

Abstract
Cruzain, an essential cysteine protease of the parasitic protozoan, Trypanosoma cruzi, is an important drug target for Chagas disease. We describe here a new series of reversible but time-dependent inhibitors of cruzain, composed of a dipeptide scaffold appended to vinyl heterocycles meant to provide replacements for the irreversible reactive “warheads” of vinyl sulfone inactivators of cruzain. Peptidomimetic vinyl heterocyclic inhibitors (PVHIs) containing Cbz-Phe-Phe/homoPhe scaffolds with vinyl-2-pyrimidine, vinyl-2-pyridine, and vinyl-2-(N-methyl)-pyridine groups conferred reversible, time-dependent inhibition of cruzain (Ki* = 0.1–0.4 μM). These cruzain inhibitors exhibited moderate to excellent selectivity versus human cathepsins B, L, and S and showed no apparent toxicity to human cells but were effective in cell cultures of Trypanosoma brucei brucei (EC50 = 1–15 μM) and eliminated T. cruzi in infected murine cardiomyoblasts (EC50 = 5–8 μM). PVHIs represent a new class of cruzain inhibitors that could progress to viable candidate compounds to treat Chagas disease and human sleeping sickness.
Graphical Abstract
INTRODUCTION
Chagas disease, caused by the American parasitic protozoan Trypanosoma cruzi (T. cruzi), affects nearly 8 million people in Mexico and Central and South America, resulting in 50,000 annual deaths.1–3 It is estimated that more than 300,000 U.S. residents are infected with T. cruzi, and cases have been reported in California, Texas, and other parts of the Southwestern U.S. in which the infections were acquired locally.4–6 Approximately 20–30% of people infected with T. cruzi will develop debilitating and potentially fatal heart dilation, arrhythmias, apical aneurysms,1 and/or a dilated colon and esophagus.6 The related African trypanosomes Trypanosoma brucei rhodesiense (T. b. rhodesiense) and Trypanosoma brucei gambiense (T. b. gambiense)7 cause human sleeping sickness. Sleeping sickness is endemic in 36 African countries, threatening an estimated 60 million people, with more than 60,000 reported cases.8–10 With the absence of vaccines, the standard treatment for Chagas disease is limited to nifurtimox and benznidazole, which are so replete with side effects that abandonment of treatment is common.11,12
Cysteine proteases of the papain family are of similar structures13 and substrate specificities14,15 and are implicated in numerous human diseases including cancer (cathepsin L),16 chronic obstructive pulmonary disorder (cathepsins C17 and S18) and osteoporosis (cathepsin K19). Likewise, the highly homologous, cathepsin L-like cysteine proteases, cruzain (E.C. 3.4.22.51; UniProt ID: P25779),20–22 rhodesain,23 and brucipain (TbCatL),24 found in the respective parasitic species T. cruzi, T. b. rhodesiense, and T. b. brucei, are essential to the establishment and maintenance of host infection by trypanosomes.20–24 The roles of protozoal cysteine proteases in disease include the scavenging of iron from metalloproteins24 and the evasion of immune surveillance by proteolysis of NF-κB.25 Gene deletion of cruzain demonstrated its role in undermining the prophylactic role of macrophages during infection.26 The work of McKerrow and colleagues demonstrated the biochemical essentiality of cruzain in the pathology of T. cruzi.26–30 They first expressed and characterized cruzain,21 including its crystal structure,22 and identified the drug candidate K11777, a dipeptide vinyl sulfone (Figure 1), which undergoes irreversible thia-Michael addition to active site Cys25.20,31–33 The progression of K11777 to human trials includes demonstration of a cure of T. cruzi infected mammalian cells in the culture29 and also elimination of parasites from acutely infected mice.28 However, K11777 has stalled in preclinical evaluation due to liver toxicity, possibly arising from its irreversible mechanism.13,34,35
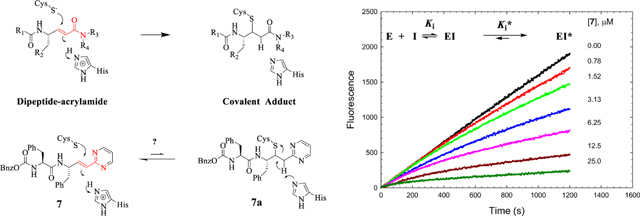
Figure 1.

Structures of K11777, GSK2793660, and Cbz-Phe-Phe-vinyl-2Pyrmd 7 with thia-Michael addition of Cys25 to the vinyl groups in these compounds, including the putative adduct 7a, which reverts to the fully conjugated 7 upon the reverse of adduct formation. The common bioisosteric atoms of K11777, GSK2793660, and 7 are highlighted in red. X is an electron-donating or electron-withdrawing substituent.
Inactivators that form reversible covalent adducts with cysteine groups on enzymes have received recent attention.36–38 Such reversible covalent inactivators demonstrate time-dependent inactivation, like irreversible inactivators, but may exert greater selectivity for the intended target rather than homologous “off-target” enzymes. This is because while the initial–collision complexes of irreversible inactivators with a panel of related enzymes may have variable affinities, over time, the establishment of permanent covalent bonds may render this initial selectivity inconsequential. However, in the case of a reversible covalent inhibitor, their residence times on these enzymes are likely to be variable,38 leading ultimately to “relief” from covalent inactivation for off-targets.
The design of dipeptide vinyl heterocyclic inhibitors is predicated on existing irreversible covalent inactivators of cysteine proteases such as the vinyl sulfone of K11777 and the acrylamide of GSK2793660, an inactivator of human cathepsin C that reached phase I clinical trials.17 Both compounds form irreversible covalent adducts with the active site cysteines of the respective enzymes via a thia-Michael reaction (Figure 1), and both have encountered either toxicity issues or adverse events in, respectively, either animals39 or humans.17 Accordingly, we sought replacements of the vinyl sulfone and acrylamide “warheads” with less electrophilic moieties that would undergo reversible thia-Michael addition, to develop inactivators of high potency for cruzain, but also with suitable selectivity for trypanosomal over human cysteine proteases. One approach is the replacement of the vinyl sulfone and carboxamide group of the acrylamide with a bioisosteric heterocyclic group, which is conjugated to the reactive vinyl group. A vinyl-2-pyrimidine is one such bioisosteric replacement (7, Figure 1) in which one of the ring nitrogens mimics the amide carbonyl and sulfone oxygen while the other substitutes for the amide nitrogen (red atoms in Figure 1). As the pyrimidine is conjugated to the vinyl group, the expected addition of the thiol group of the cysteine to the β-carbon of the vinyl group and attending protonation of the α-carbon would eliminate this conjugation. Subsequent re-establishment of conjugation via reversal of the thia-Michael reaction would provide reversible covalent inactivation of the enzyme. Another feature afforded by this vinyl heterocycle is the ability to modify the reactivity of the vinyl group by the substitution of the heterocycle with electron-donating or electron-withdrawing groups. In this study, we designed, synthesized, and evaluated a panel of dipeptide compounds containing a vinyl group replacing the scissile amide group of the substrate, which is conjugated to a phenyl group or a collection of heterocycles. For some, we investigated the ability of glutathione to form covalent adducts with their vinyl groups to explore the electronic nature of the heterocycle required for facile addition of thiols. Many of these compounds displayed potent, time-dependent inhibition of cruzain and antitrypanosomal activity in cell cultures.
RESULTS AND DISCUSSION
Evaluation of Dipeptide Substrates.
We previously conducted a detailed investigation of the catalytic mechanism of cruzain utilizing three dipeptide substrates and by using solvent kinetic isotope effects in both presteady-state and steady-state kinetic modes.40 As with other cysteine proteases, our kinetic studies supported a double-displacement chemical mechanism in which a thioester intermediate of Cys25 and substrate is first formed (enzyme acylation) concurrent with the release of the amine product. This half-reaction is followed by enzyme-catalyzed hydrolysis of the thioester (enzyme deacylation) to afford the carboxylate product. We determined that for Cbz-Phe-Arg-AMC (S1, Table 1), the most optimal substrate in terms of values of kcat and kcat/Km, and Cbz-Arg-Arg-AMC (S6), enzyme-catalyzed deacylation comprised the slow steps of the reaction (kcat = kdac), while the rates of the acylation of active site Cys25 (described by kac) were 10-fold and 4-fold, respectively, more rapid than kcat.40 The parameter kcat/Km contains the individual rate constants found in the expression for kac and therefore reports on the rates of substrate binding and catalysis up to and including the acylation of active site Cys25. Accordingly, this kinetic parameter comprises a useful guide for the selection of optimal dipeptide scaffolds to be incorporated into inhibitors that are meant to form reversible covalent complexes with Cys25 upon binding, a reaction that chemically mimics acylation. If our dipeptidic vinyl heterocyclic inhibitors do indeed inhibit cruzain by the addition of the thiolate of Cys25 to their olefinic replacements for the scissile amide bond, then the values of kcat/Km (the specificity constants) for the fluorogenic substrates should provide an approximate linear correlation with values of kinact/KI (which is the “specificity constant” of inactivation).41,42
Table 1.
Kinetic Parameters of Peptide Substances for Cruzaina
| substrate | structure | kcat (s−1) | Km (μM) | kcat/Km (μM−1 s−1) | kcat/Km rel | kcat rel |
|---|---|---|---|---|---|---|
| S1 | Cbz-Phe-Arg-AMC | 9.6 ± 0.2 | 0.89 ± 0.09 | 11 ± 1 | 1.00 | 1.00 |
| S2 | Cbz-Phe-hPhe-AMC | 2.0 ± 0.2 | 0.26 ± 0.06 | 8 ± 2 | 0.73 | 0.21 |
| S3 | Cbz-Leu-hPhe-AMC | 3.0 ± 0.2 | 0.8 ± 0.2 | 4 ± 1 | 0.36 | 0.21 |
| S4 | Cbz-Leu-Arg-AMC | 8.4 ± 0.4 | 2.2 ± 0.3 | 3.8 ± 0.7 | 0.35 | 0.88 |
| S5 | Cbz-Phe-(4-Pyr)Ala-AMC | 7.5 ± 0.5 | 3.7 ± 0.5 | 2.0 ± 0.4 | 0.18 | 0.76 |
| S6 | Cbz-Arg-Arg-AMCb | 7.2 ± 0.1 | 3.7 ± 0.4 | 1.9 ± 0.2 | 0.17 | 0.75 |
| S7 | NMePip-Phe-hPhe-AMC | 4.0 ± 0.1 | 3.1 ± 0.3 | 1.3 ± 0.1 | 0.12 | 0.42 |
| S8 | Cbz-(4-Pyr)Ala-hPhe-AMC | 5.3 ± 0.2 | 4.6 ± 0.5 | 1.1 ± 0.1 | 0.10 | 0.55 |
| S9 | Cbz-Phe-Phe-AMC | 0.26 ± 0.01 | 0.34 ± 0.06 | 0.8 ± 0.2 | 0.07 | 0.03 |
| S10 | Cbz-Arg-hPhe-AMC | 4.9 ± 0.1 | 6.8 ± 0.4 | 0.7 ± 0.04 | 0.06 | 0.51 |
| S11 | NMePip-Phe-Phe-AMC | 2.8 ± 0.06 | 14 ± 4 | 0.2 ± 0.04 | 0.02 | 0.29 |
| S12 | Cbz-Phe-Ala-AMCb | 0.89 ± 0.06 | 38 ± 2 | 0.023 ± 0.003 | 0.00 | 0.09 |
Data obtained at pH 7.5, 25 °C, and 10% DMSO (v/v).
Data obtained at 2% DMSO (v/v) from ref 40.
We have kinetically characterized 12 dipeptide fluorogenic substrates of cruzain, which are of the form Cbz/NMePip-P2-P1-AMC, for which Cbz is benzyloxycarbonyl; NMePip is N-methyl-piperazinyl; P2 = Phe, Leu, Arg, or 4-pyridyl-alanine ((4-Pyr)Ala); P1 = Phe, hPhe (homoPhe), Arg, Ala, and (4-Pyr)Ala; and AMC is 7-amino-4-methylcoumarin (Table 1). The largest specificity constant (kcat/Km) measured was that of Cbz-Phe-Arg-AMC (11 μM−1 s−1, relative kcat/Km = 1.0). Substrates S2–S4 have values of kcat/Km, which are 73–35% of that of Cbz-Phe-Arg-AMC, indicating that Cbz-Phe-hPhe-AMC, Cbz-Leu-hPhe-AMC, and Cbz-Leu-Arg-AMC all comprise highly competent substrates likely effecting rapid acylation of cruzain. The poorest substrate, Cbz-Phe-Ala-AMC (S12), demonstrated that a small side chain in the P1 position is less preferable than more bulky hydrophobic moieties. Conversely, the dipeptide Cbz-Phe-hPhe-AMC (S2) exhibited a value of kcat/Km = 8 μM−1 s−1, indicating that a peptide substrate with the same P2 and P1 amino acids as K11777 is an excellent substrate of cruzain and likely involves rapid acylation. Interestingly, when the Cbz group of substrate S2 is replaced with NMePip (S7), a substrate mimic of K11777, kcat/Km is 8-fold lower (1.3 μM−1 s−1). However, its value of kcat exceeded that of Cbz-Phe-hPhe-AMC (S2), suggesting that the NMePip N-terminus of S7 acts to retard cruzain acylation in comparison to the Cbz group of S2. While Cbz-Phe-Phe-AMC (S9) had a value of kcat/Km that was only 7% of Cbz-Phe-Arg-AMC, its Km value of 0.34 μM indicated potent binding, albeit with slow turnover. This may indicate that a P1 Phe substitution leads to favorable binding, but this side chain impedes either the acylation or deacylation step in catalysis. This was observed for dipeptide–AMC substrates for the papain-like protease, human cathepsin C.43
We explored the viability of the (4-Pyr)Ala residue as a potential mimic of both Phe and Arg, for the latter residue when the pyridine is protonated. Cbz-Phe-(4-Pyr)Ala-AMC (S5) was found to be a good substrate (relative kcat/Km = 0.10) but with a higher value of Km (3.7 μM) compared to substrates containing Phe and hPhe in the P1 residue. To leverage the ability of cruzain to tolerate both charged basic and hydrophobic residues in the P2 position, we prepared Cbz-(4-Pyr)Ala-hPhe-AMC and Cbz-Arg-hPhe-AMC (S8 and S10). These substrates displayed efficient turnover numbers (kcat = 5.3 and 4.9 s−1, respectively) but poor values of Km (4.6 and 6.8 μM) in comparison to Cbz-Phe-Phe/hPhe-AMC (0.26–0.34 μM). This demonstrates that the enzyme has a strong preference for substrates that contain a hydrophobic P2 residue. Together, these results indicated that dipeptide scaffolds in which P2 = Cbz-Phe or NMePip-Phe and P1 = Arg, hPhe, or (4-Pyr)Ala may be among the best to incorporate into the vinyl heterocyclic framework. Accordingly, for our new peptidomimetic vinyl heterocyclic inhibitors, we have primarily utilized the Cbz-Phe-Phe and NMePip/Cbz-Phe-hPhe dipeptide scaffolds.
Computer-Assisted Inhibitor Design.
To aid in the rational design of our vinyl heterocyclic inhibitors, we employed molecular docking of these compounds to a model constructed from the crystal structure of K11777-cruzain (PDB accession code: 2OZ2), which contains a covalent bond between the inactivator and Cys25.31 Owing to our hypothesis that the vinyl heterocyclic inhibitors have the ability to undergo a reversible thia-Michael addition with the active site Cys25 of cruzain, it is necessary to consider scenarios of both noncovalent and covalent binding. To this end, we first predicted the binding patterns for NMePip-Phe-hPhe-vinyl-2Pyrmd (9), which has the same scaffold as K11777 using Glide44–46 and CovDock47 modules embedded in the Schrodinger software package. In the covalent model, the binding of 9 with cruzain was highly conserved when compared to that of K11777 (Figure 2A). Pyrimidine N1 of 9 was within hydrogen bonding distance of Gln19 and Trp184, allowing the stabilization of the vinyl heterocycle in a nearly analogous fashion to the sulfone moiety in K11777. In addition, the α-carbon of the inhibitor is positioned within 2.4 Å of His162, an interatomic distance that would easily allow facile proton transfer between this carbon and the imidazole nitrogen, supporting our hypothesis that a reversible adduct could be formed with cruzain. The noncovalent model (Figure 2B) shared similar shape complementarity with the covalent binding pose, except that it was slightly shifted away from the binding site as militated by the docking algorithm to avoid clashing with Cys25. This suggested that covalent bond formation would only slightly perturb the noncovalent binding conformation. Overall, these data suggested that the binding of our newly designed compounds containing a vinyl heterocyclic warhead have the ability to interact with cruzain in a very similar fashion to the characterized, irreversible inactivators of the enzyme. Similarly, we carried out docking for five other PVH compounds (7, 11, 12, 13, and 15 in Figure S1), and their covalent-docking affinity values (Cdock affinity) are summarized in Table S1. The corresponding inhibition constants (predicted Ki) converted from these affinity values ranged from 0.79 to 6.1 μM, with the exception of compound 7, were similar with the experimental values found in Table 2. In agreement with our dipeptide substrate kinetic data, we observed an increased Cdock affinity for 13 and 15 for which each contained an hPhe in the P1 position instead of Phe. In addition, the substitution of a pyridine ring at the P1′ positions of 11, 12, 13, and 15 may subtly improve the binding compared to the pyrimidine substituent of 7. Further, the N-methylation of pyridine resulted in a fairly large shift in Cdock affinity, possibly resulting from an additional ion–ion/dipole interaction. On the basis of these docking analyses, our inhibitor design focused on using a dipeptidic scaffold containing Phe in the P2 position and either Phe or hPhe in the P1 position. We varied the identity of the heterocycle to modify the electrophilicity of the olefin bond but generally maintained functional groups that possibly could afford hydrogen bonding with Gln19 to stabilize the binding of the compounds near Cys25 of cruzain.
