

Abstract
Studies have reported associations between environmental manganese (Mn) exposure and impaired cognition, attention, impulse control, and fine motor function in children. Our recent rodent studies established that elevated Mn exposure causes these impairments. Here, rats were exposed orally to 0, 25, or 50 mg Mn/kg/day during early postnatal life (PND 1–21) or lifelong to determine whether early life Mn exposure causes heightened behavioral reactivity in the open field, lasting changes in the catecholaminergic systems in the medial prefrontal cortex (mPFC), altered dendritic spine density, and whether lifelong exposure exacerbates these effects. We also assessed astrocyte reactivity (glial fibrillary acidic protein, GFAP), and astrocyte complement C3 and S100A10 protein levels as markers of A1 proinflammatory or A2 anti-inflammatory reactive astrocytes. Postnatal Mn exposure caused heightened behavioral reactivity during the first 5 – 10 minute intervals of daily open field test sessions, consistent with impairments in arousal regulation. Mn exposure reduced the evoked release of norepinephrine (NE) and caused decreased protein levels of tyrosine hydroxylase (TH), dopamine (DA) and NE transporters, and DA D1 receptors, along with increased DA D2 receptors. Mn also caused a lasting increase in reactive astrocytes (GFAP) exhibiting increased A1 and A2 phenotypes, with a greater induction of the A1 proinflammatory phenotype. These results demonstrate that early life Mn exposure causes broad lasting hypofunctioning of the mPFC catecholaminergic systems, consistent with the impaired arousal regulation, attention, impulse control, and fine motor function reported in these animals, suggesting that mPFC catecholaminergic dysfunction may underlie similar impairments reported in Mn-exposed children.
Keywords: manganese, dopamine, norepinephrine, developmental exposure, prefrontal cortex, neuroinflammation
Graphical Abstract
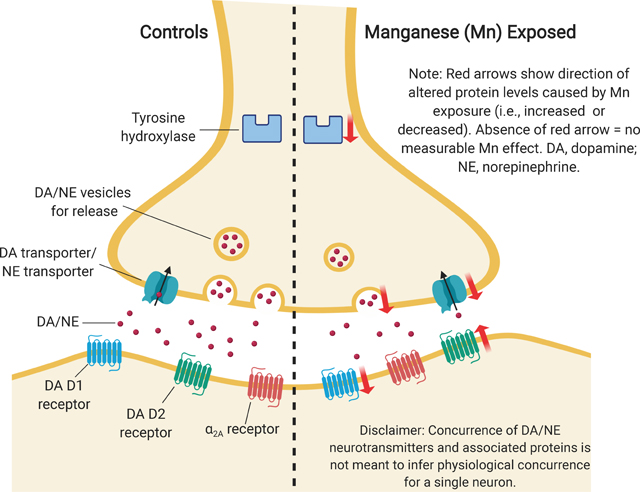
Manganese is an essential micronutrient, but elevated environmental exposures have been associated with executive function impairments in children. Here, we used our established rodent model to determine whether early postnatal or lifelong Mn exposure caused lasting changes in the catecholaminergic systems and altered astrocyte reactivity, along with other outcomes, in the prefrontal cortex that may underlie the neurological deficits reported in children. Mn exposure caused decreased protein levels of tyrosine hydroxylase (TH), dopamine (DA) and norepinephrine (NE) transporters, and DAD1 receptors, along with increased DAD2 receptors and astrocyte reactivity. This catecholaminergic hypofunctioning may underlie similar impairments in Mn-exposed children.
1.0. Introduction
Elevated environmental Mn exposure is emerging as a substantial public health problem in the U.S. and elsewhere, where vulnerable children may be exposed to elevated levels of Mn from drinking water (Wasserman et al. 2006; Bouchard et al. 2007; Bouchard et al. 2011; Ljung and Vahter 2007), soil and dust (Gunier et al. 2014a; Gunier et al. 2014b; Lucas et al. 2015; Lucchini et al. 2012), and dietary sources (Crinella 2003; Crinella 2012). Epidemiological studies have reported associations between environmental Mn exposure and impaired cognition, attention, impulse control, and fine motor function in children and adolescents (Oulhote et al. 2014; Ericson et al. 2007; Bouchard et al. 2011). Our recent rodent studies recapitulated these functional impairments in children, demonstrating that developmental Mn exposure causes highly specific and lasting impairments in selective and focused attention, impulse control, arousal regulation, and fine motor function (Beaudin et al. 2017a; Beaudin et al. 2013; Beaudin et al. 2015; Beaudin et al. 2017b). These impacts of Mn on attention and impulse control are particularly concerning, considering that impairments in these areas of functioning, including attention deficit hyperactivity disorder (ADHD), are among the most prevalent neurodevelopmental disorders in children (Willcutt 2012; Feldman and Reiff 2014; Xu et al. 2018).
The pattern of neurobehavioral dysfunction caused by developmental Mn exposure implicates disrupted function of the medial prefrontal cortex (mPFC), given that the mPFC plays an important role in mediating arousal regulation and executive function, including attentional function and cognitive flexibility, among others (Maddux and Holland 2011; Bissonette et al. 2008). The catecholaminergic systems within the mPFC has been shown to be a critical mediator for regulating executive function, in which the dopamine (DA) D1 and α2A adrenergic receptors are especially important (Arnsten and Dudley 2005; Arnsten and Pliszka 2011). However, the neurobiological mechanisms underlying these Mn impairments are poorly understood. Converging evidence suggests that altered catecholaminergic activity in the mesocorticolimbic circuit due to elevated Mn may play an important underlying role (Kern et al. 2010; Kern and Smith 2011; McDougall et al. 2008; Reichel et al. 2006), consistent with the important role of the catecholaminergic systems in regulating mPFC function (Arnsten and Pliszka 2011; Arnsten 2009a). Studies in mammals have shown that developmental Mn exposure reduces striatal DA release (Reichel et al. 2006; McDougall et al. 2008), as well as DA D1 receptor and DA transporter (DAT) protein levels in the striatum and nucleus accumbens (McDougall et al. 2008; Reichel et al. 2006; Kern et al. 2010; Kern and Smith 2011), but relatively few studies have examined the effects of developmental Mn exposure on the catecholaminergic systems in the mPFC. Limited available evidence has shown that developmental Mn exposure increases D2 receptor levels in the mPFC of weanling rodents that may last into adulthood (Kern et al. 2010; Kern and Smith 2011). Our recent studies have revealed lasting reductions in evoked DA and norepinephrine (NE) release in the mPFC in adulthood in animals exposed only during early postnatal development (Beaudin et al. 2015; Lasley et al. 2019), with lasting changes in PFC DA D2 protein levels in adults (Kern and Smith 2011). However, the extent to which developmental and lifelong Mn exposure impacts the mPFC catecholaminergic systems in adulthood is not well known.
Developmental Mn exposure has also been shown to cause neuroinflammation, manifesting in reactive astrocytes, increased expression of glial fibrillary acidic protein (GFAP), and increased release of proinflammatory cytokines (Moreno et al., 2009; Kern and Smith, 2011; Popichak et al., 2018). Astrocytes and microglia are important mediators of neuroinflammation (Prinz and Priller 2014; Liddelow and Barres 2017), and astrocytes in particular play an important role in the dynamic restructuring of synapses during neurodevelopment (Dallérac et al. 2018; Farhy-Tselnicker and Allen 2018). Recent evidence has demonstrated that environmental toxicants can induce a proinflammatory A1 phenotype in astrocytes and lead to altered synaptic function (Liddelow et al. 2017). However, little is known about whether developmental Mn exposure induces a proinflammatory A1 phenotype in mPFC astrocytes, and whether this effect is associated with Mn-induced changes in the mPFC catecholaminergic systems.
Here we used our rodent model of early childhood oral Mn exposure to systematically investigate whether Mn causes enduring disruption in the catecholaminergic system in the mPFC, using (1) quantitative protein immunohistochemistry measures of tyrosine hydroxylase (TH), DAT and NE transporter (NET), DA D1 and D2 receptors, and the α2A adrenergic receptor, (2) microdialysis for DA and NE release, and (3) quantification of dendritic spine density on pyramidal mPFC neurons. We also assessed whether changes in the mPFC catecholaminergic systems were associated with heightened behavioral reactivity in an open field behavioral paradigm. Finally, we assessed astrocyte reactivity based on protein levels of GFAP, complement C3, and S100A10, the latter two as markers of reactive astrocytes expressing an A1 proinflammatory or A2 anti-inflammatory phenotype, respectively. Given our recent findings that lifelong oral Mn exposure into adulthood did not worsen the enduring attentional and fine motor deficits of exposure restricted to early postnatal life (Beaudin et al. 2017a; Beaudin et al. 2017b), we also tested whether continued oral Mn exposure throughout postnatal life exacerbated the catecholaminergic systems and astrocyte reactivity effects of the early postnatal exposure.
2.0. Materials and Methods
2.1. Subjects
The data reported here arose from three different, but identically treated, cohorts of male Long-Evans rats. Cohorts were generated, treated, and assessed in sequential fashion. The open field data are generated from cohorts 1 and 2. The immunohistochemistry (IHC), Mn biomarker, and spine density data are from cohort 2 littermates to the behaviorally tested (open field) animals, whereas the neurochemistry data are from cohort 3 (Figure 1). All subjects were born in-house from nulliparous timed-pregnant Long Evans rats (obtained from Charles River on gestational day 18, RRID: RGD_2308852). Twelve to 24 hours after parturition (designated PND 1, birth = PND 0), litters were sexed, weighed, and culled to eight pups per litter such that each litter was composed of five to six males and the remainder females. Only one male per litter was assigned to a particular Mn treatment condition. Animals (dams and weaned pups) were fed Harlan Teklad rodent chow #2920 (reported by the manufacturer to contain 80 mg Mn/kg) and housed in polycarbonate cages at a constant temperature of 21 ± 2°C. At PND 22, all pups were weaned and pair-housed with an animal of the same Mn treatment group and maintained on a reversed 10:14 hr light/dark cycle. All aspects of behavioral testing and feeding were carried out during the active (dark) phase of the animals’ diurnal cycle. Males were exclusively used because attentional dysfunction is two to three times more prevalent in boys than girls (Feldman and Reiff 2014; Willcutt 2012), and because our prior studies have established that early postnatal Mn exposure causes lasting impairments in learning, attention, and fine motor function in male rats (Kern et al. 2010; Beaudin et al. 2017a; Beaudin et al. 2017b; Beaudin et al. 2015; Beaudin et al. 2013). All animal procedures were approved by the institutional IACUC (protocols Smitd0912 and 234193) and adhered to National Institutes of Health guidelines set forth in the Guide for the Care and Use of Laboratory Animals. Criteria for exclusion of animals from the study were based on overt signs of poor animal health, including loss of body weight, absence of grooming, impaired function, and death; no animals were excluded from the study based on these criteria. This study was not pre-registered.
Figure 1. Time-line of experimental procedures and outcome measures, indicating timing of early life (PND 1–21, top red bar), and lifelong (PND 1 – end of study for particular outcome) Mn exposure relative to the outcome measures.

Sample sizes per experimental procedure were: open field, n = 21–23 animals/treatment group; tissue Mn concentrations, n = 5–12 animals/group and timepoint; microdialysis of DA, NE, n = 8–12 animals/group; quantitative IHC of catecholamine systems and astrocyte proteins, n = 5–6 animals/group; spine density, n = 5–10 animals/group. No animals were excluded from the study based on exclusion criteria of poor health (see Materials and Methods).
