

Abstract
Delayed graft function (DGF) in renal transplantation is associated with reduced graft survival and increased immunogenicity. The complement-driven inflammatory response after brain death (BD) and post-transplant reperfusion injury play significant roles in the pathogenesis of DGF. In a non-human primate model, we tested complement-blockade in BD donors to prevent DGF and improve graft survival. BD donors were maintained 20-hours, kidneys were procured and stored at 4°C for a 43–48 hours prior to implantation into ABO-compatible, non-sensitized, MHC-mismatched recipients. Animals were divided into three different donor-treatment groups: G1-Vehicle, G2-rhC1INH+heparin, and G3-heparin only. G2 donors showed significant reduction in classical complement pathway activation and decreased levels of TNFα and MCP-1. DGF was diagnosed in 4/6 (67%) G1-recipients, 3/3 (100%) of G3-recipients, and 0/6 (0%) of G2-recipients (p=0.008). In addition, G2-recipients showed superior renal function, reduced sC5b-9, and reduced urinary NGAL in the first week post-transplant. We observed no differences in incidence or severity of graft rejection between groups. Collectively, the data indicate that donor-management targeting complement activation prevents the development of DGF. Our results suggest a pivotal role for complement activation in BD-induced renal injury and postulate complement-blockade as a promising strategy for the prevention of DGF after transplantation.
1. INTRODUCTION
Delayed graft function (DGF) manifests as a consequence of ischemia-reperfusion injury (IRI) and is characterized by acute kidney injury (AKI) within 7 days of transplant, requiring life-sustaining dialysis.1 The incidence of DGF in kidney transplants from brain dead (BD) donors is approximately 26% in the United States, and this rate can reach as high as 37% in kidneys from older donors and those subjected to extended cold ischemia >36 hours.2–4 In addition to complications related to AKI in the peri-transplant period, development of DGF is an important risk factor for acute cellular rejection (ACR), antibody-mediated rejection (AMR) and reduced graft survival.2, 5–9 Inflammatory injury secondary to IRI is also key to mechanisms leading to DGF and subsequent graft rejection.5 During BD, the donor experiences neuro-hormonal changes known to trigger a systemic inflammatory response characterized by the release of pro-inflammatory cytokines from both innate and adaptive immune cells, including interleukin-1-beta (IL-1β), tumor necrosis factor alpha (TNFα), interferon gamma (IFNγ), and interleukin-17 (IL-17), chemokines and reactive oxygen species (ROS) and reactive nitrogen species including Nitric Oxide (NO). This inflammatory response promotes recruitment of activated immune cells affecting vascular tone and exacerbating the degree of injury while enhancing graft immunogenicity.10–12
Activation of complement, whether through the classic (CP), mannose-binding lectin (LP) or alternative (AP) pathway, has gained special attention due to its role in the pathogenesis of renal IRI, transplant rejection and acute tubular injury.13–17 Recent reports suggest that systemic complement activation reduces renal allograft quality starting at the time of BD and progressing through cold-storage and reperfusion.18–21
Recombinant human C1 esterase inhibitor (rhC1INH) is a serine protease inhibitor that inactivates proteases of the complement, contact, fibrinolytic and coagulation systems. It acts as a major regulator by inhibiting the CP and LP of complement activation and preventing amplification of the inflammatory response.22–23 In renal IRI and kidney transplantation models, C1 inhibition has shown protective effects on vessel/organ integrity, and reduced IRI and progression to AMR after renal transplantation.16, 24–26 The objective of this study was to determine the impact of rhC1INH as a donor treatment strategy in BD conditions for the prevention of early post-transplant kidney dysfunction and modulation of immune responses. We utilized a non-human primate (NHP) model of BD in older animals, prolonged cold ischemia, and transplantation into non-sensitized, fully-mismatched recipients to investigate the potential protective effect of this donor-management strategy to improve outcomes in kidney transplant recipients.
2. MATERIALS AND METHODS
2.1. Animals and animal care
Rhesus macaques were used in this study (Table 1). Donor animals (n=8, aged 15–22 years) and transplant recipients (n=15, aged 3–7 years) were obtained from the University of Wisconsin Primate Center (WNPRC) and Alpha Genesis Inc.(Yemassee, SC) . All animals were pre-screened negative for tuberculosis, Herpes B, SRV, SIV, and STLV-1. Each donor–recipient pair was ABO blood compatible, non-sensitized, and fully mismatched for major histocompatibility complex (MHC) class I and II alleles identified using microsatellite analysis as previously described (data not reported).27 Animals were housed in accordance with NIH and USDA animal welfare guidelines; all protocols were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Madison.
Table 1. Experimental design, kidney function and survival data.
Individual values presented along with group averages ±SD, significance calculated by one-way ANOVA (*p<0.05 G1 vs G2; †p<0.05 G1 vs G3; ‡p<0.05 G2 vs G3).
| Donor ID | Donor Age (years) | Donor Treatment group | rhC1-INH Dose | Recovered kidney | Cold ischemia time (hours) | Recipient ID | Age (years) | Weight (Kg) |
Graft Survival (Days) | Meet DGF criteria | Peak creatinine level (mg/dL) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Group 1 | |||||||||||
| Rhas84 | 20.8 | Vehicle | N/A | Right kidney | 44.5 | R11051 | 4.3 | 8.3 | 3 | Yes | 14.8 |
| Left kidney | 47.9 | R11107 | 4.2 | 4.7 | 90 | No | 6.7 | ||||
| Rh2602 | 16.6 | Vehicle | N/A | Left kidney | 46.7 | Rh2612 | 4.0 | 4.0 | 39 | Yes | 10.9 |
| Rh2030 | 21.2 | Vehicle | N/A | Right kidney | 44.1 | Rh2636 | 5.6 | 5.4 | 29 | Yes | 15.7 |
| Left kidney | 47.2 | R13020 | 3.6 | 5.9 | 10 | Yes | 18.4 | ||||
| Average | 19.5 ± 1.8 | N/A | N/A | N/A | 46.1 ± 1.7 | N/A | 4.3 ± 0.7 | 5.7 ± 1.6 † | 34.2 ± 34.4 | 4/5 (80%) | 13.3 ± 4.6 |
| Group 2 | |||||||||||
| Rhat64 | 21.2 | rhC1INH + heparin | 500U/kg per time point | Right kidney | 45.0 | R13009 | 3.8 | 7.3 | 26 | No | 4.3 |
| rhC1INH + heparin | 500U/kg per time point | Left kidney | 47.8 | R12063 | 4.3 | 5.4 | 26 | No | 3.8 | ||
| Rh2241 | 18.8 | rhC1INH + heparin | 500U/kg per time point | Left kidney | 45.2 | Rh2622 | 5.9 | 9.7 | 67 | No | 7.3 |
| rhC1INH + heparin | 500U/kg per time point | Right kidney | 46.8 | Rh2625 | 4.8 | 7.1 | 32 | No | 13.0 | ||
| Rhav29 | 20.1 | rhC1INH + heparin | 500U/kg per time point | Right kidney | 44.0 | Rh2616 | 6.2 | 8.6 | 90 | No | 8.4 |
| rhC1INH + heparin | 500U/kg per time point | Left kidney | 46.0 | Rh2623 | 5.1 | 8.0 | 46 | No | 14.8 | ||
| Average | 20.0 ± 1.2 | N/A | N/A | N/A | 45.8 ± 1.4 | N/A | 5.0 ± 0.9 | 7.4 ± 1.4 | 47.8 ± 25.8 | 0/6 (0%) | 8.6 ± 4.5 ‡ |
| Group 3 | |||||||||||
| Rh1966 | 20.7 | Heparin | N/A | Left kidney | 45.7 | Rh2615 | 6.1 | 13.9 | 4 | Yes | 15.9 |
| R01070 | 15.7 | Heparin | N/A | Right kidney | 45.2 | Rh2627 | 6.0 | 11.4 | 5 | Yes | 20.0 |
| Heparin | N/A | Left kidney | 47.5 | Rh2629 | 6.1 | 11.1 | 5 | Yes | 18.7 | ||
| Average | 18.2 ± 3.5 | N/A | N/A | N/A | 46.1 ± 1.2 | N/A | 6.0 ± 0.05 | 12.1 ± 1.5 † | 4.6 ± 0.5 | 3/3 (100%) | 18.2 ± 2.1 ‡ |
| P-value | ns | N/A | N/A | N/A | ns | N/A | ns | † p < 0.05 | ns | p= 0.0045 | ‡ p < 0.05 |
G1 vs G2,
G1 vs G3
G2 vs G3
2.2. Study drug and experimental design
Recombinant human C1 Inhibitor (rhC1INH) was provided by Pharming Technologies B.V. (Leiden, The Netherlands).
Donor animals were randomly assigned to treatment groups (Figure 1a). Donors were maintained for a 20-hour period. BD donor management was performed following previously published guidelines in order to maintain hemodynamic stability and adequate oxygen delivery.11,28 Briefly, donor animals were anesthesized, ventilated and monitored. A 16F Foley catheter was placed in the extradural space of the cranial fossa (i.e. the intra-cranial space) and gradually inflated until hemodynamic and neurologic signs of brain-stem herniation were documented. Animals were monitored and received standard donor management based on IV fluid resuscitation and vasopressor support to achieve a stable mean arterial pressure and urinary output. Twenty hours after brain death induction, both kidneys were recovered after cannulation and retrograde infusion of UW preservation solution (Organ Recovery Systems, IL) supplemented with heparin (5 U/ml). Recovered kidneys were preserved in UW solution at 4°C for a 43–48 hours prior to implantation into the recipient. G1 donors received vehicle treatment (0.9% normal saline) by intravenous bolus injection given at t= 180, 360, 540, 720, 900, and 1080 minutes. G2 donors received rhC1INH (500 U/kg/dose) treatment by intravenous bolus injection at the indicated time points in combination with continuous intravenous heparin infusion t=180 → t=1080 minutes titrated to a partial thromboplastin time (PTT) of 80–120 seconds. G3 donors received only continuous intravenous heparin infusion as described with dosing titrated to PTT 80–120 seconds (Figure 1a). Heparin was used in recipients of G2 to potentiate the activity of rhC1INH as described previously.29–31
Figure 1. Experimental design.
