

Summary
Brain-resident microglia and bone marrow-derived macrophages represent the most abundant non-cancerous cells in the brain tumor microenvironment with critical functions in disease progression and therapeutic response. To date little is known about genetic programs that drive disease-associated phenotypes of microglia and macrophages in brain metastases. Here we used cytometric and transcriptomic analyses to define cellular and molecular changes of the myeloid compartment at distinct stages of brain metastasis and in response to radiotherapy. We demonstrate that genetic programming of tumor education in myeloid cells occurs early during metastatic onset and remains stable throughout tumor progression. Bulk and single cell RNA sequencing revealed distinct gene signatures in brain-resident microglia and blood-borne monocytes/macrophages during brain metastasis and in response to therapeutic intervention. Our data provide a framework for understanding the functional heterogeneity of brain metastasis-associated myeloid cells based on their origin.
Subject Areas: Immunology, Systems Biology, Transcriptomics
Graphical Abstract
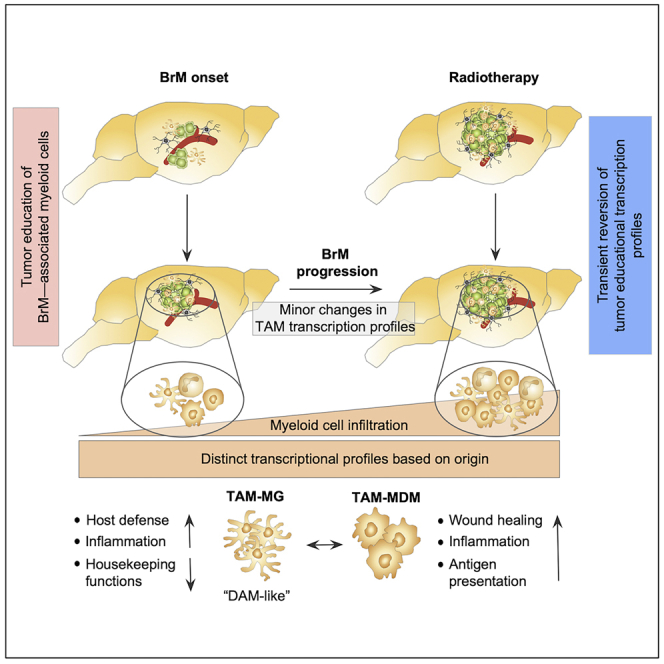
Highlights
-
•
Tumor education gene signatures arise early during tumor onset and remain stable
-
•
TAM-MG and TAM-MDM induce distinct genetic programs in response to tumor education
-
•
Radiotherapy enhances the influx of blood-borne myeloid cells
-
•
Radiotherapy transiently reverses tumor education signatures in TAM-MDM
Immunology; Systems Biology; Transcriptomics
Introduction
Brain metastases (BrM) represent the most common intracranial tumor in adults and affect approximately 20%–40% of all patients with cancer. Patients with melanoma and breast and lung cancer show the highest incidence (Ostrom et al., 2018, Stelzer, 2013, Tabouret et al., 2012). Owing to improved management of the localized primary disease, numbers of patients with cancer who develop BrM are increasing (Tabouret et al., 2012). The development of BrM is associated with poor patient prognosis and limited efficacy of standard of care as well as targeted- or immunotherapies (Kalsi et al., 2015, Valiente et al., 2018). In addition to tumor cell intrinsic traits, effects of the tumor microenvironment (TME) are known to affect tumor progression and significantly contribute to therapy resistance (Quail and Joyce, 2013, Schulz et al., 2019). The brain represents a unique tissue environment harboring a range of specialized cell types including neurons and neuroglia (Jakel and Dimou, 2017, Zeng and Sanes, 2017). Detailed insight into the cellular composition of the immune landscape of the central nervous system (CNS) has recently led to a paradigm shift challenging the traditional view of the CNS as an immune privileged organ (Mrdjen et al., 2018). The presence of innate and adoptive immune cells within border-associated regions including the meninges, choroid plexus, and perivascular space suggest their active involvement in CNS immunity. In contrast to the highly specialized areas of the brain that represent an interface between the CNS and the systemic immune system, yolk sac-derived microglia represent the major immune cell type in the brain parenchyma with critical roles in immune surveillance, host defense, and housekeeping functions such as synaptic pruning (Butovsky and Weiner, 2018, Hagemeyer et al., 2017, Norris and Kipnis, 2019, Paolicelli et al., 2011). A series of studies shed further light onto the heterogeneity of distinct microglial subsets and border-associated macrophages (BAMs) within different homeostatic states (Masuda et al., 2019, Thion et al., 2018, Utz et al., 2020) or disease conditions (Keren-Shaul et al., 2017, Krasemann et al., 2017, Mathys et al., 2017, Tay et al., 2018). Large-scale transcriptomic analyses of microglia and BAMs in neurodegenerative disorders and neuroinflammation demonstrated a striking similarity of gene signatures across different malignancies. Microglia that acquire a distinctive molecular activation signature have recently been denoted as “disease-associated microglia” (DAM) (Keren-Shaul et al., 2017) or “microglia neurodegenerative phenotype” (MgND) (Krasemann et al., 2017). The overlap of gene signatures within DAMs from different neurodegenerative or neuroinflammatory conditions suggests that genetic programs defining the switch from homeostatic to disease-associated phenotypes might represent a general microglial priming or sensing of pathogen-associated pattern rather than unique responses to specific stimuli (Deczkowska et al., 2018). Single cell analyses of neurodegenerative disorders (e.g., Huntington disease, HD) in comparison with neuroinflammatory conditions such as different stages of experimental autoimmune encephalomyelitis (EAE) revealed that disease-associated myeloid cells are predominately derived from microglia pools, whereas recruitment of monocyte populations is almost completely absent in HD and occurs at relatively low rates (below 5%–10%) during the course of EAE (Ajami et al., 2018). In contrast, the presence of brain tumors induces significant recruitment of myeloid cells from the peripheral immune system including inflammatory monocytes, monocyte-derived macrophages (MDMs), and granulocytes. In glioma models, MDMs constitute up to 50%–75% of the of tumor-associated macrophages (TAMs) population (Bowman et al., 2016, Chen et al., 2017). Given the technological challenges to discriminate TAMs derived from brain-resident microglia from recruited MDM controversy remained on the functional contribution of each subpopulation based on their origin. The use of lineage tracing models and the identification of cell type-restricted markers allowed the evaluation of disease-associated roles of macrophages and microglia in brain malignancies. Intriguingly, analyses of the molecular identity of TAMs in glioma likewise suggest the induction of disease-associated signatures (Bowman et al., 2016). Functional annotation revealed that microglia-derived TAMs (TAM-MG) display signatures associated with pro-inflammatory responses, host defense mechanisms, and synaptic pruning. In contrast, gene signatures identified in monocyte-derived macrophages (TAM-MDM) in glioma were associated with immune suppression, antigen presentation, and wound healing (Bowman et al., 2016).
However, to date little is known about the functional contribution of TAM-MG and TAM-MDM in BrM. Here, we characterized the myeloid cell compartment in a lung-to-brain metastases model to investigate cellular and molecular changes during tumor progression and in response to radiotherapy. We demonstrate that BrM-induced TAM programming occurs early during metastatic colonization and remains stable during tumor progression. Application of radiotherapy has the potential to transiently reverse tumor education signatures in TAMs in BrM.
