacement, to further weaken the dielectrophoretic force at a given driving electrode).
To demonstrate the capability of our SADA, we constructed a SADA-based fluorescence-activated droplet sorter (the SADA sorter) as shown in Fig. 1 (B and C). The functionality of the SADA sorter is as follows (also see movie S1). On a microfluidic chip, droplets are generated by the droplet generator and accelerated by the droplet accelerator to secure sufficient spacing between consecutive droplets to sort target droplets from adjacent nontarget droplets (fig. S4). Each droplet is then evaluated by the optical droplet analyzer, which consists of a continuous-wave laser and a photomultiplier tube [PMT; see Materials and Methods and fig. S5 (A and B)]. The laser light for fluorescence excitation is incident on each droplet at a distance of ~200 μm upstream of the first driving electrode. If a fluorescent material (e.g., a cell labeled with a fluorescent probe) is encapsulated within a passing droplet, the fluorescence emission is detected by the PMT, which converts the fluorescence signal into an electrical signal (fig. S5C). The electrical signal triggers a control circuit on a field-programmable gate array (FPGA) board (see Materials and Methods; fig. S6 for details of the electronics, including the FPGA), which generates a sequence of control signals that trigger the driving electrodes via electrical boosters (~2 kVpp at 80 kHz). The control signals are amplified by ~20 dB in order for the electrodes to exert sufficient dielectrophoretic forces on the droplets. The FPGA was designed such that the timing of activating and deactivating the driving electrodes maximizes the droplet displacement. The microfluidic divider is composed of a collection channel and a waste channel for the separation of target and nontarget droplets. Fluid resistance is tuned such that nontarget droplets flow straight into the waste channel by default (as do all droplets when the SADA is not activated).
Our evaluation of the SADA sorter is shown in Fig. 2 (A and B). Figure 2A shows a sequence of image frames captured by a high-speed camera (complete movie available in movie S2) that reveals the process of displacing and sorting a single 140-pl droplet, which contains a cell of large-sized microalgal species Euglena gracilis (~50 μm in length), from a mixed population of cell-containing and numerous empty droplets. The images show that the target droplet gradually deviates from the path of the other droplets due to the sequential activation and deactivation of the driving electrodes. Furthermore, Fig. 2B shows the average trajectory of 125 sorted droplets observed by a high-speed camera (Phantom v2640, Vision Research; frame rate, 18,000 frame/s; spatial resolution, ~3 μm). At the fifth driving electrode, the total displacement of the target droplet reaches 50 μm, a sufficient amount for reliable sorting. It is important to note that although some degree of structural deformation of droplets is observed, they remain unbroken during SADA’s sequential displacement process. Meanwhile, nontarget droplets are unaffected by the force and thus remain intact in the central streamline because the dielectrophoretic force applied to the target droplets is localized (Fig. 1A, note S1, and fig. S7, A and B). Bright-field images of the 140-pl droplets in the collection and waste outlets sorted at a throughput of 2384 droplets/s (Fig. 2C) show that the SADA sorter has a high sort purity of 98.8% (calculated from the true-positive and false-positive rates of 99.6 and 1.4%, respectively). The ranges of the sorting throughout and droplet volume covered by the SADA sorter are between ~850 and ~4400 droplets/s and between ~100 pl and ~1 nl, respectively (fig. S7, C to F; movies S3 and S4; and data file S1). To validate the device-to-device reproducibility, we further performed sorting of 1-nl droplets using three replicated devices (movie S5) and verified that the high-throughput sorting performance was also replicated among the devices.
Fig. 2. Performance of the SADA sorter.
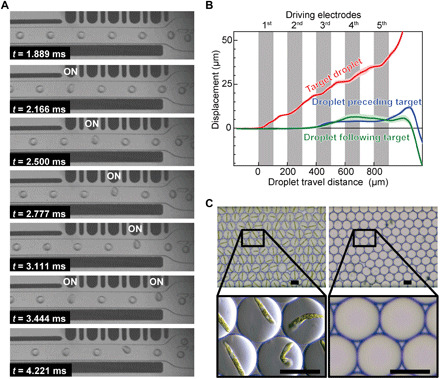
(A) Demonstration of sorting a cell-encapsulating droplet (140 pl in volume) with the SADA sorter. See movie S2 for a complete movie. (B) Accumulated displacement of target droplets sorted by the SADA sorter, in comparison with traces from droplets immediately preceding or following the target droplet. The traces indicate the average trajectories of 125 droplets. Shading indicates SDs. (C) Bright-field images of SADA-sorted and SADA-unsorted droplets with a high sort purity of 98.8% (calculated from 247 droplets in the collect channel and 216 droplets in the waste channel). The SADA-sorted droplets contain E. gracilis cells (a ~50-μm large-sized microalgal species). Scale bars, 50 μm.
Comparison with previous droplet sorters
The SADA sorter opens a new operational regime of larger droplet volumes and throughputs that has not been available in previously reported droplet sorters (13, 19, 21–25), as shown in Fig. 3 (a complete version available in fig. S7G). While the performance of previously reported droplet sorters (20) is constrained by the trade-off between droplet volume and sorting throughput, the SADA sorter occupies a whole new region achieving ~20 times higher performance in the product of droplet volume and sorting throughput as a figure of merit when compared to the previous droplet sorters (data file S1). This capability allows encapsulation and sorting of large cells and cell clusters. The inset of Fig. 3 shows images of sorted ~110-pl droplets containing large cells such as microalgal cells, including Haematococcus lacustris NIES-4141 cells (microalgal cells that produce astaxanthin), clusters of Chlamydomonas sp. JSC4 (C. sp. JSC4) cells (26) (microalgal cells with high lipid productivity), human non–small cell lung cancer (NSCLC) cells, and human induced pluripotent stem cells (iPSCs). Using larger droplet volumes compatible with the SADA sorter enables us to keep cells intact during cell sorting, resulting in reduced cellular damage to provide a notable advantage in studies that require multicellular structures to maintain activity or study cell-cell interactions. These results reveal the robust and broad utility of the SADA sorter.
Fig. 3. Performance of the SADA sorter in comparison with the prior art.
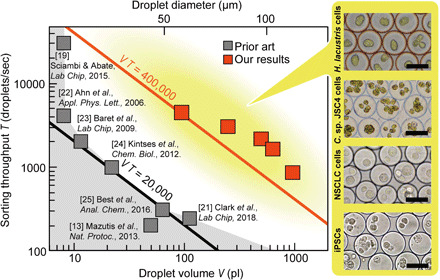
The performance of previously reported droplet sorters is constrained by the trade-off between sorting throughput and droplet volume (black line). The SADA sorter breaks this barrier and consequently achieved ~20 times higher performance in the product of sorting throughput and droplet volume as a figure of merit (red line). The inset shows images of sorted droplets containing large cells and cell clusters. Scale bars, 50 μm. A complete version of the comparison is available in fig. S7G.
