

Abstract
Muscular dystrophies are debilitating disorders that result in progressive weakness and degeneration of skeletal muscle. Although the genetic mutations and clinical abnormalities of a variety of neuromuscular diseases are well known, no curative therapies have been developed to date. The advent of genome editing technology provides new opportunities to correct the underlying mutations responsible for many monogenic neuromuscular diseases. For example, Duchenne muscular dystrophy, which is caused by mutations in the dystrophin gene, has been successfully corrected in mice, dogs, and human cells through CRISPR/Cas9 editing. In this Review, we focus on the potential for, and challenges of, correcting muscular dystrophies by editing disease-causing mutations at the genomic level. Ideally, because muscle tissues are extremely long-lived, CRISPR technology could offer a one-time treatment for muscular dystrophies by correcting the culprit genomic mutations and enabling normal expression of the repaired gene.
Muscular dystrophies (MDs) comprise a heterogeneous group of disorders that disrupt the function of different muscle groups, with manifestations including muscle weakness, muscle atrophy, spasm, muscle hypertonia, and myalgias (Figure 1A). Additionally, most MDs involve cardiac failure and respiratory dysfunction, leading to premature death. Over 800 monogenic mutations have been associated with human MDs, with many of the associated genes encoding structural muscle proteins (ref. 1 and Figure 1B). Our increased understanding of the causative genetic mutations has benefited MD classification and clinical practice by enabling a precise DNA-based diagnosis (2).
Figure 1. MD muscles and genes.
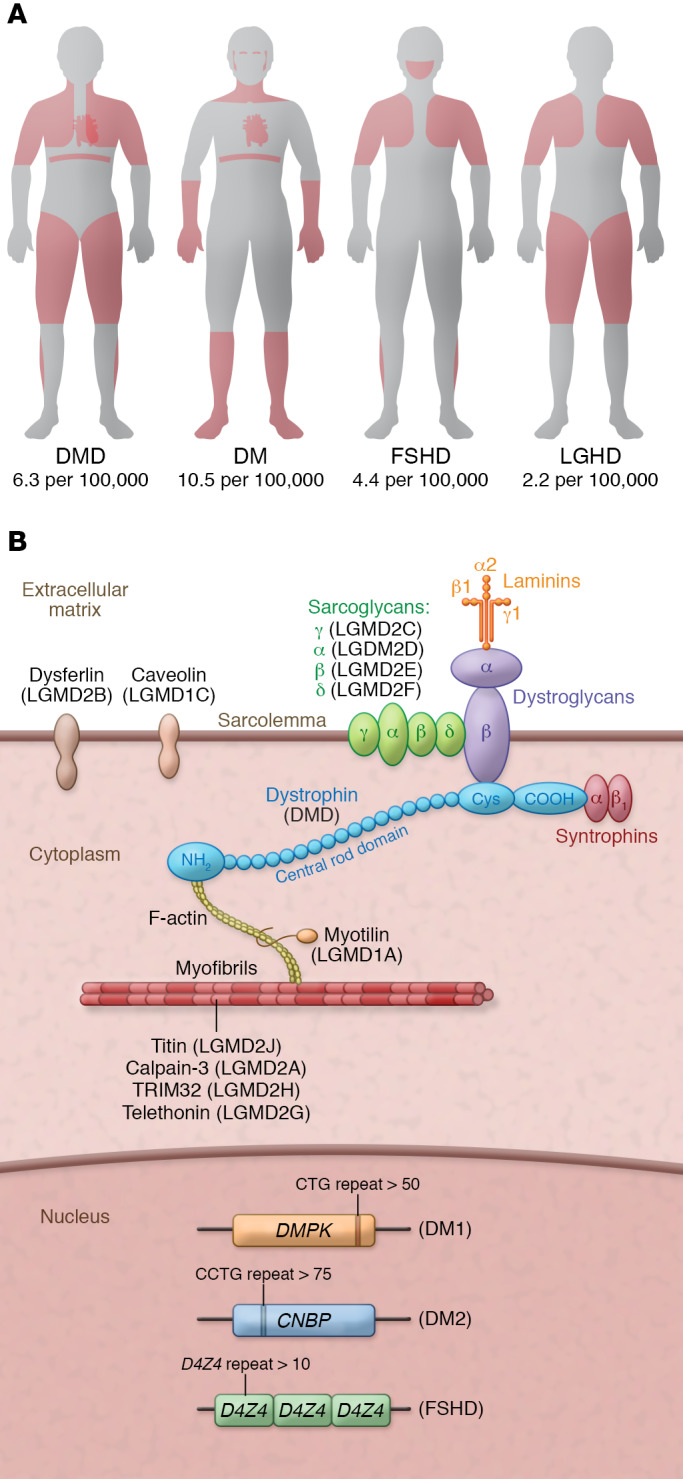
(A) Muscle areas (red) affected in the most common MDs. In Duchenne muscular dystrophy (DMD), progressive muscle weakness and atrophy are observed in major muscle groups, including heart and diaphragm. In myotonic dystrophy (DM), muscle degeneration leads to weakness and myotonia in distal muscles with progression to proximal muscles. DM also involves multiple organ systems, including cataracts, cardiac conduction defects, and other endocrine disorders. Facioscapulohumeral muscular dystrophy (FSHD) is characterized by progressive weakness and wasting of muscles of the face, shoulder, and upper arm. Limb-girdle muscular dystrophy (LGMD) shows progressive proximal muscle weakness, plus different pathogenic variants depending on the type of genetic mutation. Numbers indicate prevalence of disease. (B) Muscle proteins involved in MDs. Dystrophic muscle is caused by mutations in genes encoding proteins in the sarcolemma (e.g., dystrophin, sarcoglycans, dysferlin) and sarcomere (e.g., myotilin), as well as expansion/contraction of different genomic locations. Dystrophin protein (mutated in DMD) has four functional domains: the first actin-binding domain (ABD-1) at the amino terminus (NH2); the central rod domain, containing 24 spectrin-like repeats and ABD-2; the cysteine-rich domain (Cys); and the carboxyl-terminal domain (COOH).
To date, there are no cures for any MDs; however, different treatments have been developed to ameliorate disease symptoms. Treatments for MDs can be categorized into three classes: (a) disease-modifying therapies, (b) gene expression therapies, and (c) gene replacement therapies.
