

Abstract
Small cell lung cancer patient outcomes have not yet been significantly impacted by the revolution in precision oncology, primarily due to a paucity of genetic alterations in actionable driver oncogenes. Nevertheless, systemic therapies that include immunotherapy are beginning to show promise in the clinic. While these results are encouraging, many patients do not respond to or rapidly recur after current regimens, necessitating alternative or complementary therapeutic strategies. In this review, we discuss ongoing investigations into the pathobiology of this recalcitrant cancer and the therapeutic vulnerabilities that are exposed by the disease state. Included within this discussion is a snapshot of the current biomarker and clinical trial landscapes for small cell lung cancer. Finally, we identify key knowledge gaps that should be addressed in order to advance the field in pursuit of reduced small cell lung cancer mortality. This review largely summarizes work presented at the Third Biennial IASLC Small Cell Lung Cancer Meeting.
INTRODUCTION
Small cell lung cancer (SCLC) accounts for ~13% of all new lung cancer diagnoses [1]. SCLC exhibits many of Hanahan and Weinberg’s hallmarks of cancer to an exaggerated degree, including propensity for early metastasis, rapid cell division, high levels of replication stress, the ability to cope with certain oxidative and metabolic stresses, and evasion of apoptosis and the effector cells of the immune system [2, 3]. Together, these factors contribute to an exceedingly poor prognosis with patient survival measured in months, not years, that has led to a recalcitrant cancer designation for SCLC by the National Cancer Institute (NCI).
A meeting summary of the 2017 IASLC SCLC Workshop posed the question, “Can recent advances in our understanding of tumor biology be translated into improved outcomes?” [4]. This question has been answered affirmatively by the recent addition of immune checkpoint blockade to first-line chemotherapy in extensive stage SCLC, which constituted the first significant improvement to systemic therapy in several decades [5]. However, the magnitude of the treatment effect, while encouraging, was modest and highlights a clear need to improve the effectiveness of immunotherapy in SCLC.
While the addition of immunotherapy to the treatment of SCLC is rapidly reaching a relatively mature stage, the exploration of underlying disease mechanisms and the development of candidate predictive biomarkers remains in its comparative infancy. It is increasingly appreciated that there are discrete molecular subtypes of SCLC that can differ in their response to different therapies in preclinical models of the disease, providing a rich and untapped vein to mine for new therapeutic liabilities (reviewed in Rudin & Poirier et al. [6]). In light of the limited durability of benefit from current therapies, it is of critical need to continue to explore new biomarker-directed therapeutic strategies and treatment combinations in the laboratory and in the clinic (Figure 1). Further exploration of the processes that drive different molecular subtypes of SCLC and the therapeutic liabilities induced by these states is warranted.
Figure 1: Some of the many areas of current therapeutic interest in small cell lung cancer.
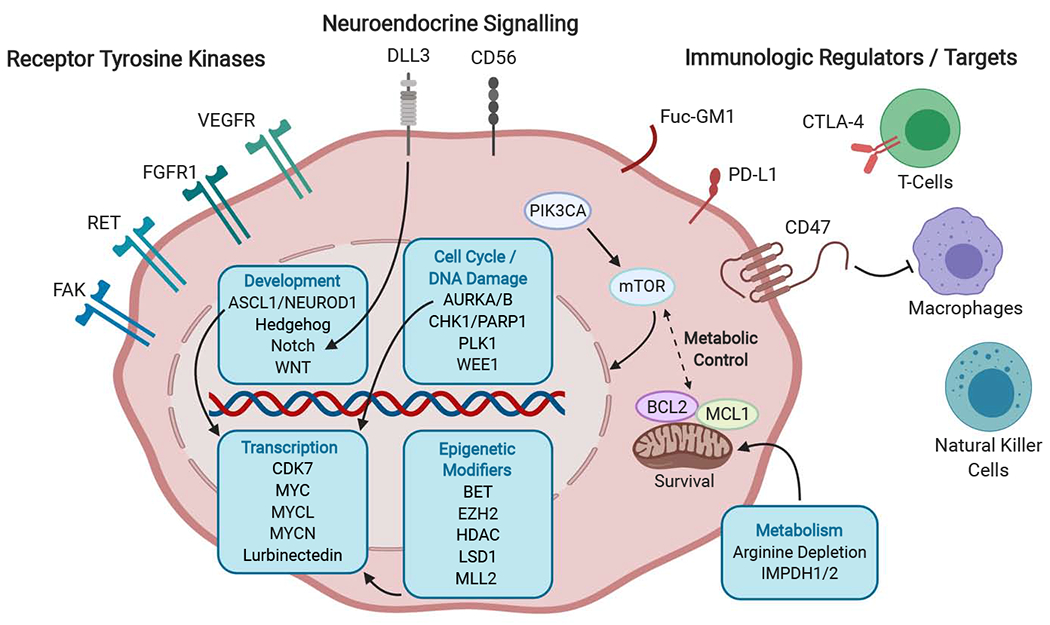