Figure 2.

Molecular models of compound 9 bound to cruzain. (A) K11777 (gray) is superimposed with a binding pose (green) in which a covalent bond is formed between the β-carbon of the vinyl group of 9. (B) Binding pose (cyan) of 9 in which no covalent bond is formed with Cys25. Yellow and white dashed lines represent hydrogen bonds with surrounding residues for 9 and K11777, respectively. Red dashed lines are measurements between the catalytic dyad and the vinyl moiety of 9.
Table 2.
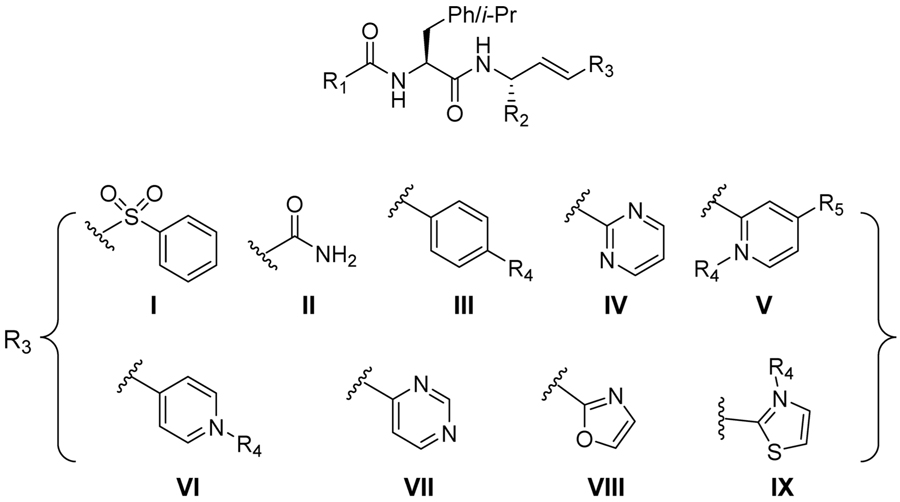
Kinetic Data of Peptidomimetic Vinyl Heterocyclic Inhibitors of Cruzaina
 |
| cruzain inhibition or inactivation |
|||||||
|---|---|---|---|---|---|---|---|
| compound | structure | R1 | R2 | R3 | Ki (μM) | Ki* (μM) | kinact/Kinact (M−1 s−1) |
| K11777 | NMePip-Phe-hPhe-VSPh | NMePip | CH2Bn | I | NA | 0.002b | 234,000b,c |
| 1 | Cbz-Phe-Phe-VSPh | BnO | Bn | I | ND | 0.0036 ± 0.0001 | ND |
| 2 | Cbz-Phe-Phe-vinyl-CONH2 | BnO | Bn | II | 37 ± 2 | NA | 21.7 ± 0.8 |
| 3 | Cbz-Phe-hPhe-vinyl-CONH2 | BnO | CH2Bn | II | 3 ± 1 | NA | 1700 ± 500 |
| 4 | NMePip-Phe-hPhe-vinyl-CONH2 | NMePip | CH2Bn | II | 3.4 ± 0.4 | NA | 1900 ± 200 |
| 5 | Cbz-Phe-Phe-vinyl-Ph | BnO | Bn | III | 1.8 ± 0.1 | 0.87 ± 0.05 | NA |
| 6 | Cbz-Phe-Phe-vinyl-(4-NO2)Ph | BnO | Bn | III, R4 = NO2 | ND | 0.37 ± 0.02 | NA |
| 7 | Cbz-Phe-Phe-vinyl-2Pyrmd | BnO | Bn | IV | 28 ± 1 | 0.364 ± 0.004 | NA |
| 8 | Cbz-Phe-hPhe-vinyl-2Pyrmd | BnO | CH2Bn | IV | >35 | NA | NA |
| 9 | NMePip-Phe-hPhe-vinyl-2Pyrmd | NMePip | CH2Bn | IV | >10 | 2.2 ± 0.1 | NA |
| 10 | Cbz-Phe-Ala-vinyl-2Pyrmd | BnO | Me | IV | 58 ± 6 | 25 ± 1 | NA |
| 11 | Cbz-Phe-Phe-vinyl-2Pyr | BnO | Bn | V | 5.5 ± 0.4 | 0.31 ± 0.01 | NA |
| 12 | Cbz-Phe-Phe-vinyl-2PyrNMe | BnO | Bn | V, R4 = Me | 3.8 ± 0.4 | 0.28 ± 0.08 | NA |
| 13 | Cbz-Phe-hPhe-vinyl-2Pyr | BnO | CH2Bn | V | 1.06 ± 0.07 | 0.171 ± 0.004 | NA |
| 14 | NMePip-Phe-hPhe-vinyl-2Pyr | NMePip | CH2Bn | V | ND | 3.4 ± 0.1 | NA |
| 15 | Cbz-Phe-hPhe-vinyl-2PyrNMe | BnO | CH2Bn | V, R4 = Me | 0.76 ± 0.04 | 0.126 ± 0.004 | NA |
| 16 | Cbz-Phe-hPhe-vinyl-2-(4-OMe)-Pyr | BnO | CH2Bn | V, R5 = OMe | >5 | NA | NA |
| 17 | Cbz-Phe-hPhe-vinyl-2-(4-CF3)-Pyr | BnO | CH2Bn | V, R5 = CF3 | NA | 0.57 ± 0.05 | NA |
| 18 | Cbz-Leu-hPhe-vinyl-2Pyr | BnO | CH2Bn | V | 7.8 ± 0.6 | 1.42 ± 0.09 | NA |
| 19 | Cbz-Phe-Ala-vinyl-2Pyr | BnO | Me | V | ND | 4.8 ± 0.2 | NA |
| 20 | Cbz-Phe-Lys-vinyl-2Pyr | BnO | (CH2)4NH2 | V | 17.3 ± 0.3 | 0.87 ± 0.02 | NA |
| 21 | Cbz-Phe-Phe-vinyl-4Pyr | BnO | Bn | VI | ND | 5.5 ± 0.2 | NA |
| 22 | Cbz-Phe-Phe-vinyl-4PyrNMe | BnO | Bn | VI, R4 = Me | 92 ± 5 | 4.0 ± 0.1 | NA |
| 23 | Cbz-Phe-Phe-vinyl-4Pyrmd | BnO | Bn | VII | 10.8 ± 1.4 | 1.14 ± 0.07 | NA |
| 24 | Cbz-Phe-Phe-vinyl-2Oxz | BnO | Bn | VIII | 10 ± 1 | 0.71 ± 0.01 | NA |
| 25 | Cbz-Phe-Phe-vinyl-2Thz | BnO | Bn | IX | ND | 1.71 ± 0.09 | NA |
| 26 | Cbz-Phe-Phe-vinyl-2ThzNMe | BnO | Bn | IX, R4 = Me | ND | 0.94 ± 0.06 | NA |
Synthesis of PVHIs.
The general synthetic routes employing either Wittig48 or Horner–Wadsworth–Emmons49 reactions shown in Scheme 1 were used to synthesize peptidomimetic vinyl heterocyclic compounds from aldehydes and halomethyl heterocycles.
Scheme 1.

General Synthetic Route to PVHIsa
a Conditions and reagents (General Procedures are detailed in Experimental Section): GP1, N,O-dimethylhydroxylamine hydrochloride, T3P, DIPEA, DCM, and 0 °C; GP2, LAH, THF, and −10 °C; GP3, (1) NaBH4, EtOH, and 0 °C and (2) SOCl2 and DCM; GP4, PPh3, benzene, and reflux; GP5, P(OEt)3 and 150 °C; GP6, LHMDS, THF, −70 to 0 °C; GP7, LHMDS, THF, −70 to 0 °C; GP8, (1) TFA, DCM, and 0 °C and (2) R1-Xaa-OH, T3P, DIPEA, DCM, and 0 °C; GP9, MeI, MeCN, and reflux; GP10, (1) LiOH and H2O and (2)ClCOOEt and NH4Cl.
Commercially available Boc-protected L-amino acids phenylalanine, homophenylalanine, and alanine (a) were converted to Weinreb amides50 by T3P-catalyzed coupling to N,O-dimethylhydroxylamine hydrochloride to afford b (GP1, General Procedure 1 in Experimental Section). Reduction of the Weinreb amide using LAH at −10 °C in anhydrous THF provided the Boc-amino acid aldehyde (c, GP2), generally in overall yields of ~80% (a–c).
Phosphonium salts of methyl heterocycles were, in general, prepared by derivatization of either the 2-methylcarboxy or 2-hydroxymethyl heterocycle (d–f, Scheme 1). Methyl 2-carboxy-pyrimidine (or pyridine, oxazole, and thiazole) was reduced using sodium borohydride to the primary alcohol, followed by conversion of the alcohol to the 2-chloromethylpyrimidine (e) using SOCl2 or POCl3 in DCM or CHCl3 (GP3). The reaction of e with triphenylphosphine provided the Wittig reagent phosphonium salt (f) at overall yields of 28–80% (GP4). Wittig coupling of f with a peptide aldehyde (c) using LHMDS in anhydrous THF or sodium methoxide in benzene as the base provided the peptide vinyl heterocyclic product h (GP6), with general overall yields of 13–54%. Typically, the ratio of E/Z was 4:1, and the separation of these regioisomers was readily achieved using silica gel column chromatography.
Alternatively, the 2-chloromethyl-heterocyclic group e was converted to its phosphonate g by use of the Arbuzov reaction with triethylphosphite (~80% yields, GP5). The resulting phosphonate was deprotonated with LHDMS in THF and then coupled with aldehyde c to provide the peptide vinyl heterocycle h at 20–80% yield (GP7). The Boc group was removed quantitatively by treatment with TFA in DCM, and then the free amine was coupled with the P3–P2 fragment (R1-Xaa-OH) using T3P to give the inhibitor i (GP8).
In addition, some of the PVHIs underwent N-methylation of the heterocycle (j, GP9). Further, we also prepared several acrylamides (k) through hydrolysis of the corresponding acrylate ester and subsequent treatment with ethyl chloroformate and NH4Cl (GP10). Final products were confirmed structurally by NMR and LCMS, as described in the Experimental Section and Supporting Information. It is important to note that proton NMR analysis of the products (i– k) indicated negligible epimerization at the α-carbon in these products, as evidenced by the absence of diastereomers.
Electrophilicity of Vinyl Heterocycles.
To evaluate the chemical reactivity of the vinyl group in our PVHIs and K11777, we treated selected compounds with glutathione (GSH) at pH 8.0 to determine their reactivity in the thia-Michael addition of the sulfhydryl group of GSH with the vinyl group of the inhibitors. Normally, the addition of glutathione to an enzyme inhibitor is to be avoided, but, here, this serves as a means to evaluate the electrophilicity of these inhibitors. K11777 and compounds 7, 11, 12, 15, 17, 25, and 26 (Table 2), which respectively contain vinyl sulfone (R3 = I), vinyl-2-pyrimidine (R3 = IV), vinyl-2-pyridine (R3 = V), vinyl-2-N-methylpyridine (R3 = V; R4 = Me), vinyl-2-(4-trifluoromethyl)-pyridine (R3 = V, R5 = CF3), vinyl-2-thiazole (R3 = IX), and vinyl-2-N-methylthiazole (R3 = IX; R4 = Me). As previously reported,51 the formation of a glutathione adduct with K11777 was very slow (k = 0.00028 s−1, Table S2; Figure 3), and we were unable to ascertain an equilibrium constant for the K11777-GSH adduct. For the PVHIs, the reaction between GSH and vinyl-2-pyrimidine (7), vinyl-pyridine (11), vinyl-2-(4-trifluoromethyl)-pyridine (17), and vinyl-thiazole (25) was negligible as no adduct was observed after 90 min of incubation with either a 2:1 or 10:1 molar ratio of GSH/inhibitor. The electron-withdrawing 4-trifluoromethyl group on the pyridine of 17 had no effect on the electrophilicity of 11. In contrast, the addition of GSH to the vinyl group of vinyl-2-N-methylpyridine (12 and 15) and vinyl-2-N-methylthiazole (26) in a 2:1 molar ratio resulted in the rapid formation of GSH adducts at respective rates of 0.037, 0.054, and 0.015 mM−1 s−1, and apparent equilibrium was achieved for these compounds in 90 min (Keq = 7400, 2400, and 930 M−1, respectively). This demonstrated that the N-methylation of the PVHs afforded a significant increase in the electrophilicity of the vinylic position, enabling rapid addition to thiols, owing to the strong electron-withdrawing effect of the methylpyridinium moiety. For example, compounds 11 and 12 are identical except for the N-methylpyridine group of compound 12; compound 12 readily forms an adduct with GSH (97% conversion of 12 to its GSH adduct in 20 min at a 10:1 molar ratio of GSH to compound), whereas compound 11 is unreactive toward GSH. Interestingly, the rate of thiolation of compound 12 is 50% that of 15, while the values of Keq indicated that the 12-GSH adduct is three times more abundant than that of 15-GSH. This suggested that the phenylalanyl side chain of 12 may retard the addition of GSH to its vinyl group and also slowed the presumed base-catalyzed elimination of GSH from its adduct with 12. Overall, these results demonstrated that the reactivity of vinyl heterocycles with GSH and presumably Cys25 vary with the nature of the heterocycles. Hence, it is possible to tune the electronic properties of the vinyl bond in the PVHIs, thereby allowing for development of modifiable electrophilic inhibitors of other enzymes that have an active site cysteine or other nucleophile.
Figure 3.

Time courses of depletion of 0.5 mM K11777 (red circles), compound 12 (blue circles), and compound 15 (black circles) upon formation of adducts (red, blue, and black squares, respectively) with 1 mM glutathione. Lines drawn through the curves for substrate depletion and adduct formation were respectively [substrate] = (0.5 mM − A)(1 − exp(−k ×t)) + C and [adduct] = A × (1 − exp(−k × t)) + C, with resulting kinetic parameters found in Table S2.
Kinetic Analysis of Cruzain Inhibitors and Inactivators.
Scheme 2 is a kinetic depiction of inhibition and inactivation of cruzain and the relevant kinetic parameters.52 The initial, and usually rapid, formation of EI is characterized by the inhibition constant Ki. For time-dependent inhibitors, EI progresses to a second, tighter complex EI*, generally over the course of minutes, characterized by Ki*, for which Ki* < Ki when k4 < k3. For irreversible covalent inactivators, k4 and Ki* ≈ 0, and the kinetic parameter kinact/KI is generally reported. For reversible time-dependent inhibitors, initiation of the reaction by adding enzyme to the substrate and inhibitor leads to concave-downward, curvilinear time courses of product formation in which reaction rates demonstrably decrease as the EI* complex forms. Typical data, as exemplified for compound 15, are shown in Figure 4A. Alternatively, extended preincubation of the enzyme and inhibitor, followed by dilution of the inhibitor and initiation of the reaction with high concentrations of the substrate, leads to concave-upward curvilinear plots of product formation as E reforms from EI* (Figure 4B). Results of this analysis for cruzain inhibitors and inactivators are collected in Table 2.
Scheme 2.

Kinetic Depiction of Inhibition and Inactivation
Figure 4.

Time-dependent inhibition of cruzain by 15. (A) Reaction initiated by addition of cruzain (100 pM) with Cbz-Phe-Arg-AMC (10 μM) and 0–20 μM 15 (pH 7.5). Lines drawn through the experimental data points were from fitting of each inhibitor concentration to eq 3, from which the replot of kobs vs [15] is shown in the inset (fitting to eq 4: Ki = 2.00 ± 0.9 μM, k3 = 0.004 ± 0.001 s−1, and k4 ≈ 0). (B) Following 1 h of preincubation of cruzain (100 pM) with 0–6 μM 15, the reaction was initiated by addition of Cbz-Phe-Arg-AMC (10 μM). (C) Fitting of cruzain inhibition by compound 15 for vi/v0 (black) and vs/v0 (red) using eq 5 with results of this found in Table 2.
K11777 comprises a useful benchmark compound despite the fact that it is an irreversible inactivator of cruzain (reported kinetic data: apparent IC50 of 2 nM, kinact/KI = 234,000 M−1 s−1).32,53 We replaced the P1 hPhe group of K11777 with a Phe side chain to provide vinyl sulfone 1, which had apparently equivalent potency (Ki* = 3.6 nM) to that of K11777, but which, interestingly, exhibited kinetically reversible inhibition of cruzain. However, a crystal structure we obtained for 1 bound to cruzain indicated the formation of a C–S bound between Cys25 and 1 (Figure S4). This may indicate that a phenylalanyl group at the P1 position partly impedes the ability of an adjacent vinyl electrophile to access Cys25, as was observed with the solution phase GSH addition studies to our PVH compounds.
We next evaluated three C-terminal acrylamides (R3 = II) within the Cbz-Phe-Phe, Cbz-Phe-hPhe, and NMePip-Pheh-Phe scaffolds (2–4). The acrylamides within the Cbz-Phe-hPhe and NMePip-Phe-hPhe scaffolds afforded apparently irreversible covalent inactivation (kinact/KI = 1700–1900 M−1 s−1), while Cbz-Phe-Phe-acrylamide (2) was less effective (kinact/KI = 22 M−1 s−1). Comparing the values of kinact/KI for K11777 and 4 indicated that the vinyl sulfone is over-whelmingly more effective as a covalent inactivator than its acrylamide counterpart, possibly owing to hydrogen bond contacts of the sulfone oxygen with Gln19, which position the vinyl group proximal to Cys25 of cruzain. As with 1, a Phe rather than an hPhe group at the P1 position may retard covalent formation over the time course of kinetic analysis when one compares the rates of apparent inactivation of 2 versus 3 and 4, as was also seen with peptide substrates.