Several disease-modifying therapies have been developed for Duchenne muscular dystrophy (DMD), a fatal disease caused by mutations in the dystrophin gene (DMD). DMD patients on corticosteroid treatment benefit from prolonged ambulation; however, they experience substantial health problems due to the side effects of steroids. Other disease-modifying therapies for DMD seek to overcome the lack of dystrophin by upregulating utrophin, a protein structurally and functionally similar to dystrophin that could compensate for the lack of dystrophin (3). Additional therapies focus on improving muscle mass and strength by using myostatin inhibitors (4). Different gene expression therapy approaches have also been developed to “mask” nonsense mutations or to induce exon skipping using an RNA-based approach to restore dystrophin expression (5). For example, eteplirsen allows skipping of exon 51, which is applicable to approximately 14% of DMD-affected boys; however, the efficacy of eteplirsen is extremely low (6).
Gene replacement therapy for DMD is challenging because of the large size of full-length dystrophin. The focus of DMD gene replacement therapy has been on the development of microdystrophin, a truncated form of dystrophin that contains the minimal functional regions of the protein (7). An alternative approach of gene replacement uses micro-utrophin, which in preclinical studies prevented most of the deleterious histological and physiological aspects of DMD (8). Treatment of limb-girdle muscular dystrophy type 2B (LGMD2B) myopathy by introduction of dysferlin cDNA also showed restoration of muscle function in a mouse model (9).
Although these therapeutic approaches have produced promising in vitro and in vivo results, some leading to ongoing clinical trials, they all have noteworthy limitations. First, they do not eliminate the disease-causing mutation, and the mutated nonfunctional protein is still present. Second, their effectiveness is temporary, so patients require lifelong treatment. Third, for gene replacement therapies, the expression of the exogenous protein is restricted to an exogenous promoter that could lead to undesired issues regarding protein localization and expression level (7, 10).
The lack of effective therapies to cure MDs emphasizes the need for new discoveries and treatment strategies. The past two decades have seen the rapid development of new genome editing technologies, which allow precise and efficient introduction of a variety of genetic alterations into mammalian cells, from a single-nucleotide modification to insertion of genes and deletion of chromosomal regions (11). Among the tools available for genome editing, clustered regularly interspaced short palindromic repeats–mediated (CRISPR-mediated) genome editing has revolutionized the field by offering a relatively simple, accurate, and effective method to alter the genome (12). This Review highlights current applications and the future potential of CRISPR technologies in the treatment of MDs.
CRISPR genome editing components
CRISPR/Cas system
CRISPR was initially discovered as an adaptive immune system in bacteria that cleaves invasive phage DNA elements (13). The CRISPR system has been adapted to eukaryotic cells and consists of two components: a CRISPR-associated (Cas) protein and a single-guide RNA (sgRNA). The Cas protein can be adapted to cleave any DNA sequence by changing the 20-bp protospacer of the sgRNA, which contains the complementary sequence of the DNA to be targeted (14). Based on its sequence, the sgRNA directs the Cas protein to bind and cleave a specific DNA sequence. Moreover, there is a specific “protospacer adjacent motif” (PAM) sequence in the DNA that is recognized by the Cas protein (ref. 15 and Figure 2A). The preferential PAM sequence of Streptococcus pyogenes Cas9 (SpCas9), a commonly used Cas, is 5′-NGG-3′, which occurs at a high frequency in the human genome (approximately every 40 bp), conferring high targetability.
Figure 2. Components and outcomes of CRISPR genome editing.

(A) DNA double-strand breaks generated by CRISPR/Cas9 and sgRNA are repaired by nonhomologous end joining (NHEJ) or, in the presence of a donor template, by homology-directed repair (HDR). The Cas protein recognizes the DNA by the protospacer adjacent motif (PAM). Base editors are generated by fusion of nCas9 or dCas9 with the cytidine deaminase APOBEC1 for cytidine base editors (CBEs) to convert nucleotide C/G to T/A within a 5-bp activity window located in the spacer sequence (108). Similarly, fusion with adenosine deaminase TadA produces adenine base editors (ABEs) to convert nucleotide A/T to G/C (109). Prime editors (PEs) are generated by fusion of nCas9 or dCas9 with reverse transcriptase to perform targeted small insertions, deletions, and base changing in a precise way (28). (B) DMD patients with deletion of exon 50 in the DMD gene have an out-of-frame mutation. (i) Double-cut myoediting results in single- or multi-exon deletion. (ii) Single-cut sgRNA myoediting precisely designed at splice sites (e.g., splice acceptor site [SAS]) restores the correct ORF by both exon skipping and exon reframing events. (iii) Nucleotide editing can be accomplished by BEs or PEs, correcting nonsense mutations. Shaded area highlights different myoediting approaches. Exons encoding ORF are shown in blue. Exons with stop codon are shown in red. The corrected exon is shown in green.
In eukaryotic cells, a DNA double-strand break (DSB) mediated by Cas9 nuclease can be repaired through nonhomologous end joining (NHEJ) or homology-directed repair (HDR) (Figure 2A). NHEJ is error prone and generates small insertions or deletions (INDELs) of nucleotides at the cut site. Recent studies have shown that the error patterns of NHEJ are strongly biased by the DNA sequence context (16, 17). In contrast, HDR repairs the DSB by precisely inserting a DNA template, either a double-stranded DNA fragment or a single-stranded oligodeoxynucleotide (ssODN), into the genome, potentially repairing a genetic mutation (18). However, quiescent cells and postmitotic cells, such as muscle stem cells and myofibers, have very low levels of HDR machinery, thereby requiring these cells and tissues to rely on NHEJ for DSB repair.
The CRISPR system possesses remarkable diversity, with new Cas proteins continuously being discovered from different bacterial strains (19). The different attributes of the various Cas proteins expand the range of genome editing options by offering different PAM sequences, reducing protein size for easier vector packaging, and cleaving different substrates, including RNA (20). Nevertheless, some Cas proteins have long, complex PAM sequences, restricting the number of target genes. Other Cas proteins are large and pose packaging dilemmas.
Engineered CRISPR/Cas systems
In addition to the endogenous Cas proteins, engineered Cas proteins offer new genome editing tools and expand therapeutic possibilities. For example, different high-fidelity SpCas9 variants (such as eSpCas9, SpCas9-HF1, HypaCas9, and evoCas9) (21–24) increase the specificity and fidelity of Cas9 without losing the sgRNA on-target activity, thus enhancing safety for future clinical use. By mutation of specific sites in the Cas9 nuclease domains, Cas9 proteins with partial or complete loss of cleavage activity have also been generated, such as Cas9 nickase (nCas9) or deactivated/dead Cas9 (dCas9). Since these engineered Cas9 variants cannot introduce DSBs in the genome, they can be fused to different effectors for genome modification purposes (25). Specifically, both nCas9 and dCas9 have been fused with specific deaminase proteins to generate base editors (BEs) that allow precise conversion of a single nucleotide base into another base without causing DSBs (ref. 26 and Figure 2A). These BEs may be instrumental in genome editing therapy, since it has been estimated that more than 50% of human genetic variants associated with diseases are caused by a single point mutation (27). Recently, nCas9 was fused with a reverse transcriptase to generate prime editors (PEs). Remarkably, these engineered proteins in combination with the prime editing extended guide RNA (pegRNA) can directly write new genetic information into a target DNA site (Figure 2A). Although the PE CRISPR technology is new and in need of further testing, it has the potential to correct many known pathogenic mutations (28).
