

Abstract
Lyme disease is a tick-borne bacterial illness that occurs in areas of North America, Europe, and Asia. Early infection typically presents as generalized symptoms with an erythema migrans (EM) skin lesion. Dissemination of the pathogen Borrelia burgdorferi can result in multiple EM skin lesions or in extracutaneous manifestations such as Lyme neuroborreliosis. Metabolic biosignatures of patients with early Lyme disease can potentially provide diagnostic targets, as well as highlight metabolic pathways that contribute to pathogenesis. Sera from well-characterized patients diagnosed with either early localized Lyme disease (ELL) or early disseminated Lyme disease (EDL), plus healthy individuals (HC), from the United States were analyzed by liquid chromatography-mass spectrometry (LC-MS). Comparative analyses were performed between ELL, or EDL, or ELL combined with EDL, and the HC to develop biosignatures present in early Lyme disease. A direct comparison between ELL and EDL was also performed to develop a biosignature for stages of early Lyme disease. Metabolic pathway analysis and chemical identification of metabolites with LC-tandem mass spectrometry (LC-MS/MS) demonstrated alterations of eicosanoid, bile acid, sphingolipid, glycerophospholipid, and acylcarnitine metabolic pathways during early Lyme disease. These metabolic alterations were confirmed using a separate set of serum samples for validation. The findings demonstrated that infection of humans with B. burgdorferi alters defined metabolic pathways that are associated with inflammatory responses, liver function, lipid metabolism and mitochondrial function. Additionally, the data provide evidence that metabolic pathways can be used to mark the progression of early Lyme disease.
Keywords: Borrelia burgdorferi, Lyme disease, Metabolomics, Mass spectrometry, eicosanoid, bile acid, sphingolipid, glycerophospholipid, acylcarnitine
For TOC only
Introduction
Lyme disease is caused by transmission of infectious spirochetes belonging to Borrelia burgdorferi sensu lato through the feeding of Ixodes spp. ticks, and occurs in areas of North America, Europe and Asia 1−3. B. burgdorferi sensu stricto is the principal cause of Lyme disease in the United States, and early stage human infection typically presents as generalized symptoms, that include fatigue, fever and malaise, in association with an erythema migrans (EM) skin lesion. Dissemination of the bacteria through the blood to other sites can result in what is referred to as disseminated early Lyme disease and can manifest as multiple EM skin lesions. More severe extracutaneous presentations of disseminated early Lyme disease (Lyme neuroborreliosis or Lyme carditis) can occur, but are less common. The early manifestations of Lyme borreliosis typically occur several days to weeks following infection. Late Lyme borreliosis, occurring several months from onset of disease, manifests as arthritis in the United States 4.
The pathology of Lyme disease results from the outcome of complex vector-pathogen-host interactions. B. burgdorferi does not produce toxins, thus tissue pathology and the different manifestations of Lyme disease result from host immune responses to pathogen invasion, dissemination and persistence 1. When ingested during a blood-meal by a feeding tick, B. burgdorferi is primed for infection of a mammalian host by differential production of the outer surface proteins OspA and OspC 1, 5. Establishment of infection in a mammalian host is aided by activities of tick salivary proteins that include coagulation, fibrinolysis, and modulation of immune responses 2. Additionally, the metabolism of the spirochete adapts to the environments of the vector and the mammalian host 6. However, the mechanisms by which the host’s responses influence the heterogeneous presentations and symptoms of Lyme disease are poorly understood 1, 2.
The majority of studies directed at elucidating the molecular mechanisms of Lyme disease pathogenesis have focused on the immunological response of the mammalian host. Of note, application of functional genomics revealed that IL-10, IL-22 and HIF-1α are important factors in controlling Lyme borreliosis 7, 8. A separate study of peripheral blood mononuclear cells (PBMCs) isolated from human blood found suppression of the eIF2 signaling pathway and activation of TREM1 and TLR signaling during active Lyme disease 9. Investigation of the transcriptional response in human skin biopsies of EM lesions identified an IFN-inducible gene signature that included factors involved in host defense as well as immune modulation 10; and measurement of cytokines and chemokines following B. burgdorferi infection demonstrated elevated serum levels of CXCL9, CXCL10, CCL19 and CRP in Lyme disease patients 11. These studies underscore the role of inflammation and the complexity of the immune response to B. burgdorferi infection. However, they also provide evidence for the involvement of host metabolic responses [e.g., the involvement of HIF-1α 8 and the identification of tryptophan–kynurenine metabolism 10].
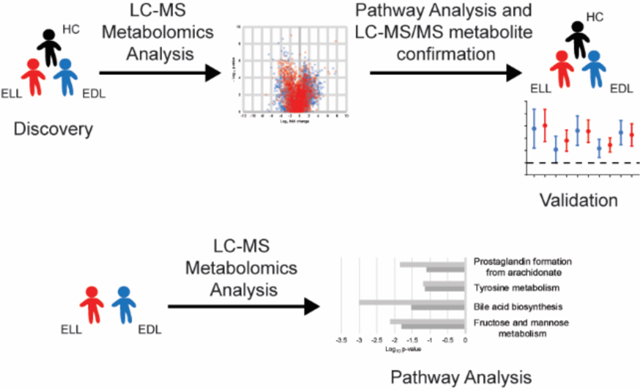
Recent efforts to define the molecular drivers of the immunopathology of Lyme disease in patients have revealed the interplay of host cell metabolism with innate and adaptive immunity 8, 10. Metabolomics provides a picture of a biological systems’ metabolic state and complements transcriptomics and proteomics analyses. In recent years liquid chromatography-mass spectrometry (LC-MS) based metabolomics has been applied to study metabolic perturbations during Lyme disease 12–14. Molins and colleagues demonstrated that serum metabolic profiles can distinguish Lyme disease patients from patients with other confounding diseases and healthy controls, as well as identify specific metabolic pathways that biochemically distinguish Lyme disease and Southern Tick- Associated Rash Illness (STARI) 12, 13. A metabolomics evaluation of urine samples from early Lyme disease patients also revealed alterations of multiple metabolic pathways including tryptophan metabolism 15. Lastly, Kerstholt et al. observed an alteration of glutathione metabolism in B. burgdorferi stimulated PBMCs, findings that were confirmed using Lyme disease patient sera 14. However, a more comprehensive evaluation of metabolic pathways altered during Lyme disease has not been reported. The present study applied serum metabolomics to define the metabolic profiles of early Lyme disease patients with an EM skin lesion and healthy controls (HC). Additionally, to assess metabolic differences in the progression of early disease, serum metabolomics analyses were applied to patients with early localized Lyme disease (ELL) and to patients with early disseminated Lyme disease (EDL) who did not have the more severe presentations of Lyme neuroborreliosis or Lyme carditis. These analyses detected differential metabolite abundances and metabolic pathways that were altered during early Lyme disease and/or at different stages of early disease.
Experimental Section
Clinical Samples.
Well characterized retrospective serum samples were procured from specimen repositories at New York Medical College, CDC, and Tufts University. It is noted that there are differences in the sites with respect to sex, age and sample collection date. The samples from New York Medical College were collected from 1992–2012 and consisted of 65% males with ages ranging from 16–81. A subset of the New York Medical College samples did not include individual patient data, these samples were collected from 2009–2010 and consisted of 49% male with ages ranging from 18–74. The samples from Tufts University were collected from 2011 and 2013 and consisted of 50% males with the ages ranging from 27–51. The samples from CDC were collected in 2012 and consisted of 29% males; however no age data were collected. The serum specimens from 166 Lyme disease patients that were used in this study were collected at the time of diagnosis before treatment. All Lyme disease patients had an EM skin lesion and were seropositive for antibody to B. burgdorferi using a first-tier serologic assay on either an acute or convalescent phase serum sample. Patients with a single EM skin lesion and who had a negative blood culture for B. burgdorferi were classified as ELL (n = 77). The majority (93.5%) of ELL patients were skin culture positive for B. burgdorferi. Patients presenting with multiple EM skin lesions (n = 37), or a single EM skin lesion and a positive blood culture for B. burgdorferi (n = 52), were classified as EDL (n = 89). Of the EDL patients with multiple EM skin lesions, 18 were also blood culture positive for B. burgdorferi. ELL patients had an age distribution of 23 to 81 years and a mean age of 47 years and EDL patients ranged from 16 to 80 years with an average age of 48 years. HC sera (n = 96) were obtained from donors without a previous history of Lyme disease in endemic (n = 68) and nonendemic regions (n = 28) for Lyme disease. A subset of these samples overlap with those utilized previously 16. The early Lyme disease samples and HC were not age and sex matched. Sera from ELL, EDL and HC patients were divided into discovery and validation sets. Specifically, 40 of the 77 ELL, 44 of the 89 EDL, and 50 of the 96 HC samples were arbitrarily selected as the discovery samples.
The samples used in this study were stored at −80°C following collection with no additives. All participating institutions obtained Institutional Review Board approval and all patients gave informed consent. All methods were carried out in accordance with the guidelines and regulations of human subjects.
Chemical Standards:
Chemical standards utilized for the confirmation of identified metabolites included glycocholic acid (sc-218574), glycoursodeoxycholate (sc-211567), and glycolithocholate (sc-396741) (Santa Cruz Biotechnology, Inc., Dallas, TX, USA); glycochenodeoxycholate (16942), taurodeoxycholate (15935), acetylcarnitine (16948), octanoylcarnitine (15048), prostaglandin B2 (11210), 12-oxo-leukotriene B4 (20140), arachidonic acid (90010), 12(R)-HETE (34560), 8(S)-HETE (34360) and lyso-PAF (C18) (60916) from Cayman Chemical (Ann Arbor, Michigan, USA); glycodeoxycholate (G9910), taurochenodeoxycholate (T6260), lysophosphatidylcholine (L4129) and from Sigma-Aldrich (St. Louis, MO, USA); and Sphingomyelin mixture (860061,2,3, Avanti Polar Lipids, Inc., Alabaster, Alabama, USA).
