

Abstract
Antisense transcription of protein-coding genes has been increasingly recognized as an important regulatory mechanism of gene expression. However, less is known about the extent and importance of antisense transcription of noncoding genes. Here, we investigate the breadth and dynamics of antisense transcription of miRNAs, a class of important noncoding RNAs. Because the antisense transcript of a miRNA is likely to form a hairpin suitable as the substrate of ADARs, which convert adenosine to inosine in double-stranded RNAs, we used A-to-I RNA editing as ultrasensitive readout for antisense transcription of the miRNAs. Through examining the unstranded targeted RNA-seq libraries covering all miRNA loci in 25 types of human tissues, we identified 7275 editing events located in 81% of the antisense strand of the miRNA loci, thus uncovering the previously unknown prevalent antisense transcription of the miRNAs. We found that antisense transcripts are tightly regulated, and a substantial fraction of miRNAs and their antisense transcripts are coexpressed. Sense miRNAs have been shown to down-regulate the coexpressed antisense transcripts, whereas the act of antisense transcription, rather than the transcripts themselves, regulates the expression of sense miRNAs. RNA editing tends to decrease the miRNA accessibility of the antisense transcripts, therefore protecting them from being degraded by the sense-mature miRNAs. Altogether, our study reveals the landscape of antisense transcription and editing of miRNAs, as well as a previously unknown reciprocal regulatory circuit of sense–antisense miRNA pairs.
One of the greatest surprises of recent transcriptome analysis is the discovery that the mammalian genome is pervasively transcribed (Kapranov et al. 2002; Carninci et al. 2005) and many of the protein-coding genes have antisense transcripts expressed in both physiological (Yelin et al. 2003; Katayama et al. 2005) and pathological contexts (Balbin et al. 2015). These antisense transcripts, which were initially considered as transcriptional noise, are later recognized as important regulators of gene expression (Faghihi and Wahlestedt 2009; Magistri et al. 2012; Castelnuovo et al. 2013; Pelechano and Steinmetz 2013; Canzio et al. 2019). A number of studies have helped to unveil the regulatory roles of antisense transcription of protein-coding genes (Hongay et al. 2006; Xu et al. 2011; Zheng et al. 2014; Murray et al. 2015; Brophy and Voigt 2016; Brown et al. 2018). Antisense transcripts, or the act of their transcription, can affect all stages of the protein-coding gene expression process, including transcriptional initiation, cotranscriptional, and post-transcriptional processes. In contrast, not as much is known about antisense transcription of noncoding genes. The presence of antisense transcripts of noncoding genes has been shown previously, despite less frequently than that of protein-coding genes (Katayama et al. 2005). However, with a few exceptions (Lee et al. 1999; Krol et al. 2015; Zong et al. 2016; Mondal et al. 2018), both the dynamics and functions of antisense transcription of noncoding genes were poorly understood.
MicroRNAs (miRNAs) are a class of short, endogenous, noncoding RNAs that play crucial roles in diverse biological processes (Bartel 2004; Li and He 2012; Olive et al. 2015). miRNA genes are transcribed as primary miRNAs (pri-miRNAs), each of which contains a local hairpin structure named precursor miRNA (pre-miRNA) where mature miRNA sequences are embedded (Kim et al. 2009). Mature miRNAs mediate post-transcriptional gene repression by guiding Argonaute (AGO) proteins to mRNA targets. Each animal miRNA potentially targets hundreds of protein-coding genes via partially complementary base pairing (Bushati and Cohen 2007; Bartel 2009). Despite the importance of miRNAs, their antisense transcription has not been systematically investigated. Stranded RNA-seq, which preserves transcript strandedness information, has been used to identify antisense transcripts. However, because of the overall weak expression of antisense transcripts (Pelechano and Steinmetz 2013), stranded RNA-seq has limited power to fully characterize the landscape of antisense transcription. Moreover, the strand specificity varies depending on the quality of the stranded RNA-seq libraries, further affecting the identification of genuine antisense transcripts. Therefore, we sought to find an alternative way to profile the miRNA antisense transcripts (miRNA-ATs) genome-wide. Pre-miRNA hairpins are known to be bound by adenosine deaminases acting on RNA (ADARs), which mediate adenosine to inosine editing (Blow et al. 2006; Nishikura 2010; Alon et al. 2012). We previously revealed that nearly all pre-miRNA hairpins are edited, and the phenomenon of miRNA editing is prevalent in all tissue types investigated in human (Li et al. 2018). We hypothesized that if the antisense strand of a miRNA is transcribed, the region that is opposite to the pre-miRNA sequence is likely to form a hairpin structure and be edited by ADARs as well. Thus, we could generate unstranded targeted RNA-seq data and use A-to-I RNA editing as readout of gene expression to survey human miRNA-ATs in an ultrasensitive and cost-effective manner.
Results
miRNA-ATs identified using RNA editing–based analysis
To test our idea, we profiled 100 unstranded targeted RNA-seq libraries generated by the miR-mmPCR-seq method, which covered all high-confidence pre-miRNA and flanking regions (Li et al. 2018). These libraries were constructed from 25 human adult and fetal tissues (Supplemental Table S1). In brief, the RNAs of these samples were reverse transcribed using random hexamer primers, amplified using a microfluidics-based multiplex PCR approach, and deeply sequenced. Both miRNAs and miRNA-ATs could be sequenced if they were expressed. RNA variants were identified as we previously described (Ramaswami et al. 2012, 2013). We required a 2% editing level cutoff, which allowed us to remove most low-frequency sequencing errors or somatic mutations. We next inferred the editing type of each site based on the strand information of the miRNAs. The T-to-C variants should be attributed to A-to-I editing from miRNA-ATs (Fig. 1A). With the 2% editing level cutoff, we found that the average A-to-G/T-to-C mismatches, indicative of A-to-I editing in both strands, compose ∼90% of all mismatches (Fig. 1B), and the false discovery rates were estimated to be ∼2.5%–5.2% (Supplemental Fig. S1A). This result suggests that the majority of the editing sites identified are real (Ramaswami et al. 2012, 2013). A substantial percentage of the variants, ranging from 24% to 34%, were inferred as T-to-C sites in different samples (Fig. 1B). In total, 7275 putative editing sites that were located in 579 miRNA-ATs were identified (Supplemental Table S2). These editing events prefer to occur in the positions opposite to the stem region of the pre-miRNAs (Fig. 1C), suggesting that the antisense sequence might form a hairpin by itself and was edited by ADARs. Consistently, structure prediction suggests that miRNA-ATs tend to form hairpin structures as pre-miRNAs (Supplemental Fig. S1B). When comparing the editing frequencies between sense–antisense pairs, we found that overall miRNAs tended to have higher editing frequencies than miRNA-ATs. In addition, miRNAs that had high-level editing tended to have a paired miRNA-AT with low-level editing, and vice versa (Supplemental Fig. S2).
Figure 1.
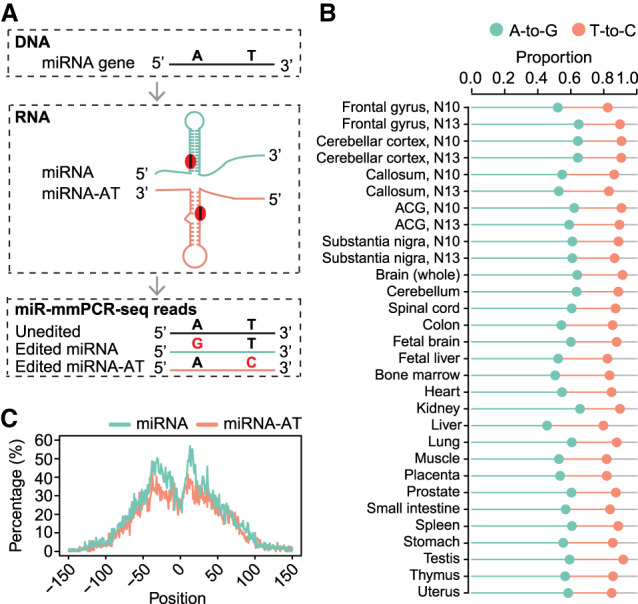
Extensive editing of miRNA-ATs. (A) Schematic of miRNA-AT identification via A-to-I RNA editing detection using miR-mmPCR-seq. (B) The percentage of A-to-G and T-to-C mismatches among all 12 mismatch types in miRNA loci in different samples. Sites with editing level ≥2% were used for analysis. Orange and green lines represent the proportions of T-to-C and A-to-G variants, respectively. (C) Metagene profiles depicting the editing site distribution surrounding pre-miRNAs and miRNA-ATs. Position 0 of a miRNA is defined as the central position of the loop region of a pre-miRNA. Position 0 of a miRNA-AT is defined as the position opposite to the central position of the loop region of a pre-miRNA. The negative value is the distance between an upstream position and the central position, and the positive value is the distance between a downstream position and the central position. Percentage of As that were edited at each position was calculated. Sites with editing level ≥2% were used for analysis.