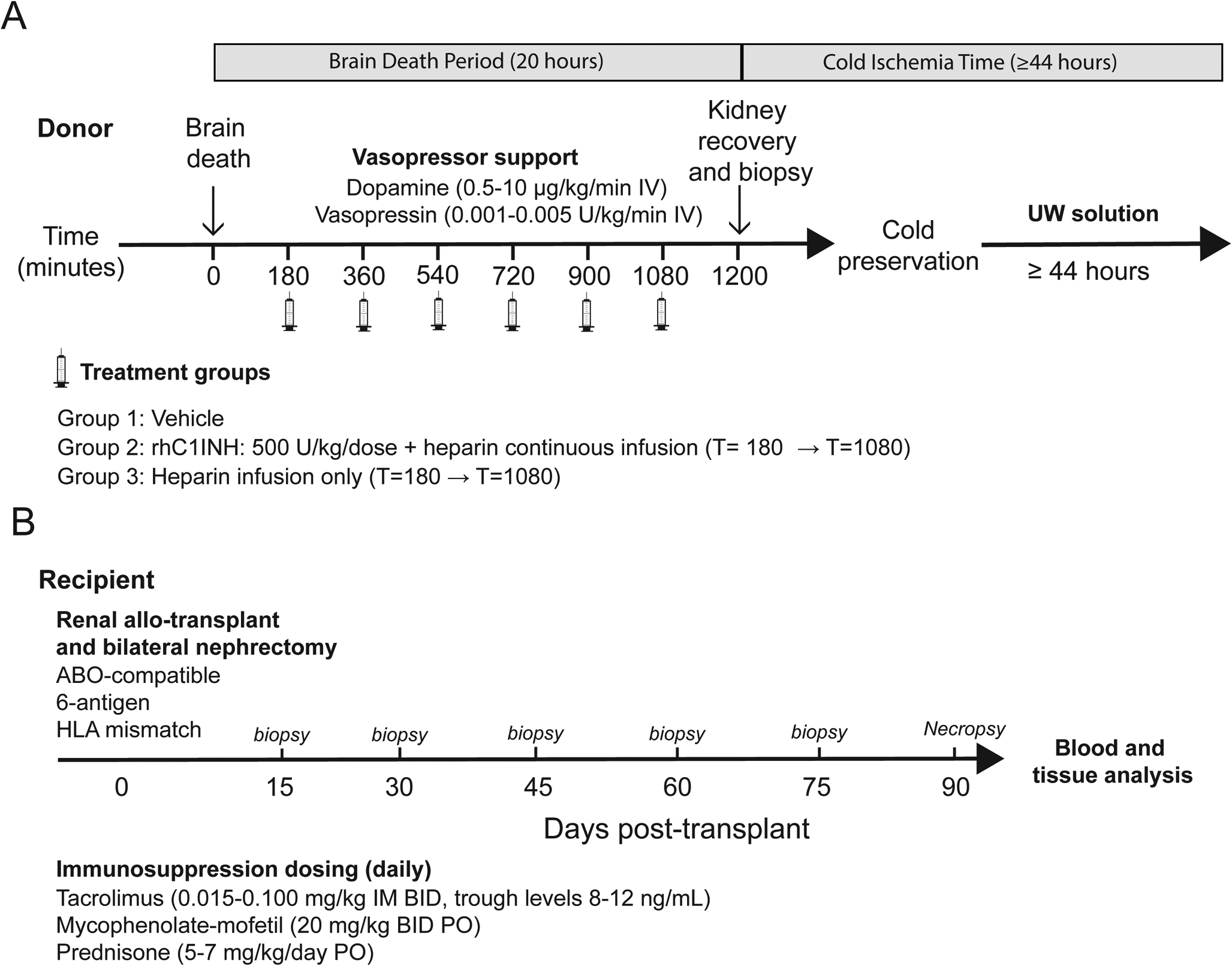
All recipients underwent bilateral native nephrectomy at the time of transplant and heterotopic kidney transplantation was performed as described previously (Figure 1b).32 Recipients were monitored for acid/base and electrolyte balance, serum BUN (blood urea nitrogen), creatinine, urinary output, proteinuria and behavioral abnormalities. No induction therapy was used, but maintenance therapy included mycophenolate-mofetil (MMF), tacrolimus, and prednisone. Tacrolimus levels were dose-adjusted bi-weekly to maintain 8–12 ng/mL trough levels. Criteria for termination of the study were defined as 1) survival for 90 days or 2) progressive acute kidney failure and severe azotemia not responsive to medical management.
2.3. Definition of delayed graft function
DGF was defined as a failure of a fall in serum creatinine of at least 10% on 3 consecutive days in the first post-transplant week and/or serum creatinine at post-transplant day 7 >2.5 mg/dL.1, 33
2.4. Circulating Cytokines
Circulating levels of IL-6, IL-8, MCP-1, and TNFα in EDTA-plasma were measured with Biolegend LEGENDplex™ NHP Mix-and-Match Subpanel according to manufacturer recommended protocols.
2.5. Complement assessment
Blood was collected into Vacuettes® (Greiner Bio-One, Austria), serum tubes were allowed to clot 15 minutes prior to centrifugation for 10 minutes at 3000xg, then serum and K3-EDTA-plasma were aliquoted and stored at −80°C. Each assay was tested for cross-reactivity with rhesus macaque and to establish a linear range. Assays were performed according to manufacturer instructions. C1 inhibitor (C1INH) was measured in rhesus serum using the C1INH ELISA Pair (Sino Biological SEK10995–5). CP, AP, and LP activation were tested using the Wieslab Complement Kits (CP310, AP330, MP320, EuroDiagnostica, Sweden). Circulating levels of sC5b-9 (membrane attack complex, MAC) were measured using a commercially available sC5b-9 enzyme immunoassay (ELISA kit; Quidel, San Diego, CA). Measured values for each assay were normalized to serum albumin (VetTest Analyzer, Idexx, Westbrook, ME) to account for hemodilution observed over the course of BD.
2.6. Histology and microscopic evaluation
At the discretion of the veterinary staff and attending surgeons, kidney core biopsies were collected from grafts prior to cold ischemia after the 20-hour BD period, as well as 60 minutes and also 7 days post-reperfusion, and finally at necropsy. Biopsies were also collected from the naïve native kidneys removed from recipients during the operation prior to graft reperfusion. Tissue was fixed in 10% formalin and embedded in paraffin or frozen in optimal cutting temperature (OCT) compound. 4μm slices were mounted onto slides and stained for histological assessment. Stains included hematoxylin and eosin (H/E), periodic acid–Schiff (PAS), Picro Sirius Red for estimation of fibrosis, as well as antibodies against CD68 (KP1, DAKO-Agilent), myeloperoxidase (MPO, Abcam), malondialdehyde (MDA, Abcam), and C4d, C3b, and C5b-9 (Ventana-Roche, AZ). The HIER method was used for antigen retrieval (BioGenex, San Ramon, CA). Slides for histopathology were interpreted by a renal pathologist. For immunohistochemistry, images were acquired from 6–12 random fields within each slide at appropriate magnification using an Olympus BX51 microscope (Olympus, Tokyo, Japan) and processed using ImageJ software (NIH, Bethesda, MD) according to internal laboratory protocols. Cell counts or area fraction measurements for each image were quantified using color-separation, automatic thresholding, and particle-analysis algorithms.
2.7. Urinary neutrophil gelatinase-associated lipocalin (NGAL) measurement
Post-transplant urine was stored at −80˚C until analysis. Urinary NGAL level was quantified using the NHP NGAL ELISA kit (Bioporto, Copenhagen, Denmark) according to the manufacturer protocol.
2.8. Statistical analysis
Statistical analyses were performed using GraphPad Prism V5.04. All data are shown as mean ± standard error of the mean (SEM) or standard deviation (SD). DGF incidence and resistive indices were analyzed by chi-square test. Comparisons of two groups were tested by two-tailed Student’s T-test. Differences between 3 or more groups were tested by one-way ANOVA and Bonferroni’s post-test correction or Kruskal-Wallis test and Dunn’s post-test correction in data sets with non-normal distribution. Data sets with two independent variables were tested by two-way ANOVA followed by Bonferroni’s correction. Differences between treatment groups were considered significant at p<0.05.
3. RESULTS
3.1. rhC1INH treatment results in sustained elevation of circulating C1INH, inhibits complement activation, and reduces C3b/C5b-9 deposition in BD donors.
Over a 20-hour BD period, donor animals received six bolus intravenous administrations of either vehicle (Group 1, G1) or rhC1INH with continuous heparin infusion (Group 2, G2), or continuous heparin infusion only (Group 3, G3) starting at 3 hours after induction of BD, until 2 hours prior to organ recovery (Figure 1a, Table 1). To confirm the BD state, we monitored the hemodynamics of BD donors throughout this 20-hour period and observed the Cushing reflex as a result of increased intracranial pressure characterized by hypertensive response followed by hypotension, tachycardia, then bradycardia (Figure 2). We did not detect clinically significant differences for any parameters between groups at any of the time points investigated (Table 2).
Figure 2.