We therefore sought to explore the effects of replacement of both the vinyl-phenylsulfone and acrylamide groups with phenyl and heterocyclic groups conjugated to the vinyl group. The Cbz-Phe-Phe-vinyl-benzene compound 5 is a time-dependent inhibitor of cruzain (Ki* = 0.87 μM), but substitution of the para position of the phenyl ring with an electron-withdrawing nitro group (compound 6) led to a nearly 3-fold improvement in potency (Ki* = 0.34 μM), suggesting that the vinyl group of 6 is more capable of thiolation by the cruzain. As seen with 1, these compounds also demonstrated reversible inhibition of cruzain, possibly due to the P1 phenylalanine. Due to poor aqueous solubility (solubility of 5 and 6, ≤2 μM in 10% DMSO), the inhibitors containing vinyl-benzene were not explored further.
Subsequently, six heterocyclic groups (R3 = IV–IX) conjugated to the presumed electrophilic vinyl group were evaluated within several dipeptide scaffolds. The vinyl-2-pyrimidine (R3 = IV), vinyl-2-pyridine (R3 = V), vinyl-2-oxazole (R3 = VIII), and vinyl-2-thiazole (R3 = IX) groups, unlike the vinyl-4-pyridine (R3 = VI) and vinyl-4-pyrimidine (R3 = VII), maintain bioisosteric similarity to the reactive acrylamides and vinyl sulfones, which is reflected in their more potent inhibition of cruzain as detailed below. Most of these compounds induced time-dependent inhibition on cruzain and were found to be kinetically reversible with residence times (τ) of 6–20 min.
The vinyl-2-pyrimidine moiety (R3 = IV) in the Cbz-Phe-Phe scaffold afforded compound 7, which exerted time-dependent inhibition of cruzain with an initial value of Ki = 5 μM and subsequent tight-binding inhibition of Ki* = 0.38 μM. Substitution of the phenyl group of 5 by a pyrimidine group greatly improved the solubility of 7 (≥100 μM in 10% DMSO). Extended preincubation with 7, followed by dilution, and addition of an excess of substrate, resulted in slow recovery of cruzain activity, indicating that any covalent reaction between cruzain and 7 was kinetically reversible (k4 = 0.0018 ± 0.0003 s−1; τ = 9 min). Interestingly, when the 2-pyrimidinyl moiety is appended to Cbz-Phe-hPhe (8), the resulting compound is a poor inhibitor of cruzain (Ki > 35 μM); however, when the 2-pyrimidinyl group is attached to afford the same scaffold as K11777, we obtained an inhibitor of low micromolar potency (9, Ki* = 2.2 μM). Substitution of the P1 Phe with Ala (10, Ki = 25 μM) produced a poor inhibitor of cruzain, indicating the essentiality of a larger side chain in the P1 position, as was observed with dipeptide substrates. To probe the importance of the vinyl group for the inhibition of cruzain, we prepared an analogue in which the vinyl group of 7 was reduced (compound 27). This inhibitor lacked time-dependent behavior (Ki = 22 μM) and was 100-fold less potent than its vinyl analogue 7, which demonstrated the importance of the vinyl group for the inhibition of cruzain. We prepared inhibitor 23, which contains a vinyl-4-pyrimidinyl (R3 = VII) group that does not maintain bioisosteric similarity to the acrylamides. 23 exhibited 3-fold less potency than the bioisosteric vinyl-2-pyrimidine (7). Similarly, inhibitors containing the vinyl-4-pyridyl (R3 = VI) (21 and 22) lack bioisosteric equivalence to the acrylamides and were found to be only modest inhibitors of cruzain.
Inhibitors containing a vinyl-2-pyridinyl group (R3 = V) were explored more widely. Cbz-Phe-Phe-vinyl-2-pyridine 11 exhibited time-dependent inhibition of cruzain with an initial value of Ki = 5.5 μM and subsequent tight-binding inhibition of Ki* = 0.31 μM (k4 = 0.0012 ± 0.0002 s−1; τ = 13 min), and solubility of 11 was ≤30 μM in 10% DMSO. Unlike the vinyl-pyrimidinyl group of 7, placement of the vinyl-2-pyridinyl group in the Cbz-Phe-hPhe scaffold improved inhibition by 3-fold (13, Ki* = 0.17 μM), while the vinyl-2-pyridinyl group was much less effective in the NMePip-Phe-Phe scaffold (14, Ki* = 3.4 μM). Substitution of an electron-donating methoxy group on the pyridine ring (16) of the Cbz-Phe-hPhe scaffold diminished the inhibitory activity of the vinyl-2-pyridinyl heterocycle compared to its unsubstituted counterpart 13 by >50-fold, suggesting that the methoxy group is large enough to create a steric barrier to inhibitor binding. In contrast, the substitution at C-4 of the pyridine with the electron withdrawing trifluoromethyl group resulted in better inhibition (17, Ki* = 0.57 μM), but nonetheless was less potent than the unsubstituted pyridine 13. Apparently, this result arises from steric crowding as OMe > CF3 > H, implicating that substitution at the C-4 position of the pyridine heterocycles are not well tolerated.
We next investigated how the P1 and P2 side chains of these vinyl-2-pyridinyl inhibitors affect inhibition. The replacement of the P2 Phe with Leu resulted in diminished potency (18, Ki* = 1.42 μM) compared to the Cbz-Phe-Phe and Cbz-Phe-hPhe scaffolds, overall demonstrating that inhibitors with bulky hydrophobic substituents in P1 and P2 enhanced binding to cruzain. To analyze how short alkyl and charged groups effected inhibition, we prepared Cbz-Phe-Ala-vinyl-2-pyridine (19) and Cbz-Phe-Lys-vinyl-2-pyridine (20). We found that the Cbz-Phe-Lys scaffold, which mimics our most optimal substrate, Cbz-Phe-Arg-AMC, exhibited good inhibition (Ki* = 0.87 μM), whereas 19 was a poor inhibitor (Ki* = 4.8 μM), in concert with the poor substrate activity of Cbz-Phe-Ala-AMC.
Seeking to improve the electrophilicity of the vinyl-2-pyridinyl group, we prepared N-methylated analogues 12 and 15. This modification resulted in improved aqueous solubility (≥ 50 μM in 10% DMSO) and provided potent time-dependent inhibition of cruzain (12, Ki* = 0.28 μM; 15, Ki* = 0.126 μM) comparable to, or exceeding, the inhibition exerted by their unmethylated counterparts (11 and 13).
Inhibition data for compound 15 were fitted by all methods outlined in Experimental Section, as shown in Figure 4. We fitted each curve in Figure 4A to eq 3, and the resulting values of kobs were replotted versus [15] (Figure 4A, inset), which demonstrated a hyperbolic dependence of the inhibitor (fitting to eq 4: Ki = 2.0 ± 0.9 μM, k3 = 0.004 ± 0.001 s−1, and k4 ≈ 0). Alternatively, global fitting of these curves to eq 6 provided values of Ki = 4.3 ± 0.1 μM, k3 = 0.0012 ± 0.0004 s−1, and k4 = 0.00019 ± 0.00005 s−1, from which was calculated a value of Ki* = 0.6 ± 0.2 μM. Fitting of the data globally for 15 and other potent PVHIs using KinTek Explorer is found in the Supporting Information. Preincubation of cruzain and variable concentrations of 15, followed by initiation of the reaction by the addition of substrate, produced time courses like that shown in Figure 4B. These data demonstrated a significant lag phase for recovery of cruzain activity, indicative of the slow desorption of the inhibitor, with or without the formation of a covalent bond with Cys25. Finally, analysis of inhibition of cruzain by 15 at early and late phases of the time courses in Figure 4A by fitting to eq 5 provided values of Ki = 0.76 ± 0.04 μM and Ki* = 0.126 ± 0.004 μM (Table 2). Of note, in preincubation studies, all PVHIs, which contain the vinylpyridinyl substituent, displayed kinetic reversibility.
We investigated five-membered ring heterocycles that are bioisosteric with acrylamide inactivators. The syntheses of vinyl-2-oxazole (24), vinyl-2-thiazole (25), and its N-methylated counterpart (26) into the Cbz-Phe-Phe scaffold proved facile and provided useful inhibitors. Vinyl-2-oxazole 24 was a submicromolar inhibitor of cruzain (Ki* = 0.71 μM). Vinyl-2-thiazole inhibitors 25 and 26 were inhibitors of similar potency (Ki* = 1.71 and 0.94 μM, respectively), for which N-methylation of the thiazole improved potency by nearly 2-fold.
Cruzain inhibitors 5–26 allowed the evaluation of six heterocyclic groups (R3 = IV–IX) appended to the presumed electrophilic vinyl group within several dipeptide scaffolds. The vinyl-2-pyrimidine, vinyl-2-pyridine, vinyl-2-N-methylpyridinium, vinyl-2-oxazole, and vinyl-2-thiazole substituents, unlike the vinyl-4-pyrimidine and vinyl-4-pyridine heterocycles, maintained bioisosteric similarity to the reactive acrylamides and provided potent, time-dependent inhibitors in accord with our hypothesis. Of these PVHIs, 2-pyridine, charged 2-N-methylpyridine, and vinyl-2-pyrimidine presented the most interesting heterocycles for further exploration. The inhibition of cruzain displayed by these PVHIs may be due to the reversible formation of an adduct with active site Cys25, as is supported by the loss of time-dependent inhibition when the vinyl group is saturated. Importantly, we have no evidence that such a reversible covalent bond is formed, and ongoing studies are underway to address this point.
Selectivity of PVHIs for Cruzain over Homologous Human Cathepsins.
Cruzain has 25, 15, and 23% amino acid identity with human cathepsins L, B, and S respectively.31,55 It is preferable to proceed with cruzain inhibitors that do not readily inhibit these human lysosomal cathepsins, which might engender cellular toxicity. We evaluated selected cruzain inhibitors versus the human cysteine proteases cathepsins L, B, and S (Table 3). For this selectivity comparison, all inhibition data were obtained at pH 5.5 for which Ki* values were invariant for all inhibitors except compound 15 (Ki* = 88 nM). The cruzain inhibitors demonstrated moderate selectivity versus cathepsins L and S (generally, 3-fold or greater), while all of these inhibitors displayed 40-fold or higher selectivity versus cathepsin B. The vinyl-2-pyridine inhibitors 13 and 15 are particularly selective as their Ki values are over 10-fold lower than the corresponding values with the three human cathepsins. In contrast, K11777 showed potent inactivation at nanomolar concentrations for all three human cathepsins; this apparent lack of selectivity possibly arising from its irreversible mode of inactivation. These results suggest that suitable selectivity for reversible cruzain inhibitors may be more easily attained than for irreversible ones.
Table 3.
Enzymatic Selectivity of Cruzain Inhibitorsa
|
Ki* (μM) |
|||||
|---|---|---|---|---|---|
| compound | structure | cruzain | human cathepsin L | human cathepsin B | human cathepsin S |
| K11777 | NMePip-Phe-hPhe-VSPh | IC50 = 0.2 nMb | IC50 = 0.2 nMb | IC50 = 5.7 nMb | IC50 = 0.6 nMb |
| 7 | Cbz-Phe-Phe-vinyl-2Pyrmd | 0.28 ± 0.01 | 1.1 ± 0.1 | 32 ± 3 | 0.37 ± 0.02 |
| 11 | Cbz-Phe-Phe-vinyl-2Pyr | 0.28 ± 0.02 | 4.3 ± 0.5 | 28 ± 4 | 1.8 ± 0.3 |
| 12 | Cbz-Phe-Phe-vinyl-2PyrNMe | 0.30 ± 0.02 | 0.70 ± 0.04 | 19 ± 4 | 0.87 ± 0.07 |
| 13 | Cbz-Phe-hPhe-vinyl-2Pyr | 0.123 ± 0.004 | 1.9 ± 0.2 | 6.5 ± 0.9 | 1.41 ± 0.06 |
| 15 | Cbz-Phe-hPhe-vinyl-2PyrNMe | 0.088 ± 0.002 | 0.88 ± 0.06 | 37 ± 6 | 0.32 ± 0.04 |
Inhibition data obtained at pH 5.5 and 25 °C in 10% DMSO (v/v).
Reported as apparent IC50 in ref 32.
Effects of PVHIs in Axenic Cultures of T. cruzi and in a Cell Model of T. cruzi Infection.
Initially, we tested selected compounds against epimastigotes of T. cruzi (strain Y, ATCC 50832GFP) in axenic cultures. As is observed here (Table 4) and has been shown previously, K11777 weakly inhibited the growth of T. cruzi epimastigotes (EC50 ≈ 60 μM).54 PVHIs 7, 12, and 15 inhibited the growth of epimastigotes of T. cruzi (EC50 = 2–20 μM), while 11, 13, and 24 were poorly effective. Compounds 12 and 15 were comparably potent against cultures of T. cruzi (EC50 = 8.6 and 2.1 μM, respectively), and were, at a minimum, 10-fold more active than K11777.
Table 4.
Effects of Cruzain Inhibitors on Trypanosome and Human Cell Growtha
| compound | cruzain Ki* (μM) |
T. cruzi axenic culture EC50 (μM) | T. cruzi-infected cardiomyoblasts (C2C12) EC50 (μM) | T. brucei brucei PCFs EC50 (μM) | T. brucei brucei BSFs EC50 (μM) | human cell cytotoxicity CC50 (μM) | C2C12 Cytotoxicity CC50 (μM) | selectivity index CC50/EC50 |
|---|---|---|---|---|---|---|---|---|
| K11777 | IC50 = 2 nMb | >20 | 0.7 ± 0.2 | 1.7 ± 0.5 | 0.09 ± 0.06 | 60–100 | >10 | 140 |
| 7 | 0.364 ± 0.004 | 20 | 9.0 ± 0.5 | 7.1 ± 0.9 | 10.4 ± 0.2 | >100 | >10 | >10 |
| 9 | 2.2 ± 0.1 | 20 | ND | 15 ± 2 | >20 | ND | ND | ND |
| 11 | 0.31 ± 0.01 | >20 | 4.9 ± 0.2 | 5 ± 1 | 5 ± 4 | >100 | >10 | >20 |
| 12 | 0.28 ± 0.08 | 8.7 ± 0.1 | 9.9 ± 0.5 | 13 ± 3 | 6.6 ± 0.6 | >100 | >10 | >10 |
| 13 | 0.171 ± 0.004 | >20 | 5.9 ± 0.3 | >10 | 4 ± 2 | >100 | >10 | >20 |
| 15 | 0.126 ± 0.004 | 2.1 ± 0.1 | 5.4 ± 0.9 | 5.9 ± 0.2 | 2.8 ± 0.1 | >100 | >10 | >20 |
| 24 | 0.71 ± 0.01 | >20 | ND | 27 ± 5 | ND | >100 | ND | ND |
Effects of inhibitors were evaluated as EC50 for axenic T. cruzi, PCFs/BSFs of T. b. brucei and T. cruzi-infected murine cardiomyoblasts. The selectivity index is the ratio of inhibitor cytotoxicity in human dermal fibroblasts (CC50)/trypanocidal activity (EC50) in infected cardiomyoblasts. ND, not determined.
Ref 32.
Selected cruzain inhibitors were further evaluated in a more relevant cellular model of Chagas disease: T. cruzi-infected murine cardiomyoblasts (C2C12 cells) (Table 4 and Figure 5). Inhibitors 7, 11, 12, 13, and 15 exhibited antiparasitic efficacy at values of EC50 = 5–10 μM while displaying no cytotoxicity against the host cardiomyoblasts (CC50 > 10 μM). These EC50 values demonstrated that the antitrypanosomal activities of the reversible PVHIs are within an order of magnitude of potency of the irreversible inactivator, K11777 (EC50 = 0.7 μM), despite the large difference in activity versus cruzain. Accordingly, the PVHIs, while reversible in action and with no apparent mammalian or human cytotoxicity, are nearly as effective as the potent, irreversible inactivator K11777. Further, the best of the PVHIs is less than 3-fold less potent than the currently used antichagasic drug benznidazole (LD50 = 1.5 μM),55 suggesting that a second generation of PVHIs may provide clinical candidates.
Figure 5.

(A–F) Effects of cruzain inhibitors on growth of T. cruzi-infected murine cardiomyoblasts in which growth of inhibition of T. cruzi (red) is superimposed with the viability of the cardiomyoblasts (blue).
Effects of PVHIs in Axenic Cultures of T. b. brucei.
We additionally tested our cruzain inhibitors in axenic cultures of the related protozoan T. b. brucei owing to the high structural similarity and reported essentiality of the cysteine protease brucipain (TbCatL) in T. b. brucei.24,34,57 It has been demonstrated that the cruzain inhibitor K11777 is active in cellular cultures of both T. b. brucei and T. cruzi, supporting the notion that our PVHIs could be effective in growth inhibition of both species of parasite. For insect procyclic forms (PCFs) of T. b. brucei (ATCC PRA-381), compounds 7, 9, 11, 12, and 15 demonstrated growth inhibition at EC50 values of 5–15 μM (Table 4 and Figure 6). When compared to K11777 (EC50 = 1.7 μM), these PVHIs exhibited potent cell growth inhibition. For example, compound 15 (EC50 = 5.9 μM) was only 3-fold less potent versus T. b. brucei than K11777. Values of EC50 for these PVHIs roughly correlated with their values of Ki*, with the exception of compound 13.