2.2. Manganese exposure protocol
Neonatal rats were orally exposed to Mn doses of 0, 25, or 50 mg Mn/kg/day starting on PND 1 through weaning on PND 21 (early postnatal Mn exposure), or throughout life until PND 100 (Figure 1). The early window of postnatal Mn exposure in rats corresponds to important frontal-cortical-striatal developmental events, including formation of DA and NE projections to the PFC, that are comparable to the gestational third trimester through adolescence in humans, whereas the continued lifelong exposure window additionally coincides with late adolescence to early adulthood in humans (Workman et al. 2013; Clancy et al. 2007; Nagarajan and Jonkman 2013; Posner and Rothbart 1998; Ruff and Rothbart 2010). For dosing over PND 1–21, Mn was delivered once daily directly into the mouth of each pup (~20 μL/dose) via a micropipette fitted with a flexible polyethylene pipet tip (Fisher Scientific, Santa Clara, CA, USA). Control animals received the vehicle solution. For this, a 225 mg Mn/mL stock solution of MnCl2 was prepared by dissolving MnCl2·4H2O with Milli-Q™ water; aliquots of the stock solution were diluted with a 2.5% (wt/vol) solution of the natural sweetener stevia to facilitate oral dosing of the pups. Oral Mn exposure post-weaning (PND 22 – end of study) occurred via the animals’ drinking water. For this, a 42 mg Mn/mL stock Mn solution was prepared fresh weekly as above and diluted with tap water to a final concentration of 420 μg Mn/mL in a polycarbonate carboy. The stock solutions were made fresh weekly, and water bottles were refilled with fresh water two to three-times per week. Water bottle weights were recorded at refilling to determine water intake per cage, and daily Mn intake per kg body weight was estimated based on daily measured body weights of the two rats housed per cage. Drinking water Mn concentrations were adjusted weekly as needed to maintain target daily oral Mn intake levels of 25 or 50 mg/kg/day based on measured water consumption. This Mn exposure regimen is relevant to children exposed to elevated Mn via drinking water, diet, or both; pre-weaning exposure to 50 mg Mn/kg/day produces a relative increase in Mn intake that approximates the increase reported in infants and young children exposed to Mn-contaminated water or soy-based formulas (Beaudin et al. 2017a; Beaudin et al. 2015; Beaudin et al. 2013; Kern et al. 2010; Kern and Smith 2011). For lifelong Mn exposure groups, oral exposure to the same daily Mn dose was maintained after weaning via drinking water to model the situation where children may continue to suffer chronic elevated Mn exposures from a variety of environmental sources (e.g., contaminated well water, dust, etc.) (Bouchard et al. 2011; Lucas et al. 2015; Oulhote et al. 2014).
2.3. Open field testing
The animals’ spontaneous exploration in a novel open field environment was tested 30 minutes each day for 5 consecutive days beginning on PND 24 (n = 21–23 animals/treatment group, 111 animals total). This age of testing corresponds to a critical developmental period of the mesocortical system involved in mediating open field locomotor activity in rodents (Kalsbeek et al. 1989). The open field apparatus consisted of enclosed arenas made of white opaque polypropylene plastic and measuring 60 cm x 60 cm x 30 cm. Each arena was featureless and placed in a darkened testing room. A ceiling-mounted digital video camera recorded the locomotor activity of individual rats under infrared light. For each daily session the animals were gently lowered and placed in the same corner of the arena facing the walls. Digital video recordings were analyzed using an automated video tracking system (SMART System, San Diego Instruments, San Diego USA). Individual activity tracks were analyzed for two-dimensional distance and time traveling with the SMART software. Open field testing was carried out at the same time of day, in the same open arena apparatus, and by the same experimenter each day for each rat. Experimenters were blinded to the animals’ Mn treatment condition and groups of animals tested by each experimenter were balanced by treatment.
2.4. Microdialysis measurement of extracellular brain DA and NE
Brain extracellular DA, NE, and DA metabolites were measured in the PFC of a separate cohort of PND 29 – 35 male Long-Evans rats by microdialysis (n = 8–12 animals/treatment group, 30 animals total). The average age of testing for each treatment group was 30.2 ± 1.1 days for control animals (mean ± SD), and 31.5 ± 1.5 days and 31.6 ± 1.9 days for the 25 and 50 mg Mn/kg/day groups, respectively. Animals were orally exposed to 0, 25, or 50 mg Mn/kg/day over PND 1 – 21 as described above. The 30 test subjects were obtained from 11 litters, with only one animal per litter assigned to a particular Mn treatment group
Intracerebral dialysis.
Rats were anesthetized with ketamine (80 mg/kg, i.p.) and xylazine (2 mg/kg, intraperitoneal injection, i.p.) supplemented with 2% (vol/vol) isoflurane in O2, mounted in a stereotaxic frame with flat skull surface, and plastic guide cannulae implanted into the right mPFC (from bregma, AP +2.5 mm, ML +1.4 mm, and DV −2.0 mm to the skull surface at a 12° angle to the vertical plane, lateral to medial approach; Paxinos and Watson, 1998). Cannulae were secured with dental acrylic and machine screws. Meloxicam (2 mg/kg, subcutaneous injection) was administered for post-operative pain. All anesthesia protocols were based on established standards for rodent surgery and were IACUC approved.
CMA12 dialysis probes (CMA Microdialysis, Kista, Sweden) with 3 mm active lengths of polyarylethersulfone membrane (concentric tube design, OD = 0.5 mm, MW cutoff = 20 kDa) were inserted into each cannula 3–4 days later, and the awake animal immediately placed into a plexiglas chamber which allowed freedom of movement. This probe location resulted in an area of dialyzed tissue that essentially comprised the dorsal to ventral extent of the mPFC region. The probe inlet was connected by fluorinated ethylene propylene (FEP) tubing to a syringe pump through a liquid switch and dual channel quartz-lined liquid swivel (Instech Labs, Plymouth Meeting, Pennsylvania). The probe outlet was connected to the swivel by the same tubing and to a collection vial in a fraction collector maintained at 4°C (CMA170, CMA Microdialysis). Between test sessions the dialysis system was flushed extensively with high purity water.
Microdialysis experimental design.
A modified Ringer’s solution (in mM: Na+ 145, K+ 4.0, Ca2+ 1.3, Cl− 152) was perfused through the probes during baseline sample collection. Two and one-half hours after probe insertion baseline extracellular fluid concentrations of analytes were assessed by two 40-min collections prior to switching for 100 min to modified Ringer’s with 120 mM K+ (K+ replaced Na+ to maintain isotonicity). Flow was maintained at 2.0 μL/min, and sample fractions were collected in tubes containing 5 μL of 0.1 M HCl and stored at −20°C until analysis. Desipramine (100 μM) was added to the perfusion medium in test sessions to inhibit reuptake of NE and DA and produce measurable amounts of the transmitters. Under these flow conditions, extraction efficiency of the perfusate from the dialysis probe is approximately 15%, indicating that the maximal extracellular fluid K+ concentration produced in vivo is in the range of 18 mM. Employing a reuptake inhibitor in the perfusion medium in order to elicit a quantifiable effect of K+ stimulation on extracellular NE/DA in PFC has been utilized by others (e.g., Bymaster et al. 2002; Higashino et al. 2014). During the period of elevated extracellular fluid transmitter concentrations, 40-min sample collections were made followed by one additional 40-min collection for the return to baseline
Catecholamine analysis.
For determination of NE, DA, and their metabolites (3, 4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA)), collected samples were loaded into an autosampler maintained at 4°C (Waters 717 Plus, Waters Chromatography Division, Milford, Massachusetts) and analyzed by isocratic liquid chromatography with electrochemical detection (LC-4C Amperometric Detector, BASi, West Lafayette, Indiana) at an oxidation potential of +700mV. Flow rate was 1.6 mL/min. The mobile phase consisted of 0.15 M monochloroacetate, pH 2.95, containing 1.3% acetonitrile (vol/vol), 1.7% tetrahydrofuran (vol/vol) (including 250 ppm butylated hydroxytoluene as an inhibitor), 0.86 mM sodium octylsulfate, and 0.18 mM EDTA, and was pumped through a 250 × 4.6mm, 5 μm Biophase ODS analytical column (PerkinElmer, Waltham, Massachusetts). Chromatographic peaks were quantified with EZChrom Elite software (Agilent Technologies, Pleasanton, California).
2.5. Quantitative immunohistochemistry
Immunostaining.
For immunohistochemical analyses, male littermates of the behaviorally tested animals were exposed to Mn as described above and sacrificed at PND 100 (n = 5–6/animals treatment group, 30 animals total). At sacrifice, animals were deeply anesthetized with sodium pentobarbital and perfused intracardially with ice cold 0.9% (wt/vol) saline, followed by perfusion with ice cold 4% (wt/vol) paraformaldehyde (PFA). Whole brains were extracted, bisected into hemispheres, and cryopreserved as described elsewhere (Kern and Smith 2011). The PFA-fixed right hemisphere was sectioned (Leica CM3050 S) into 20 μm coronal sections at −20°C in preparation for protein immunostaining for TH, DAT and NET, DA D1 and D2 receptors, adrenergic α2A receptor, GFAP, and complement 3 (C3) and S100A10; the latter two were used as protein markers of reactive A1 or A2 astrocyte phenotype (Liddelow et al. 2017; Liddelow and Barres 2017). Brain sections were stored in 30% (wt/vol) sucrose in 0.01 M phosphate buffered saline (PBS) cryoprotectant solution at −20°C until immunostaining. From each brain, 2–3 sections within the mPFC were selected for staining, ranging from bregma AP +2.16–1.56 mm (Paxinos and Watson 2006). Prior to immunostaining, brain sections underwent antigen retrieval for 15 minutes in a 10 mM sodium citrate buffer solution composed of sodium citrate dihydrate in MilliQ water, and heated in a hot water bath at 80 °C. Following antigen retrieval, sections were washed three times in 0.01 M PBS for 10 min each.
Brain sections for the catecholaminergic and GFAP proteins were double-stained for two of the proteins per section, while labeling of A1 and A2 astrocyte phenotypes used a triple stain for C3, S100A10, and GFAP. For staining, brain sections were free-floated in a blocking solution containing 1% (vol/vol) normal donkey serum (Jackson Immunoresearch), 0.3% (vol/vol) Triton X-100 (Sigma Aldrich), and 1% (vol/vol) bovine serum albumin (Sigma Aldrich), in 0.01 M PBS for one hour. Primary antibody incubation was overnight at 4 °C using the following primary antibodies and dilutions in 0.5% (vol/vol) Triton X-100 in 0.01 M PBS: sheep polyclonal anti-TH, 1:1000 (Pel Freez Biologicals, P60101, RRID: AB_461070); rabbit polyclonal anti-DAT, 1:500 (EMD Millipore, AB1591P); mouse monoclonal anti-NET, 1:500 (MAB Technologies, NET05–2; RRID: AB_2571639); rabbit polyclonal anti-D1 receptor, 1:50 (Alomone Labs, ADR-001, RRID: AB_2039826); rabbit polyclonal anti-D2 receptor, 1:250 (EMD Millipore, AB5084P); goat polyclonal anti-α2A receptor, 1:200 (Santa Cruz Biotech, sc-1478); mouse monoclonal anti-GFAP, 1:1000 (EMD Millipore, MAB360); rabbit polyclonal anti-C3, 1:120 (MyBioSource, MBS2005172); chicken polyclonal anti-S100A10, 1:50 (Abcam, ab50737, RRID: AB_881813). Next, brain sections were washed three times and incubated in a secondary antibody solution composed of 10% (vol/vol) normal donkey serum and corresponding secondary antibodies in 0.5% (vol/vol) Triton X-100 in 0.01 M PBS for 2 hours at room temperature. Secondary antibodies were: donkey anti-sheep Alexa Fluor 488, 1:1000 (Abcam, ab150177, RRID: AB_2801320); donkey anti-rabbit Alexa Fluor 488, 1:1000 (Molecular Probes, ab150073); donkey anti-mouse Alexa Fluor 594, 1:1000 (ThermoFisher, A-21203, RRID: AB_2535789); donkey anti-rabbit Alexa Fluor 594, 1:1000 (ThermoFisher, A-21207, RRID: AB_141637); donkey anti-goat Alexa Fluor 594, 1:1000 (Abcam, 150132); donkey anti-chicken CF-350 (Millipore Sigma, SAB4600219). Brain sections were then washed three more times in 0.01 M PBS and incubated for 10 min in a 1:1000 DAPI stain in 0.01 M PBS to label cell nuclei. Finally, sections were washed three times in 0.01 M PBS and mounted on slides and coverslipped with Fluoromount G mounting media (Southern Biotech) in preparation for fluorescence microscopy. Prior to imaging, 5–6 sections were mounted per slide, balanced by Mn treatment condition.
