

Abstract
The transcriptional repressor Hairy Enhancer of Split 1 (HES1) plays an essential role in the development of many organs by promoting the maintenance of stem/progenitor cells, controlling the reversibility of cellular quiescence, and regulating both cell fate decisions. Deletion of Hes1 in mice results in severe defects in multiple organs and is lethal in late embryogenesis. Here we have investigated the role of HES1 in hematopoiesis using a hematopoietic lineage‐specific Hes1 knockout mouse model. We found that while Hes1 is dispensable for steady‐state hematopoiesis, Hes1‐deficient hematopoietic stem cells (HSCs) undergo exhaustion under replicative stress. Loss of Hes1 upregulates the expression of genes involved in PPARγ signaling and fatty acid metabolism pathways, and augments fatty acid oxidation (FAO) in Hes1 f/f Vav1Cre HSCs and progenitors. Functionally, PPARγ targeting or FAO inhibition ameliorates the repopulating defects of Hes1 f/f Vav1Cre HSCs through improving quiescence in HSCs. Lastly, transcriptome analysis reveals that disruption of Hes1 in hematopoietic lineage alters expression of genes critical to HSC function, PPARγ signaling, and fatty acid metabolism. Together, our findings identify a novel role of HES1 in regulating stress hematopoiesis and provide mechanistic insight into the function of HES1 in HSC maintenance.
Keywords: fatty acid metabolism, hairy enhancer of Split 1, hematopoietic reconstitution capacity, hematopoietic stem progenitor cells, PPARγ signaling pathway, replicative stress
While Hes1 is dispensable for steady‐state hematopoiesis, Hes1‐deficient HSCs undergo exhaustion under replicative stress. Deletion of Hes1 deregulates genes in PPARγ signaling and fatty acid oxidation (FAO), and augments FAO in Hes1 f/f Vav1Cre hematopoietic stem cells (HSCs) and progenitors. Mechanistically, HES1 plays important role in stress hematopoiesis by regulating genes in HSC function, PPARγ signaling and fatty acid metabolism pathways.
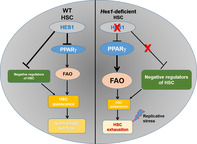
Significance statement.
The authors show that while Hes1 is dispensable for steady‐state hematopoiesis, Hes1‐deficient HSCs undergo exhaustion under replicative stress. Deletion of Hes1 deregulates genes in PPARγ signaling and fatty acid oxidation (FAO), and augments FAO in Hes1 f/f Vav1Cre hematopoietic stem cells (HSCs) and progenitors. Functionally, PPARγ targeting or FAO inhibition ameliorates the repopulating defects of Hes1 f/f Vav1Cre HSCs through improving quiescence. Transcriptome analysis reveals that disruption of Hes1 alters HSC function, PPARγ signaling, and fatty acid metabolism pathways. These results identify a novel role of HES1 in regulating stress hematopoiesis and provide mechanistic insight into the function of HES1 in HSC maintenance.
1. INTRODUCTION
The transcriptional repressor Hairy Enhancer of Split 1 (HES1) is member of hairy‐related basic helix‐loop‐helix (bHLH) family,1 and an evolutionarily conserved target of Notch signaling, which regulates several cellular processes, including cell fate decisions and proliferation in both invertebrates and mice.2, 3 There are seven described members in the mammalian HES family. Among them, HES1 and HES5 are the only members known to be involved specifically in Notch1 signaling in neural cells and in bone marrow.4, 5 HES1 is a repressor‐type bHLH that represses expression of its own gene (autoregulatory mechanism)6, 7 and antagonizes bHLH activators.8 Deletion of Hes1 in mice results in severe neural tube defects in addition to defects in the thymus, pancreas, gut, bile duct, and neural tube that are lethal in late embryogenesis.1, 9, 10 However, little is known about the role of HES1 in hematopoiesis.
Hematopoietic stem cells (HSCs) harbor the capacities of both self‐renewal and differentiation to ensure a balanced production of all blood cells throughout life. The fate decisions of HSCs (self‐renewal vs differentiation) are made through the process of cell division. In the hematopoietic system, HES1 has a major function in normal T cell development, but it is also directly involved in the maintenance of Notch‐induced T‐cell leukemias.9, 11, 12 Although Hes1 is widely expressed in the aortic endothelium and hematopoietic cluster, Hes1‐deficient mice show no overt hematopoietic abnormalities.9 However, no measurement of the activity or function of HSCs was performed in these Hes1‐deficient mice. Several studies have shown that overexpression of HES1 inhibits differentiation of bone marrow HSCs when cultured in vitro, increase HSC self‐renewal, reduce HSC cycling, and preserve the long‐term reconstitution ability of primitive hematopoietic cells.13, 14, 15, 16 How HES1 regulates in vivo hematopoiesis, especially under stress condition remains to be elucidated.
Recent studies using metabolomics technologies reveal that metabolic regulation plays an essential role in HSC maintenance. Metabolic pathways provide energy and building blocks for other factors functioning at steady state and in stress hematopoiesis.17 Altered metabolic energetics in HSCs affects HSC function and underlies the onset of most blood malignancies.18, 19, 20 Nuclear receptor superfamily members, peroxisome proliferator‐activated receptors (PPARs), classified into three isoforms, namely PPARα, β/δ, and γ, are important in whole‐body energy metabolism and collectively involved in fatty acid oxidation (FAO).21 We previously identified PPARγ as a putative negative regulator of HSCs using an in vivo RNAi screen system.22 More recently, it has been shown that inhibition of PPARγ improves ex vivo expansion of human HSCs and progenitors.23 Nevertheless, how HES1 regulates PPARγ signaling and FAO pathways in HSCs is less understood.
Here we investigated the role of Hes1 in hematopoiesis under stress condition using a hematopoietic lineage specific Hes1 knockout mouse model (Hes1 f/f Vav1Cre), and demonstrate that while Hes1 is dispensable for steady‐state hematopoiesis, Hes1‐deficient HSCs undergo exhaustion under replicative stress. Disruption of Hes1 skews the expression of a set of genes involved in hematopoietic stem cell function, PPARγ signaling pathway and fatty acid metabolism pathways. Our data identify a novel role for HES1 in regulating hematopoiesis under stress condition and provide a mechanistic insight into the function of HES1 in HSC maintenance.
2. MATERIALS AND METHODS
2.1. Mice and treatment
Heterozygous Hes1 f/+ mice24 in a C57BL/6 background were recovered from the sperm purchased at Experimental Animal Division at RIKEN BioResource Center (RBRC #: RBRC06047). The IVF procedure was performed in Transgenic Animal Core Facility at West Virginia University (WVU). Heterozygous Hes1 f/+ mice were interbred with Vav1Cre mice (Jackson Laboratory; stock # 008610) to generate Hes1 f/f Vav1Cre and Hes1 f/f littermates. This Vav1Cre strain allows reliable deletion of Hes1 throughout the entire hematopoietic compartment. Pparg f/f and Cpt1a f/f mice were purchased from Jackson laboratory (Stock #: 004584 and 032778, respectively; Jackson Laboratories, Bar Harbor, ME, https://www.jax.org/) to cross with Hes1 f/f Vav1Cre mice. Six to eight‐week‐old BoyJ mice were used as bone marrow transplant (BMT) recipients. Animals including BoyJ recipient mice were maintained in the animal barrier facility at WVU.
For treatment with PPARγ antagonist, the mice received intraperitoneal (i.p.) injections of 5 mg/kg of GW9662 (Sigma‐Aldrich, St Louis, MO, https://www.sigmaaldrich.com/united-states.html), or vehicle (5% DMSO v/v) daily from day −1 to day 7 post BMT.25 For in vivo FAO inhibition, etomoxir (50 mg/kg; Cayman Chemical, Ann Arbor, MI) was i.p. injected into the subject mice daily day −1 to day 7 post BMT.26 All experimental procedures conducted in this study were approved by the Institutional Animal Care and Use Committee of West Virginia University according to the approved guidelines.