Figure 6.

Cell growth inhibition of T. b. brucei. (A) Inhibition of bloodstream forms by 15. (B) Inhibition of bloodstream forms by 7. (C) Inhibition of procyclic forms by 15. (D) Correlation plot of values of EC50 for trypanocidal activity versus T. b. brucei BSFs (closed circles, r2 = 0.979, slope = 0.80) and T. cruzi in murine cardiomyoblasts (open circles).
We next evaluated these inhibitors in axenic cultures of human bloodstream forms (BSFs) of T. b. brucei (ATCC PRA-383). All PVHIs that were active versus procyclic forms of T. b. brucei were also trypanocidal versus the bloodstream forms but with equal or lower EC50 values compared to the procyclic forms (Table 4). Compared to PVHIs that had similar potencies in both PCFs and BSFs, K11777 was nearly 20-fold more potent in T. b. brucei BSFs than in PCFs. These results suggested that a cathepsin L-like cysteine protease in T. b. brucei, such as brucipain (or TbCatL),24 is essential for growth of procyclic and bloodstream T. b. brucei, but perhaps an additional cysteine protease, such as TbCatB, is also essential in BSFs of T. b. brucei as this enzyme is sensitive to K11777 but not to the PVHIs. This is similar to the findings of Yang et al.58 who showed using an activity-based protein probe of K11777 that TbCatB and brucipain (TbCatL) are both labeled in BSFs of T. b. brucei while only brucipain is labeled in PCFs. This could explain the exceptional trypanocidal activity of K11777 in BSFs. This will be the focus of our future studies. Nonetheless, the activity of the PVHIs versus T. b. brucei BSFs may hold promise for progression to their evaluation in models of African trypanosomiasis.
Interestingly, the values of EC50 obtained for PCFs of T. b. brucei and amastigotes of T. cruzi were nearly identical for most PVHIs despite their more modest inhibition of cruzain. Shown in Figure 6D is a correlation plot of log EC50 for antitrypanosomal activity for bloodstream forms of T. b. brucei and the amastigote forms of T. cruzi from the murine cardiomyoblast infection model. For the former, the correlation is excellent (r2 = 0.979, slope = 0.80), and the activity against parasites is nearly a 1:1 correlation with log Ki with these inhibitors. This result provided support that our PVHIs are targeting a cruzain-like protease in T. b. brucei. For T. cruzi, this correlation is not as strong, in part, due to the absence of a sufficient range of data. We have also compared the cytotoxicity of selected inhibitors in human dermal fibroblasts versus T. cruzi-infected cardiomyoblasts (selectivity index in Table 4), which demonstrates that the PVHIs are more than 10-fold selective for trypanosomes versus human cells.
CONCLUSIONS
We have developed a novel class of reversible inhibitors for the essential cysteine protease of T. cruzi, cruzain. These compounds, peptidomimetic vinyl heterocycles, contain bioisosteric replacements for the acrylamide and vinyl sulfone warheads present in irreversible, covalent inactivators such as K11777. We also demonstrated that PVHIs containing vinyl-2-N-methylpyridine or vinyl-2-N-methylthiazole groups, unlike other inhibitors, readily form Michael adducts with glutathione. Our survey demonstrated that the most optimal cruzain inhibitors contained vinyl-2-pyrimidine, vinyl-2-pyridine, and vinyl-2-N-methylpyridinium groups. These PVHIs proved to be potent, time-dependent inhibitors of cruzain, albeit fully reversible in terms of the mode of action. These PVHIs are significantly active in both axenic cultures of T. b. brucei and in a cell infection model of T. cruzi, and further optimization may produce more potent antitrypanosomal agents. Importantly, the concept of reversible covalent inactivation by vinyl heterocycles is potentially expandable to other enzymes, which contain active site cysteines, such as EGFR, G12C K-Ras, and other protein kinases for which irreversible acrylamide inactivators comprise effective drugs.56
EXPERIMENTAL SECTION
General Synthetic Chemistry Methods and Compound Characterization.
All reagents and starting materials were obtained from commercial suppliers and used without further purification unless otherwise stated. Reactions were run under an atmosphere of nitrogen or argon and at ambient temperature unless otherwise noted. Reaction progress was monitored using thin-layer chromatography and by analysis employing an HPLC–MS (UltiMate 3000 equipped with a diode array coupled to an MSQ Plus single quadrupole mass spectrometer, Thermo Fisher Scientific) using electrospray positive and negative ionization detectors. Reported liquid chromatography retention times (tR) were established using the following conditions: column: Phenomenex Luna 5 μm C18(2) 100 Å, 4.6 mm, 50 mm; mobile phase A: water with 0.1% formic acid (v/v); mobile phase B: MeCN with 0.1% formic acid (v/v); temperature: 25 °C; gradient: 0–100% B over 6 min, then a 2 min hold at 100% B; flow: 1 mL min−1; and detection: MS and UV at 254, 280, 214, and 350 nm.
Semi-preparative HPLC purification of compounds was performed on a Thermo Fisher Scientific UltiMate 3000 with a single wavelength detector coupled to a fraction collector. Purifications were conducted using the following conditions: column: Phenomenex Luna 5 μm C18(2) 100 Å, 21.2 mm, 250 mm; mobile phase A: water with 0.1% formic acid (v/v); mobile phase B: MeCN with 0.1% formic acid (v/v); temperature: room temperature; gradient: 0–100% B over 30 min, then a 5 min hold at 100% B; flow: 20 mL min−1; and detection: UV (254 nm).
1H/13C NMR magnetic resonance spectra were obtained in CDCl3, CD3OD, or DMSO-d6 at 400 MHz/100 MHz at 298 K on a Bruker AVANCE III Nanobay console with an Ascend magnet unless otherwise noted. The following abbreviations were utilized to describe peak patterns when appropriate: br = broad, s = singlet, d = doublet, q = quartet, t = triplet, and m = multiplet. All final compounds used for testing in assays and biological studies had purities that were determined to be >95% as evaluated by their proton NMR spectra and their HPLC/MS based on ultraviolet detection at 254 nm (see the Supporting Information). Similar RP-HPLC conditions were used for the experiments of GSH addition to vinyl heterocycles. Masses detected were in the range of 100–1000 Da and were detected in the positive or negative mode, depending on the ionization of the molecule.
General procedures (GP1-GP10 in Scheme 1) of synthesizing PVHIs are detailed below. Each GP described the synthesis of one representative compound. In addition, substrate synthesis and characterization are provided in the Supporting Information.
GP1: Synthesis of Weinreb Amides (a to b).
A solution of Boc-l-homophenylalanine (12.02 g, 43.03 mmol) in anhydrous DCM (200 mL) was cooled to 0 °C under a N2 atmosphere. Et3N (18.1 mL, 129.09 mmol, 3 equiv) was added slowly, followed by the addition of N,O-dimethylhydroxylamine hydrochloride (6.3 g, 64.5 mmol, 1.5 equiv) and dropwise addition of T3P (50% (w/v) in MeCN, 41.1 mL, 64.55 mmol, 1.5 equiv). The resulting mixture was stirred at 0 °C for 30 min to 1 h until TLC analysis (EtOAc/hexane = 1:1, v/v) showed the disappearance of the starting material. The reaction mixture was diluted with DCM and washed with H2O. The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo to afford the crude product. Purification of the crude product by silica gel column chromatography using a gradient of 5–50% of EtOAc in hexane as the eluent yielded the pure Weinreb amide tert-butyl (S)-(1-(methoxy(methyl)amino)-1-oxo-4-phenylbutan-2-yl)-carbamate (b, 13.3 g, 41.31 mmol, 96% yield) as a colorless gum.
GP2: LAH Reduction of Weinreb Amides (b to c).
To a solution of tert-butyl (S)-(1-(methoxy(methyl)amino)-1-oxo-4-phenylbutan-2-yl)carbamate (b, 6.7 g, 20.78 mmol) in anhydrous THF (120 mL) at −10 °C under a N2 atmosphere was added dropwise LAH (2.0 M in THF, 12.5 mL, 24.93 mmol, 1.2 equiv). The resulting mixture was stirred at −10 °C for 30 min. Upon completion of the reaction as shown by TLC analysis (EtOAc/hexane = 1:1, v/v), the reaction was quenched at the same temperature by adding dropwise 1 N HCl, followed by removal of THF by rotary evaporation. Diethyl ether (500 mL) was added to the solid residue, and the solution was washed with aqueous NaHCO3 (1 × 50 mL) and brine (1 × 50 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo to afford the crude product. Purification of the crude material by silica gel column chromatography using a gradient of 10–60% of EtOAc in hexane as the eluent yielded the pure aldehyde tert-butyl (S)-(1-oxo-4-phenylbutan-2-yl)carbamate (c, 4.89 g, 18.57 mmol, 89% yield) as a white solid.
GP3: Preparation of Chloromethyl Heterocycles (d to e).
To a suspension of methyl pyrimidine-2-carboxylate (d, 1.156 g, 8.37 mmol) in anhydrous EtOH (20 mL) at 0 °C under a N2 atmosphere was added portionwise NaBH4 (0.443 g, 11.72 mmol, 5 equiv). The reaction mixture was stirred at 25 °C for 2 h. Upon completion of the reaction as shown by TLC analysis (EtOAc/hexane = 1:1, v/v), the reaction solvents were removed by rotary evaporation. To the resultant colorless gummy residue was added ice cold H2O (20 mL) followed by extraction with DCM (5 × 50 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo to afford the crude product pyrimidin-2-yl-methanol (0.900 g, 8.17 mmol). To this pyrimidin-2-yl-methanol in CHCl3 (20 mL) at 0 °C under a N2 atmosphere was added dropwise POCl3 (1.95 mL, 3.21 g, 2.5 equiv). The reaction mixture was stirred at 25 °C for 1 h, followed by refluxing for an additional 3 h under gentle heating until TLC analysis (EtOAc/hexane = 3:1, v/v) showed the completion of the reaction. The reaction was quenched by a careful addition of aqueous NaHCO3 and further addition of solid NaHCO3 to afford a basic pH. The aqueous layer was extracted with CHCl3 (3 × 50 mL), and the organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo to afford the pure product 2-(chloromethyl)pyrimidine (e, 0.948 g, 7.43 mmol, 63% yield) as a light yellow semi-solid, which was used further without any purification.
GP4: Preparation of Heterocyclic Phosphonium Ylides (e to f, Wittig Reagents).
A mixture of 2-(chloromethyl)pyrimidine (e, 0.92 g, 7.22 mmol) and triphenylphosphine (2.1 g, 7.94 mmol, 1.1 equiv) in anhydrous benzene (25 mL) was refluxed under a N2 atmosphere for 24 h until TLC analysis (MeOH/DCM = 1:19, v/v) showed the completion of the reaction. The reaction mixture was concentrated via rotary evaporation, and the gummy residue was triturated with diethyl ether (3 × 10 mL). The solid obtained was purified by silica gel column chromatography using a gradient of 1–10% of MeOH in DCM as the eluent to afford the pure product triphenyl(pyrimidin-2-ylmethyl)phosphonium chloride (g, 0.797 g, 2.039 mmol, 28% yield).
GP5: Preparation of Heterocyclic Phosphonates (e to g, HWE Reagents).
2-(Chloromethyl)pyridine hydrochloride (e, 16.5 g, 100.6 mmol) in DCM (100 mL) was treated with aqueous NaHCO3 (20 mL), and the DCM layer was dried over anhydrous Na2SO4. The filtrate was concentrated by rotary evaporation. The alkyl halide thus obtained along with triethyl phosphite (35 mL, 201.2 mmol, 2.0 equiv) was heated at 150 °C under a N2 atmosphere for 5 h until TLC analysis (MeOH/DCM = 1:19, v/v) showed the completion of the reaction. The reaction mixture was purified by silica gel column chromatography using a gradient of 10–100% of EtOAc in hexane and later 1–10% of MeOH in DCM as the eluent to yield the pure product 2-pyridyl methyl phosphonate (g, 18.26 g, 79.66 mmol, 79% yield).
GP6: Wittig Reaction (c + f to h).
To a suspension of the Wittig reagent triphenyl(pyrimidin-2-ylmethyl)phosphonium chloride (f, 0.719 g, 1.839 mmol) in anhydrous THF (40 mL) at −70 °C under a N2 atmosphere was added dropwise LHMDS (1.0 M in THF, 2.03 mL, 2.024 mmol, 1.1 equiv), which was stirred at the same temperature for 15 min. To this mixture a solution of Boc-Phe-H (c, 0.321 g, 1.287 mmol, 0.7 equiv) in THF (10 mL) was added and stirred over 2 h until the temperature reached −40 °C. Upon completion of the reaction as revealed by TLC analysis (EtOAc/hexane = 1:1, v/v), the reaction was quenched by the addition of 0.1 mL of glacial acetic acid, followed by aqueous NaHCO3. Most of the THF was removed carefully using a rotary evaporator, and the residue was extracted with EtOAc (2×). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo to afford the crude material, which was purified by silica gel column chromatography using a gradient of 5–30% of EtOAc in hexane as the eluent, yielding the pure olefin tert-butyl (S,E)-(1-phenyl-4-(pyrimidin-2-yl)but-3-en-2-yl)carbamate (h, E isomer, 0.060 g, 14% yield). The other Z isomer (0.014 g) was isolated as a side product, and the ratio of E to Z isomers was typically 4:1.
GP7: Horner–Wadsworth–Emmons Reaction (c + g to h).
To a solution of the 2-pyridyl methyl phosphonate ester (g, 1.30 g, 5.65 mmol) in anhydrous THF (25 mL) at −70 °C under a N2 atmosphere was added dropwise LHMDS (1.0 M in THF, 6.22 mL, 6.22 mmol, 1.1 equiv). The reaction was stirred at the same temperature for 15 min, followed by the dropwise addition of a solution of Boc-hPhe-H (c, 1.34 g in 10 mL THF, 5.09 mmol, 0.9 equiv). The reaction was stirred until it reached a temperature of −20 °C over 2 h. Upon completion of the reaction as revealed by TLC analysis (EtOAc/hexane = 1:1, v/v), to the reaction mixture at 0 °C was added glacial acetic acid (0.5 mL), followed by the addition of 20 mL of saturated NaHCO3. The aqueous layer was extracted with EtOAc (3 ×100 mL). Extracts were washed with brine (1 × 50 mL), and the organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo to afford the crude product, which was purified by silica gel column chromatography using a gradient of 10–50% of EtOAc in hexane as the eluent to yield the pure product tert-butyl (S,E)-(5-phenyl-1-(pyridin-2-yl)pent-1-en-3-yl)carbamate (h, 0.344 g, 1.016 mmol, 20% yield).
GP8: Amide Coupling with P3–P2 Fragment (h to j).
To a solution of tert-butyl (S,E)-(5-phenyl-1-(pyridin-2-yl)pent-1-en-3-yl)carbamate (h, 0.143 g, 0.423 mmol) in anhydrous DCM (5 mL) at 0 °C was added dropwise TFA (1.5 mL in 1 mL DCM) with stirring at the same temperature for 1 h. Upon completion of the reaction as revealed by TLC analysis (EtOAc/hexane = 1:1, v/v), the reaction solvent was removed by a rotary evaporator. The resulting oil was co-evaporated on a rotary evaporator with CHCl3 (3×) and ether (3×). The solid product was dried on high vacuum to yield the TFA salt (S,E)-5-phenyl-1-(pyridin-2-yl)pent-1-en-3-aminium trifluoroacetate (0.149 g, 0.423 mmol), which was used in subsequent synthetic steps without further purification. To a solution of the above TFA salt in anhydrous DCM (5 mL) at −10 °C under a N2 atmosphere was added dropwise DIPEA (0.6 mL, 0.3.44 mmol, 8 equiv), followed by the addition of Cbz-Phe-OH (0.13 g, 0.43 mmol, 1 equiv) and T3P (50% in EtOAc, 0.41 mL, 1.5 equiv). The reaction was stirred at 0 °C for an additional 1 h. Upon completion of the reaction as revealed by TLC analysis (EtOAc/hexane = 1:1, v/v), the reaction mixture was diluted with DCM (50 mL) and then washed with H2O (3×) and brine (3×). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo to afford the crude product, which was purified by silica gel column chromatography using a gradient of 10–50% of EtOAc in hexane as the eluent to yield the pure product benzyl ((S)-1-oxo-3-phenyl-1-(((S,E)-5-phenyl-1-(pyridin-2-yl)pent-1-en-3-yl)amino)propan-2-yl)carbamate (i, 0.113 g, 0.217 mmol, 51% yield).
GP9: N-Methylation Using Methyl Iodide (i to j).
To a suspension of benzyl ((S)-1-oxo-3-phenyl-1-(((S,E)-1-phenyl-4-(pyridin-2-yl)-but-3-en-2-yl)amino)propan-2-yl)carbamate (i, 0.049 g, 0.098 mmol) in anhydrous MeCN (5 mL) under a N2 atmosphere was added MeI (0.03 mL, 0.490 mmol, 5 equiv), and the reaction mixture was heated under reflux for 9 h. Upon completion of the reaction as revealed by TLC analysis (EtOAc/hexane = 1:1, v/v), the solvents were removed by rotary evaporation. The resulting gummy residue was dissolved in CHCl3 (1 mL) and precipitated with ether (5 mL). The solvents were decanted, and this procedure was repeated twice. The solid obtained was dried under high vacuum to give pure product 2-((S,E)-3-((S)-2-(((benzyloxy)carbonyl)amino)-3-phenylpropanamido)-4-phenylbut-1-en-1-yl)-1-methylpyridin-1-ium iodide as a yellow solid (j, 0.039 g, 61% yield).