To further confirm the T-to-C variants identified are authentic A-to-I editing in miRNA-ATs, we performed five analyses. First, we randomly selected eight miRNA-ATs, used miRNA-AT-specific primers for reverse transcription, and performed Sanger sequencing verification; 89 out of 113 (79%) selected sites were validated (with ≥10% editing level) (Supplemental Fig. S3A; Supplemental Table S3). Second, we examined the triplet preferences of the editing sites. We found that the nucleotides neighboring both miRNAs and miRNA-ATs editing sites showed a pattern consistent with known ADAR preference (Eggington et al. 2011), as shown by the underrepresentation of G base in position −1 of the edited site and overrepresentation of G base in position +1 of the edited site (Supplemental Fig. S3B). Third, we analyzed stranded mRNA-seq data of ADAR1 knockout HEK293 and HeLa cells. For both miRNAs and miRNA-ATs, of the sites that were edited in wild-type cells, most showed no RNA editing in the knockout cells (Supplemental Fig. S3C). Fourth, we examined the unstranded targeted RNA-seq libraries generated by mmPCR-seq method (Methods; Supplemental Table S1), which covered several hundred editing sites in protein-coding genes. Most of these mRNA editing sites are nonrepetitive editing sites located in the CDS regions or 3′ UTR regions and reside in nonperfectly matched dsRNA structures. Therefore, their opposite strands are unlikely to form local secondary structures, and we predict that very few editing sites would be observed in their antisense strand, even when they were expressed. As expected, most observed RNA variants in these editing loci are A-to-G type (Supplemental Fig. S3D–F). Finally, we analyzed stranded mRNA-seq data of seven cell lines from the ENCODE Project (Djebali et al. 2012) and called RNA variants in 3′ UTR Alu regions. Most Alu editing sites in 3′ UTRs are derived from inverted Alu repeats. Thus, once transcribed, both DNA strands may form dsRNA structures. We found that, for these regions without antisense transcription, only 4%–7% of the RNA variants were T-to-C variants (Supplemental Fig. S3G). This result suggests that most T-to-C variants we observed in miRNA loci should be related to antisense transcription. Altogether, these observations suggest that A-to-I RNA editing is widespread and unique for miRNA-ATs, owing to their local hairpin structures.
The characterization of miRNA-ATs
The prevalence of RNA editing in miRNA-ATs enables us to use RNA editing as readout for gene expression of miRNA-ATs, which helped to identify an average of 183 expressed miRNA-ATs per tissue (Fig. 2A). To ask whether the miRNA-ATs can be detected by other approaches, we investigated FANTOM5 data sets, which provide a human transcriptome atlas (Hon et al. 2017). The miRNA-ATs that have been assembled by the FANTOM5 Consortium were extracted (Methods), and their expression was examined using FANTOM5 CAGE data sets (Hon et al. 2017). We found that miRNA-ATs can be identified in the FANTOM5 data sets, but with an average of only 51 miRNA-ATs detected per sample (Fig. 2A; Supplemental Table S4). This result highlights the sensitivity of our approach. Next, we inferred the expression prevalence of miRNA-ATs across different tissue types, using editing as readout. The prevalence of a miRNA-AT was defined as the proportion of tissues with the editing events occurring. We found that a few miRNA-ATs were expressed in multiple tissue types, whereas most miRNA-ATs were expressed in only limited tissue types (Fig. 2B). Compared with miRNAs, miRNA-ATs displayed more tissue-specific expression (Fig. 2B). We also used FANTOM5 CAGE data sets (Supplemental Fig. S4) to assess the tissue-specific expression pattern of protein-coding genes, miRNAs and miRNA-ATs. The tissue specificity index (TSI), a widely used tissue specificity metric (Kryuchkova-Mostacci and Robinson-Rechavi 2017), was applied. TSI varies from 0 to 1, where 0 means broadly expressed, and 1 is specific. Consistent with the editing-based analysis, we found that miRNA-ATs showed slightly higher TSI values than miRNAs, and protein-coding genes showed the lowest TSI values (Fig. 2C). To further understand the expression pattern of sense–antisense miRNA pairs, we analyzed stranded mRNA-seq data of seven cell lines from the ENCODE Project (Methods; Djebali et al. 2012). We found that miRNAs and their paired miRNA-ATs can be independently expressed in a given cell type (Supplemental Fig. S5). Moreover, an average of several dozen coexpressed sense–antisense miRNA pairs were observed per sample (Supplemental Fig. S5). Because we found some coexpressed sense–antisense miRNA pairs from the ENCODE data set, to better characterize the coexpression pattern, we used editing as readout to investigate the coexpression pattern between miRNAs and their antisense transcripts. We found that on average about 116 miRNAs and miRNA-ATs were coexpressed per sample (Fig. 2D,E). Notably, because the editing levels of both miRNAs and miRNA-ATs may vary and be affected by potential competing RNA-binding proteins of ADARs, this number is a conservative estimate of coexpressed sense–antisense miRNA pairs.
Figure 2.

The characterization of miRNA-ATs. (A) Comparison of the number of miRNA-ATs detected using miR-mmPCR-seq data sets or FANTOM5 data sets. In miR-mmPCR-seq data sets, a miRNA-AT was defined as transcribed if it has at least 50 reads covered and one A-to-I editing site with editing levels ≥2%. Liver and bone marrow were excluded because fewer than 260 loci were amplified in these two tissues. In the FANTOM5 data sets, a miRNA-AT was defined as expressed if it has at least 10 reads covered in the CAGE data. The numbers of miRNA-ATs that were present in the FANTOM5 transcript atlas and expressed in each of the 1897 FANTOM5 samples were shown. (B) Prevalence of the expression of miRNA-ATs or miRNAs across different human tissue types. Prevalence of a miRNA-AT or miRNA is defined as the percentage of tissues with the miRNA-AT or miRNA expressed. A miRNA-AT or miRNA was defined as transcribed if it has at least 50 reads covered and one A-to-I editing site with editing levels ≥2%. The numbers of miRNA-ATs or miRNAs were shown in the y-axis. (C) Tissue specificity comparison between protein-coding genes, miRNAs, and miRNA-ATs using FANTOM5 data. The tissue specificity index (TSI) was used. TSI varies from 0 to 1, where 0 means broadly expressed, and 1 is specific. To avoid the confounding effect of protein-coding genes, miRNAs and miRNA-ATs that are overlapped with protein-coding genes were excluded from this analysis. Moreover, we required that genes were with counts per million (CPM) ≥1 in at least one FANTOM5 sample. (D) The number of coexpressed sense–antisense miRNA pairs inferred from coediting data in each sample. A sense–antisense miRNA pair was defined as coexpressed if both have edited sites (≥2%) detected in a given sample. The same samples as in A were used for analysis. (E) UCSC Genome Browser view of two miRNA loci. The genomic features and stranded mRNA-seq and small RNA-seq read coverages of the sense–antisense miRNA pairs were shown from top to bottom: (1) the annotation of the full-length pri-miRNA from FANTOM5, (2) the read coverage of the pri-miRNA calculated using stranded mRNA-seq, (3) the annotation of the mature miRNA, (4) the read coverage of the mature miRNA, (5) the annotation of the full-length miRNA-AT from FANTOM5, (6) the read coverage of the miRNA-AT calculated using stranded mRNA-seq. Stranded mRNA-seq of nuclear RNA and small RNA-seq of total cell RNA of GM12878 cells from the ENCODE Project were used for analysis. (F) Expression levels of mature miRNAs or putative mature miRNAs processed from miRNA-ATs. The top 200 genes were shown.
Because the secondary structure prediction suggested that miRNA-ATs tended to form hairpin structures, we asked whether they were processed into mature miRNAs. Small RNA-seq generated from the same 16 samples used for miR-mmPCR-seq experiments were analyzed. We found that in each sample only several miRNA-ATs were processed into mature miRNAs (Fig. 2F). Moreover, we compared miRNA-AT expression levels between nuclear and cytoplasmic RNAs of seven cell lines using stranded RNA-seq data generated from the ENCODE Project (Djebali et al. 2012). We found that most miRNA-ATs had similar expression levels in the two subcompartments (Supplemental Fig. S6). These results together suggest that most miRNA-ATs are not processed into mature miRNAs. This finding is consistent with the propensity of miRNA gene predictions to identify the correct strand; because of the contribution of noncanonical base pairs, particularly G:U pairs that become less favorable A:C in the antisense strand, antisense strand of most miRNAs will not fold into hairpin structures suitable for miRNA biogenesis (Lim et al. 2003).
