

Mre11 exonuclease activity removes gemcitabine from the nascent strand during DNA replication.
Abstract
The Mre11 nuclease is involved in early responses to DNA damage, often mediated by its role in DNA end processing. MRE11 mutations and aberrant expression are associated with carcinogenesis and cancer treatment outcomes. While, in recent years, progress has been made in understanding the role of Mre11 nuclease activities in DNA double-strand break repair, their role during replication has remained elusive. The nucleoside analog gemcitabine, widely used in cancer therapy, acts as a replication chain terminator; for a cell to survive treatment, gemcitabine needs to be removed from replicating DNA. Activities responsible for this removal have, so far, not been identified. We show that Mre11 3′ to 5′ exonuclease activity removes gemcitabine from nascent DNA during replication. This contributes to replication progression and gemcitabine resistance. We thus uncovered a replication-supporting role for Mre11 exonuclease activity, which is distinct from its previously reported detrimental role in uncontrolled resection in recombination-deficient cells.
INTRODUCTION
Error-free DNA replication is essential for maintaining genome stability and for preventing the accumulation of mutations, which drive carcinogenesis. Replication fork blockage interferes with cell division and contributes to genome instability. Different sources of replication stress can impede fork progression, e.g., a lack of deoxynucleotide triphosphates (dNTPs), or obstacles (e.g., DNA lesions) in the template strand. Various mechanisms have evolved to deal with these sources of replication stress (1).
Homologous recombination (HR) is a well-studied mechanism involved in repair of DNA double-strand breaks (DSBs). In HR, the DNA is repaired through invasion of a broken DNA strand into a homologous DNA duplex followed by copy synthesis. Before strand invasion takes place, 5′ DSB ends are resected to create 3′ single-strand overhangs (2). One of the proteins responsible for end resection is Mre11, part of the Mre11/Rad50/Nbs1 (MRN) complex.
MRN is highly conserved among eukaryotes and is, together with CtIP, involved in a wide range of early responses to DNA damage, often mediated by its role in DNA end processing (3). Central to DNA end processing are Mre11 single-strand endonuclease and 3′ to 5′ exonuclease activities. The role of Mre11 exonuclease activity in HR has long been enigmatic, as the 3′ to 5′ polarity is in the opposite direction of the 5′ to 3′ resection required for HR. More recently, several studies suggest that the Mre11 endonuclease activity creates single-strand nicks, which serve as entry points for the Mre11 3′ to 5′ exonuclease activity directed toward the break, while other (nuclease) activities are responsible for resection away from the break (4–6). After resection, invasion of the 3′ end of the DSB into homologous DNA is mediated by Rad51, aided in humans by BRCA1, BRCA2, and several RAD51 paralogues. The invading 3′ end acts as a primer for copy synthesis (2).
Several Mre11 functions are independent of its nuclease activities. Saccharomyces cerevisiae mre11 nuclease-deficient mutants are only partially sensitive to ionizing radiation and are proficient for several other phenotypes observed in mre11 null mutants (7). In Schizosaccharomyces pombe, resection of the C-rich strand at telomere ends requires Mre11 but is independent of its nuclease activities (8), while an mre11 nuclease mutant is defective in Rec12Spo11 removal and shows sensitivity to topoisomerase poisons but not to methyl methanesulfonate and ionizing radiation (9).
MRN/CtIP and other proteins involved in HR have also been implicated in the restart of stalled replication forks after they encounter an obstacle in the template strand (10–16). The precise role of the MRN complex, and especially the role of the Mre11 nuclease activity in replication fork restart, is not understood. S. cerevisiae Mre11 is recruited to paused replication forks and stabilizes their association with replisome components, but this function does not depend on the Mre11 nuclease activity (17). In S. pombe, enrichment of the HR protein Rad52 at stalled forks is dependent on the MRN complex (14) but is independent of Mre11 nuclease activity (18). In absence of some HR proteins in human cells (e.g., when RAD51 or BRCA2 are mutated/depleted), MRE11 exonuclease activity is responsible for uncontrolled deleterious degradation of stalled and reversed forks (19). However, it remains unknown whether and how MRE11 nuclease activity supports replication progression in recombination-proficient cells.
While DNA damage response mechanisms that deal with fork stalling caused by nucleotide pool depletion or obstacles on the template strand have been widely studied (20), less is known about mechanisms that deal with the replication blockage of the nascent strand, e.g., caused by incorporation of chain-terminating nucleotides. Nucleoside analogs are frequently used in cancer therapy. They interfere with DNA replication by inhibiting nucleotide metabolism and can act as DNA replication chain terminators after incorporation into nascent DNA (21). The nucleoside analog gemcitabine [2′,2′-difluoro 2′-deoxycytidine (dFdC)], used to treat a range of cancers (22), is a deoxycytidine analog that contains two fluorine atoms at the 2′ carbon of the sugar ring. It is a prodrug that, once transported into a cell, is phosphorylated into dFdC monophosphate (dFdCMP) by deoxycytidine kinase (dCK) and subsequently to dFdC diphosphate (dFdCDP) and dFdC triphosphate (dFdCTP) (22). Competing with deoxycytidine triphosphate, dFdCTP is incorporated into the nascent DNA strand during replication, which leads to chain termination after incorporation of an additional dNTP (23). In addition, dFdCDP acts as an inhibitor of ribonucleotide reductase (RNR), depleting the dNTP pools, thus increasing the likelihood of dFdCTP integration (22).
Little is known about DNA repair pathways resisting treatment with dFdC. Chinese hamster ovary (CHO) cell nucleotide excision repair (xpd and ercc1), nonhomologous end joining (DNA-pkcs), base excision repair (xrcc1), and HR (xrcc3) mutants are not sensitive to dFdC (24). In contrast, mutation of BRCA2 in CHO cells and small interfering RNA (siRNA) knockdown of human RAD51 increase dFdC resistance (25).
For a cell to be able to resist treatment with dFdC, the chain-terminating nucleoside analog must be removed to allow replication restart. Little is known about mechanisms that remove replication-terminating dFdC (or other chain-terminating nucleoside analogs) from nascent DNA. In vitro, the proofreading exonuclease activity of DNA polymerase ε is able to remove the chain-terminating nucleoside analog cytarabine from DNA ends (23), and this activity contributes to cellular cytarabine tolerance (26). However, this activity is not able to efficiently remove dFdCMP from a 3′ DNA end or from the penultimate (masked by another nucleotide) position (23). Similarly, the human-base excision repair nuclease Ape1 was shown to be able to remove l-configuration nucleoside analogs (e.g., troxacitabine) from DNA but had little activity against dFdC or other d-configuration nucleoside analogs (27). TDP1 is able to remove cytarabine from 3′ DNA ends in vitro, and TDP1−/− DT40 cells and mouse embryonic fibroblasts are hypersensitive to this drug, while they are not sensitive to dFdC (28). Hence, the identity of the enzyme(s) involved in dFdC removal has remained elusive.