GP10: Preparation of Peptide Acrylamide (i to k).
A solution of ethyl (S,E)-4-((S)-2-(((benzyloxy)carbonyl)amino)-3-phenylpropanamido)-5-phenylpent-2-enoate (i, 0.346 g, 0.69 mmol) in THF (6 mL) at 0 °C was treated with LiOH (1 N in H2O, 0.83 mL, 0.83 mmol, 1.2 equiv) and stirred overnight. The reaction was concentrated by rotary evaporation, and the aqueous layer was added to water, acidified to pH 1–2, and extracted with EtOAc (3×). The combined organic layers were dried and concentrated to yield the crude acrylic acid. To a solution of this acrylic acid (0.124 g, 0.262 mmol) in THF (6 mL) at −15 °C were added Et3N (0.11 mL, 0.787 mmol, 3 equiv) and the dropwise addition of ClCO2Et (0.035 mL, 0.367 mmol), which resulted in a white precipitate. The reaction mixture was stirred at the same temperature for an additional 30 min, and then aqueous 1 M NH4Cl (0.4 mL) was added dropwise with continuous stirring over 3 h until a temperature of 25 °C was attained. Upon completion of the reaction as revealed by TLC analysis (EtOAc/hexane = 1:1, v/v), most of the reaction solvent was removed using a rotary evaporator, and the solid residue was extracted with EtOAc. The organic layer was washed with aqueous NaHCO3 (2×), H2O (1×), and brine (1×), dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated in vacuo to afford the crude product. Purification of the crude product by silica gel chromatography using a gradient of 20–100% of EtOAc in hexane as the eluent yielded the pure product benzyl ((S)-1-(((S,E)-5-amino-5-oxo-1-phenylpent-3-en-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (0.027 g, 0.057 mmol, 22% yield).
4-Methyl-N-((S)-1-oxo-3-phenyl-1-(((S,E)-1-phenyl-4-(phenylsulfonyl)but-3-en-2-yl)amino)propan-2-yl)piperazine-1-carboxamide (1, Cbz-Phe-Phe-VSPh).
White solid, 0.115 g, 0.202 mmol, 56% yield. 1H NMR (400 MHz, CDCl3): δ 2.79 (d, J = 6.8 Hz, 2H), 2.85–3.12 (m, 2H), 4.26 (q, J = 7.3 Hz, 1H), 4.79–4.95 (m, 1H), 5.04 (s, 2H), 5.13 (s, 1H), 5.75 (s, 1H), 5.96 (dd, J1 = 1.8 Hz, J2 = 15.1 Hz, 1H), 6.78 (dd, J1 = 4.8 Hz, J2 = 15.1 Hz, 1H), 6.95–7.03 (m, 2H), 7.05–7.11 (m, 2H), 7.12–7.23 (m, 6H), 7.27–7.39 (m, 5H), 7.47–7.56 (m, 2H), 7.57–7.67 (m, 1H), 7.72–7.84 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 38.4, 40.3, 50.4, 56.7, 67.4, 127.3, 127.4, 127.8, 128.2, 128.5, 128.7 (2C), 128.8, 129.0, 129.3, 129.4, 131.1, 133.6, 135.5, 136.1, 136.2, 140.2, 144.6, 156.0, 170.5. LC–MS m/z: 569.31 [M + H]+ (calcd for C33H32N2O5S+, 569.21); tR = 7.27 min.
Benzyl ((S)-1-(((S,E)-5-Amino-5-oxo-1-phenylpent-3-en-2-yl)-amino)-1-oxo-3-phenylpropan-2-yl)carbamate (2, Cbz-Phe-Phe-vinyl-CONH2).
White solid, 0.027 g, 0.057 mmol, 22% yield. 1H NMR (400 MHz, DMSO-d6): δ 2.64–2.77 (m, 1H), 2.84 (d, J = 7.2 Hz, 2H), 2.96 (dd, J1 = 3.9 Hz, J2 = 13.7 Hz, 1H), 4.19–4.31 (m, 1H), 4.62 (pentet, J = 6.8 Hz, 1H), 4.95 (s, 1H), 5.85 (d, J = 15.5 Hz, 1H), 6.56 (dd, J1 = 5.9 Hz, J2 = 15.5 Hz, 1H), 6.93 (s, 1H), 7.12–7.46 (m, 17H), 8.25 (d, J = 8.2 Hz, 1H). 13C NMR (100 MHz, DMSO-d6): δ 28.7, 37.6, 50.9, 56.07, 65.1, 124.1, 126.2, 126.4, 127.4, 127.6, 127.9, 128.1, 128.2, 129.2, 129.5, 137.0, 137.8, 138.0, 142.0, 155.6, 166.2, 170.7. LC–MS m/z: 472.46 [M + H]+ (calcd for C28H29N3O4+, 472.22); tR = 4.71 min.
Benzyl ((S)-1-(((S,E)-6-Amino-6-oxo-1-phenylhex-4-en-3-yl)-amino)-1-oxo-3-phenylpropan-2-yl)carbamate (3, Cbz-Phe-hPhe-vinyl-CONH2).
White solid, 0.013 g, 0.027 mmol, 15% yield. 1H NMR (400 MHz, DMSO-d6): δ 1.64–1.92 (m, 2H), 2.54–2.72 (m, 2H), 2.81 (dd, J1 = 10.6 Hz, J2 = 13.6 Hz, 1H), 3.03 (dd, J1 = 4.0 Hz, J2 = 13.6 Hz, 1H), 4.25–4.33 (m, 1H), 4.35–4.43 (m, 1H), 4.85–5.04 (m, 2H), 5.90 (d, J = 15.5 Hz, 1H), 6.54 (dd, J1 = 5.7 Hz, J2 = 15.5 Hz, 1H), 6.94 (s, 1H), 7.15–7.35 (m, 15H), 7.41 (s, 1H), 7.49 (d, J = 8.5 Hz, 1H), 8.22 (d, J = 8.2 Hz, 1H). LC–MS m/z: 486.24 [M + H]+ (calcd for C29H31N3O4+, 486.24); tR = 4.89 min.
N-((S)-1-(((S,E)-6-Amino-6-oxo-1-phenylhex-4-en-3-yl)amino)-1-oxo-3-phenylpropan-2-yl)-4-methylpiperazine-1-carboxamide (4, NMePip-Phe-hPhe-vinyl-CONH2).
White solid, 0.022 g, 0.048 mmol, 18% yield. 1H NMR (400 MHz, CDCl3): δ 1.66–1.91 (m, 2H), 2.23 (s, 3H), 2.25–2.34 (m, 4H), 2.50–2.64 (m, 2H), 2.99–3.15 (m, 2H), 3.25–3.41 (m, 4H), 4.44–4.57 (m, 1H), 4.62 (q, J = 7.4 Hz, 1H), 5.37 (d, J = 7.6 Hz, 1H), 5.60 (dd, J1 = 1.3 Hz, J2 = 15.3 Hz, 1H), 5.75 (s, 1H), 6.01 (s, 1H), 6.62 (dd, J1 = 5.5 Hz, J2 = 15.3 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 7.04–7.31 (m, 11H). 13C NMR (100 MHz, DMSO-d6): δ 32.0, 36.1, 38.5, 43.9, 46.1, 49.9, 54.6, 56.2, 122.9, 126.2, 126.9, 128.5, 128.6, 128.8, 129.7, 137.3, 141.1, 144.4, 157.3, 167.5, 172.0. LC–MS m/z: 478.36 [M + H]+ (calcd for C27H35N5O3+, 478.28); tR = 2.54 min.
Benzyl ((S)-1-(((S,E)-1,4-Diphenylbut-3-en-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (5, Cbz-Phe-Phe-vinyl-Ph).
Off-white solid, 0.054 g, 0.107 mmol, 35% yield. 1H NMR (400 MHz, CDCl3): δ 2.87 (dt, J = 2.8, 6.3 Hz, 2H), 2.96–3.16 (m, 2H), 4.35 (q, J = 7.6 Hz, 1H), 4.80–4.96 (m, 1H), 5.29 (d, J = 9.6 Hz, 1H), 5.69 (d, J = 9.9 Hz, 1H), 5.94 (ddt, J = 3.0, 6.3, 15.9 Hz, 1H), 6.27 (d, J = 15.9 Hz, 1H), 7.03–7.14 (m, 2H), 7.14–7.28 (m, 10H), 7.28–7.40 (m, 9H). 13C NMR (100 MHz, CDCl3): δ 29.7, 38.6, 41.3, 51.8, 67.1, 126.4, 126.6, 127.1, 127.6, 128.0, 128.2, 128.3, 128.4, 128.5, 128.7, 129.3, 129.4, 130.9, 136.5, 136.8, 169.8. LC–MS m/z: 505.31 [M + H]+ (calcd for C33H32N2O3+, 505.25); tR = 6.20 min.
Benzyl ((S)-1-(((S,E)-4-(4-Nitrophenyl)-1-phenylbut-3-en-2-yl)-amino)-1-oxo-3-phenylpropan-2-yl)carbamate (6, Cbz-Phe-Phe-vinyl-(4-NO2)Ph).
White fluffy solid, 0.530 g, 0.964 mmol, 55% yield. 1H NMR (400 MHz, DMSO-d6): δ 2.76 (dd, J1 = 9.7 Hz, J2 = 13.4 Hz, 1H), 2.85–2.97 (m, 3H), 4.18–4.35 (m, 1H), 4.68 (pentet, J = 6.7 Hz, 1H), 4.99 (s, 2H), 6.41 (d, J = 16.1 Hz, 1H), 6.50 (dd, J1 = 5.4 Hz, J2 = 16.1 Hz, 1H), 7.13–7.37 (m, 15H), 7.42 (d, J = 8.5 Hz, 1H), 7.58 (d, J = 8.5 Hz, 2H), 8.14–8.25 (m, 3H). 13C NMR (100 MHz, DMSO-d6): δ 37.7, 39.0, 51.9, 56.2, 65.2, 123.9, 126.1, 126.2, 127.0, 127.4, 127.6, 127.9, 128.0, 128.1, 128.2, 129.2, 129.3, 135.6, 137.0, 137.8, 138.0, 143.3, 146.2, 155.6, 170.6. LC–MS m/z: 550.28 [M + H]+ (calcd for C33H31N3O5+, 550.23); tR = 6.12 min.
Benzyl ((S)-1-Oxo-3-phenyl-1-(((S,E)-1-phenyl-4-(pyrimidin-2-yl)-but-3-en-2-yl)amino)propan-2-yl)carbamate (7, Cbz-Phe-Phe-vinyl-2Pyrmd).
White solid, 0.026 g, 0.0513 mmol, 33% yield. 1H NMR (400 MHz, CDCl3): δ 2.73–2.96 (m, 2H), 3.01 (d, J = 4.8 Hz, 2H), 4.23–4.50 (m, 1H), 4.91–5.02 (m, 1H), 5.05 (s, 2H), 5.30 (s, 1H), 6.05 (s, 1H), 6.48 (d, J = 15.7 Hz, 1H), 7.00 (dd, J1 = 5.6 Hz, J2 = 15.7 Hz, 1H), 7.05–7.37 (m, 16H), 8.63 (d, J = 4.0 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 38.6, 41.0, 51.6, 56.5, 67.3, 119.0, 126.9, 127.2, 128.2, 128.3, 128.6, 128.7, 128.9, 129.4, 129.5, 130.3, 136.3. 136.5, 136.7, 139.2, 156.1, 157.1, 164.2, 170.3. LC–MS m/z: 507.26 [M + H]+ (calcd for C31H30N4O3+, 507.24); tR = 5.14 min.
Benzyl ((S)-1-Oxo-3-phenyl-1-(((S,E)-5-phenyl-1-(pyrimidin-2-yl)-pent-1-en-3-yl)amino)propan-2-yl)carbamate (8, Cbz-Phe-hPhe-vinyl-2Pyrmd).
Off-white solid, 0.280 g, 0.538 mmol, 29% yield. 1H NMR (400 MHz, CDCl3): δ 1.76–2.03 (m, 2H), 2.63 (t, J = 7.9 Hz, 2H), 3.10 (t, J = 8.4 Hz, 2H), 4.47 (s, 1H), 4.74 (h, J = 7.3 Hz, 1H), 5.10 (d, J = 7.9 Hz, 2H), 5.53 (s, 1H), 6.27 (s, 1H), 6.56–6.72 (m, 1H), 6.96–7.05 (m, 1H), 7.06–7.14 (m, 3H), 7.19 (d, J = 7.3 Hz, 3H), 7.21–7.34 (m, 10H), 8.66 (dd, J = 4.9, 15.8 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 32.0, 36.2, 38.6, 50.5, 56.6, 67.1, 118.9, 126.0, 127.0, 128.0, 128.1, 128.3, 128.4, 128.5, 128.7, 128.8, 129.3, 129.4, 130.1, 136.4, 139.6, 141.2, 156.9, 164.1, 170.3. LC–MS m/z: 521.24 [M + H]+ (calcd for C32H32N4O3+, 521.26); tR = 5.56 min.
4-Methyl-N-((S)-1-oxo-3-phenyl-1-(((S,E)-5-phenyl-1-(pyrimidin-2-yl)pent-1-en-3-yl)amino)propan-2-yl)piperazine-1-carboxamide (9, NMePip-Phe-hPhe-vinyl-2Pyrmd).
Off-white gum, 0.054 g, 0.105 mmol, 54% yield. 1H NMR (400 MHz, CDCl3): δ 1.82–1.99 (m, 2H), 2.29 (s, 3H), 2.34–2.46 (m, 4H), 2.61 (t, J = 7.5 Hz, 1H), 3.09 (d, J = 7.5 Hz, 2H), 3.39 (s, 4H), 4.58–4.74 (m, 2H), 5.47 (d, J = 6.3 Hz, 1H), 6.61 (d, J = 15.7 Hz, 1H), 6.87 (d, J = 8.2 Hz, 1H), 6.98 (dd, J1 = 6.3 Hz, J2 = 15.7 Hz, 1H), 7.06–7.31 (m, 11H), 8.66 (d, J = 4.8 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 32.1, 36.5, 38.8, 43.5, 45.6, 50.6, 54.3, 56.1, 118.9, 126.0, 126.9, 128.4, 128.5 (2C), 128.6, 129.6, 130.1, 137.2, 140.1, 141.3, 157.0, 164.3, 171.8. LC–MS m/z: 513.17 [M + H]+ (calcd for C30H36N6O2+, 513.30); tR = 2.96 min.
Benzyl ((S)-1-Oxo-3-phenyl-1-(((S,E)-4-(pyrimidin-2-yl)but-3-en-2-yl)amino)propan-2-yl)carbamate (10, Cbz-Phe-Ala-vinyl-2Pyrmd).
White fluffy solid, 0.017 g, 0.039 mmol, 14% yield. 1H NMR (400 MHz, CDCl3 + MeOD): δ 1.32 (d, J = 6.6 Hz, 3H), 2.96 (dd, J1 = 8.0 Hz, J2 = 13.6 Hz, 1H), 3.10 (dd, J1 = 6.4 Hz, J2 = 13.6 Hz, 1H), 4.36–4.44 (m, 4H), 4.71 (pentet, J = 6.4 Hz, 1H), 4.97–5.12 (m, 2H), 6.52 (d, J = 15.7 Hz, 1H), 6.98 (dd, J1 = 5.8 Hz, J2 = 15.7 Hz, 1H), 7.13–7.36 (m, 11H), 8.70 (d, J = 4.9 Hz, 2H). 13C NMR (100 MHz, CDCl3 + MeOD): δ 19.6, 38.7, 46.1, 56.1, 66.7, 119.0, 126.7, 127.6, 127.9, 128.1, 128.3 (2C), 129.2, 136.3, 141.6, 156.9 (2C), 157.0, 163.9, 171.0. LC–MS m/z: 430.91 [M + H]+ (calcd for C25H26N4O3+, 431.21); tR = 4.68 min.
Benzyl ((S)-1-Oxo-3-phenyl-1-(((S,E)-1-phenyl-4-(pyridin-2-yl)but-3-en-2-yl)amino)propan-2-yl)carbamate (11, Cbz-Phe-Phe-vinyl-2Pyr).
White solid, 0.554 g, 1.096 mmol, 81% yield. 1H NMR (400 MHz, CDCl3): δ 2.87 (dq, J1 = 6.8 Hz, J2 = 13.7 Hz, 1H), 3.01 (d, J = 7.1 Hz, 2H), 4.35 (q, J = 7.0 Hz, 1H), 4.91 (pentet, J = 6.8 Hz, 1H), 5.06 (s, 2H), 5.24 (s, 1H), 5.88 (d, J = 8.4 Hz, 1H), 6.32 (d, J = 15.7 Hz, 1H), 6.58 (dd, J1 = 6.1 Hz, J2 = 15.7 Hz, 1H), 7.08–7.36 (m, 17H), 7.59 (dt, J1 = 1.6 Hz, J2 = 7.7 Hz, 1H), 8.53 (d, J = 4.3 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 38.4, 41.1, 51.7, 56.4, 67.1, 122.1, 122.2, 126.7, 127.0, 128.0, 128.2, 128.4, 128.5 (2C), 128.8, 129.4 (2C), 130.5, 133.0, 136.1, 136.4, 136.8, 149.5, 154.8, 155.9, 170.0. LC–MS m/z: 506.24 [M + H]+ (calcd for C32H31N3O3+, 506.24); tR = 4.39 min.
