

Abstract
A majority of mammalian genes exhibit daily fluctuations in expression levels, making circadian expression rhythms the largest known regulatory network in normal physiology. Cell-autonomous circadian clocks interact with daily light-dark and feeding-fasting cycles to generate approximately 24-hour oscillations in the function of thousands of genes. Circadian expression of secreted molecules and signaling components transmits timing information between cells and tissues. Such intra- and intercellular daily rhythms optimize physiology both by managing energy use and by temporally segregating incompatible processes. Experimental animal models and epidemiological data indicate that chronic circadian rhythm disruption increases the risk of metabolic diseases. Conversely, time-restricted feeding, which imposes daily cycles of feeding and fasting without caloric reduction, sustains robust diurnal rhythms and can alleviate metabolic diseases. These findings highlight an integrative role of circadian rhythms in physiology and offer a new perspective for treating chronic diseases in which metabolic disruptionis a hallmark.
A transient rise in blood sugar after a meal indicates metabolic health. A larger meal produces a larger spike, whereas a fator protein-rich meal produces a muted spike (compared with a normal meal of equivalent caloric content). Physiological responses to what and how much we eat represent the foundation for basic and translational science aimed at preventing and treating obesity, diabetes, and metabolic diseases, which together afflict close to a billion people worldwide. However, the timing of food consumption independent of total caloric intake and macronutrient quality has emerged as a critical factor in maintaining metabolic health. For instance, when healthy adults eat identical meals at breakfast, lunch, or dinner, the postprandial glucose rise is lowest after breakfast and highest after dinner (1), as if the dinner were twice the size of the breakfast. In addition, when healthy adults are given a constant glucose infusion over 24 hours, glycemia rises at night and falls around dawn (1), indicating that in addition to what and how much we eat, when we eat helps determine the physiological response to nutrient availability.
Daily rhythms in nutrient use were first documented almost 40 years ago in cells of the master circadian pacemaker located in the hypothalamic suprachiasmatic nucleus (SCN). Experiments in rats fed 14C-labeled deoxyglucose during their habitual (nighttime) feeding period showed that entry of glucose into the SCN was almost negligible, whereas during the day, radio-labeled glucose was readily detected (2). Such glucose uptake rhythms were sustained even in the absence of light cues. In all, this elegant experiment proved the existence of a circadian rhythm in nutrient demand and/or uptake in tissues. Over the next decades, research into circadian rhythms has shown that daily rhythms in the function of numerous genes prime the organism to assimilate nutrients, to mobilize these nutrients for various functions, and to discard metabolic waste at specific times of the 24-hour day (3, 4). Whereas circadian rhythms generally refer to ~24-hour oscillations that occur in the absence of external timing cues, daily or diurnal rhythms apparent during normal living conditions emerge from interactions between the internal circadian clock and timing cues, which include light and food. Accordingly, a consistent daily pattern of eating and fasting maintains normal circadian physiology, whereas frequent disruptions in daily activity-rest and eating-fasting rhythms (as occurs in shiftwork) (5) or genetic disruption of circadian clock in rodents predisposes to metabolic diseases (6). Certain diet regimens (e.g., the frequent eating of energy-dense food) and aging can dampen these daily oscillations and predispose one to metabolic diseases. Therefore, understanding the diurnal physiology of metabolism at a mechanistic level could potentially reveal lifestyle and therapeutic interventions for preventing and treating metabolic diseases.
Cell-autonomous circadian oscillator
In animals, the core mechanism that gives rise to circadian oscillations is a cell-autonomous transcriptional-translational feedback loop (TTFL) present in most cells. The transcription factors CLOCK (or NPAS2) and BMAL1 bind as heterodimers to cis-acting E boxes in the promoters of their own repressors—Cryptochrome (Cry1 and Cry2) and Period (Per−1, −2, and −3)—and of the nuclear hormone receptors Rev-erb (−α and −β), and Ror (−α, −β, and −γ). ROR and REVERB drive rhythmic Bmal1 gene expression by respectively acting so as to activate and repress its expression through RRE elements present in its promoter (Fig. 1) (7). REV and ROR proteins also affect the expression of Cry1, delaying its expression several hours relative to Cry2. Regulated transcriptional and posttranscriptional events involving a growing list of nuclear and cytoplasmic proteins generate endogenous ~24-hour rhythms in the mRNA and protein levels of most of these 13 transcriptional regulators (7). In addition to controlling each other’s expression, these regulators also drive rhythmic expression of thousands of target genes by binding cis-regulatory sites or through downstream transcriptional regulators. The transcriptional basis of circadian rhythms enables a set of transcriptional regulators to temporally couple their activity with the synchronous rhythmic expression of hundreds or even thousands of genes, with peak expression at distinct times of the day (phase). Such extensive and coordinated gene expression and function would be difficult to achieve with a timing mechanisms based entirely on protein-protein interactions.
Fig. 1. Schematics of cell-autonomous transcription-translation feedback loop (TTFL), constituting the core mechanism of mammalian circadian oscillator.
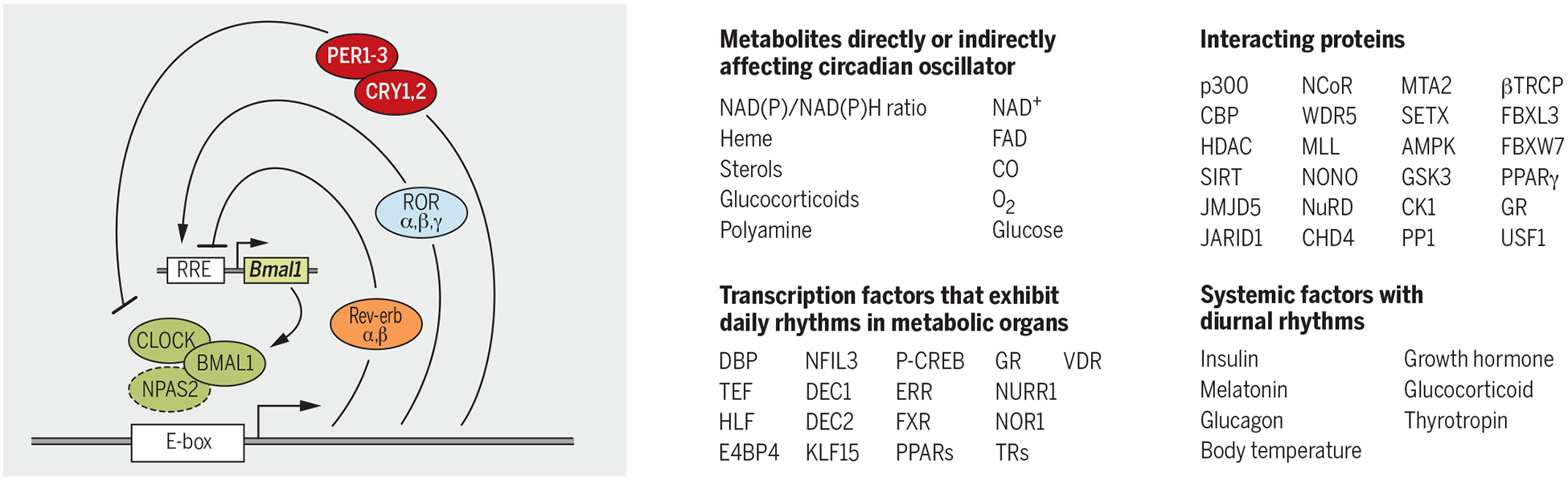
Some of the cellular metabolites and proteins that interact with the clock components are listed. Examples of circadian-regulated transcription factors or systemic factors with daily oscillations further propagate circadian timing to distant genomic and cellular targets.