We have previously shown that the nuclease activity of fission yeast Mre11 is involved in removing covalently bound Spo11 (29), topoisomerase I, and topoisomerase II (9) from DNA. After dFdC treatment, MRN proteins form nuclear foci at stalled forks in the absence of detectable DNA breaks, while siRNA knockdown of Mre11, Rad50 (30), and mirin treatment (31) causes a slight dFdC sensitivity in human cells. A recent study (32) in DT40 cells showed that some other nucleoside analogs (dFdC was not tested) retard replication in Mre11 nuclease mutants. However, removal of these nucleoside analogs from genomic DNA was not assessed, and it has remained unknown whether the observed phenotypes in these mutants were due to a role of Mre11 in resisting nucleotide pool imbalances (caused by treatment with nucleoside analogs) or whether Mre11 nuclease activity is involved in removal of chain-terminating nucleoside analogs. In addition, as the MRE11 mutants used for this study were defective for both endo- and exonuclease activity, it has remained unclear whether replication delay was caused by an endo- or exonuclease defect. We thus decided to test the potential roles for the Mre11 endo- and exonuclease activities in removing the chain-terminating nucleoside analog dFdC from genomic DNA during DNA replication in several model organisms.
RESULTS
Creation of a nucleotide salvage pathway in S. pombe
As S. pombe does not have a functional nucleotide salvage pathway that would allow the uptake and phosphorylation of dFdC, we inserted the genes encoding for the human equilibrative nucleoside transporter 1 (hENT1) (33) and the human deoxycytidine kinase (dCK) (34), both under the control of the constitutive adh promoter, into the S. pombe genome. This facilitates the import of dFdC into S. pombe cells and the subsequent phosphorylation of dFdC into dFdCMP and, subsequently, through two phosphorylation steps by endogenous nucleoside monophosphate and nucleoside diphosphate kinases (35) into dFdCTP (see Fig. 1A).
Fig. 1. Creation of a nucleotide salvage pathway in S. pombe.
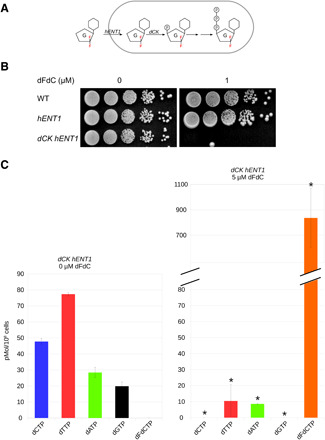
(A) Integration of the genes encoding for the hENT1 and the dCK into the S. pombe genome. This facilitates the import of dFdC into S. pombe cells and the subsequent phosphorylation of dFdC into dFdCMP and through two subsequent phosphorylation steps into dFdCTP. (B) Wild-type (WT) cells without hENT1 and dCK, as well as cells expressing only hENT1, are not sensitive to 1 μM dFdC; cells expressing both hENT1 and dCK are sensitive. (C) Treating hENT1 dCK strains with dFdC led to a reduction in dNTP levels, and the appearance of dFdCTP, resulting from the triple phosphorylation of dFdC. This shows that dFdC is transformed into dFdCTP in cells containing hENT1 and dCK. Asterisk indicates a statistically significant difference with untreated cells (t test, P < 0.05; n = 3). G, gemcitabine; F, fluorine; P, Phosphate.
As shown in Fig. 1B, strains without hENT1 and dCK, as well as cells with only hENT1 but not dCK, were resistant to 1 μM dFdC, while cells expressing both hENT1 and dCK were sensitive. We measured intracellular dNTP and dFdCTP levels, as shown in Fig. 1C, treating hENT1 dCK strains with dFdC led to the appearance of dFdCTP, resulting from the triple phosphorylation of dFdC. Similar to the effects observed in human cells (22), S. pombe dNTP pools were reduced after dFdC treatment in hENT1 dCK cells. We could detect dFdCMP, dFdCDP, and dFdCTP in hENT1 dCK cells treated with dFdC but not in dFdC-treated cells, which do not contain hENT1 and dCK (fig. S1). These results show that insertion of the hENT1 transporter and dCK kinase successfully creates a nucleotide salvage pathway in S. pombe, which sensitizes cells to dFdC.
MRN and Ctp1 promote resistance to dFdC and removal from genomic DNA
To study whether the MRN complex and the S. pombe Sae2/CtIP homolog Ctp1 (36) contribute to dFdC resistance, we tested the sensitivity of deletion mutants to dFdC and found that these were all sensitive (Fig. 2A). Next, to assess whether these mutants were deficient in removing dFdC from genomic DNA after incorporation, we quantified dFdC in genomic DNA. Briefly (for details, see Materials and Methods), wild-type (WT) and mutant cells were treated for 3 hours with dFdC and a stable “heavy” isotope of deoxycytidine, 15N3dC (hdC). Genomic DNA was isolated and subsequently enzymatically digested and dephosphorylated into single nucleosides. Using liquid chromatography–tandem mass spectrometry (LC-MS/MS), the amounts of dFdC and hdC in genomic DNA were quantified. To correct for differences in replication progression between the strains, we calculated the dFdC/hdC ratio, which reflects the amount of dFdC in genomic DNA corrected for the total amount of replication taken place during dFdC/hdC treatment. As shown in Fig. 2B, the dFdC/hdC ratios were significantly increased ~2.5- to 3-fold in the deletion mutants compared to WT.
Fig. 2. MRN and Ctp1 promote resistance to dFdC and removal from genomic DNA.
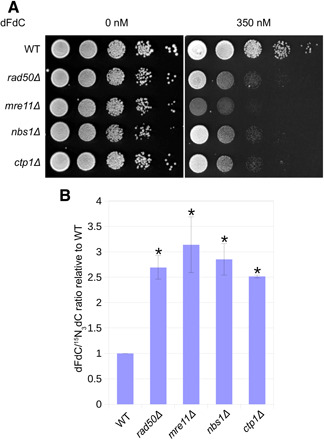
(A) mrn/ctp1 deletion mutants are sensitive to dFdC. Representative example of three experiments. (B) The dFdC/hdC ratio is significantly increased in mrn/ctp1 deletion mutants compared to WT. Error bars depict SE; asterisk indicates statistically significant difference with WT (t test, P < 0.05; n = 3).