Myoediting of DMD
We coined the term “myoediting” to refer to CRISPR-mediated genome editing in muscle to permanently correct genomic mutations of MDs and restore muscle function (29). In this section, we will describe various myoediting strategies using DMD, a severe dystrophinopathy, as a model MD. Additionally, we will describe how myoediting approaches might be expanded to other common MDs.
Dystrophinopathies
DMD, located on the X chromosome, is the largest gene in the human genome, consisting of 79 exons that encode the dystrophin protein. In muscle cells, dystrophin is one of the main components of the dystrophin-glycoprotein complex (DGC), which connects the sarcomere with the sarcolemma (Figure 1B). In the DGC, dystrophin functions as a shock absorber, reducing the mechanical stress induced by muscle contraction. Mutations in the DMD gene causing loss or dysfunction of dystrophin cause two major phenotypes: DMD or Becker muscular dystrophy (BMD). DMD is the most common and lethal muscular dystrophy, caused by mutations that disrupt the open reading frame (ORF) of dystrophin, leading to truncated, dysfunctional, and unstable protein products. DMD mutations include exon deletions (68.8% of DMD patients), exon duplications (11.2%), point mutations (10.4%), and small deletions/insertions (9.6%) (30). Although over 7000 DMD mutations have been associated with DMD patients, the mutations cluster in hotspot areas of DMD, located primarily in the regions that encode actin-binding domain-1 (ABD-1) and the central rod domain (30, 31).
Strategies for DMD myoediting are inspired by the observation that BMD patients carrying in-frame DMD mutations usually present with a less severe muscle disorder or are asymptomatic (32). Whereas DMD patients do not express dystrophin, BMD patients have mutations in DMD that preserve the ORF and produce a shortened but functional dystrophin protein. BMD severity is usually correlated to the quantity of dystrophin protein produced, ranging from a severe form (with <10% expression of dystrophin in comparison with healthy subjects) to a mild form (with >50% of dystrophin expressed) (33). Additionally, there is a subset of in-frame mutations (usually encoding a truncated form of the ABD-1 region) that phenotypically resemble DMD patients and show diminished amounts of dystrophin protein, most likely due to protein instability and degradation (34, 35).
DMD myoediting strategies
DMD myoediting is designed, in most cases, to change the DMD out-of-frame mutation to generate a BMD in-frame form of dystrophin, albeit generating a truncated but functional form of the protein. Myoediting of exon duplication mutations and pseudo-exon mutations has the potential to restore full-length dystrophin. However, there are also DMD mutations that cannot be corrected by current iterations of myoediting — for example, large deletions of essential portions of the N- or C-terminal domains. An important consideration of myoediting is that the CRISPR/Cas system in muscle, a postmitotic tissue, uses NHEJ repair to rejoin the DSB and generates unpredictable INDELs at the cleavage site. Attempts have been made in postmitotic cells to insert an exogenous DNA template into the cut site using homology-independent targeted integration (HITI) or intercellular linearized single–homology arm donor-mediated intron-targeting integration (SATI); however, these genome editing approaches are still in need of further improvement of efficiency (36, 37).
The genetic modifications needed to correct DMD mutations by myoediting can be grouped into four categories: exon deletion, exon skipping, exon reframing, and base modification events. These genetic alterations can be accomplished using myoediting with careful selection of the Cas and thoughtful design of the sgRNAs to optimize the genome editing outcome. The myoediting strategies discussed below produce genetic outcomes that can correct DMD at the genome level and restore dystrophin function (summarized in Figure 2B).
Double-cut myoediting.
One potential means to remove a DMD mutation is by exon deletion. This myoediting strategy requires two sgRNAs, flanking the mutated DMD exon, to simultaneously cut in the presence of Cas and excise the exon (Figure 2B). The location of the two sgRNAs is critical to ensure that the remaining exons will splice correctly to yield truncated but functional dystrophin. This strategy is effective when deleting exons in DMD that encode regions of dystrophin that tolerate deletions. Exon deletion approaches using two sgRNAs have been reported to excise multi-exonic genomic regions encompassing a mutational hotspot region (exons 45–55) in DMD, thereby deleting a region of the central rod domain and producing a truncated but functional form of dystrophin (38, 39). However, when numerous exons are being deleted, caution must be taken not to disrupt dystrophin function, especially when the mutations are within the essential domains, such as those that interact with actin or dystroglycan. Moreover, when two sgRNAs are used, exon deletions can generate diverse and unpredictable genome modifications, including exogenous DNA integration or aberrant splicing at the cut sites (40). Another weakness of the double-cut myoediting approach is the low efficiency of editing, which is most likely due to the necessity of simultaneous cutting of both sgRNAs separated by vast genomic distances and the religation of free DNA ends. Additionally, using two sgRNAs increases the likelihood of generating off-target effects, making it necessary to test the putative off-target sites for each sgRNA (40).
Single-cut sgRNA myoediting.
Single-cut gene editing overcomes many of the limitations of the double-cut approach and offers several advantages to correct DMD mutations. In this technology, only one sgRNA is used to generate a single cut in the genome. The sgRNA is designed to target a genomic sequence in the close vicinity of the intron-exon region of the out-of-frame exon and to produce one DSB. Since NHEJ introduces INDELs at the DSB site, it offers the ability to destroy the splicing consensus sequence and cause exon skipping to restore the dystrophin ORF. Since exon skipping has the potential to restore dystrophin expression in approximately 70% of DMD patients, the single-cut sgRNA strategy represents an especially promising approach for DMD therapy (41).
Another outcome of single-cut sgRNA myoediting is exon reframing. This occurs when the splice consensus sequence is retained and the INDELs generate a frameshift in the exon, restoring the ORF of dystrophin. Theoretically, one-third of INDELs should be in frame; however, it was reported that NHEJ repair outcomes can be predicted based on the fourth nucleotide upstream of the PAM sequence (42). Interestingly, in the event that the INDELs restore the original uncorrected DNA sequence, the DNA remains recognizable by the sgRNA, enabling Cas to cut again until the PAM site or DNA target sequence is eliminated.