Circadian transcription factors also interact with a number of coactivators, corepressors, and chromatin-associated factors that read, write, or erase chromatin histone modification marks to activate or repress transcription (Fig. 1). CLOCK/BMAL1 complexes are often associated with the histone acetyl transferase p300 and CREB-binding protein (CBP) (8). CRY/PER repressors are found in complexes with histone deacetylase (HDAC) (9). Additionally, MLL1, MLL3, WDR5, and EZH2 form complexes with circadian transcriptional factors (10). Interactions between REV-ERB and the N-CoR/HDAC3 corepressor are essential for repressive function of REV-ERB (11). A histone lysine demethylase, JARID1a (12), and a bHLHPAS protein, USF1 (13), help transition between daily cycles of activation and repression by interacting with CLOCK/BMAL1 and PER/CRY complexes. In addition to rhythms in histone modifications, some circadian clock proteins also undergo acetylation and deacetylation. Additional cis-acting promoter elements (e.g., CRE and HSE) mediate rapid adjustment of circadian clock components in response to sudden changes in cellular state. Altogether, circadian clock–mediated transcriptional regulation involves a large number of proteins and functional interactions.
Plasticity of the circadian system
The elaborate circadian transcriptional mechanism has many advantages, given that each component of the circadian system can serve as a node for integrating cellular physiology with the circadian function or to transmit circadian timing information to nonclock proteins. Cellular concentration of certain metabolites–including but not limited to heme, nicotinamide adenine dinucleotide/reduced form of nicotinamide adenine dinucleotide (NAD/NADH), nicotinamide adenine dinucleotide phosphate/reduced form of NADP (NADP/NADPH), adenosine monophosphate/adenosine triphosphate (AMP/ATP), acetyl co-enzyme A (AcCoA), alpha keto glutarate (α-KG), S-adenosyl methionine (SAM), CO, and polyamine-can affect the function of several circadian transcriptional regulators by modulating his-tone modifications, protein modifications, protein-protein interactions, protein-DNA interactions, or protein turnover (14). Extracellular factors such as temperature, hormones, and metabolites can also affect the clock and thereby constitute mechanisms for local synchrony of cellular clocks or for adjusting the phase of an organ’s clock in response to systemic signals. Hence, the circadian physiology of any given cell emerges from integrating the cell-autonomous TTFL, cellular metabolism, and extracellular systemic signals (Fig. 1). Thus, clock proteins can sense daily changes in cellular metabolism and systemic signals and make predictive changes to the circadian transcriptome.
Many circadian clock proteins interact with and modify the function of proteins that are not part of the core clock TTFL. Coupled with their own cycling levels, these clock proteins can “impose” rhythmic functions to nonclock proteins. For example, CRY and REV-ERB proteins interact with the glucocorticoid receptor (GR) to inhibit its transcriptional activity (15, 16). CRY proteins also inhibit signaling downstream of the glucagon receptor, thereby imposing a time-of-day-specific effect of glucagon on gluconeogenesis (17). Similarly, REV-ERB and HNF6 interact to regulate lipid metabolism in adult mouse liver (18). Additionally, several homologous proteins within the core TTFL (or their interacting partners) are not fully redundant but rather have specific functions. For example, the CLOCK/BMAL complex interacts with SIRT1 or SIRT6 in a locus-specific manner to target different subsets of the circadian transcriptome in the liver (19). In many cases, these homologs are not expressed in all tissues. Ror-α is expressed in neural tissue, whereas Ror-γ expression dominates in peripheral tissues (20). In summary, the molecular constituents of the clock, partial redundancy among clock components, interactions between clock and nonclock components, and the cyclic expression of downstream tissue-specific components all contribute to tissue-specific molecular circadian physiology, which functions to integrate cell type–specific intra- and extracellular signals to regulate tissue function.
Tissue organization of circadian clocks
The SCN plays a central, high-order role in the circadian regulation of metabolism by sustaining ~24-hour rhythms in activity-rest and feeding-fasting, even under constant darkness. This is achieved through both synaptic and diffusible factors that couple the SCN oscillator with cell type–specific circadian clocks in different brain regions and endocrine cells (7). Through a poly-synaptic connection, the SCN ensures that the pineal gland produces melatonin in a rhythmic fashion (peak levels at night) to promote sleep in diurnal animals. Similarly, through the paraventricular nucleus (PVN) and the pituitary gland, the SCN drives a circadian rhythm in adrenocorticotropic hormone (ACTH) release, which in turn drives a morning rise in corticosterone release from the adrenal gland. Under natural light-dark (LD) conditions, bright light strongly suppresses the production of melatonin (21) and promotes corticosterone production in the adrenal gland through an ACTH-independent sympathetic pathway (22). Corticosteroids promote arousal and alertness and drive catabolic metabolism in adipose tissue and muscle. Both melatonin and cortisol rhythms are detectable in the blood, and both hormones have pleiotropic effects on multiple tissues.
Local SCN outputs are intimately integrated with centers in the hypothalamus involved in hunger-satiety, sleep-arousal, thermoregulation, and osmolarity, as well as a forebrain oscillator that mediates an anticipatory drive for food (23). Mechanisms underlying these synaptic interactions are unclear, and both cellular and genetic phenotypes paint a complicated picture. Npas2−/− mice show normal overall diurnal rhythms in activity and rest under a LD cycle, yet they lack a siesta-type rest period in the middle of the active period and cannot adjust normally when meal timing is abruptly changed (24). On the other hand, hypomorphic Clock mutant mice show normal circadian behavior under a LD cycle, yet, owing to low amplitude of the SCN clock, rapidly adjust activity-rest rhythms in response to abrupt changes in the LD cycle, which mimic jet lag or rotating shift work (25). Similarly, mutation of a casein kinase 1 (CK1) phosphorylation site in Per2 advances the sleep onset time (26) but does not affect feeding time. A paralogous mutation in Per1 advances the daily onset of feeding without affecting the timing of activity-rest (27). It is unclear whether the different phenotypes seen in these paralogous mutants arise from shared neural mechanisms.
The SCN also communicates with peripheral tissues, including the gut and pancreas, which have their own autonomous circadian clocks. These local clocks mediate responses to nutrient intake and control the release of systemic factors. For example, a circadian clock in secretory cells of the gut drives rhythmic expression of SGLT1, accounting for increased glucose uptake at times of anticipated food intake (28). Glucose influx triggers the release of GLP1 incretin that, along with the direct effect of glucose on pancreatic islets, promotes insulin release. In pancreatic islet cells, like in many other cell types, exo-cytosis is modulated in a circadian manner (29). Accordingly, insulin release has a circadian component (Fig. 2A). Overall, the central circadian clock, through direct or indirect effects, generates systemic rhythms in several signaling molecules, including melatonin, glucocorticoids, growth hormones, insulin, glucagon, and GLP1, whose rhythms are further accentuated by LD or feeding-fasting cycles.
Fig. 2. Examples of circadian regulation of metabolic pathways and metabolic pathways affecting clock components.