Several studies have shown that dFdC is incorporated into genomic DNA during S phase and that this is responsible for its cytotoxic effect, rather than dNTP pool depletion (23, 37, 38). In our study, under the treatment conditions used to measure the dFdC/hdC ratio (50 μM dFdC), cells are arrested in early S phase (as shown in Fig. 4; cells are arrested when exposed to 2 μM dFdC). As dFdC is not incorporated outside of S phase and cells do not progress through S phase at high dFdC concentrations, the increase in the dFdC/hdC ratio in the deletion mutants is best explained with a deficiency in removing dFdC from the nascent strand during DNA replication, suggesting that dFdC removal is promoted by the MRN complex and Ctp1.
Fig. 4. Mre11 nuclease activity supports replication progression in the presence of dFdC.
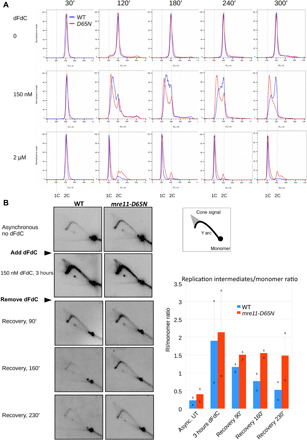
(A) Flow cytometric analysis of WT (blue) and mre11-D65N (red) cells, synchronized and released in G2 in the presence of 0, 150 nM, and 2 μM dFdC. This is an alternative presentation of data shown in fig. S3, allowing direct comparison between WT and mre11-D65N cells. Flow cytometric profiles in the absence of dFdC and in the presence of 2 μM dFdC show little difference between WT and mre11-D65N cells. In the presence of 150 nM dFdC, WT cells (blue) slowly progress through S phase from 1C to 2C, whereas the great majority of mre11-D65N cells (red) fail to progress and display a 1C content throughout the time course. Representative example of three experiments. (B) Quantification of the combined replication intermediates (Y arc and cone structures) relative to the (nonreplicating) monomer spot shows that replication intermediates persist in the mre11-D65N mutant, suggesting that Mre11 nuclease activity is required for recovery from dFdC treatment. Left: Representative example of two experiments. Right: Quantification of replication intermediate signal/monomer spot ratio, relative to the ratio in untreated asynchronous cells. Average of two experiments, “x” depicts the values from individual experiments from which the average was calculated. The replication intermediates (RI)/monomer ratio is significantly higher in mre11-D65N compared to WT (pairwise comparison using paired t test, P = 0.034).
Mre11 nuclease activity contributes to dFdC resistance and removal
To assess a potential contribution of Mre11 nuclease activity to dFdC resistance, we tested the sensitivity of two different mre11 nuclease mutants (impaired for both endo- and exonuclease activity), mre11-D65N (9, 39) and mre11-H134S (40), to dFdC. As shown in Fig. 3A, both nuclease mutants are sensitive to dFdC.
Fig. 3. Mre11 nuclease activity contributes to dFdC resistance and removal.
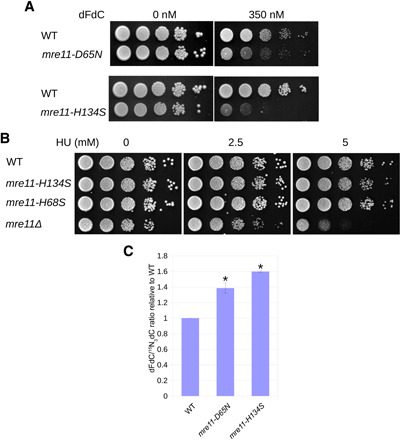
(A) The mre11 nuclease mutants mre11-D65N and mre11-H134S are sensitive to dFdC. Representative example of three experiments. (B) mre11 nuclease mutants are not sensitive against hydroxyurea (HU) concentrations, which sensitize an mre11 deletion mutant. (C) The dFdC/hdC ratio is significantly increased in mre11 nuclease mutants compared to WT. Error bars depict SE; asterisk indicates statistically significant difference with WT (t test, P < 0.05; n = 3 for WT and mre11-H134S; n = 6 for mre11-D65N).
dFdC acts as a chain terminator but also as an inhibitor of RNR, thus decreasing dNTP pools. To test whether the sensitivity of S. pombe mre11 nuclease dead mutants can be (partially) attributed to a potential role in resisting dNTP depletion, we tested mre11-H134S and the exonuclease mutant mre11-H68S (see also below) (40) for sensitivity against hydroxyurea (HU), another RNR inhibitor. As shown in Fig. 3B, these mre11 mutants were not hypersensitive to HU at concentrations that substantially sensitize mre11Δ cells. While this confirms that the MRN complex is involved in resisting dNTP pool depletion (41), it suggests that this function is not mediated by mre11 nuclease activity and that the sensitivity of the mre11 nuclease mutants to dFdC is not due to a role of this activity in resisting dNTP depletion but is more likely explained by its dFdC removal defect. We measured the dFdC/hdC ratios and found that these are increased in both mre11-D65N and mre11-H134S mutants (Fig. 3C), suggesting that the Mre11 nuclease activity removes dFdC from genomic DNA during replication.
The increase in genomic dFdC/hdC ratio in mre11 nuclease mutants compared to WT shows that the Mre11 nuclease activity preferentially removes replication-blocking dFdC over hdC (which is chemically indistinguishable from dC). We also found that an S. pombe deletion of exo1, encoding a nuclease that has been implicated in long-range resection at the replication fork (42), does not show an increase in the dFdC/hdC ratio (see fig. S2). This suggests that indiscriminate (having no preference for dFdC, hdC, or natural deoxynucleotides) long-range resection of stalled forks does not affect the dFdC/hdC ratio in our assay. These observations indicate that the increased dFdC/hdC ratio in the mre11 nuclease mutants is not due to a role of Mre11 in uncontrolled resection of nascent DNA strands at the replication fork.
While the dFdC/hdC ratio in mre11Δ shows a ~3-fold increase compared to WT (Fig. 2B), the increase in the two mre11 nuclease mutants is reduced to ~1.5-fold. This might reflect that some dFdC is removed by an alternative (MRN-dependent) activity or that the mre11 nuclease mutants are only partially nuclease deficient.