Demonstration of enhanced cell survival and secretion
We also observed increased capacity for cell growth and activity in large droplets in comparison to small droplets. We performed this comparison using E. gracilis cells (a large-sized microalgal species), Jurkat cells (an immortalized human T lymphocyte cell line), and B5F6 (27) cells [a hybridoma clone that produces anti-mouse interleukin-17RB (IL-17RB) monoclonal antibody (mAb)]. These cell types were encapsulated in both large (~110 pl) and small (~26 pl) droplets and sorted. As shown in Fig. 4A, after 7 days of incubation, the population of motile cells per droplet of E. gracilis cells in large droplets was found to be larger than that in small droplets by a factor of 9.4. The inset of Fig. 4A shows typical encapsulated cells in droplet-trap devices (28), which enables long-term monitoring of the same cell-encapsulating droplets (see fig. S8). Furthermore, as shown in Fig. 4B, after 18 and 12 hours of incubation, Jurkat cells and B5F6 cells in large droplets demonstrated, respectively, 4.7 and 4.9 times greater viability than the corresponding cells in small droplets. Last, as shown in Fig. 4C, we demonstrated that B5F6 cells have a higher secretion rate (antibody production rate) in large droplets than in small droplets. The increased incubational capacity is presumably because more space and nutrients are available for cells than in smaller droplets (17). These results firmly show the advantages of large droplets over small droplets for in-droplet cell incubation and thus open up the possibility of subsequent sorting based on biotechnologically important phenotypes.
Fig. 4. Demonstration of substantially increased capacities to support cell growth and activity in larger droplets encapsulating SADA-sorted cells.
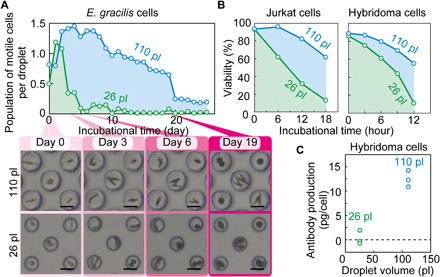
(A) After 7 days of incubation, a factor of 9.4 greater population of motile E. gracilis cells per droplet was identified in large SADA-sorted droplets (110 pl) than in small SADA-sorted droplets (26 pl). Insets show photos of typical trapped large and small droplets (110 and 26 pl) containing E. gracilis cells. The droplets shown are exactly the same droplets across days. Scale bars, 50 μm. (B) After 18 and 12 hours of incubation, 4.7 and 4.9 times higher viability is observed for Jurkat cells and a B5F6 hybridoma clone, respectively, in large SADA-sorted droplets (110 pl) than in small SADA-sorted droplets (26 pl). The incubation time began when the sorting process was finished. The sample size (n) for each dataset is available in data file S2. (C) Higher antibody production rate for the B5F6 clone in large SADA-sorted droplets (110 pl) than in small SADA-sorted droplets (26 pl). Each dot indicates one measurement taken from a different sample.
Isolation of cells with low proliferative activity
Identification and isolation of cells with low proliferative activity, or simply slow-growing cells, is important for basic research in microbiology (e.g., genetic screening) (29), pharmacology (e.g., antimicrobial drug discovery) (30), and cancer biology (e.g., anticancer drug discovery) (31) but remains challenging because fast-growing cells often outcompete slow-growing cells and dominate a cell population when cultured in bulk (32). We used the SADA sorter to demonstrate isolation of slow-growing Saccharomyces cerevisiae (budding yeast) cells from a mixture consisting of slow-growing (adh1Δ with pRS315-GFP-Ras2 plasmid, 280.43 ± 14.94–min doubling time) and fast-growing (YEF8045 with pRS315-GFP-Ras2 plasmid, 100.94 ± 1.33–min doubling time) strains at a ratio of 1:49 (see Materials and Methods and Fig. 5A). Cells in the mixture were encapsulated in larger 1-nl droplets and incubated for 12 hours to allow sufficient time to amplify the signal from the difference in growth rate between these two strains. The large volume (1 nl) of the droplets prevents saturation of growth and was effective for accurately differentiating cells with various growth rates. After incubation, remaining slow-growing clonal colonies were enriched by sorting out fast-growing clones into the nontarget channel (called “collect” in Fig. 1) based on their fluorescence signals (the stronger the fluorescence intensity, the faster the cell growth). Bright-field images of the droplets in the target and nontarget channels (Fig. 5, B and C) show that the droplets that contain the slow-growing cells are the dominant occupant of the target channel with a sort purity as high as 93%, resulting in an enrichment factor of 46.5 (Fig. 5D). In this sorting experiment, taking the data shown in Fig. 5 required a sorting run for ~30 min to collect ~1600 target droplets and thereby produce statistically meaningful results. It is important to note the merit of the high throughput in this application because if the throughput was 20 times lower, it would take ~600 min, leading to large biological fluctuations in measurements (because it allows the proliferation of the cells during the measurements). These results indicate that the SADA sorter can sort populations based on differences in proliferation rate and can contribute to screening larger libraries for drug discovery and research applications focused on this critical phenotype.
Fig. 5. Demonstration of sorting budding yeast cells with low proliferative activity.
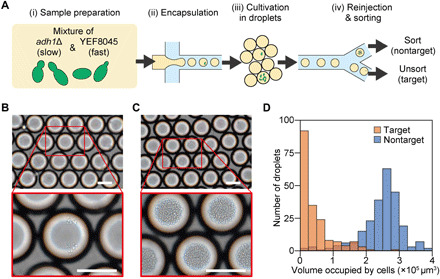
(A) Experimental procedure. (i) Strains of adh1Δ and YEF8045 were mixed at a ratio of 1:49, (ii) encapsulated in ~1-nl droplets, (iii) incubated for 12 hours, and (iv) reinjected and sorted with the SADA sorter. (B and C) Bright-field images of SADA-unsorted (target) and SADA-sorted (nontarget) droplets, respectively. (D) Histogram of the volume occupied by budding yeast cells within each droplet [n = 182 droplets for unsorted (target) droplets and n = 240 droplets for sorted (nontarget) droplets]. Defining the threshold for the low proliferative activity cutoff to be 1.5 × 105 μm3, the purities of the target and nontarget droplets in the corresponding channels were both found to be 93%. Scale bars, 100 μm.