Sample preparation and LC-MS.
Small molecule metabolites were extracted from sera and analyzed by LC-MS as previously described 12, 13. Specifically, cold methanol (60 μL) was added to 20 μL of sera, vortexed and incubated at −20°C for 1 h. Samples were warmed to room temperature for ten minutes, vortexed and centrifuged at 18,000 x g for 30 min. The supernatant (65 μL) was transferred to a new microcentrifuge tube and dried under vacuum. The dried metabolite extract was suspended in 50% methanol (40 μL) centrifuged at 18,000 x g for 15 min. An aliquot (35 μL) was transferred to an autosampler vial for LC-MS analysis.
All samples were processed and analyzed in duplicate by LC-MS in an arbitrary manner. Metabolite extracts (10 μL) were applied to a Poroshell 120, EC-C8, 2.1 × 100 mm, 2.7 μm LC column (Agilent Technologies) and eluted with a nonlinear gradient of acetonitrile using Agilent 1200 Series LC system 13. Effluent was introduced directly into an Agilent 6520 quadrupole-time-of-flight (Q-TOF) mass spectrometer equipped with an electrospray ionization source. LC-MS data were collected in the positive ionization mode under the following parameters: gas temperature, 310°C; drying gas at 10 L/min; nebulizer at 45 lb/in2; capillary voltage, 4000 V; fragmentation energy, 120 V; skimmer, 65 V; and octapole RF setting, 750 V. MS data for a mass range of 75 to 1700 Da were acquired at a rate of 2 scans/s. Data were collected in both centroid and profile modes in 2-GHz extended dynamic range. To monitor instrument performance, quality control samples consisting of a metabolite extract of HC sera (BioreclamationIVT) were analyzed in duplicate at the beginning of each analysis day and every 20 samples during the analysis day. MS/MS spectra for the targeted molecular features (MFs) and commercial standards were obtained using the above parameters, except MS/MS data were acquired at a rate of 1 scan/s and precursor ions were fragmented via collision-induced dissociation with nitrogen at a collision energy of 15 or 20 V. MS/MS spectra for selected molecules were also evaluated in the negative ionization mode using the parameters listed above.
Prediction and verification of MFs.
Experimental accurate masses from each developed biosignature were searched in the Human Metabolome Database (HMDB) with a tolerance of 15 ppm to obtain putative metabolite identifications 17. Non-endogenous metabolites were excluded from the results. Confirmation of the chemical structure of selected MFs was performed by LC-MS/MS. The level of structural identification followed refined Metabolomics Standards Initiative guidelines proposed by Schymanski et al. 18.
Data analyses.
LC-MS data files from discovery samples of ELL (n = 40), EDL (n = 44), and HC (n = 50) were processed for MF extraction as previously described 12, 13. MFs detected in at least 50% of samples in a single group using the Mass Profiler Professional software were utilized for downstream analyses (n = 4491). Duplicate MFs were identified and removed using the default parameters for MS Feature List Optimizer (MS-FLO) 19 with the retention time tolerance set to 0.25 and minimum peak match ratio set to 0.70, as well as manual assessment of extracted ion chromatograms for potential duplicates highlighted by MS-FLO using the Agilent MassHunter Qualitative Analysis software. Abundance data for individual MFs were normalized as follows: 1) all missing values were imputed with a value of 10; 2) abundances of each MF in each sample were normalized by dividing by the median intensity of all MFs in that sample; 3) the median fold change of each stable MF (MFs present in at least 50% of all sample data files) between the initial quality control sample and subsequent quality control samples were calculated; 4) the median fold change calculated for the quality control sample that directly followed each series of 20 clinical samples was multiplied by the normalized MF abundances in the clinical samples of that series; and 5) resulting MF abundances were then multiplied by a scalar (10000). MFs that differed between patient groups were selected based on having a ≥ 2-fold abundance difference between the medians of the two comparator groups in either of the replicate LC-MS analyses. Additionally, MFs that had a significant difference (p <0.05, unadjusted) between the two comparator groups in either of the replicate LC-MS analyses regardless of fold change were also selected. Statistical analyses were performed on log2 transformed data. An F-test was performed to determine whether the variance was equal and to establish whether significance was determined using a student’s t-test (equal variance) or a Welch’s t-test (unequal variance) in excel (Microsoft). Those MFs with > 20% missing values in both comparator groups, MFs that failed to produce a fold change of ≥ 2 in at least 50% of samples within a patient group compared to the median of the other patient group, and MFs with inconsistent fold change directions between replicates were removed from further analyses. The selection process for MF differences between the patient groups resulted in a total of four metabolic biosignatures (EDL vs HC, ELL vs HC, ELL/EDL vs HC, and ELL vs EDL) (Figure S1).
Metabolic pathway analyses were performed via Mummichog pathway and network analysis software 20. Specifically, biosignature data files generated by group comparisons containing m/z value, retention time, p-value, t-score and identifier for each MF were used as input for metabolic pathway analysis using Mummichog software version 2. Mummichog software was operated under python program version 2.7.14. The default parameters were used for pathway analysis except the mass error was set at 20 ppm, the p-value cutoff was 0.05, and the analytical mode was set for positive ionization.
Directed semi-quantitative analyses of confirmed metabolites were performed. Experimental m/z and retention time (RT) values obtained from authentic standards were used to create methods for quantitation in Mass Hunter Quantitative analysis software (Agilent Technologies). The relative abundances (normalized peak area) of the corresponding confirmed metabolites were obtained for each of the discovery and validations samples. Normalized log2 transformed abundances were plotted using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA).
The associations of MF with age, sex and sample collection date were evaluated using all samples for which these meta-data were recorded. Sample collection date was defined as “Pre 2000” for collections made prior to 2000 and “Post 2000” for collections made in 2000 and later. Each replicate LC-MS analysis was evaluated separately using the limma package version 3.40.6 21 for sample collection date and sex (group classification) and in linear models for age (continuous variable) using R version 3.6.1. All p-values were adjusted for false discovery rate.
Results
Development of early Lyme disease vs HC biosignatures.
Small molecule metabolic biosignatures based on LC-MS data were developed for group comparisons of 1) ELL vs HC; 2) EDL vs HC; and 3) ELL and EDL combined (ELL/EDL) vs HC (Figure S1). These biosignatures were comprised of MFs with a 2-fold abundance change and/or a significant difference (p < 0.05) in abundance between the two comparator groups in at least one of replicate LC-MS analyses. MFs consist of detected signals with a determined mass and retention time. Structure confirmation was required for a MF to be designated as a particular metabolite. The number of differentiating MFs identified for ELL vs HC, EDL vs HC, and ELL/EDL vs HC were 1633, 1771, and 1750 MFs, respectively (Figure 1A and Table S1). It was noted that there were 24 MFs in the ELL/EDL vs HC comparison that were not present in either the ELL vs HC or the EDL vs HC comparisons. These 24 MFs were close to the 2 fold change and/or p < 0.05 cut-offs in the individual comparisons (ELL vs HC and EDL vs HC), and when the data for the ELL and EDL groups were combined (ELL/EDL vs HC), these MFs met the selection criteria. Of these MFs unique to the ELL/EDL vs HC list, 17 and six met the selection criteria in replicate 1 and 2, respectively, only one MF met the selection criteria in both replicates.
Figure 1. Metabolic changes between early Lyme disease patients and HC.

Venn diagram depicting the unique and overlapping MFs among the three different comparisons performed (ELL vs HC, EDL vs HC, and ELL/EDL vs HC). These MFs comprise the combined early Lyme disease vs HC biosignature consisting of 2193 MFs (A). Evaluation of the number of MFs (y-axis) and the magnitude of fold-change (x-axis) in each of the three comparisons (ELL vs HC, EDL vs HC, and ELL/EDL vs HC) using the average fold-change of the replicate analyses (B).
When comparing ELL vs HC, EDL vs HC and ELL/EDL vs HC, 906, 1105, and 974 MFs, respectively, achieved an averaged abundance change of at least two-fold for the replicate analyses. Over half of these MFs (75%, 63%, and 72%, respectively) displayed absolute abundance fold changes between 2 and 4 in each of the comparisons (Figure 1B). The number of MFs with an absolute abundance fold change ≥ 4 were 224, 409, and 274 for the ELL vs HC, EDL vs HC and ELL/EDL vs HC comparisons, respectively. There were a total of 1511, 1646, and 1662 MFs that were significant (p < 0.05) for the ELL vs HC, EDL vs HC and ELL/EDL vs HC comparisons, respectively. The unique and overlapping differentiating MFs obtained from each of the three comparisons were combined, resulting in an early Lyme disease vs HC biosignature consisting of 2193 MFs (Figure 1 and Table S1).
Metabolic changes occur between ELL and EDL patients
Further analyses of the early Lyme disease vs HC biosignature (2193 MFs) initially assessed whether the MFs significantly differed between ELL and EDL patients relative to HC. Because it is not possible to average statistical significance (p-values), the following analyses were performed by evaluating the LC-MS data of replicate analyses separately (the data from the first replicate are presented in Figure 2 and the second replicate are presented in Figure S2). Volcano plot analyses (Figure 2A and Figure S2A) to compare how EDL and ELL patient groups differed from HCs demonstrated that the EDL group yielded a greater number MFs with a fold change ≥ 2 and a significance of p < 0.05. Specifically, in the first replicate there were 924 and 779 MFs that met the ≥ 2-fold abundance change and significance criteria in the EDL vs HC and ELL vs HC analyses, respectively. This same trend was observed in the second replicate. The mean abundance fold change of MFs that met the ≥ 2- fold abundance change and significance criteria in the EDL vs HC was higher than the mean abundance fold change of the same MFs in the ELL vs HC comparison, in both replicates (Figure 2B and Figure S2B). The same analysis for the MFs that met the ≥ 2-fold abundance change and significance criteria in the ELL vs HC comparison, demonstrated a higher mean abundance fold change or no difference for the EDL vs HC comparison in the two replicate experiments (Figure 2C and Figure S2C). These data derived from the early Lyme disease vs HC biosignature provided evidence that the ELL and EDL stages of Lyme disease are distinguishable based on their metabolic profiles, and that alterations in the EDL patients vs HCs were greater than those observed for the ELL patients vs HCs.