Mre11 nuclease activity supports replication progression in the presence of dFdC
Data presented so far suggest that Mre11 nuclease activity contributes to dFdC resistance by removing chain-terminating dFdC from nascent DNA, thus supporting replication progression. We therefore assessed replication progression using flow cytometry in synchronized WT and mre11-D65N strains. Temperature sensitive nda3-KM311 cells were arrested at G2-M and, subsequently, released after which they performed synchronous mitosis followed by replication (43).
As shown in fig. S3A, after release from the G2 block, the shift to G1 (due to mitosis) was barely visible in untreated WT and mre11-D65N cells. This is because S. pombe cells have a very short G1 phase and will initiate DNA replication directly after mitosis, before cytokinesis (see fig. S3B for schematic). When WT and mre11-D65N cells were treated with 2 μM dFdC, replication was inhibited and cells were blocked in early S phase. As shown in Fig. 4A (a different representation of the data shown in fig. S3A), in both untreated cells and cells treated with 2 μM dFdC, there was little difference in replication progression between WT and mre11-D65N cells. However, in cells treated with 150 nM dFdC, we observed delayed S phase progression in WT and pronounced replication arrest in mre11-D65N cells.
We also compared the replication fork progression between WT and mre11-D65N using two-dimensional (2D) gel electrophoresis, which allows the analysis of replication intermediates (44). Asynchronous WT and mutant cells were treated with 150 nM dFdC for 3 hours, after which dFdC was washed out to allow recovery. As shown in Fig. 4B, replication intermediates (including Y arc and cone signal) accumulated in both strains upon dFdC treatment, consistent with a global slowdown in S phase progression. During recovery, DNA replication intermediates persisted longer in mre11-D65N compared to WT, indicating that replication fork progression was delayed in the absence of Mre11 nuclease activity. Overall, the replication intermediates/monomer ratio is significantly higher in mre11-D65N compared to WT (P = 0.034, pairwise comparison using paired t test). These results show that Mre11 nuclease activity supports replication progression after dFdC treatment.
The S. pombe mre11-H68S mutant is mildly sensitive to dFdC
Mre11 has both single-strand endonuclease and 3′ to 5′ exonuclease activities. As the polarity of the 3′ to 5′ exonuclease activity is ideally suited to remove replication blocking nucleoside analogs from the growing 3′ end of the nascent DNA strand, we tested whether an S. pombe mre11-H68S mutant is sensitive to dFdC. This mutant was previously assumed to be defective for the 3′ to 5′ exonuclease activity based on the absence of exonuclease activity in an equivalent Pyrococcus furiosus mre11 mutant in vitro (40). As shown in Fig. 5A, the mre11-H68S mutant was mildly sensitive to dFdC compared to WT but less sensitive than the mre11-H134S mutant, which is deficient for both endo- and exonuclease activities. We also determined the dFdC/hdC ratio in this mutant and found that it showed a small insignificant increase compared to WT (see Fig. 5B). However, it was previously shown that the equivalent S. cerevisiae mutant protein Mre11-H59S showed reduced, but not abolished, 3′ to 5′ exonuclease activity in biochemical assays (4). Thus, while the slight sensitivity of the mre11-H68S mutant suggests that the exonuclease activity contributes to dFdC resistance, the extent of this role might be masked by residual exonuclease activity in the mre11-H68S mutant.
Fig. 5. The S. pombe mre11-H68S mutant is mildly sensitive to dFdC.
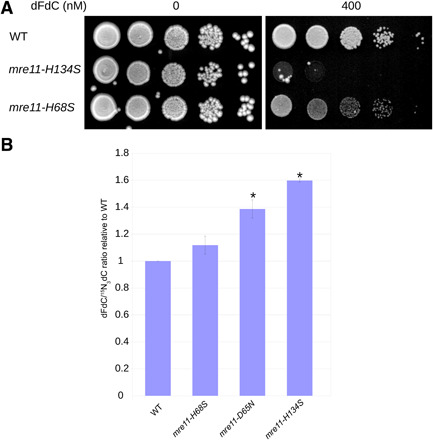
(A) While the mre11-H134S nuclease mutant, which is impaired for both endo- and exonuclease activity, is sensitive to 400 nM dFdC, the mre11-H68S mutant is only mildly sensitive. Representative example of three experiments. (B) The dFdC/hdC ratio is significantly increased in mre11-D65N and mre11H134S (same data as shown in Fig. 3C to allow direct comparison) compared to WT, mre11-H68S shows a small but insignificant increase. Error bars depict SE. Asterisk indicates a statistically significant difference with WT (t test, P < 0.05; n = 3 for WT and mre11-H134S, n = 6 for mre11-D65N, and n = 3 for mre11-H68S).
Mre11 3′ to 5′ exonuclease activity contributes to dFdC resistance and removal in DT40 and MRC-5 cells
As results obtained using the S. pombe mre11-H68S mutant were not conclusive and to determine whether the role of the Mre11 nuclease activity has been conserved in higher eukaryotes, we decided to study dFdC resistance and removal in chicken DT40 and human MRC-5 cells. First, to assess the contribution of Mre11 nuclease activity to dFdC removal and resistance in vertebrate cells, we created an MRE11H129N/− nuclease mutant (defective for both endo- and exonuclease activity) in DT40 cells, expressing the mutant protein from its endogenous promotor, preserving the intron/exon structure of the gene (see Materials and Methods for details). As shown in Fig. 6A (blue lines), this mutant was sensitive to dFdC compared to MRE11+/− cells. We also measured genomic dFdC incorporation in this mutant and found that the dFdC/hdC ratio (Fig. 6B) increased approximately fourfold in MRE11H129N/− cells compared to MRE11+/− cells.
Fig. 6. Mre11 3′ to 5′ exonuclease activity contributes to dFdC resistance and removal in DT40 and MRC-5 cells.