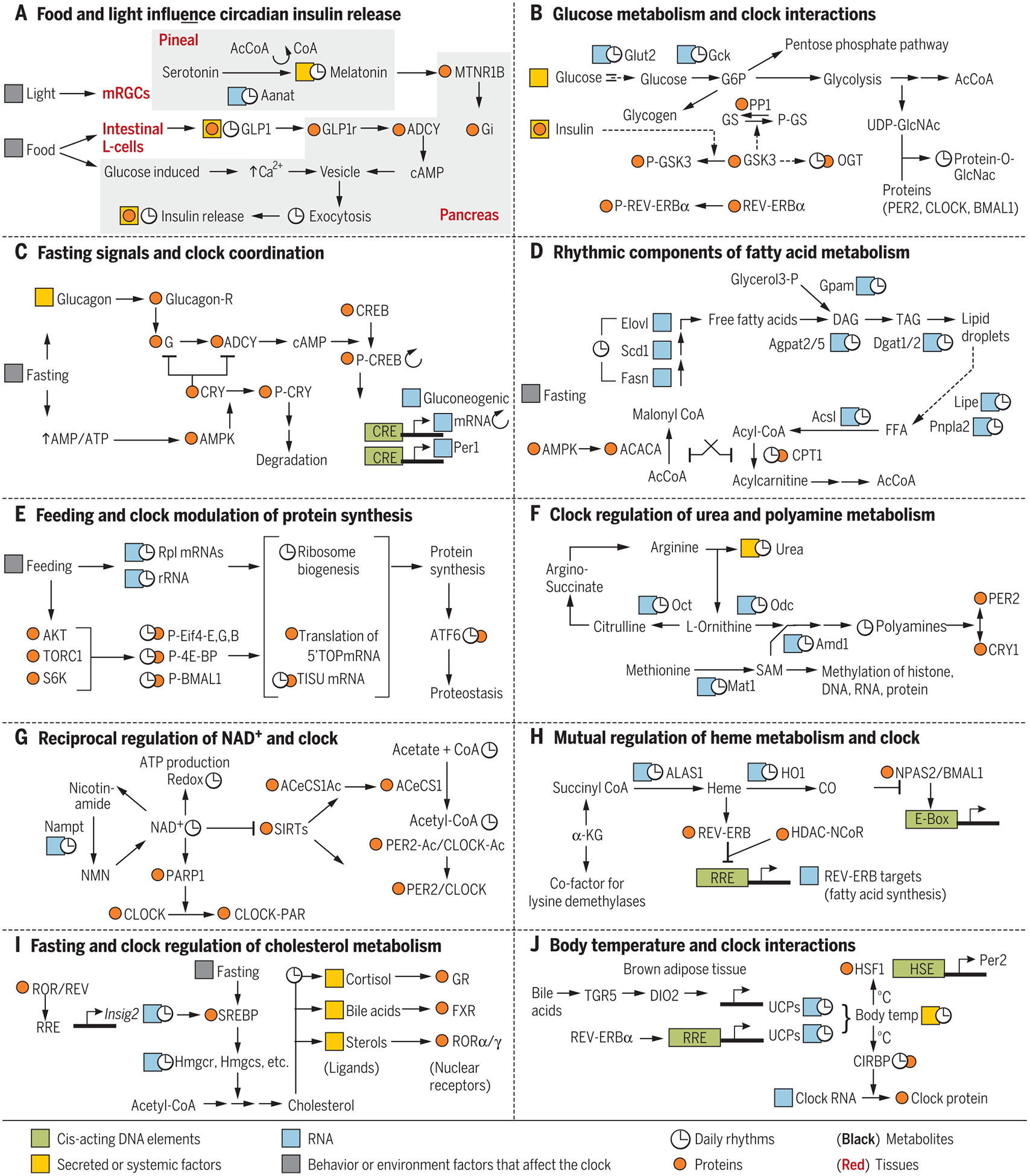
Cis-acting DNA elements are in green, RNA in blue, proteins in orange; metabolites are shown in black letters, and tissues are underlined. Any RNA, protein, or metabolite (other than clock components) known to show daily rhythms are marked with ↻. Secreted or systemic factors are highlighted in yellow, and behavior or environment factors that can affect the clock are highlighted in gray. (A) Light and food intake can interact through multiple tissues to modulate insulin release from pancreatic islet cells. (B) Feeding-induced glucose metabolism in the liver affects clock components. (C) During fasting, activation of glucagon receptor and AMPK impinge on clock components. (D) Fatty acid synthesis and degradation are under feeding-fasting and circadian regulation. (E) Circadian clock and feeding signals act together to produce a daily rhythm in protein synthesis. (F) Circadian regulation of urea cycle, SAM synthesis, and polyamine production. Polyamines affect interaction between PER2 and CRY1. (G) Reciprocal regulation between circadian clock and NAD production. (H) Circadian production of meme and CO affect the function of core circadian clock components. (I) Fasting and circadian clock regulate cholesterol metabolism and production of several ligands for nuclear hormone receptors. (J) Reciprocal regulation between circadian clock and body-temperature rhythm.
Global circadian regulation in the liver
Because the liver plays a central role in nutrient metabolism, is composed of relatively homogeneous cell types, and is easy to access, a number of ≥24-hour time-course “-omics” studies have been conducted using mouse liver tissue. These have included chromatin immunoprecipitation sequencing (ChIP-seq), RNA-seq (and comparable microarray hybridization), ribo-seq, nascentseq, proteomics, and metabolomics. The majority of these studies used young male mice (<20 weeks old) entrained to a 12-hour light/12-hour dark cycle for several days and fed a standard diet (calories from fat <18%, protein ~25%, carb ~60%) ad libitum. Tissue samples have usually been collected under circadian conditions (constant darkness). Such an approach allows the removal of confounding effects derived from light cues. Normally, the majority of a mouse’s daily food intake occurs during the subjective night, but ≥15% of food intake occurs during the subjective day in the form of frequent small snacks. Thus, under the ad libitum–fed condition, mice rarely fast a few contiguous hours. Under such conditions, at least 20% of expressed protein coding genes in the liver show circadian rhythms in transcription, mature mRNA, active translation, or protein levels. Rhythmic gene expression has several advantages. First, bioenergetic modeling of gene expression in yeast demonstrates that rhythmic gene expression is more energy efficient than maintaining constant levels of expression. This form of energy conservation is observed in a range of mammalian and insect tissues (30). Conversely, when a mouse is subjected to continuous 24-hour fasting, the number of rhythmic transcripts is reduced by almost 80%. This largely results from reduced peak levels of expression, rather than elevated trough level of rhythmic expression (30). Second, rhythmic expression helps to temporally separate incompatible biochemical processes, thereby preventing futile cycles (e.g., the simultaneous biosynthesis and degradation of a given molecule). Because almost 20% of liver transcripts show daily rhythms, it is logical that at least some of the enzymes and regulators in every metabolic pathway are likely to display circadian rhythmicity. More important, these rhythmically regulated pathway components often mediate rate-limiting steps, with peak levels of expression coinciding with substrate availability or metabolic need. Several key rhythmic metabolites exert effects on circadian clock components, thereby integrating the metabolic state with the regulatory mechanism. Sometimes, posttranslational modifiers (e.g., kinases and phosphatases) that regulate key enzymes of metabolic pathways also act on clock components, thus coupling metabolic and circadian regulatory processes. Some of these examples will be highlighted in the following section.
Metabolic pathways, metabolites, and their integration with the circadian clock
Functional annotation of the liver circadian cistrome, transcriptome, and proteome show that these are enriched with metabolic regulators. To maintain energy homeostasis, the liver stores nutrients during feeding periods and taps into this stored energy reserve during fasting periods. Intermediates from these cycles of anabolism and catabolism are used for cellular components and signaling. As feeding and fasting naturally alternate between day and night, interactions among feeding-fasting–driven regulation, metabolism, and circadian clocks have evolved to maintain normal physiology.