Figure 2. Differential metabolic changes between ELL and EDL patients vs HC.

22Volcano plot depicting the log2 fold change (x-axis) and –log10 p-value (y-axis) for the 2193 MFs (from the combined early Lyme disease vs HC biosignature) as compared to HC. MFs in EDL and ELL are plotted in blue and red, respectively, for replicate 1. The upper left quadrant contains 319 and 401 MFs and the upper right quadrant contains 605 and 378 MFs for EDL and ELL, respectively (A). A plot of |log2 fold change from HC| (x-axis) and –log10 p-value (y-axis) for the 924 MFs with >1 log2 fold change and p < 0.05 in the EDL vs HC comparison plotted for EDL (blue) and ELL (red) for replicate 1 (B). A plot of |log2 fold change| from HC (x-axis) and –Log10 p-value (y-axis) for the 779 MFs with > 1 log2 fold change and p < 0.05 in the ELL vs HC comparison plotted for EDL (blue) and ELL (red) for replicate 1 (C). The vertical blue lines depict the mean fold change (2.24 +/− 1.07 and 2.39 +/– 1.17 for B and C respectively) for EDL vs HC and the vertical red lines depict the mean fold change (1.55 +/– 0.97 and 2.03 +/– 0.86 for B and C respectively) for ELL vs HC.
A direct comparison of LC-MS data for ELL and EDL patients’ sera (Figure S1) resulted in 671 differentiating MFs. Of these, 566 MFs overlapped with those in the early Lyme disease vs HC biosignature. Thus, 105 MFs were unique to the ELL vs EDL biosignature (Figure 3A). As expected, the 1627 MFs that were unique to the early Lyme disease vs HC biosignature were strongly correlated when the abundance fold change between ELL and HC was plotted against the fold change between EDL and HC, R2 = 0.82 in the first replicate (Figure 3B) and R2 = 0.78 in the second replicate (Figure S3A). In contrast, the same analysis performed with the 105 MFs unique to the ELL vs EDL biosignature revealed weaker correlation between the abundance fold change of ELL vs HC and the abundance fold change of EDL vs HC, R2 = 0.65 in the first replicate (Figure 3C) and R2 = 0.53 in the second replicate (Figure S3B). It is noted that 39 MFs in Figure 3C (34 MFs in Figure S3B) are plotted with a 2 fold change or greater from HC for either EDL or ELL (regions not boxed in blue). These MFs were not included in the early Lyme disease vs HC biosignature because they did not meet the inclusion criterion of an abundance fold change in the same direction in the replicate analyses when compared to HCs. However, this criterion was achieved in the replicate EDL vs ELL comparisons. A correlation analysis of the 566 MFs that overlapped between the ELL vs EDL biosignature and the early Lyme disease vs HC biosignature resulted in an R2 = 0.64 in the first replicate (Figure 3D) and R2 = 0.47 in the second replicate (Figure S3C). While these 566 MFs were determined to differ in abundance between early Lyme disease patients and HC, the plotting of each MF abundance fold change to HCs revealed that the MF abundances of ELL patients were closer to HCs than those of EDL patients. Only 147 and 183 of the 566 MFs had an absolute fold change for ELL vs HC that was greater than that of EDL vs HC in replicate 1 and 2, respectively (Figure S4). Additionally, 256 of the 566 MFs that were shared between the ELL vs EDL biosignature and the early Lyme disease vs HC biosignature had a ≥ 2- absolute fold abundance change between EDL and HC, but not ELL and HC.
Figure 3. Metabolic changes between EDL and ELL patients.

A Venn diagram depicting the unique and overlapping features between the EDL vs ELL biosignature (671 MFs) and the early Lyme disease vs HC biosignature (2193 MFs) (A). Scatter plots for replicate 1 depicting log2 fold change from HC for EDL (x-axis) and log2 fold change from HC for ELL (y-axis) using the 1627 MFs unique to the EL vs HC biosignature (B); or the 105 MFs unique to the ELL vs EDL biosignature (C); or the 566 MFs in common between the early Lyme disease vs HC and the ELL vs EDL biosignatures (D). The red dotted line is the line of best fit. Blue shaded box indicates 66 out of 105 MFs with a |log2 fold change| of ≤ 1 from HC for EDL and ELL.
Pathways altered during early Lyme disease
2To investigate the underlying biology of the early Lyme disease biosignatures, the MFs were interrogated against the HMDB to provide putative metabolite identifications, and each biosignature was subjected to pathway analyses. For the 2193 MFs of the early Lyme disease vs HC biosignature, 998 MFs (46%) were matched to putative metabolites in HMDB and 705 MFs (71%) of these were annotated as endogenous (Table S1). Similarly, 317 MFs (47%) of the 671 MF ELL vs EDL biosignature were assigned a putative metabolite identification and 226 (71%) of these were annotated as endogenous (Table S1).
Metabolic pathways that differed between the comparator groups were predicted with the Mummichog pathway analysis tool 20. This was performed for the individual biosignatures and data from each of the replicate sample analyses (Figure 4, and Figure S4). The dominant enriched pathway identified in the ELL vs HC, EDL vs HC, and ELL/EDL vs HC biosignatures was linoleate metabolism. Six additional pathways common among these three biosignatures were identified; however, their level of enrichment differed (Figure 4). These included arachidonic acid metabolism, retinol metabolism, prostaglandin formation from arachidonic acid, de-novo fatty acid biosynthesis, omega-3 fatty acid metabolism, and leukotriene metabolism. Two pathways (glycerophospholipid metabolism and fatty acid activation) were common between the EDL vs HC and ELL/EDL vs HC biosignatures. The inclusion of EDL patients in the generation of these two biosignatures suggested that they were driving the enrichment of glycerophospholipid metabolism and fatty acid activation pathways. The enriched pathways in the ELL vs EDL biosignature were largely different from those of the ELL/EDL vs HC, ELL vs HC, and EDL vs HC biosignatures. The only pathway that overlapped was that of prostaglandin formation from arachidonic acid. The other dominant pathways were fructose and mannose metabolism, bile acid biosynthesis, and tyrosine metabolism (Figure 4D). It should be noted that in the Mummichog analyses of the replicate data sets, there were pathways that were enriched for only one of the replicate data sets (Figure S4), and thus were not considered to be robust pathway identifications.
Figure 4. Metabolic pathways altered during early Lyme disease.

Mummichog enriched pathways and level of enrichment (Log10 p-value) for replicate 1 (solid) and replicate 2 (checkered) for ELL vs HC (A), EDL vs HC (B), ELL/EDL vs HC (C), and ELL vs EDL (D).
Metabolite Confirmation
The pathway analyses described were performed based on presumptive chemical identifications obtained with accurate mass data (Table S1). Thus, to confirm pathway enrichment data, specific metabolites putatively identified in the biosignatures were targeted for analyses by MS/MS and compared to chemical standards when possible (Table 1). Several of the metabolic pathways identified indicated alterations in polyunsaturated fatty acid (PUFA) metabolism in early Lyme disease. Thus, eicosanoids and other PUFA metabolites were targeted for structural identification. The early Lyme disease vs HC biosignature contained five MFs putatively identified and confirmed as eicosanoids (Table 1 and Table S1). Three MFs, MF#1241 (m/z 335.2208 @ 16.84 min), MF# 2298 (m/z 335.2214 @ 15.94 min), and MF# 1269 (m/z 327.2285 @ 20.57 min) were confirmed to level 1 based on retention time (RT) alignment and MS/MS as 12-oxo-leukotriene B4 (LTB4), prostaglandin B2 (PGB2), and arachidonic acid, respectively (Figures S7 – 12). Two metabolites, MF# 1823 (m/z 279.2327 @ 18.49 min) and MF# 1841 (m/z 321.2396 @ 19.31 min) were identified to level 2 (Figures S6 and S13). The first, MF# 1823, had similar MS/MS fragmentation as γ-linolenic acid, a polyunsaturated fatty acid. MF# 1841 was also identified to a level 2 as a hydroxyeicosatetraenoic acid (HETE) based on similar MS/MS fragmentation; however, it was not possible to determine the exact site of hydroxylation. The abundances of these metabolites in EDL and ELL patients were increased with respect to HC in the discovery set samples; MF# 1241, 2298, 1823, 1841, and 1269 were present on the early Lyme disease vs HC biosignature list due to having a 2-fold abundance change and being significantly different (Table S1). The abundances of these five PUFA structures were further evaluated using a validation sample set, all five MFs maintained increased abundance in EDL and ELL patients with respect to HC (Figure 5A). Additionally, all five MFs maintained a 2-fold abundance change and significance when comparing EDL or ELL patients to HC in the validation sample set for each replicate analysis, except for MF#2298 when comparing EDL to HC in replicate 1 (Figure 5A).
Table 1.