DISCUSSION
Our SADA sorter combines the advantages of high-throughput single-cell sorting with those of large-droplet microfluidics, overcoming a critical limitation that hindered the performance of previous microfluidic droplet sorting techniques. In addition to the above demonstrations, the use of large droplets enables long-term viable cell incubation and clonal expansion, increased production of secreted products, and co-encapsulation of cells (e.g., T cells, dendritic cells, cancer cells, and microorganisms). Sorting based on these phenotypes can open up rapid approaches to select the most productive clones to produce therapeutic antibodies, separate out cells most effective in tumor cell killing for developing optimal cell therapies (33), or select algal clones with the highest growth rate and biomass accumulation. These approaches ultimately create new opportunities in biologic and cell therapy manufacturing, precision medicine, regenerative medicine, and green biotechnology.
The capabilities of the SADA sorter can be further extended in multiple directions. First, its throughput can be boosted by making a few modifications to it. For instance, the number of electrodes that constitute the electrode array can be increased as discussed above to increase accumulated dielectrophoretic force. While we used five driving electrodes in the SADA sorter in this work, there is no physical limitation on the number of electrodes. In addition, three-dimensional (3D) electrodes with larger thickness (34) than 2D electrodes used in this work can be implemented to provide larger dielectrophoretic force on droplets. Second, the SADA sorter can be combined with other droplet-microfluidic techniques (e.g., adding reagents to droplets, mixing reagents in droplets, and on-chip droplet incubation) (35) to broaden its applicability, depending on the type of applications. Last, other optical interrogation methods [e.g., multicolor detection, high-speed imaging (36) for morphology-based cell sorting, and high-speed Raman detection (37) for label-free cell sorting] can be implemented instead of the simple fluorescence detection demonstrated in this work. With these improvements, the SADA sorter has the potential to cover a wider range of cell types in diverse biomedical fields, thus paving the way of droplet microfluidics toward applications that have not been feasible with small droplets.
MATERIALS AND METHODS
Dielectrophoretic force
Time-averaged dielectrophoretic force FDEP acting on a spherical dielectric particle is given by (38)
where εm is the absolute permittivity of the surrounding medium, r is the radius of the particle, Re(CM) is the real part of the Clausius-Mossotti factor, and E is the root mean square value of the amplitude of the electric field at the position of the particle. The Clausius-Mossotti factor is given by CM = (εp − εm)/(εp + 2εm), where εp is the absolute permittivity of the particle. Note that εm is a real number because the conductivity of the surrounding medium is zero, while εp is a real number independent of the applied AC signal if the imaginary part of the permittivity given by the ratio of the conductivity of the particle to the frequency of the AC signal is much smaller than the real part, which is the case in this article (conductivity, 5.5 × 10−6 S/m; AC signal frequency, 80 kHz). Therefore, FDEP can be considered as a constant value independent of the frequency of the applied AC signal, while the strength of the constant force varies, depending on the permittivity of the droplets and the surrounding medium. Note that free charges do not accumulate in droplets during droplet sorting with the SADA sorter because the frequency of the AC signal is much higher than the reciprocal of the typical charge relaxation time of an aqueous droplet [0.125 ms (39)].
Simulation of the SADA
To obtain an in-depth understanding of the SADA’s functionality, we performed computational simulations based on the finite element method (FEM), using a commercial simulation software package (COMSOL Multiphysics version 4.4, COMSOL, USA). We evaluated the distribution patterns of the electric field within the microchannel and the distribution patterns of the dielectrophoretic forces on droplets. Figure S1A shows a static simulation model. Nontarget droplets were placed at the center of the microchannel (y = 0), while target droplets were placed at various positions in the microchannel along the y direction. The material properties of droplets, microchannel walls, and electrodes are shown in the legend in figs. S1 and S2. The permittivity of the carrier oil is either 2 or 6, which corresponds to the permittivities of two typical carrier oils, Novec 7300 (3M) and FC-40 (3M), respectively. In this simulation, we applied an 80-kHz AC signal to the driving electrode(s) closest to the target droplet(s), while the other electrodes were grounded. The voltage applied to each electrode in the simulation is 2.33 kVpp for the first electrode, 2.18 kVpp for the second electrode, 2.10 kVpp for the third electrode, 1.97 kVpp for the fourth electrode, and 1.93 kVpp for the fifth electrode. Figure S1B shows the simulation results for single-droplet sorting, specifically the distribution patterns of the electric fields in the whole microchannel, with various droplet y positions, and the distribution patterns of the dielectrophoretic forces on the target droplet and adjacent droplets, the droplets preceding (right) and following (left) the target droplet. The spatial gradient of the electric field surrounding the target droplet is large, which allows the activated electrode to attract, via dielectrophoretic forces, only the target droplet, leaving nontarget droplets unaffected. This functionality enables us to sort target droplets from nontarget droplets with minimal interdroplet spacing in the flow, yielding higher throughputs. Figure S1C shows the simulation results for multiple-droplet sorting, which corresponds to the case of target droplets flowing in tandem. This figure shows the distribution of the electric field along the microchannel, with the y positions of the droplets ranging from −30 to 30 μm from the center of the channel. The figure also shows the distribution patterns of the dielectrophoretic forces on the droplets at x = 0 mm. The spatial gradient of the electric field surrounding all of the target droplets in the microchannel is large such that all of the droplets are attracted to each electrode. The direction of the dielectrophoretic force distribution on each droplet is in the positive-y direction, regardless of the carrier oil permittivity.
Importance of the grounded electrodes
Another FEM simulation was conducted, using the simulation model shown in fig. S2A, to study the effect of the absence of the grounded electrodes. In the case of single-droplet sorting, as shown in fig. S2B, there are no notable differences compared to the simulation results of the model with the grounded electrodes (fig. S1B). However, in the case of multiple-droplet sorting, as shown in fig. S2C, there are notable differences. First, the electric flux lines are almost parallel to each other in the microchannel, thus exerting a much weaker dielectrophoretic force on the flowing droplets compared to the case with the grounded electrodes. Consequently, the resulting force is too weak to displace the flowing target droplets by an amount necessary for sorting. Second, a droplet at y ≤ 0 in the carrier oil with permittivity of 2 has a dielectrophoretic force in the negative-y direction as well as in the positive-y direction, resulting in the droplets being stretched vertically instead of being drawn toward the upper microchannel wall. When a droplet’s y position is >0, there are dielectrophoretic forces that compress in the horizontal direction, resulting in droplet deformation. This simulation verifies that the presence of the grounded electrodes between the driving electrodes plays an essential role in the SADA by facilitating the generation of effective dielectrophoretic force regions that can independently displace the flowing droplet. In other words, without the grounded electrodes, it is difficult to generate spatially nonuniform electric fields around each of the driving electrodes when they are activated simultaneously because neighboring driving electrodes act as pseudo-large–driving electrodes. By placing grounded electrodes between the driving electrodes, each of the driving electrodes acts as an independent driving electrode that generates localized high electric field gradients to pull droplets.