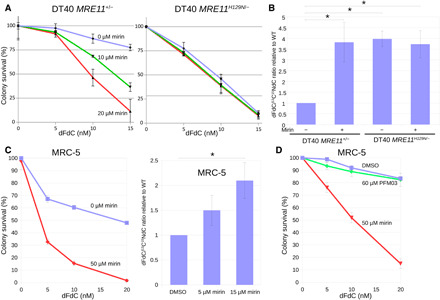
(A) MRE11H129N/− DT40 cells are sensitive to dFdC compared to MRE11+/− cells in the absence of the exonuclease inhibitor mirin (blue line). In MRE11+/− cells, the addition of mirin (green and red lines) increases dFdC sensitivity, whereas in MRE11H129N/− cells, mirin does not sensitize against dFdC beyond the sensitivity caused by the nuclease mutation. Error bars depict SE; n = 3. (B) Mirin treatment increases the dFdC/hdC ratio in MRE11+/− cells. MRE11H129N/− cells also show an increased dFdC/hdC ratio, but the addition of mirin to the nuclease mutant cells does not further increase the dFdC/hdC ratio. Error bars depict SE. Asterisk indicates statistically significant difference with MRE11+/− cells (t test, P < 0.05; n = 7). (C) Mirin treatment increases dFdC sensitivity (left; error bars depict SE; n = 3) and increases the dFdC/hdC ratio in MRC-5 cells (right; error bars depict SE; asterisk indicates statistical significance; t test, P < 0.05; n = 7). (D) The Mre11 endonuclease inhibitor PFM03 does not sensitize MRC-5 cells to dFdC. Error bars depict SE; n = 3. DMSO, dimethyl sulfoxide.
To assess whether the Mre11 3′ to 5′ exonuclease activity, rather than the single strand endonuclease activity, contributes to dFdC resistance and removal, we tested dFdC sensitivity of both DT40 MRE11+/− and MRE11H129N/− cells treated with different concentrations of mirin, which has been shown to specifically inhibit the Mre11 3′ to 5′ exonuclease but not the endonuclease activity (6, 40). Increasing concentrations of mirin (Fig. 6A, green and red lines) sensitized MRE11+/− cells to dFdC, whereas MRE11H129N/− cells were not sensitized beyond the sensitivity caused by the MRE11H129N/− mutation. We obtained similar results using another Mre11 exonuclease inhibitor, PFM39 (see fig. S4). After mirin treatment, the dFdC/hdC ratio increased approximately fourfold in MRE11+/− cells compared to untreated cells, while mirin treatment did not further increase the dFdC/hdC ratio in MRE11H129N/− cells (Fig. 6B). The observation that mirin treatment does not increase dFdC sensitivity or the dFdC/hdC ratio in MRE11H129N/− cells confirms that mirin specifically targets Mre11 (exo)nuclease activity and that the effects of mirin on dFdC sensitivity and the dFdC/hdC ratio in MRE11+/− cells are not due to interference of mirin with other (nuclease) activities.
We also measured the amount of constituent dC in genomic DNA for a subset of experiments which formed the basis for Fig. 6B. As shown in fig. S5, which depicts the number of genomic dFdC and hdC molecules per 104 dC molecules in genomic DNA, the increased dFdC/hdC ratio in mirin-treated MRE11+/− cells and in MRE11H129N/− cells results from an increase in dFdC accompanied by a smaller increase in hdC. This suggests that the Mre11 nuclease activity removes (chain-terminating) dFdC together with a stretch of newly synthesized DNA.
We also tested the effect of mirin on dFdC sensitivity and the dFdC/hdC ratio in human lung fibroblast MRC-5 cells. As shown in Fig. 6C, mirin treatment increased the sensitivity of MRC-5 cells to dFdC (Fig. 6C, left) and also increased the dFdC/hdC ratio (Fig. 6C, right). Last, to assess whether the Mre11 endonuclease activity contributes to dFdC resistance, we treated MRC-5 cells with 60 μM Mre11 endonuclease inhibitor PFM03 (6) and found that it does not increase dFdC sensitivity (Fig. 6D). Our observations suggest that the Mre11 3′ to 5′ exonuclease activity, but not the endonuclease activity, contributes to dFdC resistance and removal in DT40 and MRC-5 cells.
DISCUSSION
Treatment with DNA damaging and replication inhibiting agents is one of the mainstays of cancer therapy. While defects in DNA repair pathways associated with carcinogenesis can sensitize cancer cells to treatment, it remains largely unknown which (often redundant) repair pathways/activities are responsible for repairing DNA damage caused by commonly used cancer drugs. Identification of and mechanistic insight into these pathways is important to improve treatment efficacy by tailoring the choice of drug to DNA repair defects present in cancer cells (personalized medicine) or by exploiting synthetic lethality with novel DNA repair–inhibiting drugs (45).
Artificial chain-terminating nucleoside analogs (e.g., dFdC and cytarabine) are extensively used not only in cancer treatment but also in antiviral therapy (46). Whereas nucleoside analogs used in cancer treatment target the replication machinery in the cancer cell, antiviral nucleoside analogs are designed to inhibit viral replication; integration into the genomic DNA of the host cell causes unwanted side effects (47). Therefore, identification of cellular activities that resist treatment with these drugs is also important to understand the off-target effects of (novel) antiviral nucleoside analogs.
While recent studies in yeast and human cells have implied a role for the Mre11 3′ to 5′ exonuclease activity in DSB end resection in meiotic and nonmeiotic cells, its role during replication has remained elusive. The 3′ to 5′ exonuclease activity of recombinant yeast Mre11 acts on blunt-ended double-strand DNA or a 3′ DNA end with a 5′ overhang but is not active toward a 3′ overhang (19). This activity is thus ideally suited to remove nucleotide analogs from the nascent 3′ end during replication. In this study, we have shown that inhibition of the Mre11 3′ to 5′ exonuclease activity sensitizes cells to dFdC and leads to an increase in dFdC in genomic DNA. This suggests that Mre11 exonuclease activity removes dFdC from genomic DNA during DNA replication, contributing to the innate resistance of cells against this drug.
Most previous studies on replication fork stalling have concentrated on stalling caused by nucleotide pool depletion or obstacles on the template strand (20). Under treatment conditions used in our study to quantify the dFdC/hdC ratio in genomic DNA, cells arrest at the first opportunity for dFdC to be integrated into genomic DNA, which is S phase. To the best of our knowledge, our study is the first to identify an activity that removes a chain-terminating nucleoside analog from the nascent (rather than the template) strand during DNA replication in vivo.