Glucose metabolism
Glucose enters hepatocytes where it is phosphorylated. Phosphoglucose is then (i) used for energy production via glycolysis, (ii) stored as glycogen for further use (glucogenesis), or (iii) used in the pentose phosphate pathway (PPP) (Fig. 2B). Expression of the hepatic glucose transporter GLUT2 and glucokinase (GCK) show daily rhythms with peak levels coinciding with periods of feeding (31). In the fed state, insulin activates glycogenesis through a signaling cascade that leads to the inhibition of glycogen synthase kinase (GSK3), thereby releasing the activity of glycogen synthase (GS). GSK3 has daily rhythms of phosphorylation and activity and acts on some circadian clock components (e.g., affecting the stability of REV-ERB) (32). β-linked N-acetylglucosamine (i.e., O- β -GlcNAc or O-GlcNAc), the attachment of UDP-GlcNAc to Ser/Thr residues of proteins, is yet another link between the circadian clock and glucose metabolism. The activity of the enzyme O-GlcNac transferase (OGT) is regulated by GSK3 (33) and, accordingly, a number of hepatic proteins show circadian rhythms in O-GlcNAcylation (34), including PER2, CLOCK, and BMAL1. The balance between O-GlcNacylation, glycosylation, CK1 phosphorylation, and protein phosphatase 1 (PP1)–mediated dephosphorylation of PER2 determines its stability (33). In parallel, O-GlcNacylation of CLOCK and BMAL1 interferes with their ubiquitination and degradation (35). Glucose use through the PPP is also connected to the circadian clock. The PPP is essential for nucleotide and amino acid biosynthesis, as well for replenishing the pool of cytoplasmic NADPH. Low NADPH levels activate the transcription factor NRF2, which can drive transcription of Rev-erb, thereby affecting the molecular clock (36).
In the fasted state, the circadian clock also influences glucose metabolism by interacting with glucagon signaling (Fig. 2C). Glucagon signals through its G-protein–coupled receptor and adeny-late cyclase to activate protein kinase A (PKA), which in turn promotes glycogenolysis and gluconeogenesis to supply glucose (37). PKA phosphorylates and thereby activates the bZIP transcription factor cyclic AMP response element–binding protein (CREB) so that it binds to cis-acting CRE sites at the Per1 and several gluconeogenic promoters, thereby stimulating their transcription (38). Additionally, CRY1 inhibits the activation of PKA by negatively regulating the G protein or adenylate cyclase (17, 39). On the other hand, prolonged fasting also increases the ratio of AMP/ATP (adenosine triphosphate) and activates AMP-activated kinase (AMPK), which phosphorylates CRYs and targets them for degradation (40). Together, this suggests a mechanism by which CRY1 acts as a balance point between the short- and long-term responses to nutrient deficit.
Lipid metabolism
Fatty acid synthesis and b oxidation are tightly controlled in the liver (Fig. 2D). Mitochondrial acetyl CoA is exported to the cytoplasm via a citrate/palmitate shuttle, where ATP citrate lyase (ACLY) is a rate-limiting enzyme. The circadian peak of ACLY expression coincides with feeding (31). The first committed step of fatty acid synthesis is the carboxylation of acetyl CoA by ACACA (acetyl CoA carboxylase) to produce malonyl CoA. ACACA is inactivated via phosphorylation by fasting-induced AMPK. The rate of mitochondrial b oxidation is limited by the entry of fatty acyl groups into the mitochondria by carnitine palmitoyl transferase 1 (CPT1) and CPT2. The levels of L-carnitine, CPT1, and CPT2 show daily rhythms (41). Furthermore, high levels of malonyl CoA, which are produced during fatty acid synthesis and peak during feeding, inhibit CPT activity. Such circadianand product-mediated regulation generates a daily rhythm in fatty acid synthesis and oxidation, which peak during feeding and fasting, respectively. This also gives rise to daily rhythms in several liver lipids (42). Rhythmic repression by REV-ERB α generates daily rhythms in many transcripts involved in the fatty acid synthesis pathway. Accordingly, Rev-erb α −/− mice exhibit fatty liver disease (43, 44).
Protein
Ingested proteins are degraded to amino acids in the small intestine and transported to the liver. Amino acids rarely remain free in cells, because they can be (i) used for protein synthesis during the fed state; (ii) used for gluconeogenesis during fasting; (iii) metabolized to bioactive molecules (e.g., methionine is adenylated to produce SAM); or (iv) degraded to liberate ammonia, which is fed into the urea cycle. During the fed state, insulin receptor substrate (IRS) downstream kinase AKT activates the mTOR-S6kinase pathway to promote protein translation. AKT or S6K1 also phosphorylates BMAL1 and recruits it to translation complexes, where it promotes complex activity (Fig. 2E) (45, 46). In combination with circadian rhythms in ribosome biogenesis (47) and preferential translation of specific subsets of mRNAs (48), this general rhythm in protein synthesis is specifically important for liver function, because it is a major source of critical secreted proteins, including albumin, retinol-binding protein, transthyretin, and proteins of the complement pathway.
During overnight fasting, circadian regulation of the transcription factor KLF15 in muscle and liver cells mediates circadian expression of downstream enzymes implicated in amino acid mobilization from muscle and their reuse in the liver for gluconeogenesis and the production of ammonia for the urea cycle (49). Accordingly, plasma levels of total amino acids, branched chain amino acids, and urea show circadian rhythms in humans, with peak levels at night (49). In the urea cycle, use of mitochondrial L-ornithine (derived from glutatmate) by ornithine carbamoyl transferase (OCT) serves as a critical step in the clearance of CO2 (Fig. 2F). Circadian regulation of Oct is imposed by KLF15. The importance of amino acid metabolism by Klf15 is demonstrated by the acute metabolic disruption seen in Klf15−/− mice when they are fed a protein-rich diet. These mice exhibit hypoglycemia, hyperammonemia, and impaired ureagenesis, which together lead to severe morbidity (49). In addition, circadian rhythms in several metabolites, Ca2+ [at least in the SCN (50)], and Mg2+ can affect the activity of Ca2+-activated protein kinases, as well as Mg2+ ATP- or Mg2+ uridine triphosphate- (glycogen synthesis) using enzymes (51).
Intermediate metabolites and the circadian clock
Intermediate products of nutrient metabolism give rise to several small molecules that can affect clock function in the cytoplasm or nucleus. Cytoplasmic ornithine is decarboxylated by ornithine decarboxylase (ODC) as the initial step in polyamine biosynthesis. Several genes involved in polyamine production are transcriptionally regulated by the circadian clock components, giving rise to a daily rhythm in polyamine (Fig. 2F) (52, 53). Circadian expression of the Odc gene is directly driven by CLOCK/BMAL (53). A derivative of methionine plays an important role in this pathway. mRNAs encoding regulatory proteins in methionine production and its adenylation to SAM (Bhmt, Mtrr, Mat1, and Mat2) show daily rhythms (31). SAM is a reactive methyl carrier that is used in methyl-group transfer reactions, including histone methylation, as well as the biosynthesis of polyamine, phosphatidylcholine, and the energy-rich phosphocreatine. SAM also inhibits the activity of methyl tetrahydrofolate reductase (MTHFR) and thereby is tightly linked to tetrahydrofolate or one-carbon metabolism, which is required for purine metabolism and RNA synthesis in non-dividing cells (54). Several genes involved in tetrahydrofolate metabolism exhibit a circadian rhythm (31, 55). Both reactive methyl-group metabolism and one-carbon metabolism likely signal to the circadian clock through polyamines. Polyamines modulate many protein-protein and protein-DNA interactions, regulating the circadian clock by promoting the interaction between PER2 and CRY1. Age-related dampening of the circadian clock may lead to reductions in polyamine, because supplementing mouse food with polyamine improves circadian rhythms in older mice (53).