2.2. Bone marrow transplantation
For competitive transplantation, 106 BM cells from Hes1 f/f Vav1Cre mice or their wild‐type littermates (Hes1 f/f;CD45.2+), along with an equal number of BM cells from congenic BoyJ mice (CD45.1+), were transplanted into lethally irradiated (11.75 Gy) BoyJ recipients (CD45.1+). Donor‐derived hematopoietic reconstitution in the recipient mice at 4 and 16 weeks post‐transplantation was determined by staining for CD45.1‐PE and CD45.2‐FITC markers followed by flow cytometry analysis with a FACSCanto I (BD Biosciences, San Jose, CA). For secondary BM transplantation, 3 × 106 BM cells from primary recipients were injected to lethally irradiated BoyJ recipients. Donor‐derived chimera were determined 16 weeks post BMT.
Serial BMT was performed to evaluate the engraftment of long‐term HSCs.27 Briefly, 106 CD45.2+ BM cells from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates were transplanted into lethally irradiated BoyJ (CD45.1+, Jackson Laboratories) recipients. For secondary and tertiary transplantation, recipient mice were sacrificed and 3 to 5 × 106 BM cells were transplanted into recipient BoyJ mice. Donor reconstitution (CD45.2+ cells) was assessed 16 weeks after each BMT.
2.3. Competitive repopulating unit assays
Graded numbers of BM cells from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates (CD45.2+), along with 2 × 105 radio‐protector BM cells, into lethally irradiated congenic recipients (CD45.1+). The competitive repopulating unit (CRU) frequencies were then calculated from the proportions of negative mice (<1% donor engraft) with L‐calc software (StemCell Technologies, Vancouver, BC, Canada), which uses Poisson statistics.
2.4. Flow cytometry
The lineage marker (Lin) mixture (BD Biosciences) for BM cells from treated or untreated mice included the following biotinylated antibodies: CD3ε (145‐2C11), CD11b (M1/70), CD45R/ B220 (RA3‐6B2), mouse erythroid cells Ly‐76 (Ter119), Ly6G, and Ly‐6C (RB6‐8C5). Other conjugated Abs (BD BioSciences) used for surface staining included: CD45.1 (A20), CD45.2 (A104), Sca1 (D7), c‐kit (2B8), CD34 (RAM34), Flt3 (A2F10.1), CD48 (HM48‐1), CD150 (9D1), IL‐7Rα (hIL‐7R‐M21). A two‐step staining procedure was performed by using biotinylated primary antibodies followed by the incubation of antibody coated cells with streptavidin‐PerCP Cy5.5 or FITC (BD Biosciences).
For apoptosis staining, surface marker stained cells were incubated with Annexin V and 7AAD using the BD ApoAlert Annexin V Kit (BD Pharmingen, San Jose, CA) in accordance with the manufacturer's instruction. Flow cytometry analysis was then performed to determine the proportion of Annexin V‐positive cells.
For cell cycle analysis, cells stained for surface markers were fixed and permeabilized with Cytofix/Cytoperm buffer (BD Pharmingen) followed by intensive wash using Perm/Wash Buffer (BD Pharmingen). Anti‐mouse Ki67 antibody (BD Pharmingen) and DAPI (Sigma‐Aldrich) were then used to incubate the cells followed by flow cytometry analysis. For the BrdU incorporation assay, Bromodeoxyuridine (BrdU, 150 μL of 10 mg/mL) were i.p. injected to subjected mice followed by BM cells isolation 14 hours later. BrdU incorporated cells (S phase) were analyzed with the APC BrdU Flow Kit (BD Biosciences), following the manufacturer's instructions. Briefly, cells were surface stained then fixed and permeabilized using BD Cytofix/Cytoperm Buffer. After 1 hour incubation with DNase at 37°C, cells were stained with APC‐conjugated anti‐BrdU monoclonal antibody. 7‐aminoactinomycin (7‐AAD) was added to each sample right before flow cytometry analysis (BD Biosciences).
To determine the quiescence of donor‐derived cells in the transplanted recipients, surface marker stained cells were fixed and permeabilized using Cytofix/Cytoperm buffer (BD PharMingen) followed by intensive wash using Perm/Wash Buffer (BD PharMingen). Cells were then labeled with Pyronin Y staining buffer (150 ng/mL Pyronin Y in Perm/Wash buffer; Sigma‐Aldrich) at 37°C for 1 hour followed by flow cytometry analysis on CD45.2+ signaling lymphocyte activation molecule (SLAM; Lin−Sca1+c‐kit+CD150+CD48−) gated population.
2.5. Measurement of fatty acid oxidation
FAO was determined by palmitate oxidation method.28 Briefly, metabolism of 1‐14C‐palmitic acid (60 mCi/mmol; PerkinElmer, Waltham, MA) was determined as the formation of 14C‐acid‐soluble β‐oxidation products in LSK cells isolated from indicated mice. Cells were permeabilized (10 μg digitonin/million cells), incubations contained 2 mM 1‐14C‐palmitate (10 nCi/assay) and the incubation lasted 15 minutes.
2.6. Quantitative PCR and RNA sequencing
Total RNA were extracted from BM LSK cells of each mouse genotype and treated with RNase‐free DNase to remove contaminating genomic DNA. Reverse transcription was performed with random hexamers and Superscript II RT (Invitrogen, Grand Island, NY) and was carried out at 42°C for 60 minutes and stopped at 95°C for 5 minutes. First‐strand cDNA was used for real‐time PCR using primers listed in Table S1. Samples were normalized to the level of GAPDH mRNA.
For RNA sequencing, total RNA was extracted from LSK cells isolated from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates following standard protocol with TRIZol reagent (Life Technologies, Carlsbad, CA) followed by RNA library preparation with the Ilumina TreSeq strand‐specific mRNA sample preparation system. All RNA‐seq libraries were sequenced with a read length of single‐end 75 bp using the Illumina NextSeq 500 and final of over 45 million reads per sample.
Pair‐end RNA‐seq reads were aligned to the mouse genome (mm10) with the subread aligner.29 The number of reads for RefSeq genes were summarized by using the FeatureCounts function within the Rsubread R package.30 Gene expression level was quantified by Reads Per Kilobase of transcript, per million mapped reads (RPKM) by using Excel with the summarized read counts exported from Rsubread. Differentially expressed genes were predicted by EdgeR 3 by controlling for batch effects and by considering the following criteria: FC > 1.5, P < .01, and an average of RPKM across replicates >2 in at lease on condition.31 Only protein‐coding genes were included for downstream functional inference. The online Panther gene list analysis (http://www.pantherdb.org/)32 was used for gene‐ontology enrichment analysis on biological processes for genes upregulated in Hes1 f/f Vav1Cre LSK cells as compared to Hes1 f/f cells; the analysis included all expressed genes as background. GSEA 4.0.2 was used for gene set enrichment analysis (GSEA) by including all expressed genes, ranked by FC of expression (Hes1 f/f Vav1Cre vs. Hes1 f/f cells), against interested gene sets from C2.CPG (chemical and genetic perturbations) and C2.CP.KEGG from MSigDB.33 RNA‐Seq read distribution across the mouse genome was visualized by the WashU epigenome browser.34
2.7. Histopathology
Bone tissue was fixed in 4% paraformaldehyde in PBS (BioRad, Hercules, CA), decalcified in 14% EDTA (Sigma‐Aldrich) and embedded in paraffin (Sigma‐Aldrich). Sections were then stained with hematoxylin and eosin (H&E; Sigma‐Aldrich) and examined at ×400 by microscope.
2.8. Statistical analysis
Student's t test was performed using GraphPad Prism v8 (GraphPad software). Comparison of more than two groups was analyzed by one‐way ANOVA test. Values of P < .05 were considered statistically significant. Results are presented as mean ± SD. * indicates P < .05; ** indicates P < .01.
3. RESULTS
3.1. Hes1f/fVav1Cre mice exhibit normal steady‐state hematopoiesis
To elucidate the role of HES1 in hematopoiesis, we recently generated a constitutive and hematopoietic‐specific Hes1 deleted mouse strain Hes1 f/f Vav1Cre, by crossing a conditional Hes1 knockout strain24 with the hematopoietic‐specific Vav1Cre deleter. Expression of Cre recombinase under the promoter of Vav1, a guanine nucleotide exchange factor for Rho‐GTPases, induces deletion of Hes1 alleles, specifically in the fetal and adult hematopoietic system.35, 36, 37 The genotypes of offspring from Hes1 f/+ Vav1Cre breeders followed predicted Mendelian frequencies, indicating that no embryonic lethality or perinatal lethality was associated with the hematopoietic Hes1 deletion (data not shown). Genotyping PCR (Figure S1A) and an inspection of the distribution of RNA‐seq read cross the Hes1 locus (Figure S1B) indicate a successful deletion of Hes1 in mouse hematopoietic cells.