Our study demonstrates a role for Mre11 nuclease activity in dFdC removal in the model eukaryote S. pombe, in chicken DT40 cells, and in human MRC-5 cells, suggesting that this role in removing chain-terminating nucleotides from DNA has been conserved during evolution. While cells are unlikely to encounter dFdC in the natural environment, some recent studies suggest that chain-terminating nucleosides are present within the cellular free nucleoside/nucleotide pool, which is prone to endogenous damaging modifications; free nucleotides are much more susceptible to modifications than nucleotides already incorporated into DNA (48, 49). While the effect of integration of naturally occurring noncanonical nucleotides into genomic DNA is an underresearched area of study, there is evidence from biochemical assays with human proteins that incorporation of the oxidized nucleotide 8-oxo-dGTP into genomic DNA delays or terminates chain elongation (50, 51). It is, thus, possible that the Mre11 nuclease activity has evolved to remove noncanonical, endogenously damaged chain-terminating nucleotides from DNA during replication.
Previous studies have shown involvement of MRE11 exonuclease activity in uncontrolled deleterious degradation of DNA at the replication fork in cells that are deficient for HR (e.g., when RAD51 or BRCA2 are mutated/depleted); however, these studies have not revealed a beneficial role of Mre11 exonuclease activity in supporting progression of stalled forks in recombination-proficient cells. Our observations that the Mre11 exonuclease activity preferentially removes dFdC over hdC (which is chemically indistinguishable from dC) and that deletion of exo1 does not lead to an increase in the dFdC/hdC ratio show that the Mre11-dependent dFdC removal activity we uncovered is not the result of indiscriminate resection and is, thus, distinct from the previously described role of Mre11 nuclease activity in the uncontrolled deleterious resection resulting from defective HR.
We have thus uncovered a previously unidentified role for the Mre11 exonuclease activity in supporting replication fork progression, which has major implications for our understanding of the evolutionary conserved role of Mre11 in unblocking stalled replication forks. These findings will form a basis for future studies into the role of Mre11 and other (nuclease) activities in the removal of naturally occurring endogenously damaged nucleosides and artificial nucleoside analogs used in cancer and antiviral therapy.
MATERIALS AND METHODS
S. pombe strains and techniques
For strain construction and propagation, standard genetic methods and media were used (52). Strains used and constructed in this study are listed in table S1. For spot tests, cultures were diluted to 107 cells/ml and 10-fold diluted to 102 cells/ml. Of these dilutions, 10 μl were spotted for each culture on each plate.
The dCK gene (34) under control of the S. pombe adh promoter, was integrated into the S. pombe genome, replacing ura4. Subsequently, the hENT1 gene (33), under control of the adh promoter, coupled to a nourseothricin resistance marker, was integrated adjacent to dCK. Strains containing dCK and hENT1 were grown on minimal media (Edinburgh Minimal Medium and glutamate), as they display slow growth and elongated cell phenotype on yeast extract. For the high-performance liquid chromatography (HPLC)–based quantification of free cellular dNTP and dFdCTP pools, the procedure described in (53) was followed.
To assess replication progression using flow cytometry, asynchronous nda3-KM311 cells were arrested in G2 by a 6-hour incubation at restrictive temperature (16°C) and released at permissive temperature (30°C) in the presence of dFdC. Samples were taken every 30 min and were processed for flow cytometry according to the standard procedures (54). 2D gel electrophoresis (in the absence of trimethylpsoralen) was performed, as described (42), using the AseI/BamHI ura4+ fragment.
For mass spectrometric detection of genomic dFdC and hdC, a 100-ml S. pombe culture at a density of 4 × 106 cells/ml was treated for 3 hours with 50 μM dFdC and 50 nM hdC. Cells were harvested and genomic DNA was isolated, as previously described (55), with minor modifications (detailed protocol available on request).
DT40 and MRC-5 cell lines and techniques
DT40 (56) and MRC-5 (SV40 transformed; provided by A. R. Lehmann, University of Sussex) cells were maintained using standard methods and media. The DT40 MRE11H129N/− nuclease mutant was created from MRE11+/− cells (57) provided by S. Takeda (Faculty of Medicine, Kyoto University) by a targeted knock-in, leaving MRE11 under control of its own promoter, and apart from the H129N mutation in exon 4 and integration of a puromycin resistance marker in the intron downstream of this mutation, leaving the overall intron/exon structure intact.
For DT40 colony survival assays, cells were exposed to drugs for 24 hours in liquid medium, before being plated in methylcellulose-containing media (56). For MRC-5 colony survival assays, cells were exposed to drugs for 24 hours before being processed (58).
The MTS assay was carried out using a CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega). DT40 cells (50,000 per well) were incubated for 4 hours at 39°C, after which drugs and compounds were applied. Cells were incubated for an additional 48 hours at 39°C. MTS reagent (20 μl) was added to each well, and cells were incubated for 4 hours.
Absorbance was read at 490 nm using a microplate reader and corrected for the absorbance of wells containing medium without cells or drugs. Rates of metabolic activity were expressed as a percentage relative to untreated control cells.
For mass spectrometric detection of genomic dFdC and 13C15NdC, 5 ml of DT40 (1 × 106 cells/ml) were treated with 500 nM dFdC and 10 nM 13C15NdC and varying doses of Mirin for 24 hours. MRC-5 cells were grown to 50% confluence in a 100-mm cell culture dish and treated with 10 nM 13C15NdC and 1 μM dFdC for 24 hours. Genomic DNA was isolated using the QIAGEN DNeasy Blood and Tissue Kit.
Preparation of genomic DNA and nucleoside quantification by LC-MS/MS
Genomic DNA obtained from S. pombe, DT40, or MRC-5 cells was hydrolyzed and dephosphorylated by a combination of nuclease P1 (Sigma-Aldrich, N8630), phosphodiesterase I (Sigma-Aldrich, P3243), and alkaline phosphatase (Sigma-Aldrich, P5931) into single nucleosides, as previously described (59). Nucleosides were separated using a C18 reverse-phase HPLC column and quantified by an in-line triple quadrupole mass spectrometer. The mass/charge ratio transitions used for detecting the various nucleosides were as follows: dFdC, 264.1 → 112.1; hdC, 231.1 → 115.0; 13C15NdC, 240.1 → 119.1; dC, 228.1 → 112.1; deoxyadenosine, 252.1 → 136.1; deoxyguanosine, 268.1 → 152.1; and thymidine, 243.1 → 127.1. A detailed protocol is available on request. Using this methodology, we typically detect ~1 dFdC per 16,000 dC in WT DT40 cells not treated with mirin; this is well above the dFdC detection limit, which we estimate to be ~1 dFdC per 70,000 dC.
Statistics
Statistical significance was tested using the (paired or unpaired) Student’s t test. P < 0.05 was considered to be significant. Analysis was based on at least three independent experiments, for details see Results and figure legends.