The hypothesis of reciprocal regulation of sense–antisense miRNA pairs
The observed widespread expression of miRNA-ATs and the considerable number of coexpressed sense–antisense pairs suggest that the antisense miRNAs might interact with sense miRNAs to form an elaborate regulatory network. First, a unique characteristic of sense–antisense miRNA pair is that the antisense transcript could be directly down-regulated by the coexpressed sense miRNA via complementary base pairing; miRNAs could regulate miRNA-ATs regardless of whether miRNA-ATs function dependently or independently of their sense miRNA counterparts. Second, the act of the transcription of miRNA-ATs could regulate the expression of sense miRNAs. Third, antisense transcripts of miRNAs might regulate the expression or processing of their sense miRNAs.
To verify this network, we first analyzed the genic locations of miRNA-ATs by overlapping the genic locations of regions in the miRNA-ATs that are opposite to the pre-miRNA hairpin with known gene models. We found that these regions span various classes of genic regions (Fig. 3A; Supplemental Table S5). Of the 579 miRNA-ATs, 72% are located at the intergenic regions. Similar to miRNAs, prevalently edited miRNA-ATs prefer to be within protein-coding genes, whereas the rarely edited miRNA-ATs tend to be within ncRNAs or located at intergenic regions. For example, miRNA-215-AT, which is edited in most of the tissues we examined (Supplemental Fig. S7A), is located in the intron of a protein-coding gene (Supplemental Fig. S7B). miRNA-517b-AT, which is specifically edited in testis (Supplemental Fig. S7C), is located at an intergenic region (Supplemental Fig. S7D). Taken together, miRNA-ATs can be transcribed from intergenic DNA, ncDNA, and protein-coding gene, and may function dependently or independently of their sense miRNAs.
Figure 3.

miRNAs regulate the expression of miRNA-ATs. (A) Annotation of the regions of miRNA-ATs that are opposite to pre-miRNAs: (intron) intronic region of both protein-coding and noncoding genes; (ncRNA) noncoding RNAs; (downstream) 1 kb downstream from a gene. All edited miRNA-ATs, as well as the prevalently and rarely edited miRNA-ATs, were separately annotated: (prevalently edited) edited in >70% of the normal tissues; (rarely edited) edited in <10% of the normal tissues. (B) Dual-luciferase reporter assays to detect the effect of sense miRNAs on miRNA-ATs. Wild-type or mutant (a deletion of a 7 nt sequences complementary to miRNA seed region) miRNA-AT sequences were inserted into the Renilla luciferase 3′ UTR. Firefly luciferase was used as a reference reporter. Luciferase reporters with either wild-type or mutant miRNA-ATs were cotransfected with either miRNA or empty vector, and 48 h later the luciferase activities were measured: (relative activity) average Renilla luciferase activity normalized to firefly luciferase in four biological replicates; (error bars) SD based on four biological replicates. P-values were calculated using student's t-test: (***) P < 0.001; (NS) not significant. (C–E) Comparison of expression levels of pri-miRNAs (C), mature miRNAs (D), and miRNA-ATs (E) between DROSHA knockdown and control HeLa cells. HeLa cells were selected because DROSHA was highly expressed (https://www.proteinatlas.org/ENSG00000113360-DROSHA/cell). pri-miRNA and miRNA-AT expressions were calculated using stranded mRNA-seq data. Mature miRNA expression was calculated using small RNA-seq data. Only pri-miRNAs and miRNA-ATs with FPKM ≥1 in at least one sample were used. Only mature miRNAs with CPM ≥1 in at least one sample were used. P-values were calculated using paired Wilcoxon signed-rank test: (***) P < 0.001; (*) P < 0.05; (CPM) counts per million. (F,G) Comparison of the expression levels of sense-mature miRNAs (F) or miRNA-ATs (G) between control and DROSHA knockdown HeLa cells. Five miRNAs were not significantly down-regulated on DROSHA knockdown, which may be because these miRNAs can still be effectively processed with reduced DROSHA protein level. (Error bars) SD based on three biological replicates. P-values were calculated using student's t-test: (*) P < 0.05; (**) P < 0.01.
Sense miRNAs regulate the expression of miRNA-ATs
To answer whether sense miRNAs down-regulate their paired miRNA-ATs, we selected 17 miRNA-ATs, of which the regions that are opposite to the pre-miRNAs are located in intergenic regions, ncRNA, or 3′ UTR of protein-coding genes, for investigation. Each of the miRNA-AT fragments that are opposite to the sense pre-miRNAs and flanking sequences was then cloned into a reporter gene (Methods). Each reporter gene and its paired sense miRNA were cotransfected into HEK293T cells. We found that 15 out of the 17 reporter genes were down-regulated by the sense miRNAs (Fig. 3B). To validate if the sense miRNAs directly bind the complementary antisense sequences, we generated deletion mutants that interfere in the binding for 10 of the 17 miRNA-ATs that were strongly repressed by the sense miRNAs. As expected, the mutated reporter genes showed increased expression levels compared with that of the wild-type miRNA-ATs (Fig. 3B).
To further test if the endogenously expressed miRNAs regulate their paired miRNA-ATs, we knocked down DROSHA using siRNA and verified the knockdown efficiency by real-time PCR (RT-PCR) (Supplemental Fig. S8). We then measured the expression of pri-miRNAs, mature miRNAs, and miRNA-ATs using stranded mRNA-seq and small RNA-seq. Because only a very limited number of sense–antisense miRNA pairs were well-annotated, we examined the expression changes of pri-miRNAs, mature miRNAs, and miRNA-ATs, respectively. As expected, we found that, on DROSHA knockdown, overall pri-miRNAs showed an increased abundance and mature miRNAs showed a decreased abundance (Fig. 3C,D). Moreover, miRNA-ATs showed an overall increased abundance on DROSHA knockdown (Fig. 3E), supporting the proposed role of miRNAs in down-regulating their paired miRNA-ATs. We further confirmed our finding using RT-PCR. Eleven sense–antisense miRNA pairs with both miRNAs and miRNA-ATs expressed in HeLa cells were selected for quantification (Supplemental Fig. S9). We found that the miRNA-ATs were up-regulated with the down-regulation of their sense miRNAs (Fig. 3F,G). Moreover, we analyzed the published Ago CLASH data (Supplemental Fig. S10A; Helwak et al. 2013) and found that eight mature miRNAs bind to their paired miRNA-ATs (Supplemental Fig. S10B). Taken together, these results suggest that sense miRNAs down-regulate the coexpressed miRNA-ATs.
RNA editing modulates sense miRNA–mediated regulation of miRNA-ATs
It is known that mismatches in the miRNA seed-matching region may disrupt miRNA targeting. In addition, secondary structures of miRNA binding regions affect miRNA target recognition, because there is an energetic cost to free base-pairing interactions within mRNA to make the target accessible for miRNA binding (Kertesz et al. 2007). As we have shown that sense miRNAs target miRNA-ATs, the ample editing events, which were located in both miRNA seed-matching region and other regions (Fig. 4A), might modulate target recognition through introducing mismatches or altering secondary structures. To answer whether RNA secondary structures were different between the unedited and edited sequences, we compared the free energy (ΔG) of the predicted structures between unedited and edited forms of miRNA-ATs. We used guanosines, with closest properties to inosines, to replace inosines in prediction as previously described (Liddicoat et al. 2015). We found that the edited forms of miRNA-ATs tend to have a more stable secondary structure (Fig. 4B). Consistently, 27% of the unpaired editing site A become paired after editing, whereas only 4% of the paired A become unpaired after editing (Fig. 4C). To examine the impact of the editing on modulating sense miRNA targeting, we experimentally tested six miRNA-ATs in which the editing events altered predicted secondary structures and did not locate in the seed-matching region (Supplemental Fig. S11). These six miRNA-ATs have been proven to be targeted by the sense miRNAs (Fig. 3B), and their sense miRNAs are expressed in HEK293T cells based on small RNA-seq data. For each miRNA-AT, the unedited (one harboring the A nucleotide) or edited form (one harboring the G nucleotide) was cloned into the reporter gene. These two types of reporter genes were transfected into HEK293T cells separately. We found that, compared with the unedited reporters, five edited reporters were less reduced by the coexpressed sense miRNAs, and one was more reduced (Fig. 4D). Last, to test if RNA editing modulates miRNA-mediated regulation of miRNA-ATs in vivo, we generated ADAR1 knockout cells and measured the expression of miRNA-ATs via stranded mRNA-seq. The successful knockout of ADAR1 was confirmed by the observed loss of editing for most known mRNA editing sites (Fig. 4E). For miRNA-ATs of which the regions that are opposite to pre-miRNAs are located in the exonic regions and have high-level editing, an overall decreased expression in knockout cells was observed (Fig. 4F). Although for miRNA-ATs with low-level editing or miRNA-ATs of which the regions that are opposite to pre-miRNAs are located in the intronic regions that were not targeted by the sense miRNAs, no significant expression changes between wild-type and knockout cells was observed (Fig. 4F). Altogether, we revealed that the editing events in the miRNA-ATs may modulate miRNA targeting, and in most cases protect miRNA-ATs from being degraded by coexpressed sense miRNAs.