Identified MFs
| MF # | Mass | RT | Metabolite ID | Method of ID |
|---|---|---|---|---|
| 2 | 129.0429 | 1.37 | 5-oxo-L-proline | MS and RT alignment with 5-oxo- proline standard |
| 53 | 465.31 | 14.72 | Glycocholic acid | MS/MS and RT alignment with glycocholic acid standard |
| 66 | 449.313 | 16.00 | Glycodeoxycholic acid | MS/MS and RT alignment with glycodeoxycholic standard |
| 106 | 523.365 | 19.17 | 2-octadecanoyl-sn-glycero-3- phosphocholine (lysoPC (18:0)) | MS/MS and RT alignment with lysoPC standard mixture |
| 134 | 702.566 | 22.87 | Sphingomyelin (d18:1/16:0) | MS/MS and RT alignment with sphingomyelin mixture |
| 189 | 271.251 | 16.04 | Sphingosine (d16:1) | MS/MS diagnostic ions |
| 253 | 523.365 | 19.22 | 1-octadecanoyl-sn-glycero-3- phosphocholine (lysoPC (18:0)) | MS/MS and RT alignment with lysoPC standard mixture |
| 923 | 551.395 | 20.24 | 1-eicosanoyl-sn-glycero-3- phosphocholine (lysoPC (20:0)) | MS/MS and RT alignment with lysoPC standard mixture |
| 996 | 287.209 | 13.56 | Octanoylcarnitine | MS/MS and RT alignment with Octanoylcarnitine standard |
| 1006 | 315.24 | 15.01 | Decanoylcarnitine | MS/MS diagnostic ion |
| 1086 | 217.130 | 1.51 | Propionylcarnitine | MS/MS diagnostic ion |
| 1182 | 259.178 | 11.33 | Hexanoylcarnitine | MS/MS diagnostic ion |
| 1241 | 334.213 | 16.84 | 12-oxo-leukotriene b4 | MS/MS and RT alignment with 12-oxo-leukotriene b4 standard |
| 1264 | 509.386 | 19.51 | 1-octadecylglycero-3- Phosphocholine (lyso-PAF (C18)) | MS/MS and RT alignment with 1- octadecylglycero-3- Phosphocholine standard |
| 1269 | 304.239 | 20.57 | Arachidonic acid | MS/MS and RT alignment with Arachidonic acid standard |
| 1460 | 203.114 | 1.19 | Acetylcarnitine | MS/MS and RT alignment with Acetylcarnitine standard |
| 1601 | 449.314 | 14.75 | Glycoursodeoxycholic acid | MS/MS and RT alignment with glycoursodeoxycholic acid standard |
| 1780 | 499.297 | 15.16 | Taurodeoxycholic acid | MS/MS and RT alignment with taurodeoxycholic acid standard |
| 1823 | 278.226 | 18.49 | Polyunsaturated fatty acid (PUFA) | MS/MS spectra similar to γ - linoleic acid |
| 1827 | 415.308 | 17.39 | glycolithocholic acid | MS/MS and RT alignment with glycolithocholic acid standard |
| 1841 | 320.473 | 19.31 | hydroxyeicosatetraenoic acid (HETE) | MS/MS and RT similar to hydroxyeicosatetraenoic acid (HETE) standards |
| 2237 | 551.395 | 20.13 | 2-eicosanoyl-sn-glycero-3- phosphocholine (lysoPC (20:0)) | MS/MS and RT alignment with lysoPC standard mixture |
| 2298 | 334.214 | 15.94 | Prostaglandin B2 | MS/MS and RT alignment with Prostaglandin B2 standard |
Figure 5. Directed Analysis of Pathways in Validation Samples.

Log2 abundances normalized to HC in ELL (red) and EDL (blue) patients for early Lyme disease vs HC biosignature MFs classified as eicosanoids (A), bile acids (B), lyso-PAF and lysoPCs (C), acylcarnitines (D), sphingolipids (E), and 5-oxo-L-proline (F) for replicate 1, left and replicate 2, right. Metabolites that were present on the ELL vs EDL biosignature list are noted with #. Data is presented as the mean +/− 95% confidence interval. Dotted line at zero indicates HC baseline. Significance between the patient cohort and HCs is indicated by: *, p < 0.05; **, p < 0.01; ***, p < 0.001, ****, p < 0.0001. Panel B significance (p < 0.05) between ELL and EDL is indicated by * over a solid line between the groups.
Bile acid biosynthesis was noted as a pathway that was altered in ELL vs EDL patients (Figure 4D). Specifically, four putative bile acids, MF# 53 (m/z 466.3149 @ 14.7173), MF# 66 (m/z 450.3209 @ 16.0022), MF# 1601 (m/z 450.3209 @ 14.7493), and MF# 1780 (m/z 500.3045 @ 15.1581) were detected in higher abundance in EDL patients relative to ELL patients in the discovery set samples (Table S1). The structure of these metabolites were respectively confirmed as glycocholic acid, glycodeoxycholic acid, glycoursodeoxycholic acid, and taurodeoxycholic acid to level 1 based on RT alignment and MS/MS (Table 1 and Figures S14 – 16, S18 and S22). The increased abundance of glycocholic acid, glycoursodeoxycholic acid, and taurodeoxycholic acid in EDL patients, as compared to ELL patients was maintained in the validation sample set data for replicate analyses; however, this increase was not 2-fold and was only significant for glycocholic acid in the second replicate (Figure 5B).
Evaluation of the early Lyme disease vs HC biosignature also indicated alterations in bile acid biosynthesis. Glycocholic acid, glycodeoxycholic acid, and taurodeoxycholic acid along with an additional putative bile acid MF# 1827 (m/z 416.3153 @ 17.3911) were significantly increased in abundance in EDL patients relative to HC in the discovery set (Table S1). This additional bile acid was confirmed as glycolithocholic acid (Figure S19 and S20). Evaluation of these MFs in the validation sample set showed that glycodeoxycholic acid, glycolithocholic acid, and taurodeoxycholic acid were decreased in abundances in EDL patients vs HC (Figure 5B); a trend opposite of that observed for the discovery data. However, the increased levels of glycocholic acid in EDL patients in comparison with HC was maintained for the validation set though it was not quite 2-fold nor significant (Figure 5B). Additionally, directed analysis of two other bile acids confirmed as glycochenodeoxycholic acid and taurochenodeoxycholic acid (Figures S17 and S21), that were not included in the early Lyme disease vs HC biosignature revealed that their abundances are increased in EDL patients relative to ELL patients and HC in the validation set of samples. The most notable change was the significantly increased levels of glycochenodeoxycholic acid in EDL patients (Figure 5B).
The enrichment for glycerophospholipid metabolism was based primarily on the putative identification of lysophosphatidylcholine (lysoPC) structures, including ether linked forms of lysoPC, that displayed increased abundances in ELL and/or EDL patients in comparison to HCs. Of the ether linked structures, 1-octadecylglycero- 3-phosphocholine (lyso-PAF (C18)) was confirmed by MS/MS and RT alignment with an authentic standard (Table 1 and Figure S23 and S24). Several other lysoPC molecules that were increased in abundance in the Lyme disease patient sera in comparison to that of HCs were confirmed as lysoPC (16:0), lysoPC (18:0), and lysoPC (20:0) by use of a lysoPC standard mixture (Table 1, Figure S25–S28, Table S1). A double peak was present for all of these metabolites, presumably due to acylation of glycerol at different positions (sn-1 or sn-2) 22.
2Lyso-PAF (C18) levels were increased in both EDL and ELL patients as compared to HC in the discovery set (Table S1). This metabolite maintained a > 2-fold and significant increase in EDL and ELL patients vs HC using the validation data set in both replicates (Figure 5C). There was no significant difference in Lyso-PAF (C18) levels between ELL and EDL patients. The abundances for all six lysoPCs were increased in EDL and ELL patients compared to HC in the discovery set of samples (Table S1). Excluding lyso-PC (16:0), the increased abundance in EDL and ELL patients was maintained in the validation samples for the first replicate; however, not a 2-fold change (Figure 5C).
While acylcarnitine metabolism is not a pathway annotated in Mummichog, these molecules are linked with fatty acid activation, a pathway that is annotated and was enriched in the Mummichog analysis. Additionally, acylcarnitines are documented as being altered in other diseases, as well as Lyme disease 23–27. Five MFs had putative identities as acylcarnitines according to HMDB (Table S1). Thus, we evaluated these metabolites that were presumptively identified in the early Lyme disease vs HC biosignature. Five MFs, MF# 1460 (m/z 204.1200 @ 1.1936), MF# 996 (m/z 288.2172 @ 13.5559), MF# 1006 (m/z 316.2491 @ 15.0075), MF# 1182 (m/z 260.1854 @ 11.3300), and MF# 1086 (m/z 218.1369 @ 1.5101) were respectively confirmed as acetylcarnitine, octanoylcarnitine, decanoylcarnitine, hexanoylcarnitine, and propionylcarnitine by MS/MS based on the signature fragment ion of m/z 85.0292 (Figures S29 – S31). Two of the metabolites (acetylcarnitine and octanoylcarnitine) were also confirmed using authentic chemical standards (Table 1 and Figure S29 and S30). The abundances of these short and medium chain acylcarnitines were significantly lower in both EDL and ELL patients as compared with HC in the discovery set (Table S1). The significantly decreased abundance in EDL and ELL patients in comparison to HC was maintained in the validation set for all acylcarnitines, except decanoylcarnitine which was only significant for the ELL vs HC comparison in replicate 1 (Figure 5D).
Sphingolipid metabolism was not identified as an altered metabolic pathway via the Mummichog analyses; however, two MFs; MF# 134 (m/z 703.5769 @ 22.8701) and MF# 189 (m/z 272.2582 @ 16.0437) of the early Lyme disease vs HC biosignature had putative identities as the sphingomyelin (SM) (d18:1/16:0) and sphingosine (d16:1), respectively (Table S1). The SM (d18:1/16:0) structure was confirmed by MS/MS and RT alignment with a mixture of sphingomyelins (Table 1 and Figure S32). The identification of sphingosine (d16:1) was confirmed to level 2 based on diagnostic ions corresponding to [M+H – H2O]+, [M+H – 2H2O]+, and [M+H – H2O – CH2O],+ as well as eluting at an earlier RT than that of sphingosine (d18:1) (Table 1 and Figure S33).