Results
Cellular Composition of the Tumor Microenvironment in Brain Metastases
We first analyzed changes of the cellular composition of the TME in the xenograft lung-to-brain metastasis model H2030 (Nguyen et al., 2009) (Figure 1A, S1A, and S1B) following metastatic seeding and outgrowth. Histological assessment of distinct stages of brain metastasis (BrM) revealed that early-stage metastases induce the recruitment and activation of glial cells including astrocytes and microglia (Lorger and Felding-Habermann, 2010, Valiente et al., 2014). Astrogliosis remains prominent at later stages with GFAP+ reactive astrocytes forming a glial scar that delineates the tumor from the brain parenchyma (Figure 1B, left panel). Astrocytes are located at the tumor-stroma interface, whereas tumor-associated macrophages/microglia (TAMs) infiltrate into BrM lesions (Figure 1B). TAMs in close vicinity to tumor cells show strong induction of Iba1 expression and a morphological change from a ramified phenotype typical for resting microglia toward an ameboid morphology of activated microglia (Figure 1C, left). Co-staining with the microglia marker Tmem119 (Bennett et al., 2016) showed Tmem119+Iba1+ cells in the adjacent brain parenchyma, whereas Tmem119+Iba1+ as well as Tmem119-Iba1+ TAMs are present within the tumor lesion indicating the infiltration of monocyte-derived macrophages (MDMs) (Figure 1C, right) and/or a downregulation of the expression of homeostatic microglia marker in TAMs as previously observed in the context of different brain malignancies (Jordao et al., 2019, Keren-Shaul et al., 2017, Sankowski et al., 2019). Other brain resident cell types such as neurons are rather displaced by the growing tumor (Seano et al., 2019) and do not infiltrate BrM lesions (Figure 1B, middle panel). Upon extravasation, H2030-BrM tumors grow along pre-existing blood vessels (Figure 1B, right panel). However, the vasculature within large H2030-BrM lesions is abnormal with highly irregular and enlarged vessels (Figure 1B) and hemorrhages are frequently observed in H2030-BrM (Figure S1A) indicating a leaky blood-tumor barrier (BTB).
Figure 1.

Cellular Composition of the Tumor Microenvironment of H2030-BrM
(A) Schematic overview of the experimental design and methodology.
(B) Representative immunofluorescence images of tumor-free control brain and small and large metastatic lesions in the H2030-BrM model stained with indicated markers. Scale bar, 200 μm.
(C) Representative immunofluorescence image of a brain metastatic lesion and adjacent normal brain parenchyma stained for Iba1 and Tmem119. Dotted line depicts the tumor margin. Overview is split into Iba1 staining (left panel) and an overlay of Iba1 and TMEM119 staining (right panel). Scale bar, 100 μm.
(D) Quantification of myeloid cell populations in normal brain and in the H2030-BrM model by flow cytometry (n = 3 for each condition). p values were obtained by one-way ANOVA.
(E) Quantification of the concentration of secreted factors in tumor cell-conditioned media from H2030BrM cells in vitro (n = 3 replicates).
(F) Principle component analysis (PCA) of different cell types in tumor-free versus BrM condition. N-MG, normal microglia; N-Mono, normal blood monocytes; TA, tumor-associated cell type. n = 4 for cells from tumor-free mice and n = 9 for cells isolated from BrM.
(G) DEGs in small versus large BrM in the individual cell types. n = 4 for tumor-free mice, n = 4–5 for mice with small BrM, and n = 5 for mice with large BrM.
Peripheral Monocyte-Derived Cells Contribute to the Myeloid Compartment in Lung-to-Brain Metastases
To gain further insight into the dynamics of the recruitment from the periphery and the cellular identity of blood-borne immune cells in BrM, we analyzed the myeloid cell compartment at distinct stages of tumor progression by flow cytometry. Given the previous observation that microglia and blood-borne monocytes/macrophages associated to different CNS pathologies show assimilation of microglial marker expression including P2ry12 and Tmem119, we employed an alternative gating strategy to discriminate brain-resident microglia and macrophages (MDMs) based on CD49d expression as previously described (Bowman et al., 2016). We focused our analysis on the four most abundant myeloid immune cell populations in BrM, namely, inflammatory monocytes (CD45+CD11b+Ly6ChighLy6G−; TA-Mono), granulocytes (CD45+CD11b+Ly6CmedLy6Ghigh; TA-Granu), MDM (CD45+CD11b+Ly6ClowLy6G−CD49d+; TAM-MDM), and microglia (CD45+CD11b+Ly6ClowLy6G−CD49d−; TAM-MG) (Figure S1D). BrM-bearing mice were stratified into mice with small and large lesions based on bioluminescent intensity (BLI) with a mean BLI output of 4x107 photons x sec−1 (ranging from 3x105 to 7.5x107 photons x sec−1) or 2.15x108 photons x sec−1 (ranging from 1x108 to 5.3x108 photons x sec−1), respectively. Flow cytometry of macro-dissected BrM lesions revealed continuous infiltration of inflammatory monocytes, granulocytes, and MDMs during tumor progression compared with tumor-free control mice that only showed minimal amounts of non-microglial myeloid cells (Figure 1D). TAMs constitute the largest cell population within the myeloid cell compartment comprising 85% and 75% of the CD45+CD11b+ cell population in small and large BrM lesions, respectively (Figure 1D). In small BrM lesions, only 10% of the TAM population was derived from MDM (TAM-MDM), whereas the remaining 90% originated from brain-resident, yolk sac-derived microglia (TAM-MG) (Figure 1D). An increase of the TAM-MDM population to approximately 20% of the TAM population was observed in large BrM lesions (Figure 1D). This increase is likely due to influx from the periphery rather than local proliferation of TAMs in BrM lesions given the low percentage of Ki67+Iba1+ cells that are present in the tumor core and at the tumor-stroma interface (Figure S1C). We did not observe differences in the amount of Ki67+Iba1+ cells depending on the tumor size. Next, we analyzed the secretome from the H2030-BrM cell line to identify factors that are implicated in the recruitment and activation of TAMs using a multiplex assay. We found a range of chemokines in tumor cell-conditioned media from H2030-BrM cells that were previously associated to monocyte/macrophage recruitment including CCL2, CCL4, CCL5, and CXCL10 (Shi and Pamer, 2011) and cytokines with known roles in macrophage polarization/activation including the interleukins (IL)-4, IL-6, IL-8, IL-17, as well as VEGF, IFN-β, TNF-α, and GM-CSF (Palma et al., 2018) (Figure 1E).
Tumor Education Induces Distinct Inflammatory Gene Signatures in Myeloid Cells
Next, we purified myeloid cells by fluorescence-activated cell sorting (FACS) for RNA sequencing (RNA-seq) analysis to gain insight into the molecular identity of different BrM-associated myeloid subpopulations. The purity of the sorted cell populations was validated by the expression of cell type-restricted markers for each population based on RNA-seq data (Figure S1E). Principal component analysis (PCA) of BrM-associated cells (TAM-MG, TAM-MDM, and TA-Mono) and their respective normal cellular counterparts (normal microglia from tumor-free brain [N-MG] and blood monocytes [N-Mono]) revealed distinct cell type-specific clustering (Figure 1F, Table S1). Surprisingly, we did not observe major changes in gene expression depending on the tumor stage with only a few differentially expressed genes (DEGs) in distinct BrM stages (small versus large BrM-associated cells; Figures 1F and 1G, Table S1). Among the four analyzed tumor-associated myeloid cell populations, granulocytes segregated most strongly from the remaining populations, whereas monocytes and macrophages clustered closely together but clearly separated from microglia (Figure S1F). Granulocytes displayed the highest variation in gene expression depending on the tumor stage with 21 DEGs, whereas TA-Mono and TAM-MDM only showed 8 and 7 DEGs, respectively. The lowest impact of tumor progression was found in TAM-MG with only 2 DEGs between small and large BrM lesions (Figure 1G, Table S1). In summary, our data indicate that metastatic colonization induces disease-associated transcriptional profiles in myeloid subpopulations that significantly differ from genetic programs of their normal cellular counterparts. Each BrM-associated myeloid cell population clustered separately indicating the maintenance of lineage-specific gene signatures despite their recruitment to the brain and education by metastatic tumor cells. Strikingly, transcriptional profiles induced by tumor education within the myeloid compartment occurred at early stages of metastatic colonization and remained stable during tumor progression.