Cell surface targets include a number of receptor tyrosine kinases implicated in proliferative signaling, invasion, and angiogenesis; factors regulating neuroendocrine differentiation that are being explored as targets for antibody drug conjugates; immunologic regulators; and targets for tumor-specific vaccine strategies. Intracellular pathways of particular interest include metabolic and apoptotic regulators, cell cycle and DNA damage checkpoint controls, developmental signaling pathways, transcriptional regulators including the MYC family of transcriptional factors, and epigenetic modifiers of histones that affect chromosomal accessibility and gene expression. FAK, focal adhesion kinase; RET, ret proto-oncogene; FGFR1, fibroblast growth factor receptor 1; VEGFR, vascular endothelial growth factor receptor; DLL3, delta-like 3 (Drosophila); CD56, neural cell adhesion molecule 1; Fuc-GM1, fucosyl-monosialotetrahexosylganglioside; PD-L1, programmed death ligand-1; CTLA4, cytotoxic T-lymphocyte associated protein 4; MHC 1, Major Histocompatibility Complex 1; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; mTOR, mammalian target of rapamycin; BCL2, B-cell lymphoma 2; MCL1, MCL1 apoptosis regulator, BCL2 family member ; ASCL1, achaete-scute family bHLH transcription factor 1; NEUROD1, neuronal differntiation 1; WNT, wingless-type MMTV integration site family member; AURKA/B, Aurora kinase A/B; PLK1, Polo-like Kinase 1; WEE1, WEE1 G2 checkpoint kinase; CHK1, checkpoint kinase 1; PARP1, poly-ADP ribose polymerase 1; CDK7, Cyclin Dependent Kinase 7; MYCL, MYCL proto-oncogene, BHLH transcription factor; MYCN, MYCN proto-oncogene, BHLH transcription factor; MYC, MYC proto-oncogene, BHLH transcription factor; EZH2, enhancer of zeste 2 polycomb repressive complex 2 subunit; LSD1, lysine (K)-specific demethylase 1A; MLL2, myeloid/lymphoid or mixed-lineage leukemia 2; HDAC, Histone Deacetylase; BET, bromodomain and extra-terminal domain; IMPDH1/2, inosine monophosphate dehydrogenase 1/2
In this review, we highlight recent advances in SCLC research from the literature as well as unpublished data from a variety of researchers in the field as presented at the 2019 IASLC SCLC Workshop.
DIAGNOSTIC AND MOLECULAR PATHOLOGY
The World Health Organization classification schema for pulmonary neuroendocrine tumors was updated in 2015 and includes three primary histologic classes: small cell lung carcinoma (SCLC), large cell neuroendocrine carcinoma (LCNEC), and pulmonary carcinoid tumors [7]. Histologically, SCLC appears as a “small round blue cell” tumor under hematoxylin and eosin staining (Figure 2A) and is characterized by high proliferative index as assessed by Ki67 immunohistochemistry (Figure 2B). Carcinoid tumors have a much lower proliferative index. Additionally, histologic subcategories within SCLC and LCNEC include combined SCLC and combined LCNEC, which refer to mixed histology tumors containing other non-small cell components (Figure 2C,D). Within SCLC, combined SCLC represents a substantial fraction of cases: in one series, 16% of SCLC tumors had combined large cell carcinoma components, 9% had combined adenocarcinoma, and 3% had combined squamous cell carcinoma [8].
Figure 2: Small cell carcinoma.
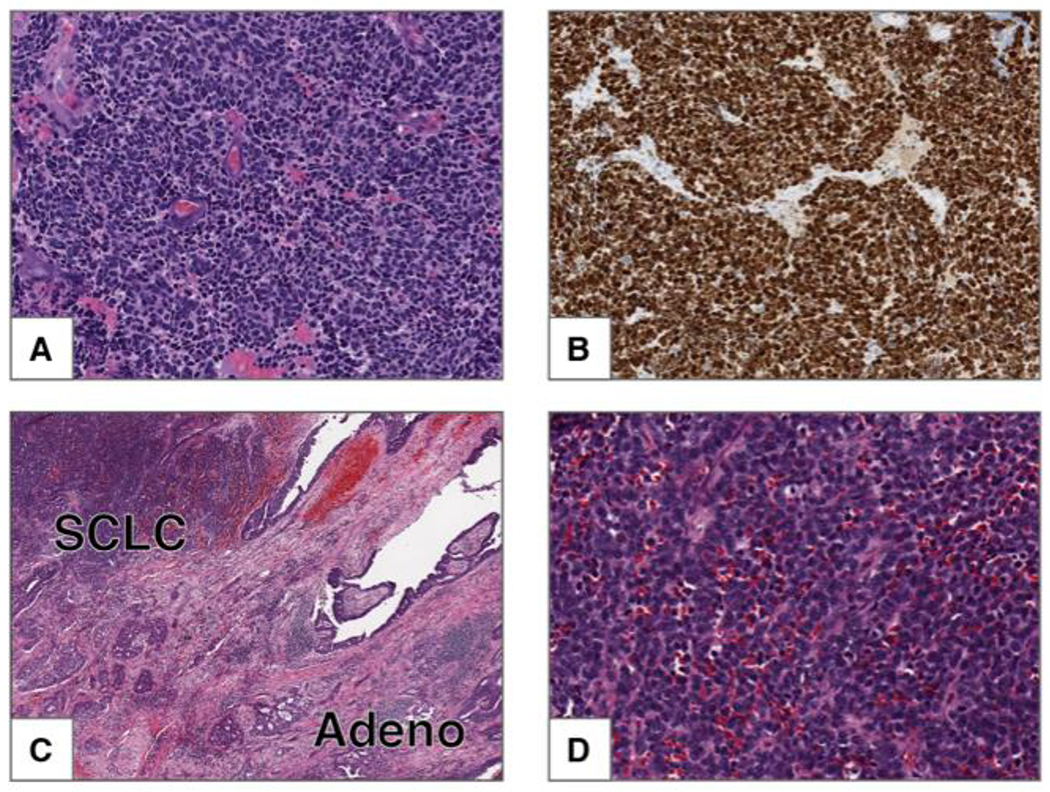
A) This tumor is composed of small cells with scant cytoplasm, finely granular chromatin and frequent mitoses. Nucleoli are absent. B) Ki-67 shows strong nuclear staining in 100% of the tumor cells. Combined small cell carcinoma and adenocarcinoma. C) Low power image of tumor composed of two components: small cell carcinoma (upper left) and adenocarcinoma with acinar pattern (lower right). D) High power image of the SCLC component from C.