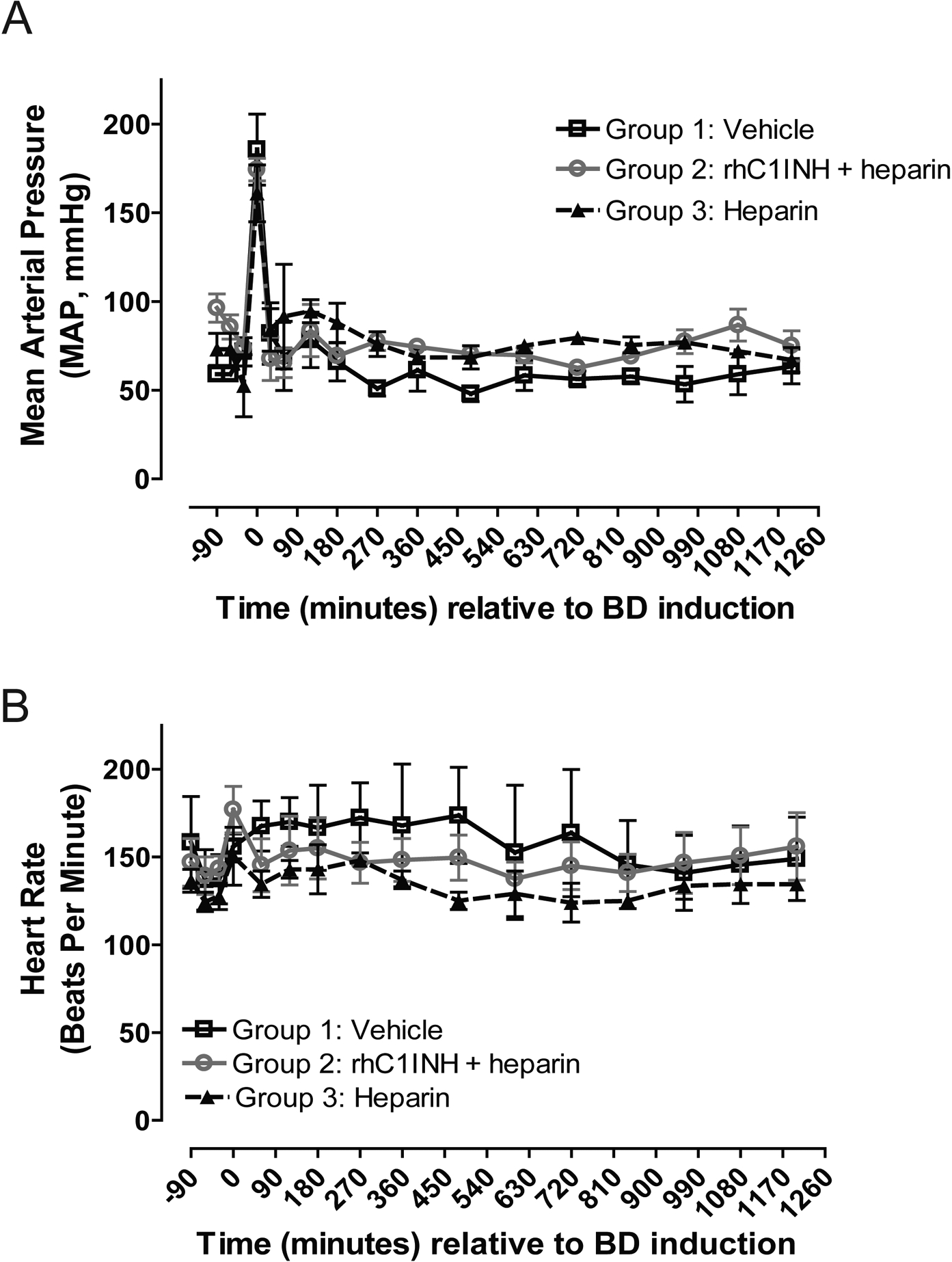
Hemodynamic assessment of brain-dead donors over the course of the experimental period. (a) Mean arterial pressure (MAP) measured in mmHg by continuous invasive intra-arterial monitor. (b) Heart rate in beats per minute.
Table 2. Kidney Donor Vital Signs and Laboratory Values at Baseline and 20 hours after Brain Death.
Data presented as average values ± SD. Vital signs were monitored throughout the BD period, no clinically significant differences were detected for any parameters between groups at any of the time points investigated. Body temperature was monitored in degrees Farenheit (°F).
| Baseline | T=1200 minutes (20 hours) | |||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Group 1 Vehicle | Group 2 rhC1INH + heparin | Group 3: Heparin | P Value | Group 1 Vehicle | Group 2 rhC1INH + heparin | Group 3: Heparin | P Value |
| Vitals, mean ± SD | ||||||||
| Temperature, ◦F | 98.7 ± 1.8 | 95.8 ± 1.7 | 97.9 ± 1.8 | 0.226 | 98.7 ± 1.4 | 98.6 ± 1.8 | 99.9 ± 0.2 | 0.614 |
| Pulse, bpm | 138.3 ± 24.7 | 139.3 ± 18.6 | 125.0 ± 14.1 | 0.726 | 156.7 ± 28.0 | 150.7 ± 33.6 | 134.0 ± 8.5 | 0.686 |
| MAP, mmHg | 70.0 ± 6.2 | 78.3 ± 8.5 | 52.5 ± 24.7 | 0.184 | 64.0 ± 16.1 | 77.7 ± 6.4 | 59.5 ± 0.7 | 0.234 |
| UOP, mL/kg/h | NA | 3.3 ± 0.8 | 4.9 ± 3.9 | 12.2 ± 7.6 | 0.149 | |||
| Metabolic panel , mean ± SD | ||||||||
| Na, mmol/L | 147.3 ± 2.3 | 146.3 ± 1.5 | 148.5 ± 0.7 | 0.465 | 143.7 ± 2.9 | 150.0 ± 4.4 | 157.5 ± 0.7 | 0.016 |
| K, mmol/L | 3.5 ± 0.4 | 3.5 ± 1.3 | 3.3 ± 0.0 | 0.945 | 3.6 ± 0.5 | 2.9 ± 0.3 | 3.2 ± 0.1 | 0.151 |
| Cl, mmol/L | 111.3 ± 2.3 | 109.3 ± 1.2 | 111.0 ± 1.4 | 0.406 | 107.7 ± 2.9 | 112.0 ± 5.2 | 119.5 ± 2.1 | 0.053 |
| CO2, mmol/L | 23.7 ± 0.6 | 26.0 ± 1.0 | 24.0 ± 2.8 | 0.211 | 22.3 ± 2.5 | 23.7 ± 3.1 | 23.0 ± 2.8 | 0.849 |
| BUN, mg/dL | 18.0 ± 1.7 | 14.7 ± 4.0 | 21.5 ± 0.7 | 0.107 | 16.7 ± 5.5 | 13.3 ± 8.6 | 8.5 ± 0.7 | 0.446 |
| Cr, mg/dL | 0.7 ± 0.2 | 0.4 ± 0.1 | 0.6 ± 0.0 | 0.064 | 0.8 ± 0.1 | 0.5 ± 0.2 | 0.5 ± 0.1 | 0.031 |
| Glc, mg/dL | 68.3 ± 5.5 | 84.3 ± 16.9 | 61.0 ± 8.5 | 0.167 | 117.0 ± 37.3 | 106.7 ± 41.7 | 135.5 ± 2.1 | 0.690 |
| CBC , mean ± SD | ||||||||
| Hb, g/dL | 12.6 ± 0.4 | 13.0 ± 1.2 | 11.7 ± 4.1 | 0.775 | 9.3 ± 2.0 | 10.6 ± 0.9 | 11.6 ± 1.9 | 0.382 |
| Hct, % | 37.0 ± 1.0 | 38.3 ± 3.5 | 34.5 ± 12.0 | 0.782 | 27.3 ± 5.9 | 31.0 ± 2.6 | 34.0 ± 5.7 | 0.374 |
| ABG, mean ± SD | ||||||||
| pH | 7.4 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.1 | 0.468 | 7.3 ± 0.1 | 7.4 ± 0.1 | 7.3 ± 0.1 | 0.181 |
| pCO2, mmHg | 49.7 ± 14.1 | 49.1 ± 6.8 | 49.3 ± 3.9 | 0.997 | 50.2 ± 11.8 | 35.7 ± 10.7 | 47.3 ± 11.7 | 0.344 |
| pO2, mmHg | 478.7 ± 115.0 | 323.0 ± 138.3 | 401.5 ± 41.7 | 0.336 | 288.7 ± 169.4 | 258.3 ± 115.5 | 272.5 ± 40.3 | 0.961 |
| HCO3, mmol/L | 26.0 ± 1.2 | 26.3 ± 1.1 | 25.4 ± 0.9 | 0.675 | 23.6 ± 3.1 | 27.7 ± 9.8 | 22.6 ± 2.5 | 0.661 |
| Lac, mmol/L | 0.5 ± 0.2 | 0.3 ± 0.0 | 0.4 ± 0.2 | 0.639 | 0.5 ± 0.2 | 0.8 ± 0.4 | 0.6 ± 0.0 | 0.629 |
ABG, arterial blood gas; BPM, beats per minute; BUN, blood urea nitrogen; CBC, complete blood count; Cr, creatinine; Glc, glucose; Hb, hemoglobin; Hct, hematocrit; Lac, lactate; MAP, mean arterial pressure; UOP, urine output.
We evaluated circulating levels of C1INH as well as the activity of the CP, AP, and LP of complement activation to confirm the therapeutic range of the drug after systemic delivery. Endogenous C1INH levels measured prior to initiation of treatment (i.e. at −30 and +30 minutes) showed no difference between groups. G2 donors received rhC1INH as described, circulating C1INH measured at 720 and 1200 minutes post-induction showed significantly higher levels when compared to donors in G1 and G3 (Figure 3a). G2 donors also showed significant suppression of the CP at 720 and 1200 minutes after BD, in contrast to the G1 and G3 donors (Figure 3b), in parallel to the increased circulating C1INH levels. Activation of LP (Figures 3c) showed wide variation and was not statistically different between groups. Activation of AP (Figures 3d) was significantly lower in G3 compared to G1 at both 720 (p<0.05) and 1200 minutes (p<0.01). We investigated complement activation in donors by analyzing C3b/C5b-9 deposition in kidney biopsies obtained immediately after the 20-hour BD period. Immunofluorescence analysis of G2 donor kidneys revealed minimal C5b-9 and C3b deposition compared to increased staining of both markers in G1 and G3 kidneys prior to cold storage (Figure 4). Renal pathology did not differ between treatment groups based on evaluation of H/E and PAS stains (not shown).
Figure 3. rhC1INH treatment results in sustained elevation of circulating C1INH and inhibits complement activation in the BD donor.