We first examined the effect of Hes1 deletion on steady state hematopoiesis. By using HemaVet 950, we first analyzed peripheral blood (PB) of 6 to 8‐week‐old mice and found a slight increase in white blood cell (WBC) counts in Hes1 f/f Vav1Cre mice than Hes1 f/f control animals. We observed no significant difference in the hemoglobin and hematocrit values between Hes1 f/f Vav1Cre and the control Hes1 f/f mice, although the platelet count was somewhat reduced in the Hes1 f/f Vav1Cre group (Table 1). All other hematological parameters, including total erythrocyte counts, appeared to be normal in Hes1 f/f Vav1Cre mice, as compared to their Hes1 f/f littermates. Therefore, there is no indication of anemia in these mutant animals under steady state.
Table 1.
Hematopoietic parameters
| Absolute and differential WBC counts | Characterization or red blood cells | Plt (×109/L) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| WBC count (cells/μL) | % Lymphocytes | % Neutrophils | % Monocytes | RBC count (×1012/L) | HCT, % | MCV, fL | Hb (g/dL) | ||
| Hes1 f/f | 7.07 ± 0.72 | 80.46 ± 3.94 | 11.63 ± 1.19 | 2.37 ± 0.69 | 11.08 ± 1.29 | 53.1 ± 2.31 | 56.29 ± 3.45 | 15.94 ± 1.49 | 728 ± 84.54 |
| Hes1 f/f Vav1Cre | 7.14 ± 0.93 | 83.06 ± 4.47 | 12.39 ± 2.47 | 3.09 ± 0.78 | 11.82 ± 1.31 | 52.682 ± 2.9622 | 50.38 ± 2.4917 | 16.76 ± 1.37 | 579.5 ± 95.1 |
| P | .41 | .12 | .43 | .08 | .39 | .22 | .17 | .15 | .037 |
Note: P values were determined using Student's t test. For all tests on wild‐type mice, the sample size was 10. For all tests on Hes1 f/f Vav1Cre mice, the sample size was 9.
Abbreviations: % lymphocytes, percentage of WBC count that are lymphocytes; % neutrophils, percentage of WBC count that are neutrophils; % of monocytes, percentage of WBC count that are monocytes; Hb, hemoglobin concentration; HCT, hematocrit (percentage of whole blood volume); MCV, mean cell volume; Plt, Platelet count.; RBC count, red blood cell count; WBC count, white blood cell count.
3.2. Hes1 is dispensable for HSC maintenance at steady state
We then analyzed different cell compartments in the BM of Hes1 f/f Vav1Cre mice and found a comparable total BM cellularity of Hes1 f/f Vav1Cre mice and their Hes1 f/f littermates (Figure 1A). Further analysis of the mice showed no effect of Hes1 deletion on the relative frequencies of hematopoietic progenitor cells (LSK; Lin−Sca1+c‐kit+) and the phenotypic HSCs (Lin−Sca1+c‐kit+CD150+CD48−; Signaling lymphocyte activation molecules, SLAM)38 compartment (Figure 1B), suggesting that Hes1 may be not mandatory for steady‐state HSC homeostasis.
Figure 1.
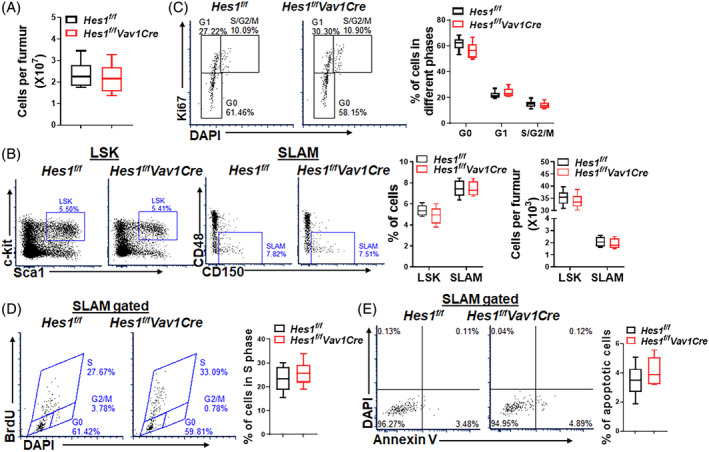
Hes1 is dispensable for steady‐state hematopoiesis. A, Normal bone marrow cellularity in Hes1 f/f Vav1Cre mice. Whole bone marrow cells (WBMCs) from one femur of Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates were enumerated (n = 8/group). B, Hes1 deficiency does not affect the frequencies of HSCs and progenitors. WBMCs isolated from 8‐week‐old Hes1 f/f Vav1Cre mice and their Hes1 f/f littermates were subjected to flow cytometry analysis for LSK (Lin−Sca1+c‐kit+) and SLAM (LSKCD150+CD48−) cells. Representative flow lots (left), quantification of frequencies (middle), and absolute cell numbers (right) are shown (n = 6‐8/group). C, Loss of Hes1 does not affect HSC quiescence. WBMCs described in B were subjected to Ki67/DAPI staining. SLAM cells were gated for analysis. Representative lots (left) and quantification (right) are shown (n = 6‐8/group). D, Hes1 deletion does not increase HSC cycling. Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates were i.p. injected with BrdU (150 μL of 10 mg/mL). Whole bone marrow cells (WBMCs) were subjected to BrdU incorporation assay 14 hours later. SLAM cells were gated for analysis. Representative flow lots (left) and quantification (right) are shown (n = 6‐8/group). E, Loss of Hes1 does not increase HSC apoptosis. WBMCs described in B were subjected to apoptosis analysis by Annexin V and 7AAD staining. SLAM cells were gated for analysis. Representative flow plots (left) and quantification (right) are shown. Results are mean ± SD of three independent experiments (n = 6‐8/group). *P < .05; **P < .01
Quiescence is known to be an important feature of HSC homeostasis.39 We next analyzed the cell cycle profile of HSCs deficient for Hes1. Ki67/DAPI staining revealed a slight decrease, albeit not statistically significant, in the proportion of quiescent (G0) and an slight increase in the proportion of cycling (S/G2/M) SLAM cells in Hes1 f/f Vav1Cre mice compared to Hes1 f/f control animals (Figure 1C). In line with the cell cycle data, the percentage of SLAM cells in S phase was comparable in Hes1 f/f Vav1Cre mice compared to their Hes1 f/f littermates by an in vivo BrdU incorporation assay (Figure 1D). Moreover, Annexin V/7AAD staining revealed that loss of Hes1 did not affect the viability of SLAM cells at the steady state (Figure 1E). These results suggest that the Hes1 protein is dispensable for steady‐state hematopoiesis.
3.3. Hes1‐deficient HSCs undergo exhaustion under transplant stress
HSCs possess multilineage differentiation and self‐renewal activities that maintain the entire hematopoietic system during an organism's lifetime. We next conducted competitive BMT to determine the hematopoietic repopulating ability of HSCs deficient for Hes1 by transplanting equal numbers of BM cells from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates (CD45.2+), along with equal number of BM cells from congenic BoyJ mice (CD45.1+) into lethally irradiated BoyJ recipients. While recipients transplanted with Hes1 f/f Vav1Cre cells showed comparable donor‐derived chimera (CD45.2+) to those transplanted with the control Hes1 f/f cells at 4 weeks post‐transplant; Hes1 deficiency led to significantly reduced donor chimera at 16 weeks post‐transplant (Figure 2A), indicating a progressive decline of hematopoietic repopulating ability of the Hes1‐deficient HSCs. Consistent with previous report that HES1 has a major function in normal T‐cells development,9, 11, 12 Hes1 deficiency caused a significant reduction of T cells and increase of B cells in the transplanted recipients (Figure 2B). We also found a significantly decreased proportion of quiescent cells in donor‐derived Hes1 f/f Vav1Cre LSK cells as compared to that of Hes1 f/f donor LSK cells (Figure 2C), suggesting that the Hes1 f/f Vav1Cre hematopoietic stem and progenitor cells (HSPCs) were extensively cycling in the transplanted recipients under replicative stress.