Fluorescence microscopy and image quantification.
Images were collected within the mPFC at 40x magnification for catecholaminergic proteins and GFAP, while C3 and S100A10 images were collected at 63x. To avoid bias in image acquisition, a box was drawn in both subregions of interest using the Zeiss ZEN imaging software, and a pseudorandomization tool determined three non-overlapping fields of view for collection. This yielded a total of six images per brain section and 12–18 images per animal across proteins, with the exception of C3 and S100A10, for which only four images per section were collected. All images per protein were captured under identical microscopy imaging settings, including exposure time and gain, using a Zeiss AxioImager microscope. Each image was collected in a z-stack format at 0.5 μm intervals between each z-focal plane over a total imaging range of 13 μm in the 20 μm brain slice. Following image collection, the number of z-plane images were reduced to 20 (10 μm z-plane distance) to remove out-of-focus z-planes, and then deconvolved using AutoQuant X3 software (version 3.1). Deconvolved images were imported into Imaris (software version 9.2) for fluorescence intensity quantification.
Fluorescence quantification was performed using the Imaris “Surfaces” tool, with unique quantification algorithms applied to each fluorescence channel and protein. Algorithms were customized to each protein, first by applying automated thresholds for absolute fluorescence intensity to determine whether quantified surfaces matched the amount of fluorescent objects in the image based on visual inspection. If the automated thresholding appeared incongruous with the staining pattern, the “Background Subtraction” tool was used to improve specificity of the algorithm. The primary quantification outcomes used for analysis were the sum total fluorescence per three dimensional object summed across all objects per image, the total number of objects per image, and the total volume of all objects per image.
For astrocyte A1 and A2 phenotype analysis, a colocalization algorithm was developed in Imaris using GFAP as an identifier for reactive astrocytes, and then fluorescence intensity of GFAP-colocalized C3 (A1 phenotype) and S100A10 (A2 phenotype)-positive astrocytes was determined. For this, imaging channels for C3 and S100A10 were separately colocalized with GFAP, first by generating a GFAP surface algorithm, followed by a “Spots” algorithm for the more punctate staining patterns of C3 and S100A10 proteins. A “Distance to Surfaces” transformation tool was used to separately isolate C3 and S100A10 objects that were less than 0.2 μm from a GFAP surface object to include for quantification. Primary outcomes for quantification data were total fluorescence per image of all GFAP-colocalized C3 and S100A10 objects, respectively. In all cases of image acquisition and quantification, the experimenter was blinded to the treatment condition.
2.6. Dendritic spine density analysis
Spine density on PFC layer III pyramidal cell dendrites was quantified in separate cohorts of PND 24 or PND 145 animals treated with 0 or 50 mg Mn/kg/day over PND 1 – 21 (n = 10 cells/animal, 5–10 animals/treatment group and age, 28 animals total), as described previously (Beaudin et al. 2015). Briefly, rats were deeply anesthetized with sodium pentobarbital and perfused intracardially with 0.9% (wt/vol) saline. The brains were extracted, rinsed with Milli-Q water, and then prepared for Golgi–Cox staining using the rapid Golgi stain kit (FD Neuroethologies, Inc., Ellicott City, MD). For this, the brains were placed in a Golgi–Cox solution provided by the manufacturer and stored at room temperature in the dark for 14 days followed by 3 days in a 30% sucrose solution (wt/vol). Brains were cut into 250 μm coronal sections using a vibratome, mounted on gelatin-coated slides, and allowed to dry naturally at room temperature before staining within the next 12–24 hours. To be included in the analysis of spine density, the dendritic branch of a given neuron had to be well-impregnated and free of stain precipitations, blood vessels, and astrocytes. The mPFC brain region of interest was identified at 10x magnification using a Leica DM5500B widefield microscope fitted with a motorized stage and multi-point image acquisition. In the mPFC, five layer III pyramidal cells were selected from each hemisphere in the cortical areas (from bregma, AP + 2.16–1.56 mm, ML +0.2–1.5 mm, DV +0.9–3.6 mm; n =10 cells total) (Paxinos and Watson 2006). For the PND 24 animals, dendritic spines were counted live at 100x magnification from a ~30–50 μm segment of a single basal dendrite, and individual second-order and terminal tip (third-order) apical dendrites were defined from each pyramidal cell, with the condition that the entire dendritic segment was within the focal plane of the microscope. Similarly, second-order apical dendrites on 10 mPFC layer III pyramidal cells were counted in the PND 145 animals. The exact length of counted dendrite was determined using the length measurement tool function of the microscope. Spine density was calculated as the number of spines per 10 μm of dendrite length. All neuronal cell selection and spine counting were done by individuals blind to the treatment conditions of the rats.
2.7. Blood and brain Mn levels, and blood hematocrit
Blood and brain Mn concentrations were determined in littermates of the immunostaining study animals that were retained specifically for tissue Mn analyses (PND 24 and PND 66) or that were sacrificed following behavioral testing and evoked neurotransmitter measurement by microdialysis (~PND 590; n = 6 – 12/treatment group and time point, 104 animals total), as reported in Beaudin et al. (2017a; 2015; 2013) and Lasley et al. (2019). Animals were heavily anesthetized with sodium pentobarbital overdose (75 mg/kg intraperitoneal injection), and whole blood (2 – 3 mL) was collected from the left ventricle of the surgically-exposed heart and stored in EDTA vacutainers at −20 °C for analyses. Whole brain was immediately removed, bisected into hemispheres, and hind-brain regions of each hemisphere were collected and stored at −80 °C for Mn concentration determinations (forebrain was dedicated to other outcome measures). Aliquots of whole blood were digested overnight at room temperature with 16 M HNO3 (Optima grade, Fisher Scientific), followed by addition of H2O2 and Milli-Q water. Digestates were centrifuged (13,000 x g for 15 min.) and the supernatant collected for Mn analysis. For brain, aliquots of homogenized hind-brain tissue (~200 mg wet weight) were dried and digested with hot 16 M HNO3, evaporated and redissolved in 1 M HNO3 for analyses. Rhodium was added to sample aliquots as an internal standard. Manganese levels were determined using a Thermo Element XR inductively coupled plasma – mass spectrometer, measuring masses 55Mn and 103Rh (the latter for internal standardization). External standardization for Mn used certified SPEX standards (Spex Industries, Inc., Edison, NJ). National Institutes of Standards and Technology SRM 1577b (bovine liver) was used to evaluate procedural accuracy. The analytical detection limit for Mn in blood and brain was 0.04 and 0.015 ng/mL, respectively. Finally, whole blood hematocrit was measured at PND 24 and 66 in cohorts 1 and 2 to assess whether Mn exposure overtly impacted body iron status.
2.8. Statistical analyses
The open field activity data were modeled by way of structured covariance mixed models. Fixed effects included in the model were Mn treatment as a between-subjects factor (five levels corresponding to the five treatment groups), and test day (five levels corresponding to the 5 days of testing) and test interval (six levels corresponding to the six 5-minute intervals per 30 minute test session) as within-subjects factors. In all models, animal was included as a random effect to account for correlations within observations from the same animal. Statistical tests used a Satterthwaite correction. Plots of residuals by experimental condition were used to examine the assumption of homogeneity of variance. The distribution of the random effect was inspected for approximate normality and presence of influential outliers. Significant main effects or interaction effects were followed by single-degree of freedom contrasts in order to clarify the nature of the interactions, using the Student’s t-test for pairwise comparisons of least squared means. Immunohistochemistry data were analyzed using a standard least squares mixed model analysis of variance that included Mn treatment group (five levels) as the between subjects factor and animal as a random effect. Pair-wise treatment group comparisons were performed using Tukey’s post hoc test. Tukey outlier box plots were used to identify possible outliers, and frequency distribution plots were used to assess data normality. The DA and NE microdialysis data were analyzed by ANOVA for each neurotransmitter with Mn exposure group as the between subjects factor (three levels) and time after K+ stimulation as the within subjects factor. Concentrations determined in individual animals were averaged at each baseline time point, then collapsed across time to yield a single group baseline value. Significant main effects or Mn x time interactions after K+ initiation for neurotransmitter concentrations were followed by Tukey’s post hoc test to make pair-wise comparisons between individual exposure groups. The ROUT and Grubb’s test were used to identify outliers in the microdialysis data, and the most conservative outcome was accepted. Spine density data were analyzed similarly, with Mn treatment (two levels) as the main effect and animal as a random effect; data for the three dendrite locations (i.e., second-order, terminal tip, basal) and two ages of animals were analyzed separately. Data for blood and brain Mn levels were analyzed using the Wilcoxon/Kruskal-Wallis test. Data were log transformed before analysis if necessary to achieve normal distribution and homogeneity of variance. In all cases, the significance level was set at p ≤ 0.05. Power analyses were applied to determine the sample size for open field behavioral outcomes (n = 21–23 rats in each of the five treatment groups), based on the ability to detect small differences in group behavioral performance (Beaudin et al. 2013; Beaudin et al. 2015; Beaudin et al. 2017a; Beaudin et al. 2017b; Kern et al. 2010; Kern and Smith 2011). For the IHC (n = 5–6 animals/treatment), Mn biomarker (n = 5–12 animals/treatment group), and spine density (n = 5–10 animals/treatment group) outcomes, the group sizes were based on power analyses indicating the number of animals per group needed to statistically detect differences between treatments, taking into account plausible effect sizes and group variances reported in our published studies (Kern and Smith 2011; Beaudin et al. 2013; Beaudin et al. 2015; Beaudin et al. 2017a; Beaudin et al. 2017b). Analyses were conducted using SAS (version 9.4) for Windows on a mainframe computer (open field data), GraphPad Prism v.6.04 for Windows (GraphPad Software, San Diego, CA, microdialysis data), or JMP (version 13.0; SAS Institute, Inc. for all other data).