Supplementary Material
Acknowledgments
We would like to thank S. Takeda, N. Rhind, A. R. Lehmann, B. S. Mitchell, and E. Petricci for the gift of reagents. Funding: This work was funded by CRUK (C20600/A11809 and C52465/A19145), NWCR (CR870 and CR894), the Life Sciences Research Network Wales (NRN20130034), the Charity 8Q03 Cancer Research Fund, and a donation from the Hannah Mary Michael Fund. We acknowledge the support of the Centre for Environmental Biotechnology Project part-funded by the European Regional Development Fund (ERDF) through the Welsh Government. Author contributions: Conceived and designed the study: E.H. Method development and data collection: L.B., R.K., A.K., M.U.G., E.G.V., R.B., C.B.V., D.C., and E.H. Supervision: S.G., H.E.K., S.A.E.L., B.P., and E.H. Manuscript writing: E.H. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaaz4126/DC1
REFERENCES AND NOTES
- 1.Cortez D., Replication-coupled DNA repair. Mol. Cell 74, 866–876 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright W. D., Shah S. S., Heyer W.-D., Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 293, 10524–10535 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oh J., Symington L. S., Role of the Mre11 complex in preserving genome integrity. Genes (Basel) 9, E589 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia V., Phelps S. E. L., Gray S., Neale M. J., Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479, 241–244 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reginato G., Cannavo E., Cejka P., Physiological protein blocks direct the Mre11–Rad50–Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev. 31, 2325–2330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shibata A., Moiani D., Arvai A. S., Perry J., Harding S. M., Genois M.-M. M., Maity R., van Rossum-Fikkert S., Kertokalio A., Romoli F., Ismail A., Ismalaj E., Petricci E., Neale M. J., Bristow R. G., Masson J.-Y. Y., Wyman C., Jeggo P. A., Tainer J. A., DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 53, 7–18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreau S., Ferguson J. R., Symington L. S., The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol. Cell. Biol. 19, 556–566 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomita K., Kibe T., Kang H.-Y., Seo Y.-S., Uritani M., Ushimaru T., Ueno M., Fission yeast Dna2 is required for generation of the telomeric single-strand overhang. Mol. Cell. Biol. 24, 9557–9567 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartsuiker E., Neale M. J., Carr A. M., Distinct requirements for the Rad32Mre11 nuclease and Ctp1CtIP in the removal of covalently bound topoisomerase I and II from DNA. Mol. Cell 33, 117–123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aze A., Zhou J. C., Costa A., Costanzo V., DNA replication and homologous recombination factors: Acting together to maintain genome stability. Chromosoma 122, 401–413 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Bryant H. E., Petermann E., Schultz N., Jemth A.-S., Loseva O., Issaeva N., Johansson F., Fernandez S., Mcglynn P., Helleday T., PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carr A. M., Lambert S. A. E., Replication stress-induced genome instability: The dark side of replication maintenance by homologous recombination. J. Mol. Biol. 425, 4733–4744 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Costanzo V., Robertson K., Bibikova M., Kim E., Grieco D., Gottesman M., Carroll D., Gautier J., Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol. Cell 8, 137–147 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Lambert S. A. E., Mizuno K. K., Blaisonneau J., Martineau S., Chanet R., Fréon K., Murray J. M., Carr A. M., Baldacci G., Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell 39, 346–359 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Przetocka S., Porro A., Bolck H. A., Walker C., Lezaja A., Trenner A., von Aesch C., Himmels S.-F., D’Andrea A. D., Ceccaldi R., Altmeyer M., Sartori A. A., CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol. Cell 72, 568–582.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Trenz K., Smith E., Smith S., Costanzo V., ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 25, 1764–1774 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tittel-Elmer M., Alabert C., Pasero P., Cobb J. A., The MRX complex stabilizes the replisome independently of the S phase checkpoint during replication stress. EMBO J. 28, 1142–1156 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsang E., Miyabe I., Iraqui I., Zheng J., Lambert S. A. E., Carr A. M., The extent of error-prone replication restart by homologous recombination is controlled by Exo1 and checkpoint proteins. J. Cell Sci. 127, 2983–2994 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolinjivadi A. M., Sannino V., De Antoni A., Zadorozhny K., Kilkenny M., Técher H., Baldi G., Shen R., Ciccia A., Pellegrini L., Krejci L., Costanzo V., Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 andstable Rad51 nucleofilaments. Mol. Cell 67, 867–881.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeman M. K., Cimprich K. A., Causes and consequences of replication stress. Nat. Cell Biol. 16, 2–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galmarini C. M., Mackey J. R., Dumontet C., Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 3, 415–424 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Mini E., Nobili S., Caciagli B., Landini I., Mazzei T., Cellular pharmacology of gemcitabine. Ann. Oncol. 17, v7–v12 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Huang P., Chubb S., Hertel L. W., Grindey G. B., Plunkett W., Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 51, 6110–6117 (1991). [PubMed] [Google Scholar]
- 24.Crul M., van Waardenburg R. C. A. M., Bocxe S., van Eijndhoven M. A. J., Pluim D., Beijnen J. H., Schellens J. H. M., DNA repair mechanisms involved in gemcitabine cytotoxicity and in the interaction between gemcitabine and cisplatin. Biochem. Pharmacol. 65, 275–282 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Jones R. M., Kotsantis P., Stewart G. S., Groth P., Petermann E., BRCA2 and RAD51 promote double-strand break formation and cell death in response to gemcitabine. Mol. Cancer Ther. 13, 2412–2421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsuda M., Terada K., Ooka M., Kobayashi K., Sasanuma H., Fujisawa R., Tsurimoto T., Yamamoto J., Iwai S., Kadoda K., Akagawa R., Huang S.