2-((S,E)-3-((S)-2-(((Benzyloxy)carbonyl)amino)-3-phenylpropanamido)-4-phenylbut-1-en-1-yl)-1-methylpyridin-1-ium Iodide (12, Cbz-Phe-Phe-Vinyl-2PyrNMe).
Yellow solid, 0.039 g, 0.060 mmol, 61% yield. 1H NMR (400 MHz, CDCl3): δ 3.01–3.15 (m, 3H), 3.19 (dd, J1 = 7.7 Hz, J2 = 13.6 Hz, 1H), 4.19 (s, 3H), 4.51 (q, J = 7.0 Hz, 1H), 4.90–5.02 (m, 2H), 5.06 (s, 1H), 5.81 (s, 1H), 6.69 (d, J = 15.7 Hz, 1H), 6.86 (dd, J1 = 4.6 Hz, J2 = 15.7 Hz, 1H), 7.08–7.28 (m, 14H), 7.65–7.79 (m, 2H), 7.89–8.01 (m, 1H), 8.21–8.31 (m, 1H), 8.97–9.09 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 38.1, 39.8, 47.5, 52.6, 57.1, 66.8, 119.9, 126.1, 126.3, 126.9, 127.1, 127.5, 127.6, 127.9, 128.5, 128.6, 128.8, 129.6, 129.7, 136.7, 136.9, 144.9, 146.0, 147.6, 152.8, 156.2, 171.5. LC–MS m/z: 520.32 [M + H]+ (calcd for C33H34N3O3+, 520.26); tR = 3.25 min.
Benzyl ((S)-1-Oxo-3-phenyl-1-(((S,E)-5-phenyl-1-(pyridin-2-yl)pent-1-en-3-yl)amino)propan-2-yl)carbamate (13, Cbz-Phe-hPhe-vinyl-2Pyr).
White solid, 0.113 g, 0.217 mmol, 51% yield. 1H NMR (400 MHz, CDCl3): δ 1.73–1.95 (m, 2H), 2.59 (t, J = 7.9 Hz, 2H), 3.04 (d, J = 7.0 Hz, 2H), 4.38–4.52 (m, 1H), 4.65 (pentet, J = 7.1 Hz, 1H), 5.03 (s, 2H), 5.59 (d, J = 6.4 Hz, 1H), 6.34 (d, J = 5.1 Hz, 1H), 6.43 (d, J = 15.7 Hz, 1H), 6.53 (dd, J1 = 6.2 Hz, J2 = 15.7 Hz, 1H), 7.02–7.32 (m, 17H), 7.57 (dt, J1 = 1.4 Hz, J2 = 7.7 Hz, 1H), 8.51 (d, J = 4.4 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 32.1, 36.4, 38.7, 50.8, 56.6, 67.1, 122.1, 122.3, 126.0, 127.0, 128.0, 128.2, 128.4, 128.5 (2C), 128.7, 129.5, 130.5, 133.8, 136.2, 136.5 (2C), 141.4, 149.5, 155.0, 156.1, 170.4. LC–MS m/z: 518.74, 520.41 [M + H]+ (calcd for C33H33N3O3+, 520.26); tR = 4.68 min.
4-Methyl-N-((S)-1-oxo-3-phenyl-1-(((S,E)-5-phenyl-1-(pyridin-2-yl)pent-1-en-3-yl)amino)propan-2-yl)piperazine-1-carboxamide (14, NMePip-Phe-hPhe-vinyl-2Pyr).
Off-white solid, 0.090 g, 0.176 mmol, 44% yield. 1H NMR (400 MHz, CDCl3): δ 1.77–1.94 (m, 2H), 2.21 (s, 3H), 2.22–2.27 (m, 4H), 2.59 (t, J = 8.0 Hz, 2H), 3.08 (d, J = 7.0 Hz, 2H), 3.28 (m, 4H), 4.61 (pentet, J = 7.3 Hz, 1H), 4.7 (q, J = 7.3 Hz, 1H), 5.39 (d, J = 7.7 Hz, 1H), 6.46 (d, J = 15.8 Hz, 1H), 6.56 (dd, J1 = 6.2 Hz, J2 = 15.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 7.06–7.24 (m, 12H), 7.59 (dt, J1 = 1.8 Hz, J2 = 7.7 Hz, 1H), 8.53 (d, J = 4.3 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 32.1, 36.6, 39.0, 43.8, 46.1, 50.7, 54.6, 55.9, 122.0, 122.2, 125.9, 126.8, 128.4 (2C), 128.5, 129.6, 130.3, 134.2, 136.4, 137.2, 141.4, 149.5, 155.1, 157.0, 171.6. LC–MS m/z: 512.28 [M + H]+ (calcd for C31H37N5O2+, 512.30); tR = 2.60 min.
2-((S,E)-3-((S)-2-(((Benzyloxy)carbonyl)amino)-3-phenylpropanamido)-5-phenylpent-1-en-1-yl)-1-methylpyridin-1-ium (15, Cbz-Phe-hPhe-vinyl-2PyrNMe).
Yellow solid, 0.029 g, 0.044 mmol, 89% yield. 1H NMR (400 MHz, CDCl3): δ 1.98–2.10 (m, 1H), 2.11–2.25 (m, 1H), 2.56–2.85 (m, 2H), 3.04–3.35(m, 2H), 4.22 (s, 3H), 4.62 (d, J = 5.5 Hz, 1H), 4.79 (s, 1H), 4.92–5.09 (m, 2H), 5.88 (s, 1H), 6.70 (d, J = 15.8 Hz, 1H), 6.77 (dd, J1 = 3.6 Hz, J2 = 15.8 Hz, 1H), 7.09–7.34 (m, 14H), 7.66–7.79 (m, 2H), 7.90 (d, J = 6.0 Hz, 1H), 8.17–8.30 (m, 1H), 8.98 (d, J = 4.5 Hz, 1H). LC–MS m/z: 534.25 [M + H]+ (calcd for C34H36N3O3+, 534.28); tR = 3.48 min.
Benzyl ((S)-1-(((S,E)-1-(4-Methoxypyridin-2-yl)-5-phenylpent-1-en-3-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (16, Cbz-Phe-hPhe-vinyl-2-(4-OMe)-Pyr).
Pale yellow solid, 0.072 g, 0.131 mmol, 33% yield. 1H NMR (400 MHz, CDCl3): δ 1.73–2.01 (m, 2H), 2.59 (dt, J = 7.9, 37.9 Hz, 2H), 3.11 (dd, J = 7.0, 11.7 Hz, 2H), 3.87 (d, J = 9.2 Hz, 3H), 4.50 (dd, J = 7.6, 23.1 Hz, 1H), 4.68 (t, J = 7.1 Hz, 1H), 5.08 (d, J = 4.6 Hz, 2H), 5.74 (dd, J = 8.1, 65.8 Hz, 1H), 6.39–6.58 (m, 1H), 6.67–6.81 (m, 2H), 7.11–7.15 (m, 2H), 7.16– 7.26 (m, 8H), 7.27–7.34 (m, 7H), 8.37 (dd, J = 5.8, 23.3 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 32.0, 36.3, 38.7, 50.6, 55.2, 56.5, 67.1, 108.3, 108.5, 126.0, 126.6, 127.0, 127.9, 128.0, 128.1, 128.1, 128.3, 128.4, 128.4, 128.4, 128.5, 128.5, 128.7, 129.4, 129.4, 134.7, 135.1, 136.5, 141.3, 150.0, 156.1, 166.6, 170.3. LC–MS m/z: 550.16 [M + H]+ (calcd for C34H35N3O4+, 549.26); tR = 3.57 min.
Benzyl ((S)-1-Oxo-3-phenyl-1-(((S,E)-5-phenyl-1-(4-(trifluoromethyl)pyridin-2-yl)pent-1-en-3-yl)amino)propan-2-yl)-carbamate (17, Cbz-Phe-hPhe-vinyl-2-(4-CF3)-Pyr).
White solid, 0.260 g, 0.442 mmol, 79% yield. 1H NMR (400 MHz, CDCl3): δ 1.76–1.95 (m, 2H), 2.59 (t, J = 7.8 Hz, 2H), 3.06 (d, J = 7.1 Hz, 2H), 4.36–4.51 (m, 1H), 4.65 (pentet, J = 7.1 Hz, 1H), 5.05 (s, 2H), 5.49 (s, 1H), 6.13 (s, 1H), 6.35 (d, J = 15.7 Hz, 1H), 6.59 (dd, J1 = 6.0 Hz, J2 = 15.7 Hz, 1H), 7.06–7.11 (m, 2H), 7.14–7.33 (m, 15H), 8.67 (d, J = 4.9 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 32.1, 36.4, 38.7, 50.7, 56.8, 67.3, 117.5, 121.6, 124.3, 126.2, 127.0 (C–F), 127.2, 128.1, 128.3, 128.5, 128.6 (3C), 128.8, 129.2, 129.5, 136.2, 136.6, 138.7, 139.1, 141.2, 150.5, 156.2 (C–F), 156.4, 170.4. LC–MS m/z: 587.95 [M + H]+ (calcd for C34H32F3N3O3+, 588.25); tR = 5.16 min.
Benzyl ((S)-4-Methyl-1-oxo-1-(((S,E)-5-phenyl-1-(pyridin-2-yl)-pent-1-en-3-yl)amino)pentan-2-yl)carbamate (18, Cbz-Leu-hPhe-vinyl-2Pyr).
White solid, 0.038 g, 0.078 mmol, 26% yield. 1H NMR (400 MHz, CDCl3): δ 0.92 (t, J = 6.4 Hz, 6H), 1.45–1.57 (m, 1H), 1.59–1.73 (m, 2H), 1.79 (s, 1H), 1.86–2.05 (m, 2H), 2.68 (t, J = 7.9 Hz, 2H), 4.03–4.27 (m, 1H), 4.71 (pentet, J = 6.9 Hz, 1H), 5.10 (s, 2H), 5.14 (s, 1H), 6.26 (d, J = 6.2 Hz, 1H), 6.58 (d, J = 15.8 Hz, 1H), 6.69 (dd, J1 = 6.0 Hz, J2 = 15.8 Hz, 1H), 7.10–7.34 (m, 12H), 7.60 (dt, J1 = 1.7 Hz, J2 = 7.7 Hz, 1H), 8.54 (d, J = 4.6 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 23.1, 24.9, 32.3, 36.7, 41.2, 50.9, 54.0, 67.3, 122.3, 122.4, 126.1, 128.2, 128.4, 128.6 (2C), 128.7, 130.6, 134.0, 136.3, 136.7, 141.6, 149.7, 155.1, 156.5, 171.6. LC–MS m/z: 484.46, 485.49, 486.38 [M + H]+ (calcd for C30H35N3O3+, 486.28); tR = 4.58 min.
Benzyl ((S)-1-Oxo-3-phenyl-1-(((S,E)-4-(pyridin-2-yl)but-3-en-2-yl)amino)propan-2-yl)carbamate (19, Cbz-Phe-Ala-vinyl-2Pyr).
White solid, 0.590 g, 1.374 mmol, 75% yield. 1H NMR (400 MHz, CDCl3): δ 1.58 (s, 3H), 2.96–3.23 (m, 2H), 4.36 (q, J = 7.4 Hz, 1H), 4.63–4.80 (m, 1H), 5.10 (d, J = 1.4 Hz, 2H), 5.33 (s, 1H), 5.67 (d, J = 8.3 Hz, 1H), 6.40 (dd, J = 1.3, 15.8 Hz, 1H), 6.51 (dd, J = 5.5, 15.8 Hz, 1H), 7.13 (ddd, J = 1.1, 4.8, 7.4 Hz, 1H), 7.16–7.21 (m, 3H), 7.21–7.37 (m, 8H), 7.62 (td, J = 1.8, 7.7 Hz, 1H), 8.45–8.61 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 20.0, 38.8, 46.4, 56.2, 66.8, 121.8, 122.3, 126.7, 127.7, 128.0, 128.3, 128.7, 129.3, 135.6, 136.4, 137.0, 148.8, 155.0, 170.8. LC–MS m/z: 430.35 [M + H]+ (calcd for C26H27F3N3O3+, 430.21); tR = 3.49 min.
(S,E)-5-((S)-2-(((Benzyloxy)carbonyl)amino)-3-phenylpropanamido)-7-(pyridin-2-yl)hept-6-en-1-aminium Chloride (20, Cbz-Phe-Lys-vinyl-2Pyr).
White solid, 0.027 g, 0.048 mmol, 51% yield. 1H NMR (400 MHz, CDCl3): δ 1.44–1.59 (m, 2H), 1.65–1.84 (m, 4H), 2.89–3.02 (m, 3H), 3.09–3.19 (m, 1H), 4.43 (t, J = 7.5 Hz, 1H), 4.64–4.70 (m, 1H), 5.01–5.11 (m, 2H), 6.54 (d, J = 16.1 Hz, 1H), 6.92 (dd, J1 = 5.7 Hz, J2 = 16.1 Hz, 1H), 7.11 (t, J = 7.5 Hz, 1H), 7.21 (t, J = 7.5 Hz, 2H), 7.25–7.40 (m, 7H), 7.91 (t, J = 6.8 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 8.54 (t, J = 8.0 Hz, 1H), 8.69 (d, J = 5.2 Hz, 1H). 13C NMR (100 MHz, MeOD): δ 23.8, 27.9, 34.0, 39.1, 40.6, 51.9, 58.2, 67.6, 121.6, 125.9, 126.5, 127.7, 128.5, 128.9, 129.5, 129.7, 130.5, 138.1, 138.4, 142.0, 146.3, 147.8, 151.2, 158.3, 174.1. LC–MS m/z: 486.98 [M + H]+, 508.94 [M + Na]+ (calcd for C29H35N4O3+, 487.27 and calcd for C29H35N4O3Na+, 509.25, respectively); tR = 3.16 min.
Benzyl ((S)-1-Oxo-3-phenyl-1-(((S,E)-1-phenyl-4-(pyridin-4-yl)-but-3-en-2-yl)amino)propan-2-yl)carbamate (21, Cbz-Phe-Phe-vinyl-4Pyr).
Off-white solid, 0.051 g, 0.109 mmol, 50% yield. 1H NMR (400 MHz, CDCl3): δ 2.83 (d, J = 6.8 Hz, 2H), 3.02 (d, J = 7.8 Hz, 2H), 4.36 (s, 1H), 4.86 (s, 1H), 5.08 (s, 2H), 5.90 (s, 1H), 6.13 (s, 1H), 7.10 (dd, J = 6.5, 31.0 Hz, 6H), 7.18–7.27 (m, 6H), 7.31 (dd, J = 3.6, 6.4 Hz, 5H), 8.50 (d, J = 6.0 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 38.4, 41.0, 51.5, 52.1, 67.1, 105.0, 120.9, 126.9, 127.1, 127.9, 128.3, 128.4, 128.5, 128.6, 128.8, 129.3, 129.4, 133.4, 136.4, 150.0, 170.1. LC–MS m/z: 506.29 [M + H]+ (calcd for C32H31N3O3+, 506.24); tR = 3.49 min.
4-((S,E)-3-((S)-2-(((Benzyloxy)carbonyl)amino)-3-phenylpropanamido)-4-phenylbut-1-en-1-yl)-1-methylpyridin-1-ium Iodide (22, Cbz-Phe-Phe-vinyl-4PyrNMe).
Yellow solid, 0.020 g, 0.030 mmol, 87% yield. 1H NMR (400 MHz, MeOD + CDCl3): δ 2.75–3.09 (m, 4H), 4.28–4.43 (m,5H), 4.87 (q, J = 6.3 Hz, 1H), 5.08 (s, 2H), 6.28 (d, J = 15.9 Hz, 1H), 6.83 (dd, J1 = 5.0 Hz, J2 = 15.9 Hz, 1H), 7.16–7.35 (m, 15H), 7.82 (d, J = 6.5 Hz, 2H), 8.71 (d, J = 6.5 Hz, 2H). 13C NMR (100 MHz, CDCl3 + MeOD): δ 38.2, 40.1, 47.8, 52.2, 56.3, 66.8, 124.5, 126.8 (2C), 127.6, 128.0, 128.3, 128.4 (3C), 128.5, 129.1, 129.2, 136.1, 136.4, 143.9, 144.7, 153.1, 156.2, 171.3. LC–MS m/z: 521.33 [M + H]+ (calcd for C33H34N3O3+, 521.27); tR = 3.36 min.
Benzyl ((S)-1-Oxo-3-phenyl-1-(((S,E)-1-phenyl-4-(pyrimidin-4-yl)-but-3-en-2-yl)amino)propan-2-yl)carbamate (23, Cbz-Phe-Phe-vinyl-4Pyrmd).
Off-white solid, 0.800 g, 1.579 mmol, 64% yield. 1H NMR (400 MHz, CDCl3): δ 2.79–2.93 (m, 2H), 3.01 (d, J = 6.9 Hz, 2H), 4.35–4.48 (m, 1H), 4.93 (pentet, J = 6.7 Hz, 1H), 5.04 (s, 2H), 5.49 (s, 1H), 6.16 (d, J = 15.6 Hz, 1H), 6.29 (s, 1H), 6.88 (dd, J1 = 5.8 Hz, J2 = 15.6 Hz, 1H), 6.97 (d, J = 4.6 Hz, 1H), 7.05–7.33 (m, 15H), 8.56 (d, J = 5.2 Hz, 1H), 9.06 (s, 1H). 13C NMR (100 MHz, CDCl3): δ 38.5, 40.9, 51.6, 56.6, 67.2, 118.8, 126.9, 127.1, 128.0, 128.2, 128.3, 128.6 (3C), 128.8, 129.4 (2C), 136.2, 136.5, 138.6, 156.1, 157.4, 158.8, 161.5, 170.4. LC–MS m/z: 507.35 [M + H]+ (calcd for C31H30N4O3+, 507.24); tR = 5.27 min.