3.0. Results
Overall, early postnatal Mn exposure led to increased behavioral reactivity in the novel open field environment, and produced lasting changes in the catecholaminergic systems of the mPFC. Specifically, in the open field, Mn exposure increased total distance traveled, but only in the initial minutes of each daily test session. With respect to the mPFC catecholaminergic systems, Mn exposure produced lasting alterations in the synaptic proteins TH, D1, D2, DAT, and NET, and reductions in evoked outflow of extracellular NE. In contrast, protein levels of the α2A adrenergic receptor were unchanged by Mn. These catecholaminergic systems changes were accompanied by a lasting increase in astrocyte GFAP protein levels, and a predominantly proinflammatory A1 astrocyte phenotype, but no measurable change in mPFC layer III pyramidal cell dendritic spine density was observed. These findings are detailed below.
3.1. Early postnatal Mn exposure caused increased behavioral reactivity in the open field
To determine the effect of early and lifelong postnatal Mn exposure on behavioral reactivity and habituation to a novel environment, the distance traveled in an open field was measured in daily 30 minute test sessions over 5 consecutive days, starting on PND 24. The main effect of Mn exposure was not significant [F(4, 102) = 1.13, p = 0.34], nor was the interaction of Mn exposure x test session day [F(16, 776) = 1.27, p = 0.21]. The three-way interaction of Mn x test session day x within-session time interval [F(80, 2724) = 0.82, p = 0.86] was also not significant. However, the effects of test session day [F(4, 776) = 12.36, p < 0.0001] and within-session time interval [F(5, 975) = 988.13, p < 0.0001] were significant, as was the interaction of Mn exposure x within-session time interval [F(20, 974) = 2.03, p = 0.0047] (Figure 2). Specifically, early postnatal Mn exposure over PND 1 – 21 increased the total distance travelled within the first 5-minute interval of the daily 30-minute testing session across the 5 days of testing in both the early 25 (p = 0.0042) and early 50 Mn groups (p = 0.0022), relative to controls (Figure 2a). Lifelong Mn exposure (PND 1 – 29) also caused a significant increase in total distance travelled over the first two 5-minute intervals for the lifelong 25 (p = 0.038) and lifelong 50 Mn (p = 0.041) groups, as well as a trending increase for both treatment groups during the third within-session interval (p = 0.078 and 0.076 for the lifelong 25 and 50 groups, respectively), relative to controls (Figure 2b). A direct comparison of the lifelong and early life exposure groups showed that the longer exposure did not exacerbate the Mn effect (contrasts between the lifelong vs. early life Mn groups for each within-session time interval yielded p’s > 0.3 and p’s > 0.18 for the 25 and 50 Mn groups, respectively). This latter result is not surprising, given that the lifelong Mn groups received only one additional week of exposure versus the early life Mn groups during open field testing over PND 24 – 29.
Figure 2. Early postnatal Mn exposure caused increased behavioral reactivity in the open field.

Total distance (cm) for the early (a) and lifelong (b) postnatal Mn exposure groups as a function of time in the open field, shown in 5 minute within-session intervals in a daily 30-min open field test sessions conducted over 5 consecutive days; data are collapsed across the 5 daily test sessions because there was no main effect or interaction involving test session day. * and ** indicate p < 0.05 and p < 0.01 versus controls, respectively. † indicates 0.05 ≤ p < 0.1 versus controls. Data are least squares means ± SEM (n = 21–23 animals/group).
3.2. Early postnatal Mn exposure caused dose-dependent reductions in evoked NE outflow in the mPFC
In order to determine the effect of early postnatal Mn exposure on catecholaminergic systems function and to elucidate the alterations that may underlie the Mn-induced impairments in attention, impulse control, and arousal regulation reported previously (Beaudin et al. 2013; Beaudin et al. 2015; Beaudin et al. 2017a; Beaudin et al. 2017b), we assessed the impact of early postnatal Mn exposure over PND 1 – 21 on the evoked release of DA and NE in the mPFC of PND 29–35 animals. For extracellular NE there was a significant main effect of exposure [F(2, 27) = 5.10, p = 0.0133], as well as a significant Mn exposure x time interaction [F(14, 189) = 1.91, p = 0.028]. These effects were observed as statistically significant decreases from control values at the 40 minute time point after initiation of K+ stimulation in both Mn groups (p < 0.01 and p < 0.0001 for the early 25 and 50 Mn groups, respectively; Figure 3a). Moreover, NE in the early 50 Mn group at this time point was found to be significantly less than the early 25 Mn group (p < 0.05), establishing a dose dependence. Analogous effects of early life Mn exposure on extracellular DA were not apparent – neither a main effect of Mn exposure [F(2, 27) = 1.76, p = 0.191], nor an exposure x time interaction [F(14, 189) = 1.37, p = 0.173] were present, even though a dose-dependent ordering of NE responses was noted (Figure 3b). We also measured the evoked release of the neurotransmitter metabolites, DOPAC and HVA, to assess whether Mn exposure altered DA and NE clearance. For DOPAC, there was no significant main effect of Mn exposure [F(2, 27) = 1.16, p = 0.327], or an exposure x time interaction [F(14, 189) = 1.11, p = 0.353] (Supplementary Figure S1a). Similarly for HVA, there was no main effect of Mn exposure [F(2, 27) = 0.0369, p = 0.964], or an exposure x time interaction [F(14, 189) = 0.956, p = 0.500] (Supplementary Figure S1b).
Figure 3. Mn exposure caused a significant reduction in the stimulated release of NE in the mPFC of young adolescent animals.

Concentrations of (a) NE and (b) DA in nM as a function of time after administration of a high K+ stimulus. Statistical analysis reflects differences in NE at 40 min after initiation of high K+ perfusion based on Tukey’s multiple comparison test. ** and *** indicate p < 0.01 and p < 0.0001 versus controls, respectively. + indicates p < 0.05 vs. the early 25 Mn dose. Data are means ± SEM (n = 8–12 animals/group).
Additionally, we measured the basal concentrations of catecholamine neurotransmitters and their metabolites to further determine the impacts of Mn exposure prior to the introduction of the high K+ stimulus. There was a trending, but non-significant, main effect of Mn on baseline NE [F(2, 27) = 2.892, p = 0.073], and no main effect of Mn on DA [F(2, 27) = 1.468, p = 0.248], DOPAC [F(2, 27) = 0.111, p = 0.895], or HVA [F(2, 27) = 0.408, p = 0.669] concentrations (Supplementary Table 1).
3.3. Quantitative Immunohistochemistry of PFC Catecholaminergic Synaptic Proteins
To further determine the impact of early and lifelong postnatal Mn exposure on the neuronal synaptic environment, we measured protein levels of the catecholaminergic systems proteins TH, DAT, NET, DA D1 and D2 receptors, and the adrenergic α2A receptor in the mPFC of PND 100 animals. Many of these proteins have been shown to play a role in neurobehavioral functions mediated by the mPFC, including arousal regulation, attention, and impulse control functions (Arnsten and Dudley 2005; Arnsten 2009b; Arnsten and Pliszka 2011; Schmeichel and Berridge 2013; Logue and Gould 2014) and have been shown to be disrupted by early postnatal Mn exposure (Kern et al. 2010; Kern and Smith 2011; Mcdougall et al. 2011; McDougall et al. 2008; Anderson et al. 2009). Notably, based on DAPI-labeling there was no effect of Mn exposure on total cell numbers collected in the immunofluorescence images [F(4, 22.53) = 1.209, p = 0.33; overall mean number of DAPI-labeled cells/image = 185 ± 1.0 SEM, n = 540 images from 30 animals x 18 images/animal; the range across images = 125 – 236 cells/image].
3.3.1. Early postnatal Mn exposure caused lasting reductions in presynaptic catecholaminergic protein levels
The impact of early and lifelong Mn exposure on TH protein levels was assessed to determine whether the reduction in evoked NE outflow in PND 29 – 35 animals was consistent with changes in TH levels in mPFC neurons in PND 100 animals. Overall, postnatal Mn exposure led to a significant reduction in mPFC TH levels [F(4, 24.79) = 88.85, p < 0.0001]. Specifically, early postnatal Mn exposure to the higher 50 mg Mn/kg/day dose caused a significant reduction in TH protein levels to ~58% of controls (p = 0.019), while no measurable change was found in the lower dose early 25 group (p = 0.77). By comparison, lifelong Mn exposure over PND 1 – 100 caused a significant decrease in TH levels for both the lifelong 25 and lifelong 50 groups to ~54% and ~45% of controls, respectively (p’s <0.0001 for both). Moreover, TH protein levels in the both the lifelong 25 and 50 exposure groups were significantly lower than their early life counterparts (p < 0.0001 and p = 0.019, respectively), indicating that lifelong Mn exposure further exacerbated the impacts of early life Mn exposure on TH protein levels in the mPFC (Figure 4a).
Figure 4. Postnatal Mn exposure caused lasting reductions in PFC TH, DAT, and NET protein levels.

Representative immunohistochemistry images of (a) TH, (b) DAT and (c) NET staining from mPFC brain sections of control and early life 50 Mn treatment groups. Representative images at 63x magnification for clarity (scale bars, 10 μm). Bar charts show quantified fluorescence intensity reflecting 12 images/animal (at 40x magnification) and n = 6 animals/treatment group; data are least squares means ± SEM, shown as percent of control values generated from the statistical model that included all five treatment groups. Bars with different superscripts are statistically different (p < 0.05) based on Tukey’s multiple comparisons test.
DAT and NET protein levels were measured in the mPFC of PND 100 animals to determine whether catecholamine reuptake transporters were impacted by postnatal Mn exposure in a manner consistent with the effect of Mn exposure to reduce TH protein levels and evoked release of NE. Levels of DAT were significantly reduced by Mn exposure [F(4, 24.64) = 20.72, p < 0.0001], with the early 50 Mn group reduced to ~23% of controls (p < 0.0001), while the early 25 group trended towards a decrease to ~60% of controls (p = 0.075). Similarly, lifelong exposure to the 25 and 50 Mn doses caused a significant reduction in DAT to ~40% (p = 0.0008) and ~22% (p < 0.0001) of controls, respectively. However, the lifelong Mn exposure groups were not different from their early life counterparts (p’s > 0.34), indicating that lifelong Mn exposure did not worsen the effects of early life exposure on DAT protein levels in the mPFC (Figure 4b).
Similarly, NET protein levels were also significantly reduced by Mn exposure [F(4, 25.16) = 42.47, p < 0.0001], with the early life 25 and 50 exposures reducing NET levels to ~57% and ~36% of controls, respectively (p < 0.0001 for both). Lifelong Mn exposure caused similar reductions in mPFC NET levels to ~63% (p = 0.0002) and ~47% (p < 0.0001) of controls for the lifelong 25 and 50 Mn groups, respectively (Figure 4c). As with DAT protein above, lifelong Mn exposure did not worsen the effects of early life Mn on NET protein levels (p’s > 0.74 for the lifelong vs. early life Mn group contrasts).
To assess whether the effect(s) of Mn exposure on catecholaminergic protein levels based on immunofluorescence intensity reported above could be accounted for by changes in the number or volume of Imaris-rendered immunofluorescent objects, we determined the average number and volume of Imaris-rendered objects in the collected images. With this, we assumed that changes in average object number and volume reflects changes in the number and size of spatially separate puncta of the expressed protein. Results show that both TH and NET, but not DAT object number and volume, were affected by Mn exposure. Specifically, early postnatal Mn exposure caused reductions in both TH total object number [F(4, 25) = 108.30, p < 0.0001; Supplementary Figure S2a] and TH object volume [F(4, 25.09) = 39.82, p < 0.0001; Supplementary Figure S2b], whereas for NET, only the object volume was significantly reduced from controls across all Mn groups [F(4, 23.63) = 8.98, p = 0.0001; p’s < 0.008 for all specific contrasts relative to control; Supplementary Figures S2f, S2e]. By comparison, Mn had no effect on DAT object number [F(4, 24.84) = 1.64, p = 0.19; Supplementary Figure S2c] or object volume [F(4, 25.02) = 1.14, p = 0.36; Supplementary Figure S2d].