Several studies demonstrated high-efficiency editing with the single-cut sgRNA strategy to restore dystrophin expression (43–46). Other advantages of the single-cut sgRNA approach are that it requires minimal modification of the genome and decreases the likelihood of off-target mutations since only one sgRNA is used. Although any Cas protein can perform the single-cut sgRNA approach, SpCas9 has a distinct advantage, since the PAM sequence is NAG or NGG, which represents exon splicing consensus sites.
There are some limitations to the single-cut approach. First, to augment efficiency of the single-cut strategy, both exon skipping and reframing outcomes should occur using one sgRNA. For example, an sgRNA targeting exon 44 in the ΔEx45 DMD mouse model efficiently restores dystrophin by exon skipping and reframing events, but the same sgRNA shows reduced dystrophin restoration in the ΔEx43 DMD mouse model, as only exon skipping occurs in this correction (46). Second, the single-cut strategy precisely targets specific DMD mutations, so consequently each sgRNA is limited to specific DMD patients, as opposed to the double-cut approach, which eliminates a large region of genome and can be applied to many DMD mutations (39). Third, there remains the possibility of adeno-associated virus (AAV) integration into the cut site (47). However, the advantages of single-cut supersede the minor limitations.
Nucleotide myoediting.
The new engineered CRISPR technologies, BE and PE, expand the breadth of myoediting strategies. Point mutation correction by BEs could play a meaningful role in correcting DMD, as 10% of DMD cases are caused by point mutations (30). For gene therapy, where a single nucleotide base change is required and editing of adjacent bases is not tolerated, using newly engineered BEs for site-specific single nucleotide replacement is desirable (48). nCas9 and dCas9 do not create DSBs, thereby minimizing AAV vector integration after AAV delivery, and circumventing the production of undesired INDELs by NHEJ. However, a current major weakness of BEs is their limited target specificity, which can result in many off-target changes to the genome (49). Since BEs offer a powerful approach to correct genomic mutations, much effort has gone toward developing new BEs that minimize both RNA and DNA off-target editing activity (50, 51). Additionally, newly engineered PEs can also function as BEs, allowing for all possible base conversions at a specific position of the genome. BEs are versatile and can also be targeted to mutate splice acceptor or donor sites to induce exon skipping (52). Theoretically, both exon skipping and exon reframing can be accomplished using PEs, by mutation of the splicing consensus sequences or introduction of a specific number of nucleotides to restore the dystrophin ORF.
Myoediting human induced pluripotent stem cells and DMD animal models
For preclinical studies of DMD, hundreds of patient-derived induced pluripotent stem cell (iPSC) lines have been established, and more than 60 spontaneous or engineered DMD animal models have been reported (53, 54). These DMD models offer valuable platforms to evaluate efficacy and efficiency of myoediting for future clinical applications.
Myoediting human iPSC models of DMD
Human iPSCs can be derived from patient samples, such as peripheral blood mononuclear cells, and directed to differentiate into relevant cell types for testing, such as cardiomyocytes and myotubes (39, 55). Moreover, instead of being derived from DMD patients, iPSCs can be obtained from healthy individuals and modified with CRISPR technology to generate mutations in DMD, producing induced DMD (iDMD) iPSCs. This strategy provides an isogenic iPSC control line that differs from the iDMD iPSC line only at the DMD locus, minimizing intrinsic variations between iPSC lines (56).
Patient-derived DMD and iDMD iPSCs with exon deletions, duplications, point mutations, or pseudo-exons have been corrected by myoediting to restore dystrophin (Table 1 and refs. 39, 40, 43, 45, 46, 56–72). To assess optimization of sgRNAs and evaluate the efficiency of correction with different CRISPR systems (such as SaCas9, Cpf1, and BEs), various myoediting strategies, including double-cut or single-cut sgRNA and nucleotide base editing, have been performed on DMD and iDMD iPSCs. To evaluate the function of the restored dystrophin in corrected DMD and iDMD iPSCs, calcium transient assays and 3D engineered heart muscle contraction assays have been performed (56, 58). These studies performed in human cells are instrumental in identifying sgRNAs that can be translated to the clinic.
Table 1. DMD mutation corrections in human iPSC and mouse models.

Myoediting animal models of DMD
Although germline editing is not permissible or acceptable in humans, it was successfully used as a proof of concept for myoediting in mdx mice, which harbor a stop mutation causing loss of dystrophin (59, 73). Although the mdx mouse is a widely used DMD model, it does not reflect the common mutations reported in human DMD patients. To test postnatal myoediting strategies, several DMD mouse models representing the most commonly deleted exons in DMD patients (including deletion of exon 43, 44, 45, 50, or 52) were generated by CRISPR technology to optimize myoediting strategies (43, 45, 46, 54, 69).
Clearly, one of the biggest challenges of postnatal, somatic myoediting is efficient delivery of the CRISPR components, Cas and sgRNA, to skeletal muscle and cardiac tissue. Both nonviral and viral delivery systems have been developed and used for myoediting (Table 1). Nonviral delivery systems, including electroporation and gold nanoparticles, have been reported to deliver Cas9 and sgRNAs into skeletal muscle of DMD mice and showed genomic correction of the dystrophin mutation (67, 68). However, because high-efficiency body-wide delivery of CRISPR components is required for effective DMD therapy, improvements are needed to enhance distribution of CRISPR components by nonviral delivery systems.
Adeno-associated virus (AAV) is an effective viral delivery system for myoediting, especially since AAV serotypes (1, 6, 8, 9, rh10, or rh74) have tropism for skeletal muscle and cardiac tissue (74). However, the small packaging size limitations (<4.7 kb) of the AAV delivery system pose a substantial challenge, especially because the size of SpCas9 is approximately 4.2 kb. One way to circumvent the cargo size limitation is to use a dual-AAV system, with one AAV system encoding Cas (AAV-Cas) and the second AAV expressing sgRNAs (AAV-sgRNA). In initial studies of postnatal myoediting of mdx mice, three independent groups used single- or dual-AAV systems for intramuscular or systemic injection (62, 63, 65). In these original studies, Cas9 expression was regulated by a constitutive CMV or mini-CMV promoter, so tissue specificity was determined primarily by the AAV serotype used for delivery. In subsequent studies, a muscle-specific promoter was used to express Cas9 to enhance myoediting specificity (43). Using myoediting, several DMD mouse models expressing common human DMD mutations were corrected, and high levels of dystrophin were restored. Most impressively, the dual-AAV delivery system was used to restore dystrophin expression and function in a canine model of spontaneous DMD (44). For nucleotide base myoediting, the BEs and PEs are too large to be packaged into the AAV delivery system. This necessitates splitting the BE or PE constructs in half and using a trans-splicing AAV vector system (75, 76). This split-construct delivery system substantially decreases editing efficiency and consequently restores less dystrophin protein (71). Recently, intein-split SpCas9 was shown to restore dystrophin expression in a pig DMD model (77, 78).