Figure 4.

RNA editing modulates miRNA-mediated regulation of miRNA-ATs. (A) The percentage of miRNA-AT editing sites that occur in the positions opposite to the sense miRNAs. (Outside) Outside of the region opposite to the mature miRNA. The number of editing sites is indicated above the bar. miRNA-ATs with their intronic regions targeted by the paired sense-mature miRNAs were excluded. (B) Comparison of the free energy (ΔG) between unedited and edited regions of miRNA-ATs that are opposite to the paired pre-miRNAs. For a miRNA-AT with multiple editing sites, a fully edited form was considered. miRNA-ATs with their intronic regions targeted by the paired sense-mature miRNAs were excluded. (C) The proportion of editing sites with different types of pairing patterns before and after editing. For each editing site, we predicted the secondary structure of the miRNA-ATs before and after editing and compared the pairing patterns of this site. (D) Dual-luciferase reporter assays to compare the effect of sense miRNAs on unedited and edited forms of miRNA-ATs. For each miRNA-AT, the predicted RNA secondary structures of unedited and edited versions were shown in Supplemental Figure S11. (Relative activity) Average Renilla luciferase activity normalized to firefly luciferase in four biological replicates; (error bars) SD based on four biological replicates. P-values were calculated using student's t-test. (E) Editing level comparison between control and ADAR1 knockout HEK293 cells. All known human editing sites obtained from RADAR database version 2 were used for analysis. Sites that were edited in control sample and covered by at least 30 reads in both samples were selected. P-values were calculated using paired Wilcoxon signed-rank test. (F) Comparison of miRNA-AT expression levels between control and ADAR1 knockout HEK293 cells. We required that miRNAs were expressed in HEK293 cells. P-values were calculated using the paired Wilcoxon signed-rank test. (Level ≥ 10%) The regions of miRNA-ATs that are opposite to pre-miRNAs are located in the exonic regions and have at least one site with editing level ≥ 10%; (level < 10%) control set 1, the regions of miRNA-ATs that are opposite to pre-miRNAs are located in the exonic regions and have all sites with editing level < 10%; (intronic) control set 2, the regions of miRNA-ATs that are opposite to pre-miRNAs are located in the intronic regions.
Antisense transcription regulates the expression of sense miRNAs
Finally, we investigated whether miRNA-ATs can regulate their sense miRNAs. To do so, we first classified the sense–antisense pairs into five categories, based on the orientation of pri-miRNAs and miRNA-ATs: head-to-head (HTH; 5′-regions overlap), tail-to-tail (TTT; 3′-regions overlap), embedded (EMB; antisense transcript is fully contained within the sense transcript), and included (INC; sense transcript is fully contained within the antisense transcript), and undetermined (annotation was not available for pri-miRNAs and/or miRNA-ATs) (Fig. 5A). The annotation of pri-miRNAs was obtained from previous studies (Chang et al. 2015; Bouvy-Liivrand et al. 2017; de Rie et al. 2017). The annotation of full-length miRNA-ATs was obtained from FANTOM5 data sets (Methods). Of the 579 sense–antisense pairs identified via miR-mmPCR-seq, ∼13% pairs are with known gene models and all four categories were identified (Fig. 5B).
Figure 5.

Global pattern of expression of sense–antisense miRNA pairs. (A) Schematic representation of different types of sense–antisense miRNA pairs. Depending on the positions of the transcripts involved, the pairs are classified as follows: (HTH) head-to-head (5′-regions overlap), (TTT) tail-to-tail (3′-regions overlap), (EMB) embedded (antisense transcript is fully contained within the sense transcript), and (INC) included (sense transcript is fully contained within the antisense transcript). The arrow indicates transcriptional direction. (Red) pri-miRNA; (blue) miRNA-AT; (slashed lines) pre-miRNA or the region opposite to the pre-miRNA. (B) The proportion of different types of pairs classified in A. (Undefined) no annotation was found for either pri-miRNA or miRNA-AT. (C) Distribution of Spearman's correlation coefficient between sense–antisense miRNA expression. A total of 130 sense–antisense miRNA pairs with measured expression levels in the FANTOM5 CAGE data were analyzed. Correlation between random pairs of miRNA and miRNA-AT genes is represented by a gray dashed line (n = 130).
The diverse orientations of sense–antisense pairs indicate the possible different models of miRNA-AT-mediated regulation. Previous studies suggested that antisense transcripts, or the act of transcription, may modulate gene expression levels of the protein-coding genes. To ask whether miRNA-ATs may function in a similar way, we first examined the relationship of expression between sense–antisense miRNA pairs, using FANTOM5 CAGE data. We found an excess of the positive correlation between sense–antisense pairs (Fig. 5C), which is greater than what would be expected by chance. Next, we randomly selected nine sense–antisense pairs for functional assays. CRISPR activation (CRISPRa) (Chavez et al. 2015) was used to induce the transcription of each miRNA-AT. Four gRNAs were designed to target the putative promoter regions, which were annotated based on FANTOM5 and ENCODE resources (Methods; Fig. 6A; Supplemental Fig. S12). The expression of three out of the nine miRNA-ATs were successfully induced >50-fold (Fig. 6B). We then focused on these three miRNA-ATs and compared the expression of sense-mature miRNAs between induced cells and control cells. We found that two sense-mature miRNAs (miR-135b and miR-184) had increased expression levels (>twofold) (Fig. 6C). The pri-miRNAs of these two miRNAs also had increased expression levels (Fig. 6D). The observed effects of CRISPRa are a combination of the effects of antisense transcription and antisense transcripts. To distinguish the effect of antisense transcription from that of antisense transcripts, we performed overexpression experiments. We cloned each of the two miRNA-ATs into a construct with a CMV promoter for ectopic expression (Fig. 6E) and then examined the expressions of mature miRNAs. In both cases the mature miRNAs had subtle expression changes upon strong overexpression of miRNA-ATs (Fig. 6F; Supplemental Fig. S13). Thus in these two cases, the antisense transcription promotes sense miRNA expression, and the antisense transcript had a very weak effect. Last, to further investigate the possible effect of miRNA-AT transcripts on sense miRNA expression, we randomly selected seven miRNA-ATs (Supplemental Fig. S14), cloned each of them into a construct with a CMV promoter for ectopic expression, and examined sense miRNA expression. We found that none of them had a strong effect (Fig. 6G). Taken together, our observations reveal that the act of antisense transcription, rather than the transcripts themselves, has a great effect to regulate the expression of sense miRNAs.
Figure 6.