Origin of TAMs Affects Transcriptomic Profiles Associated with Tumor Education in Brain Metastases
To evaluate whether TAMs in BrM show differences in their transcriptomes depending on their origin (bone marrow derived versus yolk sac derived), we analyzed gene expression in TA-Mono, TAM-MDM, and TAM-MG compared with their respective normal counterparts. This analysis revealed a total of 3,279 upregulated and 26,175 downregulated genes in tumor-associated myeloid cells compared with control conditions across all analyzed cell types (Figure 2A). The majority of DEGs were unique for each cell type. By comparison, TA-Mono and TAM-MDM shared a higher number of common DEGs than TAM-MG with TA-Mono or TAM-MDM. Moreover, we identified 239 and 777 genes that were up- or downregulated in all three cell types compared with normal controls (Figures 2A and S2A). Among the DEGs that were concomitantly upregulated in all analyzed BrM-associated myeloid populations, we found several key regulators of CNS inflammation including Axl, Il1b, C3ar1, and Apoe (Figures 2B–2D and S2A) (Albulescu et al., 2013, Bouwens van der Vlis et al., 2018, Krasemann et al., 2017, Ray et al., 2017). Although the gene signatures revealed a considerable overlap of DEGs, our data also indicated the induction of different modes of inflammation in brain-resident versus recruited TAM populations. Several DEGs in TAM-MG were associated with type-1 interferon signaling (e.g., Irf7 and Zbp1) indicating a pro-inflammatory phenotype. Analysis of DEGs in TAM-MG from H2030-BrM revealed striking similarities to previously described DAM markers (Keren-Shaul et al., 2017) (Figures S3A and S3B), further supporting the hypothesis that different pathological stimuli induce similar core signatures in disease-associated microglia (Deczkowska et al., 2018). Moreover, we observed down-regulation of homeostatic marker, including Cx3cr1, P2ry12, and Tmem119 (Figures 2B and S3A), as previously demonstrated for disease-associated microglia in neurodegenerative disorders and brain cancer (Jordao et al., 2019, Keren-Shaul et al., 2017, Sankowski et al., 2019), as well as upregulation of those markers in TAM-MDM (Figure S3A). In contrast, genes involved in antigen presentation as well as responses to hypoxia and induction of angiogenesis were induced in TAM-MDM and TA-Mono (Figures 2C and 2D). Gene expression changes were further validated by qRT-PCR for each cell type (Figures S2B–S2D). Functional annotation of the DEGs identified in TAMs confirmed that TAM-MG, TAM-MDM, and TA-Mono collectively induce inflammatory responses, whereas the loss of housekeeping functions was predominately represented in the TAM-MG population with four of five gene ontology (GO) terms being related to CNS processes (Figures 2E–2G). The induction of different modes of inflammatory responses was further supported by the analysis of the 50 most differentially expressed genes. Unsupervised clustering revealed that TAM-MG show a high representation of genes that are associated with innate immune sensing and host defense mechanism including chemokines (e.g., Cxcl13, Cxcl16), components of the complement cascade (e.g., C3, C4b), Toll-like receptor signaling (Tlr2), and Tyro-Axl-Mer receptor signaling (Axl) (Figure 2H). In contrast, we identified gene signatures in TAM-MDM that are rather associated with antigen presentation and adaptive immunity (H2-Eb1, H2-Oa, Cd83, Cd86) as well as alternative macrophage activation (Ms4a7, Mrc1) known to be involved in wound healing and tumor promotion (Figures 2F and 2I). TA-Mono displayed a considerable overlap of DEGs with TAM-MDM (Figures 2A, 2G, and 2J).
Figure 2.

Tumor Education Signatures of TAMs in BrM
(A) Euler diagrams depict the number of unique and shared upregulated (left panel) or downregulated (right panel) DEGs in control versus TAM-MG, or TAM-MDM, or TA-Mono.
(B–D) Volcano plot of control cells versus BrM-associated cells in (B) microglia, (C) MDM, and (D) monocytes. Cutoffs: ± 1 log2 fold change and adjusted p value <0.05.
(E–G) Functional gene annotation of altered cellular pathways in (E) TAM-MG, (F) TAM-MDM, and (G) TA-Mono compared with control cell types from tumor-free animals. Cutoffs: ± 1, log2 fold change and adjusted p value <0.05. All DEGs or top 3,000 DEGs based on adjusted p values were subjected to analysis.
(H–J) Unsupervised clustering of the top 50 DEGs in control versus BrM-associated (small and large stage) TAMs for (H) microglia, (I) MDM, and (J) monocytes. n = 4–5 replicates for each condition.
Given the enrichment of complement cascade components in TAM-MG in response to tumor activation, we queried our dataset for the expression of a range of members. We observed a significant induction of gene expression for the majority of analyzed complement components indicating that activation of the complement cascade represents a central pathway in instigating an inflammatory response upon interaction with cancer cells (Figures 3A and 3B). Since central mediators of the complement cascade act paracrine, it is not surprising that on protein level distinct complement components were present at the interface of Iba1+ and H2030 tumor cells (Figure 3C). Interestingly, we found the complement inhibitor Factor H in tumor cells in close vicinity to Iba1+ TAMs, indicating a protective mechanism to block complement-mediated anti-tumor responses (Figure 3C). Another protective mechanism of tumor cells that we observed in H2030-BrM is the expression of the “don't eat me-signal” CD47 (Figure S4A) that protects cells from phagocytic uptake and is likely induced in response to extracellular TNF-α (Betancur et al., 2017). Based on our RNA-seq and cytokine array data, TAMs (Figure S4B) as wells as tumor cells (Figure 1E) represent a potential source for TNF-α expression in H2030-BrM. Similar to the induction of complement cascade components, we noticed a high representation of solute carrier (Slc) family members among the 500 most differentially expressed genes in TAM-MG (Figure S5A). The Slc family comprises a range of different transporters with known roles in regulating cellular physiology (Lin et al., 2015). Interestingly, we found 66 genes that belong to the Slc family to be differentially expressed in TAM-MG isolated from small and large BrM compared with N-MG indicating that the acquisition of the tumor-educated phenotype requires a profound modulation of the cellular machinery regulating the sensing and transport of nutrients and metabolites suggesting altered energy consumption by TAM-MG.
Figure 3.

The Complement System in TAMs and Their Polarization States in BrM
(A) Expression of complement members in TAM-MG
(B) Expression of complement members in TAM-MDM; for (A) and (B): Values are based on variance-stabilized data (vst (≈log2)) of RNA-seq data. N-Mono: n = 4, N-MG: n = 4, small BrM-associated cells, all other groups: n = 5. Significance based on padj.
(C) Representative immunofluorescence images of control and H2030-BrM sections stained for the indicated markers. Scale bars, 100 and 25 μm.
(D) Heatmaps display the relative expression level of different activation/polarization marker within both TAM populations. Genes are grouped into M1-like (green, left panel) and M2-like (red, right panel). Values display the average per group based on vst values from the comparison of control (N-MG or N-Mono with n = 4 for each group) versus BrM-associated TAM-MG or TAM-MDM (n = 10 for each group). Significance based on padj.
Based on our results that TAM-MDM display characteristics of alternative activation, we next queried the expression of a range of markers to evaluate activation/polarization states of TAM-MDM and TAM-MG in small and large BrM lesions. We chose markers associated to key macrophage functions including inflammatory pathways, phagocytosis, and chemotaxis. Among the analyzed markers (M1-like = 19 genes, left panel and M2-like = 13 genes, right panel) we found several to be significantly induced in distinct stages of tumor progression in TAM-MG and TAM-MDM indicating a mixed activation/polarization state rather than the acquisition of an M1-like or M2-like phenotype in TAMs (Figure 3D). Interestingly, we observed that TAM-MG rather induced expression of markers associated with pro-inflammatory (M1-like) phenotypes (e.g., Ccl2, Ccl3, Ccl4, Ccl5, Ifnb1, Il12b, Tlr2, Tlr4, Tnf), whereas markers of alternative activation (M2-like) were induced in TAM-MDM (e.g., Ccl17, Ccl22, Cd163, Tlr1) (Figure 3D). Collectively, our data indicate that TAM-MG in H2030-BrM induce transcriptional states associated with host defense mechanisms, whereas TAM-MDM rather display signatures of antigen presentation and wound healing.