The serum levels of both sphingolipid metabolites were higher in EDL and ELL patients vs HC in the discovery set (Table S1). The increased levels of sphingosine (d16:1) in EDL and ELL patients as compared to HC was maintained in the validation set and was significant except for ELL patients vs HC in replicate 2 (Figure 5E).
A final metabolic pathway interrogated was that of glutathione metabolism. Kerstholt et al. noted an increase in glutathione metabolism in B. burgdorferi stimulated PBMCs, a finding confirmed in Lyme patient sera 14. In order to see if glutathione metabolism was altered in our patient samples, we looked for metabolites identified by Kerstholt et al. as well as other metabolites in the glutathione metabolic pathway. MF# 2 (m/z 152.0327 @ 1.37 min) was putatively identified as 5-oxo-L-proline and was confirmed by MS and RT alignment with 5-oxo-L-proline standard (Figure S34). 5-oxo-L-proline was increased in ELL and EDL patients compared to HC in the discovery set, and this increase was maintained in the validation sets of samples (Figure 5F, Table S1).
The abundance data for the MFs that comprised each of the biosignatures were analyzed to see whether associations occurred with sex, age or sample collection date. Of the samples for which sex was recorded, no significant differences were found between groups in either of the replicate LC-MS analyses. Of the samples for which age was recorded, no significant differences were found between groups in either of the replicate LC-MS analyses. The majority of HC samples (81%) did not have age data and were thus excluded from this analysis. Several MFs produced abundance differences that were significant with respect to sample collection date (Pre 2000 vs Post 2000); however, these differences were driven mainly by the HC samples. Therefore, the differences associated with sample collection date were analyzed separately for the early Lyme disease vs HC biosignature, and the ELL vs EDL biosignature. For the early Lyme disease vs HC biosignature, 90 MFs showed evidence of differences due to sample collection date in both replicate LC-MS analyses. These 90 MFs did not include any of the MFs associated with the altered metabolic pathways described in this manuscript (Table S1). In the ELL vs EDL biosignature, no MFs showed evidence of differences due to sample collection date in either of the replicate analyses of the early Lyme disease samples.
Discussion
To develop an understanding of host biochemical alterations associated with early Lyme disease, metabolomics approaches have been applied 12–15. These studies demonstrated that the altered metabolic profiles of Lyme disease patients can be measured and used to differentiate Lyme disease from confounding diseases and from HC. However, a broad survey of human serum metabolites that are altered in response to B. burgdorferi infection during early Lyme disease has not been performed. Additionally, the potential metabolic differences that occur between the two well-defined manifestations of early Lyme disease (ELL and EDL) has not been investigated. In this current study, the metabolomics methods applied detected 2193 MFs that differed in serum samples collected from early Lyme disease patients compared with HC. The selection of these MFs was based on multiple group comparisons, as well as on biosignatures that divided the early Lyme disease patients into EDL and ELL groups. Overall, a greater number of MFs that differed in relative abundance were observed in the EDL vs HC biosignature as compared to the ELL vs HC biosignature. This, along with the observation that the abundance fold changes for MFs in EDL vs HC comparisons were generally greater than those of the ELL vs HC comparisons, was consistent with EDL being a more severe presentation of early Lyme disease. Additionally, inflammation at multiple sites in response to bacterial dissemination would be hypothesized to cause EDL patients to be more distinguishable from HC 2. A direct comparison of serum metabolites between ELL and EDL patients resulted in a smaller biosignature consisting of 671 MFs. It was not surprising that this biosignature was about 1/3 of the size of the early Lyme disease vs HC biosignature since a more subtle metabolic shift would be expected between two presentations of early Lyme disease.
2Pathway analysis enables a better understanding of the specific host metabolic states that occur during early Lyme disease. However, pathway analyses performed in our studies were based on the match of MF monoisotopic masses to metabolites annotated in databases interrogated by Mummichog. Thus, all enriched pathways and altered metabolites were based on putative structure identifications. Through the application of LC- MS/MS in combination with available databases and authentic chemical standards, we confirmed the alteration of eicosanoid, bile acid, sphingolipid, glycerophospholipid, and carnitine metabolic pathways during early Lyme disease.
Eicosanoids are diverse inflammatory mediators derived from arachidonic acid by three main processes; cyclooxygenase, lipoxygenase, and cytochrome P450 resulting in over 100 species of eicosanoids 28, 29. In this study, metabolic pathways corresponding to eicosanoid metabolism (arachidonic acid, linoleic, and alpha- linolenic) were highlighted as being altered in all of the biosignatures resulting from the comparison of early Lyme disease groups to HCs, as well as the direct comparison of the ELL and EDL patients. We confirmed the identity of five metabolites (PGB2, 12-oxo-LTB4, arachidonic acid, a HETE species, and a PUFA) associated with eicosanoid metabolism. All five were increased in Lyme disease patients with respect to HC, which is in concordance with previous reports of increased eicosanoid metabolism in Lyme arthritis 30–32. Similar to our findings, Kerstholt et al. reported arachidonic acid metabolism to be one of the top pathways perturbed during infection of cell cultures with B. burgdorferi and in serum samples from acute Lyme disease patients 14. The metabolite PGB2 is a non-enzymatic isomerization product of PGA2 that results from enzymatic dehydration of PGE2, an eicosanoid associated with proinflammatory responses 28. PGB2 has been noted as a driver of mesenchymal stem cell immunosuppression of T-cells, as well as causing pulmonary hypertension, both of which were mediated through its binding of the thromboxane A2 receptor 33, 34. The increased levels of PGB2 in early Lyme disease could reflect increased PGE2 production. Another metabolite identified (12-oxo-LTB4) is a degradation product of LTB4, an important proinflammatory lipid mediator and chemotactic factor 35. However, 12-oxo-LTB4 is associated with anti-inflammatory mechanisms 35, 36. Thus, the increased abundance of PGB2 and 12-oxo-LTB4 in early Lyme disease could result from the hosts’ attempt to dampen the inflammatory response in this disease 1, 2. Interestingly, higher levels of inflammation-initiating mediators were observed in sepsis non- survivors as compared to patients that did survive 37. Since eicosanoids can be involved in pro- and anti- inflammatory responses, the inability to directly identify the PUFA and HETE metabolite limits specific insight into the inflammatory status of early Lyme disease patients; however, our findings provide additional confirmation of increased eicosanoid metabolism during early Lyme disease.
More comprehensive and directed analyses of eicosanoids in various stages and presentations of Lyme disease, such as that used by Brown and colleagues in experimental Lyme arthritis 31, 32, are needed to obtain a complete understanding of eicosanoid metabolism involvement during Lyme disease and its progression.
Additionally, given the pleotropic outcomes of altered eicosanoid metabolism 38, an evaluation of the eicosanoid receptors and cell types that contribute to the eicosanoid response of B. burgdorferi infection should be included in future studies.
2Primary bile acids are the end products of cholesterol catabolism in the liver and can be converted to secondary bile acids by the intestinal microbiota 39. In humans, the primary bile acids consist of cholic acid, chenodeoxycholic acid and their taurine or glycine conjugates, while the secondary bile acids are based on the core structures of deoxycholic acid and lithocholic acid 39. Bile acids function as detergents in the intestinal lumen to facilitate digestion and absorption of fat and fat-soluble vitamins 40. The enterohepatic circulation, facilitated by bile acid transporters, allows for the migration of bile acids and conjugates between the liver and the intestine 39.
In cases of hepatocyte damage, bile acid synthesis is perturbed. This can include restricted bile flow (cholestasis) and the retention of compounds normally excreted in bile such as conjugated bilirubin 40. Acute hepatitis has been reported in early stages of Lyme disease 41, 42, and patients with early disseminated Lyme disease were significantly more likely to have one or more liver function abnormalities than patients with localized disease 43. The detection of increased bile acid levels in early Lyme disease and in EDL vs ELL sera is consistent with previous reports of altered hepatic function in early Lyme disease. Glycocholic acid, glycoursodeoxycholic acid, glycodeoxycholic acid, taurodeoxycholic acid, taurochenodeoxycholic acid, and glycochenodeoxycholic acid were observed as the bile acids with trends of increased abundance in EDL sera of the discovery sample set. It is noted that this trend was only replicated for glycocholic acid and glycochenodeoxycholic acid in the validation sample set; thus, a more targeted extraction and analysis method for bile acid metabolites is needed to fully understand bile acid perturbations during early Lyme disease. Borrelial invasion has been suggested to cause direct hepatocyte damage in the severe combined immunodeficiency mouse model of Lyme disease 44 and elevated liver enzymes as well as transient hepatitis have been described in humans 45, 46. During sepsis, LPS mediated inflammation leads to downregulation of hepatocellular transporters yielding liver dysfunction and dysregulated bile acid homeostasis 47. Bile acids can also actively suppress the pro-inflammatory effects of macrophages possibly as a protective mechanism against liver damage 48. It has also been demonstrated that an antigenic component of the B. burgdorferi flagella protein is shared by several human tissues, including hepatocytes 49, thus altered hepatic function driven by immune responses in early Lyme disease is also possible. Whether hepatic dysfunction associated with Lyme disease results from a direct interaction between the hepatic cells and the spirochete or its products, or whether it is an immune mediated event requires further investigation. Such investigations should consider not only the regulation of bile acid synthesis, but also the function and regulation of the bile acid transporters 39.
Fatty acid activation was a pathway enriched in the EDL vs HC and ELL/EDL vs HC biosignatures. An important downstream process of fatty acid activation is fatty acid oxidation via the transport of fatty acids into the mitochondria as acylcarnitines 27. Acylcarnitine metabolism was not a specific pathway reported via the Mummichog analyses; however, the altered abundance of five acylcarnitines can be linked to the enriched fatty acid activation pathway in the EDL vs HC and ELL/EDL vs HC biosignatures. These products were observed at reduced levels in the sera of early Lyme disease patients. This suggests impaired energy metabolism due to perturbations in transport of fatty acids into the mitochondria for β-oxidation 27. However, acylcarnitines are also known to have secondary roles as mediators of oxidative stress, apoptosis and inflammation 50–52. Lowered acylcarnitine levels are observed in patients with impaired immune reactions resulting from sepsis, HIV, chronic fatigue syndrome, and tick-borne encephalitis 23, 26, 53–55. Our findings support a previous report of lower serum levels of free carnitine, acyl carnitine and total carnitine in European patients with Lyme disease 26.