Myoediting of other MDs
Myotonic dystrophy
Myotonic dystrophy (DM) is caused by nucleotide expansion and is categorized into two major types, DM1 and DM2, each caused by mutations in a different gene. DM1 results from CTG trinucleotide repeat expansion in the noncoding region of the DMPK gene, while DM2 is caused by CCTG tetranucleotide repeat expansion in the CNBP gene (encoding ZNF9) (Figure 1B). In both forms of DM, the expanded RNA is toxic, folding into hairpin-like structures and forming RNA plus protein accumulations that cause ribonuclear foci to form. This causes misregulation of crucial RNA-binding proteins involved in alternative splicing, resulting in splicing deregulation of many genes (79–81).
The goal of myoediting of DM is deletion of the nucleotide expansions (82). A commonly used correction strategy is double-cut myoediting to excise the repeated region using Cas9 and two sgRNAs flanking the repeat expansion in both DM1 cell and animal models (83–85). A single DSB near a long repeat expansion was shown to potentially induce loss of the entire repeat, offering another genomic editing approach for DM (86). However, this approach’s efficiency is extremely low. Additionally, it was reported that CRISPR/Cas9–mediated HDR efficiently introduced a polyadenylation signal into the 3′-UTR upstream of the CTG repeats, reverting the DM1 phenotype and serving as a practical strategy for advancing therapeutic genome editing for DM1 (87).
Facioscapulohumeral muscular dystrophy
Facioscapulohumeral muscular dystrophy (FSHD) is characterized by the contraction of the D4Z4 region in the genome that leads to the aberrant expression of the transcription factor DUX4 and, eventually, misregulation of hundreds of genes (Figure 1B). To date, no CRISPR-based genome editing technologies have been applied to the different DUX4-transgenic mouse models of FSHD. To decrease expression of full-length DUX4 mRNA in primary FSHD myoblasts, dCas9 was fused to the transcriptional repressor KRAB in order to epigenetically repress DUX4 gene expression (88). However, this approach does not permanently modify the genome, so repeated administration would likely be required for therapeutic benefit.
Limb-girdle muscular dystrophy
Limb-girdle muscular dystrophy (LGMD) pertains to a group of muscle diseases caused by mutations in many different genes (Figure 1B). LGMD2B is caused by mutations in the dysferlin gene (DYSF), and LGMD2D is caused by mutations in the α-sarcoglycan gene (SGCA). Using patient-derived iPSCs, CRISPR genome editing with a DNA template (ssODN) induced HDR to correct two of the most common point mutations, the c.5713C>T mutation in DYSF and the c.229C>T mutation in SGCA (89). Although this method worked in cells, HDR is inefficient in postmitotic skeletal muscle, so different approaches based on new genome editing technologies such as BEs and PEs should be explored to correct this class of MDs.
Future challenges of myoediting
CRISPR genome editing technology has revolutionized the field of MDs, offering the anticipation of a therapeutic cure (Figure 3). Successful clinical trials of CRISPR technology with other monogenic diseases such as sickle cell disease and Leber congenital amaurosis have paved the way for future clinical application of CRISPR (90). Here we delineate challenges that need to be overcome and possible solutions to make therapeutic genome editing a reality for MD patients.
Figure 3. Overview of correction of DMD by myoediting.

Sample is collected from DMD patients (or healthy individuals) for genotyping and generation of DMD-iPSCs. sgRNA will be designed based on genotyping information. DMD-iPSCs are transduced with Cas9 and sgRNA and differentiated to cardiomyocytes to assess myoediting by restoration of dystrophin. AAV delivery system is generated using optimized sgRNA and Cas9, and used to infect mice (to test in vivo myoediting), dogs (to scale up the animal size), or eventually humans (as a therapeutic approach) to correct DMD.
Efficiency of editing
It is clear that the higher the efficiency of editing, the greater the functional recovery of muscle. Therefore, optimization of CRISPR technology by careful design of the editing strategies and efficient delivery of CRISPR components to muscle is key to generating an effective CRISPR therapy. In DMD mouse models, the single-cut myoediting approach for exon skipping and exon reframing dramatically increases editing efficiency over that observed with the double-cut myoediting approach (43). Additionally, increasing the ratio of AAV-sgRNA to AAV-Cas highly improves the genomic editing events and consequently enhances dystrophin protein recovery (45, 91).
Since myofibers are multinucleated, it may not be necessary to correct 100% of nuclei to achieve clinical benefit. Findings from patients indicate that restoring dystrophin protein expression between 4% and 50% can convert a DMD phenotype to a BMD phenotype, increasing quality of life and prolonging lifespan (33). However, the minimal level of dystrophin per myofiber and the number of nuclei that need to be corrected remain uncertain. Future transcriptomic and proteomic analyses at the single-myofiber level, in conjunction with single-nucleus RNA sequencing, can assess the efficiency of editing needed in MDs.
Delivery of CRISPR components
As muscle comprises about 40% of the body mass, the major challenge for CRISPR therapy in MDs is delivery. To date, three different approaches have been applied: (a) engineered nanoparticles, (b) muscle progenitor engraftment, and (c) AAV delivery. The only in vivo study in which engineered nanoparticles were used achieved editing efficiency of less than 1% (68), making it unlikely that nanoparticles, at least in current formulations, will be sufficiently efficient for systemic delivery for myoediting. Additionally, muscle progenitor cells were corrected by CRISPR genome editing and then engrafted in muscle of mdx mice by intramuscular injection (92). Major weaknesses of this approach include low efficiency of engraftment and the inability to generate muscle stem cells with long-term repopulation potential.
Currently, AAV is the most promising delivery system for CRISPR genome editing therapies and has many advantages, including low immunogenicity, minimal integration risk, long-term transgene expression, and approval by the Food and Drug Administration for human clinical use. The skeletal and cardiac muscle tropism of some serotypes can be further enhanced using specific promoters for these tissues. For efficient in vivo genome editing, high doses of AAV are required, posing challenges for clinical application. A recent study demonstrated that the use of self-complementary AAV for the expression of the sgRNA, instead of the single-stranded AAV, enhances the in vivo delivery efficiency, allowing a lower viral dose to achieve efficient myoediting (72). Most encouragingly, the first studies using CRISPR technology in a spontaneously generated DMD dog model and in an induced DMD pig model, involving systemic administration of AAV9, allowed efficient restoration of dystrophin protein production, thereby validating therapeutic genome editing in large animals and auguring future clinical application (44, 78).