In summary, our data reveal that lung-to-brain metastases induce distinct disease-associated transcriptional programs in TAM-MG and TAM-MDM suggesting non-redundant functions in tumor progression. To further interrogate cellular responses of TAM-MG and TAM-MDM upon different stimuli, we extended our analysis to effects of therapeutic intervention on TAM phenotypes. Given recent reports on immune modulatory functions of ionizing radiation (IR) (Vanpouille-Box et al., 2017, Wilkins et al., 2019) and the significance of radiotherapy as standard of care for patients with brain metastases (Suh et al., 2020), we next asked how therapeutic intervention would modulate tumor-associated inflammation in BrM.
Fractionated Whole-Brain Radiotherapy of Lung Cancer-BrM-Bearing Mice Enhances Infiltration of Myeloid Cells
IR was applied as fractionated whole-brain radiotherapy (WBRT) with 2 Gy on 5 consecutive days (5 × 2 Gy WBRT) or as one single dose of 10 Gy (1 × 10 Gy) in 1 arc using the Small Animal Radiation Research Platform (SARRP) (Wong et al., 2008) (Figure 4A) as previously described (Chae et al., 2019). Histological assessment of Iba1+ macrophages indicated an increase in the number of TAMs in response to WBRT (Figure 4B). To distinguish general effects of IR on myeloid cell infiltration into the CNS from tumor-induced effects, we analyzed the amount of blood-borne immune cells in tumor-free brains at d3 and d5 after IR compared with non-irradiated mice by flow cytometry (Figure 4C). This analysis revealed that IR with the chosen dosage and fractionation alone does not induce influx of peripheral immune cells into the brain in the absence of BrM (Figure 4C). In contrast, we observed a continuous increase in the infiltration of blood-borne myeloid cells over time after WBRT in BrM-bearing mice (Figure 4C). Infiltration of granulocytes and inflammatory monocytes peaked at d5 after fractionated WBRT with a relative decrease of the granulocytes and inflammatory monocyte content at d10 after WBRT, due to an expansion of the macrophage population between d3 and d10 after WBRT (Figure 4C). Interestingly, application of WBRT as one single dose of 1 × 10 Gy resulted in diminution of the TAM-MDM population at d3 after WBRT compared with 5 × 2 Gy but showed less prominent effects on the other BrM-associated myeloid populations. In summary, our data show that different IR regimens regulate the influx or ablation of blood-borne myeloid cells in BrM in a time- and dose-dependent manner supporting its applicability as an immune modulatory agent.
Figure 4.

Effects of Whole-Brain Radiotherapy on the Myeloid Cell Compartment in BrM
(A) Schematic representation of the application of whole-brain radiotherapy (WBRT) in two different radiation regimens, 5 × 2 and 1 × 10 Gy.
(B) Representative immunofluorescence images of control and irradiated H2030-BrM stained for the indicated markers. Scale bars, 100 μm.
(C) Relative quantification of myeloid cell populations in normal brain at d3 and 5 after 5 × 2 Gy WBRT and in H2030-BrM at d3, d5, and d10 after 5 × 2 Gy WBRT or d3 after 1 × 10 Gy compared with untreated BrM (n = 3 for each condition). Data are represented as mean ± SD. p values were obtained by one-way ANOVA.
(D) PCA plot shows clustering of individual samples (all non-irradiated versus all irradiated samples) indicating that clustering is mostly driven by cell type with minor effect of IR. PCA plots per cell type indicate heterogeneous clustering across conditions (see small box).
(E) Amount of significant DEGs (cutoff: BM > 20, adj. p value <0.05) in different cell types, by comparing RNA-seq data of large stage BrM-associated samples (n = 5) with its irradiated BrM-associated cellular counterparts at different days upon WBRT (5 × 2 Gy/d3 and d5 n = 4, 1 × 10 Gy/d3 and 5 × 2 Gy/d10 n = 3).
(F) Venn diagram depicts shared and unique DEGs (cutoff: BM > 20, adj. p value <0.05) in each cell type from RNA-seq data of H2030-BrM-associated immune cells, comparing all non-irradiated (no IR) versus all irradiated (IR) groups; note: granulocytes did not reveal any DEG (5 × 2 Gy/d3 and d5 n = 4, 1 × 10 Gy/d3 and 5 × 2 Gy/d10 n = 3).
WBRT Induces Transcriptomic Changes in BrM-Associated Myeloid Cells
To evaluate the effects of WBRT on transcriptional profiles of H2030-BrM-associated myeloid cells, we performed RNA-seq on FACS-purified cell populations at distinct time points after WBRT (Figure 4A). PCA analysis across all samples from BrM revealed that clustering was mostly driven by cell type with no pronounced sub-clustering based on tumor size or in response to treatment at different time points (Figure 4D). This effect was also evident in the comparison of the number of DEGs that were identified in the analysis of untreated large BrM and BrM with IR (Figure 4E). Interestingly, for TAM-MG, TA-Mono, and TA-Granu we found the highest amount of DEGs in response to 1 × 10 Gy compared with 5 × 2 Gy WBRT, whereas this effect was inversed in the case of TAM-MDM (Figure 4E). Moreover, we only observed few common DEGs shared between different cell types by comparing all non-irradiated versus all irradiated (5 × 2 Gy) samples (Figure 4F) indicating high heterogeneity in cellular responses upon IR in different cell types (Table S2).
To further investigate cell type-specific radio-responses, we focused our analysis on the most abundant myeloid cells in BrM, TAM-MG, and TAM-MDM. Unsupervised clustering of the top 50 DEGs in TAM-MG and TAM-MDM at d3 and d10 after 5 × 2 Gy WBRT revealed cell type- and time point-dependent changes in their transcriptomes (Figure 5A). In TAM-MG, we predominately observed induction of transcription profiles at d3 after WBRT that are associated with an immediate danger response to cytotoxic effects of ionizing radiation (Figure 5A). At d10 after IR, not only danger responses are still evident in TAM-MG but also innate immune responses that we observed in response to tumor activation emerge (Figure 5A). In contrast, we found a loss of tumor education signatures in TAM-MDM at d3 after WBRT, followed by induction of immune effector processes and inflammatory responses (e.g., IFN type-1 responses) at d10 after WBRT (Figure 5A). We next sought to compare effects of conventionally fractionated (5 × 2 Gy) to one single dose of WBRT (1 × 10 Gy) on myeloid cells in BrM to evaluate potential dose-dependent differences in immune modulation (Figures 5B and S5B–S5E). Functional annotation of combined DEGs of all TAMs isolated from mice 3 days after 1 × 10 Gy and 5 × 2 Gy WBRT revealed the induction of stress response pathways upon DNA damage or oxidative stress in both experimental settings. Single application of 10 Gy led to a further induction of host defense mechanisms (Figure 5B). Interestingly, although 1 × 10 Gy WBRT led to an overall lower amount of peripheral myeloid cells at 3 days after IR, effects on gene signatures were more pronounced in response to single high doses (Figure 4E) and functional annotation across all four analyzed cell types suggests a more efficient induction of pro-inflammatory host defense responses (Figure 5B). To further evaluate the extent of IR-mediated modulation of transcriptional states of TAM-MG and TAM-MDM, we compared the number of DEG in each cell type in response to the different stimuli analyzed in this study. This comparison illustrated the dominant effect of tumor education of BrM-associated TAMs compared with tumor-naive cell types, whereas effects of tumor progression from small to large lesions are insignificant in both cell types. Effects of IR are evident in both TAM populations; however, they are less pronounced than effects of tumor education (Figure 5C). Interestingly, the relative amount of DEGs in TAM-MG in response to IR is higher than in TAM-MDM (Figure 5C). This effect is most likely due to the fact that the entire TAM-MG population is exposed to IR, whereas the TAM-MDM population is constantly replenished by newly recruited cells that have not directly been exposed to IR. Consequently, the TAM-MDM population is expected to show higher heterogeneity than TAM-MG.