Fabrication of the SADA
Figure S3 (A to C) shows the fabrication process of the microfluidic chip. The microfluidic chip was fabricated by a standard photolithography process, while the electrodes were fabricated by a liftoff process. Photoresist (PFR7790G-27cp, JSR) was patterned onto a glass substrate, onto which a 10-nm-thick Cr layer and a 70-nm-thick Au layer were deposited. Excess metal was removed by a liftoff process, revealing the electrodes. The microchannel was fabricated using a conventional soft lithography process. A photoresist (KMPR-1035, MicroChem, USA) master structure was patterned onto a Si wafer. The thickness of the photoresist was 60, 110, and 130 μm for devices of designs A, B, and C, respectively. Uncured polydimethylsiloxane (PDMS) solution consisting of base and curing agent at a 10:1 ratio was poured onto the master structure, degassed in a vacuum chamber, and baked at 80°C for 14 hours. The cured PDMS device was peeled off from the master mold, and three inlet and two outlet holes were made with a hole puncher. The PDMS slab and the glass substrate were exposed to oxygen plasma for 180 s at 450 mtorr (PDC-001-HP, Harrick plasma, USA) and bonded by manually aligning the electrode array and the microchannel under a microscope to create a closed channel. After the alignment, the device was heated on a hotplate at 100°C for at least 10 min. The fabricated channel was then washed with 0.1% perfluorodecyl dimethylchlorosilane solution to make the surface of the microchannel hydrophobic, thereby enabling stable droplet generation. Last, the microchannel was washed with ethanol and baked in an oven at 80°C for at least 12 hours to remove excess perfluorodecyl dimethylchlorosilane solution.
Optical detection and monitoring systems of the SADA sorter
The optical detection and monitoring systems are schematically shown in fig. S5A. As summarized in fig. S5B, some optical components of these systems were selected so that fluorescence signals from target objects were detectable. Figure S5A shows the optical systems of setup A listed in fig. S5B. The optical detection module consists of a continuous-wave laser diode (see fig. S5B), a cylindrical lens (f = −200 mm), a short-pass dichroic beamsplitter (cutoff wavelength of 550 nm), an objective lens (6.3×), a long-pass filter (filter 1; see fig. S5B), a spherical lens (f = 175 mm), an iris, and a PMT. The laser light from the laser diode is cylindrically shaped by the cylindrical lens, passes through the short-pass dichroic beamsplitter, and is focused onto the microfluidic chip via the objective lens. Fluorescence or autofluorescence (centered at 700 nm) from a cell encapsulated in a droplet is collected by the same objective lens and directed to the PMT via the short-pass dichroic beamsplitter, the long-pass filter, the spherical lens, and the iris. The PMT converts the detected fluorescent signal into an electrical signal that triggers the SADA. The optical monitoring module consists of a halogen lamp (OSL2, Thorlabs, USA), a band-pass filter (filter 2; see fig. S5B), a 70:30 beamsplitter, two notch filters (filter 3; see fig. S5B), a spherical lens, a high-speed camera (Phantom Miro M110 or Phantom v2640, Vision Research), and the same objective lens shared with the optical detection module. Broadband light from the lamp is filtered by the band-pass filter and is incident on the microfluidic chip. The diffracted light from the chip is collected by the objective lens and detected, via the beamsplitter, the notch filter (used to remove the 488-nm laser light), and the spherical lens, by the high-speed camera to monitor the flow of the droplets and the SADA operation in the microfluidic chip.
Electrical control circuit of the SADA sorter
Figure S6A shows the schematic of the electrical control system, which consists of a low-pass filter, a comparator, an FPGA board (MAX 10 - 10M08 Evaluation Kit, Intel, USA), and five booster circuits (Fig. 1B and fig. S6B). The electrical signals from the optical detection system are sent to the FPGA board, which sends control signals to the booster circuits with a constant time delay that can be tuned according to the flow speed, as shown in fig. S6C. The control signals are square waves (3.3 V). The square waves are amplified to the desired application voltage, which is tunable from ~0.7 kV to ~3.2 kVpp by a transformer in the booster circuits. The shape of the boosted signal is shown in fig. S6 (D and E). The boosted signals are applied to the electrodes in the SADA. The capacitance of the electrodes is 0.25 pF, which is negligible when operating the booster circuits.
Preparation of microalgal cells
Three microalgal strains were used: E. gracilis NIES-48 and H. lacustris NIES-4141 obtained from the Microbial Culture Collection at the National Institute for Environmental Studies (NIES), and Chlamydomonas sp. JSC4 (26) isolated from a brackish water area of southern Taiwan. E. gracilis and H. lacustris cells were cultured in 10 ml of Koren-Hutner medium with a pH of 3.5 and AF-6 medium, respectively, in 50-ml flasks (690195, Greiner, Austria). These cells were subcultured every 3 to 4 days at a cell concentration of log growth phase. E. gracilis cells were incubated in a custom-made growth chamber with a temperature of 25°C and the lighting inside the chamber by warm-white light-emitting diode lighting at 120 μmol photons/m2 per second with a 12-hour/12-hour light/dark cycle. H. lacustris cells were incubated in a plant growth chamber (TG-180-5L, NKsystem, Japan) with a temperature of 25°C and the lighting by cool-white fluorescent lamps at 120 μmol photons/m2 per second with a 14-hour/10-hour light/dark cycle. E. gracilis cells were encapsulated 3 days after subculturing when the concentration was >1 × 106 cells/ml, which is when the cell concentration is in the mid-logarithmic phase. H. lacustris cells were encapsulated 2 days after subculturing when the concentration was ~5 × 104 cells/ml. To make the concentration >1× 106 cells/ml, H. lacustris cells were concentrated by a centrifuge (100g, 3 min). C. sp. JSC4 cell line was maintained by subculturing every 2 months on a BG-11 agar plate under an illumination of 30 μmol photons/m2 per second. Before the encapsulation, C. sp. JSC4 cells were transferred into 70 ml of Modified Bold (MB) 6N medium with 3% (w/v) sea salt (Sigma-Aldrich Co., USA) and cultured in a double-deck photobioreactor (26) with the following culture conditions: 250 μmol photons/m2 per second illumination with natural-white fluorescent lamps, 2% (v/v) CO2 aeration, and 30°C. No staining process is necessary because the autofluorescence of the microalgal cells functions as the cell-sorting triggers.