Figure 2.
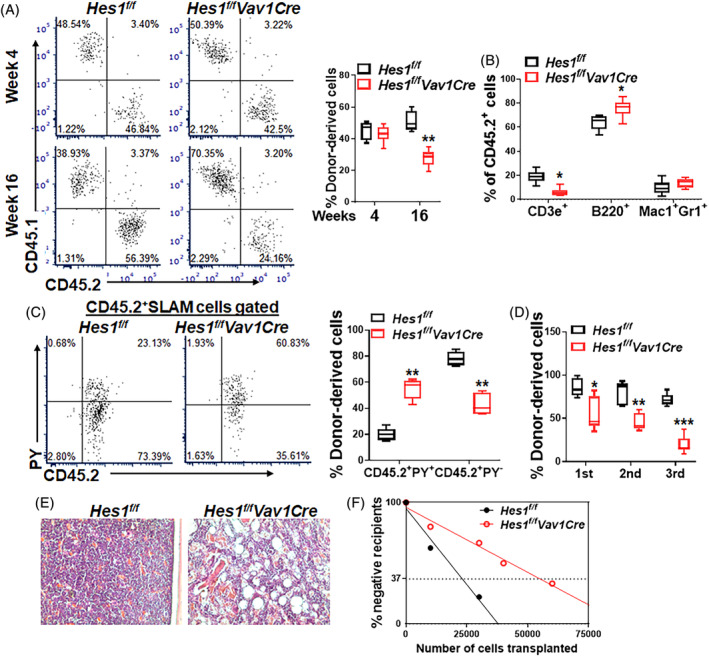
Hes1‐deficient HSCs undergo exhaustion under transplant stress. A, Hes1 deficiency impairs the repopulating ability of HSCs. One million WBMCs isolated from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates (CD45.2+), along with equal numbers of congenic WBMCs from BoyJ mice (CD45.1+), were transplanted into lethally irradiated BoyJ recipients (CD45.1+). Donor‐derived chimera were assessed at 4 weeks and 16 weeks post BMT. Representative flow lots (left) and quantification (right) are shown (n = 8‐10). B, Hes1 deficiency impairs T lineage reconstitution in the recipients. Peripheral blood (PB) from recipients described in A was subjected to lineage differentiation analysis in the donor‐derived compartment (CD45.2+) 16 weeks post BMT. Quantification are shown (n = 8‐10). C, Donor‐derived Hes1 f/f Vav1Cre HSCs loss quiescent in the transplanted recipients. WBMCs from the recipients described in B were subjected to cell cycle staining using Pyronin Y (PY). Donor‐derived (CD45.2+) SLAM cells were gated for analysis. Representative flow lots (left) and quantification (right) are shown (n = 8‐10). D, Hes1‐deficient HSCs undergo exhaustion under serial transplant stress. One million WBMCs from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates were transplanted into lethally irradiated BoyJ recipients. After 16 weeks, primary recipients were sacrificed and 106 WBMCs were used for secondary transplantation into lethally irradiated BoyJ recipients. The same protocol was employed for the tertiary transplantation. Donor reconstitution (CD45.2+ cells) were monitored in PB was accessed at week 16 of each transplantation. Results are mean ± SD of three independent experiments (n = 6‐9/group). E, Recipient mice of Hes1‐deficient BM cells exhibit BM failure‐like phenotype. Femurs from the secondary recipients described in D were subjected to histologic examination. Representative H&E stained bone sections are shown. F, Analysis of replication‐induced HSC exhaustion by limiting dilution assay. Graded numbers of low density BM cells from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates were transplanted into lethally irradiated recipients. Plotted are the percentages of recipients containing less than 1% donor (CD45.2+) blood nucleated cells at 16 weeks post‐transplantation. The frequency of functional HSCs was calculated according to Poisson statisticic stem cells (HSCs) harbor the capacitis. *P < .05; **P < .01
The observation that Hes1‐deficiency induced hyper‐proliferation but progressively decreased repopulation of HSCs, a phenotype characteristic of HSC exhaustion,40, 41 prompted us to perform serial BMT to determine whether Hes1‐deficient HSCs undergo exhaustion under replicative stress. Indeed, we found a progressive decline of hematopoietic repopulating ability of the Hes1‐deficient HSCs during three rounds of BMT (Figure 2D). Furthermore, histology of BM from the secondary recipients transplanted with the Hes1 f/f Vav1Cre donor cells showed significantly decreased cellularity 10‐week post‐transplantation (Figure 2E). These results suggest that Hes1‐deficient HSCs may undergo replicative exhaustion in the transplanted recipients.
To substantiate these findings, we performed a limiting dilution assay,42, 43 in which we transplanted graded numbers of low‐density BM cells (LDBMCs) from Hes1 f/f or Hes1 f/f Vav1Cre mice, along with 2 × 105 radio‐protector BM cells, into lethally irradiated congenic recipients, and analyzed the frequency of the functional HSCs in the tested BM cells. Poisson statistical analysis at 16 weeks post‐transplantation showed a 2.5‐fold reduction in the frequency of competitive repopulating units (CRUs) (1/37 885 in LDBMCs of Hes1 f/f donor and 1/89 647 in LDBMCs of Hes1 f/f Vav1Cre donors; P = .0016; Figure 2F and Table 2). Thus, these results further demonstrate that replicative stress induces stem cell exhaustion in Hes1‐deficient HSCs.
Table 2.
Competitive repopulating units
| Genotype | Hes1 f/f | Hes1 f/f Vav1Cre |
|---|---|---|
| CRU frequency | 1/37 855 | 1/89 647 |
Note: Graded numbers of low density BM cells from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates were transplanted into lethally irradiated recipients. Frequency of competitive repopulating units (CRUs) was calculated according to Poisson statistics. P = .0016.
3.4. Hes1 deficiency upregulates genes in PPARγ signaling and fatty acid metabolism
Since HES1 suppresses expression of PPARG, which encodes the peroxisome proliferator‐activated receptor PPARγ and regulates fatty acid storage and glucose metabolism,44, 45 we then attempted to examine whether Hes1 loss affects expression of PPARγ target genes. Quantitative RT‐PCR analysis identified a set of upregulated PPARγ target genes, including Fabp4,46 Ncoa4,47 Pparg, and Zfml 48 in Hes1 f/f Vav1Cre LSK cells compared to those from Hes1 f/f mice (Figure 3A). Since PPARγ is the master regulator in FAO metabolic pathway,49, 50 we also observed a panel of upregulated FAO related genes, such as Slc25a20,51 Hadha,52 Cpt1a,53 Cpt2 54 (Figure 3B). We next assessed FAO rates and found that Hes1 deletion significantly increased FAO in LSK cells isolated from Hes1 f/f Vav1Cre mice compared to those from Hes1 f/f mice as determined by the palmitate oxidation method (Figure 3C). Together, these data indicate that deletion of Hes1 deregulated the expression of genes involved in PPARγ signaling and fatty acid metabolism pathways, and consequently augmented FAO in Hes1 f/f Vav1Cre HSCs.
Figure 3.