Figure 5.

Effects of Whole-Brain Radiotherapy on Distinct TAM populations in BrM
(A) Unsupervised hierarchical clustering heatmaps depicting Top 50 DEGs in TAM-MG (upper panel) and TAM-MDM (lower panel) of large stage BrM (each n = 4) versus d3 (left, both cell types n = 4) or d10 (right, both cell types n = 3), respectively.
(B) Top five functional gene annotation of DEG in the indicated conditions (cutoff: BM > 20, adj. p value <0.05; log2 fold change).
(C) Pie chart depicting the amount of significantly DEGs in different comparisons of distinct MG or MDM conditions, reflecting BrM onset, progression, and radiotherapy, respectively. Cutoffs: BM > 20, adj. p value < 0.05. n = 3–5 per condition.
Single-Cell RNA-Seq Reveals Cellular Heterogeneity within Individual TAM Populations and in Response to IR
To further explore the heterogeneity of TAM populations in H2030-BrM under treatment-naive conditions and in response to WBRT, we performed single-cell RNA-seq of index-sorted cells (Figure 6A). Combined tSNE analysis revealed the existence of 16 discrete clusters across all conditions (Figures 6B–6D). We observed an overall lower contribution of untreated and irradiated TAM-MG to the individual clusters indicating that TAM-MG represent a more homogeneous population compared with TAM-MDM (Figures 6B–6D). Although TAM-MG were found predominately in cluster 9 (TAM-MG; no WBRT) and cluster 14 (TAM-MG; 5 × 2 Gy/d3), clustering of TAM-MDM spread across more clusters (Figure 6D) suggesting a higher cellular heterogeneity within the TAM-MDM population. Interestingly, we detected only minimal overlap between untreated TAM-MG and TAM-MDM, whereas irradiated TAM-MG and TAM-MDM were found in overlapping clusters (i.e., cluster 10, 14, and 15) thus likely reflecting shared radio-response states (Figures 6D, S6D, and S6E). The three clusters, however, showed only little overlap of similarly regulated genes. For example, clusters 14 and 15 revealed upregulation of Lyz1 and Lyz2, whereas Cd74 was downregulated in clusters 10, 14, and 15.
Figure 6.

Single-Cell RNA-Seq Reveals Heterogeneous TAM Populations
(A) Schematic overview of the experimental design for the single-cell RNA-seq analyses.
(B) tSNE plot of all analyzed cells reveal the existence of 16 clusters within the four analyzed experimental groups (untreated TAM-MG, 5 × 2 Gy/d3 TAM-MG, untreated TAM-MDM, 5 × 2 Gy/d3 TAM-MDM).
(C) tSNE plot of all analyzed cells with color coding for each experimental condition indicates representation of individual conditions in multiple cluster.
(D) Heatmap depicts the number of cells per condition contributing to the individual cluster.
(E) Top 10 upregulated genes in TAM-MG versus TAM-MDM based on log2 fold change.
(F) tSNE plots indicate the expression of representative genes in TAM-MG cluster.
(G) Top 10 upregulated genes in TAM-MDM versus TAM-MG based on log2 fold change.
(H) tSNE plots indicate the expression of representative genes in TAM-MDM cluster.
(I and J) Functional gene annotation of up- (red) and downregulated (blue) genes for (I) untreated TAM-MG (cl.9) versus 5 × 2Gy/d3 TAM-MG (cl.11) and (J) untreated TAM-MDM (cl.8) versus (cl.12) 5 × 2Gy/d3 TAM-MDM. A maximum of five pathways is represented.
We further explored the expression of the 10 most differently expressed genes in TAM-MG versus TAM-MDM (Figures 6E–6H). In line with our bulk RNA-seq data, we observed high expression of pro-inflammatory mediators in TAM-MG including Cxcl13, Ccl3, Ccl4, and C1qb as well as the microglial marker Tmem119 and Hexb (Figures 6E and 6F). In contrast to the pro-inflammatory signature found in TAM-MG, scRNA-seq data further pointed toward the involvement of TAM-MDM in antigen presentation (e.g., Cd74, H2-Aa, H2-Ab1, H2-Eb1) (Figures 6G and 6H). Functional annotation of DEGs further supported transcriptional dichotomy of TAM-MG and TAM-MDM in H2030-BrM across different clusters (Figures 6I, 6J, and S6C). Moreover, we observed the presence of three additional clusters (clusters 3, 5, and 6) that segregated from the main TAM populations and were largely represented by untreated TAM-MDM (Figures 6B–6D). Although the identity of cells in clusters 3, 5, and 6 remains elusive, the respective clusters might represent either border-associated macrophages (BAMs) or migratory dendritic cells (migDCs) (Van Hove et al., 2019). Especially, genes highly expressed in cluster 6 (Ccr7, Fscn, Socs2, Tmem123) showed a considerable overlap of enriched genes previously shown for migDC (Figure S6D).
Taken together, data from the scRNA-seq analyses support our findings from bulk RNA-seq and independently confirm cell type-specific transcriptional states of TAM-MG and TAM-MDM. Importantly, scRNA-seq illustrated heterogeneity in the individual cell populations and supported our hypothesis that TAM-MG represent a more homogeneous population than TAM-MDM likely due to differences in the population turnover rates. Our data underpin the applicability of IR as an immune modulatory agent that can be used to transiently revert the immunosuppressive milieu in BrM by the recruitment of naive inflammatory cells that replenish the pool of tumor-educated TAM-MDM.
Discussion
TAMs represent an integral part of the TME in many different cancer types. Owing to their high functional plasticity, it is not surprising that known TAM functions can range from anti-tumor activity to tumor promotion in a cancer type- and stage-dependent manner (Cassetta and Pollard, 2018, Yang et al., 2018). Recent studies shed further light onto the functional diversity of TAMs in brain cancers based on their ontogenetic origin as yolk sac-derived microglia or bone marrow-derived macrophages (Bowman et al., 2016, Chen et al., 2017). However, to date a detailed characterization of the myeloid immune landscape in BrM is lacking. Here we used bulk and single-cell RNA-seq to dissect the molecular identity of the myeloid cell compartment in different stages of lung-to-brain metastasis and in response to radiotherapy. Although BrM recruit lower percentages of myeloid cells from the periphery compared with glioma, we observed a striking similarity of tumor-associated gene signatures of TAMs. In both glioma and BrM, a functional dichotomy was observed with TAM-MG being associated with host defense mechanisms and housekeeping functions such as synaptic pruning, whereas TAM-MDM are implicated in antigen presentation, immune suppression, and wound healing processes (Bowman et al., 2016). Given the central role of TAMs in regulating tumor progression, targeted therapies are emerging as a tool to disrupt tumor-promoting TAM functions. Current strategies often rely on the blockade of central macrophage differentiation and survival pathways such as CSF1R signaling (Pyonteck et al., 2013, Quail et al., 2016). Pre-clinical testing confirmed the efficacy of TAM-targeted therapies against different tumor types including brain cancers as mono-therapies and in combination with standard of care (Pyonteck et al., 2013, Quail et al., 2016, Stafford et al., 2016). However, a major limitation arises from the inability of those approaches to selectively target tumor promoting TAM populations while preserving physiologically critical functions. Analyses of tumor education gene signatures in TAM-MG and TAM-MDM in BrM presented herein provide a comprehensive resource to identify potential candidates as molecular targets for future functional validation and pre-clinical testing. In this regard two strategies appear most feasible: (1) TAM-MDM-directed strategies to block immune suppression and tumor promoting wound healing processes and (2) TAM-MG-directed approaches to maintain or induce pro-inflammatory host defense mechanisms against tumor cells. This view can even be further extended when taking the contribution of non-parenchymal CNS-associated macrophage populations (CAMs; also known as border-associated macrophages [Utz et al., 2020]) as well as potential differences of microglia subpopulations into account (Masuda et al., 2020). Based on our bulk and single-cell sequencing data it is tempting to speculate that dominant effects of tumor education could overwrite potential transcriptional differences in microglia subpopulations. Indeed, a previous study by Wingrove et al. did not observe differences in bulk sequencing data from BrM-associated stromal cells isolated from tumors at different anatomical sites within the brain (Wingrove et al., 2019). The heterogeneity of parenchymal and non-parenchymal CNS macrophage populations together with the influx of monocyte-derived macrophage populations into brain tumor lesions emphasizes the need for unbiased single cell analyses to capture the complexity of CNS inflammation in the context of different brain malignancies including brain metastasis. Moreover, the existence of distinct myeloid subpopulations as described, for example, in EAE in the brain (Jordao et al., 2019) or spinal cord (Giladi et al., 2020) could extend the spectrum of potential therapeutic targets.