3.3.2. Postnatal Mn exposure caused lasting alterations in dopaminergic, but not α2A, receptor protein levels
To determine whether the behavioral changes reported here and in our previous studies (Beaudin et al. 2017a; Beaudin et al. 2017b; Beaudin et al. 2015; Beaudin et al. 2013; Kern and Smith 2011; Kern et al. 2010) reflected alterations in the abundance of catecholaminergic neurotransmitter receptors, we quantified the protein levels of DA D1 and D2 receptors, along with adrenergic α2A receptors in the mPFC of PND 100 animals. Notably, the presence and direction of effects of postnatal Mn exposure were different for the dopaminergic versus α2A adrenergic receptors, and for the direction of the effect on the excitatory D1 (decreased) versus inhibitory D2 (increased) receptors (Figure 5). Specifically, D1 receptor levels were reduced by Mn exposure [F(4, 25.47) = 117.96, p < 0.0001], with a measurable effect of both Mn exposure dose and duration. Early postnatal exposure to the 25 and 50 mg/kg/day Mn doses caused significant reductions in D1 protein levels to ~78% and ~47% of controls, respectively (p < 0.0001 for both). Lifelong Mn exposure caused similar reductions in D1 to ~70% and ~38% of controls for the lifelong 25 and 50 groups, respectively (p < 0.0001 for both). Moreover, lifelong exposure to the higher 50 Mn dose worsened the effects of Mn exposure restricted to early postnatal life on mPFC D1 protein levels (p = 0.026), while lifelong exposure to the lower 25 Mn dose led to a trending reduction compared to early life exposure (p = 0.085) (Figure 5a).
Figure 5. D1 and D2 protein levels, but not α2A, were altered by early postnatal Mn exposure.

Representative immunohistochemistry images of (a) D1, (b) D2 and (c) α2A receptor staining from mPFC brain sections of control and early life 50 Mn treatment groups. Bar charts show quantified fluorescence intensity reflecting 12–18 images/animal (40x magnification), where n = 6 animals/treatment group. Representative images at 63x magnification for clarity (scale bars, 10 μm). Data are least squares means ± SEM shown as percent of control values generated from the statistical model that included all five treatment groups. Bars with different superscripts are statistically different (p < 0.05) based on Tukey’s multiple comparisons test.
Postnatal Mn exposure also led to significant lasting changes in mPFC D2 receptor levels, but the effects were directionally opposite to those observed for D1 receptor protein. Specifically, early life exposure to the higher 50 Mn dose caused a significant increase in D2 protein levels to ~240% of controls (p < 0.0001), while the lower early life 25 dose had no measurable effect (p = 0.54) (Figure 5b). By comparison, both lifelong Mn exposure doses significantly increased mPFC D2 protein levels to ~115% (p = 0.011) and ~247% (p < 0.0001) of controls for the lifelong 25 and 50 doses, respectively. However, there were no measurable differences in D2 protein levels between the lifelong Mn groups and their early life Mn counterparts (p’s >0.28 for group contrasts), indicating that lifelong Mn exposure did not worsen the effects from Mn exposure restricted to early postnatal life (Figure 5b).
In contrast to the Mn effects reported above, there was no measurable effect of Mn on α2A receptor levels [F(4, 23.94) = 0.119, p = 0.97] (Figure 5c). In addition, regarding assessment of Imaris-rendered object number and volume for D1, D2, and α2A, only D1 object number was significantly affected by Mn exposure [F(4, 25.16) = 3.40, p = 0.024], reflecting a lower number of D1 objects in the lifelong 50 Mn group versus controls (p = 0.013; Supplementary Figure S2g). Mn exposure also caused a trending reduction in D1 object volume (p = 0.072; Supplementary Figure S2h), but had no effect on object number or volume for D2 and α2A (p’s > 0.153; Supplementary Figures S2i – S2k).
3.3.3. Early postnatal Mn exposure caused lasting increases in astrocyte reactivity
We measured astrocyte GFAP protein levels in order to determine whether postnatal Mn exposure led to heightened astrocyte reactivity as an indicator of neuroinflammation, which could alter the synaptic environment and possibly contribute to changes in catecholaminergic synaptic proteins within the mPFC. Postnatal Mn exposure caused lasting increases in astrocyte GFAP levels [F(4, 25.09) = 40.93, p < 0.0001], with increases of ~242% and ~215% of controls in the early life and lifelong 50 mg/kg/day Mn dose groups, respectively (p’s < 0.0001 for both; Figure 6). In contrast, astrocyte GFAP levels in the early life and lifelong 25 mg/kg/day Mn dose groups were not different from controls (p’s >0.70). Moreover, astrocyte GFAP levels were not measurably different between the lifelong versus early life 50 groups, indicating that lifelong Mn exposure did not alter the lasting effects caused by Mn exposure restricted to early life (p = 0.67). Finally, there was no effect of Mn exposure on GFAP object number in the mPFC (p = 0.78; Supplementary Figure S2l), though there was an effect of Mn on GFAP total object volume [F(4, 28.97) = 17.05, p < 0.0001; Supplementary Figure S2m], with an increase in GFAP object volume specifically in the early life 50 group versus controls (p = 0.0004; Supplementary Figure S2m).
Figure 6. GFAP protein levels varied in a dose-dependent manner in response to early postnatal Mn exposure.

Representative immunohistochemistry images of GFAP staining from mPFC brain sections of control and early life 50 Mn treatment groups. Bar charts show quantified fluorescence intensity reflecting 12 images/animal (40x magnification), where n = 6 animals/treatment group. Representative images at 63x magnification for clarity (scale bars, 10 μm). Data are least squares means ± SEM shown as percent of control values generated from the statistical model that included all five treatment groups. Bars with different superscripts are statistically different (p < 0.05) based on Tukey’s multiple comparisons test.
3.3.4. A1 reactive astrocytes were induced in greater proportions than A2 astrocytes by early postnatal Mn exposure
In order to further investigate the inflammatory phenotype of the reactive astrocytes in the mPFC following Mn exposure, we co-immunostained mPFC brain sections with GFAP and complement C3 or S100A10 protein-specific antibodies – the latter two as markers for proinflammatory A1 and anti-inflammatory A2 astrocyte phenotypes, respectively (Liddelow et al. 2017; Liddelow and Barres 2017). Given that astrocyte reactivity (i.e., increased GFAP) in PND 100 animals was most evident in the early life 50 Mn group, we restricted analyses to the early life 25 and 50 Mn groups versus controls. Results show that early life Mn exposure caused significant increases in both GFAP co-localized C3 [F(2, 12) = 23.73, p < 0.0001], and S100A10 [F(2, 12) = 45.91, p < 0.0001]. Specifically, early life exposure to the higher 50 mg/kg/day Mn dose increased GFAP co-localized C3 to ~570% of controls (p < 0.0001), and increased GFAP co-localized S100A10 levels to 200% of controls (p < 0.0001) (Figures 7c, d). In contrast, there was no effect of the lower early life 25 mg/kg/day Mn dose on either GFAP co-localized C3 or S100A10 (p’s ≥ 0.90 for both).
Figure 7. C3 and S100A10 protein levels are increased in response to early postnatal Mn exposure.

Representative immunohistochemistry images of (a) GFAP, C3, and S100A10 staining from mPFC brain sections of control and early life 50 Mn treatment groups at 63x magnification; white boxes are enlarged (b) for clarity (scale bars, 10 μm). (c, d) Bar charts show quantified fluorescence intensity of GFAP-colocalized puncta for C3 and S100A10, respectively; data reflect 4 images/animal, where n = 5 animals/treatment group. Data are least squares means ± SEM shown as percent of control values generated from the statistical model that included three treatment groups. Bars with different superscripts are statistically different (p < 0.05) based on Tukey’s multiple comparisons test.
3.4. Dendritic Spine Density
Early postnatal Mn exposure did not alter mPFC dendritic spine density
To determine whether the lasting changes in catecholaminergic protein levels and increased inflammatory reactive astrocytes were accompanied by changes in dendritic spine density, we quantified spine density on mPFC layer III pyramidal cell dendrites (# spines/10 μm dendrite length) in PND 24 and PND 145 animals exposed over early postnatal life to the higher 50 mg/kg/day Mn dose. In postweaned PND 24 animals, there was no effect of early life Mn exposure on spine density on apical (2nd order + terminal tip) dendrites [F(1, 7.76) = 0.73, p = 0.42], or on basal dendrites [F(1, 6.91) = 1.04, p = 0.34] of mPFC layer III pyramidal neurons. Similarly, there was no effect of early life Mn on spine density on apical 2nd order dendrites in PND 145 animals [F(1, 16.39) = 2.74, p = 0.12] (Figure 8).
Figure 8. Dendritic spine density was not measurably altered in the mPFC following Mn exposure.

Data are least squares mean spine density (spines/10 μm dendrite length ± SEM) on mPFC layer III pyramidal neuron dendrites in the control and early 50 mg/kg/day Mn-exposed groups across dendrite type and animal age (n = 10 neurons/animal, 5–10 animals/treatment group).
3.5. Tissue Mn and blood hematocrit levels
Mn exposure resulted in body Mn levels consistent with environmental exposures
Early postnatal Mn exposure led to a dose-dependent increase in blood and brain Mn levels across age groups at PND 24, 66, and ~590, though levels were significantly higher in the PND 24 weanlings compared to their older adolescent and adult counterparts. Notably, tissue Mn levels in the latter two age groups were very comparable to each other across Mn treatment condition, and within the adolescent and adult ages tissue Mn levels in the Mn exposed groups were only slightly higher than their age-matched controls (Table 1). Finally, there were no measurable differences in blood hematocrit levels between Mn exposure groups at age PND 24 (hematocrit range 39.4 – 41.0 % between treatment groups, F(2, 15) = 1.49, p = 0.26) or PND 66 (range 45.6 – 46.4 % between treatment groups, F(4, 35) = 0.18, p = 0.95).
Table 1.
Blood and brain Mn concentrations of animals at PND 24, 66, and ~590.
| Age (PND) | Control | 25 mg Mn/kg/day | 50 mg Mn/kg/day | |||
|---|---|---|---|---|---|---|
| Early life | Lifelong | Early life | Lifelong | |||
| Blood | 24 | 24.6 ± 1.21 (6)A, a | NA | 131 ± 22.7 (7) B, a | NA | 231 ± 33 (7) C, a |
| 66 | 9.3 ± 0.59 (7) A, b | 12.2 ± 0.69 (8) B | 12.9 ± 0.86 (9) B, b | 11.5 ± 0.57 (8) B | 18.6 ± 1.9 (7) C, b | |
| 590 | 5.66 ± 0.57 (5) A, c | 6.98 ± 0.95 (11) A | 21.9 ± 8.66 (9) B, c | 8.28 ± 1.32 (8) AB | 16.8 ± 1.83 (10) C, b | |
| Brain | 24 | 3.62 ± 0.13 (6) A, a | NA | 6.49 ± 0.56 (7) AB, a | NA | 11.5 ± 2.57 (6) B, a |
| 66 | 2.12 ± 0.060 (7) A, b | 2.19 ± 0.031 (8) A | 2.41 ± 0.045 (9) BC, b | 2.26 ± 0.062 (8) AB | 2.51 ± 0.068 (7) C, b | |
| 590 | 1.82 ± 0.11 (6) A, b | 2.20 ± 0.17 (12) A | 2.19 ± 0.049 (10) AB, b | 2.00 ± 0.086 (7) A | 2.68 ± 0.11 (11) C, b | |
Data are mean ± SEM with group sizes in parentheses; blood Mn in ng/mL, brain Mn in μg/g dry weight. Uppercase superscripts: within an age group and tissue, treatment groups with different capital letters are statistically different from one another (p < 0.05), based on Wilcoxon / Kruskal-Wallis test. Lowercase superscripts: within a treatment group and tissue, values across ages with different lowercase superscripts are statistically different from one another. PND, postnatal day.