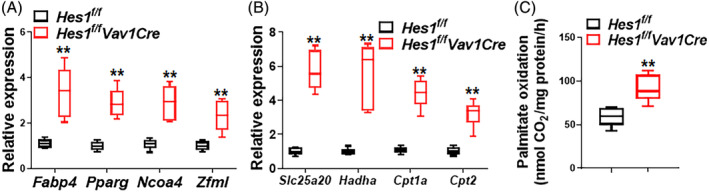
Deletion of Hes1 de‐regulates PPARγ and FAO. A, B, Upregulated PPARγ target genes (A) and fatty acid metabolism‐related genes (B) in Hes1 f/f Vav1Cre LSK cells. RNA were extracted from LSK cells of Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates followed by qPCR analysis using primers listed in Table S1. Samples were normalized to the level of GAPDH mRNA (n = 6‐9). C, Loss of Hes1 augments FAO. LSK cells from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates were subjected to palmitate oxidation rates measurement as captured 14CO2 using the isolated mitochondria and 1‐14C‐palmitate as substrate. Quantification are shown. Results are mean ± SD of three independent experiments. *P < .05; **P < .01
3.5. Genetic and pharmacological inhibition of PPARγ or FAO improves hematopoietic repopulation of Hes1‐deficient HSCs
We next asked whether inhibition of PPARγ or FAO can improve the function of Hes1‐deficient HSCs in vivo. We employed both genetic and pharmacological approaches to inhibit PPARγ or FAO. For genetic inhibition of PPARγ, we crossed Hes1 f/f Vav1Cre mice with a conditional Pparg f/f strain and generated Pparg +/+ Hes1 f/f Vav1Cre and Pparg f/f Hes1 f/f Vav1Cre isogenic lines. Equal numbers of BM cells from Pparg +/+ Hes1 f/f Vav1Cre and Pparg f/f Hes1 f/f Vav1Cre mice (CD45.2+) were transplanted into lethally irradiated recipient mice (CD45.1+). The repopulating capacity of the donor HSCs were determined by analyzing the percentage of CD45.2+ cells in the recipient mice 16 weeks post‐transplantation. We observed a markedly increased proportion of CD45.2+ cells in the BM of the recipient mice transplanted with Pparg f/f Hes1 f/f Vav1Cre cells compared with those transplanted with Pparg +/+ Hes1 f/f Vav1Cre cells (Figure 4A). For pharmacological inhibition of PPARγ, we treated the recipients transplanted with cells from Hes1 f/f Vav1Cre or their Hes1 f/f littermates with the well‐characterized PPARγ antagonist GW9662.23 Similar to genetic inhibition, in vivo administration of GW9662, albeit to less extend compared to Pparg deletion, also increased donor‐derived chimera in the recipients transplanted with Hes1 f/f Vav1Cre HSCs compared to vehicle controls (Figure 4B). Thus, PPARγ inhibition improves hematopoietic repopulation of Hes1‐deficient HSCs.
Figure 4.
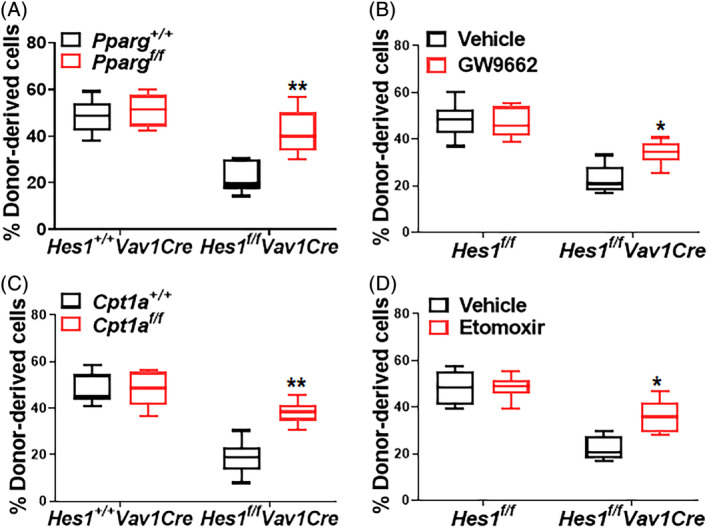
Inhibition of PPARγ or FAO improves repopulation of Hes1‐deficient HSCs. A, Genetic inhibition of Pparg improves repopulation of Hes1 f/f Vav1Cre HSCs. One million whole bone marrow cells (WBMCs) from Pparg f/f Hes1 f/f Vav1Cre mice or their Pparg +/+ Hes1 +/+ Vav1Cre littermates, along with equal numbers of congenic BM cells, were transplanted into lethally irradiated congenic mice. Donor‐derived chimera were determined by flow cytometry 16 weeks post BMT (n = 6‐8). B, Pharmacological inhibition of PPARγ improves repopulation of Hes1 f/f Vav1Cre HSCs. One million WBMCs from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates, along with equal numbers of congenic BM cells, were transplanted into lethally irradiated congenic mice. Recipients were treated with PPARγ antagonist GW9662 (5 mg/kg) daily for 8 days (day −1 to day 7). Donor‐derived chimera were determined by flow cytometry 16 weeks post BMT (n = 9). C, Cpt1a targeting improves repopulation of Hes1 f/f Vav1Cre HSCs. One million WBMCs from Cpt1a f/f Hes1 f/f Vav1Cre mice or their Cpt1a +/+ Hes1 +/+ Vav1Cre littermates, along with equal numbers of congenic BM cells, were transplanted into lethally irradiated congenic mice. Donor‐derived chimera were determined by flow cytometry 16 weeks post BMT (n = 7‐9). D, FAO inhibition improves repopulation of Hes1 f/f Vav1Cre HSCs. One million WBMCs from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates, along with equal numbers of congenic BM cells, were transplanted into lethally irradiated congenic mice. Recipients were i.p. administrated with FAO inhibitor Etomoxir (50 mg/kg) daily for 8 days (day −1 to day 7). Donor‐derived chimera were determined by flow cytometry 16 weeks post BMT (n = 9). *P < .05; **P < .01
Next, we performed genetic and pharmacological inhibition of FAO. We crossed Hes1 f/f Vav1Cre mice with mice carrying a conditional Cpt1a allele, which encodes the mitochondrial carnitine palmitoyltransferase‐1 (CPT‐1; a rate‐limiting enzyme in mitochondrial FAO),28, 55 to generate Cpt1a +/+ Hes1 f/f Vav1Cre and Cpt1a f/f Hes1 f/f Vav1Cre mice. We found that deletion of Cpt1a significantly increased donor‐derived chimera in Hes1 f/f Vav1Cre cell transplanted recipients (Figure 4C). Similarly, treatment of the recipients transplanted with BM cells from Hes1 f/f Vav1Cre or the Hes1 f/f control mice with the FAO inhibitor Etomoxir54 significantly increased donor‐derived chimera in the recipients transplanted with Hes1 f/f Vav1Cre HSCs (Figure 4B). Therefore, genetic and pharmacological inhibition of FAO improves hematopoietic repopulation of Hes1‐deficient HSCs.
3.6. Inhibition of PPARγ and FAO increases quiescence of Hes1‐deficient HSCs
To explore the underlying mechanism how Hes1 deficiency could cause HSC exhaustion under transplant stress, we first examined whether inhibition of PPARγ and FAO affected apoptosis of Hes1‐deficient HSCs. Analysis of donor‐derived HSCs (CD45.2+SLAM) cells in the transplanted recipient mice showed that deletion of Hes1 marginally increased apoptosis of Hes1 f/f Vav1Cre HSCs, and that genetic or pharmacological inhibition of PPARγ by Pparg f/f deletion or antagonist (GW9662) treatment, respectively, did not have significant effect on apoptosis of Hes1 f/f Vav1Cre HSCs (Figure 5A). Similarly, FAO targeting by either deleting Cpt1a or in vivo administration of Etomoxir to the transplanted recipients did not show much effect on apoptosis of CD45.2+SLAM cells in the recipients of donor Hes1 f/f Vav1Cre cells (Figure 5B). Thus, apoptosis does not appear to be a causal factor in transplant stress‐induced HSC exhaustion in Hes1‐deficient HSCs.
Figure 5.