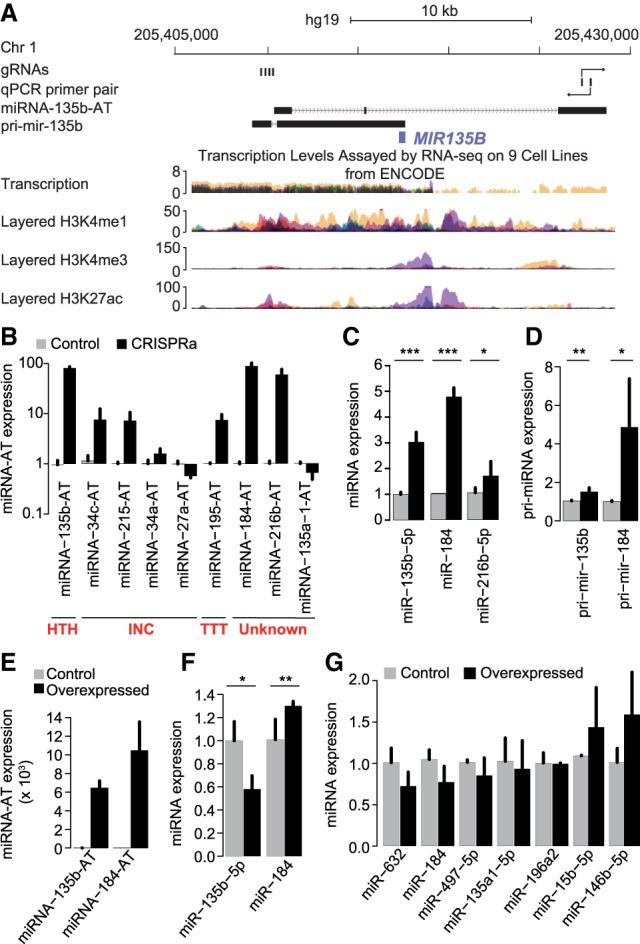
The act of antisense transcription, rather than the transcripts themselves, regulates the expression of sense miRNAs. (A) The various genomic and chromatin features of a sense–antisense miRNA pair were shown from top to bottom: (1) the positions of the four gRNAs used for miRNA-135b-AT activation; (2) the location of the primer pair used to measure miRNA-135b-AT expression; (3) the annotation of the full-length miRNA-135b-AT from FANTOM5; (4) the annotation of the pri-mir-135b transcript from previous studies; (5) transcription levels assayed by RNA-seq on nine cell lines from ENCODE; (6) three histone marks associated with regulatory elements and promoter from the ENCODE. (B–D) RNA expression of miRNA-ATs (B), sense-mature miRNAs (C), and sense pri-miRNAs (D) in HEK293T cells transfected simultaneously with four gRNAs targeting the indicated miRNA-ATs along with the dCas9-activator construct. Control cells were transfected with the indicated guide RNAs alone. (Error bars) SD based on three biological replicates. P-values were calculated using student's t-test. (E,F) RNA expression of miRNA-ATs (E) and sense-mature miRNAs (F) in HEK293T cells transfected with expression vectors harboring individual miRNA-ATs. Control cells were transfected with the empty expression vectors. (Error bars) SD based on three biological replicates. P-values were calculated using student's t-test. The expression level of miR-135b-5p in F was quantified using Qsep1 but not real-time PCR (Supplemental Fig. S13). (G) RNA expression of sense-mature miRNAs in HEK293T cells transfected with expression vectors harboring individual miRNA-ATs. Control cells were transfected with the empty expression vectors. (Error bars) SD based on three biological replicates. P-values were calculated using student's t-test, and no significant changes were observed between overexpressing and control cells for all miRNAs studied.
To reveal the potential biological relevance of miRNA-ATs, we selected three miRNA-ATs of which their paired miRNAs have been shown to be involved in cell proliferation and tumor progression. CRISPR interference (CRISPRi) was used to repress the transcription of each miRNA-AT. Of the three miRNA-ATs, the transcription of miRNA-27a-AT was successfully repressed (Supplemental Fig. S15A), and we found that miRNA-27a-AT negatively regulated the expression of its paired miRNA in HEK293T cells (Supplemental Fig. S15B). The repression of miRNA-27a-AT expression increased cell proliferation (Supplemental Fig. S15C). Consistently, inhibition of miRNA-27a by the miRNA inhibitor led to decreased cell proliferation (Supplemental Fig. S15D). These results were consistent with the known oncogenic role of miRNA-27a (Chhabra et al. 2010) and suggested that miRNA-ATs may play a role in regulating cell proliferation.
Discussion
In this study, using A-to-I RNA editing as readout of gene expression, we uncovered the prevalence and dynamics of miRNA-ATs and the coexpression pattern of sense–antisense miRNA pairs genome-wide. These data serve as a resource that will be leveraged by the community to guide focused experiments to understand the regulation of a particular sense–antisense miRNA pair. For example, the reciprocal imprinting of protein-coding genes and their antisense transcripts in imprinted clusters suggests a regulatory role for antisense RNAs in epigenetic control (Moore et al. 1997; Sleutels et al. 2002; Nagano et al. 2008). A few novel miRNA-ATs that were located in imprinted loci and regulated by their sense miRNAs were identified in this study (Supplemental Fig. S16), which will be of great interest for the imprinting community.
Based on our observations, we propose that sense–antisense miRNA pairs form an ingenious regulatory network (Fig. 7). On the one hand, miRNAs regulate the expression of coexpressed miRNA-ATs via complementary base pairing. Moreover, RNA editing of sequences opposite to the pre-miRNA regions in miRNA-ATs modulates the sense miRNA targeting. Among the 6158 editing sites of miRNA-ATs located at nonintronic regions, 893 sites have editing levels >10%, which can exert a considerable effect on the regulation of sense miRNA targeting. Notably, because the editing levels measured are from the unstranded RNA-seq and both strands may be expressed, the actual editing levels in miRNA-ATs are likely higher than we reported. On the other hand, the act of antisense transcription, rather than the transcripts themselves, regulate the sense miRNAs, probably via modulating the chromatin architectures of the resided genomic loci (Murray et al. 2015; Brown et al. 2018). Last, the observed antisense expression of miRNAs raises the possibility that, analogous to protein-coding genes, antisense transcription of noncoding RNAs could be a contributing factor for shaping the tissue-specific expression of their sense noncoding RNAs.
Figure 7.
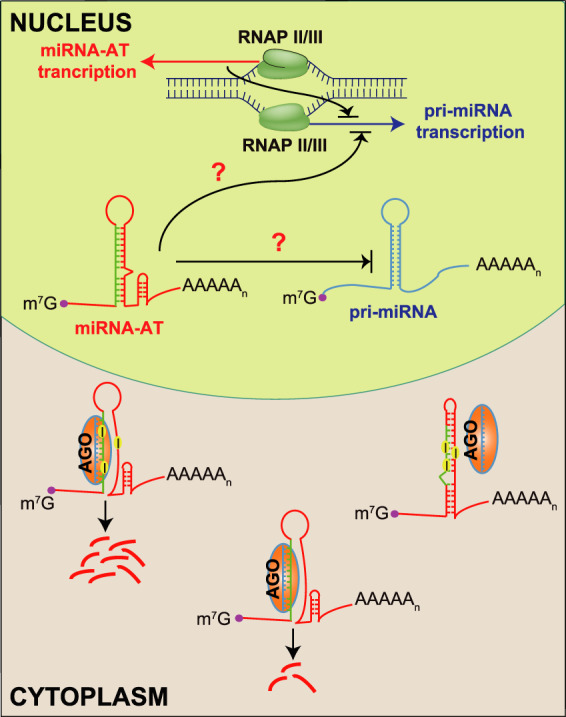
Schematic model of the regulatory network of sense–antisense miRNA pairs. In a cell, a few miRNAs and their antisense transcripts are coexpressed. miRNA-ATs can be protein-coding genes or ncRNAs that function dependently or independently of their sense miRNAs. Because of the mode of miRNA-mediated regulation, the sense-mature miRNAs may bind to the perfectly complementary sequences in the miRNA-ATs that are opposite to the pre-miRNA regions and repress their expression. RNA editing in miRNA-ATs further modulates the repression effect of sense miRNAs and in most cases, it decreases the accessibility of miRNA-ATs and protects them from being degraded by the sense-mature miRNAs. Meanwhile, the act of the transcription and/or the transcripts of miRNA-ATs may regulate the expression of the sense miRNAs. The reciprocal interactions between miRNAs and miRNA-ATs may ultimately determine the expression landscape of a substantial fraction of miRNAs and miRNA-ATs in different types of human tissues or cells.
Methods
miR-mmPCR-seq
Total RNA was extracted with TRIzol and Direct-zol RNA Kits (Zymo Research). After two rounds of DNase I treatment, 1 μg of total RNA was used to synthesize the cDNA. cDNA was purified with Axygen AxyPrep Mag PCR clean-up beads (Axygen). Each cDNA library (300–500 ng) was preamplified using a primer pool (50 μM each) covering all human miRNAs (Li et al. 2018). Preamplified product was purified using Axygen AxyPrep Mag PCR clean-up beads. Preamplified cDNAs and primer pools were loaded into the 48.48 Access Array IFC (Fluidigm) and amplified as described before (Li et al. 2018). PCR products of each sample were subject to 15 cycle barcoding PCR and pooled together. All pools were combined at equal volumes and purified via Axygen AxyPrep Mag PCR clean-up beads. All libraries were sequenced on Illumina MiSeq or NextSeq 500 to produce 150-bp paired-end reads.
mmPCR-seq
Total RNA was treated with DNase I, and 1 μg of total RNA was used to synthesize the cDNA. cDNA was purified with Axygen AxyPrep Mag PCR clean-up beads (Axygen). Next, 500 ng cDNAs and primer pools that cover 570 sites in protein-coding genes (Zhang et al. 2014) were loaded into the 48.48 Access Array IFC (Fluidigm) and amplified as described before (Zhang et al. 2014). PCR products of each sample were subject to 15 cycle barcoding PCR and pooled together. All pools were combined at equal volumes and purified via Axygen AxyPrep Mag PCR clean-up beads. All libraries were sequenced on Illumina MiSeq or NextSeq 500 to produce single-end 150-bp reads.