Our data suggest that the complement system plays a central role in BrM-associated inflammation indicated by pronounced expression of different complement components in TAMs such as C1q and C3 in TAM-MG as well as C3ar1 in TA-Mono and TAM-MDM. Likewise, transcriptomes that indicate inflammasome activation (e.g., expression of Nlrp3 and Il1b) were evident in TAMs compared with normal microglia or monocytes. Both pathways are expected to modulate tumor immunity in multiple ways including the regulation of immune responses (e.g., by opsonizing tumor cells) as well as enhancing inflammation by further increasing the permeability of the blood-brain barrier (BBB) or BTB and additional recruitment of peripheral immune cells (Jacob and Alexander, 2014, Khan et al., 2015). However, the production of pro-inflammatory mediators such as C1q, IL1β, or TNF-α is expected to induce neurotoxic effects likely with the involvement of other cell types such as astrocytes (Liddelow et al., 2017). Therefore, future functional studies should address the question whether the induction of pro-inflammatory signatures in TAM-MG reflects an anti-tumor immune response or rather represents stromal cooption by the tumor cells in which host defense mechanisms are exploited to foster brain metastatic colonization and outgrowth as previously demonstrated (Chuang et al., 2013).
Synergistic effects of TME-targeted therapies in combination with standard of care including chemo- and radiotherapy represent a frequently observed phenomenon (Ding et al., 2019). Here we focused our analysis on the effects of WBRT on myeloid cells in BrM. Although immune-modulating effects of radiotherapy were neglected for a long time, recent studies established a link between ionizing radiation and the induction of pro-inflammatory responses that were shown to synergize with immunotherapies (Vanpouille-Box et al., 2017). As part of our study we sought to evaluate if WBRT could be used as an immune modulatory agent to revert immunosuppressive TAM functions. We found that WBRT further enhances the recruitment of myeloid cells from the periphery. This effect was more pronounced in response to 5 × 2 Gy WBRT compared with 1 × 10 Gy WBRT. The observed differences at early time points upon irradiation can at least in part be explained by a higher degree of genotoxicity of single high doses on radio-sensitive blood-borne myeloid cells, dose-dependent differences in the impairment of the BBB/BTB, or different profiles of secreted cytokines. Although the recruitment of blood-borne cells was stronger after classically fractionated WBRT, single high doses induced more pronounced effects on gene expression changes based on the number of DEGs. Moreover, single high doses might have a greater potential to enhance pro-inflammatory responses compared with classically fractionated regimens.
Taken together, our data support the applicability of IR as an immune modulator. Given the cell type and dose-dependent differences of radio-responses, it will be critical to evaluate the optimal regimen for the desired outcome (e.g., rate of recruitment versus transcriptomic changes) for individual tumor types. Importantly, our data indicate that IR has the potential to transiently lift the immunosuppressive capacity of TAM-MDM either by direct effects on the TAM-MDM population or by enhancing the recruitment of tumor- and treatment-naive monocytes/macrophages to the CNS that have not yet acquired tumor education signatures. Single-cell RNA-seq analyses suggest that irradiated TAM-MDM represent a more heterogeneous population than TAM-MG, which could be due to the infiltration of naive monocytes/macrophages that replenish the cell pool. However, regardless of the underlying mechanism, reversion of immune suppression could be exploited to transiently generate a milieu that allows for higher efficacy of TME-targeted or immunotherapies that are otherwise blunted by the immune-suppressive BrM environment.
In summary, our data represent a comprehensive resource on transcription profiles of different tumor-associated myeloid cells during BrM progression and in response to radiotherapy. Differences of transcriptional states in TAMs underscore the importance of detailed mechanistic insight in order to develop therapeutic strategies that target tumor-promoting functions with minimal risk in inducing adverse effects by either blocking physiologically important functions or inducing neurotoxic tissue damage within the CNS. Further investigation of patient BrM will be required to evaluate to which extent the pre-clinical data can be translated into the clinical situation.
Limitations of the Study
In this study, cellular and molecular changes in the myeloid cell compartment were characterized using the xenograft lung to brain metastasis model H2030-BrM. The aim was to examine effects of tumor progression and ionizing radiation on transcriptomic profiles of BrM-associated immune cells. Although we focused our analysis on the myeloid cell compartment as the most abundant immune cell population in brain metastases, it has to be acknowledged that the lack of mature T cells in the immunocompromised Athymic nude mice and potential incompatibilities of human cytokines and murine cytokine receptors can affect myeloid cell biology and thereby alter transcriptomic profiles in myeloid cells. Follow-up studies using different syngeneic and xenograft brain metastasis models are underway to address the question of lymphoid-myeloid cross talk in brain metastasis and to evaluate the degree of T cell-mediated effects on myeloid cells in brain metastases. Moreover, future studies will be critical to evaluate the generalizability of the findings described herein based on one model and broaden the knowledge on core transcriptomic profiles of TAMs in BrM derived from different primary tumor entities that metastasize to the brain. In this context it will be important to include spontaneous BrM models to study the effects of the primary tumor on monocyte trafficking to the brain and its impact on TAM phenotypes in BrM. Given the complication of choosing the optimal marker combination to discriminate different myeloid subpopulations without using lineage tracing models, single cell-omics techniques will become indispensable to fully capture the complexity of the cellular composition and transcriptional repertoire of the BrM TME.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lisa Sevenich (sevenich@gsh.uni-frankfurt.de).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Tables S1 and S2 provide direct access to the main results derived from transcriptomic analysis presented in this study. In addition, all sequencing data have been deposited to the Gene Expression Omnibus (GEO) under the superseries accession number GSE137797. Single-cell RNA-seq data can be found under GSE137512. All bulk RNA-seq data are deposited under GSE137762.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Petra Dinse, Annette Trzmiel, Julius Oppermann, Stephanie Hehlgans, Jeannie Peifer, Judith Bergs, Sophie Giess, and Ewelina Czlonka for excellent technical support and members of the Sevenich lab and the Georg-Speyer-Haus for insightful discussion. We thank Joan Massague for providing the H2030-BrM cell line. Research in the lab of L.S. is supported by institutional funds from the Georg-Speyer-Haus jointly funded by the German Federal Ministry of Health and the Ministry of Higher Education, Research and the Arts of the State of Hesse (HMWK), as well as grants from the LOEWE Center Frankfurt Cancer Institute (FCI), the German Cancer Consortium (DKTK partner site Frankfurt/Mainz), the German Cancer Aid (Max-Eder Junior Group Leader Program 70111752), German Research Foundation (SE2234/3-1), and the Beug Foundation for Metastasis Research.
Author Contributions
M.S. and L.S. designed all experiments, M.S. performed most of the experiments and bioinformatic analyses, B.M. and H.F. assisted in the bioinformatics analysis, K.N. and S.S. assisted in the flow cytometric analysis, F.R. assisted and advised on application of radiotherapy, and M.S. and L.S. wrote the manuscript. L.S. conceived and supervised the project. All authors edited and commented on the manuscript.
Declaration of Interests
The authors declare no conflict of interest.
Published: June 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101178.