-Y. N., Pommier Y., Sale J. E., Takeda S., Hirota K., The dominant role of proofreading exonuclease activity of replicative polymerase ε in cellular tolerance to cytarabine (Ara-C). Oncotarget 8, 33457–33474 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou K. M., Kukhanova M., Cheng Y. C., A novel action of human apurinic/apyrimidinic endonuclease: Excision of L-configuration deoxyribonucleoside analogs from the 3’ termini of DNA. J. Biol. Chem. 275, 31009–31015 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Huang S. Y. N., Murai J., Dalla Rosa I., Dexheimer T. S., Naumova A., Gmeiner W. H., Pommier Y., TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res. 41, 7793–7803 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartsuiker E., Mizuno K. K., Molnar M., Kohli J., Ohta K., Carr A. M., Ctp1CtIP and Rad32Mre11 nuclease activity are required for Rec12Spo11 removal, but Rec12Spo11 removal is dispensable for other MRN-dependent meiotic functions. Mol. Cell. Biol. 29, 1671–1681 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ewald B., Sampath D., Plunkett W., ATM and the Mre11-Rad50-Nbs1 complex respond to nucleoside analogue–Induced stalled replication forks and contribute to drug resistance. Cancer Res. 68, 7947–7955 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raoof M., Zhu C., Cisneros B. T., Liu H., Corr S. J., Wilson L. J., Curley S. A., Hyperthermia Inhibits recombination repair of gemcitabine-stalled replication forks. J. Natl. Cancer Inst. 106, dju183 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohiuddin M., Rahman M. M., Sale J. E., Pearson C. E., CtIP-BRCA1 complex and MRE11 maintain replication forks in the presence of chain terminating nucleoside analogs. Nucleic Acids Res. 47, 2966–2980 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sivakumar S., Porter-Goff M., Patel P. K., Benoit K., Rhind N., In vivo labeling of fission yeast DNA with thymidine and thymidine analogs. Methods 33, 213–219 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chottiner E. G., Shewach D. S., Datta N. S., Ashcraft E., Gribbin D., Ginsburg D., Fox I. H., Mitchell B. S., Cloning and expression of human deoxycytidine kinase cDNA. Proc. Natl. Acad. Sci. U.S.A. 88, 1531–1535 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Rompay A. R., Johansson M., Karlsson A., Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol. Ther. 87, 189–198 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Limbo O., Chahwan C., Yamada Y., de Bruin R. A. M., Wittenberg C., Russell P., Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell 28, 134–146 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang P., Plunkett W., Fludarabine- and gemcitabine-induced apoptosis: Incorporation of analogs into DNA is a critical event. Cancer Chemother. Pharmacol. 36, 181–188 (1995). [DOI] [PubMed] [Google Scholar]
- 38.Ostruszka L. J., Shewach D. S., The role of DNA synthesis inhibition in the cytotoxicity of 2’,2’-difluoro-2’-deoxycytidine. Cancer Chemother. Pharmacol. 52, 325–332 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Krogh B. O., Llorente B., Lam A., Symington L. S., Mutations in Mre11 phosphoesterase motif I that impair Saccharomyces cerevisiae Mre11-Rad50-Xrs2 complex stability in addition to nuclease activity. Genetics 171, 1561–1570 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams R. S., Moncalian G., Williams J. S., Yamada Y., Limbo O., Shin D. S., Groocock L. M., Cahill D., Hitomi C., Guenther G., Moiani D., Carney J. P., Russell P., Tainer J. A., Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135, 97–109 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartsuiker E., Vaessen E., Carr A. M., Kohli J., Fission yeast Rad50 stimulates sister chromatid recombination and links cohesion with repair. EMBO J. 20, 6660–6671 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teixeira-Silva A., Ait Saada A., Hardy J., Iraqui I., Nocente M. C., Fréon K., Lambert S. A. E., The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nat. Commun. 8, 1982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hiraoka Y., Toda T., Yanagida M., The NDA3 gene of fission yeast encodes β-tubulin: A cold-sensitive nda3 mutation reversibly blocks spindle formation and chromosome movement in mitosis. Cell 39, 349–358 (1984). [DOI] [PubMed] [Google Scholar]
- 44.Friedman K. L., Brewer B. J., Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 262, 613–627 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Liao H., Ji F., Helleday T., Ying S., Mechanisms for stalled replication fork stabilization: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 19, e46263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitsuya H., Yarchoan R., Broder S., Molecular targets for AIDS therapy. Science 249, 1533–1544 (1990). [DOI] [PubMed] [Google Scholar]
- 47.Sommadossi J. P., Carlisle R., Zhou Z., Cellular pharmacology of 3’-azido-3’-deoxythymidine with evidence of incorporation into DNA of human bone marrow cells. Mol. Pharmacol. 36, 9–14 (1989). [PubMed] [Google Scholar]
- 48.Rudd S. G., Valerie N. C. K., Helleday T., Pathways controlling dNTP pools to maintain genome stability. DNA Repair 44, 193–204 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Topal M. D., Baker M. S., DNA precursor pool: A significant target for N-methyl-N-nitrosourea in C3H/10T1/2 clone 8 cells. Proc. Natl. Acad. Sci. U.S.A. 79, 2211–2215 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fouquerel E., Lormand J., Bose A., Lee H. T., Kim G. S., Li J., Sobol R. W., Freudenthal B. D., Myong S., Opresko P. L., Oxidative guanine base damage regulates human telomerase activity. Nat. Struct. Mol. Biol. 23, 1092–1100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whitaker A. M., Smith M. R., Schaich M. A., Freudenthal B. D., Capturing a mammalian DNA polymerase extending from an oxidized nucleotide. Nucleic Acids Res. 45, 6934–6944 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreno S., Klar A., Nurse P., Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194, 795–823 (1991). [DOI] [PubMed] [Google Scholar]
- 53.Kumar D., Viberg J., Nilsson A. K., Chabes A., Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res. 38, 3975–3983 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sabatinos S. A., Forsburg S. L., Measuring DNA content by flow cytometry in fission yeast. Methods Mol. Biol. 521, 449–461 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Wright A. P. H., Maundrell K., Shall S., Transformation of Schizosaccharomyces pombe by non-homologous, unstable integration of plasmids in the genome. Curr. Genet. 10, 503–508 (1986). [DOI] [PubMed] [Google Scholar]
- 56.J.-M. Buerstedde, S. Takeda, Reviews and Protocols in DT40 Research (Dordrecht, Springer Netherlands, 2006) vol. 40. [Google Scholar]
- 57.Yamaguchi-Iwai Y., Sonoda E., Sasaki M. S., Morrison C., Haraguchi T., Hiraoka Y., Yamashita Y. M., Yagi T., Takata M., Price C., Kakazu N., Takeda S., Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J. 18, 6619–6629 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Franken N. A. P., Rodermond H. M., Stap J., Haveman J., van Bree C., Clonogenic assay of cells in vitro. Nat. Protoc. 1, 2315–2319 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Crain P. F., Preparation and enzymatic hydrolysis of DNA and RNA for mass spectrometry. Methods Enzymol. 193, 782–790 (1990). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaaz4126/DC1