Preparation of Jurkat cells
Jurkat cells [American Type Culture Collection, TIB-152] were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS; MP Biomedicals), penicillin (100 U/ml), and streptomycin (100 μg/ml; Wako Chemicals) in a cell incubator at 5% CO2 and 37°C. The cells were subcultured every 3 to 4 days at 1 × 105 cells/ml in 10 ml of medium in dishes or flasks and used for sorting experiments 3 days after the last passage in the mid-logarithmic phase. The cells were gently suspended in RPMI 1640 medium without FBS and stained with CellTracker Orange CMRA (Thermo Fisher Scientific) to a final concentration of 15 μM at a concentration of 7 × 106 cells/ml according to the manufacturer’s instruction. After staining in a cell incubator for 45 min, the cells were washed with phosphate-buffered saline (PBS; Wako Chemicals) to remove excess dye, which could generate unwanted signals during cell detection. Stained cells were resuspended in the cell culture medium at a final concentration of 7 × 106 cells/ml for sorting experiments.
Preparation of hybridoma cells
A hybridoma cell line (B5F6, anti-mouse IL-17RB mAb-producing line) has been established as described previously (27) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS (MP Biomedicals), 10% BM Condimed H1 (Roche), and penicillin (100 U/ml) and streptomycin (100 μg/ml; Wako Chemicals) in a cell incubator at 37°C and 5% CO2. The cells were subcultured every 3 days at 8 × 104 cells/ml in 10 ml of medium in dishes or flasks and used for sorting experiments 3 days after the last passage in the mid-logarithmic phase. To sort the hybridoma cells, the cells were stained by the same procedure as the staining of Jurkat cells and resuspended in the cell culture medium containing 10% FBS at a final concentration of 7 × 106 cells/ml.
Preparation of tumor cells
PC9 (human non–small cell lung cancer) cells (1 × 105 cells) were cultured with 3 ml of DMEM (Wako Chemicals) supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml) on PrimeSurface 35-mm dish (Sumitomo Bakelite, Japan) in a cell incubator at 5% CO2 and 37°C for overnight. Through the overnight culture, most of the PC9 cells were clustered. Before sorting, the cells were stained with 500× diluted calcein AM solution (Dojindo, Japan) in the cell incubator for 30 min and fixed with 0.25% of glutaraldehyde (Nacalai Tesque, Japan) for 10 min at room temperature, followed by washing with PBS (Wako Chemicals).
Preparation of iPSCs
iPSCs were generated using a virus-free method from IMR-90 fibroblasts, which are commonly used for cellular reprogramming, and characterized as described previously (40). Mycoplasma tests were conducted using MycoAlert (Lonza) when the iPSC line (#IM-E1-5) was generated. Human iPSCs were cultured with Essential 8 medium with penicillin (100 U/ml) and streptomycin (100 μg/ml) on plates or dishes coated with Geltrex (#A14133-022, from Life Technologies) following the manufacturer’s instruction. The iPSC line was maintained and passaged following the previously reported protocol using dispase (Life Technologies) (40). To sort iPSCs, the cells were dissociated with Accutase (StemPro Accutase Cell Dissociation Reagent, Thermo Fisher Scientific) for 5 min at 37°C and then washed with prewarmed Essential 8 medium, stained by the same procedure as with the Jurkat cells, and then resuspended in the cell culture medium containing 10% FBS, at a final concentration of 7 × 106 cells/ml.
Preparation of budding yeast cells
We used two strains of budding yeast (S. cerevisiae) in this study: adh1Δ, which was purchased from EUROSCARF (http://euroscarf.de), and YEF8045 with pRS315-GFP-Ras2 plasmid, which was described previously (41). The pRS315-GFP-Ras2 plasmid was recovered and purified using the FastGene Plasmid Mini Kit (NIPPON Genetics Co. Ltd., Japan), and used to transform adh1Δ by the LiAc/SS carrier DNA/polyethylene glycol (PEG) method (42). Both strains of budding yeast cells were stocked at −80°C in 16.7% glycerol stock solution. Cells were activated on a selection agar plate, which contained synthetic complete medium without leucine, and were then incubated at 25°C for 1 week for colony formation. The colonies were precultured by inoculating the colonies into 5 ml of the selection liquid medium for 24 hours. The precultured cells were inoculated into 20 ml of yeast extract peptone dextrose (YPD) medium with 400 mM sorbitol overnight and were then harvested during their log phase. Cells were resuspended in YPD medium with 400 mM sorbitol at 1 × 105 cells/ml before encapsulation.
Operation of the SADA
Before the sorting experiments, several conditions and parameters of the SADA were checked and tuned using movies taken by the high-speed camera. First, we aligned the position of the excitation laser spot within the SADA. Second, we tuned the flow rates of the dispersed phase, continuous phase, and spacing phase to generate droplets of the desired size at the desired throughput. The dispersed phase is medium containing cells, and the continuous phase and spacing phase are fluorinated oil (Novec 7300, 3M) with 0.5% (w/w) surfactant (Neat 008-FluoroSurfactant, RAN Biotechnologies). Third, we checked fluorescent signals from cells detected by the PMT and tuned the signal threshold for activating the electrodes. Fourth, we measured the flow speed of droplets using the high-speed camera. Fifth, we input the timing to apply voltage to each driving electrode, according to the measured flow speed of droplets. Sixth, we manually optimized the timing to apply the voltage by monitoring the motion of the droplets actuated by the SADA. Last, we tuned the amplitude of the applied voltage to optimize the dielectrophoretic force applied to target droplets. If the applied dielectrophoretic force is too small, droplets are insufficiently displaced from the central streamline, whereas if the applied dielectrophoretic force is too large, droplets are ruptured (see note S2 and movie S6 for details of the droplet breakage). Either case results in sorting errors, thus lowering the purity and/or yield of sorting results. Note that the intensity of the applied dielectrophoretic force varies, depending on the distance between the electrode and the droplet; the larger the distance (typically, the distance is the largest when a droplet is located by the first electrode), the smaller the applied dielectrophoretic force. Once these setting parameters were optimized, the SADA stably performed droplet sorting for several hours without additional tuning. Typical parameters are shown in table S1. After disposing of the droplets generated during the first 5 min of an experiment, to avoid unwanted carryovers, the sorted and unsorted droplets were collected. The typical signals for hybridoma cells are shown in fig. S5C. The droplets were sorted and collected continuously for approximately 3 hours to obtain approximately 3 × 107 droplets containing cells for a typical experiment.