Benzyl ((S)-1-(((S,E)-4-(Oxazol-2-yl)-1-phenylbut-3-en-2-yl)-amino)-1-oxo-3-phenylpropan-2-yl)carbamate (24, Cbz-Phe-Phe-vinyl-2Oxz).
White solid, 0.020 g, 0.040 mmol, 29% yield. 1H NMR (400 MHz, CDCl3): δ 2.72–2.88 (m, 2H), 2.93–3.11 (m, 2H), 4.35 (q, J = 7.0 Hz, 1H), 4.88 (pentet, J = 6.7 Hz, 1H), 5.05 (s, 2H), 5.30 (s, 1H), 5.99 (d, J = 5.8 Hz, 1H), 6.14 (d, J = 16.1 Hz, 1H), 6.48 (dd, J1 = 5.9 Hz, J2 = 16.1 Hz, 1H), 7.04–7.35 (m, 16H), 7.54 (s, 1H). 13C NMR (100 MHz, CDCl3): δ 38.6, 40.9, 51.5, 56.7, 67.3, 117.3, 127.1, 127.4, 128.2, 128.4, 128.7 (3C), 129.0, 129.4 (2C), 136.2, 136.4 (2C), 137.1, 138.3, 156.1, 160.8, 170.3. LC–MS m/z: 496.25 [M + H]+ (calcd for C30H29N3O4+, 496.22); tR = 5.41 min.
Benzyl ((S)-1-Oxo-3-phenyl-1-(((S,E)-1-phenyl-4-(thiazol-2-yl)-but-3-en-2-yl)amino)propan-2-yl)carbamate (25, Cbz-Phe-Phe-vinyl-2Thz).
Light yellow solid, 0.206 g, 0.403 mmol, 54% yield. 1H NMR (400 MHz, CDCl3): δ 2.75–2.89 (m, 2H), 2.99 (d, J = 7.1 Hz, 2H), 4.28–4.46 (m, 1H), 4.87 (d, J = 6.4 Hz, 1H), 4.96–5.11 (m, 2H), 5.44 (s, 1H), 6.19 (s, 1H), 6.39 (dd, J1 = 5.1 Hz, J2 = 16.0 Hz, 1H), 6.45 (d, J = 16.0 Hz, 1H), 7.01–7.39 (m, 16H), 7.71 (d, J = 3.2 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 38.7, 41.0, 51.5, 56.6, 67.2, 118.5, 123.9, 126.9, 127.2, 128.1, 128.3, 128.6 (2C), 128.8, 129.4 (2C), 135.0, 136.2, 136.5 (2C), 143.4, 156.0, 166.0, 170.4. LC–MS m/z: 512.18 [M + H]+ (calcd for C30H29N3O3+, 512.20); tR = 5.64 min.
2-((S,E)-3-((S)-2-(((Benzyloxy)carbonyl)amino)-3-phenylpropanamido)-4-phenylbut-1-en-1-yl)-3-methylthiazol-3-ium Iodide (26, Cbz-Phe-Phe-vinyl-2ThzNMe).
Yellow solid, 0.003 g, 0.005 mmol, 15% yield. 1H NMR (400 MHz, CDCl3): δ 2.51 (s, 1H), 2.81–3.96 (m, 1H), 2.98–3.18 (m, 3H), 3.71 (s, 3H), 3.93 (s, 2H), 4.36–4.49 (m, 1H), 4.99 (s, 2H), 5.85–6.11 (m, 1H), 6.81 (d, J = 15.6 Hz, 1H), 6.91 (dd, J1 = 3.4 Hz, J2 = 15.6 Hz, 1H), 7.06–7.33 (m, 15H), 7.76 (s, 1H), 7.96 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 38.1, 39.8, 52.5, 54.7, 57.3, 66.7, 115.1, 121.6, 126.9, 127.1, 127.6, 128.0 (2C), 128.5, 128.7, 128.8, 129.5 (2C), 136.9, 137.0, 138.8, 150.6, 156.4, 168.7, 171.9. LC–MS m/z: 526.18 [M + H]+ (calcd for C31H32N3O3S+, 526.22); tR = 3.31 min.
Benzyl ((S)-1-Oxo-3-phenyl-1-(((S)-1-phenyl-4-(pyrimidin-2-yl)-butan-2-yl)amino)propan-2-yl)carbamate (27, Cbz-Phe-Phe-(CH2)2-2Pyrmd).
To a solution of Boc-Phe-vinyl-2Pyrmd (h, prepared following GP1–GP7, 0.05 g, 0.15 mmol) in anhydrous EtOAc (8 mL) was added Pd/C (10%wt, 0.016 mg) under a H2 atmosphere and stirred overnight. The reaction was filtered, and the filtrate was concentrated. The product was coupled with the P3–P2 fragment following GP8 to give compound 27. White solid, 0.031 g, 0.060 mmol, 48% yield. 1H NMR (400 MHz, CDCl3): δ 1.63–1.82 (m, 1H), 1.85–2.01 (m, 1H), 2.63–2.77 (m, 2H), 2.78–2.94 (m, 2H), 2.99 (d, J = 6.8 Hz, 2H), 4.05–4.23 (m, 1H), 4.31 (pentet, J = 7.1 Hz, 1H), 5.07 (s, 2H), 5.34 (s, 1H), 6.43 (d, J = 6.4 Hz, 1H), 7.04–7.38 (m, 16H), 8.59 (d, J = 4.9 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 31.5, 35.8, 38.8, 41.1, 51.1, 56.8, 67.2, 118.6, 126.6, 127.0, 128.2, 128.3, 128.5, 128.7, 128.8, 129.4, 129.6, 136.4, 136.7, 137.9, 155.9, 157.0, 170.5, 170.9. LC–MS m/z: 509.15, 509.28 [M + H]+ (calcd for C31H32N4O3+, 509.26); tR = 5.10 min.
Solubility of Cruzain Inhibitors.
Synthetic PVHIs such as 5–16 and 22–24 at final concentrations of 1–200 μM were added to 0.25 mL solutions of 10% DMSO (v/v) in clear 96-well Greiner plates, and absorbance at 620 nm was measured using a BioTek M2 Synergy plate reader at t = 0 and 120 min to evaluate increased light scattering and precipitated inhibitor.
Evaluation of Covalent Adducts of Glutathione and Cruzain Inhibitors.
The compounds 7, 11, 12, 15, 25, 26, and K1777 (0.5 mM) were added to 100 mM Tris (pH 8.0), 10% (v/v) DMSO, and 1 mM or 5 mM reduced glutathione to a final volume of 0.2 mL at room temperature. Samples were analyzed by HPLC–MS (as described above) by injecting 0.01 mL aliquots onto a Luna 5 mm C18(2) 100 Å, 4.6 mm, 50 mm column (Phenomenex) using the HPLC method described in the chemistry section at 0–6 h time points. The chromatographic peaks for each cruzain inhibitor and its covalent adduct with glutathione were characterized by their values of m/z using electrospray positive ionization detection and UV absorbance at 254 nm: K11777, retention time: 4.75 min, m/z: 575.05; K11777-GSH, retention time: 4.64 min, m/z: 882.30; 7, retention time: 6.71 min, m/z: 507.11; 11, retention time: 5.48 min, m/z: 506.06; 12, retention time: 4.77 min, m/z: 520.10; 15, retention time: 4.81 min, m/z: 535.23; 15-GSH, retention time: 4.72 min, m/z: 842.42; 17, retention time: 5.03 min, m/z: 587.95; 25, retention time: 4.43 min, m/z: 511.87; 26, retention time: 4.42 min, m/z: 525.90; 26-GSH, retention time: 3.61 min, m/z: 833.97. Integration of the chromatographic peaks of the inhibitors and their GSH adducts at each time point was used to determine the rate of GSH adduct formation.
Enzyme Expression and Purification.
Recombinant human cathepsins B, L, and S were purchased from Millipore Sigma and used without further treatment. General procedures for cruzain expression, purification, and activation were performed according to published protocols59 with modifications as described.40 Activated cruzain at >95% purity was stored either as MMTS-conjugated samples, as described, at −80 °C in a buffer containing 50 mM Tris (pH 8.0) and 20% glycerol, at protein concentrations of 5 mg mL−1. Prior to use, MMTS was removed by successive dialysis in the presence of 5 mM DTT. Activated samples of cruzain were used immediately or stored after use at 4 °C for a period of 1–2 months.
Enzyme Assays and Evaluation of Inhibitors.
All enzyme assays were performed at 25 °C. Initial rates of the peptidolytic reaction catalyzed by cruzain were measured by monitoring the fluorescence generated by cleavage of the dipeptide–AMC bond. Assays were conducted in 96-well plates (Greiner; flat-bottom, clear black plates) in a total volume of 250 μL, containing either 50 mM MES (pH 7.5), 50 mM TAPSO, 100 mM DEA, 1 mM CHAPS, 1 mM Na2EDTA, 5 mM DTT, and 10% DMSO (v/v) or 50 mM sodium acetate (pH 5.5), 50 mM MES, 100 mM TEA, 1 mM CHAPS, 1 mM Na2EDTA, 5 mM DTT, and 10% DMSO (v/v). Substrates were dissolved in 100% DMSO and were then diluted 10-fold such that when added to reaction mixtures, final DMSO concentration was 10% (v/v). Reactions were initiated with the addition of 1–10 μL of cruzain (final concentrations: 0.1–3.0 nM (preincubation studies)). Fluorescence was measured on either a SpectraMax M5 (Molecular Devices) or a Synergy HTX (BioTek, Winooski, VT) microplate reader (λex = 360 nm, λem = 460 nm). Initial rates were determined from continuous kinetic time courses and calculated from the earliest time points, typically at less than 10 min.
Compounds were evaluated as inhibitors or inactivators of cruzain in two ways: (1) enzyme was added to reaction mixtures containing the substrate (typically, 10 μM Cbz-Phe-Arg-AMC) and inhibitor, and reaction time courses were measured for 0–40 min. In addition to other methods, the effects of all inhibitors on reaction rates were determined at t = 0–200 s (vi) and at longer incubation times (t > 1000 s; vs), to ascertain the respective inhibition constants Ki and Ki*. (2) Enzyme and compound were preincubated over extended periods of time, and then aliquots were removed and diluted 50-fold to 100-fold into reaction mixtures containing 10 μM Cbz-Phe-Arg-AMC, followed by the assessment of the resulting time courses.
For assays of cathepsin B, L, and S, cruzain inhibitors were evaluated in reaction mixtures containing a buffer of sodium acetate (pH 5.5), 1 mM CHAPS, 1 mM Na2EDTA, and 5 mM DTT at 25 °C. The substrate Cbz-Phe-Arg-AMC was dissolved in 100% DMSO, as were all inhibitors, and aliquots of both substrates and inhibitors were added to 0.25 mL reaction mixtures to final concentrations of 10% DMSO (v/v). Michaelis constants for Cbz-Phe-Arg-AMC were determined for all three human cathepsins as cathepsin L (2.9 μM), cathepsin S (60 μM), and cathepsin B (150 μM), and fixed concentrations of Cbz-Phe-Arg-AMC of 1 or 2 Km were used to evaluate inhibitors. Cruzain inhibitors were added at seven concentrations and one fixed concentration of Cbz-Phe-Arg-AMC, and time courses of AMC formation were analyzed as with cruzain.
Evaluation of Cruzain Inhibitors in Axenic Cell Cultures of T. b. brucei and T. cruzi.
Selected cruzain inhibitors were evaluated in axenic cell cultures of T. b. brucei and T. cruzi. Procyclic trypomastigotes of T. b. brucei (ATCC PRA-381) were grown in the SDM-79 medium at 26 °C, and bloodstream forms (ATCC PRA-383) were grown at 37 °C in the HMI-9 medium at 5% CO2. T. cruzi (epimastigote forms, strain Y, ATCC 50832GFP) was grown in the ATCC medium (1029 LIT medium). Both forms of T. b. brucei and T. cruzi were grown in media containing 10% fetal bovine serum (FBS) and penicillin/streptomycin (50 U mL−1). Test compounds, including K11777, were dissolved in 100% DMSO and added to cell cultures at final concentrations of 0.5–20 μM (maximum DMSO = 1% (v/v)). Control samples contained equal amounts of DMSO. T. b. brucei and T. cruzi (5 mL in flask cultures) were seeded at ~3 × 106 cells and diluted daily, maintaining a mid-log growth phase for up to 120 h. Treated cells were typically grown for 4 days (T. b. brucei) or 5 days (T. cruzi). After each cell dilution, fresh compound or an equal volume of DMSO (control samples) was supplemented into the cultures, while maintaining a constant concentration of each inhibitor. Cell counts were scored using a Z2 Coulter Counter.
Evaluation of Cruzain Inhibitors in T. cruzi-Infected Murine Cardiomyoblasts.
For the evaluation of the antitrypanocidal activity of cruzain inhibitors, we infected a C2C12 mouse cardiomyoblast cell line (ATCC CRL-1772) with T. cruzi strain Ca-I/72 (a gift from James Dvorak, National Institutes of Health) in 1536-microwell plates. In each well were added 103 cells and 104 parasites in a total volume of 10 μL including the test compounds in 10-point dose–response dilutions starting at 10 μM (3-fold dilutions). The plates were incubated at 37 °C for 48 h, and the wells were fixed with 2% paraformaldehyde in PBS and stained with 5 μg mL−1 4′,6-diamidino2-phenylindole. After at least 30 min of incubation at room temperature in the dark, the plates were read in an automated microscope, ImageXpress Micro XL (Molecular Devices), and the images were analyzed by custom-built software to quantify and assess viability of the parasites, as well as the host cells independently. The compilation of data was used to calculate the antiparasitic activity (EC50) and host cytotoxicity (CC50).
Evaluation of Human Cell Toxicity.
Primary human dermal fibroblast (HDF) cells were used to evaluate human cell toxicity of cruzain inhibitors. HDF cells were plated in a 384-well plate at 2400 cells per well (62,000 cells mL−1). Inhibitors in 100% DMSO were added in duplicate to final concentrations of 0.001–0.1 mM and 1% DMSO (v/v) with 1% DMSO as a control sample, and cells were cultured at 37 °C for 48 h, followed by the addition of resazurin. Cell viability was then assessed by reading of fluorescence (excitation/emission: 544 nm/590 nm) after an additional 24 h of incubation.
Kinetic Data Analysis.
Initial velocity data for cruzain-catalyzed reactions of fluorogenic peptide substrates were determined by fitting to eq 1 using GraphPad Prism 6.0 or SigmaPlot 12.0. For eq 1, kcat is the turnover number, [E]t is the concentration of active sites of cruzain, and Ka is the Michaelis constant for the substrate A. Cruzain concentrations were determined by spectrophotometric analysis of purified sample solutions as described.44
| (1) |
Competitive inhibition was fitted to eq 2, in which A and I are concentrations of substrate and inhibitor, respectively, Vmax is the maximal velocity, and Kis is the slope inhibition constant.
| (2) |
Data for time-dependent inhibition were fitted by several methods. All time-course data were fitted to eq 3 for studies in which the reaction was initiated by the addition of enzyme, wherein P is the fluorescence generated by AMC formation, C is a nonzero constant, vs and vi are respectively the steady-state and initial enzymatic rates, t is time, and kobs is the observed rate of conversion of the initial inhibited rate to the final inhibited rate.52 In cases for which the reaction was initiated with an excess of substrate, following preincubation of enzyme and inhibitor, for eq 3, vi = 0.
| (3) |
Values of kobs versus [inhibitor] were then replotted and fitted to eq 4 for which k3 and k4 represent the respective rates of formation and dissolution of the EI* complex as depicted in Scheme 1.
| (4) |
Inhibition constants were also obtained by fitting vi and vs data to eq 5, in which vx is the rate in the presence of inhibitor I for either early (vi) or late (vs) phases of each time course, v0 is the rate in the absence of inhibitor, Ka is the Michaelis constant of the substrate A, and Kix is the apparent inhibition constant, Ki or Ki*, obtained from fitting of vi or vs, respectively.
| (5) |
| (6) |
In addition, time-course data were fitted globally by two methods. First, expressions for vs, vi, and kobs were substituted into eq 3 to generate eq 6, which was used to fit time-course data at all concentrations of inhibitors simultaneously. Second, time-course data were fitted using KinTek Explorer as either a single-binding step model with the two kinetic parameters k3/Ki and k4 or a two-step binding mechanism as shown in Scheme 1 from which rate constants k1–k4 are determined (Supporting Information).
Supplementary Material
ACKNOWLEDGMENTS
We wish to thank Jasmin Ackermann and Ivani Pauli for assisting with testing of cruzain inhibitors in T. cruzi-infected cardiomyoblasts; Thomas Snavely, Dr. Deeann Wallis, and Kim Loesch in the laboratory of Prof. James C. Sacchettini for testing cruzain inhibitors for human cell toxicity; Prof. James Sacchettini and Dr. Su Tang for crystallography analysis; Jiyun Zhu, Charis Fernandez, and Nora McGuffey for assistance with analysis of inhibitors versus cruzain and human cathepsins; and Mireya Luna for support with synthetic chemistry. The authors thank Professor Charles S. Craik for providing a construct for the expression of cruzain and Dr. Larry Dangott for providing protein sequencing. The authors also thank Professor Ken Johnson for assistance with the use of KinTek Explorer. Financial support for the research was provided by NIH grant R21AI127634 and Texas A&M AgriLife Research.