NAD+
NAD+ (oxidized form of NAD) is emerging as a key regulator of metabolism because it (i) functions as an electron carrier, (ii) modulates protein function, and (iii) serves as a substrate for ADP ribosylation. In animal tissues, NAD+ is either synthesized de novo, or the nicotinic moiety is salvaged from nicotinamide for the synthesis of NAD+ in a reaction catalyzed by nicotinamide phosphoribosyltransferase (NAMPT). Owing to the short half-life of NAD+, this salvage pathway is essential for sustained availability of NAD+. The circadian clock drives daily rhythms in NAMPT, as well as the resulting rhythm in cellular NAD+ levels (56, 57). Direct binding of NAD+ to SIRT proteins inhibits their ability to deacetylate target proteins, including clock components (58, 59) and several metabolic enzymes. NAD+ is also used by PARP1 to add poly(ADP) ri-bose moieties to CLOCK, which increases CLOCK/BMAL1 affinity to DNA and delays repression by CRY/PER complex (Fig. 2G). This makes the hepatic clock less susceptible to abrupt changes in eating time (60).
Acetyl CoA
Acetyl CoA is a major energy metabolite that is generated by glycolysis, b-oxidation, and amino acid metabolism in the mitochondria, which is then transported out of the mitochondria by ACLY. In addition, acetyl CoA is also produced in the cytoplasm or nucleus by de novo synthesis from acetate and CoA by acetyl CoA synthase (AceCS1). The circadian oscillator exerts a dominant effect on the cytoplasm in several ways. First, key rate-limiting steps of the metabolic pathways that generate acetyl CoA are under circadian control. Second, the expression of ACLY is rhythmic. Third, the activity of AceCS1 is regulated by the NAD-dependent deacetylase SIRT1 (61, 62), and as seen above, NAD levels are under strong circadian control (Fig. 2G). Cytoplasmic acetyl CoA can diffuse into the nucleus through nuclear pores, where it can modulate the activity of lysine acetyl transferases with relatively high KD (low affinity) for acetyl CoA (63). Similarly, the concentration of acetyl CoA or the relative ratio of CoA/acetyl CoA can change the specificity of some proteins, such as p300 and CBP (63), both of which are associated with CLOCK/BMAL1 and activate histone acetylation (55).
Acetyl CoA is a crucial precursor for the synthesis of several molecule classes, including fatty acids, cholesterol, bile acids, steroid hormones, and ketone bodies (63). Many enzymes in these pathways are regulated in a circadian fashion, both at the mRNA and protein levels. In mitochondria, acetyl CoA molecules from pyruvate and fatty acid oxidation are primarily fed into the tricarboxylic acid (TCA) cycle. Two intermediates of the TCA cycle, α-KG and succinyl CoA, can have broader effects on transcription. Alpha-KG is the cofactor for several histone demethylases, and the ratio of succinyl CoA to α-KG has recently been shown to affect histone methylation state (Fig. 2H) (64).
Succinyl CoA-Aminolevulinate metabolic network
Succinyl CoA is the starting material for the synthesis of aminolevulinic acid, a reaction catalyzed by ALAS1, whose expression is circadian (65). Aminolevulinate is subsequently used for the synthesis of tetrapyrroles, including heme and cytochromes. Heme, among its many functions, is a ligand for REV-ERB. In addition, the degradation of heme is catalyzed by heme oxygenase (HO), which also shows circadian expression (66, 67). Such rhythms in these production and degradation pathways likely produce rhythms in Heme, as well as CO (a product of heme degradation). CO can presumably diffuse into the nucleus, where it can affect the DNA binding function of NPAS2 (Fig. 2H) (68). Cytochromes are also tetrapyrroles and function as cofactors for components of the mitochondrial electron transport chain and for cytochrome p450 (Cyp), which is a class of proteins that mediates xenobiotic metabolism. Interestingly, the PAR bZIP transcription factors DBP, TEF, and HLF are clock-controlled, rhythmic genes that drive the expression of a large number of Cyp genes and thereby generate rhythms in xenobiotic metabolism. (69).
Interorgan communications
Both fasting and circadian clock regulate the biosynthesis of cholesterol from acetyl CoA (Fig. 2I). Metabolism of cholesterol to produce several ligands for nuclear hormone receptors also shows daily rhythms. The Cyp7 proteins (Cyp7a and Cyp7b) mediate the first step in the metabolism of cholesterol to bile acids. Hepatic expression of Cyp7 is strongly circadian, and peak levels of Cyp7 expression correlate with a reduction in cholesterol and an increase in bile acids (Fig. 2I) (70, 71). Excess bile acids produced in liver can enter the circulation and subsequently activate UCP gene expression within brown adipose tissue (BAT), thereby contributing to thermoregulation (72). In BAT, REV-ERB α also imposes a circadian rhythm in UCP expression and contributes to daily rhythm in thermogenesis (Fig. 2J) (73). Accordingly, BAT-specific Rev-erb α−/− loses the rhythmic repression of UCP proteins and rhythmic fluctuation in body temperature (73). Both increases and decreases in temperature are thought to affect the clock throughout the body (74). Increases in body temperature can activate HSF1, which in turn activates Per2 gene expression from a cis-acting HSE site within its promoter (75). During temperature declines, expression of a cold-inducible RNA binding protein (CIRBP) can bind to CLOCK pre-mRNA and affect its processing (76).
In the pineal gland, acetyl CoA is used to produce melatonin by the enzymatic action of AANAT (77). Circadian expression of AANAT contributes to the circadian rhythm in melatonin, with peak levels during the night. Illumination in the cyan-blue spectrum can effectively suppress AANAT gene expression and melatonin production (Fig. 2A) (78).
Central integration of peripheral metabolic status
The mechanisms described above illustrate the extensive, reciprocal regulation between the circadian clock and cellular metabolism. Notably, these connections extend to the systemic level. For example, metabolic signals from the periphery also affect brain-specific circadian clocks. Although peripheral signals provide feedback to the SCN, these signals are often insufficient to override the robust influence of light. Forcing nocturnal rodents to eat during the day changes the peak phase of expression for nearly all rhythmically expressed genes in the liver without affecting phases of gene expression in the SCN, which remains tied to the LD cycle (31, 79, 80). However, the phase of circadian gene expression in the pituitary, dorsomedial hypothalamus (DMH), and PVN are affected by daytime feeding (81). Daytime access to a limited amount of food also elicits a survival strategy in nocturnal rodents by suppressing the natural circadian drive for daytime sleep and increasing food-seeking activity before the arrival of the daytime meal (82). This food anticipatory activity (FAA) seems to be mediated by ketone bodies produced by the liver that act on the dorsal striatum in a Per2-dependent manner (83). Such FAA is independent of the circadian clock in the SCN, as normal FAA can be triggered in mice lacking the SCN.
The effect of eating patterns on central nervous system clocks (e.g., the emergence of FAA, which seems to override SCN control of the activity-rest cycle) raises a larger question about the origin of nocturnal or diurnal (N-D) behavior in different species. The phase of the SCN circadian clock in both nocturnal and diurnal species is similar (84), implying that the N-D switch may not involve the SCN. The effect of food access time on extra-SCN brain clocks raises the provocative hypothesis that the N-D switch may be driven by complex interactions between the light-dark cycle, the feeding-fasting cycle, and overall energy balance. In mice, reducing the ambient temperature and restricting nutrition quantity can trigger daytime activity, suggesting that diurnality is a strategy for conserving energy (85). Understanding the mechanisms by which food, light, and ambient temperature affect the daily sleep-wake cycle and metabolism has increasing importance for humans who are living under diverse work schedules, lifestyles, and food preferences.