Evaluation of the population of motile E. gracilis cells encapsulated in droplets
The sorted cell-containing droplets were injected into droplet-trap devices with the same carrier oil as the sorting experiment (fluorinated oil, Novec 7300). The droplets, which have a lower density than the carrier oil, rose naturally and occupied the wells. Novec 7300 was replaced with another type of fluorinated oil (FC-40), which has a lower vapor pressure (287 Pa) than Novec 7300 (5986 Pa), by flowing the FC-40 with 0.5% (w/w) surfactant (Neat 008-FluoroSurfactant, RAN Biotechnologies) right after trapping the droplets to avoid the evaporation of the fluorinated oil during long-term incubation (>20 days). The inlet and the outlet of the droplet-trap devices were sealed, and the whole devices were submerged in water and placed in an incubator with a temperature of 25°C and light intensity of 120 μmol photons/m2 per second, with a 12-hour/12-hour light/dark controller (fig. S8). Microalgal cells in droplets (n = 328 droplets for large droplets and n = 302 droplets for small droplets) were observed on a daily basis under a microscope, and movies were taken. Using the movies, we manually counted motile cells in each droplet containing a single cell on day 0.
Evaluation of the viability of Jurkat cells and hybridoma cells encapsulated in droplets
Before the sorting experiments, as a preparation step for sorted-cell incubation, we prepared CO2-rich fluorinated oil and mineral oil by storing them overnight in a cell incubator at 37°C and 5% CO2. Then, we sorted the droplets containing cells and collected them in a 1-ml polyethylene syringe for incubation. To prevent evaporation, the droplets in the syringe were sandwiched between two oil layers, with the less dense mineral oil on top and the fluorinated oil below. The syringe was placed horizontally inside the cell incubator to increase the area of the contact surface between the droplets and the fluorinated oil. Every 6 hours for Jurkat cells and every 3 hours for hybridoma cells, we transferred by pipette 10 μl of the droplets to a 0.5-ml microtube. To recover the cells in droplets, we added a phase separator pico-break (Dolomite Microfluidics, UK) in a 1:1 volume ratio to the droplets. The cells in medium were then collected, and we performed live-dead assays with a dye exclusion test using trypan blue solution.
Preparation for the flow cytometry–based antibody secretion assay
We have previously generated a stable human embryonic kidney (HEK) 293T cell line, which is constitutively and bicistronically expressing mouse IL-17RB and enhanced green fluorescent protein (EGFP; mIL-17RB-EGFP/293T) by the introduction of pCMV-mIL-17RB-IRES-EGFP (27) for hybridoma screening. The reactivity of anti–mIL-17RB mAb against mIL-17RB-EGFP/293T can be quantitatively detected by phycoerythrin (PE)–conjugated anti-rat immunoglobulin G (IgG) as the secondary antibody. Twenty-four hours after the sorting experiments, cell-containing droplets were transferred into a 1.5-ml microfuge tube and broken, with the same procedure as for the viability evaluation, and the supernatant was collected. The produced antibodies present in the supernatant were incubated with mIL-17RB-EGFP/293T cells in a U-shaped 96-well plate (BD Falcon) with a concentration of ~5 × 106 cells/ml. After the incubation for 20 min at 4°C, cells were washed with Dulbecco’s PBS (DPBS) (−) containing 2% fetal calf serum (FCS) and 2 mM EDTA (MACS buffer). Cells were further stained with 10 μl of diluted PE anti-rat IgG (BioLegend) for 20 min at 4°C in a dark environment. The unbound PE anti-rat IgG was washed away with MACS buffer. The cells were resuspended with 150 μl of MACS buffer and transferred into 1.2-ml microtiter tubes (Thermo Fisher Scientific Inc.) for flow cytometry–based analysis. Standard solutions (i.e., mIL-17RB mAb) of known concentrations, ranging from 0 to 1000 ng/ml, were simultaneously prepared for obtaining a standard curve.
Flow cytometric analysis of antibody production from hybridoma cells
A cell analyzer (BD FACSCalibur) was used for quantitative analysis. The laser wavelength was set to 488 nm, and four detectors were used: a 488/10-nm forward scatter light (FSC) detector, a 488/10-nm side scatter light (SSC) detector, a 530/30-nm fluorescent light detector (for detecting the EGFP), and a 585/42-nm fluorescent light detector (for detecting the PE). Live cells were gated by the scatter plots of FSC and SSC intensities, and EGFP+ cells were further analyzed by signal intensities of PE, which reflects the staining level of antibody (see fig. S9A for the gating strategy). The average signal intensities of the gated PE were used as the representative of the quantified antibody production from hybridoma cells. Figure S9B shows the standard curve, while fig. S9C shows the histograms of the signal intensities of the gated PE depicting the difference between the antibody production of hybridoma cells encapsulated in small droplets (26 pl) and large droplets (110 pl).
Sorting of budding yeast cells
We used a droplet-generating chip and the SADA chip with a droplet reinjector (13) instead of the droplet generator for the budding yeast sorting experiment. adh1Δ and YEF8045 both tagged with pRS315-GFP-Ras2 plasmid were mixed at a ratio of 1:49. Single cells in the mixture were encapsulated in ~1-nl droplets with a throughput of 450 droplets/s by the droplet-generating chip. The cell-encapsulating droplets were incubated at 25°C for 12 hours. Then, the incubated droplets were reinjected into the SADA chip and sorted on the basis of the fluorescence intensity of the droplets, where we assumed the fluorescence intensity of each fluorescent droplet to be higher as cells inside the droplet proliferate. Specifically, a droplet with higher fluorescence intensity than the sorting threshold was expected to contain fast-growing cells and thus sorted into the nontarget channel (called collect in Fig. 1), whereas a droplet with lower fluorescence intensity than the sorting threshold was expected to be empty or contain slow-growing cells and thus flowed into the target channel (called “waste” in Fig. 1). In other words, slow-growing cells were purified in the target channel by sorting out fast-growing cells to the nontarget channel. The sorting operation was performed with a throughput of ~500 droplets/s for at least 60 min. Some of the SADA-sorted and SADA-unsorted droplets were collected via glass capillaries (3520-100, VitroCom) to measure the volume within the droplet occupied by budding yeast cells, which corresponds to the number of cells in a droplet. In other words, the larger the volume, the faster the growth of encapsulated cells. The volume was quantified by the following steps: (i) taking 2D bright-field images of the droplets in a glass capillary; (ii) measuring the area occupied by the cells within each droplet; (iii) calculating the radius of the measured area, assuming that its shape is circular; and (iv) estimating the volume using the calculated radius. Cells that occupied less than 1.5 × 105 μm3 of the droplets were considered as those with low proliferative activity. Given that threshold for the proliferative activity of cells, we determined the numbers of target and nontarget droplets in the target channel (Ntarget droplet, target and Nnontarget droplet, target) and those in the nontarget channel (Ntarget droplet, nontarget and Nnontarget droplet, nontarget). The sort purity of the target droplets in the target channel was calculated as the ratio of Ntarget droplet, target to the sum of Ntarget droplet, target and Nnontarget droplet, target; the sort purity of the nontarget droplets in the nontarget channel was calculated as the ratio of Nnontarget droplet, nontarget to the sum of Ntarget droplet, nontarget and Nnontarget droplet, nontarget. The enrichment factor of the slow-growing cells was calculated as the ratio of the post-sort ratio (the ratio of Ntarget droplet, target to the sum of Ntarget droplet, target and Nnontarget droplet, target) to the pre-sort ratio (the ratio of the number of slow-growing cells to the sum of the numbers of fast-growing cells and slow-growing cells = 1 to 50).