A-to-I RNA editing site validations by Sanger sequencing
To synthesize the cDNA, 1–5 μg of total RNA was used with Gene-specific primer with GoScript Reverse Transcriptase kit (Promega). Next, we performed PCR and conventional Sanger sequencing on a set of randomly selected editing sites. A 25-µL PCR reaction was assembled with 2× rTaq Supermix (Genstar), ∼50 ng of gDNA (or ∼1 ng cDNA) template, and 200 nM each of the forward and reverse primers. We used the following PCR program: 3 min at 94°C, 35 cycles of 15 sec at 94°C, 30 sec at 60°C, and 30 sec at 72°C. PCR amplicons were sequenced by Beijing TsingKe Biotech Co., Ltd.
Cell culture
HEK293T and HeLa cell lines were purchased from Cell Bank, Type Culture Collection, Chinese Academy of Sciences (CBTCCCAS). All cell lines were identity verified using STR analysis and checked for mycoplasma contamination by CBTCCCAS.
Cells were cultured in DMEM medium (Gibco) supplemented with 10% FBS and penicillin/streptomycin (Gibco).
DROSHA siRNA knockdown
DROSHA knockdown was performed as previously described with some modification (Gregory et al. 2004). siRNAs (AACGAGUAGGCUUCGUGACUU and AAGGACCAAGUAUUCAGCAAG) were synthesized by RiboBio. siRNA transfection was performed with Lipofectamine 3000 (Thermo Fisher Scientific). In brief, cells were plated in six-well plates to 40% confluence. For each well, 75 pmol of siRNA was mixed with 7.5 μL of Lipofectamine 3000 in 300 μL of Opti-MEM medium. The mixture was added to cells and incubated for 24 h. After 24 h, a second transfection was performed in the same way. Total RNA was prepared 48 h after the second transfection and was used for RT-PCR. The primers for the expression level quantification of DROSHA, mature miRNAs, and miRNA-ATs are listed in Supplemental Table S6. The locations of the primers for miRNA-AT quantification were shown in Supplemental Figure S9.
ADAR1 knockout cells
ADAR1 knockout HEK293 cells were generated via CRISPR-Cas9-induced mutagenesis. In brief, a sgRNA sequence was designed using CRISPR-ERA (http://CRISPR-ERA.stanford.edu). The sgRNA template oligonucleotide was synthesized and cloned into lentiCRISPR v2 plasmid (Addgene 52961). The plasmid was transfected into the cells using Lipofectamine 3000 (Thermo Fisher Scientific) following the manufacturer's instruction. Transfected cells were selected using puromycin (1 µg/mL, Sigma-Aldrich). The loss of ADAR1 was verified by cDNA Sanger sequencing and western blot.
mRNA and miRNA quantification via RT-PCR
Total RNA was extracted with TRIzol and Direct-zol RNA Kits (Zymo Research). Total RNAs used for RT-PCR were pretreated with DNase I. For DROSHA mRNA RT-PCR, 1–5 µg of total RNA was used to synthesize the cDNA with iScript Advanced cDNA Synthesis Kit (Bio-Rad). For miRNA-AT or pri-miRNA RT-PCR, 1–5 µg of total RNA was used to synthesize the cDNA with Gene-specific primer with GoScript Reverse Transcriptase kit (Promega). For miRNA RT-PCR, 1 µg of total RNA was used to synthesize the cDNA with Mir-X miRNA First Strand Synthesis kit (Takara). PCR by ChamQ SYBR qPCR Master Mix (Vazyme) for each cDNA was run in triplicates. Housekeeping gene GAPDH was used as an internal control for mRNA RT-PCR. RNU6-1 was used as an internal control for miRNA RT-PCR. Primers are listed in Supplemental Table S6.
Dual-luciferase reporter assay
The pri-miRNAs were inserted into pcDNA3.1 to generate miRNA expression vectors. The wild-type miRNA-AT fragments (a region that is opposite to the paired pre-miRNA and flanking 70-bp sequences) were amplified with Phusion High-Fidelity DNA Polymerase (NEB) using human genomic DNA and inserted into a multiple cloning region located downstream from the Renilla translational stop codon (psiCHECK2, Promega). The mutant alleles of miRNA-AT fragments were synthesized from Synbio Technologies LLC and inserted into the same position of the Renilla gene. Plasmids were transfected into HEK293T cells using Lipofectamine 3000 (Thermo Fisher Scientific). Renilla and Firefly luminescences were measured 48 h later using Dual-Glo Luciferase Assay System (Promega) on GloMax 96 Microplate Luminometer (Promega). All primers used to construct the reporter genes are listed in Supplemental Table S6.
Stranded mRNA-seq library construction
Total RNA was extracted with TRIzol and Direct-zol RNA Kits (Zymo Research). mRNA was selected using Poly(A) mRNA Magnetic Isolation Module (NEB E7490) and then fragmented into 150–200 nt fragments by incubation for 8 min at 94°C in fragmentation buffer (NEBNext Ultra II Directional RNA Library Prep Kit, NEB). The fragmented RNA was then used for library construction, following the manufacturer's protocol (NEBNext Ultra II Directional RNA Library Prep Kit, NEB). Libraries were sequenced on HiSeq X Ten (Illumina) to produce paired-end 150-bp reads. All libraries were summarized in Supplemental Table S1.
Small RNA-seq library construction
The small RNA-seq libraries were generated using VAHTSTM Small RNA Library Prep Kit for Illumina (Vazyme) according to the manufacturer's protocol. Libraries were sequenced on HiSeq X Ten (Illumina) to produce paired-end 150-bp reads. All libraries were summarized in Supplemental Table S1.
dCas9-VPR-mediated activation
gRNAs were selected to bind between 1 and 1000 bp upstream of the transcriptional start site (TSS). miRNA-AT annotations are from FANTOM5 data. gRNAs were designed using the CRISPR design tool (Chavez et al. 2015), and all gRNA sequences are listed in Supplemental Table S7. The locations of all gRNAs were shown in Supplemental Figure S12. All gRNAs were cloned into the gRNA expression vector (Addgene 52963).
Cells were plated in 12-well plates to 40% confluence, and 500 ng of dCas9 activator (Addgene 63798) and 40 ng mix of gRNAs were added to each well with Lipofectamine 3000. For each gene, four gRNAs were used against the target. Cells were grown 48 h after transfection before being lysed for RNA purification and quantification. All primers used for miRNA and miRNA-AT quantification are listed in Supplemental Table S6.
Cell proliferation assay
For CRISPRi, cells were plated in 96-well plates to 60% confluence, and 100 ng of dCas9 plasmid and 10 ng mix of gRNAs were added to each well with Lipofectamine 3000. For each gene, four gRNAs were used against the target. The transfected cells were incubated and collected at different time points.
miRNA inhibitor (hsa-miR-27a-3p inhibitor: GCGGAACUUAGCCACUGUGAA) was purchased from RiboBio. miRNA inhibitor transfection was performed with Lipofectamine 3000 (Thermo Fisher Scientific). In brief, cells were plated in 24-well plates to 60% confluence. For each well, 5 nmol of miRNA inhibitor was mixed with 1.5 μL of Lipofectamine 3000 in 50 μL of Opti-MEM medium. The mixture was added to cells and cells were incubated and collected at different time points.
For each well of cells, the medium was removed and the cells were frozen in the microplate and stored at −70°C until the samples were to be assayed. The numbers of cells were counted using CyQUANT Cell Proliferation Assay Kit (Thermo Fisher Scientific) following the manufacturer's instruction.
miRNA-AT cloning and ectopic expression
For each miRNA-AT, we cloned the miRNA-AT to the expression vector and transfected it into HEK293T cells using Lipofectamine 3000 (Thermo Fisher Scientific). To exclude the confounding effect from the possible leaky expression of the strand opposite to the miRNA-AT, a deletion mutation next to the pre-miRNA was introduced to prevent DROSHA cleavage and miRNA release for each miRNA-AT vector. Cells were grown 48 h after transfection before being lysed for RNA purification and quantification. All primers used for miRNA and miRNA-AT quantification are listed in Supplemental Table S6.