Health implication of metabolic and circadian integration of physiology in model organisms
Reciprocal interactions between metabolism and the circadian clock imply that nutrition quality, quantity, and daily eating pattern can affect diurnal rhythms, which in turn determines whole-body physiology. When mice are fed a standard diet ad libitum, they typically consume a majority of their daily food intake during the night. The vast majority of circadian -omics studies have been performed using mice fed a standard diet ad libitum. However, relative to these ad libitum–fed animals, the number and amplitude of rhythmic transcripts is substantially reduced in animals deprived of food or in circadian mutant mice, whereas it is increased in mice fed the same calories within an 8- to 12-hour interval [time-restricted feeding (TRF)]. TRF alone cannot sustain rhythmic expression of a vast majority of hepatic rhythmic transcripts, proteins, or metabolites in mice lacking a functional circadian clock (31, 41, 42, 48). When mice are fed a high-fat diet ad libitum, which is a widely used for diet-induced obesity (DIO), mice spread their caloric intake evenly throughout the 24-hour day (86). This eating pattern reprograms the hepatic diurnal transcriptome by dampening the oscillation of numerous circadian clock targets (compared to the diurnal transcriptome of mice fed a standard diet) (87). However, as seen in mice fed a standard diet, TRF of a high-fat diet improves molecular oscillations (70, 71, 88).
Subjecting genetically identical animals to caloric restriction or a high-fat diet (beneficial and adverse effects, respectively) has served as a powerful experimental system for understanding the roles of nutrition quantity and quality on health. Similarly, feeding isogenic animals identical, isocaloric diets ad libitum or via TRF has offered a foundation for understanding how the daily eating pattern affects diurnal rhythms and health (89). TRF can attenuate the adverse metabolic consequences (i.e., diet-induced pathologies) of high-fat, high-sucrose, or high-fructose diets in rodents and insects (Fig. 3) (70, 71, 88, 90). Comparing mice fed a normal or high-fat diet—either ad libitum or via TRF—is yielding new insights into how circadian regulation of metabolism is an integral part of physiology. DIO disrupts the temporal regulation of metabolism by tonic activation, by tonic suppression, or by mistiming the activation of several liver metabolic pathways (e.g., gluconeogenesis, fatty acid synthesis, cholesterol synthesis, bile acid production, and the pentose phosphate pathways). TRF reverses the adverse effects of a high-fat diet in the liver and other metabolic organs. Although the benefit of TRF on body weight is comparable for 8-, 9-, or 12-hour feeding intervals, several metabolic and physiological health indicators are differentially affected by these regimens, suggesting that the duration of fasting is also important (91). Similarly, TRF does not reduce body weight in mice fed a standard diet but increases lean mass at the expense of fat mass. TRF benefits are also seen in insects, where it has been shown to (i) support body-weight homeostasis, (ii) reduce age-dependent or high-fat diet-dependent deterioration of cardiac function, (iii) maintain sleep patterns, and (iv) promote flight-muscle function (90). Because subjecting model organisms to caloric restriction or DIO has revealed molecular mechanisms of metabolic health, the TRF model will likely yield new insights into how metabolism is temporally regulated. Preliminary studies in flies have shown that TRF and caloric restriction elicit different gene expression signatures and that TRF benefits on cardiac function depend on a functional circadian clock (90). However, it is still possible that fasting-induced molecular changes could contribute to TRF benefits. Similarly, it is not known whether TRF has beneficial effects in the absence of a functional clock in rodents. It is also worth mentioning that for many caloric-restriction experiments (both in rodents and in higher animals), the restricted group is often given food at a fixed time of the day and the animals consume the daily ration within a few hours, similar to TRF. In contrast, the control animals are given ad libitum access to food. Because of this experimental design, some of the health benefits seen with caloric restriction may have resulted from TRF. Altogether, these experiments stress the importance of eating patterns in metabolic regulation and have begun to inspire researchers to examine the contribution of daily eating patterns on metabolic outcomes in experimental animals and humans. For example, efforts to improve metabolic homeostasis in mice (or hepatocytes) have revealed two promising strategies: (i) behavioral intervention to improve circadian rhythm and (ii) pharmacological agents that target CRY, REV-ERB, or CLOCK (92–94). Targeting the interface between circadian rhythms and metabolism may therefore prove effective in alleviating the effect of metabolic disorders.
Fig. 3. Chronic circadian rhythm disruption by erratic lifestyle or high-fat-diet–induced obesity compromises physiology, whereas time-restricted feeding can restore daily rhythms and improve health.
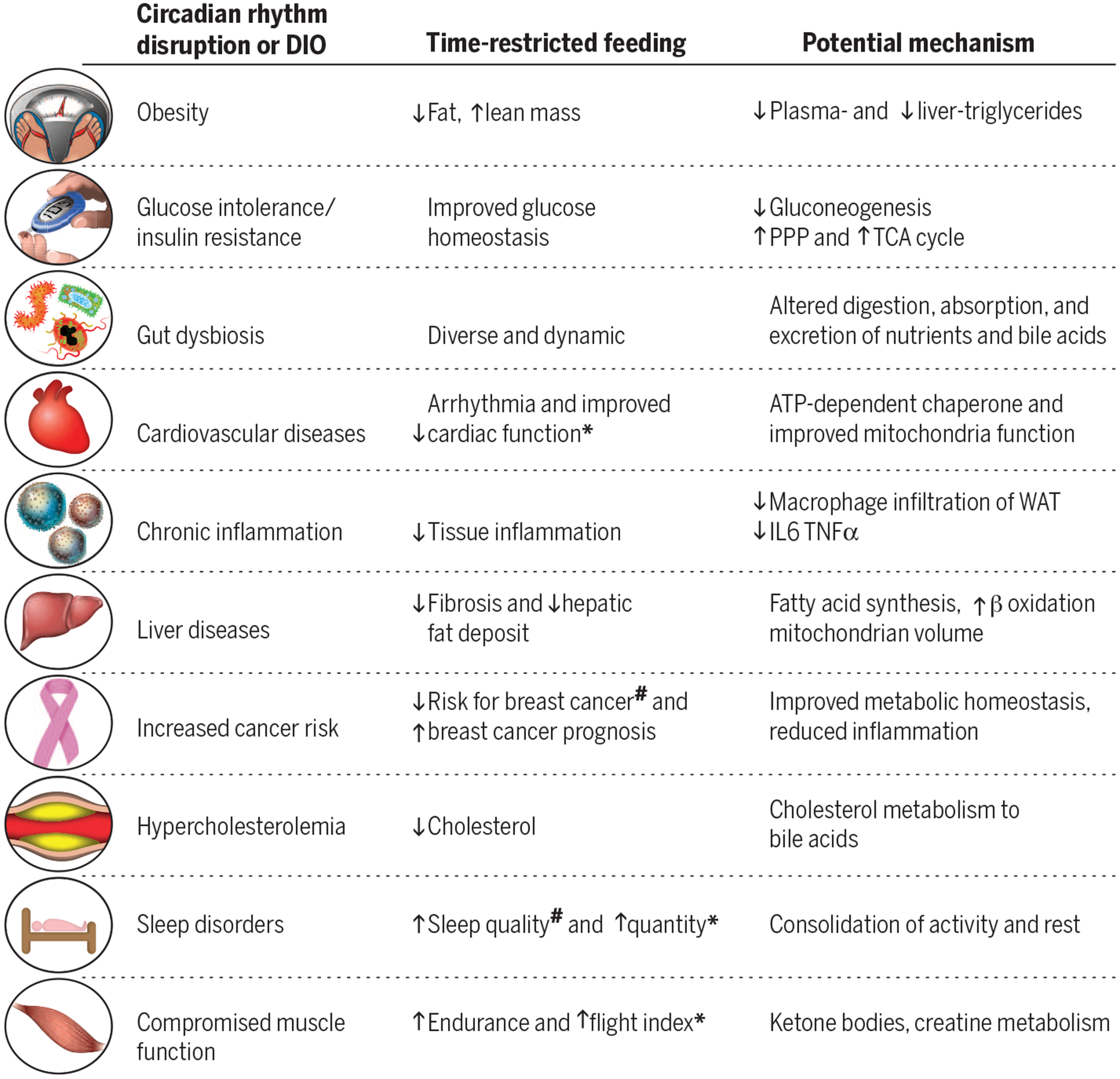
The potential mechanisms are largely based on rodent studies. Few observations have been made in insects (*) and in humans (#). IL, interleukin; TNF, tumor necrosis factor.