Supplementary Material
Acknowledgments
We thank N. T. Ishii for assistance with the cell analysis. Funding: This work was funded by the ImPACT Program of the Council for Science, Technology, and Innovation (Cabinet Office, Government of Japan). This work was also partially supported by JSPS KAKENHI (JP18K14101), JSPS Core-to-Core Program, JST Adaptable and Seamless Technology transfer Program (JPMJTM19Y9), Kanagawa Institute of Industrial Science and Technology (KISTEC), White Rock Foundation, Murata Science Foundation, and Cross-ministerial Strategic Innovation Promotion Program (SIP). Author contributions: A.I. and K.G. conceived the concept of the SADA. A.I. carried out the computational analysis. A.I., Y. Shib., S.U., and Y.Shir. designed the SADA sorter. Y. Shib., N.T., D.L.S., J.-W.P., M.H.L., Y.N., and D.H. constructed the sorter. A.I. and T.O. designed the electrical control circuit. Y.Shib. and H.Mik. designed the optical setup for the SADA sorter. T.O. constructed the electrical control circuit. H.Mik. and M.H.L. constructed the evaluation platform of the fluorescent signals from cells. Y.N. and M.H.L. constructed the reinjection platform for the SADA sorter. S.O., Y.O., K.N., H.W., K.Y., O.I., T.I., Y. Kan., Y. Kat., T.H., S.M., M. Yam., M. Yaz., and K.S. prepared the cells. K.N., H.T., and H.W. constructed the evaluation platform of the antibody production of the hybridoma. Y. Shib., N.T., M.H.L., Y.N., H.Miw. C.T.R., and D.L.S. performed the sorting experiments. A.I. led the experimental work. K.G. supervised the project. A.I., D.D.C., and K.G. participated in writing the manuscript. Competing interests: K.G., A.I., Y. Shib., S.U., Y. Shir., and D.H. are inventors of a patent application covering the SADA sorter. K.G. is a shareholder of K.K. CYBO. K.S. is a shareholder of Euglena Co. Ltd. The authors declare that they have no other competing interests. Data and materials availability: The raw data in this manuscript are available in data files (data file S1 or S2) or from the corresponding author upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba6712/DC1
REFERENCES AND NOTES
- 1.Obexer R., Godina A., Garrabou X., Mittl P. R. E., Baker D., Grif A. D., Hilvert D., Emergence of a catalytic tetrad during evolution of a highly active artificial aldolase. Nat. Chem. 9, 50–56 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Abalde-Cela S., Taladriz-Blanco P., De Oliveira M. G., Abell C., Droplet microfluidics for the highly controlled synthesis of branched gold nanoparticles. Sci. Rep. 8, 1–6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macosko E. Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A. R., Kamitaki N., Martersteck E. M., Trombetta J. J., Weitz D. A., Sanes J. R., Shalek A. K., Regev A., McCarroll S. A., Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rane T. D., Zec H. C., Puleo C., Lee A. P., Wang T.-H., Droplet microfluidics for amplification-free genetic detection of single cells. Lab Chip 12, 3341–3347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallina M. E., Kim T. J., Shelor M., Vasquez J., Mongersun A., Kim M., Tang S. K. Y., Abbyad P., Pratx G., Toward a droplet-based single-cell radiometric assay. Anal. Chem. 89, 6472–6481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abatemarco J., Sarhan M. F., Wagner J. M., Lin J.-L., Liu L., Hassouneh W., Yuan S.-F., Alper H. S., Abate A. R., RNA-aptamers-in-droplets (RAPID) high-throughput screening for secretory phenotypes. Nat. Commun. 8, 332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hindson C. M., Chevillet J. R., Briggs H. A., Gallichotte E. N., Ruf I. K., Hindson B. J., Vessella R. L., Tewari M., Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 10, 1003–1005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller O. J., El Harrak A., Mangeat T., Baret J.-C., Frenz L., El Debs B., Mayot E., Samuels M. L., Rooney E. K., Dieu P., Galvan M., Link D. R., Griffiths A. D., High-resolution dose–response screening using droplet-based microfluidics. Proc. Natl. Acad. Sci. U.S.A. 109, 378–383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oxnard G. R., Paweletz C. P., Kuang Y., Mach S. L., O’Connell A., Messineo M. M., Luke J. J., Butaney M., Kirschmeier P., Jackman D. M., Jänne P. A., Noninvasive detection of response and resistance in EGFR-Mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer Res. 20, 1698–1705 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C.-H., Miller M. A., Sarkar A., Beste M. T., Isaacson K. B., Lauffenburger D. A., Griffith L. G., Han J., Multiplexed protease activity assay for low-volume clinical samples using droplet-based microfluidics and its application to endometriosis. J. Am. Chem. Soc. 135, 1645–1648 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi N., Lee J., Ko J., Jeon J. H., Rhie G.-E., DeMello A. J., Choo J., Integrated SERS-based microdroplet platform for the automated immunoassay of F1 antigens in Yersinia pestis. Anal. Chem. 89, 8413–8420 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Guo M. T., Rotem A., Heyman J. A., Weitz D. A., Droplet microfluidics for high-throughput biological assays. Lab Chip 12, 2146–2155 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Mazutis L., Gilbert J., Ung W. L., Weitz D. A., Griffiths A. D., Heyman J. A., Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 8, 870–891 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang B. L., Ghaderi A., Zhou H., Agresti J., Weitz D. A., Fink G. R., Stephanopoulos G., Microfluidic high-throughput culturing of single cells for selection based on extracellular metabolite production or consumption. Nat. Biotechnol. 32, 473–478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sart S., Tomasi R. F.-X., Amselem G., Baroud C. N., Multiscale cytometry and regulation of 3D cell cultures on a chip. Nat. Commun. 