Identification of RNA editing sites from miR-mmPCR-seq, mmPCR-seq data, and ENCODE RNA-seq data
For each sample of miR-mmPCR-seq data, we merged all reads from technique replicates into a single FASTQ file and called RNA variants as we previously described (Zhang et al. 2014). In brief, we trimmed the first 20-bp primer sequences and last 10 bp of the reads using FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). We used BWA (Li and Durbin 2010) to align reads to the reference genome (BWA –n 6). We took variant positions in which the mismatch was supported by at least two mismatch reads with base quality score ≥20 and located at targeted loci. We further removed all known human genomic variants, including all SNPs present in dbSNP (except SNPs of molecular type “cDNA”; v150), the 1000 Genomes Project, and gDNA of the individuals used for the editing call. Additional filters were used to remove false positive mismatches, and separate filtering criteria were used for variants occurring in Alu and non-Alu regions as we previously described (Ramaswami et al. 2012). We excluded sites with an extreme degree of variation (>97%), which are likely genomic SNPs. We removed sites in homopolymer runs of ≥5 bp. Finally, we removed sites in regions that were highly similar to other parts of the genome using BLAT. We inferred the editing type of each site based on the strand of annotated miRNA genes.
For each sample of mmPCR-seq data, we used the same pipeline for RNA variant call with two modifications. First, we mapped the reads to a combination of the reference genome and exonic sequences surrounding known splicing junctions from available gene models. We chose the length of the splicing junction regions to be slightly shorter than the RNA-seq reads to prevent redundant hits. Gene models were obtained from the UCSC Genome Browser. Second, we inferred the editing type of each site based on the strand of annotated protein-coding genes.
To calculate the A-to-G and T-to-C proportions of RNA variants identified in the 3′ UTR Alu regions without antisense transcription, stranded mRNA-seq reads were first mapped to the human reference genome (hg19). featureCounts (Version 2.0.0) was then used to calculate the read counts of the antisense loci of each of the 3′ UTR Alu regions. The regions with read count = 0 in the antisense strand were selected, and RNA variants in these regions were then identified using the same pipeline for RNA variant call in mmPCR-seq data.
miRNA-AT loci annotation
To obtain the annotation of miRNA-AT, we searched for overlaps between the regions opposite to the pre-miRNA region and the 5′ complete transcripts that were constructed by FANTOM5 (Hon et al. 2017). This FANTOM5 5′ complete transcriptome atlas integrated transcript models from GENCODE release 19, Human BodyMap 2.0, miTranscriptome, ENCODE, and an RNA-seq assembly from 70 FANTOM5 samples. We also integrated the alternative polyadenylation information from a previous study (Wang et al. 2018) to define the 3′ end of miRNA-ATs.
Small RNA-seq data analysis
Adapter sequences, low-quality bases, and Ns were first trimmed using cutadapt (Martin 2011). Clean reads were next mapped to the human reference genome (hg19) with STAR (Version 2.5.2b) (Dobin et al. 2013). Read numbers were counted by featureCounts (Version 2.0.0) (Liao et al. 2014), and the CPM values were calculated using a custom Perl script. To visualize the read coverage, BEDTools (Quinlan and Hall 2010) genomecov was used to calculate the coverage, and the results were stored in bedGraph format. The bedGraph files were uploaded to the UCSC Genome Brower for visualization.
ENCODE RNA-seq data analysis
RNA-seq reads were mapped to the human reference genome (hg19) with STAR (Version 2.5.2b). The FPKM values were calculated by Cufflinks (Trapnell et al. 2010). The FANOM5 raw annotation GTF file was used as the gene model. To visualize the read coverage, the BEDTools genomecov was used to calculate the coverage, and the results were stored in bedGraph format. The bedGraph files were uploaded to UCSC Genome Brower for visualization.
FANTOM data acquisition
FANTOM5 data (Hon et al. 2017) were downloaded from fantom.gsc.riken.jp/5/suppl/Hon_et_al_2016/data/; transcript annotation file was from https://fantom.gsc.riken.jp/5/suppl/Hon_et_al_2016/data/assembly/lv1_raw/FANTOM_CAT.lv1_raw.gtf.gz; and gene expression profile was from https://fantom.gsc.riken.jp/5/suppl/Hon_et_al_2016/data/expression/expression_atlas/FANTOM_CAT.expression_atlas.gene.lv1_raw.count.tsv.gz.
CLASH data analysis
Raw CLASH data generated from human HEK293 cells was obtained from a previous study (Helwak et al. 2013). The analysis was performed as previously described (Helwak et al. 2013) with some modifications (Supplemental Fig. S10A). In brief, reads were first cleaned and aligned to both the transcriptome and genome. Unmapped reads were then extracted and mapped to the mature miRNA sequences using BLAST. Reads that matched a miRNA were retained (E-value <0.1; no mismatch allowed; the best hit selected; named as “miR hit”). The sequence downstream from or upstream of the miR hit part of a read was next mapped to the genome using BLAST (E-value <0.1; no mismatch allowed; the best hit selected; named as “non-miR hit”). We considered a hybrid read as an authentic sense–antisense miRNA interaction read only if (1) the miR hit and non-miR hit were mapped to a sense–antisense miRNA pair; (2) the two hits were either directly adjacent in the read, or with up to 4 nt gap or overlap between hits; (3) the non-miR hit did not match the pre-miRNA region; and (4) the miRNA loci was the best hit when the hybrid read was mapped to both the genome and the transcriptome.
RNA-seq data analysis
RNA-seq data were mapped to the human reference genome (hg19) using TopHat2 (Kim et al. 2013). Gene expression levels were calculated by Cufflinks (Cufflinks v2.2.1) (Trapnell et al. 2010). Genes with FPKM larger than 5 were used for analysis. RNA editing level was quantified using RNA-seq data as we previously described (Ramaswami et al. 2013). All known human editing sites obtained from RADAR database version 2 (Ramaswami and Li 2014) were used for analysis.
Free energy analysis
Free energy was calculated using the RNAfold program provided in the Vienna RNA package (Lorenz et al. 2011). The thermodynamics of inosine base pairing is not defined, so we modeled secondary structures by replacing I with G.
Statement for GRCh37 (hg19) used in this study
Because the miRNA information (miRBase v20) and the FANTOM5 data we used are based on GRCh37 Reference Genome (hg19), we used hg19 for all our analysis. An updated version of reference genome (GRCh38/hg38) has been available, and the main updates in hg38 are the inclusion of model centromere sequences and the addition of alternate loci. Thus our choice of hg19 would not significantly affect the conclusions.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE146947. All codes used in this study are available as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank Xin Hong and Jin Billy Li for critical reading and discussion of the manuscript. We thank Sun Yat-Sen University (SYSU) Ecology and Evolutionary Biology Sequencing Core Facility for sequencing service. This study was supported by grants from the National Natural Science Foundation of China (91631108 and 31571341 to R.Z., 31900437 to Y.S.), Guangdong Major Science and Technology Projects (2017B020226002 to R.Z.), Guangdong Innovative and Entrepreneurial Research Team Program (2016ZT06S638 to R.Z.), and the Fundamental Research Funds for the Central Universities (R.Z. and Y.S.).
Author contributions: R.Z., N.G., and W.L. conceived the project. Y.S. performed bioinformatics analysis. L.L., W.Y., Q.F., Z.F., and W.C. conducted the experiments. Y.S., L.L., N.G., W.L., and R.Z. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.257121.119.