Supplemental Information
RNA-seq data of analyzed cell types are summarized by listing all differently expressed genes in each cell type, in the control versus BrM situation (Tab1), as well as within the comparison during BrM progression (small versus large BrM-associated cell types, Tab2). Total up- and downregulated genes as well as overlaps are represented in Tab1. Changes during BrM progression are represented by the corresponding gene as well as the log2 fold change.
RNA-seq data of the myeloid compartment of irradiated H2030-BrM-bearing mice. In Tab1 differently expressed genes in all non-irradiated versus all irradiated samples are displayed. Within each cell type, large BrM samples versus different irradiated conditions are displayed in the following Tabs (Tab2-5). Represented are gene names, log2 fold change and adjusted p-value. Positive log2 fold change value indicates upregulation in the corresponding IR condition.
References
- Ajami B., Samusik N., Wieghofer P., Ho P.P., Crotti A., Bjornson Z., Prinz M., Fantl W.J., Nolan G.P., Steinman L. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci. 2018;21:541–551. doi: 10.1038/s41593-018-0100-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albulescu R., Codrici E., Popescu I.D., Mihai S., Necula L.G., Petrescu D., Teodoru M., Tanase C.P. Cytokine patterns in brain tumour progression. Mediators Inflamm. 2013;2013:979748. doi: 10.1155/2013/979748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett M.L., Bennett F.C., Liddelow S.A., Ajami B., Zamanian J.L., Fernhoff N.B., Mulinyawe S.B., Bohlen C.J., Adil A., Tucker A. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. U S A. 2016;113:E1738–E1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur P.A., Abraham B.J., Yiu Y.Y., Willingham S.B., Khameneh F., Zarnegar M., Kuo A.H., McKenna K., Kojima Y., Leeper N.J. A CD47-associated super-enhancer links pro-inflammatory signalling to CD47 upregulation in breast cancer. Nat. Commun. 2017;8:14802. doi: 10.1038/ncomms14802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwens van der Vlis T.A.M., Kros J.M., Mustafa D.A.M., van Wijck R.T.A., Ackermans L., van Hagen P.M., van der Spek P.J. The complement system in glioblastoma multiforme. Acta Neuropathol. Commun. 2018;6:91. doi: 10.1186/s40478-018-0591-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman R.L., Klemm F., Akkari L., Pyonteck S.M., Sevenich L., Quail D.F., Dhara S., Simpson K., Gardner E.E., Iacobuzio-Donahue C.A. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. 2016;17:2445–2459. doi: 10.1016/j.celrep.2016.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O., Weiner H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018;19:622–635. doi: 10.1038/s41583-018-0057-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L., Pollard J.W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018;17:887–904. doi: 10.1038/nrd.2018.169. [DOI] [PubMed] [Google Scholar]
- Chae W.H., Niesel K., Schulz M., Klemm F., Joyce J.A., Prummer M., Brill B., Bergs J., Rodel F., Pilatus U., Sevenich L. Evaluating magnetic resonance spectroscopy as a tool for monitoring therapeutic response of whole brain radiotherapy in a mouse model for breast-to-brain metastasis. Front. Oncol. 2019;9:1324. doi: 10.3389/fonc.2019.01324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Feng X., Herting C.J., Garcia V.A., Nie K., Pong W.W., Rasmussen R., Dwivedi B., Seby S., Wolf S.A. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 2017;77:2266–2278. doi: 10.1158/0008-5472.CAN-16-2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang H.N., van Rossum D., Sieger D., Siam L., Klemm F., Bleckmann A., Bayerlova M., Farhat K., Scheffel J., Schulz M. Carcinoma cells misuse the host tissue damage response to invade the brain. Glia. 2013;61:1331–1346. doi: 10.1002/glia.22518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A., Keren-Shaul H., Weiner A., Colonna M., Schwartz M., Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. 2018;173:1073–1081. doi: 10.1016/j.cell.2018.05.003. [DOI] [PubMed] [Google Scholar]
- Ding A.S., Routkevitch D., Jackson C., Lim M. Targeting myeloid cells in combination treatments for glioma and other tumors. Front. Immunol. 2019;10:1715. doi: 10.3389/fimmu.2019.01715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giladi A., Wagner L.K., Li H., Dorr D., Medaglia C., Paul F., Shemer A., Jung S., Yona S., Mack M. Cxcl10(+) monocytes define a pathogenic subset in the central nervous system during autoimmune neuroinflammation. Nat. Immunol. 2020;21:525–534. doi: 10.1038/s41590-020-0661-1. [DOI] [PubMed] [Google Scholar]
- Hagemeyer N., Hanft K.M., Akriditou M.A., Unger N., Park E.S., Stanley E.R., Staszewski O., Dimou L., Prinz M. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017;134:441–458. doi: 10.1007/s00401-017-1747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A., Alexander J.J. Complement and blood-brain barrier integrity. Mol. Immunol. 2014;61:149–152. doi: 10.1016/j.molimm.2014.06.039. [DOI] [PubMed] [Google Scholar]
- Jakel S., Dimou L. Glial cells and their function in the adult brain: a journey through the history of their ablation. Front. Cell Neurosci. 2017;11:24. doi: 10.3389/fncel.2017.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordao M.J.C., Sankowski R., Brendecke S.M., Sagar, Locatelli G., Tai Y.H., Tay T.L., Schramm E., Armbruster S., Hagemeyer N. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. 2019;363:eaat7554. doi: 10.1126/science.aat7554. [DOI] [PubMed] [Google Scholar]
- Kalsi R., Feigenberg S., Kwok Y., Tkaczuk K., Mehta M., Chumsri S. Brain metastasis and response to ado-trastuzumab emtansine: a case report and literature review. Clin. Breast Cancer. 2015;15:e163–e166. doi: 10.1016/j.clbc.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T.K., David E., Baruch K., Lara-Astaiso D., Toth B. A unique microglia type Associated with restricting development of Alzheimer's disease. Cell. 2017;169:1276–1290.e1. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- Khan M.A., Assiri A.M., Broering D.C. Complement and macrophage cross talk during process of angiogenesis in tumor progression. J. Biomed. Sci. 2015;22:58. doi: 10.1186/s12929-015-0151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S., Madore C., Cialic R., Baufeld C., Calcagno N., El Fatimy R., Beckers L., O'Loughlin E., Xu Y., Fanek Z. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–581.e9. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow S.A., Guttenplan K.A., Clarke L.E., Bennett F.C., Bohlen C.J., Schirmer L., Bennett M.L., Munch A.E., Chung W.S., Peterson T.C. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L., Yee S.W., Kim R.B., Giacomini K.M. SLC transporters as therapeutic targets: emerging opportunities. Nat. Rev. Drug Discov. 2015;14:543–560. doi: 10.1038/nrd4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorger M., Felding-Habermann B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am. J. Pathol. 2010;176:2958–2971. doi: 10.2353/ajpath.2010.090838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T., Sankowski R., Staszewski O., Bottcher C., Amann L., Sagar, Scheiwe C., Nessler S., Kunz P., van Loo G. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388–392. doi: 10.1038/s41586-019-0924-x. [DOI] [PubMed] [Google Scholar]
- Masuda T., Sankowski R., Staszewski O., Prinz M. Microglia heterogeneity in the single-cell era. Cell Rep. 2020;30:1271–1281. doi: 10.1016/j.celrep.2020.01.010. [DOI] [PubMed] [Google Scholar]
- Mathys H., Adaikkan C., Gao F., Young J.Z., Manet E., Hemberg M., De Jager P.L., Ransohoff R.M., Regev A., Tsai L.H. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 2017;21:366–380. doi: 10.1016/j.celrep.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrdjen D., Pavlovic A., Hartmann F.J., Schreiner B., Utz S.G., Leung B.P., Lelios I., Heppner F.L., Kipnis J., Merkler D. High-Dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. 2018;48:380–395.e6. doi: 10.1016/j.immuni.2018.01.011. [DOI] [PubMed] [Google Scholar]