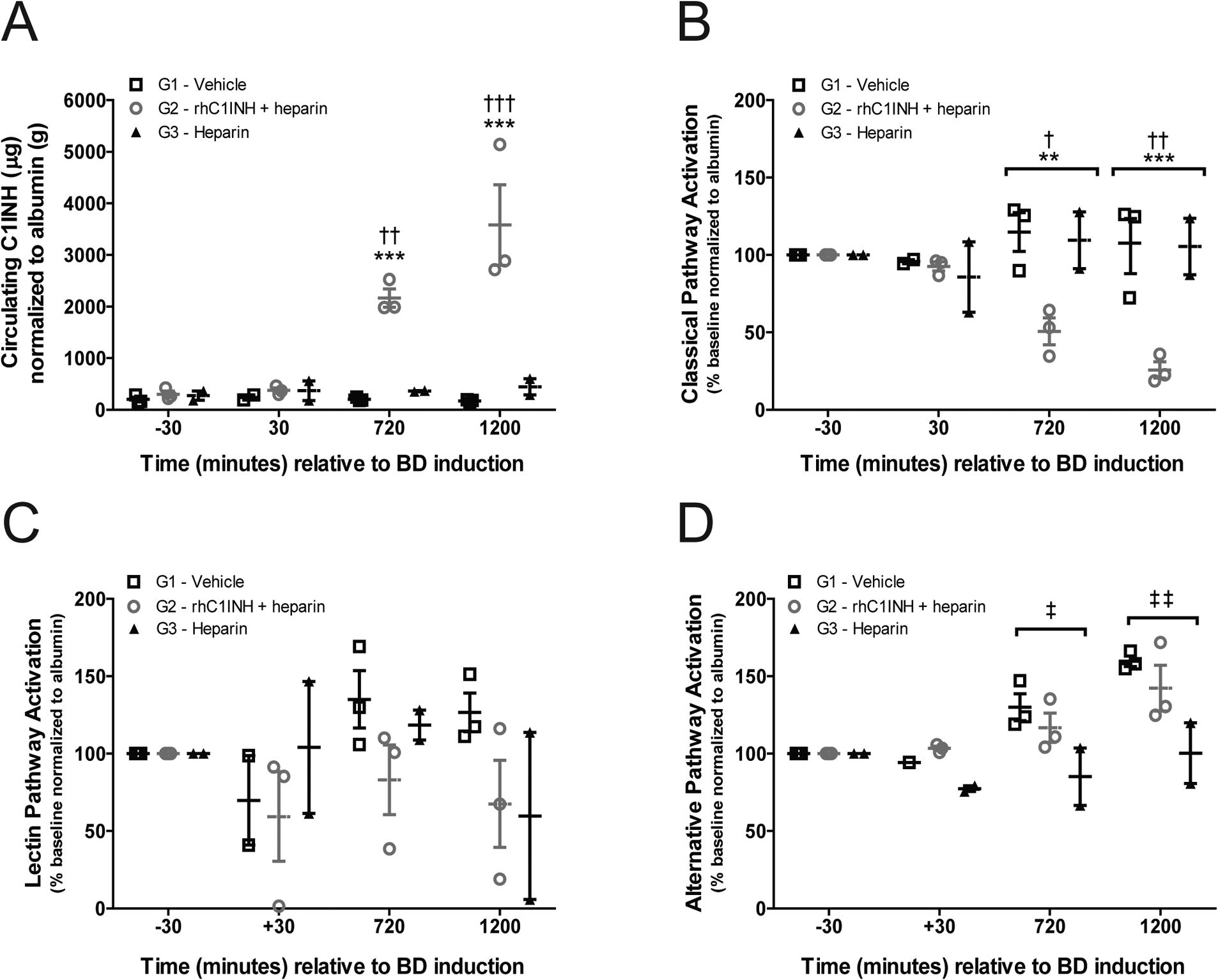
(a) Levels of serum C1INH in G1 (vehicle, n=3), G2 (rhC1INH+heparin, n=3), and G3 (heparin, n=2) donors at −30, 30, 720, and 1200 minutes relative to BD induction, five minutes after bolus injection of drug or vehicle where applicable. Data are presented as sample C1INH (μg) normalized to serum albumin (g) to compensate for dilution effects. (b-d) Complement activation determined by the complement system screen assay of the (b) CP, (c) LP, and (d) AP in G1, G2 and G3 donors. Data are expressed as percent activation normalized to albumin, relative to baseline (30 minutes before induction of BD). Data in a – d presented as mean values ±SEM, significance calculated by two-way ANOVA and Bonferroni’s post-hoc correction (**p<0.01 G1 vs G2; ***p<0.001 G1 vs G2; †p<0.05 G2 vs G3; ††p<0.01 G2 vs G3; †††p<0.001 G2 vs G3).
Figure 4. rhC1INH treatment reduces C3b/C5b-9 deposition in the BD donor graft.
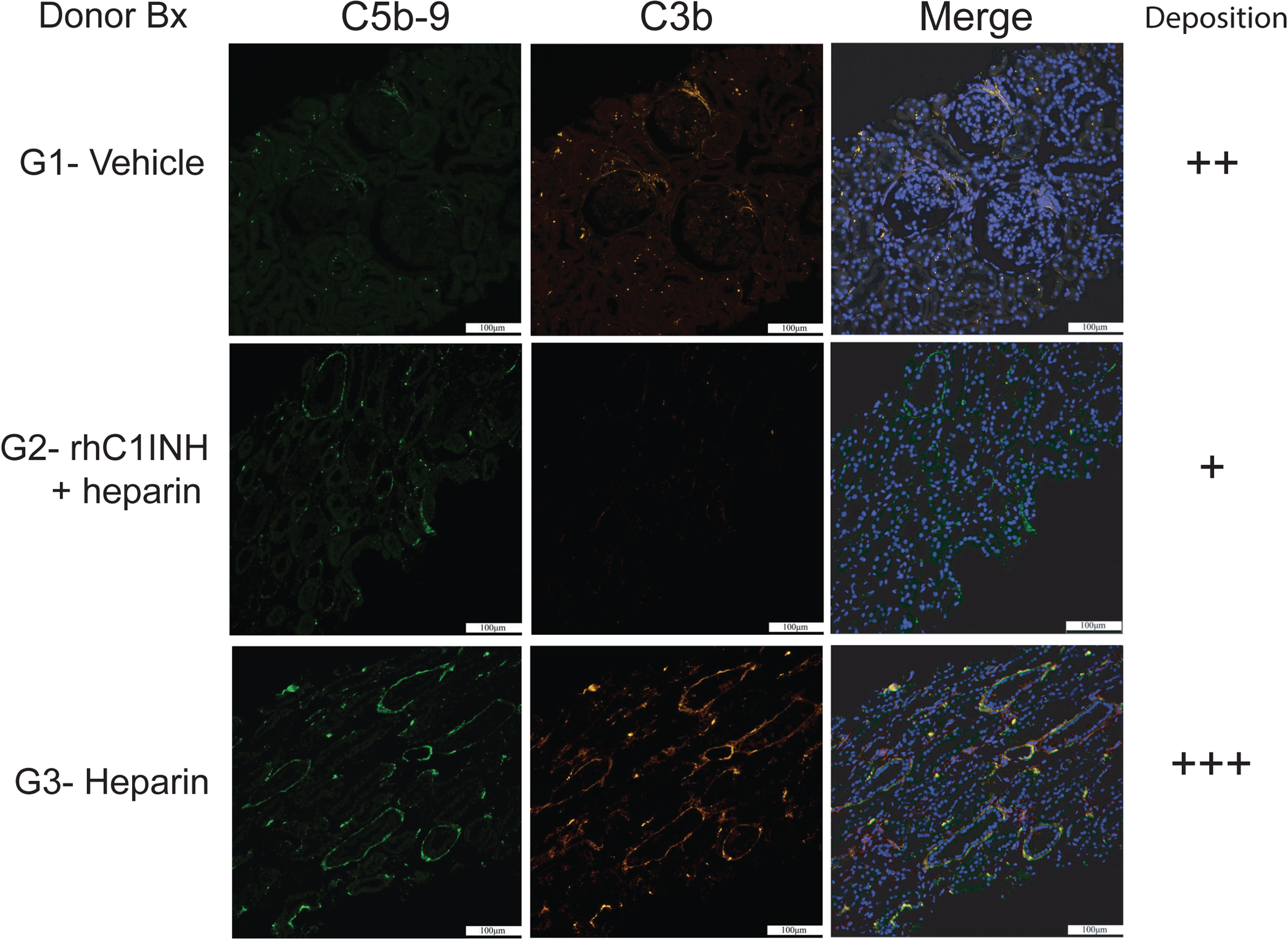
(a-b) Representative micrographs at 200X magnification depicting C5b-9 (green) and C3b (orange) deposition by immune-fluorescent staining in kidney biopsies obtained from G1 (vehicle), G2 (rhC1INH+heparin) and G3 (heparin) donor grafts at the time of organ recovery; semiquantitative assessment of combined complement deposition.
3.2. rhC1INH treatment limits systemic levels of TNFα and MCP-1 in BD donors.
We assessed levels of interleukin-6 (IL-6), TNFα, interleukin-8 (IL-8), and monocyte chemoattractant protein 1 (MCP-1) as pro-inflammatory cytokines and chemokines implicated in the acute inflammatory response to tissue injury and trauma during the BD period. We documented significantly lower levels of TNFα and MCP-1 in G2 donors compared to G1 and/or G3 donors (Figure 5a,b). No significant differences were observed in the circulating levels of IL-6 or IL-8 between groups (Figure 5c,d).
Figure 5. rhC1INH treatment limits systemic levels of TNFα and MCP-1 in BD donors.
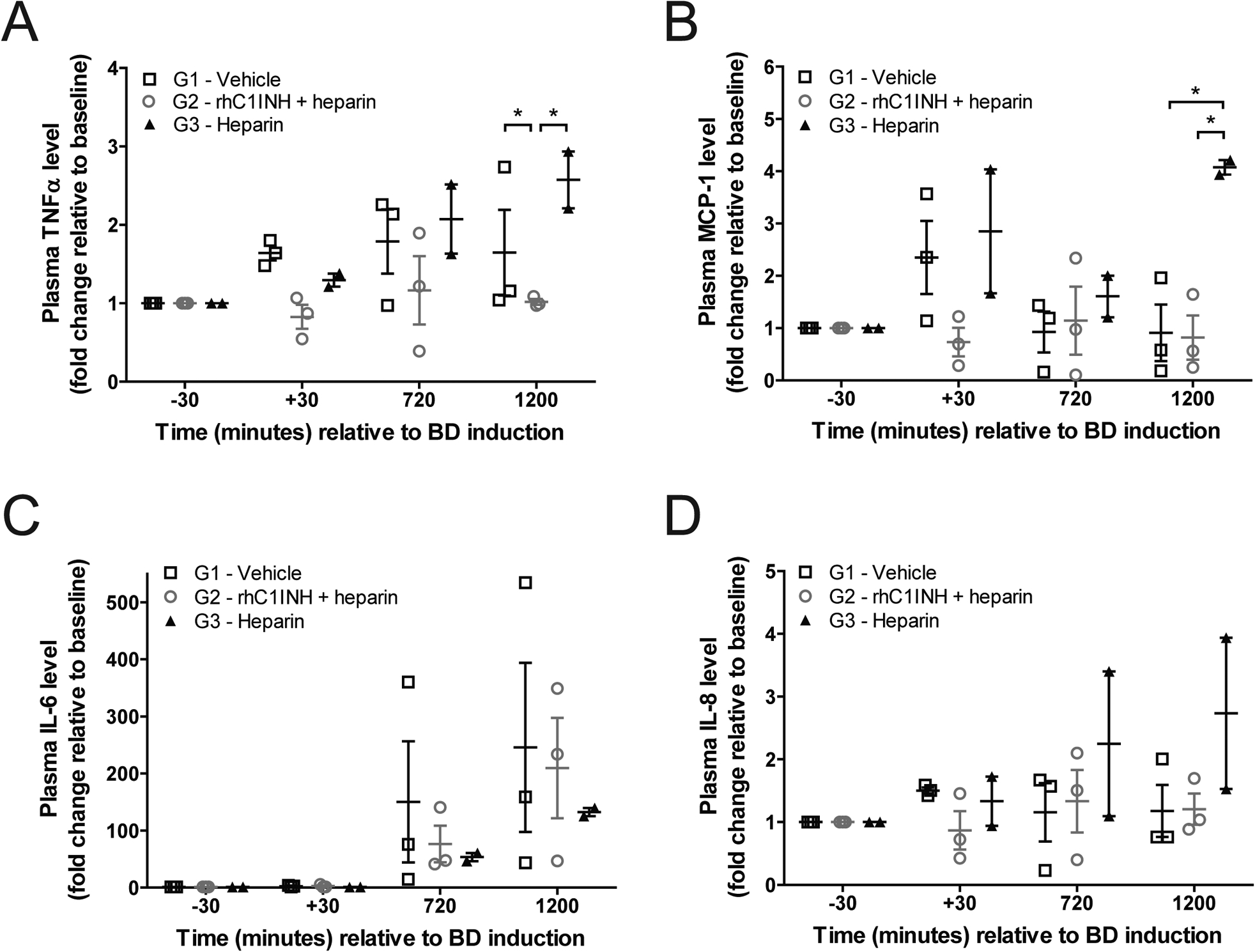
Plasma levels of (a) IL-6, (b) IL-8, (c) TNFα, and (d) MCP-1 in donors from G1 – vehicle (n=3), G2 – rhC1INH + heparin (n=3) and G3 – heparin (n=2). Data expressed as fold change relative to baseline value, significance is calculated by one-way ANOVA and Bonferroni’s post-hoc correction (*p<0.05).