References
- Alon S, Mor E, Vigneault F, Church GM, Locatelli F, Galeano F, Gallo A, Shomron N, Eisenberg E. 2012. Systematic identification of edited microRNAs in the human brain. Genome Res 22: 1533–1540. 10.1101/gr.131573.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbin OA, Malik R, Dhanasekaran SM, Prensner JR, Cao X, Wu YM, Robinson D, Wang R, Chen G, Beer DG, et al. 2015. The landscape of antisense gene expression in human cancers. Genome Res 25: 1068–1079. 10.1101/gr.180596.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297. 10.1016/S0092-8674(04)00045-5 [DOI] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow MJ, Grocock RJ, van Dongen S, Enright AJ, Dicks E, Futreal PA, Wooster R, Stratton MR. 2006. RNA editing of human microRNAs. Genome Biol 7: R27 10.1186/gb-2006-7-4-r27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvy-Liivrand M, Hernández de Sande A, Pölönen P, Mehtonen J, Vuorenmaa T, Niskanen H, Sinkkonen L, Kaikkonen MU, Heinaniemi M. 2017. Analysis of primary microRNA loci from nascent transcriptomes reveals regulatory domains governed by chromatin architecture. Nucleic Acids Res 45: 12054 10.1093/nar/gkx909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brophy JA, Voigt CA. 2016. Antisense transcription as a tool to tune gene expression. Mol Syst Biol 12: 854 10.15252/msb.20156540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown T, Howe FS, Murray SC, Wouters M, Lorenz P, Seward E, Rata S, Angel A, Mellor J. 2018. Antisense transcription-dependent chromatin signature modulates sense transcript dynamics. Mol Syst Biol 14: e8007 10.15252/msb.20178007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N, Cohen SM. 2007. microRNA functions. Annu Rev Cell Dev Biol 23: 175–205. 10.1146/annurev.cellbio.23.090506.123406 [DOI] [PubMed] [Google Scholar]
- Canzio D, Nwakeze CL, Horta A, Rajkumar SM, Coffey EL, Duffy EE, Duffié R, Monahan K, O'Keeffe S, Simon MD, et al. 2019. Antisense lncRNA transcription mediates DNA demethylation to drive stochastic protocadherin α promoter choice. Cell 177: 639–653.e15. 10.1016/j.cell.2019.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. 2005. The transcriptional landscape of the mammalian genome. Science 309: 1559–1563. 10.1126/science.1112014 [DOI] [PubMed] [Google Scholar]
- Castelnuovo M, Rahman S, Guffanti E, Infantino V, Stutz F, Zenklusen D. 2013. Bimodal expression of PHO84 is modulated by early termination of antisense transcription. Nat Struct Mol Biol 20: 851–858. 10.1038/nsmb.2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Pertea M, Lee S, Salzberg SL, Mendell JT. 2015. Genome-wide annotation of microRNA primary transcript structures reveals novel regulatory mechanisms. Genome Res 25: 1401–1409. 10.1101/gr.193607.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Iyer EPR, Lin S, Kiani S, Guzman CD, Wiegand DJ, et al. 2015. Highly efficient Cas9-mediated transcriptional programming. Nat Methods 12: 326–328. 10.1038/nmeth.3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra R, Dubey R, Saini N. 2010. Cooperative and individualistic functions of the microRNAs in the miR-23a∼27a∼24-2 cluster and its implication in human diseases. Mol Cancer 9: 232 10.1186/1476-4598-9-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rie D, Abugessaisa I, Alam T, Arner E, Arner P, Ashoor H, Åström G, Babina M, Bertin N, Burroughs AM, et al. 2017. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat Biotechnol 35: 872–878. 10.1038/nbt.3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108. 10.1038/nature11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington JM, Greene T, Bass BL. 2011. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2: 319 10.1038/ncomms1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghihi MA, Wahlestedt C. 2009. Regulatory roles of natural antisense transcripts. Nat Rev Mol Cell Biol 10: 637–643. 10.1038/nrm2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. 2004. The Microprocessor complex mediates the genesis of microRNAs. Nature 432: 235–240. 10.1038/nature03120 [DOI] [PubMed] [Google Scholar]
- Helwak A, Kudla G, Dudnakova T, Tollervey D. 2013. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153: 654–665. 10.1016/j.cell.2013.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J, et al. 2017. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543: 199–204. 10.1038/nature21374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongay CF, Grisafi PL, Galitski T, Fink GR. 2006. Antisense transcription controls cell fate in Saccharomyces cerevisiae. Cell 127: 735–745. 10.1016/j.cell.2006.09.038 [DOI] [PubMed] [Google Scholar]
- Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SP, Gingeras TR. 2002. Large-scale transcriptional activity in chromosomes 21 and 22. Science 296: 916–919. 10.1126/science.1068597 [DOI] [PubMed] [Google Scholar]
- Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, et al. 2005. Antisense transcription in the mammalian transcriptome. Science 309: 1564–1566. 10.1126/science.1112009 [DOI] [PubMed] [Google Scholar]
- Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. 2007. The role of site accessibility in microRNA target recognition. Nat Genet 39: 1278–1284. 10.1038/ng2135 [DOI] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. 2009. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10: 126–139. 10.1038/nrm2632 [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Krol I, Alvarez CP, Fiscella M, Hierlemann A, Roska B, Filipowicz W. 2015. A network comprising short and long noncoding RNAs and RNA helicase controls mouse retina architecture. Nat Commun 6: 7305 10.1038/ncomms8305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryuchkova-Mostacci N, Robinson-Rechavi M. 2017. A benchmark of gene expression tissue-specificity metrics. Brief Bioinform 18: 205–214. 10.1093/bib/bbw008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Davidow LS, Warshawsky D. 1999. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet 21: 400–404. 10.1038/7734 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26: 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MA, He L. 2012. microRNAs as novel regulators of stem cell pluripotency and somatic cell reprogramming. Bioessays 34: 670–680. 10.1002/bies.201200019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Song Y, Shi X, Liu J, Xiong S, Chen W, Fu Q, Huang Z, Gu N, Zhang R. 2018. The landscape of miRNA editing in animals and its impact on miRNA biogenesis and targeting. Genome Res 28: 132–143. 10.1101/gr.224386.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120. 10.1126/science.aac7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, Rhoades MW, Burge CB, Bartel DP. 2003. The microRNAs of Caenorhabditis elegans. Genes Dev 17: 991–1008. 10.1101/gad.1074403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26 10.1186/1748-7188-6-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistri M, Faghihi MA, St Laurent G III, Wahlestedt C. 2012. Regulation of chromatin structure by long noncoding RNAs: focus on natural antisense transcripts. Trends Genet 28: 389–396. 10.1016/j.tig.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17: 10–12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Mondal T, Juvvuna PK, Kirkeby A, Mitra S, Kosalai ST, Traxler L, Hertwig F, Wernig-Zorc S, Miranda C, Deland L, et al. 2018. Sense-antisense lncRNA pair encoded by locus 6p22.3 determines neuroblastoma susceptibility via the USP36-CHD7-SOX9 regulatory axis. Cancer Cell 33: 417–434.e7. 10.1016/j.ccell.2018.01.020 [DOI] [PubMed] [Google Scholar]
- Moore T, Constancia M, Zubair M, Bailleul B, Feil R, Sasaki H, Reik W. 1997. Multiple imprinted sense and antisense transcripts, differential methylation and tandem repeats in a putative imprinting control region upstream of mouse Igf2. Proc Natl Acad Sci 94: 12509–12514. 10.1073/pnas.94.23.12509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray SC, Haenni S, Howe FS, Fischl H, Chocian K, Nair A, Mellor J. 2015. Sense and antisense transcription are associated with distinct chromatin architectures across genes. Nucleic Acids Res 43: 7823–7837. 10.1093/nar/gkv666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P. 2008. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322: 1717–1720. 10.1126/science.1163802 [DOI] [PubMed] [Google Scholar]
- Nishikura K. 2010. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79: 321–349. 10.1146/annurev-biochem-060208-105251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive V, Minella AC, He L. 2015. Outside the coding genome, mammalian microRNAs confer structural and functional complexity. Sci Signal 8: re2 10.1126/scisignal.2005813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Steinmetz LM. 2013. Gene regulation by antisense transcription. Nat Rev Genet 14: 880–893. 10.1038/nrg3594 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Li JB. 2014. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res 42: D109–D113. 10.1093/nar/gkt996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Lin W, Piskol R, Tan MH, Davis C, Li JB. 2012. Accurate identification of human Alu and non-Alu RNA editing sites. Nat Methods 9: 579–581. 10.1038/nmeth.1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB. 2013. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10: 128–132. 10.1038/nmeth.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleutels F, Zwart R, Barlow DP. 2002. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 415: 810–813. 10.1038/415810a [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zheng D, Yehia G, Tian B. 2018. A compendium of conserved cleavage and polyadenylation events in mammalian genes. Genome Res 28: 1427–1441. 10.1101/gr.237826.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Clauder-Münster S, Smolik M, Huber W, Steinmetz LM. 2011. Antisense expression increases gene expression variability and locus interdependency. Mol Syst Biol 7: 468 10.1038/msb.2011.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A, Diber A, Biton S, Tamir Y, Khosravi R, et al. 2003. Widespread occurrence of antisense transcription in the human genome. Nat Biotechnol 21: 379–386. 10.1038/nbt808 [DOI] [PubMed] [Google Scholar]
- Zhang R, Li X, Ramaswami G, Smith KS, Turecki G, Montgomery SB, Li JB. 2014. Quantifying RNA allelic ratios by microfluidic multiplex PCR and sequencing. Nat Meth 11: 51–54. 10.1038/nmeth.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Valakh V, Diantonio A, Ben-Shahar Y. 2014. Natural antisense transcripts regulate the neuronal stress response and excitability. eLife 3: e01849 10.7554/eLife.01849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong X, Nakagawa S, Freier SM, Fei J, Ha T, Prasanth SG, Prasanth KV. 2016. Natural antisense RNA promotes 3′ end processing and maturation of MALAT1 lncRNA. Nucleic Acids Res 44: 2898–2908. 10.1093/nar/gkw047 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.