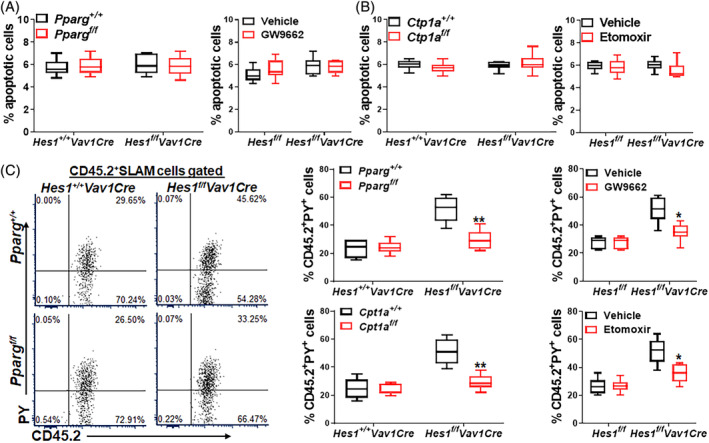
Inhibition of PPARγ or FAO improves quiescence of Hes1‐deficient HSCs. A, Effect of genetic or pharmacological inhibition of PPARγ on apoptosis. One million whole bone marrow cells (WBMCs) from Pparg f/f Hes1 f/f Vav1Cre mice or their Pparg +/+ Hes1 +/+ Vav1Cre littermates (left), or Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates (right), along with an equal number of BM cells from congenic BoyJ mice (CD45.1+), were transplanted into lethally irradiated congenic mice. Recipients transplanted with WBMCs from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates were then administered with PPARγ antagonist GW9662 daily for 8 days (day −1 to day 7). Apoptotic (Annexin V‐positive) donor‐derived HSCs (CD45.2+SLAM) cells were determined by flow cytometry (n = 6‐9). B, Effect of genetic or pharmacological inhibition of FAO on apoptosis. One million WBMCs from Cpt1a f/f Hes1 f/f Vav1Cre mice or their Cpt1a +/+ Hes1 +/+ Vav1Cre littermates (left), or Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates (right), along with an equal number of BM cells from congenic BoyJ mice (CD45.1+), were transplanted into lethally irradiated congenic mice. Recipients transplanted with WBMCs from Hes1 f/f Vav1Cre mice or their Hes1 f/f littermates were then administered with Etomoxir daily for 8 days (day −1 to day 7). Apoptotic (Annexin V‐positive) donor‐derived (CD45.2+) cells were determined by flow cytometry (n = 7‐9). C, Inhibition of PPARγ or FAO improves quiescence of Hes1‐deficient HSCs. WBMCs from recipients described in A and B were subjected to Pyronin Y staining. CD45.2+SLAM cells were gated for analysis. Representative lots (left) and quantification (right) are shown (n = 6‐9). *P < .05; **P < .01
Quiescence is an important feature of HSC homeostasis,56 and increased HSC cycling may lead to HSPC exhaustion.57 We next examined whether inhibition of PPARγ or FAO affected quiescence of Hes1‐deficient HSCs. Flow cytometry‐based cell cycle analysis revealed that deletion of Hes1 significantly decreased quiescence of Hes1 f/f Vav1Cre HSCs in the transplanted recipients, as marked significantly reduced Pyronin Y+CD45.2+SLAM cells (Figure 5C). Genetic and pharmacological inhibition of PPARγ by Pparg f/f deletion or antagonist (GW9662) treatment, respectively, improved quiescence as evidenced by significantly reduced Pyronin Y+CD45.2+SLAM cells in the recipients transplanted with Hes1 f/f Vav1Cre donor cells (Figure 5C, upper). Similarly, FAO targeting by Cpt1a deletion or Etomoxir treatment also significantly reduced Pyronin Y+CD45.2+SLAM cells in the recipients transplanted with Hes1 f/f Vav1Cre donor cells (Figure 5C, lower). These data indicate that compromised stem cell quiescence plays a major role in the observed exhaustion of Hes1‐deficient HSCs.
3.7. Hes1 deficiency upregulates genes involved in PPAR signaling and fatty acid metabolism pathways
To explore the molecular roles of HES1 in the homeostasis of HSCs and progenitor cells, we performed RNA sequence (RNA‐Seq) analysis using LSK cells isolated from Hes1 f/f Vav1Cre mice and their Hes1 f/f littermates. Inspection of the RNA‐Seq read distribution across specific genes confirmed the successful deletion of Hes1 in the Hes1 f/f Vav1Cre LSK cells (Figure S1B). Differentially expressed gene analysis revealed that Hes1 deletion upregulated 131 genes and repressed 22 genes in the Hes1 f/f Vav1Cre LSK cells (Figure 6A). Panther gene‐ontology enrichment analysis revealed a positive association of expression upregulation with biological processes such as fatty acid metabolism and hemopoiesis (Figure 6B). GSEA demonstrated that the upregulated genes were enriched for those related to negative regulators of HSC (Figure 6C).58 Consistent with the gene‐ontology enrichment analysis, the GSEA analysis also revealed that classic adipogenetic targets of Pparγ (Figure 6D) and genes from the KEGG PPAR signaling pathway (Figure 6E)59 were preferentially upregulated in the absence of Hes1, confirming that HES1 regulates HSC homeostasis possibly through suppression of PPARγ‐related metabolic pathways.
Figure 6.
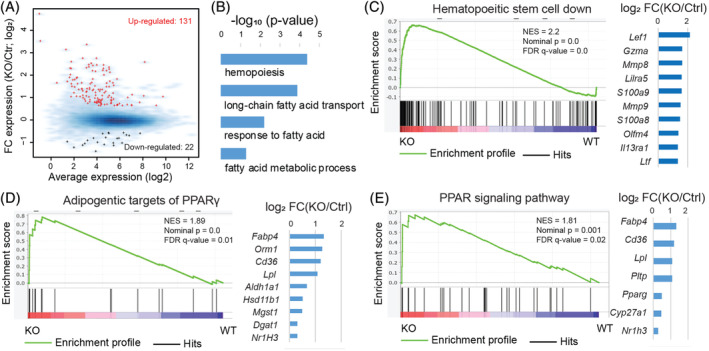
Hes1 deficiency alters the expression of genes involved in PPARγ signaling and fatty acid metabolism pathways. A, Scatter plot for average expression vs fold change of expression between Hes1 f/f Vav1Cre and Hes1 f/f LSK cells. Red: upregulated genes; black: downregulated genes; blue background: all expressed genes. B, Panther gene‐ontology enrichment analysis for biological processes related to fatty acid metabolism and hemopoiesis for the 131 upregulated genes. C, Gene set enrichment analysis (GSEA) of expressed genes, ranked by the FC of expression (Hes1 f/f Vav1Cre vs Hes1 f/f), against the MSigDB gene set “Jaatinen hematopoietic stem cell dn,” which includes genes downregulated in hematopoietic stem cells (HSC; CD133+) compared to CD133− cells. Right panel: fold changes of expression for the top 10 leading genes (out of 51). D, GSEA analysis against the MSigDB gene set “Wang classic adipogenic targets of PPARγ”, which includes classic adipogenic genes induced by PPARγ during adipogenesis in three T3‐L1 predipocytes. Right panel: fold changes of expression for all leading genes. E, GSEA analysis against the MSigDB gene set “KEGG PPAR signaling pathway.” Right panel: fold changes of expression for all leading genes. FDR, false discovery rate; NES, normalized enrichment score
4. DISCUSSION
The maintenance of HSCs depend on both intrinsic and extrinsic elements. Understanding the signaling pathways that govern the homeostasis of HSCs is fundamental to both normal and malignant hematopoiesis. Recent studies have identified the Notch pathway as a principal player in stem cells regulation and differentiation.60 As a Notch target, overexpression of HES1 increases HSC self‐renewal and reduces HSC cycling, thereby preserving long‐term reconstitution ability.13, 14, 15, 16 In this work, we have identified a novel role of HES1 in regulating stressed hematopoiesis through suppressing PPARγ and regulating fatty acid metabolism pathway. There are several findings that highlight the significance of our study: (a) Hes1 is dispensable for steady‐state hematopoiesis; (b) Hes1‐deficient HSCs undergo exhaustion under transplant stress; (c) Deletion of Hes1 deregulates PPARγ signaling and FAO; (d) Genetic and pharmacological inhibition of PPARγ or FAO improves hematopoietic repopulation of Hes1‐deficient HSCs; (e) PPARγ targeting or FAO inhibition ameliorates the repopulating defects of Hes1‐deficient HSCs through improving quiescence in HSCs; (f) Loss of Hes1 upregulates genes involved in PPARγ signaling and fatty acid metabolism pathways.
One interesting observation of our study is that Hes1 is dispensable for steady‐state hematopoiesis. Hes1 is known to play an essential role in the development of many organs by promoting the maintenance of stem/progenitor cells, by controlling the reversibility of cellular quiescence, and by regulating cell fate decision.1, 61, 62 Deletion of Hes1 in mice results in severe defects in multiple organs and is lethal in late embryogenesis.1, 9, 10 However, our hematopoietic lineage‐specific deletion of Hes1 in mice exhibited normal steady‐state hematopoiesis with comparable BM cellularity and HSC/progenitor pool to WT control animals, suggesting that HES1 is not required for HSC maintenance at steady state. These observations are consistent with previous report using an inducible Hes1 knockout strain (Hes1 f/f Mx1Cre),12 and lend support to the notion that HES1 is dispensable for steady‐state hematopoiesis.