Perspective and conclusion
Although TRF results in health benefits irrespective of nutrition quality and quantity, numerous questions remain. How is “energy balance” explained in TRF? How do different macronutrients, micronutrients, supplements, and medications affect the clock? If timing of food intake can determine metabolic outcomes, can timing of medication be optimized for efficacy? Does TRF during the day versus night have different effects? How can we translate these findings to clinical practice or standard of care?
Close examination of TRF reveals that diurnal rhythms may affect components of energy balance (energy intake = absorption + storage + expenditure). In addition to circadian rhythms in the gut epithelium that affect nutrient absorption (95, 96), the composition of the gut microbiome, with respect to nutrient metabolism, also shows diurnal rhythms (97). Compared with ad libitum feeding, TRF does not affect the composition of the predominant cecal microbiome, but TRF mice excrete more simple sugars, which are microbially derived from complex carbohydrates in food (98). This implies that complex sugars are digested in the lower intestine (where absorption is relatively low) or that TRF somehow reduces overall sugar absorption. TRF also changes energy storage by increasing metabolically active lean mass and preventing the accumulation of fat mass. Even fat mass in TRF mice has a higher mitochondrial content (70, 71). TRF increases the peak level of Cyp7a/b expression, an effect that correlates with reduced cholesterol and increases in bile acids. Bile acids can act through TGR5 and DIO2 to increase BAT thermogenesis (72) and contribute to higher energy expenditure and increased O2 consumption in TRF mice. However, it is unclear why a considerable amount of bile acids are excreted in the feces of TRF mice. Nevertheless, inhibition of bile acid reabsorption in the gut protects against fatty liver disease (99).
Beyond the simple effects of nutrition on energy balance, some food components may affect the circadian clock even when consumed in moderation (i.e., at small caloric levels typically ignored for energy balance). For example, caffeine itself can change the phase of the circadian clock (100). As the mere presence of the gut microbiome is necessary for a robust liver circadian rhythm (101), noncaloric artificial sweeteners (102), as well as antibiotics known to change the gut microbiome composition, are likely to affect gut or hepatic circadian rhythms. Similarly, the absorption, target function, and clearance of many drugs are likely circadian (103). Therefore, systematic analyses of the timing of drug activities (specifically for those with a short half-life or those provided in small doses), as well the resulting prognosis, are warranted.
How might we translate these results into the standard of care? Epidemiological studies have repeatedly shown that sleep deprivation and shift work correlate with higher incidence of metabolic diseases in humans (104, 105). Conversely, overnight fasting (≥13 hours after controlling for sleep and activity) both prevents breast cancer and improves the prognosis of breast cancer patients (106, 107). These observations suggest that daily patterns of activity, sleep, and food intake may dramatically affect human heath and that these patterns should be systematically dissected to determine their roles in health. Preliminary studies using a smart-phone app have shown that nearly 50% of nonshift workers distribute their food intake over greater than 15 hours and, therefore, implementing a 10-hour TRF may promote weight loss and improve sleep (108). Retrospective analyses of a weight-loss study showed that eating earlier may lead to increased weight loss (109), suggesting that the relationship between the eating interval and the day-night cycle may affect metabolism. As stated above, hyperglycemia is sustained for a longer period of time after the evening meal than after the morning meal. This is likely because melatonin inhibits insulin release from pancreatic islets through the melatonin receptor 1B (Fig. 2A). Therefore, the evening rise in melatonin likely causes hyperglycemia (110). Because light in the blue spectrum strongly suppresses plasma melatonin level, it raises the interesting possibility that adjusting spectral quality and quantity in the indoor environment may affect metabolism. [However, melatonin action on metabolism extends beyond the pancreas (111), and its rise during nighttime feeding in nocturnal rodents adds further complexity to generalization of melatonin effects in both diurnal and nocturnal animals.] The rise of consumer markets for wearable sensors, ubiquity of smartphones, and their increasing use in research offer an unprecedented opportunity to longitudinally measure human feeding behavior, sleep patterns, activity levels, ambient light, body temperature, heart rate, and blood glucose to understand how these factors interact in free-living conditions. These data may reveal how the environment and diet may be manipulated to optimize the circadian physiology of metabolism.
ACKNOWLEDGMENTS
The author thanks A. Chaix, E. Manoogian, L. DiTacchio, and G. Benegiamo for their scientific input, and D. O’Keefe for copyediting the manuscript. Research in the author’s laboratory is partially supported by NIH grants EY016807, CA014195, EY019005, American Federation of Aging Research grant M14322, Leona M. and Harry B. Helmsley Charitable Trust’s grant 2012-PG-MED002, and Glenn Center for Aging Research.