3.3. Donor rhC1INH treatment reduces circulating sC5-b9 in recipients.
After prolonged cold preservation (43–48 hours), donor grafts were transplanted into ABO-compatible, non-sensitized, MHC fully-mismatched recipients who underwent bilateral nephrectomy of their native kidneys and received post-transplant maintenance immunosuppression (Figure 1b). We obtained biopsies from the grafts at 60 minutes and day 4 post-transplant and analyzed these for innate immune cell infiltration, complement deposition, and oxidative damage, and compared these to biopsies collected from recipient native kidneys. Due to concern for the well-being of the animals, biopsies were limited to only 2–3 recipients per group. In biopsies collected 60 minutes post-reperfusion, renal pathology evaluated by H/E and PAS did not differ between treatment groups, nor were differences observed in the level of complement deposition (C3b/C5b-9), immune cell infiltration (CD68+ macrophages and MPO+ neutrophils), or oxidative damage (MDA), although all were higher than was observed in naïve native kidneys (data not shown). Day 4 post-transplant, biopsies from recipients of G2 donor kidneys seemed to display lower levels of C3b/C5b-9 deposition compared to recipients of G1 and G3 donors, however the limited number of specimens lacked statistical power to establish significance of the results (Figure 6a,b). We further analyzed levels of soluble C5b-9 (sC5b-9) in circulation as a measure of complement activation at 60 minutes and 4 days post-transplant and found that values at 60 minutes were again equivalent for all experimental groups, while day 4 levels demonstrated significantly less sC5b-9 in recipients of G2 grafts compared to recipients of G1 and G3 grafts (Figure 6c, p<0.05). Day 4 levels of oxidative damage and immune cell infiltration by immunohistochemistry were equivalent between experimental groups (Figure S1).
Figure 6. rhC1INH treatment in BD donors reduces C3b/C5b-9 deposition and circulating sC5-b9 in transplant recipients during the first post-operative week.
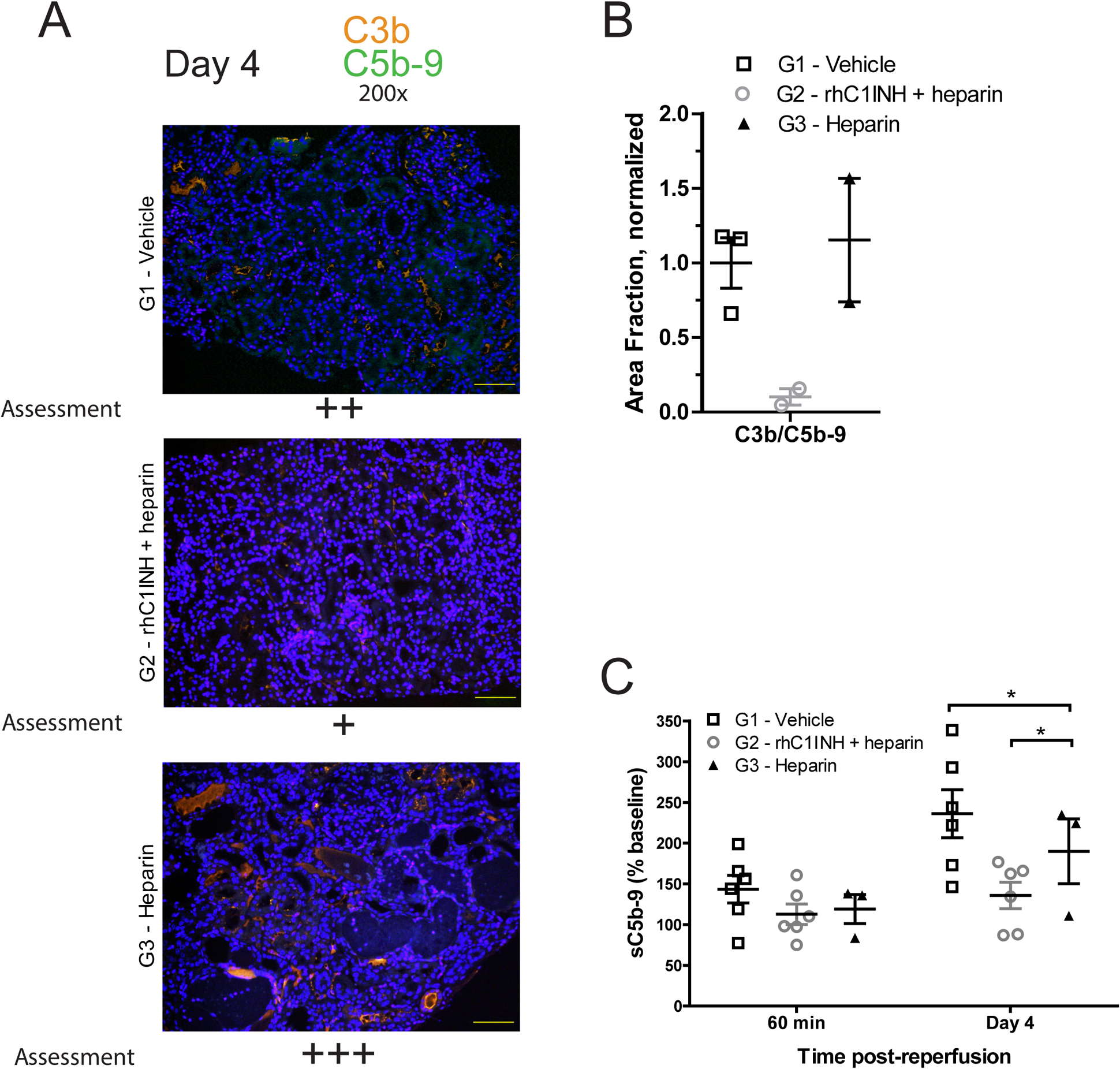
(a) Representative micrographs at 200X magnification depicting C5b-9 (green) and C3b (orange) combined deposition by immune-fluorescent staining in kidney biopsies obtained from G1 (vehicle), G2 (rhC1INH+heparin) and G3 (heparin) grafts at day 4 post-transplant; semiquantitative assessment. (b) Quantitative assessment of C3b/C5b9 deposition at day 4 post-transplant - G1 (vehicle, n=3), G2 (rhC1INH+heparin, n=3), and G3 (heparin, n=2), one G3 biopsy was excluded by outlier test. Data expressed as area fraction normalized to G1-Vehicle average ±SEM. (c) Serum levels of sC5b-9 in recipients of kidney grafts from donors in G1 (vehicle, n=6), G2 (rhC1INH+heparin, n=6), and G3 (heparin, n=3) at 60 minutes and day 4 post-transplant, analyzed by ELISA, data expressed as percent value relative to baseline (pre-transplant). Significance is calculated by one-way ANOVA and Bonferroni’s post-hoc correction (*p<0.05).
3.4. Donor rhC1INH treatment improves post-transplant renal function, reduces injury and incidence of DGF, and improves graft survival
Recipients of kidneys from G1 and G3 donors exhibited significant kidney dysfunction within the first week after surgery. DGF was diagnosed in 4/6 recipients of G1 kidneys and in 3/3 G3 graft recipients. None (0/6) of the recipients of G2 donor grafts met criteria for DGF (p=0.0081) (Table 1). Post-transplant serum creatinine was elevated in recipients of G1 and G3 donors; in contrast, recipients of G2 donor kidneys displayed lower serum creatinine at days 4–6 post-transplant (p<0.05, Figure 7a) and lower peak serum creatinine vs. G3 (p<0.05, Table 2) indicating superior renal function. In addition, G2 kidney recipients presented lower levels of urinary neutrophil gelatinase-associated lipocalin (NGAL) than G1 kidney recipients, indicating reduced renal injury (Figure 7b). Recipients in G3 remained anuric until euthanasia criteria were met, preventing measurement of urinary NGAL.
Figure 7. rhC1INH treatment in BD donors improves graft function and reduces kidney injury and incidence of DGF in transplant recipients.