8, 469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Köster S., Angilè F. E., Duan H., Agresti J. J., Wintner A., Schmitz C., Rowat A. C., Merten C. A., Pisignano D., Griffiths A. D., Weitz D. A., Drop-based microfluidic devices for encapsulation of single cells. Lab Chip 8, 1110–1115 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Rajeswari P. K. P., Joensson H. N., Andersson-svahn H., Droplet size influences division of mammalian cell factories in droplet microfluidic cultivation. Electrophoresis 38, 305–310 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Klein A. M., Macosko E., InDrops and Drop-seq technologies for single-cell sequencing. Lab Chip 17, 2540–2541 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Sciambi A., Abate A. R., Accurate microfluidic sorting of droplets at 30 kHz. Lab Chip 15, 47–51 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beneyton T., Wijaya I. P. M., Postros P., Najah M., Leblond P., Couvent A., Mayot E., Griffiths A. D., Drevelle A., High-throughput screening of filamentous fungi using nanoliter-range droplet-based microfluidics. Sci. Rep. 6, 27223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark I. C., Thakur R., Abate A. R., Concentric electrodes improve microfluidic droplet sorting. Lab Chip 18, 710–713 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahn K., Kerbage C., Hunt T. P., Westervelt R. M., Link D. R., Weitz D. A., Dielectrophoretic manipulation of drops for high-speed microfluidic sorting devices. Appl. Phys. Lett. 88, 024104 (2006). [Google Scholar]
- 23.Baret J.-C., Miller O. J., Taly V., Ryckelynck M., El-Harrak A., Frenz L., Rick C., Samuels M. L., Hutchison J. B., Agresti J. J., Link D. R., Weitz D. A., Griffiths A. D., Fluorescence-activated droplet sorting (FADS): Efficient microfluidic cell sorting based on enzymatic activity. Lab Chip 9, 1850–1858 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Kintses B., Hein C., Mohamed M. F., Fischlechner M., Courtois F., Lainé C., Hollfelder F., Picoliter cell lysate assays in microfluidic droplet compartments for directed enzyme evolution. Chem. Biol. 19, 1001–1009 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Best R. J., Lyczakowski J. J., Abalde-Cela S., Yu Z., Abell C., Smith A. G., Label-free analysis and sorting of microalgae and cyanobacteria in microdroplets by intrinsic chlorophyll fluorescence for the identification of fast growing strains. Anal. Chem. 88, 10445–10451 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Kato Y., Ho S.-H., Vavricka C. J., Chang J.-S., Hasunuma T., Kondo A., Bioresource Technology Evolutionary engineering of salt-resistant Chlamydomonas sp. strains reveals salinity stress-activated starch-to-lipid biosynthesis switching. Bioresour. Technol. 245, 1484–1490 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Terashima A., Watarai H., Inoue S., Sekine E., Nakagawa R., Hase K., Iwamura C., Nakajima H., Nakayama T., Taniguchi M., A novel subset of mouse NKT cells bearing the IL-17 receptor B responds to IL-25 and contributes to airway hyperreactivity. J. Exp. Med. 205, 2727–2733 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu H., Eustace D., Merten C. A., Efficient cell pairing in droplets using dual-color sorting. Lab Chip 15, 3989–3993 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Boeke J. D., Trueheart J., Natsoulis G., Fink G. R., 5-fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 154, 164–175 (1987). [DOI] [PubMed] [Google Scholar]
- 30.Silver L. L., Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24, 71–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marusyk A., Almendro V., Polyak K., Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 12, 323–334 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Suwa Y., Hattori T., Suwa Y., Population dynamics of soil bacteria cultured under different nutrient conditions. Soil Sci. Plant Nutr. 33, 235–244 (1987). [Google Scholar]
- 33.Di Carlo D., Technologies for the directed evolution of cell therapies. SLAS Technol. 24, 359–372 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Li S., Li M., Hui Y. S., Cao W., Li W., Wen W., A novel method to construct 3D electrodes at the sidewall of microfluidic channel. Microfluid. Nanofluidics. 14, 499–508 (2013). [Google Scholar]
- 35.Brouzes E., Medkova M., Savenelli N., Marran D., Twardowski M., Hutchison J. B., Rothberg J. M., Link D. R., Perrimon N., Samuels M. L., Droplet microfluidic technology for single-cell high-throughput screening. Proc. Natl. Acad. Sci. U.S.A. 106, 14195–14200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lei C., Kobayashi H., Wu Y., Li M., Isozaki A., Yasumoto A., Mikami H., Ito T., Nitta N., Sugimura T., Yamada M., Yatomi Y., Di Carlo D., Ozeki Y., Goda K., High-throughput imaging flow cytometry by optofluidic time-stretch microscopy. Nat. Protoc. 13, 1603–1631 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Hiramatsu K., Ideguchi T., Yonamine Y., Lee S. W., Luo Y., Hashimoto K., Ito T., Hase M., Park J.-W., Kasai Y., Sakuma S., Hayakawa T., Arai F., Hoshino Y., Goda K., High-throughput label-free molecular fingerprinting flow cytometry. Sci. Adv. 5, eaau0241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pethig R., Dielectrophoresis: Status of the theory, technology, and applications. Biomicrofluidics. 4, 022811 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou H., Yao S., Electrostatic charging and control of droplets in microfluidic devices. Lab Chip 13, 962–969 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Song L., Awari D. W., Han E. Y., Uche-Anya E., Park S.-H. E., Yabe Y. A., Chung W. K., Yazawa M., Dual optical recordings for action potentials and calcium handling in induced pluripotent stem cell models of cardiac arrhythmias using genetically encoded fluorescent indicators. Stem Cell Transl. Med. 4, 468–475 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kubo K., Okada H., Shimamoto T., Kimori Y., Mizunuma M., Bi E., Ohnuki S., Ohya Y., Implications of maintenance of mother–bud neck size in diverse vital processes of Saccharomyces cerevisiae. Curr. Genet. 65, 253–267 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Gietz R. D., Schiestl R. H., High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba6712/DC1