4.0. Discussion
Our findings show that early postnatal Mn exposure causes heightened behavioral reactivity, reductions in evoked NE outflow, lasting alterations to catecholaminergic systems protein levels within the mPFC, and lasting heightened astrocyte reactivity that is dominated by a proinflammatory A1 astrocyte phenotype. In general, neither the behavioral nor catecholaminergic effects were exacerbated by continued Mn exposure following weaning. Given that behavioral and cognitive function are among the most important public health outcomes, understanding how developmental exposure to environmental toxicants such as Mn impacts neurobehavioral function is key in devising effective treatment strategies (Developmental Toxicology 2000; Landrigan et al. 2002). These findings and their implications for humans exposed to elevated Mn are discussed below.
Postnatal Mn exposure caused broad lasting alterations in mPFC catecholaminergic systems
Early postnatal Mn exposure over PND 1–21 caused lasting hypofunctioning of the catecholaminergic systems in the mPFC, as evidenced by the significant Mn-dose related reductions in TH, DAT, NET, and D1 receptors, and increased D2 receptor protein levels in PND 100 young adults. These effects were generally not worsened by continued exposure following weaning through PND 100. Notably, blood and brain tissue Mn levels in the early life Mn-exposed groups were comparable to control levels well before PND 100, indicating that the catecholaminergic systems disruptions were due to elevated Mn during the early postnatal life exposure period, rather than elevated Mn levels in the PND 100 young adults (Table 1). Whereas lifelong Mn exposure generally did not significantly worsen the effects of early postnatal exposure, the dose-response for effects on the catecholaminergic systems qualitatively appears to correspond to the increasing degree of exposure insult (i.e., control < early 25 Mn < lifelong 25 Mn < early 50 Mn < lifelong 50 Mn) (Figure 9). The Mn effect on the mPFC catecholaminergic systems, while broad in scope, was also somewhat specific, as there was no measurable effect of Mn on α2A receptor protein levels (Figures 5c and 9). Finally, these catecholaminergic protein changes, most notably the reduction in TH (Figure 4a and 9), are consistent with the lasting reductions in the evoked release of NE in young weanling (Figure 3a, b) and adult (Beaudin et al. 2015; Lasley et al. 2019) animals exposed to the same oral Mn doses.
Figure 9.

Mean levels of mPFC proteins across treatment groups, with levels of each protein normalized to its respective control group (error bars omitted for clarity). Symbol shape indicates the protein, and open symbols indicate significantly different from respective control (p < 0.05), based on statistical models that included all five treatment groups.
The mechanism(s) through which early life Mn exposure causes these lasting effects on the catecholaminergic systems is not well understood. One possibility is they may be driven by a Mn-induced reduction in TH expression that results in reduced DA and NE synthesis, leading to compensatory reductions in NET, DAT, and D1 expression, along with heightened D2 levels that may represent additional compensatory changes following lower synaptic catecholamine levels. Alternatively, or in addition, these lasting protein level changes may be mediated via epigenetic mechanisms, given that expression of several of them (e.g., TH, D2, DAT, and NET) are in part epigenetically regulated via DNA methylation (Hillemacher et al. 2009; Archer et al. 2011; Day et al. 2013; Groleau et al. 2014). This latter suggestion is supported by several studies in human neuroblastoma SH-SY5Y cells showing that Mn exposure led to hypermethylated TH promoter and downregulated TH gene transcription (Tarale et al. 2016; Gandhi et al. 2018). Recent evidence has also shown that TH expression can be mediated in part by the activity of c-RET kinase, and that human neuroblastoma cells exposed to 30 μM Mn experienced a c-RET mediated reduction in TH (Kumasaka et al. 2017). Finally, we believe that the reductions in TH, DAT, NET, and D1 immunofluorescence reflect reduced protein levels within Imaris-rendered objects, rather than fewer DAT and/or NET-positive cells/nerve terminals, since there was no Mn effect on total cell number or on the number of Imaris-rendered DAT or NET objects, relative to controls; DAT and NET are widely used as a markers for catecholaminergic nerve terminals (Miller et al. 1997; Stephenson et al. 2007; Moron et al. 2002).
Prior studies from our group have reported similar reductions in DAT and D1 within the striatum and nucleus accumbens, along with increased D2 in the mPFC of PND 24 rats exposed to the same 50 mg/kg/day Mn dose over PND 1–21 (Kern and Smith 2011; Kern et al. 2010). In those studies the increased mPFC D2 levels persisted into adulthood (PND 107), while the reduced DAT and D1 levels were normalized to control levels in adults. Together, these changes are consistent with our prior results showing reduced evoked release of DA and NE in the mPFC of adult rats following lifelong Mn exposure (Beaudin et al. 2015; Lasley et al. 2019). Others have reported similar, albeit more limited, effects of early life Mn exposure on brain catecholaminergic systems. For example, McDougall et al. (2008) showed that oral exposure to 750 μg Mn/day over PND 1–21 reduced DAT protein expression and [3H]DA uptake in the striatum and nucleus accumbens, along with reduced striatal DA efflux in PND 90 rats. Subsequently, McDougall et al. (2011) showed that the same Mn exposure paradigm decreased the abundance of D2 binding sites in the PFC, but increased D2 binding sites and protein levels in the dorsal striatum of PND 90 rats. Anderson et al. (2009) reported that weanling rats exposed orally to 1 mg Mn/mL via drinking water for 6 weeks post-weaning exhibited reductions in evoked release of striatal NE levels, and reduced NET and α2 receptor protein levels in the striatum. Regarding our observed reduction in TH protein levels, Peres et al. (2016) similarly found that rats exposed to 20 mg Mn/kg/day via i.p. injection over PND 8–12 exhibited a significant reduction in striatal TH protein levels at PND 70, when tissue Mn concentrations were no longer elevated, and the reduced TH protein levels correlated with TH phosphorylation at serine 40 and 19. Other rodent studies in adults treated with Mn via i.p. injection have similarly reported increased D2 binding sites and protein levels in the dorsal striatum (Seth et al. 1981; Nam and Kim 2008). More recently, Guilarte et al. (2019) showed that young adult non-human primates chronically exposed to 15–20 mg/kg MnSO4 per week via jugular catheter injection exhibited reduced DA release in the frontal cortex, relative to baseline controls, an observation not greatly different from the exposure-dependent ordering found in the DA time course in our work. Interestingly, our findings may help explain the Mn-induced effects of another protein involved in the catecholaminergic system, DARPP-32, a DA-regulated phosphoprotein that is known to play a role in DAergic protein kinase A-dependent signaling (Scheggi et al. 2018). Given that D1 receptor activation is known to stimulate phosphorylation of DARPP-32 at threonine 34, and the fact that Cordova et al. (2013) recently reported that neonatal exposure to 20 mg Mn/kg/day, i.p, over PND 8–27 caused a decrease in DARPP-32 phosphorylation at threonine 34, their finding may reflect the synaptic alterations between D1 and D2 receptor levels reported here.
Changes in the mPFC catecholaminergic systems were associated with heightened behavioral reactivity and attentional impairments
All groups of animals were significantly more active during the initial 5–10 minutes of each daily test session relative to later in each of the sessions, reflecting heightened arousal engendered by the handling, novelty, and stress of being in an open field testing environment (Prut and Belzung 2003). However, this transient period of heightened arousal was significantly greater for all four Mn exposure groups relative to controls (Figure 2a, b). This pattern of effects suggests a transient increase in arousal state in the Mn animals (relative to controls) that dissipated over the course of the daily test session. These findings suggest that Mn caused either a heightened emotional response to handling and the novel, inherently stressful environment, and/or an impaired ability to regulate this heightened arousal. This interpretation is consistent with our prior findings that exposure to 25 mg Mn/kg/day over PND 1–21 impaired arousal regulation in adulthood in a 5-Choice Serial Reaction Time task, in which animals displayed a transient impairment of response accuracy for non-distraction trials in a visual discrimination and attention test (Beaudin et al. 2017a).
These findings of impaired emotion regulation reported here further elucidate the behavioral phenotype produced by early postnatal Mn exposure. Prior animal studies have reported that oral Mn exposure caused lasting deficits in spatial learning and memory (Golub et al. 2005; Kern et al. 2010), as well as lasting impairments in executive functioning (e.g., deficits in focused and selective attention, impulse control, and fine motor function), consistent with the mPFC catecholamine systems changes reported here (Beaudin et al. 2017b; Beaudin et al. 2017a; Beaudin et al. 2015; Beaudin et al. 2013). Together, these findings have important implications for the environmental etiology of neurobehavioral disorders, such as ADHD, and their underlying neurobiology in children (Arnsten et al. 2015; Arnsten and Pliszka 2011). This is underscored by the fact that attention and impulse control dysfunctions, including ADHD, are the most prevalent neurodevelopmental disorders in children, affecting ~6–11% of all U.S. children age 6–17 years (Feldman and Reiff 2014; Kaiser et al. 2015; Willcutt 2012).
Early postnatal Mn exposure caused a proportionally greater lasting induction of A1 proinflammatory versus A2 neuroprotective astrocytes
The above lasting changes in mPFC catecholaminergic proteins due to early postnatal Mn exposure were accompanied by a lasting heightened reactivity of GFAP-positive astrocytes in the early postnatal and lifelong 50 mg Mn/kg/day groups to ~250% of controls. Moreover, in the GFAP-positive astrocytes, the relative increase in the A1 proinflammatory astrocyte marker C3 (to ~568% of controls) was ~2.8-fold greater than the anti-inflammatory A2 marker S100A10 (200% of controls), suggesting an overall proinflammatory neuroenvironment in the mPFC; this interpretation assumes that the proportional increase in C3 versus S100A10 protein levels in GFAP-positive astrocytes directly reflects the relative increase in A1 and A2 cell phenotypes. Prior in vivo and in vitro studies have previously shown that Mn exposure may promote the reactivity of astrocytes and microglia to contribute to neuroinflammation, as measured by increased proinflammatory gene and protein expression, along with higher levels of proinflammatory cytokines (Kern and Smith 2011; Liu et al. 2006; Zhao et al. 2009; Popichak et al. 2018; Jin et al. 2019). Further, Liddelow et al. (2017) recently demonstrated that reactive astrocytes exhibiting an A1 proinflammatory phenotype led to loss of synaptic function, increased synaptic pruning, impaired endocytosis of extracellular debris, and an increased risk for neurodegeneration. Given the evidence that C3 and S100A10 are distinct, non-overlapping markers of A1 and A2 phenotypic astrocytes (Liddelow and Barres 2017; Liddelow et al. 2017), our findings suggest that multiple sub-populations of GFAP-positive astrocytes are induced by early postnatal Mn exposure, with a lasting net shift towards an A1 proinflammatory phenotype. These results are consistent with other studies of neurotoxic insults. For example, Zamanian et al. (2012) showed that lipopolysaccharide via i.p. injection in mice induced a heterogeneous astrocyte response, with cortical reactive astrocytes exhibiting phenotypes similar to the A1 and A2 phenotypes described by Liddelow et al. (2017).