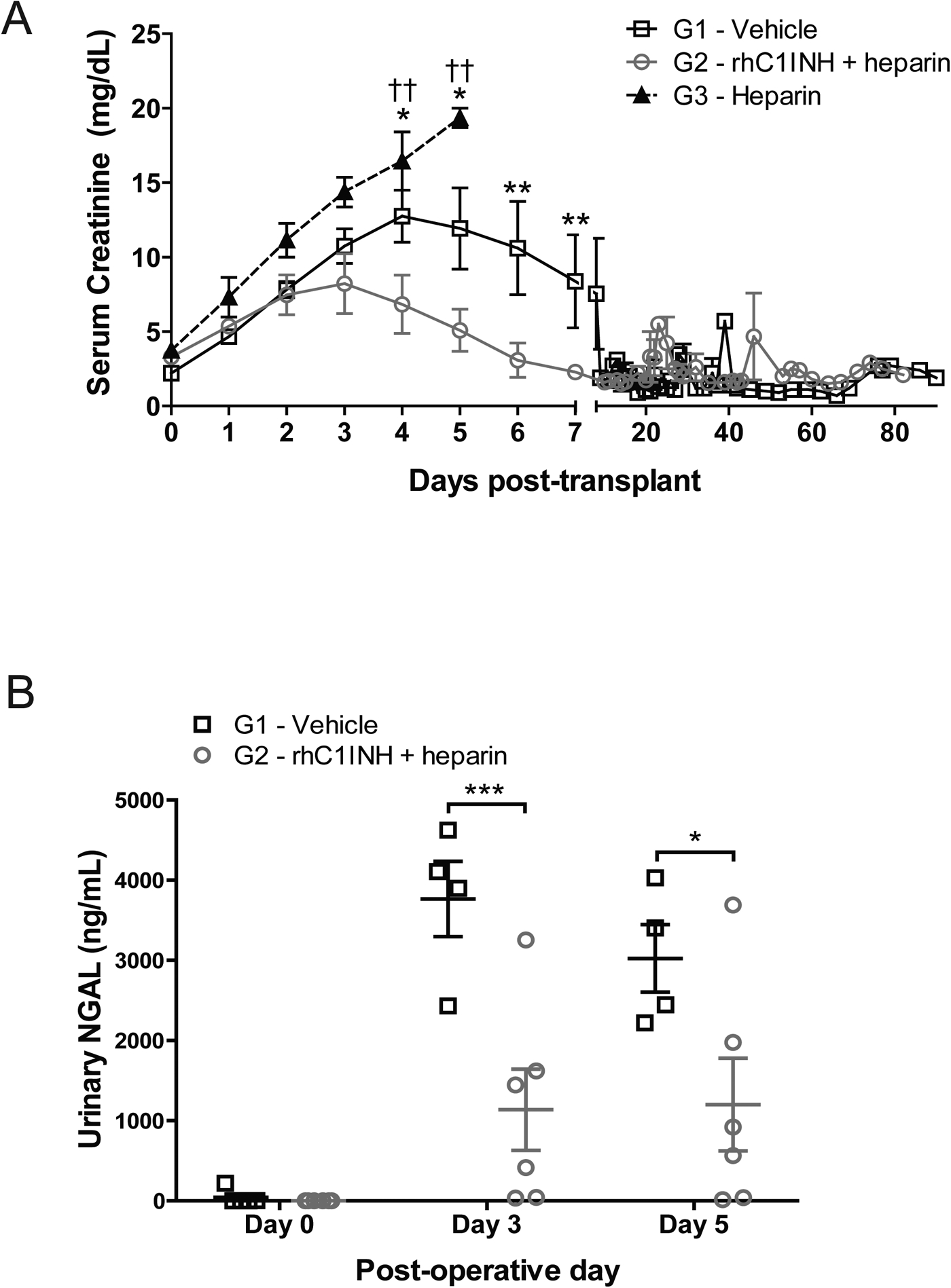
(a) Serum creatinine levels in recipients of kidney grafts from donors in G1 (vehicle, n=6), G2 (rhC1INH+ heparin, n=6), and G3 (heparin, n=3). Data are expressed as mean values ±SEM, significance is calculated by two-way ANOVA and Bonferroni’s post-hoc correction (*p<0.05, **p<0.01, G1 vs G2; ††p<0.01, G2 vs G3). (b) Urinary NGAL measured at baseline, day 3, and day 5 post-transplant in recipients of kidney grafts from donors in G1 (vehicle, n=6), G2 (rhC1INH+heparin, n=6), and G3 (heparin, n=3). Data are expressed as mean values ±SEM, significance is calculated by Student’s T-test.
To further investigate the impact of donor therapy on renal function, we performed daily ultrasounds to determine renal resistive indices in transplanted grafts. Resistive index measurement has been used to assess post-transplant renal function in the clinic, studies have shown a correlation between elevated indices and progression to DGF.34 All imaged kidneys transplanted from G1 (4/4) and G3 (2/2) donors showed elevated indices on post-transplant day 1 compared to 0/4 kidneys from G2 donors (p=0.006, Figure 7). Further, kidneys from G2 donors exhibited significantly longer graft survival compared to those from G1 and G3 donors (p<0.05, Table 1).
Altogether, these results indicate that donor treatment with rhC1INH provided a protective effect in renal grafts subjected to BD and prolonged cold-ischemia, demonstrable through improved renal function in the first week post-transplant and overall graft survival.
4. DISCUSSION
We investigated the impact of targeted complement blockade in BD organ donors using rhC1INH in combination with heparin to prevent DGF and improve graft survival. For this purpose we induced BD in older rhesus macaque donors and maintained them hemodynamically stables for 20 hours prior to organ procurement, then subjected the kidneys to prolonged cold storage (44–48 hours) with the intent of generating a translational model of clinical DGF using the definitions proposed by Boom et al.1, 33
Complement activation has gained significant attention in the context of IRI and organ donation in the last decade.35 Our approach targeting the complement-driven inflammatory response in older BD donors in the context of prolonged cold storage resulted in a significant reduction in the incidence of DGF in recipients along with superior renal function within the first 2 weeks after transplant as evinced by significantly lower serum creatinine and decreased urinary NGAL measurements. Our data indicate that C1 complement blockade and heparin treatment in the donor limits inflammation by reducing cytokine, CP activity and deposition of complement-proteins within the graft, and that this has an ameliorating effect on complement-mediated tissue injury in transplant recipients.
C1 inhibitor plays a central role in the modulation of inflammation by upstream regulation of the complement, coagulation and contact systems. While the mechanisms leading to complement activation during BD remain unclear, studies using knock-out technology and other complement-intervention strategies during IRI and BD have shown reduced tissue inflammation and improved renal function after reperfusion in multiple models.19, 24–26, 36–40 Poppelaars, et al. showed that treatment of brain-dead rats with rhC1INH resulted in reduced renal mRNA expression and serum levels of IL-6, improved renal function and reduced renal injury prior to transplantation.25 These observations are supported further by multiple studies demonstrating the protective anti-inflammatory effect of C1-blockade in models of sepsis, as well as renal, neurological, myocardial and intestinal IRI.16,41–46 Although rodent models of renal IRI indicate a predominant role for the alternative pathway in complement-mediated renal injury, recent reports suggest that CP and LP are critical in the pathogenesis of IRI, DGF and acute rejection in large animal models and humans.35,37,47–50
Interactions between C1INH and heparin have been reported to augment rhC1INH activity 5–11 fold and potentiate the inhibitory effect on C1-dependent activation of the complement cascade.30 The synergistic effect between rhC1INH and heparin has been previously shown to enhance inhibition of the CP, LP and AP in human samples in a dose-dependent fashion.31 We exploited this interaction to maximize complement inhibition in donors in our model. Heparin is known to inhibit neutrophil adhesion, chemotaxis and reactive oxygen species production.51 The use of heparin to ameliorate IRI remains controversial. Sedigh, et al. recently demonstrated utilization of a heparin conjugate during hypothermic machine perfusion to reduce cold preservation injury and improve organ function shortly after reperfusion.52 In a sheep model of IRI, Cheung Soo Shin, et al. observed that heparin therapy significantly attenuated neutrophil infiltration within the interstitium but did not affect the degree of renal damage or renal function as compared to animals that did not received treatment.53 Our findings here show that recipients of kidneys from donors treated with a high dose of heparin alone (G3) suffer similar if not worse tubular injury through complement deposition when compared to controls (G1). In addition, we did not observe differences in neutrophil or monocyte infiltration of the grafts between the three different groups at the time of organ recovery or during transplantation (Figure S1).
Remarkable in our model is the effect observed in the group treated with rhC1INH+heparin (G2) in which any possible deleterious effect of heparin is superceded by the enhanced effect of complement inhibition when combined with heparin. Our treatment with rhC1INH+heparin in BD donors led to a significant decrease in CP activity as well as a decrease in systemic release of TNFα and MCP-1, potent pro-inflammatory mediators known to enhance innate immune cell trafficking and amplify inflammatory response.54 While we did not observe a reduction in the level of neutrophil and macrophage infiltration, we did note reduced tissue deposition of C3b/C5b-9 in renal grafts from treated donors both at organ recovery and during the first week post-transplant, although our observations lacked sufficient power to demonstrate a significant difference between groups. Nevertheless, these results correlate to observations in humans and animal models of inflammatory injury and C1INH administration.16,55–56 The formation and deposition of C5b-9 has been directly linked to tubular epithelial injury and characterized by tubular thinning, protein cast formation and tubular dilation in IRI.57 Selective blockade of the CP with rhC1INH has previously been shown to prevent acute tubular damage in a porcine model of renal warm IRI.16,58 Furthermore, circulating sC5b-9 has been proposed as a biomarker of tissue injury and AKI severity.59 These data correlate to our observation of reduced circulating sC5b-9 coupled to the previously indicated superior post-transplant renal function in recipients of rhC1INH-treated donors by day 4 post-transplant. As stated previously, the ischemic and inflammatory environment recreated by this model is likely much more severe than that of marginal grafts currently used for transplantation. As such, our observations on the reduction of DGF and improved kidney function may translate in the form of an even larger advantage in standard clinical practice with non-marginal donors.
Transplant recipients who experience DGF are at increased risk of graft rejection and reduced graft survival.4 We utilized a fully-mismatched model of renal transplantation after BD to reduce the potential for immune-tolerant regulation providing accessory protection to the graft in the post-transplant period. We documented the expected onset of ACR and AMR in grafts that survived the DGF period (data not shown), however we noted a significant increase in graft survival in recipients of rhC1INH-treated donors (Table 1). While donor treatment with rhC1INH+heparin did not abrogate development of graft rejection, it did reduce the inflammatory state of rhC1INH-treated donors and led to superior post-transplant renal function and reduced incidence of DGF in their recipients. This observation matches clinical studies demonstrating that the inflammatory state of BD donors and the development of DGF are both independently associated with progression to acute rejection.2,4,6–9,60
However, clinical data on the protective effect of C1 inhibition in the context of IRI and DGF is limited. Jordan, et al. recently published the results of a phase I/II trial showing that patients receiving C1INH required fewer dialysis sessions in weeks 2–4 post-transplant and had superior renal function 12 months after surgery; this effect was most significant in those receiving low quality grafts.61 These encouraging results support complement blockade in the peri-transplant period as a valid and attractive approach to protect kidneys from IRI, prevent dysfunction and improve long-term renal function after transplantation. Our unique strategy of using rhC1INH at the level of the donor for the prevention of post-transplant DGF could be coupled to a recipient treatment-regimen which may produce a synergistic effect that could constitute a valuable strategy for prevention of DGF and also potentially reduce immunogenicity in the graft.