Interestingly fatigue and cognitive complaints experienced by Lyme disease patients are similar to neurological symptoms in other diseases with lowered acylcarnitine levels 25, 54. The brain is highly reliant on acylcarnitines for oxidative metabolism and neuroprotection 56. Short- and medium- chain fatty acids can permeate the blood-brain barrier and the five acylcarnitines detected in this study are short- and medium-chain acylcarnitines. Thus, more comprehensive analyses of acylcarnitines in Lyme neuroborreliosis and post-treatment Lyme disease syndrome, both of which have increased neurological components, may provide more insight.
An interesting aspect of the pathways associated with early Lyme disease was the observation that enrichment of glycerophospholipid metabolism was associated with phosphatidylcholine and related products. One of these, that had the greatest abundance difference between early Lyme disease and HCs, was lyso-PAF (C18). PAF is a proinflammatory phospholipid that plays a role in numerous conditions such as Lassa fever, HIV, ulcerative colitis, and Lyme disease 57–60. PAF levels are tightly regulated through an equilibrium between PAF and lyso-PAF, with lyso-PAF being an anabolic precursor and a catabolic product of PAF 61. In humans it has been shown that the catabolic conversion of PAF to lyso-PAF via PAF-acetylhydrolase is the primary regulator of PAF/lyso-PAF equilibrium 61, 62. If this is the case in Lyme disease, the increased levels of lyso-PAF may be attributable to increased levels in PAF production as well. However, our data did not indicate increased PAF levels in the sera of early Lyme disease patients. Lyso-PAF has been shown to have anti-inflammatory activity and increased levels of this product is a potential mechanism to dampen the proinflammatory effects of PAF 63. Our findings are consistent with a previous study that postulated a role for PAF in the pathogenesis of Lyme disease. Specifically, it was hypothesized that Borrelia induces polymorphonuclear leukocytes (PMNs) to make PAF and increased levels of PAF activates the binding of B. burgdorferi to platelets, facilitating pathogen dissemination 60. In addition to lyso-PAF, several lysoPCs, as well as arachidonic acid, were increased in early Lyme disease patients indicating a potential increase in phospholipase A2 activity 64.
2Sphingolipids are bioactive molecules known to play a role in cell signaling processes and immunological responses, and alterations in these lipids have been described for multiple infectious diseases 65–67. The formation of sphingolipids is initiated through the condensation of palmitoyl-CoA and serine, and results in the formation of ceramide which serves as the main substrate for the formation of complex sphingolipids ranging from glycosphingolipids, gangliosides and sphingomyelins 68. Sphingosine formation occurs with the hydrolysis of ceramide by ceramidase, and is a product of sphingolipid recycling 68. Elevated levels of sphingolipids are a common feature of the inflammatory response to infection 67, 69. In fact, in Lyme disease, sphingomyelin metabolism was observed to be altered in patients suffering from neuroborreliosis and was hypothesized to result from degeneration of the myelin sheaths of the central and peripheral nervous systems 70. B. burgdorferi was recently found to incorporate sphingolipids into its own membrane through the exchange of lipid rafts with host cells 71. Our data demonstrated an increase in sphingosine and sphingomyelin levels in the sera of early Lyme disease patients. However, only increased sphingosine was observed with the validation sample set. The accumulation of sphingosine in Lyme disease may indicate increased ceramide production and/or increased ceramidase activity. However, like the lipid mediators and the bile acids, the sphingolipids represent a large and complex family of lipids. Targeted analyses of the sphingolipids are required to fully understand which aspects of this metabolic pathway are altered in Lyme disease and at what stage of disease.
Three of the enriched pathways (prostaglandin formation from arachidonate, tyrosine metabolism and bile acid biosynthesis) identified via the Mummichog analyses of ELL vs EDL are also perturbed in patients with sepsis and alterations in these metabolic pathways can influence patient outcome 37, 72, 73. Further investigation of these pathways in additional disseminated Lyme disease patients is needed to understand whether factors influencing the outcome of sepsis are also involved in patients who develop disseminated Lyme disease.
Pathway enrichment defined via Mummichog analyses is based on the total number of MFs that map to a pathway, as well as the pathway size 20. While extremely useful for annotated pathways, poorly annotated pathways are less likely to be selected as an enriched pathway by Mummichog. This is the case for glutathione metabolism where γ-glutamyl amino acids are lumped together, and γ-glutamyl peptides and other glutathione conjugates are not present in the KEGG pathway 74. Thus, glutathione metabolism was not a major enriched pathway identified with our metabolomics data; however, Kerstholt et al. recently described glutathione metabolism as a dominant pathway altered during infection with B. burgdorferi 14. While we did not observe γ-glutamyl peptides nor glutathione as altered metabolites in our dataset, 5-oxo-L-proline was found to be significantly increased in the sera of early Lyme disease patients. This metabolite is generated from glutathione or γ-glutamyl amino acids via a γ-glutamylcyclotransferase 74, 75. The minimal concordance of differential abundances of glutathione metabolites in our datasets could be due to different methods, sample classification criteria, sample numbers, and sample location in comparison to the studies of Kerstholt et al. 14. Interestingly, decreased levels of γ-glutamyl dipeptides were predictors of outcome in non-alcoholic steatohepatitis (NASH) patients 76. Based on this and other similarities between the early Lyme disease biosignature and metabolites altered in NASH and non-alcoholic fatty liver disease (NAFLD) 77, 78,future comparisons of early Lyme disease patients and patients with NASH and NAFLD would be informative in understanding the effects of B. burgdorferi infection on the liver.
Limitations of this study included the relative quantification of metabolite or MF abundances based on the semi-quantitative LC-MS methodology. However, the use of replicate analyses allowed for the evaluation of robustness in defining the pathways that differed in the various group comparisons. Additionally, the use of validation samples allowed for confirmation of specific metabolites and metabolic pathways altered during early Lyme disease. The Lyme disease patient samples were obtained from a single clinical site, thus further studies that encompass the geographic distribution and clinical heterogeneity of Lyme disease will enable more detailed assessment of metabolic variations associated with this disease. In our current studies, the use of NSAIDs or other medications in the patient and control cohorts was not a data point available to us; however, serum of Lyme disease subjects was collected before the start of antibiotic therapy. Future studies need to address metabolic changes that are mediated or exacerbated by Lyme disease patients’ use of over-the-counter medications.
Conclusion
The discovery and identification of metabolites that are altered in early Lyme disease provides insight into the potential mechanisms utilized by the host in response to B. burgdorferi infection, and provide evidence that different manifestations of early Lyme disease may be associated with different metabolic alterations. Further research is needed to determine the involvement of altered metabolites in the pathogenesis of Lyme disease.
Supplementary Material
Acknowledgments
The work was supported by the U.S. NIH R33 AI-100228. We thank Kristofor Webb for support in the operation of LC-MS instrumentation.
Footnotes
Conflicts of Interest Disclosures: Dr. Wormser reports receiving research grants from Immunetics, Inc., Institute for Systems Biology, Rarecyte, Inc., and Quidel Corporation. He owns equity in Abbott/AbbVie; has been an expert witness in malpractice cases involving babesiosis and Lyme disease; and is an unpaid board member of the American Lyme Disease Foundation. Drs. Belisle, Molins and Wormser report: U.S. Patent Application, “High Sensitivity Method for Early Lyme Disease Detection” (Application No. 15/046,204); and U.S. Provisional Patent Application, “Use of Metabolic Biosignatures for Differentiation of Early Lyme Disease from Southern Tick-Associated Rash Illness (STARI)” (Application No. 62/277,252). This project was supported in part by an appointment to the Research Participation Program at the Centers for Disease Control and Prevention administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the Centers for Disease Control and Prevention.
2The mass spectrometry data is available at the NIH Common Fund’s National Metabolomics Data Repository (NMDR) website, the Metabolomics Workbench (https://www.metabolomicsworkbench.org) where it has been assigned Project ID PR000820 and Study ID ST001233. The data can be accessed directly via it’s Project DOI: 10.21228/M8PQ4V.
Supporting Information
Figure S–1, LC-MS metabolomics workflow; Figure S–2, volcano plots for replicate 2; Figure S–3, scatter plots for replicate 2; Figure S–4, scatter plots for 566 MFs organized by EDL vs HC or ELL vs HC for replicates 1 and 2; Figure S–5, pathway analyses for replicates 1 and 2 separately; Figure S–6 – S–34, LC-MS/MS confirmation for metabolites in Table 1; and Table S–1, 2298 MFs contained in the early Lyme disease vs HC and/or ELL vs EDL biosignature lists.