However, one important and novel finding of our study is that Hes1‐deficient HSCs undergo exhaustion under replicative stress induced by transplantation. HSC exhaustion, defined as a progressive decline in the number of functional HSCs caused by enhanced cell cycling, is considered one cellular mechanism of bone marrow failure (BMF). Transplantation‐associated replicative stress can compromise the hematopoietic potential of HSCs. As a consequence, HSCs may undergo “exhaustion” in serial transplant recipients.63 Here we show that HSCs deficient for Hes1 exhibited a progressive decline in hematopoietic repopulating capacity in serial transplanted recipients (Figure 3). Thus, we propose that HES1 is a critical regulator that prevents HSCs from replicative stress‐induced exhaustion. Our data are somewhat in disagreement with a previous report using a different conditional Hes1 knockout strain crossed with the interferon‐inducible Mx1Cre mice, in which deletion of Hes1 did not affect self‐renewal and survival of HSCs after 5‐FU challenge.12 The discrepancy between this report and our study could be due to the utility of different Hes1 knockout models with distinct delete strains and stressors, as well as the relatively large amount of donor cells (2.5‐5 × 106 BM cells) used for competitive transplantation in Reference 12.
Peroxisome proliferator‐activated receptor (PPAR) isoforms, α, β/δ and γ constitute a family of transcription factors that are members of the nuclear hormone receptor gene super family and play fundamental roles in dietary fat storage and catabolism.21 We previously identified PPARγ as a putative negative regulator of HSCs using an in vivo RNAi screen system.22 More recent studies show that inhibition of PPARγ improves ex vivo expansion of human HSCs and progenitors.23 However, less is known about how PPARγ is regulated and the identity of its downstream targets in HSCs. Our gene profiling analysis (Figures 3 and 6) indicate that Hes1 represses some of the important genes involved in PPARγ signaling and fatty acid metabolism. Furthermore, our functional studies (Figures 4 and 5) demonstrate that Hes1 plays a crucial role in regulation of FAO in HSCs. In this context, our study identifies a novel role of HES1 in regulating cellular metabolism and suggests that targeting the HES1‐PPARγ‐FAO metabolic axis may have therapeutic potential for chronic stress‐related hematological diseases.
In summary, we have employed a hematopoietic specific Hes1 knockout mouse model to study the role of Hes1 in stress hematopoiesis. Our results identify a novel role of HES1 in HSC maintenance through regulating the PPARγ‐ FAO metabolic axis and provide a mechanistic insight into the function of HES1 in HSC homeostasis.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Z.M.: performed the research, analyzed the data; J.X., L.W., J.W., Q.L., F.A.C., M.H.H.M., X.L.: performed some of the research, assisted data analysis; G.H.: performed RNA‐Seq bioinformatics analysis; W.D.: designed the research, analyzed the data, wrote the paper.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
We thank Dr. Ryoichiro Kageyama (Kyoto University) for Hes1 f/f mice. W.D. is supported by NIH Tumor Microenvironment Center of Biomedical Excellence Award (P20GM121322), West Virginia University (WVU) Health Science Center (HSC) and School of Pharmacy (SOP) startup funds, a Leukemia Research Foundation (LRF) Award, and an American Cancer Society (ACS) Institutional Research Grant. The work was partially supported by National Institute of General Medical Sciences Grant 5U54GM104942‐04 to G.H. We would like to acknowledge the WVU Transgenic Animal Core Facility, Genomics Core Facility for support provided to help make this publication possible.
Ma Z, Xu J, Wu L, et al. Hes1 deficiency causes hematopoietic stem cell exhaustion. Stem Cells. 2020;38:756–768. 10.1002/stem.3169
Funding information National Institute of General Medical Sciences, Grant/Award Numbers: P20GM121322, 5U54GM104942‐04; American Cancer Society; Leukemia Research Foundation; West Virginia University; NIH Tumor Microenvironment Center of Biomedical Excellence Award, Grant/Award Number: P20GM121322
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from corresponding author upon reasonable request.
REFERENCES
- 1. Kageyama R, Ohtsuka T, Kobayashi T. The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development. 2007;134:1243‐1251. [DOI] [PubMed] [Google Scholar]
- 2. Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian notch. Nature. 1995;377:355‐358. [DOI] [PubMed] [Google Scholar]
- 3. Nishimura M, Isaka F, Ishibashi M, et al. Structure, chromosomal locus, and promoter of mouse Hes2 gene, a homologue of Drosophila hairy and enhancer of split. Genomics. 1998;49:69‐75. [DOI] [PubMed] [Google Scholar]
- 4. Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, Kageyama R. Hes1 and Hes5 as notch effectors in mammalian neuronal differentiation. EMBO J. 1999;18:2196‐2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kawamata S, Du C, Li K, Lavau C. Overexpression of the Notch target genes Hes in vivo induces lymphoid and myeloid alterations. Oncogene. 2002;21:3855‐3863. [DOI] [PubMed] [Google Scholar]
- 6. Castella P, Sawai S, Nakao K, Wagner JA, Caudy M. HES‐1 repression of differentiation and proliferation in PC12 cells: role for the helix 3‐helix 4 domain in transcription repression. Mol Cell Biol. 2000;20:6170‐6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takebayashi K, Sasai Y, Sakai Y, Watanabe T, Nakanishi S, Kageyama R. Structure, chromosomal locus, and promoter analysis of the gene encoding the mouse helix‐loop‐helix factor HES‐1: negative autoregulation through the multiple N box elements. J Biol Chem. 1994;269:5150‐5156. [PubMed] [Google Scholar]
- 8. Kageyama R, Ohtsuka T, Hatakeyama J, Ohsawa R. Roles of bHLH genes in neural stem cell differentiation. Exp Cell Res. 2005;306:343‐348. [DOI] [PubMed] [Google Scholar]
- 9. Tomita K, Hattori M, Nakamura E, Nakanishi S, Minato N, Kageyama R. The bHLH gene Hes1 is essential for expansion of early T cell precursors. Genes Dev. 1999;13:1203‐1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ishibashi M, Ang SL, Shiota K, Nakanishi S, Kageyama R, Guillemot F. Targeted disruption of mammalian hairy and enhancer of split homolog‐1 (HES‐1) leads to up‐regulation of neural helix‐loop‐helix factors premature neurogenesis, and severe neural tube defects. Genes Dev. 1995;9:3136‐3148. [DOI] [PubMed] [Google Scholar]
- 11. Espinosa L, Cathelin S, D'Altri T, et al. The Notch/Hes1 pathway sustains NF‐κB activation through CYLD repression in T cell leukemia. Cancer Cell. 2010;18:268‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wendorff AA, Koch U, Wunderlich FT, et al. Hes1 is a critical but context‐dependent mediator of canonical Notch signaling in lymphocyte development and transformation. Immunity. 2010;33:671‐684. [DOI] [PubMed] [Google Scholar]
- 13. Stier S, Cheng T, Dombkowski D, Carlesso N, Scadden DT. Notch1 activation increases hematopoietic stem cell self‐renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood. 2002;99:2369‐2378. [DOI] [PubMed] [Google Scholar]
- 14. Kunisato A, Chiba S, Nakagami‐Yamaguchi E, et al. HES‐1 preserves purified hematopoietic stem cells ex vivo and accumulates side population cells in vivo. Blood. 2003;101:1777‐1783. [DOI] [PubMed] [Google Scholar]
- 15. Varnum‐Finney B, Xu L, Brashem‐Stein C, et al. Pluripotent, cytokine‐dependent, hematopoietic stem cells are immortalized by constitutive Notch1 signaling. Nat Med. 2000;6:1278‐1281. [DOI] [PubMed] [Google Scholar]
- 16. Yu X, Alder JK, Chun JH, et al. HES1 inhibits cycling of hematopoietic progenitor cells via DNA binding. Stem Cells. 2006;24:876‐888. [DOI] [PubMed] [Google Scholar]
- 17. Karigane D, Takubo K. Metabolic regulation of hematopoietic and leukemic stem/progenitor cells under homeostatic and stress conditions. Int J Hematol. 2017;106(1):18‐26. [DOI] [PubMed] [Google Scholar]
- 18. Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9:298‐310. [DOI] [PubMed] [Google Scholar]
- 19. Warr MR, Pietras EM, Passegué E. Mechanisms controlling hematopoietic stem cell functions during normal hematopoiesis and hematological malignancies. Wiley Interdiscip Rev Syst Biol Med. 2011;3(6):681‐701. [DOI] [PubMed] [Google Scholar]