REFERENCES AND NOTES
- 1.Van Cauter E, Polonsky KS, Scheen AJ, Endocr. Rev 18, 716–738 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Schwartz WJ, Gainer H, Science 197, 1089–1091 (1977). [DOI] [PubMed] [Google Scholar]
- 3.Asher G, Schibler U, Cell Metab. 13, 125–137 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Asher G, Sassone-Corsi P, Cell 161, 84–92 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Karlsson B, Knutsson A, Lindahl B, Occup. Environ. Med 58, 747–752 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudic RD et al. , PLOS Biol. 2, e377 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohawk JA, Green CB, Takahashi JS, Annu. Rev. Neurosci 35, 445–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Etchegaray JP, Lee C, Wade PA, Reppert SM, Nature 421, 177–182 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Duong HA, Robles MS, Knutti D, Weitz CJ, Science 332, 1436–1439 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masri S, Sassone-Corsi P, Nat. Rev. Neurosci 14, 69–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin L, Lazar MA, Mol. Endocrinol 19, 1452–1459 (2005). [DOI] [PubMed] [Google Scholar]
- 12.DiTacchio L et al. , Science 333, 1881–1885 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimomura K et al. , eLife 2, e00426 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aguilar-Arnal L, Sassone-Corsi P, Curr. Opin. Cell Biol 25, 170–176 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamia KA et al. , Nature 480, 552–556 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okabe T et al. , J. Cell Sci jcs.190959 (2016). [Google Scholar]
- 17.Zhang EE et al. , Nat. Med 16, 1152–1156 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y et al. , Genes Dev. 30, 1636–1644 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masri S et al. , Cell 158, 659–672 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato TK et al. , Neuron 43, 527–537 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Panda S et al. , Science 301, 525–527 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Ishida A et al. , Cell Metab. 2, 297–307 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Welsh DK, Takahashi JS, Kay SA, Annu. Rev. Physiol 72, 551–577 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dudley CA et al. , Science 301, 379–383 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Vitaterna MH et al. , Proc. Natl. Acad. Sci. U.S.A 103, 9327–9332 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toh KL et al. , Science 291, 1040–1043 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Liu Z et al. , Cell Reports 7, 1509–1520 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Rhoads DB, Rosenbaum DH, Unsal H, Isselbacher KJ, Levitsky LL, J. Biol. Chem 273, 9510–9516 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Perelis M et al. , Science 350, aac4250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang GZ et al. , Cell Reports 13, 1868–1880 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vollmers C et al. , Proc. Natl. Acad. Sci. U.S.A 106, 21453–21458 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin L, Wang J, Klein PS, Lazar MA, Science 311, 1002–1005 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Kaasik K et al. , Cell Metab. 17, 291–302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robles MS, Cox J, Mann M, PLOS Genet. 10, e1004047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li MD et al. , Cell Metab. 17, 303–310 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rey G et al. , Cell Metab. 24, 462–473 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mayr B, Montminy M, Nat. Rev. Mol. Cell Biol 2, 599–609 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Travnickova-Bendova Z, Cermakian N, Reppert SM, Sassone-Corsi P, Proc. Natl. Acad. Sci. U.S.A 99, 7728–7733 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Narasimamurthy R et al. , Proc. Natl. Acad. Sci. U.S.A 109, 12662–12667 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamia KA et al. , Science 326, 437–440 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neufeld-Cohen A et al. , Proc. Natl. Acad. Sci. U.S.A 113, E1673–E1682 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adamovich Y et al. , Cell Metab. 19, 319–330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng D et al. , Science 331, 1315–1319 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho H et al. , Nature 485, 123–127 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dang F et al. , Nat. Commun 7, 12696 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lipton JO et al. , Cell 161, 1138–1151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jouffe C et al. , PLOS Biol. 11, e1001455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Atger F et al. , Proc. Natl. Acad. Sci. U.S.A 112, E6579–E6588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeyaraj D et al. , Cell Metab. 15, 311–323 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ikeda M et al. , Neuron 38, 253–263 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Feeney KA et al. , Nature 532, 375–379 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Panda S et al. , Cell 109, 307–320 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Zwighaft Z et al. , Cell Metab. 22, 874–885 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Ducker GS, Rabinowitz JD, Cell Metab, 10.1016/j.cmet.2016.08.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koike N et al. , Science 338, 349–354 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P, Science 324, 654–657 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramsey KM et al. , Science 324, 651–654 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Asher G et al. , Cell 134, 317–328 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Nakahata Y et al. , Cell 134, 329–340 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Asher G et al. , Cell 142, 943–953 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E, Proc. Natl. Acad. Sci. U.S.A 103, 10224–10229 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hallows WC, Lee S, Denu JM, Proc. Natl. Acad. Sci. U.S.A 103, 10230–10235 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G, Cell Metab. 21, 805–821 (2015). [DOI] [PubMed] [Google Scholar]
- 64.TeSlaa T et al. , Cell Metab. 24, 485–493 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaasik K, Lee CC, Nature 430, 467–471 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Rubio MF, Agostino PV, Ferreyra GA, Golombek DA, Neurosci. Lett 353, 9–12 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Xu YQ et al. , PLOS ONE 7, e44237 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dioum EM et al. , Science 298, 2385–2387 (2002). [DOI] [PubMed] [Google Scholar]
- 69.Gachon F, Olela FF, Schaad O, Descombes P, Schibler U, Cell Metab. 4, 25–36 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Hatori M et al. , Cell Metab. 15, 848–860 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chaix A, Zarrinpar A, Miu P, Panda S, Cell Metab. 20, 991–1005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Watanabe M et al. , Nature 439, 484–489 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Gerhart-Hines Z et al. , Nature 503, 410–413 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buhr ED, Yoo SH, Takahashi JS, Science 330, 379–385 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reinke H et al. , Genes Dev. 22, 331–345 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morf J et al. , Science 338, 379–383 (2012). [DOI] [PubMed] [Google Scholar]
- 77.Klein DC, Weller JL, Science 169, 1093–1095 (1970). [DOI] [PubMed] [Google Scholar]
- 78.Hatori M, Panda S, Trends Mol. Med 16, 435–446 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yamazaki S et al. , Science 288, 682–685 (2000). [DOI] [PubMed] [Google Scholar]
- 80.Damiola F et al. , Genes Dev. 14, 2950–2961 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mukherji A et al. , Proc. Natl. Acad. Sci. U.S.A 112, E6691–E6698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boulos Z, Rosenwasser AM, Terman M, Behav. Brain Res 1, 39–65 (1980). [DOI] [PubMed] [Google Scholar]
- 83.Chavan R et al. , Nat. Commun 7, 10580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nunez AA, Bult A, McElhinny TL, Smale L, Biol J. Rhythms 14, 300–306 (1999). [DOI] [PubMed] [Google Scholar]
- 85.van der Vinne V, Gorter JA, Riede SJ, Hut RA, J. Exp. Biol 218, 2585–2593 (2015). [DOI] [PubMed] [Google Scholar]
- 86.Kohsaka A et al. , Cell Metab. 6, 414–421 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Eckel-Mahan KL et al. , Cell 155, 1464–1478 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sherman H et al. , FASEB J. 26, 3493–3502 (2012). [DOI] [PubMed] [Google Scholar]
- 89.Mattson MP et al. , Proc. Natl. Acad. Sci. U.S.A 111, 16647–16653 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gill S, Le HD, Melkani GC, Panda S, Science 347, 1265–1269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Longo VD, Panda S, Cell Metab. 23, 1048–1059 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hirota T et al. , Science 337, 1094–1097 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Solt LA et al. , Nature 485, 62–68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.He B et al. , Cell Metab. 23, 610–621 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hussain MM, Pan X, Curr. Opin. Clin. Nutr. Metab. Care 15, 336–341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stubblefield JJ, Terrien J, Green CB, Trends Endocrinol. Metab 23, 326–333 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thaiss CA et al. , Cell 159, 514–529 (2014). [DOI] [PubMed] [Google Scholar]
- 98.Zarrinpar A, Chaix A, Yooseph S, Panda S, Cell Metab. 20, 1006–1017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rao A et al. , Sci. Transl. Med 8, 357ra122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Burke TM et al. , Sci. Transl. Med 7, 305ra146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Leone V et al. , Cell Host Microbe 17, 681–689 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Suez J et al. , Nature 514, 181–186 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Dallmann R, Okyar A, Lévi F, Trends Mol. Med 22, 430–445 (2016). [DOI] [PubMed] [Google Scholar]
- 104.Spiegel K, Leproult R, Van Cauter E, Lancet 354, 1435–1439 (1999). [DOI] [PubMed] [Google Scholar]
- 105.Arble DM et al. , Sleep 38, 1849–1860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marinac CR et al. , JAMA Oncol. 2, 1049–1055 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marinac CR et al. , Cancer Epidemiol. Biomarkers Prev 24, 783–789 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gill S, Panda S, Cell Metab. 22, 789–798 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Garaulet M et al. , Int. J. Obes 37, 604–611 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tuomi T et al. , Cell Metab. 23, 1067–1077 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Persaud SJ, Jones PM, Engl N. J. Med 375, 1090–1092 (2016). [DOI] [PubMed] [Google Scholar]