References
- 1.Stanek G; Wormser GP; Gray J; Strle F, Lyme borreliosis. Lancet 2012, 379 (9814), 461–473. [DOI] [PubMed] [Google Scholar]
- 2.Steere AC; Strle F; Wormser GP; Hu LDT; Branda JA; Hovius JR; Li X; Mead PS, Lyme borreliosis. Nat. Rev. Dis. Primers 2016, 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hyde JA, Borrelia burgdorferi keeps moving and carries on: a review of borrelial dissemination and invasion. Front. Immunol. 2017, 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marques AR, Lyme disease: a review. Curr. Allergy Asthma Rep. 2010, 10 (1), 13–20. [DOI] [PubMed] [Google Scholar]
- 5.Adams PP; Avile CF; Popitsch N; Bilusic I; Schroeder R; Lybecker M; Jewett MW, In vivo expression technology and 5 ‘ end mapping of the Borrelia burgdorferi transcriptome identify novel RNAs expressed during mammalian infection. Nucleic Acids Res. 2017, 45 (2), 775–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corona MS, I., Borrelia burgdorferi: carbon metabolism and the tick-mammal enzootic cycle. Microbiol. Spectrum 2015, 3 (3), MBP-0011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gautam A; Dixit S; Philipp MT; Singh SR; Morici LA; Kaushal D; Dennis VA, Interleukin-10 alters effector functions of multiple genes induced by Borrelia burgdorferi in macrophages to regulate Lyme disease inflammation. Infect. Immun. 2011, 79 (12), 4876–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oosting M; Kerstholt M; ter Horst R; Li Y; Deelen P; Smeekens S; Jaeger M; Lachmandas E; Vrijmoeth H; Lupse M; Flonta M; Cramer RA; Kullberg BJ; Kumar V; Xavier R; Wijmenga C; Netea MG; Joosten LAB, Functional and genomic architecture of Borrelia burgdorferi-induced cytokine responses in humans. Cell Host Microbe 2016, 20 (6), 822–833. [DOI] [PubMed] [Google Scholar]
- 9.Bouquet J; Soloski MJ; Swei A; Cheadle C; Federman S; Billaud JN; Rebman AW; Kabre B; Halpert R; Boorgula M; Aucott JN; Chiu CY, Longitudinal Transcriptome Analysis Reveals a Sustained Differential Gene Expression Signature in Patients Treated for Acute Lyme Disease. Mbio 2016, 7 (1), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marques A; Schwartz I; Wormser GP; Wang YM; Hornung RL; Demirkale CY; Munson PJ; Turk SP; Williams C; Lee CCR; Yang J; Petzke MM, Transcriptome assessment of erythema migrans skin lesions in patients with early Lyme disease reveals predominant interferon signaling. J. Infect. Dis. 2018, 217 (1), 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soloski MJ; Crowder LA; Lahey LJ; Wagner CA; Robinson WH; Aucott JN, Serum Inflammatory Mediators as Markers of Human Lyme Disease Activity. Plos One 2014, 9 (4), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molins CR; Ashton LV; Wormser GP; Hess AM; Delorey MJ; Mahapatra S; Schriefer ME; Belisle JT, Development of a metabolic biosignature for detection of early Lyme disease. Clin. Infect. Dis. 2015, 60 (12), 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molins CR; Ashton LV; Wormser GP; Andre BG; Hess AM; Delorey MJ; Pilgard MA; Johnson BJ; Webb K; Islam MN; Pegalajar-Jurado A; Molla I; Jewett MW; Belisle JT, Metabolic differentiation of early Lyme disease from southern tick-associated rash illness ( STARI). Sci. Transl. Med. 2017, 9 (403), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerstholt M; Vrijmoeth H; Lachmandas E; Oosting M; Lupse M; Flonta M; Dinarello CA; Netea MG; Joosten LAB, Role of glutathione metabolism in host defense against Borrelia burgdorferi infection. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (10), E2320–E2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pegalajar-Jurado A; Fitzgerald BL; Islam MN; Belisle JT; Wormser GP; Waller KS; Ashton LV; Webb KJ; Delorey MJ; Clark RJ; Molins CR, Identification of urine metabolites as biomarkers of early Lyme disease. Sci. Rep. 2018, 8, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Molins CR; Ashton LV; Wormser GP; Hess AM; Delorey MJ; Mahapatra S; Schriefer ME; Belisle JT, Development of a Metabolic Biosignature for Detection of Early Lyme Disease. Clin. Infect. Dis. 2015, 60 (12), 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wishart DS; Jewison T; Guo AC; Wilson M; Knox C; Liu Y; Djoumbou Y; Mandal R; Aziat F; Dong E; Bouatra S; Sinelnikov I; Arndt D; Xia J; Liu P; Yallou F; Bjorndahl T; Perez-Pineiro R; Eisner R; Allen F; Neveu V; Greiner R; Scalbert A, HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res 2013, 41 (Database issue), D801–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schymanski EL; Jeon J; Gulde R; Fenner K; Ruff M; Singer HP; Hollender J, Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ. Sci. Technol. 2014, 48 (4), 2097–2098. [DOI] [PubMed] [Google Scholar]
- 19.DeFelice BC; Mehta SS; Samra S; Čajka T; Wancewicz B; Fahrmann JF; Fiehn O, Mass spectral feature list optimizer (MS-FLO): a tool to minimize false positive peak reports in untargeted liquid chromatography-mass spectroscopy (LC-MS) data processing. Anal. Chem. 2017, 89 (6), 3250–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li SZ; Park Y; Duraisingham S; Strobel FH; Khan N; Soltow QA; Jones DP; Pulendran B, Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9 (7), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ritchie ME; Phipson B; Wu D; Hu Y; Law CW; Shi W; Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43 (7), e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Creer MH; Gross RW, SEPARATION OF ISOMERIC LYSOPHOSPHOLIPIDS BY REVERSE PHASE HPLC. Lipids 1985, 20 (12), 922–928. [DOI] [PubMed] [Google Scholar]
- 23.Cassol E; Misra V; Morgello S; Kirk GD; Mehta SH; Gabuzda D, Altered monoamine and acylcarnitine metabolites in HIV-positive and HIV-negative subjects with depression. J. Acquir. Immune Defic. Syndr. 2015, 69 (1), 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung KP; Chen GY; Chuang TY; Huang YT; Chang HT; Chen YF; Liu WL; Chen YJ; Hsu CL; Huang MT; Kuo CH; Yu CJ, Increased plasma acetylcarnitine in sepsis is associated with multiple organ dysfunction and mortality: a multicenter cohort study. Crit. Care Med. 2019, 47 (2), 210–218. [DOI] [PubMed] [Google Scholar]
- 25.Cristofano A; Sapere N; La Marca G; Angiolillo A; Vitale M; Corbi G; Scapagnini G; Intrieri M; Russo C; Corso G; Di Costanzo A, Serum levels of acyl-carnitines along the continuum from normal to Alzheimer’s dementia. PLoS One 2016, 11 (5), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kepka A; Pancewicz SA; Janas RM; Swierzbinska R, Serum carnitine concentration is decreased in patients with Lyme borreliosis. Postepy Hig. Med. Dosw. 2016, 70, 180–185. [DOI] [PubMed] [Google Scholar]
- 27.Reuter SE; Evans AM, Carnitine and acylcarnitines pharmacokinetic, pharmacological and clinical aspects. Clin. Pharmacokinet. 2012, 51 (9), 553–572. [DOI] [PubMed] [Google Scholar]
- 28.Buczynski MW; Dumlao DS; Dennis EA, An integrated omics analysis of eicosanoid biology. J. Lipid Res. 2009, 50 (6), 1015–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stables MJ; Gilroy DW, Old and new generation lipid mediators in acute inflammation and resolution. Prog. Lipid Res. 2011, 50 (1), 35–51. [DOI] [PubMed] [Google Scholar]
- 30.Blaho VA; Buczynski MW; Brown CR; Dennis EA, Lipidomic analysis of dynamic eicosanoid responses during the induction and resolution of Lyme arthritis. J. Biol. Chem. 2009, 284 (32), 21599–21612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown CR; Dennis EA, Borrelia burgdorferi infection induces lipid mediator production during Lyme arthritis. Biochimie 2017, 141, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luczaj W; Moniuszko A; Rusak M; Pancewicz S; Zajkowska J; Skrzydlewska E, Lipid peroxidation products as potential bioindicators of Lyme arthritis. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30 (3), 415–422. [DOI] [PubMed] [Google Scholar]
- 33.van den Berk LCJ; Jansen BJH; Snowden S; Siebers-Vermeulen KGC; Gilissen C; Kogler G; Figdor CG; Wheelock CE; Torensma R, Cord blood mesenchymal stem cells suppress DC-T cell proliferation via prostaglandin B2. Stem Cells Dev. 2014, 23 (14), 1582–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu F; Orr JA; Wu JY, Prostaglandin B2-induced pulmonary hypertension is mediated by TxA2/PGH2 receptor stimulation. Am. J. Physiol. Lung Cell. Mol. Physiol. 1994, 267 (5), L602–L608. [DOI] [PubMed] [Google Scholar]
- 35.Yokomizo T; Izumi T; Takahashi T; Kasama T; Kobayashi Y; Sato F; Taketani Y; Shimizu T, Enzymatic inactivation of leukotriene B4 by a novel enzyme found in the porcine kidney: purification and properties of leukotriene B4 12-hydroxydehydrogenase. J. Biol. Chem. 1993, 268 (24), 18128–18135. [PubMed] [Google Scholar]
- 36.Wainwright SL; Powell WS, Mechanism for the formation of dihydro metabolites of 12- hydroxyeicosanoids: conversion of leukotriene B4 and 12-hydroxy-5,8,10,14-eicosatetraenoic acid to 12-oxo intermediates J. Biol. Chem. 1991, 266 (31), 20899–20906. [PubMed] [Google Scholar]