The significance of our study resides in the novel approach of donor pre-treatment targeting complement inhibition with rhC1INH and heparin as a strategy to prevent DGF in kidney transplantation recipients in a clinically relevant model of BD in older donors, prolonged cold ischemia, and allo-transplantation in NHP. Our results indicate that treatment with rhC1INH and heparin during BD limits systemic and local activation of the complement system and the inflammatory response, providing a protective effect in the host kidneys that translates to reduced risk of DGF and improved transplant outcomes. Successful clinical implementation of these findings could vastly increase the pool of acceptable donors, reduce DGF rates, improve graft life and patient survival, and decrease morbidity and cost of care associated to kidney transplantation. While our focus has been on kidney transplantation, the positive impacts may encompass other transplantable organs as well. Further investigations into the mechanism of action of donor pre-treatment with rhC1INH and heparin, particularly in regard to other organs, as well as clinical trials on the effectiveness of targeting the complement system at the donor level, are warranted to further validate these results.
Supplementary Material
Figure 8. Renal resistive indices determined by sonographic assessment of the transplanted graft on the first post-transplant day.
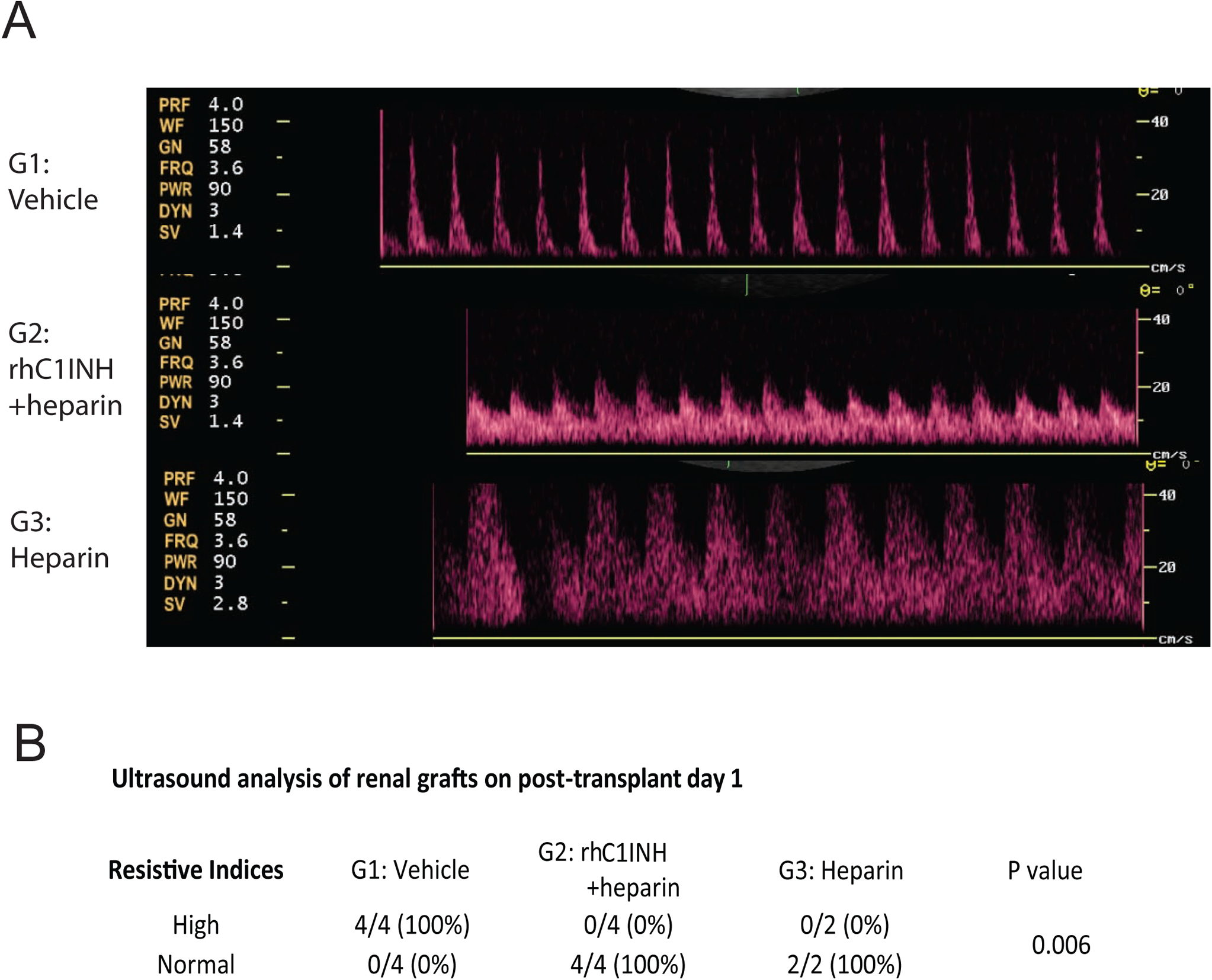
(a) Representative images of the ultrasound waveform in the arcuate and/or interlobar arteries in each group. (b) Table with results of the sonographic comparison between all tested groups.
ACKNOWLEDGMENTS
We gratefully acknowledge the veterinary and SPI staff at the WNPRC. We also thank D. Roenneburg, S. Raglin, W. Zhong, A. Mejia, H. Simmons, S. Larson, C. Boetcher, J. Rose and T. Roehling for expert technical assistance. We thank Kristy Kraemer and Julia Shaw from NIH and Isabella Lussier from Alpha-genesis for their assistance in identifying appropriate non-human primate pairs for our experimental design. This work was supported by National Institutes of Health NIH #1-R01-AI110617-01A1 (PI: Luis Fernandez), and NIH training grants T32DK007665 (Jose Reyes), T32AI125231 (Juan Danobeitia), and the 2016 American Society of Transplant Surgeons Scientist scholarship (Juan Danobeitia). The rhC1INH was kindly provided by Pharming Technologies B.V. (Leiden, The Netherlands).
Abbreviations:
- ACR
Acute cellular rejection
- AKI
Acute kidney injury
- AMR
Antibody-mediated rejection
- ANOVA
Analysis of variance
- AP
Alternative (complement) pathway
- BD
Brain death
- BUN
blood urea nitrogen
- C1INH
C1 inhibitor
- CP
Classic (complement) pathway
- DGF
Delayed graft function
- ELISA
enzyme-linked immunosorbent assay
- HAE
hereditary angioedema
- H/E
Hematoxylin and Eosin
- IFNγ
interferon gamma
- IL-1β
interleukin-1-beta
- IL-17
interleukin-17
- IL-6
interleukin-6
- IL-8
interleukin-8
- IRI
ischemia-reperfusion injury
- LP
mannose-binding lectin (complement) pathway
- MAC
membrane attack complex
- MAP
Mean arterial pressure
- MCP-1
Monocyte chemoattractant protein 1
- MHC
Major histocompatibility complex
- MMF
mycophenolate-mofetil
- NGAL
neutrophil gelatinase-associated lipocalin
- NHP
Non-human primate
- NO
Nitric oxide
- PTT
partial thromboplastin time
- rhC1INH
recombinant human C1 esterase inhibitor
- ROS
reactive oxygen species
- SD
standard deviation
- SEM
standard error of the mean
- SRV
Simian type D retrovirus
- SIV
Simian immunodeficiency virus
- STLV-1
Simian T-cell leukemia virus type 1
- TNFα
Tumor necrosis factor alpha
Footnotes
DISCLOSURE
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- 1.Mallon DH, Summers DM, Bradley JA, et al. Defining delayed graft function after renal transplantation: simplest is best. Transplantation 2013; 96: 885–889. [DOI] [PubMed] [Google Scholar]
- 2.Siedlecki A, Irish W, Brennan DC. Delayed graft function in the kidney transplant. Am J Transplant 2011; 11: 2279–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irish WD, Ilsley JN, Schnitzler MA, et al. A risk prediction model for delayed graft function in the current era of deceased donor renal transplantation. Am J Transplant 2010; 10: 2279–2286. [DOI] [PubMed] [Google Scholar]
- 4.Stewart DE, Kucheryavaya AY, Klassen DK, Turgeon NA, Formica RN, Aeder MI. Changes in Deceased Donor Kidney Transplantation 1 Year After KAS Implementation. Am J Transplant 2016; 16: 1834–1847 [DOI] [PubMed] [Google Scholar]
- 5.Lauzurica R, Pastor MC, Bayes B, et al. Pretransplant inflammation: a risk factor for delayed graft function? J Nephrol 2008; 21: 221–228. [PubMed] [Google Scholar]
- 6.Perico N, Cattaneo D, Sayegh MH, et al. Delayed graft function in kidney transplantation. Lancet 2004; 364: 1814–1827. [DOI] [PubMed] [Google Scholar]
- 7.Yarlagadda SG, Coca SG, Formica RN, et al. Association between delayed graft function and allograft and patient survival: a systematic review and meta-analysis. Nephrol Dial Transplant 2009; 24: 1039–1047. [DOI] [PubMed] [Google Scholar]
- 8.Ojo AO, Wolfe RA, Held PJ, et al. Delayed graft function: risk factors and implications for renal allograft survival. Transplantation 1997; 63: 968–974. [DOI] [PubMed] [Google Scholar]
- 9.Matas AJ, Gillingham KJ, Elick BA, et al. Risk factors for prolonged hospitalization after kidney transplants. Clin Transplant 1997; 11: 259–264. [PubMed] [Google Scholar]
- 10.Westendorp WH, Leuvenink HG, Ploeg RJ. Brain death induced renal injury. Curr Opin Organ Transplant 2011; 16: 151–156. [DOI] [PubMed] [Google Scholar]
- 11.Danobeitia JS, Sperger JM, Hanson MS, et al. Early Activation of the Inflammatory Response in the Liver of Brain-Dead Non-Human Primates. J Surg Res 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim IK, Bedi DS, Denecke C, et al. Impact of innate and adaptive immunity on rejection and tolerance. Transplantation 2008; 86: 889–894. [DOI] [PubMed] [Google Scholar]
- 13.Diepenhorst GM, van Gulik TM, Hack CE. Complement-mediated ischemia-reperfusion injury: lessons learned from animal and clinical studies. Ann Surg 2009; 249: 889–899. [DOI] [PubMed] [Google Scholar]
- 14.Sacks S, Lee Q, Wong W, et al. The role of complement in regulating the alloresponse. Curr Opin Organ Transplant 2009; 14: 10–15. [DOI] [PubMed] [Google Scholar]
- 15.Asgari E, Zhou W, Sacks S. Complement in organ transplantation. Curr Opin Organ Transplant 2010; 15: 486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castellano G, Melchiorre R, Loverre A, et al. Therapeutic targeting of classical and lectin pathways of complement protects from ischemia-reperfusion-induced renal damage. Am J Pathol 2010; 176: 1648–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Damman J, Seelen MA, Moers C, et al. Systemic complement activation in deceased donors is associated with acute rejection after renal transplantation in the recipient. Transplantation 2011; 92: 163–169. [DOI] [PubMed] [Google Scholar]
- 18.Lewis AG, Köhl G, Ma Q, et al. Pharmacological targeting of C5a receptors during organ preservation improves kidney graft survival. Clin Exp Immunol 2008; 153: 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atkinson C, Floerchinger B, Qiao F, et al. Donor brain death exacerbates complement-dependent ischemia/reperfusion injury in transplanted hearts. Circulation 2013; 127: 1290–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poppelaars F, Seelen MA. Complement-mediated inflammation and injury in brain dead organ donors. Mol Immunol 2017; 84: 77–83. [DOI] [PubMed] [Google Scholar]
- 21.Damman J, Daha MR, van Son WJ, et al. Crosstalk between complement and Toll-like receptor activation in relation to donor brain death and renal ischemia-reperfusion injury. Am J Transplant 2011; 11: 660–669. [DOI] [PubMed] [Google Scholar]
- 22.Jansen PM, Eisele B, de Jong IW, et al. Effect of C1 inhibitor on inflammatory and physiologic response patterns in primates suffering from lethal septic shock. J Immunol 1998; 160: 475–484. [PubMed] [Google Scholar]
- 23.Davis AE. Biological effects of C1 inhibitor. Drug News Perspect 2004; 17: 439–446. [DOI] [PubMed] [Google Scholar]