While our findings show evidence of lasting astrocyte reactivity skewed towards the proinflammatory phenotype, it is not known whether the heightened astrocyte reactivity is involved in the catecholaminergic protein and neurotransmitter changes reported above. For example, the fact that early postnatal exposure to the lower 25 mg/kg/day Mn dose caused lasting reductions in DAT and NET protein levels, but did not increase astrocyte reactivity (GFAP) or markers of A1 or A2 astrocyte phenotypes (Figure 7c, d), does not necessarily reflect an absence of astrocyte-related effects on the mPFC catecholaminergic system, since our analyses were limited to changes in astrocyte protein expression and not more detailed tests of astrocyte function. Additionally, the lack of an effect of early life 50 mg/kg/day Mn exposure on mPFC pyramidal neuron spine density (Figure 8) suggests that the neuroinflammatory environment present in the Mn-exposed animals did not lead to neurobiological changes that altered dendritic spine density in the mPFC.
Collectively, the lasting changes in the rat mPFC catecholaminergic systems caused by early postnatal Mn exposure reported here are consistent with, and may well underlie, the lasting impairments in arousal regulation, selective and focused attention, impulse control, and fine motor function that we have reported in Mn-exposed animals (Beaudin et al. 2017a; Beaudin et al. 2015; Beaudin et al. 2013). These findings have significant implications for human Mn exposure by suggesting that the attentional and executive function impairments associated with Mn exposure in children may be due to hypofunctioning of the PFC catecholaminergic systems (Wasserman et al. 2006; Oulhote et al. 2014; Haynes et al. 2015).
Supplementary Material
Acknowledgements
The authors would like to thank T. Jursa for analytical assistance, J. Alvarado, A. Chen, A. Cruz, J. Fee, M. Fung, R. Garcia, K. Goetz, S. Greenberg, C. Horton, I. Jing, K. Kekkonen, M. Kern, G. Kouklis, J. Sabile, T. Lau, S. Lee, L. Loh, A. Luo, C. Matysiak, D. Michue, H. Monday, M. Ngo, L. Nguyen, S. Nisam, M. Quail, C. Rew, M. Richter, K. Riffel, J. Shen, A. Smith, A. Spock, D. Tsang, R. Turk, A. Watson, F. Wu, K. Younes, S. Young, and S. Zhong for their assistance with behavioral testing. We also thank B. Abrams for assistance with microscopy and Imaris quantification, R. Jaramillo and D. McDonald for assistance in image processing, and R. Eastman, R. Cathey, E. Hiolski, C. Moyer, M. Camps, S. Mukhopadhyay, and Y. Zuo for their contributions to the study.
Funding
This research was funded by grants from the National Institutes of Health, National Institute of Environmental Health Sciences (NIEHS R01ES018990 and R01ES028369).
Abbreviations used:
- Mn
manganese
- PND
postnatal day
- mPFC
medial prefrontal cortex
- GFAP
glial fibrillary acidic protein
- NE
norepinephrine
- TH
tyrosine hydroxylase
- DA
dopamine
- DAT
dopamine transporter
- NET
norepinephrine transporter
- ADHD
attention deficit hyperactivity disorder
- RRID
research resource identifier
- i.p
intraperitoneal
- AP
anterior-posterior
- ML
medial-lateral
- DV
dorsal-ventral
- FEP
fluorinated ethylene propylene
- DOPAC
3,4-dihydroxyphenylacetic acid
- HVA
homovanillic acid
- EDTA
ethylenediaminetetraacetic acid
- PFA
paraformaldehyde
- PBS
phosphate buffered saline
- DAPI
4´,6-diamidino-2-phenylindole
Footnotes
--Human subjects --
Involves human subjects:
If yes: Informed consent & ethics approval achieved:
=> if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
ARRIVE guidelines have been followed:
Yes
=> if it is a Review or Editorial, skip complete sentence => if No, include a statement: “ARRIVE guidelines were not followed for the following reason:
“
(edit phrasing to form a complete sentence as necessary).
=> if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.” unless it is a Review or Editorial
conflict of interest disclosure
The authors have no competing or potential financial conflicts of interest to declare.
References
- Anderson JG, Fordahl SC, Cooney PT, Weaver TL, Colyer CL, Erikson KM (2009) Extracellular norepinephrine, norepinephrine receptor and transporter protein and mRNA levels are differentially altered in the developing rat brain due to dietary iron deficiency and manganese exposure. Brain Res. 1281, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer T, Berman MO, Blum K. (2011) Epigenetics in Developmental Disorder: ADHD and Endophenotypes. J. Genet. Syndr. Gene Ther 02, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF (2009a) Toward a New Understanding of Attention-Deficit Hyperactivity Disorder Pathophysiology: An Important Role for Prefrontal Cortex Dysfunction. CNS Drugs 23 Suppl 1, 33–41. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Dudley AG (2005) Methylphenidate improves prefrontal cortical cognitive function through alpha2 adrenoceptor and dopamine D1 receptor actions: Relevance to therapeutic effects in Attention Deficit Hyperactivity Disorder. Behav. Brain Funct 1, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT (2009b) The Emerging Neurobiology of Attention Deficit Hyperactivity Disorder: The Key Role of the Prefrontal Association Cortex. J. Pediatr 154, S22–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Pliszka SR (2011) Catecholamine influences on prefrontal cortical function: Relevance to treatment of attention deficit/hyperactivity disorder and related disorders. Pharmacol. Biochem. Behav 99, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Wang M, Paspalas CD (2015) Dopamine’s Actions in Primate Prefrontal Cortex: Challenges for Treating Cognitive Disorders. Pharmacol. Rev [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudin SA, Nisam S, Smith DR (2013) Early life versus lifelong oral manganese exposure differently impairs skilled forelimb performance in adult rats. Neurotoxicol. Teratol 38, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudin SA, Strupp BJ, Lasley SM, Fornal CA, Mandal S, Smith DR (2015) Oral Methylphenidate Alleviates the Fine Motor Dysfunction Caused by Chronic Postnatal Manganese Exposure in Adult Rats. Toxicol. Sci 144, 318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudin SA, Strupp BJ, Strawderman M, Smith DR (2017a) Early Postnatal Manganese Exposure Causes Lasting Impairment of Selective and Focused Attention and Arousal Regulation in Adult Rats. Environ. Health Perspect 230, 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudin SA, Strupp BJ, Uribe W, Ysais L, Strawderman M, Smith DR (2017b) Methylphenidate alleviates manganese-induced impulsivity but not distractibility. Neurotoxicol. Teratol 61, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonette GB, Martins GJ, Franz TM, Harper ES, Schoenbaum G, Powell EM (2008) Double Dissociation of the Effects of Medial and Orbital Prefrontal Cortical Lesions on Attentional and Affective Shifts in Mice. J. Neurosci 28, 11124–11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard MF, Sauvé S, Barbeau B, Legrand M, Brodeur MÈ, Bouffard T, Limoges E, Bellinger DC, Mergler D. (2011) Intellectual Impairment in School-Age Children Exposed to Manganese from Drinking Water. Environ. Health Perspect 119, 138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard M, Laforest F, Vandelac L, Bellinger D, Mergler D. (2007) Hair manganese and hyperactive behaviors: pilot study of school-age children exposed through tap water. Environ. Health Perspect 115, 122–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bymaster FP, Katner JS, Nelson DL, Hemrick-Luecke SK, Threlkeld PG, Heiligenstein JH, Morin SM, Gehlert DR, Perry KW (2002) Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat. Neuropsychopharmacology 27, 699–711. [DOI] [PubMed] [Google Scholar]
- Clancy B, Finlay BL, Darlington RB, Anand KJS (2007) Extrapolating brain development from experimental species to humans. Neurotoxicology 28, 931–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordova FM, Aguiar AS, Peres TV, Lopes MW, Gonçalves FM, Pedro DZ, Lopes SC, et al. (2013) Oxidative Stress is Involved in Striatal Neurotoxicity in Manganese-Exposed Developing Rats. Arch. Toxicol 87, 1231–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crinella FM (2003) Does soy-based infant formula cause ADHD? Expert Rev. Neurother 3, 145–148. [DOI] [PubMed] [Google Scholar]
- Crinella FM (2012) Does soy-based infant formula cause ADHD? Update and public policy considerations. Expert Rev. Neurother 12, 395–407. [DOI] [PubMed] [Google Scholar]
- Dallérac G, Zapata J, Rouach N. (2018) Versatile control of synaptic circuits by astrocytes: where, when and how? Nat. Rev. Neurosci 19, 729–743. [DOI] [PubMed] [Google Scholar]
- Day JJ, Childs D, Guzman-Karlsson MC, Kibe M, Moulden J, Song E, Tahir A, Sweatt JD (2013) DNA methylation regulates associative reward learning. Nat. Neurosci 16, 1445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Developmental Toxicology N. A. of S. C. (2000) Scientific Frontiers in Developmental Toxicology and Risk Assessment. National Academy Press, Washington, DC. [PubMed] [Google Scholar]
- Ericson JE, Crinella FM, Clarke-Stewart KA, Allhusen VD, Chan T, Robertson RT (2007) Prenatal manganese levels linked to childhood behavioral disinhibition. Neurotoxicol. Teratol 29, 181–187. [DOI] [PubMed] [Google Scholar]
- Farhy-Tselnicker I, Allen NJ (2018) Astrocytes, neurons, synapses: A tripartite view on cortical circuit development. Neural Dev. 13, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman HM, Reiff MI (2014) Attention Deficit–Hyperactivity Disorder in Children and Adolescents. N. Engl. J. Med 370, 838–846. [DOI] [PubMed] [Google Scholar]
- Gandhi D, Sivanesan S, Kannan K. (2018) Manganese-Induced Neurotoxicity and Alterations in Gene Expression in Human Neuroblastoma SH-SY5Y Cells. Biol. Trace Elem. Res 183, 245–253. [DOI] [PubMed] [Google Scholar]
- Golub MS, Hogrefe CE, Germann SL, Tran TT, Beard JL, Crinella FM, Lonnerdal B. (2005) Neurobehavioral evaluation of rhesus monkey infants fed cow’s milk formula, soy formula, or soy formula with added manganese. Neurotoxicol. Teratol 27, 615–627. [DOI] [PubMed] [Google Scholar]
- Groleau P, Joober R, Israel M, Zeramdini N, DeGuzman R, Steiger H. (2014) Methylation of the dopamine D2 receptor (DRD2) gene promoter in women with a bulimia-spectrum disorder: associations with borderline personality disorder and exposure to childhood abuse. J. Psychiatr. Res 48, 121–7. [DOI] [PubMed] [Google Scholar]
- Guilarte TR, Yeh CL, McGlothan JL, Perez J, Finley P, Zhou Y, Wong DF, Dydak U, Schneider JS (2019) PET imaging of dopamine release in the frontal cortex of manganese-exposed non-human primates. J. Neurochem 150, 188–201. [DOI] [PubMed] [Google Scholar]