- 37.Dalli J; Colas RA; Quintana C; Barragan-Bradford D; Hurwitz S; Levy BD; Choi AM; Serhan CN; Baron RM, Human Sepsis Eicosanoid and Proresolving Lipid Mediator Temporal Profiles: Correlations With Survival and Clinical Outcomes. Crit Care Med 2017, 45 (1), 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tam VC, Lipidomic profiling of bioactive lipids by mass spectrometry during microbial infections. Semin. Immunol. 2013, 25 (3), 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li T; Chiang JY, Bile acid signaling in liver metabolism and diseases. J. Lipids 2012, 2012, 754067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clayton PT, Disorders of bile acid synthesis. J. Inherit. Metab. Dis. 2011, 34 (3), 593–604. [DOI] [PubMed] [Google Scholar]
- 41.Chavanet P; Pillon D; Lancon JP; Waldnercombernoux A; Maringe E; Portier H, Granulomatous hepatitus associated with Lyme disease. Lancet 1987, 2 (8559), 623–624. [DOI] [PubMed] [Google Scholar]
- 42.Zaidi SA; Singer C, Gastrointestinal and hepatic manifestations of tickborne diseases in the United States. Clin. Infect. Dis. 2002, 34 (9), 1206–1212. [DOI] [PubMed] [Google Scholar]
- 43.Horowitz HW; Dworkin B; Forseter G; Nadelman RB; Connolly C; Luciano BB; Nowakowski J; Obrien TA; Calmann M; Wormser GP, Liver function in early Lyme disease. Hepatology 1996, 23 (6), 1412–1417. [DOI] [PubMed] [Google Scholar]
- 44.Schaible UE; Gay S; Museteanu C; Kramer MD; Zimmer G; Eichmann K; Museteanu U; Simon MM, Lyme borreliosis in the severe combined immunodeficiency (SCID) mouse manifests predominantly in the joints, heart, and liver. Am. J. Pathol. 1990, 137 (4), 811–820. [PMC free article] [PubMed] [Google Scholar]
- 45.Steere AC; Bartenhagen NH; Craft JE; Hutchinson GJ; Newman JH; Rahn DW; Sigal LH; Spieler PN; Stenn KS; Malawista SE, The early clinical manifestations of Lyme disease. Ann. Intern. Med. 1983, 99 (1), 76–82. [DOI] [PubMed] [Google Scholar]
- 46.Goellner MH; Agger WA; Burgess JH; Duray PH, Hepatitis due to recurrent Lyme disease. Ann. Intern. Med. 1988, 108 (5), 707–8. [DOI] [PubMed] [Google Scholar]
- 47.Kosters A; Karpen SJ, The role of inflammation in cholestasis: clinical and basic aspects. Semin Liver Dis 2010, 30 (2), 186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wammers M; Schupp AK; Bode JG; Ehlting C; Wolf S; Deenen R; Kohrer K; Haussinger D; Graf D, Reprogramming of pro-inflammatory human macrophages to an anti-inflammatory phenotype by bile acids. Sci Rep 2018, 8 (1), 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aberer E; Brunner C; Suchanek G; Klade H; Barbour A; Stanek G; Lassmann H, Molecular mimicry and Lyme borreliosis: a shared antigenic determinant between Borrelia burgdorferi and human tissue. Ann. Neurol. 1989, 26 (6), 732–737. [DOI] [PubMed] [Google Scholar]
- 50.Famularo G; de Simone C; Trinchieri V; Mosca L, Carnitines and its congeners: a metabolic pathway to the regulation of immune response and inflammation Ann. N. Y. Acad. Sci. 2004, 1033, 132–138. [DOI] [PubMed] [Google Scholar]
- 51.McCoin CS; Knotts TA; Adams SH, Acylcarnitines: old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 2015, 11 (10), 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schonfeld P; Wojtczak L, Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J. Lipid Res. 2016, 57 (6), 943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kępka A; Janas RM; Pancewicz SA; Świerzbińska R, Serum carnitine and acyl-carnitine in patients with meningitis due to tick-borne encephalitis virus infection. Adv. Clin. Exp. Med. 2017, 26 (2), 277–280. [DOI] [PubMed] [Google Scholar]
- 54.Reuter SE; Evans AM, Long-chain acylcarnitine deficiency in patients with chronic fatigue syndrome: potential involvement of altered carnitine palmitoyltransferase-I activity. J. Intern. Med. 2011, 270 (1), 76–84. [DOI] [PubMed] [Google Scholar]
- 55.Nanni G; Pittiruti M; Giovannini I; Boldrini G; Ronconi P; Castagneto M, Plasma carnitine levels and urinary carnitine excretion during sepsis. J. Parenter. Enteral Nutr. 1985, 9 (4), 483–490. [DOI] [PubMed] [Google Scholar]
- 56.Jones LL; McDonald DA; Borum PR, Acylcarnitines: role in brain. Prog. Lipid Res. 2010, 49 (1), 61–75. [DOI] [PubMed] [Google Scholar]
- 57.Gale TV; Horton TM; Grant DS; Garry RF, Metabolomics analyses identify platelet activating factors and heme breakdown products as Lassa fever biomarkers. PLoS Negl. Trop. Dis. 2017, 11 (9), 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelesidis T; Papakonstantinou V; Detopoulou P; Fragopoulou E; Chini M; Lazanas MC; Antonopoulou S, The role of platelet-activating factor in chronic inflammation, immune activation, and comorbidities associated with HIV infection. Aids Rev. 2015, 17 (4), 191–201. [PMC free article] [PubMed] [Google Scholar]
- 59.Guimbaud R; Izzo A; Martinolle JP; Vidon N; Couturier D; Benveniste J; Chaussade S, Intraluminal excretion of PAF, lysoPAF, and acetylhydrolase in patients with ulcerative colitis. Dig. Dis. Sci. 1995, 40 (12), 2635–2640. [DOI] [PubMed] [Google Scholar]
- 60.Isogai E; Kimura K; Fujii N; Nishikawa T; Ishii N; Postic D; Baranton G; Isogai H, Platelet- activating-factor-mediated pathogenesis in Lyme disease. Infect. Immun. 1996, 64 (3), 1026–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stafforini DM; McIntyre TM; Zimmerman GA; Prescott SM, Platelet-activating factor, a pleiotrophic mediator of physiological and pathological processes. Crit. Rev. Clin. Lab. Sci. 2003, 40 (6), 643–672. [DOI] [PubMed] [Google Scholar]
- 62.Hanahan DJ, Platelet activating factor: a biologically active phosphoglyceride Ann. Rev. Biochem. 1986, 55, 483–509. [DOI] [PubMed] [Google Scholar]
- 63.Welch EJ; Naikawadi RP; Li ZY; Lin P; Ishii S; Shimizu T; Tiruppathi C; Du XP; Subbaiah PV; Ye RD, Opposing effects of platelet-activating factor and lyso-platelet-activating factor on neutrophil and platelet activation. Mol. Pharmacol. 2009, 75 (1), 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dennis EA, Diversity of group types, regulation, and function of phospholipase A2. J. Biol. Chem. 1994, 269 (18), 13057–13060. [PubMed] [Google Scholar]
- 65.Martin-Acebes MA; Merino-Ramos T; Blazquez AB; Casas J; Escribano-Romero E; Sobrino F; Saiz JC, The composition of west nile virus lipid envelope unveils a role of sphingolipid metabolism in flavivirus biogenesis. J. Virol. 2014, 88 (20), 12041–12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chotiwan N; Andre BG; Sanchez-Vargas I; Islam MN; Grabowskia JM; Hopf-Jannasch A; Gough E; Nakayasu E; Blair CD; Belisle JT; Hill CA; Kuhn RJ; Perera R, Dynamic remodeling of lipids coincides with dengue virus replication in the midgut of Aedes aegypti mosquitoes. PLoS Pathog. 2018, 14 (2), 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim MJ; Wainwright HC; Locketz M; Bekker LG; Walther GB; Dittrich C; Visser A; Wang W; Hsu FF; Wiehart U; Tsenova L; Kaplan G; Russell DG, Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2010, 2 (7), 258–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hannun YA; Obeid LM, Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9 (2), 139–150. [DOI] [PubMed] [Google Scholar]
- 69.Chiricozzi E; Loberto N; Schiumarini D; Samarani M; Mancini G; Tamanini A; Lippi G; Dechecchi MC; Bassi R; Giussani P; Aureli M, Sphingolipids role in the regulation of inflammatory response: from leukocyte biology to bacterial infection. J. Leukoc. Biol. 2018, 103 (3), 445–456. [DOI] [PubMed] [Google Scholar]
- 70.Luczaj W; Domingues P; Domingues MR; Pancewicz S; Skrzydlewska E, Phospholipidomic analysis reveals changes in sphingomyelin and lysophosphatidylcholine profiles in plasma from patients with neuroborreliosis. Lipids 2017, 52 (1), 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Monco JCG; Villar BF; Rogers RC; Szczepanski A; Wheeler CM; Benach JL, Borrelia burgdorferi and other related spirochetes bind to galactocerebroside Neurology 1992, 42 (7), 1341–1348. [DOI] [PubMed] [Google Scholar]
- 72.Jenniskens M; Langouche L; Vanwijngaerden YM; Mesotten D; Van den Berghe G, Cholestatic liver (dys)function during sepsis and other critical illnesses. Intensive Care Med 2016, 42 (1), 16–27. [DOI] [PubMed] [Google Scholar]
- 73.Beck G; Brinkkoetter P; Hanusch C; Schulte J; van Ackern K; van der Woude FJ; Yard BA, Clinical review: immunomodulatory effects of dopamine in general inflammation. Crit Care 2004, 8 (6), 485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kanehisa M; Sato Y; Kawashima M; Furumichi M; Tanabe M, KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44 (D1), D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kumar A; Bachhawat AK, Pyroglutamic acid: throwing light on a lightly studied metabolite. Curr. Sci. 2012, 102 (2), 288–297. [Google Scholar]
- 76.Saoi M; Sasaki K; Sagawa H; Abe K; Kogiso T; Tokushige K; Hashimoto E; Ohashi Y; Britz- McKibbin P, High Throughput Screening of Serum γ-Glutamyl Dipeptides for Risk Assessment of Nonalcoholic Steatohepatitis with Impaired Glutathione Salvage Pathway. J. Proteome Res. 2019. [DOI] [PubMed] [Google Scholar]
- 77.Chow MD; Lee YH; Guo GL, The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mol. Aspects Med. 2017, 56, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Svegliati-Baroni G; Pierantonelli I; Torquato P; Marinelli R; Ferreri C; Chatgilialoglu C; Bartolini D; Galli F, Lipidomic biomarkers and mechanisms of lipotoxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.