DEDICATION
This paper is dedicated to the memory of Robert L. Stevenson.
ABBREVIATIONS
- AMC
7-amino-4-methylcoumarin
- BSF
bloodstream form
- CHAPS
3-[3-(cholamidopropyl)dimethylammonio]-1-propanesulfonate
- DIPEA
N,N-diisopropylethylamine
- hPhe
homophenylalanine
- MES
2-(N-morpholino)ethanesulfonic acid
- MMTS
S-methyl methanethiosulfonate
- Oxz
oxazolyl
- NMePip
N-methylpiperazinyl
- PCF
procyclic form
- PVHI
peptidomimetic vinylheterocyclic inhibitor
- Pyr
pyridinyl
- Pyrmd
pyrimidinyl
- Thz
thiazolyl
- T3P
propylphosphonic anhydride
- VS
vinyl sulfone
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b02078.
Molecular modeling information, fitting of time-course inhibition data using KinTek Explorer, thiolation of cruzain inhibitors by glutathione, synthesis and characterization of AMC-peptide substrates, structures of PVHIs, HPLC traces and NMR spectra of synthesized substrates and inhibitors, and unpublished crystallographic data (PDF)
Molecular formula strings of S2–S5 and S7–S11 and compounds 1–27 (CSV)
Model coordinates for cruzain bound to 7, 9, 11–13, and 15 using CovDock module(PDB)
Model coordinates for cruzain bound to 7, 9, 11–13, and 15 using Glide module (PDB)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.9b02078
The authors declare no competing financial interest.
Contributor Information
Bala C. Chenna, Department of Biochemistry & Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Linfeng Li, Department of Biochemistry & Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Drake M. Mellott, Department of Biochemistry & Biophysics, Texas A&M University, College Station, Texas 77843, United States
Xiang Zhai, Department of Biochemistry & Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Jair L. Siqueira-Neto, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California-San Diego, La Jolla, California 92093, United States
Claudia Calvet Alvarez, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California-San Diego, La Jolla, California 92093, United States.
Jean A. Bernatchez, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California-San Diego, La Jolla, California 92093, United States.
Emily Desormeaux, Department of Biochemistry & Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Elizabeth Alvarez Hernandez, Department of Biochemistry & Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Jana Gomez, Department of Biochemistry & Biophysics, Texas A&M University, College Station, Texas 77843, United States.
James H. McKerrow, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California-San Diego, La Jolla, California 92093, United States
Jorge Cruz-Reyes, Department of Biochemistry & Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Thomas D. Meek, Department of Biochemistry & Biophysics, Texas A&M University, College Station, Texas 77843, United States.
REFERENCES
- (1).Muñoz-Saravia SG; Haberland A; Wallukat G; Schimke I. Chronic Chagas’ Heart Disease: a Disease on its Way to Becoming a Worldwide Health Problem: Epidemiology, Etiopathology, Treatment, Pathogenesis and Laboratory Medicine. Heart Fail. Rev 2012, 17, 45–64. [DOI] [PubMed] [Google Scholar]
- (2).Centers for Disease Control and Prevention. https://www.cdc.gov/parasites/chagas(Accessed October 2019).
- (3).Coura JR; Viñas PA Chagas Disease: a New Worldwide Challenge. Nature 2010, 465, S6–S7. [DOI] [PubMed] [Google Scholar]
- (4).Castro Y; Marcus R. Epidemiology of Chagas Disease in the USA: High-Risk Patient Populations for Screening. Curr. Trop. Med. Rep 2019, 6, 8–12. [Google Scholar]
- (5).Montgomery SP; Parise ME; Dotson EM; Bialek SR What Do We Know About Chagas Disease in the United States? Am. J. Trop. Med. Hyg 2016, 95, 1225–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rassi A Jr.; Rassi A; Marcondes de Rezende J. American Trypanosomiasis (Chagas Disease). Infect. Dis. Clin. North Am 2012, 26, 275–291. [DOI] [PubMed] [Google Scholar]
- (7).Lejon V; Bentivoglio M; Franco JR Human African Trypanosomiasis. In Handbook of Clinical Neurology; Garcia HH; Tanowitz HB; Del Brutto OH, Eds; Elsevier: Cambridge, MA, 2013; pp 169–181. [DOI] [PubMed] [Google Scholar]
- (8).World Health Organization. Trypanosomiasis, human African (sleeping sickness) https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (Accessed October 2019).
- (9).Drugs for Neglected Diseases initiative. https://www.dndi.org/wpcontent/uploads/2019/09/Factsheet2019_SleepingSickness.pdf (Accessed October 2019).
- (10).Simarro PP; Jannin J; Cattand P. Eliminating Human African Trypanosomiasis: Where Do We Stand and What Comes Next? PLOS Medicine 2008, 5, No. e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kennedy PGE Clinical Features, Diagnosis, and Treatment of Human African Trypanosomiasis (Sleeping Sickness). Lancet Neurol. 2013, 12, 186–194. [DOI] [PubMed] [Google Scholar]
- (12).Salomon CJ First Century of Chagas’ Disease: An Overview on Novel Approaches to Nifurtimox and Benznidazole Delivery Systems. J. Pharm. Sci 2012, 101, 888–894. [DOI] [PubMed] [Google Scholar]
- (13).Cianni L; Feldmann CW; Gilberg E; Gütschow M; Juliano L; Leitão A; Bajorath J; Montanari CA Can Cysteine Protease Cross-Class Inhibitors Achieve Selectivity? J. Med. Chem 2019, 62, 10497–10525. [DOI] [PubMed] [Google Scholar]
- (14).Choe Y; Leonetti F; Greenbaum DC; Lecaille F; Bogyo M; Brömme D; Ellman JA; Craik CS Substrate Profiling of Cysteine Proteases Using a Combinatorial Peptide Library Identifies Functionally Unique Specificities. J Biol. Chem 2006, 281, 12824–12832. [DOI] [PubMed] [Google Scholar]
- (15).Lecaille F; Authié E; Moreau T; Serveau C; Gauthier F; Lalmanach G. Subsite Specificity of Trypanosomal Cathepsin L-Like Cysteine Proteases. Eur. J. Biochem 2001, 268, 2733–2741. [DOI] [PubMed] [Google Scholar]
- (16).Sudhan DR; Siemann DW Cathepsin L Targeting in Cancer Treatment. Pharmacol. Ther 2015, 155, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Miller BE; Mayer RJ; Goyal N; Bal J; Dallow N; Boyce M; Carpenter D; Churchill A; Heslop T; Lazaar AL Epithelial Desquamation Observed in a Phase I Study of an Oral Cathepsin C Inhibitor (GSK2793660). Br. J. Clin. Pharmacol 2017, 83, 2813–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wilkinson RDA; Williams R; Scott CJ; Burden RE Cathepsin S: Therapeutic, Diagnostic, and Prognostic Potential. Biol. Chem 2015, 396, 867–882. [DOI] [PubMed] [Google Scholar]
- (19).Drake MT; Clarke BL; Oursler MJ; Khosla S. Cathepsin K Inhibitors for Osteoporosis: Biology, Potential Clinical Utility, and Lessons Learned. Endocr. Rev 2017, 38, 325–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sajid M; Robertson SA; Brinen LS; McKerrow JH Cruzain In Cysteine Proteases of Pathogenic Organisms; Robinson MW; Dalton JP, Eds; Springer: Boston, MA, 2011; pp 100–115. [Google Scholar]
- (21).Eakin AE; Mills AA; Harth G; McKerrow JH; Craik CS The Sequence, Organization, and Expression of the Major Cysteine Protease (Cruzain) from Trypanosoma cruzi. J. Biol. Chem 1992, 267, 7411–7420. [PubMed] [Google Scholar]
- (22).McGrath ME; Eakin AE; Engel JC; McKerrow JH; Craik CS; Fletterick RJ The Crystal Structure of Cruzain: A Therapeutic Target for Chagas’ Disease. J. Mol. Biol 1995, 247, 251–259. [DOI] [PubMed] [Google Scholar]
- (23).Caffrey CR; Hansell E; Lucas KD; Brinen LS; Hernandez AA; Cheng J; Gwaltney SL II; Roush WR; Stierhof YD; Bogyo M; Steverding D; McKerrow JH Active Site Mapping, Biochemical Properties and Subcellular Localization of Rhodesain, the Major Cysteine Protease of Trypanosoma brucei rhodesiense. Mol. Biochem. Parasitol 2001, 118, 61–73. [DOI] [PubMed] [Google Scholar]
- (24).Steverding D; Sexton DW; Wang X; Gehrke SS; Wagner GK; Caffrey CR Trypanosoma brucei: Chemical Evidence that Cathepsin L is Essential for Survival and a Relevant Drug Target. Int. J. Parasitol 2012, 42, 481–488. [DOI] [PubMed] [Google Scholar]
- (25).O’Brien TC; Mackey ZB; Fetter RD; Choe Y; O’Donoghue AJ; Zhou M; Craik CS; Caffrey CR A Parasite Cysteine Protease is Key to Host Protein Degradation and Iron Acquisition. J. Biol. Chem 2008, 283, 28934–28943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Doyle PS; Zhou YM; Hsieh I; Greenbaum DC; McKerrow JH; Engel JC The Trypanosoma cruzi Protease Cruzain Mediates Immune Evasion. PLoS Pathog. 2011, 7, No. e1002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Harth G; Andrews N; Mills AA; Engel JC; Smith R; McKerrow JH Peptide-fluoromethyl Ketones Arrest Intracellular Replication and Intercellular Transmission of Trypanosoma cruzi. Mol. Biochem. Parasitol 1993, 58, 17–24. [DOI] [PubMed] [Google Scholar]
- (28).Doyle PS; Zhou YM; Engel JC; McKerrow JH A Cysteine Protease Inhibitor Cures Chagas Disease in an Immunodeficient-Mouse Model of Infection. Antimicrob. Agents Chemother 2007, 51, 3932–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Engel JC; Doyle PS; McKerrow JH Trypanocidal Effect of Cysteine Protease Inhibitors in vitro and in vivo in Experimental Chagas Disease. Medicina 1999, 59, 171–175. [PubMed] [Google Scholar]
- (30).Engel JC; Doyle PS; Hsieh I; McKerrow JH Cysteine Protease Inhibitors Cure an Experimental Trypanosoma cruzi Infection. J. Exp. Med 1998, 188, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kerr ID; Lee JH; Farady CJ; Marion R; Rickert M; Sajid M; Pandey KC; Caffrey CR; Legac J; Hansell E; McKerrow JH; Craik CS; Rosenthal PJ; Brinen LS Vinyl Sulfones as Antiparasitic Agents and a Structural Basis for Drug Design. J. Biol. Chem 2009, 284, 25697–25703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chen YT; Brinen LS; Kerr ID; Hansell E; Doyle PS; McKerrow JH; Roush WR In vitro and in vivo Studies of the Trypanocidal Properties of WRR-483 against Trypanosoma cruzi. PLoS Negl. Trop. Dis 2010, 4, No. e825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Mott BT; Ferreira RS; Simeonov A; Jadhav A; Ang KKH; Leister W; Shen M; Silveira JT; Doyle PS; Arkin MR; McKerrow JH; Inglese J; Austin CP; Thomas CJ; Shoichet BK; Maloney DJ Identification and Optimization of Inhibitors of Trypanosomal Cysteine Proteases: Cruzain, Rhodesain, and TbCatB. J. Med. Chem 2010, 53, 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).McKerrow JH; Doyle PS; Engel JC; Podust LM; Robertson SA; Ferreira R; Saxton T; Arkin M; Kerr ID; Brinen LS; Craik CS Two Approaches to Discovering and Developing New Drugs for Chagas Disease. Mem. Inst. Oswaldo Cruz 2009, 104, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).McKerrow JH Update on Drug Development Targeting Parasite Cysteine Proteases. PLoS Negl. Trop. Dis 2018, 12, No. e0005850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bradshaw JM; McFarland JM; Paavilainen VO; Bisconte A; Tam D; Phan VT; Romanov S; Finkle D; Shu J; Patel V; Ton T; Li X; Loughhead DG; Nunn PA; Karr DE; Gerritsen ME; Funk JO; Owens TD; Verner E; Brameld KA; Hill RJ; Goldstein DM; Taunton J. Prolonged and Tunable Residence Time Using Reversible Covalent Kinase Inhibitors. Nat. Chem. Biol 2015, 11, 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Krishnan S; Miller RM; Tian B; Mullins RD; Jacobson MP; Taunton J. Design of Reversible, Cysteine-Targeted Michael Acceptors Guided by Kinetic and Computational Analysis. J. Am. Chem. Soc 2014, 136, 12624–12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Copeland RA; Pompliano DL; Meek TD Drug–Target Residence Time and its Implications for Lead Optimization. Nat. Rev. Drug Discovery 2006, 5, 730–739. [DOI] [PubMed] [Google Scholar]
- (39).Barr SC; Warner KL; Kornreic BG; Piscitelli J; Wolfe A; Benet L; McKerrow JH A Cysteine Protease Inhibitor Protects Dogs from Cardiac Damage during Infection by Trypanosoma cruzi. Antimicrob. Agents Chemother 2005, 49, 5160–5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhai X; Meek TD Catalytic Mechanism of Cruzain from Trypanosoma cruzi As Determined from Solvent Kinetic Isotope Effects of Steady-State and Pre-Steady-State Kinetics. Biochemistry 2018, 57, 3176–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Silverman RB Mechanism-Based Enzyme Inactivators In Methods in Enzymology; Johnson ML; Brand L, Eds; Academic Press: Cambridge, MA, 1995; pp 240–283. [DOI] [PubMed] [Google Scholar]
- (42).Brady K; Abeles RH Inhibition of Chymotrypsin by Peptidyl Trifluoromethyl Ketones: Determinants of Slow-Binding Kinetics. Biochemistry 1990, 29, 7608–7617. [DOI] [PubMed] [Google Scholar]
- (43).Rubach JK; Cui G; Schneck JL; Taylor AN; Zhao B; Smallwood A; Nevins N; Wisnoski D; Thrall SH; Meek TD The Amino-Acid Substituents of Dipeptide Substrates of Cathepsin C Can Determine the Rate-Limiting Steps of Catalysis. Biochemistry 2012, 51, 7551–7568. [DOI] [PubMed] [Google Scholar]
- (44).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (45).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- (46).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes. J. Med. Chem 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- (47).Zhu K; Borrelli KW; Greenwood JR; Day T; Abel R; Farid RS; Harder E. Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model 2014, 54, 1932–1940. [DOI] [PubMed] [Google Scholar]
- (48).Wittig G; Schöllkopf U. Über Triphenyl-phosphin-methylene als Olefinbildende Reagenzien I. Mitteil. Chem. Ber 1954, 87, 1318–1330. [Google Scholar]
- (49).Wadsworth WS; Emmons WD The Utility of Phosphonate Carbanions in Olefin Synthesis. J. Am. Chem. Soc 1961, 83, 1733–1738. [Google Scholar]
- (50).Nahm S; Weinreb SM N-methoxy-N-Methylamides as Effective Acylating Agents. Tetrahedron 1981, 22, 3815–3818. [Google Scholar]
- (51).Klaus JL; Palmer JT; Rasnick D. Irreversible Cysteine Protease Inhibitors Containing Vinyl Groups Conjugated to Electron Withdrawing Groups. U.S. Patent 6,287,840, 2001. [Google Scholar]
- (52).Morrison JF; Walsh CT The Behavior and Significance of Slow-Binding Enzyme Inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol 1988, 61, 201–301. [DOI] [PubMed] [Google Scholar]
- (53).Jones BD; Tochowicz A; Tang Y; Cameron MD; McCall LI; Hirata K; Siqueira-Neto JL; Reed SL; McKerrow JH; Roush WR Synthesis and Evaluation of Oxyguanidine Analogues of the Cysteine Protease Inhibitor WRR-483 against Cruzain. ACS Med. Chem. Lett 2015, 7, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Ndao M; Beaulieu C; Black WC; Isabel E; Vasquez-Camargo F; Nath-Chowdhury M; Massé F; Mellon C; Methot N; Nicoll-Griffith DA Reversible Cysteine Protease Inhibitors Show Promise for a Chagas Disease Cure. Antimicrob. Agents Chemother 2014, 58, 1167–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Boudreau PD; Miller BW; McCall LI; Almaliti J; Reher R; Hirata K; Le T; Siqueira-Neto JL; Hook V; Gerwick WH Design of Gallinamide A Analogs as Potent Inhibitors of the Cysteine Proteases Human Cathepsin L and Trypanosoma cruzi Cruzain. J. Med. Chem 2019, 62, 9026–9044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Zhao Z; Bourne PE Progress with Covalent Small-Molecule Kinase Inhibitors. Drug Discov. Today 2018, 23, 727–735. [DOI] [PubMed] [Google Scholar]
- (57).Nkemgu NJ; Grande R; Hansell E; McKerrow JH; Caffrey CR; Steverding D. Improved Trypanocidal Activities of Cathepsin L Inhibitors. Int. J. Antimicrob. Agents 2003, 22, 155–159. [DOI] [PubMed] [Google Scholar]
- (58).Yang PY; Wang M; He CY; Yao SQ Proteomic Profiling and Potential Cellular Target Identification of K11777, a Clinical Cysteine Protease Inhibitor, in Trypanosoma brucei. Chem. Commun 2012, 48, 835–837. [DOI] [PubMed] [Google Scholar]
- (59).Lee GM; Balouch E; Goetz DH; Lazic A; McKerrow JH; Craik CS Mapping Inhibitor Binding Modes on an Active Cysteine Protease via Nuclear Magnetic Resonance Spectroscopy. Biochemistry 2012, 51, 10087–10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.