- Gunier RB, Jerrett M, Smith DR, Jursa T, Yousefi P, Camacho J, Hubbard A, Eskenazi B, Bradman A. (2014a) Determinants of manganese levels in house dust samples from the CHAMACOS cohort. Sci. Total Environ 497–498, 360–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunier RB, Mora AM, Smith D, Arora M, Austin C, Eskenazi B, Bradman A. (2014b) Biomarkers of manganese exposure in pregnant women and children living in an agricultural community in California. Environ. Sci. Technol 48, 14695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Zhang Z, Wang Q, Zhang J, Wang L, Zhang Q, Li H, Wu S. (2018) Manganese chloride induces histone acetylation changes in neuronal cells: Its role in manganese-induced damage. Neurotoxicology 65, 255–263. [DOI] [PubMed] [Google Scholar]
- Haynes EN, Sucharew H, Kuhnell P, Alden J, Barnas M, Wright RO, Parsons PJ, et al. (2015) Manganese exposure and neurocognitive outcomes in rural school-age children: The communities actively researching exposure study (Ohio, USA). Environ. Health Perspect 123, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashino K, Ago Y, Umehara M, Kita Y, Fujita K, Takuma K, Matsuda T. (2014) Effects of acute and chronic administration of venlafaxine and desipramine on extracellular monoamine levels in the mouse prefrontal cortex and striatum. Eur. J. Pharmacol 729, 86–93. [DOI] [PubMed] [Google Scholar]
- Hillemacher T, Frieling H, Hartl T, Wilhelm J, Kornhuber J, Bleich S. (2009) Promoter specific methylation of the dopamine transporter gene is altered in alcohol dependence and associated with craving. J. Psychiatr. Res 43, 388–92. [DOI] [PubMed] [Google Scholar]
- Jin H, Kanthasamy AG, Huang X, Kanthasamy A, Rokad D, Malovic E, Anantharam V, et al. (2019) Manganese activates NLRP3 inflammasome signaling and propagates exosomal release of ASC in microglial cells. Sci. Signal 12, eaat9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser M-L, Schoemaker MM, Albaret J-M, Geuze RH (2015) What is the evidence of impaired motor skills and motor control among children with attention deficit hyperactivity disorder (ADHD)? Systematic review of the literature. Res. Dev. Disabil 36, 338–357. [DOI] [PubMed] [Google Scholar]
- Kalsbeek A, Bruin J. P. de, Matthijssen MA, Uylings HB (1989) Ontogeny of open field activity in rats after neonatal lesioning of the mesocortical dopaminergic projection. Behav. Brain Res 32, 115–127. [DOI] [PubMed] [Google Scholar]
- Kern CH, Smith DR (2011) Preweaning Mn Exposure Leads to Prolonged Astrocyte Activation and Lasting Effects on the Dopaminergic System in Adult Male Rats. Synapse 65, 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern CH, Stanwood GD, Smith DR (2010) Preweaning Manganese Exposure Causes Hyperactivity, Disinhibition, and Spatial Learning and Memory Deficits Associated with Altered Dopamine Receptor and Transporter Levels. Synapse 64, 363–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumasaka MY, Yajima I, Ohgami N, Ninomiya H, Iida M, Li X, Oshino R, Tanihata H, Yoshinaga M, Kato M. (2017) Manganese-Mediated Decrease in Levels of c-RET and Tyrosine Hydroxylase Expression In Vitro. Neurotox. Res 32, 661–670. [DOI] [PubMed] [Google Scholar]
- Landrigan PJ, Schechter CB, Lipton JM, Fahs MC, Schwartz J. (2002) Environmental pollutants and disease in American children: Estimates of morbidity, mortality, and costs for lead poisoning, asthma, cancer, and developmental disabilities. Environ. Health Perspect 110, 721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasley SM, Fornal CA, Mandal S, Strupp BJ, Beaudin SA, Smith DR (2019) Early postnatal manganese exposure reduces rat cortical and striatal biogenic amine activity in adulthood. Toxicol. Sci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA (2017) Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 46, 957–967. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sullivan KA, Madl JE, Legare M, Tjalkens RB (2006) Manganese-induced neurotoxicity: The role of astroglial-derived nitric oxide in striatal interneuron degeneration. Toxicol. Sci 91, 521–531. [DOI] [PubMed] [Google Scholar]
- Ljung K, Vahter M. (2007) Time to Re-evaluate the Guideline Value for Manganese in Drinking Water? Environ. Health Perspect 115, 1533–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue SF, Gould TJ (2014) The neural and genetic basis of executive function: Attention, cognitive flexibility, and response inhibition. Pharmacol. Biochem. Behav 123, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EL, Bertrand P, Guazzetti S, Donna F, Peli M, Jursa TP, Lucchini R, Smith DR (2015) Impact of ferromanganese alloy plants on household dust manganese levels: Implications for childhood exposure. Environ. Res 138, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchini RG, Guazzetti S, Zoni S, Donna F, Peter S, Zacco A, Salmistraro M, Bontempi E, Zimmerman NJ, Smith DR (2012) Tremor, olfactory and motor changes in Italian adolescents exposed to historical ferro-manganese emission. Neurotoxicology 33, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddux JM, Holland PC (2011) Effects of dorsal or ventral medial prefrontal cortical lesions on five-choice serial reaction time performance in rats. Behav. Brain Res 221, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcdougall SA, Der-Ghazarian T, Britt CE, Varela FA, Crawford CA (2011) Postnatal manganese exposure alters the expression of D2L and D2S receptor isoforms: Relationship to PKA activity and Akt levels. Synapse 65, 583–591. [DOI] [PubMed] [Google Scholar]
- McDougall SA, Reichel CM, Farley CM, Flesher MM, Der-Ghazarian T, Cortez a. M., Wacan JJ, et al. (2008) Postnatal Manganese Exposure Alters Dopamine Transporter Function in Adult Rats: Potential Impact on Nonassociative and Associative Processes. Neuroscience 154, 848–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GW, Staley JK, Heilman CJ, Perez JT, Mash DC, Rye DB, Levey AI (1997) Immunochemical analysis of dopamine transporter protein in Parkinson’s disease. Ann. Neurol 41, 530–539. [DOI] [PubMed] [Google Scholar]
- Moreno JA, Streifel KM, Sullivan KA, Legare ME, Tjalkens RB (2009) Developmental exposure to manganese increases adult susceptibility to inflammatory activation of glia and neuronal protein nitration. Toxicol. Sci 112, 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moron JA, Brockington A, Wise RA, Rocha BA, Hope BT (2002) Dopamine Uptake Through the Norepinephrine Transporter in Brain Regions with Low Levels of the Dopamine Transporter: Evidence from Knock-Out Mouse Lines. J. Neurosci 22, 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan R, Jonkman JN (2013) A Neural Network Model to Translate Brain Developmental Events across Mammalian Species. PLoS One 8, e53255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J, Kim K. (2008) Abnormal motor function and the expression of striatal dopamine D2 receptors in manganese-treated mice. Biol. Pharm. Bull 31, 1894–7. [DOI] [PubMed] [Google Scholar]
- Oulhote Y, Mergler D, Barbeau B, Bellinger DC, Bouffard T, Brodeur M-È, Saint-Amour D, Legrand M, Sauvé S, Bouchard MF (2014) Neurobehavioral function in school-age children exposed to manganese in drinking water. Environ. Health Perspect 122, 1343–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. (2006) The Rat Brain in Stereotaxic Coordinates Sixth Edition. [DOI] [PubMed] [Google Scholar]
- Peres TV, Ong LK, Costa AP, Eyng H, Venske DKR, Colle D, Gonçalves FM, et al. (2016) Tyrosine hydroxylase regulation in adult rat striatum following short-term neonatal exposure to manganese. Metallomics 8, 597–604. [DOI] [PubMed] [Google Scholar]
- Popichak KA, Afzali MF, Kirkley KS, Tjalkens RB (2018) Glial-neuronal signaling mechanisms underlying the neuroinflammatory effects of manganese. J. Neuroinflammation 15, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner MI, Rothbart MK (1998) Attention, self-regulation and consciousness. Philos. Trans. R. Soc. B Biol. Sci 353, 1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Priller J. (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci 15, 300–12. [DOI] [PubMed] [Google Scholar]
- Prut L, Belzung C. (2003) The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. Eur. J. Pharmacol 463, 3–33. [DOI] [PubMed] [Google Scholar]
- Reichel CM, Wacan JJ, Farley CM, Stanley BJ, Crawford CA, McDougall SA (2006) Postnatal manganese exposure attenuates cocaine-induced locomotor activity and reduces dopamine transporters in adult male rats. Neurotoxicol. Teratol 28, 323–332. [DOI] [PubMed] [Google Scholar]
- Ruff HA, Rothbart MK (2010) Attention in Early Development: Themes and Variations. [Google Scholar]
- Scheggi S, Montis M. G. De, Gambarana C. (2018) DARPP-32 in the orchestration of responses to positive natural stimuli. J. Neurochem 147, 439–453. [DOI] [PubMed] [Google Scholar]
- Schmeichel BE, Berridge CW (2013) Neurocircuitry underlying the preferential sensitivity of prefrontal catecholamines to low-dose psychostimulants. Neuropsychopharmacology 38, 1078–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth PK, Jau-Shyong H, Kilts CD, Bondy SC (1981) Alteration of cerebral neurotransmitter receptor function by exposure of rats to manganese. Toxicol. Lett 9, 247–254. [DOI] [PubMed] [Google Scholar]
- Stephenson DT, Childs MA, Li Q, Carvajal-Gonzalez S, Opsahl A, Tengowski M, Meglasson MD, Merchant K, Emborg ME (2007) Differential loss of presynaptic dopaminergic markers in Parkinsonian monkeys. Cell Transplant. 16, 229–244. [DOI] [PubMed] [Google Scholar]
- Tarale P, Sivanesan S, Daiwile AP, Stöger R, Bafana A, Naoghare PK, Parmar D, Chakrabarti T, Kannan K. (2016) Global DNA methylation profiling of manganese-exposed human neuroblastoma SH-SY5Y cells reveals epigenetic alterations in Parkinson’s disease-associated genes. Arch. Toxicol [DOI] [PubMed] [Google Scholar]
- Tjalkens RB, Popichak KA, Kirkley KA (2017) Inflammatory Activation of Microglia and Astrocytes in Manganese Neurotoxicity. Adv. Neurobiol 18, 159–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman GA, Liu X, Parvez F, Ahsan H, Levy D, Factor-Litvak P, Jennie Kline, et al. (2006) Water Manganese Exposure and Children’s Intellectual Function in Araihazar, Bangladesh. Environ. Health Perspect 114, 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcutt EG (2012) The Prevalence of DSM-IV Attention-Deficit/Hyperactivity Disorder: A Meta-Analytic Review. Neurotherapeutics 9, 490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman AD, Charvet CJ, Clancy B, Darlington RB, Finlay BL (2013) Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci 33, 7368–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Strathearn L, Liu B, Yang B, Bao W. (2018) Twenty-Year Trends in Diagnosed Attention-Deficit/Hyperactivity Disorder Among US Children and Adolescents, 1997–2016. JAMA Netw. Open 1, e181471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA (2012) Genomic Analysis of Reactive Astrogliosis. J. Neurosci 32, 6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Cai T, Liu M, Zheng G, Luo W, Chen J. (2009) Manganese induces dopaminergic neurodegeneration via microglial activation in a rat model of manganism. Toxicol. Sci 107, 156–164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.