- 24.Danobeitia JS, Ziemelis M, Ma X, et al. Complement inhibition attenuates acute kidney injury after ischemia-reperfusion and limits progression to renal fibrosis in mice. PLoS One 2017; 12: e0183701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poppelaars F, Jager NM, Kotimaa J, et al. C1-Inhibitor Treatment Decreases Renal Injury in an Established Brain-Dead Rat Model. Transplantation 2018; 102: 79–87. [DOI] [PubMed] [Google Scholar]
- 26.Delpech PO, Thuillier R, SaintYves T, et al. Inhibition of complement improves graft outcome in a pig model of kidney autotransplantation. J Transl Med 2016; 14: 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Budde ML, Wiseman RW, Karl JA, et al. Characterization of Mauritian cynomolgus macaque major histocompatibility complex class I haplotypes by high-resolution pyrosequencing. Immunogenetics 2010; 62: 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zens TJ, Danobeitia JS, Chlebeck PJ, et al. Guidelines for the management of a brain death donor in the rhesus macaque: A translational transplant model. PLoS One 2017; 12: e0182552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caldwell EE, Andreasen AM, Blietz MA, et al. Heparin binding and augmentation of C1 inhibitor activity. Arch Biochem Biophys 1999; 361: 215–222. [DOI] [PubMed] [Google Scholar]
- 30.Poppelaars F, Damman J, de Vrij EL, et al. New insight into the effects of heparinoids on complement inhibition by C1-inhibitor. Clin Exp Immunol 2016; 184: 378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schoenfeld AK, Lahrsen E, Alban S. Regulation of Complement and Contact System Activation via C1 Inhibitor Potentiation and Factor XIIa Activity Modulation by Sulfated Glycans - Structure-Activity Relationships. PLoS One 2016; 11: e0165493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haustein S, Kwun J, Fechner J, et al. Interleukin-15 receptor blockade in non-human primate kidney transplantation. Transplantation 2010; 89: 937–944. [DOI] [PubMed] [Google Scholar]
- 33.Boom H, Mallat MJ, de Fijter JW, et al. Delayed graft function influences renal function, but not survival. Kidney Int 2000; 58: 859–866. [DOI] [PubMed] [Google Scholar]
- 34.Naesens M, Heylen L, Lerut E, et al. Intrarenal resistive index after renal transplantation. N Engl J Med 2013; 369: 1797–1806. [DOI] [PubMed] [Google Scholar]
- 35.de Vries B, Walter SJ, Peutz-Kootstra CJ, et al. The mannose-binding lectin-pathway is involved in complement activation in the course of renal ischemia-reperfusion injury. Am J Pathol 2004; 165: 1677–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thurman JM, Ljubanovic D, Edelstein CL, et al. Lack of a functional alternative complement pathway ameliorates ischemic acute renal failure in mice. J Immunol 2003; 170: 1517–1523. [DOI] [PubMed] [Google Scholar]
- 37.Atkinson C, Varela JC, Tomlinson S. Complement-dependent inflammation and injury in a murine model of brain dead donor hearts. Circ Res 2009; 105: 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Damman J, Hoeger S, Boneschansker L, et al. Targeting complement activation in brain-dead donors improves renal function after transplantation. Transpl Immunol 2011; 24: 233–237. [DOI] [PubMed] [Google Scholar]
- 39.Damman J, Nijboer WN, Schuurs TA, et al. Local renal complement C3 induction by donor brain death is associated with reduced renal allograft function after transplantation. Nephrol Dial Transplant 2011; 26: 2345–2354. [DOI] [PubMed] [Google Scholar]
- 40.Damman J, Schuurs TA, Ploeg RJ, et al. Complement and renal transplantation: from donor to recipient. Transplantation 2008; 85: 923–927. [DOI] [PubMed] [Google Scholar]
- 41.Liu D, Lu F, Qin G, et al. C1 inhibitor-mediated protection from sepsis. J Immunol 2007; 179: 3966–3972. [DOI] [PubMed] [Google Scholar]
- 42.Igonin AA, Protsenko DN, Galstyan GM, et al. C1-esterase inhibitor infusion increases survival rates for patients with sepsis*. Crit Care Med 2012; 40: 770–777. [DOI] [PubMed] [Google Scholar]
- 43.Singer M, Jones AM. Bench-to-bedside review: the role of C1-esterase inhibitor in sepsis and other critical illnesses. Crit Care 2011; 15: 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heydenreich N, Nolte MW, Göb E, et al. C1-inhibitor protects from brain ischemia-reperfusion injury by combined antiinflammatory and antithrombotic mechanisms. Stroke 2012; 43: 2457–2467. [DOI] [PubMed] [Google Scholar]
- 45.Begieneman MP, Kubat B, Ulrich MM, et al. Prolonged C1 inhibitor administration improves local healing of burn wounds and reduces myocardial inflammation in a rat burn wound model. J Burn Care Res 2012; 33: 544–551. [DOI] [PubMed] [Google Scholar]
- 46.Lu F, Chauhan AK, Fernandes SM, et al. The effect of C1 inhibitor on intestinal ischemia and reperfusion injury. Am J Physiol Gastrointest Liver Physiol 2008; 295: G1042–1049. [DOI] [PubMed] [Google Scholar]
- 47.van der Pol P, Schlagwein N, van Gijlswijk DJ, et al. Mannan-binding lectin mediates renal ischemia/reperfusion injury independent of complement activation. Am J Transplant 2012; 12: 877–887. [DOI] [PubMed] [Google Scholar]
- 48.Chun N, Fairchild RL, Li Y, et al. Complement Dependence of Murine Costimulatory Blockade-Resistant Cellular Cardiac Allograft Rejection. Am J Transplant 2017; 17: 2810–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vo AA, Zeevi A, Choi J, et al. A phase I/II placebo-controlled trial of C1-inhibitor for prevention of antibody-mediated rejection in HLA sensitized patients. Transplantation 2015; 99: 299–308. [DOI] [PubMed] [Google Scholar]
- 50.Montgomery RA, Orandi BJ, Racusen L, et al. Plasma-Derived C1 Esterase Inhibitor for Acute Antibody-Mediated Rejection Following Kidney Transplantation: Results of a Randomized Double-Blind Placebo-Controlled Pilot Study. Am J Transplant 2016; 16: 3468–3478. [DOI] [PubMed] [Google Scholar]
- 51.Brown RA, Lever R, Jones NA, Page CP. Effects of heparin and related molecules upon neutrophil aggregation and elastase release in vitro. Br J Pharmacol. 2003;139(4):845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sedigh A, Nordling S, Carlsson F, Larsson E, Norlin B, benow N, Lennmyr F, Tufveson G, Magnusson PU, Lorant T. Perfusion of Porcine Kidneys With Macromolecular Heparin Reduces Early Ischemia Reperfusion Injury. Transplantation. 2019. February;103(2):420–427. [DOI] [PubMed] [Google Scholar]
- 53.Shin Cheung Soo, Han Jeong Uk, Kim Jung Lyul, Schenarts Paul J, Traber Lillian D, Hawkins Haland Traber Daniel L. Heparin attenuated neutrophil infiltration but did not affect renal injury induced by ischemia reperfusion. Yonsei Med J. 1997. Jun;38(3):133–141. [DOI] [PubMed] [Google Scholar]
- 54.Skrabal C, Thompson L, Potapov E, et al. Organ-specific regulation of pro-inflammatory molecules in heart, lung, and kidney following brain death. J Surg Res 2005; 123: 118–125. [DOI] [PubMed] [Google Scholar]
- 55.de Vries DK, van der Pol P, van Anken GE, et al. Acute but transient release of terminal complement complex after reperfusion in clinical kidney transplantation. Transplantation 2013; 95: 816–820. [DOI] [PubMed] [Google Scholar]
- 56.Tillou X, Poirier N, Le Bas-Bernardet S, et al. Recombinant human C1-inhibitor prevents acute antibody-mediated rejection in alloimmunized baboons. Kidney Int 2010; 78: 152–159. [DOI] [PubMed] [Google Scholar]
- 57.Zhou W, Farrar CA, Abe K, et al. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J Clin Invest 2000; 105: 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Castellano G, Intini A, Stasi A, et al. Complement Modulation of Anti-Aging Factor Klotho in Ischemia/Reperfusion Injury and Delayed Graft Function. Am J Transplant 2016; 16: 325–333. [DOI] [PubMed] [Google Scholar]
- 59.Rodríguez E, Riera M, Barrios C, et al. Value of Plasmatic Membrane Attack Complex as a Marker of Severity in Acute Kidney Injury. Biomed Res Int 2014; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu WK, Famure O, Li Y, et al. Delayed graft function and the risk of acute rejection in the modern era of kidney transplantation. Kidney Int 2015; 88: 851–858. [DOI] [PubMed] [Google Scholar]
- 61.Jordan SC, Choi J, Aubert O, et al. A Phase I/II, Double-Blind, Placebo-Controlled Study Assessing Safety and Efficacy of C1 Esterase Inhibitor for Prevention of Delayed Graft Function in Deceased Donor Kidney Transplant Recipients. Am J Transplant 2018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.