- 20. Baumann K. Stem cells: a metabolic switch. Nat Rev Mol Cell Biol. 2013;14(2):64‐65. [DOI] [PubMed] [Google Scholar]
- 21. Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405(6785):421‐424. [DOI] [PubMed] [Google Scholar]
- 22. Sertorio M, Du W, Amarachintha S, Wilson A, Pang Q. In Vivo RNAi screen unveils PPARγ as a regulator of hematopoietic stem cell homeostasis. Stem Cell Rep. 2017;8(5):1242‐1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo B, Huang X, Lee MR, Lee SA, Broxmeyer HE. Antagonism of PPAR‐γ signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nat Med. 2018;24(3):360‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Imayoshi I, Shimogori T, Ohtsuka T, Kageyama R. Hes genes and neurogenin regulate non‐neural versus neural fate specification in the dorsal telencephalic midline. Development. 2008;135(15):2531‐2541. [DOI] [PubMed] [Google Scholar]
- 25. Martin HL, Mounsey RB, Mustafa S, Sathe K, Teismann P. Pharmacological manipulation of peroxisome proliferator‐activated receptor γ (PPARγ) reveals a role for anti‐oxidant protection in a model of Parkinson's disease. Exp Neurol. 2012;235(2):528‐538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hossain F, Al‐Khami AA, Wyczechowska D, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid‐derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3(11):1236‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xavier‐Ferrucio J, Ricon L, Vieira K, et al. Hematopoietic defects in response to reduced Arhgap21. Stem Cell Res. 2018;26:17‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huynh FK, Green MF, Koves TR, Hirschey MD. Measurement of fatty acid oxidation rates in animal tissues and cell lines. Methods Enzymol. 2014;542:391‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liao Y, Smyth GK, Shi W. The subread aligner: fast, accurate and scalable read mapping by seed‐and‐vote. Nucleic Acids Res. 2013;41:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liao Y, Smyth GK, Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288‐4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mi H, Muruganujan A, Huang X, et al. Protocol update for large‐scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc. 2019;14:703‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li D, Hsu S, Purushotham D, Sears RL, Wang T. WashU epigenome browser update 2019. Nucleic Acids Res. 2019;47(W1):W158‐W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Daria D, Filippi MD, Knudsen ES, et al. The retinoblastoma tumor suppressor is a critical intrinsic regulator for hematopoietic stem and progenitor cells under stress. Blood. 2008;111:1894‐1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ghiaur G, Ferkowicz MJ, Milsom MD, et al. Rac1 is essential for intraembryonic hematopoiesis and for the initial seeding of fetal liver with definitive hematopoietic progenitor cells. Blood. 2008;111:3313‐3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sengupta A, Duran A, Ishikawa E, et al. Atypical protein kinase C (aPKCzeta and aPKClambda) is dispensable for mammalian hematopoietic stem cell activity and blood formation. Proc Natl Acad Sci U S A. 2011;108(24):9957‐9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109‐1121. [DOI] [PubMed] [Google Scholar]
- 39. Kobayashi M, Srour EF. Regulation of murine hematopoietic stem cell quiescence by Dmtf1. Blood. 2011;118(25):6562‐6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maryanovich M, Oberkovitz G, Niv H, et al. The ATM‐BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat Cell Biol. 2012;14(5):535‐541. [DOI] [PubMed] [Google Scholar]
- 41. Ito K, Hirao A, Arai F, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12(4):446‐451. [DOI] [PubMed] [Google Scholar]
- 42. Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1–2):70‐78. [DOI] [PubMed] [Google Scholar]
- 43. Du W, Amarachintha S, Wilson AF, Pang Q. Hyper‐active non‐homologous end joining selects for synthetic lethality resistant and pathological Fanconi anemia hematopoietic stem and progenitor cells. Sci Rep. 2016;6:22167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Herzig S, Hedrick S, Morantte I, Koo SH, Galimi F, Montminy M. CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR‐gamma. Nature. 2003;426(6963):190‐193. [DOI] [PubMed] [Google Scholar]
- 45. Maniati E, Bossard M, Cook N, et al. Crosstalk between the canonical NF‐κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J Clin Invest. 2011;121(12):4685‐4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Garin‐Shkolnik T, Rudich A, Hotamisligil GS, Rubinstein M. FABP4 attenuates PPARγ and adipogenesis and is inversely correlated with PPARγ in adipose tissues. FABP4 attenuates PPARγ and adipogenesis and is inversely correlated with PPARγ in adipose tissues. Diabetes. 2014;63(3):900‐911. [DOI] [PubMed] [Google Scholar]
- 47. Heinlein CA, Ting HJ, Yeh S, Chang C. Identification of ARA70 as a ligand‐enhanced coactivator for the peroxisome proliferator‐activated receptor gamma. J Biol Chem. 1999;274:16147‐16152. [DOI] [PubMed] [Google Scholar]
- 48. Meruvu S, Hugendubler L, Mueller E. Regulation of adipocyte differentiation by the zinc finger protein ZNF638. J Biol Chem. 2011;286:26516‐26523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith SA. Peroxisome proliferator‐activated receptors and the regulation of mammalian lipid metabolism. Biochem Soc Trans. 2002;30(Pt 6):1086‐1090. [DOI] [PubMed] [Google Scholar]
- 50. Ito K, Carracedo A, Weiss D, et al. A PML‐PPAR‐delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18(9):1350‐1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vatanavicharn N, Yamada K, Aoyama Y, et al. Carnitine‐acylcarnitine translocase deficiency: two neonatal cases with common splicing mutation and in vitro bezafibrate response. Brain Dev. 2015;37(7):698‐703. [DOI] [PubMed] [Google Scholar]
- 52. Miklas JW, Clark E, Levy S, et al. TFPa/HADHA is required for fatty acid beta‐oxidation and cardiolipin re‐modeling in human cardiomyocytes. Nat Comm. 2019;10(1):4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Briant LJB, Dodd MS, Chibalina MV, et al. CPT1a‐dependent long‐chain fatty acid oxidation contributes to maintaining glucagon secretion from pancreatic islets. Cell Rep. 2018;23(11):3300‐3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee J, Choi J, Scafidi S, Wolfgang MJ. Hepatic fatty acid oxidation restrains systemic catabolism during starvation. Cell Rep. 2016;16(1):201‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vickers AE. Characterization of hepatic mitochondrial injury induced by fatty acid oxidation inhibitors. Toxicol Pathol. 2009;37:78‐88. [DOI] [PubMed] [Google Scholar]
- 56. Guiu J, Shimizu R, D'Altri T, et al. Hes repressors are essential regulators of hematopoietic stem cell development downstream of Notch signaling. J Exp Med. 2013;210(1):71‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Du W, Amarachintha S, Erden O, et al. Fancb deficiency impairs hematopoietic stem cell function. Sci Rep. 2015;5:18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jaatinen T, Hemmoranta H, Hautaniemi S, et al. Global gene expression profile of human cord blood‐derived CD133+ cells. Stem Cells. 2006;24(3):631‐641. [DOI] [PubMed] [Google Scholar]
- 59. Wang H, Qiang L, Farmer SR. Identification of a domain within peroxisome proliferator‐activated receptor gamma regulating expression of a group of genes containing fibroblast growth factor 21 that are selectively repressed by SIRT1 in adipocytes. Mol Cell Biol. 2008;28(1):188‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kumano K, Chiba S, Kunisato A, et al. Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity. 2003;18:699‐711. [DOI] [PubMed] [Google Scholar]
- 61. Harris L, Guillemot F. HES1, two programs: promoting the quiescence and proliferation of adult neural stem cells. Genes Dev. 2019;33(9–10):479‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sang L, Coller HA, Roberts JM. Control of the reversibility of cellular quiescence by the transcriptional repressor HES1. Science. 2008;321(5892):1095‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu H, Yuan Y, Shen H, Cheng T. Hematopoietic stem cell exhaustion impacted by p18 INK4C and p21 Cip1/Waf1 in opposite manners. Blood. 2006;107(3):1200‐1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
The data that support the findings of this study are available from corresponding author upon reasonable request.