- Nguyen D.X., Chiang A.C., Zhang X.H., Kim J.Y., Kris M.G., Ladanyi M., Gerald W.L., Massague J. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009;138:51–62. doi: 10.1016/j.cell.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris G.T., Kipnis J. Immune cells and CNS physiology: microglia and beyond. J. Exp. Med. 2019;216:60–70. doi: 10.1084/jem.20180199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom Q.T., Wright C.H., Barnholtz-Sloan J.S. Brain metastases: epidemiology. Handb. Clin. Neurol. 2018;149:27–42. doi: 10.1016/B978-0-12-811161-1.00002-5. [DOI] [PubMed] [Google Scholar]
- Palma A., Jarrah A.S., Tieri P., Cesareni G., Castiglione F. Gene regulatory network modeling of macrophage differentiation corroborates the continuum hypothesis of polarization states. Front. Physiol. 2018;9:1659. doi: 10.3389/fphys.2018.01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli R.C., Bolasco G., Pagani F., Maggi L., Scianni M., Panzanelli P., Giustetto M., Ferreira T.A., Guiducci E., Dumas L. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Pyonteck S.M., Akkari L., Schuhmacher A.J., Bowman R.L., Sevenich L., Quail D.F., Olson O.C., Quick M.L., Huse J.T., Teijeiro V. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail D.F., Bowman R.L., Akkari L., Quick M.L., Schuhmacher A.J., Huse J.T., Holland E.C., Sutton J.C., Joyce J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352:aad3018. doi: 10.1126/science.aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail D.F., Joyce J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A.K., DuBois J.C., Gruber R.C., Guzik H.M., Gulinello M.E., Perumal G., Raine C., Kozakiewicz L., Williamson J., Shafit-Zagardo B. Loss of Gas6 and Axl signaling results in extensive axonal damage, motor deficits, prolonged neuroinflammation, and less remyelination following cuprizone exposure. Glia. 2017;65:2051–2069. doi: 10.1002/glia.23214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankowski R., Bottcher C., Masuda T., Geirsdottir L., Sagar, Sindram E., Seredenina T., Muhs A., Scheiwe C., Shah M.J. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019;22:2098–2110. doi: 10.1038/s41593-019-0532-y. [DOI] [PubMed] [Google Scholar]
- Schulz M., Salamero-Boix A., Niesel K., Alekseeva T., Sevenich L. Microenvironmental regulation of tumor progression and therapeutic response in brain metastasis. Front. Immunol. 2019;10:1713. doi: 10.3389/fimmu.2019.01713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seano G., Nia H.T., Emblem K.E., Datta M., Ren J., Krishnan S., Kloepper J., Pinho M.C., Ho W.W., Ghosh M. Solid stress in brain tumours causes neuronal loss and neurological dysfunction and can be reversed by lithium. Nat. Biomed. Eng. 2019;3:230–245. doi: 10.1038/s41551-018-0334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C., Pamer E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford J.H., Hirai T., Deng L., Chernikova S.B., Urata K., West B.L., Brown J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. 2016;18:797–806. doi: 10.1093/neuonc/nov272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer K.J. Epidemiology and prognosis of brain metastases. Surg. Neurol. Int. 2013;4:S192–S202. doi: 10.4103/2152-7806.111296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh J.H., Kotecha R., Chao S.T., Ahluwalia M.S., Sahgal A., Chang E.L. Current approaches to the management of brain metastases. Nat. Rev. Clin. Oncol. 2020;17:279–299. doi: 10.1038/s41571-019-0320-3. [DOI] [PubMed] [Google Scholar]
- Tabouret E., Chinot O., Metellus P., Tallet A., Viens P., Goncalves A. Recent trends in epidemiology of brain metastases: an overview. Anticancer Res. 2012;32:4655–4662. [PubMed] [Google Scholar]
- Tay T.L., Sagar, Dautzenberg J., Grun D., Prinz M. Unique microglia recovery population revealed by single-cell RNAseq following neurodegeneration. Acta Neuropathol. Commun. 2018;6:87. doi: 10.1186/s40478-018-0584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thion M.S., Low D., Silvin A., Chen J., Grisel P., Schulte-Schrepping J., Blecher R., Ulas T., Squarzoni P., Hoeffel G. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. 2018;172:500–516.e6. doi: 10.1016/j.cell.2017.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utz S.G., See P., Mildenberger W., Thion M.S., Silvin A., Lutz M., Ingelfinger F., Rayan N.A., Lelios I., Buttgereit A. Early fate defines microglia and non-parenchymal brain macrophage development. Cell. 2020;181:557–573.e18. doi: 10.1016/j.cell.2020.03.021. [DOI] [PubMed] [Google Scholar]
- Valiente M., Ahluwalia M.S., Boire A., Brastianos P.K., Goldberg S.B., Lee E.Q., Le Rhun E., Preusser M., Winkler F., Soffietti R. The evolving landscape of brain metastasis. Trends Cancer. 2018;4:176–196. doi: 10.1016/j.trecan.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M., Obenauf A.C., Jin X., Chen Q., Zhang X.H., Lee D.J., Chaft J.E., Kris M.G., Huse J.T., Brogi E., Massague J. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156:1002–1016. doi: 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hove H., Martens L., Scheyltjens I., De Vlaminck K., Pombo Antunes A.R., De Prijck S., Vandamme N., De Schepper S., Van Isterdael G., Scott C.L. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019;22:1021–1035. doi: 10.1038/s41593-019-0393-4. [DOI] [PubMed] [Google Scholar]
- Vanpouille-Box C., Alard A., Aryankalayil M.J., Sarfraz Y., Diamond J.M., Schneider R.J., Inghirami G., Coleman C.N., Formenti S.C., Demaria S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017;8:15618. doi: 10.1038/ncomms15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins A.C., Patin E.C., Harrington K.J., Melcher A.A. The immunological consequences of radiation-induced DNA damage. J. Pathol. 2019;247:606–614. doi: 10.1002/path.5232. [DOI] [PubMed] [Google Scholar]
- Wingrove E., Liu Z.Z., Patel K.D., Arnal-Estape A., Cai W.L., Melnick M.A., Politi K., Monteiro C., Zhu L., Valiente M. Transcriptomic hallmarks of tumor plasticity and stromal interactions in brain metastasis. Cell Rep. 2019;27:1277–1292.e7. doi: 10.1016/j.celrep.2019.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J., Armour E., Kazanzides P., Iordachita I., Tryggestad E., Deng H., Matinfar M., Kennedy C., Liu Z., Chan T. High-resolution, small animal radiation research platform with x-ray tomographic guidance capabilities. Int. J. Radiat. Oncol. Biol. Phys. 2008;71:1591–1599. doi: 10.1016/j.ijrobp.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., McKay D., Pollard J.W., Lewis C.E. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. 2018;78:5492–5503. doi: 10.1158/0008-5472.CAN-18-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H., Sanes J.R. Neuronal cell-type classification: challenges, opportunities and the path forward. Nat. Rev. Neurosci. 2017;18:530–546. doi: 10.1038/nrn.2017.85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RNA-seq data of analyzed cell types are summarized by listing all differently expressed genes in each cell type, in the control versus BrM situation (Tab1), as well as within the comparison during BrM progression (small versus large BrM-associated cell types, Tab2). Total up- and downregulated genes as well as overlaps are represented in Tab1. Changes during BrM progression are represented by the corresponding gene as well as the log2 fold change.
RNA-seq data of the myeloid compartment of irradiated H2030-BrM-bearing mice. In Tab1 differently expressed genes in all non-irradiated versus all irradiated samples are displayed. Within each cell type, large BrM samples versus different irradiated conditions are displayed in the following Tabs (Tab2-5). Represented are gene names, log2 fold change and adjusted p-value. Positive log2 fold change value indicates upregulation in the corresponding IR condition.
Data Availability Statement
Tables S1 and S2 provide direct access to the main results derived from transcriptomic analysis presented in this study. In addition, all sequencing data have been deposited to the Gene Expression Omnibus (GEO) under the superseries accession number GSE137797. Single-cell RNA-seq data can be found under GSE137512. All bulk RNA-seq data are deposited under GSE137762.