The new CRISPR genome editing tools, such as BEs and PEs, are too large to be packaged in a single AAV, requiring split systems that are relatively inefficient (71). Another major limitation for clinical use of AAV delivery system is the preexisting anti-AAV antibodies in the human population (93, 94). Although myoediting strategies could be considered “one and done” treatments, there might be a need to repeat myoediting. Due to high antibody titers developed against the first AAV dose, high corticosteroids would need to be administered to reduce the activity of the immune system (95).
Recent studies have shown innate and adaptive cellular immunogenicity to Cas9 in mouse models and the presence of anti-Cas9 antibodies in human plasma (96, 97). However, different approaches, such as masking the immunogenic Cas9 epitopes, have the potential to circumvent the problem of immune recognition (98, 99). In addition, it was observed that sgRNAs could potentially trigger an innate immune response (100). Further studies on the specific post-transcriptional modifications of sgRNAs need to be conducted to avoid this induced immune response (101).
Off-target activity detection
One of the major intrinsic concerns regarding CRISPR genome editing is off-target activity. To reduce this potential outcome, different high-fidelity Cas enzymes have been developed, but off-target mutagenesis has not been thoroughly assessed in vivo. CRISPR/Cas off-target activity has been analyzed only at predicted sites. The use of different methods is needed to capture all potential genomic sites, and other events such as viral genome integration or aberrant splicing. These investigative methods include Digenome-seq (102), GUIDE-seq (40), and CIRCLE-seq (103). The first generation of BEs introduced a large number of off-target edits, also at the transcriptome level, so the complete sequencing of the genome and transcriptome is required to demonstrate specificity. In addition, comprehensive transcriptomic and proteomic analyses after myoediting could identify dysregulated biological pathways due to off-target activities.
Age of treatment and long-term effects
Another important variable in achieving high efficiency is the timing of the correction. When should myoediting be done in patients? Generally, all the in vivo studies that show efficient dystrophin recovery have been performed using young mice or dogs, suggesting the importance of starting the treatment as early as possible. Future applications of recently developed dystrophin reporter mice could elucidate the importance of timing of myoediting for treatment of DMD patients (104).
Two different studies were performed to test the long-term effects of CRISPR genome editing (40, 91). Both studies showed reduced skeletal muscle editing at 12 or 18 months after AAV injection of the CRISPR components and a disproportionate reduction of the sgRNA vector compared with the Cas9 vector. These studies were performed using the double-cut approach for exon deletion, and currently there are no reported studies showing the long-term effects of the single-cut approach on myoediting outcomes.
The ability to deliver CRISPR components into satellite cells or muscle stem cells could help to sustain long-term genome editing. However, AAV infectivity of satellite cells has been controversial (63, 105, 106). Encouragingly, a recent study using a muscle graft model showed that AAV9 effectively transduces satellite cells (107). Single-cell analyses and the use of a satellite cell–specific promoter (instead of a constitutive CMV promoter) to drive CRISPR component expression could advance our understanding in this field and be beneficial to DMD therapy.
Novel myoediting strategies
Although many different CRISPR/Cas systems have been engineered and developed, only Cas9 has been extensively used to correct MDs. BEs and PEs possess specific editing activities that could advance the field of genome editing of MDs, if their in vivo delivery efficiency is improved. In addition, it might be of interest to take a combinatorial approach to therapy and combine genome editing with other treatments in order to boost the effect of genome editing.
Concluding remarks
Different CRISPR-mediated technologies have the potential to be applied to many MDs. It may be argued that gene replacement therapy, which involves ectopic overexpression of missing proteins in muscles, may be a sufficient approach to treat MDs. However, the limitations of gene replacement therapy are that the disease-causing mutation is retained, many of the protein-coding transcripts are too large for gene replacement delivery, and there is no regulation of ectopic expression, so excessive amounts of protein could potentially be made. In contrast, genomic editing corrects the mutation at the genomic level and allows for endogenous regulation of the restored protein, thereby providing the appropriate amount of protein in the correct tissue.
Myoediting has successfully been applied in human iPSCs and in animal models, providing a new path for MD treatment. Because of all the effort placed on genome editing systems during the last several years, the efficacy of the technology is no longer a deterrent for treatment of MDs in the clinic. The remaining challenges before clinical translation are validation of the safety of the system and optimization of the delivery systems. Considering all the recent preclinical successes obtained with CRISPR systems, especially in DMD, it seems reasonable that these challenges will be overcome in the next few years.
Acknowledgments
We thank J. Cabrera for his creative assistance with the graphics. This work was supported by grants from the NIH (HL-130253, HL-138426, and AR-067294), the Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center (U54 HD 087351), and the Robert A. Welch Foundation (1-0025 to ENO).
Version 1. 06/01/2020
Print issue publication
Footnotes
Conflict of interest: RBD and ENO are consultants for Exonics/Vertex Therapeutics.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(6):2766–2776. https://doi.org/10.1172/JCI136873.
Contributor Information
Francesco Chemello, Email: Francesco.Chemello@utsouthwestern.edu.
Rhonda Bassel-Duby, Email: Rhonda.Bassel-Duby@UTSouthwestern.edu.
Eric N. Olson, Email: Eric.Olson@utsouthwestern.edu.
References
- 1.Benarroch L, Bonne G, Rivier F, Hamroun D. The 2020 version of the gene table of neuromuscular disorders (nuclear genome) Neuromuscul Disord. 2019;29(12):980–1018. doi: 10.1016/j.nmd.2019.10.010. [DOI] [PubMed] [Google Scholar]
- 2.Reilly MM, Hanna MG. Genetic neuromuscular disease. J Neurol Neurosurg Psychiatry. 2002;73(suppl 2):II12–II21. doi: 10.1136/jnnp.73.suppl_2.ii12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guiraud S, Davies KE. Pharmacological advances for treatment in Duchenne muscular dystrophy. Curr Opin Pharmacol. 2017;34:36–48. doi: 10.1016/j.coph.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov. 2015;14(1):58–74. doi: 10.1038/nrd4467. [DOI] [PubMed] [Google Scholar]
- 5.Koo T, Wood MJ. Clinical trials using antisense oligonucleotides in duchenne muscular dystrophy. Hum Gene Ther. 2013;24(5):479–488. doi: 10.1089/hum.2012.234. [DOI] [PubMed] [Google Scholar]
- 6.Lim KR, Maruyama R, Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des Devel Ther. 2017;11:533–545. doi: 10.2147/DDDT.S97635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duan D. Systemic AAV micro-dystrophin gene therapy for duchenne muscular dystrophy. Mol Ther. 2018;26(10):2337–2356. doi: 10.1016/j.ymthe.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song Y, et al. Non-immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat Med. 2019;25(10):1505–1511. doi: 10.1038/s41591-019-0594-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lostal W, et al. Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum Mol Genet. 2010;19(10):1897–1907. doi: 10.1093/hmg/ddq065. [DOI] [PubMed] [Google Scholar]
- 10.Asher DR, et al. Clinical development on the frontier: gene therapy for duchenne muscular dystrophy. Expert Opin Biol Ther. 2020;20(3):263–274. doi: 10.1080/14712598.2020.1725469. [DOI] [PubMed] [Google Scholar]
- 11.Gupta RM, Musunuru K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J Clin Invest. 2014;124(10):4154–4161. doi: 10.1172/JCI72992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 13.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 14.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507(7490):62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Overbeek M, et al. DNA repair profiling reveals nonrandom outcomes at Cas9-mediated breaks. Mol Cell. 2016;63(4):633–646. doi: 10.1016/j.molcel.2016.06.037. [DOI] [PubMed] [Google Scholar]
- 17.Chen W, et al. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 2019;47(15):7989–8003. doi: 10.1093/nar/gkz487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci U S A. 1994;91(13):6064–6068. doi: 10.1073/pnas.91.13.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makarova KS, Wolf YI, Koonin EV. Classification and nomenclature of CRISPR-Cas systems: where from here? CRISPR J. 2018;1(5):325–336. doi: 10.1089/crispr.2018.0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9(1):1911. doi: 10.1038/s41467-018-04252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351(6268):84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleinstiver BP, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529(7587):490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen JS, et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550(7676):407–410. doi: 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casini A, et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat Biotechnol. 2018;36(3):265–271. doi: 10.1038/nbt.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pickar-Oliver A, Gersbach CA. The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20(8):490–507. doi: 10.1038/s41580-019-0131-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19(12):770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landrum MJ, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anzalone AV, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Long C, Bassel-Duby R, Olson EN. Myoediting: toward prevention of muscular dystrophy by therapeutic genome editing. Physiol Rev. 2018;98(3):1205–1240. doi: 10.1152/physrev.00046.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bladen CL, et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395–402. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Echigoya Y, Lim KRQ, Nakamura A, Yokota T. Multiple exon skipping in the duchenne muscular dystrophy hot spots: prospects and challenges. J Pers Med. 2018;8(4):E41. doi: 10.3390/jpm8040041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- 33.Hoffman EP, Kunkel LM, Angelini C, Clarke A, Johnson M, Harris JB. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology. 1989;39(8):1011–1017. doi: 10.1212/WNL.39.8.1011. [DOI] [PubMed] [Google Scholar]
- 34.Beggs AH, et al. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am J Hum Genet. 1991;49(1):54–67. [PMC free article] [PubMed] [Google Scholar]
- 35.Henderson DM, Lee A, Ervasti JM. Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc Natl Acad Sci U S A. 2010;107(21):9632–9637. doi: 10.1073/pnas.1001517107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki K, et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540(7631):144–149. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki K, et al. Precise in vivo genome editing via single homology arm donor mediated intron-targeting gene integration for genetic disease correction. Cell Res. 2019;29(10):804–819. doi: 10.1038/s41422-019-0213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun. 2015;6:6244. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young CS, et al. A Single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell. 2016;18(4):533–540. doi: 10.1016/j.stem.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nelson CE, et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat Med. 2019;25(3):427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fletcher S, Adams AM, Johnsen RD, Greer K, Moulton HM, Wilton SD. Dystrophin isoform induction in vivo by antisense-mediated alternative splicing. Mol Ther. 2010;18(6):1218–1223. doi: 10.1038/mt.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chakrabarti AM, Henser-Brownhill T, Monserrat J, Poetsch AR, Luscombe NM, Scaffidi P. Target-specific precision of CRISPR-mediated genome editing. Mol Cell. 2019;73(4):699–713.e6. doi: 10.1016/j.molcel.2018.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amoasii L, et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci Transl Med. 2017;9(418):eaan8081. doi: 10.1126/scitranslmed.aan8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amoasii L, et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362(6410):86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Min YL, et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci Adv. 2019;5(3):eaav4324. doi: 10.1126/sciadv.aav4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Min YL, et al. Correction of three prominent mutations in mouse and human models of Duchenne muscular dystrophy by single-cut genome editing. Mol Ther. doi: 10.1016/j.ymthe.2020.05.024. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanlon KS, et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat Commun. 2019;10(1):4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan J, Zhang F, Karcher D, Bock R. Engineering of high-precision base editors for site-specific single nucleotide replacement. Nat Commun. 2019;10(1):439. doi: 10.1038/s41467-018-08034-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zuo E, et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science. 2019;364(6437):289–292. doi: 10.1126/science.aav9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grünewald J, et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569(7756):433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rees HA, Wilson C, Doman JL, Liu DR. Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci Adv. 2019;5(5):eaax5717. doi: 10.1126/sciadv.aax5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gapinske M, et al. CRISPR-SKIP: programmable gene splicing with single base editors. Genome Biol. 2018;19(1):107. doi: 10.1186/s13059-018-1482-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech. 2015;8(3):195–213. doi: 10.1242/dmm.018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wasala NB, Chen SJ, Duan D. Duchenne muscular dystrophy animal models for high-throughput drug discovery and precision medicine. Expert Opin Drug Discov. 2020;15(4):443–456. doi: 10.1080/17460441.2020.1718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi IY, et al. Concordant but varied phenotypes among Duchenne muscular dystrophy patient-specific myoblasts derived using a human iPSC-based model. Cell Rep. 2016;15(10):2301–2312. doi: 10.1016/j.celrep.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 56.Long C, et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci Adv. 2018;4(1):eaap9004. doi: 10.1126/sciadv.aap9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li HL, et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports. 2015;4(1):143–154. doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kyrychenko V, et al. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight. 2017;2(18):95918. doi: 10.1172/jci.insight.95918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, et al. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci Adv. 2017;3(4):e1602814. doi: 10.1126/sciadv.1602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuan J, et al. Genetic modulation of RNA splicing with a CRISPR-guided cytidine deaminase. Mol Cell. 2018;72(2):380–394.e7. doi: 10.1016/j.molcel.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 61.Morisaka H, et al. CRISPR-Cas3 induces broad and unidirectional genome editing in human cells. Nat Commun. 2019;10(1):5302. doi: 10.1038/s41467-019-13226-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Long C, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351(6271):400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tabebordbar M, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351(6271):407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bengtsson NE, et al. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nelson CE, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351(6271):403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.El Refaey M, et al. In vivo genome editing restores dystrophin expression and cardiac function in dystrophic mice. Circ Res. 2017;121(8):923–929. doi: 10.1161/CIRCRESAHA.117.310996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu L, et al. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol Ther. 2016;24(3):564–569. doi: 10.1038/mt.2015.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee K, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng. 2017;1:889–901. doi: 10.1038/s41551-017-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Young CS, Mokhonova E, Quinonez M, Pyle AD, Spencer MJ. Creation of a novel humanized dystrophic mouse model of duchenne muscular dystrophy and application of a CRISPR/Cas9 gene editing therapy. J Neuromuscul Dis. 2017;4(2):139–145. doi: 10.3233/JND-170218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koo T, et al. Functional rescue of dystrophin deficiency in mice caused by frameshift mutations using Campylobacter jejuni Cas9. Mol Ther. 2018;26(6):1529–1538. doi: 10.1016/j.ymthe.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ryu SM, et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat Biotechnol. 2018;36(6):536–539. doi: 10.1038/nbt.4148. [DOI] [PubMed] [Google Scholar]
- 72.Zhang Y, et al. Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci Adv. 2020;6(8):eaay6812. doi: 10.1126/sciadv.aay6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345(6201):1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16(6):1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 75.Levy JM, et al. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat Biomed Eng. 2020;4(1):97–110. doi: 10.1038/s41551-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yan Z, Zhang Y, Duan D, Engelhardt JF. Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proc Natl Acad Sci U S A. 2000;97(12):6716–6721. doi: 10.1073/pnas.97.12.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Truong DJ, et al. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015;43(13):6450–6458. doi: 10.1093/nar/gkv601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moretti A, et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat Med. 2020;26(2):207–214. doi: 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fardaei M, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11(7):805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 80.Ho TH, Savkur RS, Poulos MG, Mancini MA, Swanson MS, Cooper TA. Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J Cell Sci. 2005;118(pt 13):2923–2933. doi: 10.1242/jcs.02404. [DOI] [PubMed] [Google Scholar]
- 81.Kim EY, et al. Distinct pathological signatures in human cellular models of myotonic dystrophy subtypes. JCI Insight. 2019;4(6):122686. doi: 10.1172/jci.insight.122686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Raaijmakers RHL, Ripken L, Ausems CRM, Wansink DG. CRISPR/Cas applications in myotonic dystrophy: expanding opportunities. Int J Mol Sci. 2019;20(15):E3689. doi: 10.3390/ijms20153689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Provenzano C, et al. CRISPR/Cas9-mediated deletion of CTG expansions recovers normal phenotype in myogenic cells derived from myotonic dystrophy 1 patients. Mol Ther Nucleic Acids. 2017;9:337–348. doi: 10.1016/j.omtn.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dastidar S, et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018;46(16):8275–8298. doi: 10.1093/nar/gky548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lo Scrudato M, et al. Genome editing of expanded CTG repeats within the human DMPK gene reduces nuclear RNA foci in the muscle of DM1 mice. Mol Ther. 2019;27(8):1372–1388. doi: 10.1016/j.ymthe.2019.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Agtmaal EL, et al. CRISPR/Cas9-induced (CTG·CAG)n repeat instability in the myotonic dystrophy type 1 locus: implications for therapeutic genome editing. Mol Ther. 2017;25(1):24–43. doi: 10.1016/j.ymthe.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang Y, et al. Therapeutic genome editing for myotonic dystrophy type 1 using CRISPR/Cas9. Mol Ther. 2018;26(11):2617–2630. doi: 10.1016/j.ymthe.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Himeda CL, Jones TI, Jones PL. CRISPR/dCas9-mediated transcriptional inhibition ameliorates the epigenetic dysregulation at D4Z4 and represses DUX4-fl in FSH muscular dystrophy. Mol Ther. 2016;24(3):527–535. doi: 10.1038/mt.2015.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Turan S, Farruggio AP, Srifa W, Day JW, Calos MP. Precise correction of disease mutations in induced pluripotent stem cells derived from patients with limb girdle muscular dystrophy. Mol Ther. 2016;24(4):685–696. doi: 10.1038/mt.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schacker M, Seimetz D. From fiction to science: clinical potentials and regulatory considerations of gene editing. Clin Transl Med. 2019;8(1):27. doi: 10.1186/s40169-019-0244-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hakim CH, et al. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight. 2018;3(23):124297. doi: 10.1172/jci.insight.124297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu P, Wu F, Mosenson J, Zhang H, He TC, Wu WS. CRISPR/Cas9-mediated genome editing corrects dystrophin mutation in skeletal muscle stem cells in a mouse model of muscle dystrophy. Mol Ther Nucleic Acids. 2017;7:31–41. doi: 10.1016/j.omtn.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boutin S, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21(6):704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 94.Louis Jeune V, Joergensen JA, Hajjar RJ, Weber T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum Gene Ther Methods. 2013;24(2):59–67. doi: 10.1089/hgtb.2012.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Moreno AM, et al. Immune-orthogonal orthologues of AAV capsids and of Cas9 circumvent the immune response to the administration of gene therapy. Nat Biomed Eng. 2019;3(10):806–816. doi: 10.1038/s41551-019-0431-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chew WL, et al. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat Methods. 2016;13(10):868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Charlesworth CT, et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med. 2019;25(2):249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ferdosi SR, et al. Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat Commun. 2019;10(1):1842. doi: 10.1038/s41467-019-09693-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mehta A, Merkel OM. Immunogenicity of Cas9 Protein. J Pharm Sci. 2020;109(1):62–67. doi: 10.1016/j.xphs.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wienert B, Shin J, Zelin E, Pestal K, Corn JE. In vitro-transcribed guide RNAs trigger an innate immune response via the RIG-I pathway. PLoS Biol. 2018;16(7):e2005840. doi: 10.1371/journal.pbio.2005840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. doi: 10.1101/gr.231936.117. Kim S, et al. CRISPR RNAs trigger innate immune responses in human cells [published online February 22, 2018]. Genome Res. http://doi.org/10.1101/gr.231936.117. [DOI] [PMC free article] [PubMed]
- 102.Kim D, et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods. 2015;12(3):237–343, 1 p following 243. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- 103.Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar VV, Aryee MJ, Joung JK. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods. 2017;14(6):607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Amoasii L, et al. In vivo non-invasive monitoring of dystrophin correction in a new Duchenne muscular dystrophy reporter mouse. Nat Commun. 2019;10(1):4537. doi: 10.1038/s41467-019-12335-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Arnett AL, et al. Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol Ther Methods Clin Dev. 2014;1:14038. doi: 10.1038/mtm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Goldstein JM, et al. In situ modification of tissue stem and progenitor cell genomes. Cell Rep. 2019;27(4):1254–1264.e7. doi: 10.1016/j.celrep.2019.03.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nance ME, et al. AAV9 edits muscle stem cells in normal and dystrophic adult mice. Mol Ther. 2019;27(9):1568–1585. doi: 10.1016/j.ymthe.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gaudelli NM, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]