The histological appearance of SCLC can resemble other tumor types, notably carcinoid with crush artifact, Ewing sarcoma, desmoplastic round cell tumor, Merkel cell carcinoma, SMARCA4- or SMARCB1-deficient cancers, or basaloid squamous cell carcinoma. Differentiation from LCNEC is based primarily on tumor cell size and nuclear/cytoplasmic characteristics, SCLC being comparatively smaller in diameter and with a greater nuclear/cytoplasmic (N/C) ratio. Other distinguishing features of SCLC include finely granular uniform nuclear chromatin, absent or inconspicuous nucleoli, nuclear molding and a fusiform shape. High mitotic counts are an important differentiator of carcinoids and SCLC; however, in the setting of metastatic pulmonary carcinoids, recent data reveal that recurrent and metastatic carcinoids can shift toward more aggressive growth with a higher proliferation rate [9].
Small cell lung cancer is known for its highly metastatic nature. One newly investigated model of invasive spread that has been proposed as a putative prognostic indicator in SCLC is “Spread through air spaces” or STAS. STAS has been shown to be associated with poor outcome in non-small cell lung cancers (NSCLCs) [10, 11]. Recent data indicate that STAS is also prognostic in lung neuroendocrine tumors including SCLC, LCNEC, and atypical carcinoid [12].
Common genetic lesions in SCLC are simultaneous pathognomonic inactivation of the tumor suppressor genes TP53 and RB1, MYC family copy number gain, and inactivating mutations in epigenetic readers and writers as well as NOTCH family members [13–15]. Although current clinical pathology guidelines consider all pure SCLC as a single disease entity, recent gene expression profiling of both human SCLC patient tumors and cell lines, and representative murine models, suggests that biologically discrete subtypes of disease exist that can be distinguished based on relative expression of three or four key transcriptional regulators: ASCL1, NEUROD1, POU2F3, and YAP1 (Figure 3). ASCL1 and NEUROD1 are critical factors in normal neuroendocrine development [16, 17]. Concomitant disruption of Ascl1, Trp53, Rb1 and Rb1 family member Rbl2 in a genetically engineered mouse model (GEMM) of SCLC suggests that Ascl1 is required for SCLC tumorigenesis in this model, whereas Neurod1 is dispensable [18, 19]. In human cancers, ASCL1 and NEUROD1 bind to distinct super-enhancer loci in ASCL1-high (SCLC-A) and NEUROD1-high (SCLC-N) tumors [18, 20] respectively, and appear to drive differential gene expression. While ASCL1 and NEUROD1 define distinct molecular subtypes of disease, some tumors express both factors to different extents, and the lineage relationship between these subsets has not been fully defined. In contrast, POU2F3-positive SCLC (SCLC-P) appears to have a distinct transcriptional signature from other subsets [21]. POU2F3 biology and its role in this subtype of SCLC is discussed in detail in another section of this review. The YAP1-high subtype (SCLC-Y) has been the least extensively characterized in terms of determinants of differential gene expression; however, this subtype appears to be enriched in human cell lines with detectable RB1 protein by western blot [22]. It is unclear if YAP1 itself is a driver of this phenotype, or a marker thereof. The pathological distinctions between these transcriptionally-defined subtypes, in terms of natural history of disease and therapeutic outcome, have not been fully investigated and represent substantial unmet needs.
Figure 3. Diagram of the relative abundance, MYC status, and NE character of the four molecular subtypes of SCLC, each identified by their key transcriptional regulator.
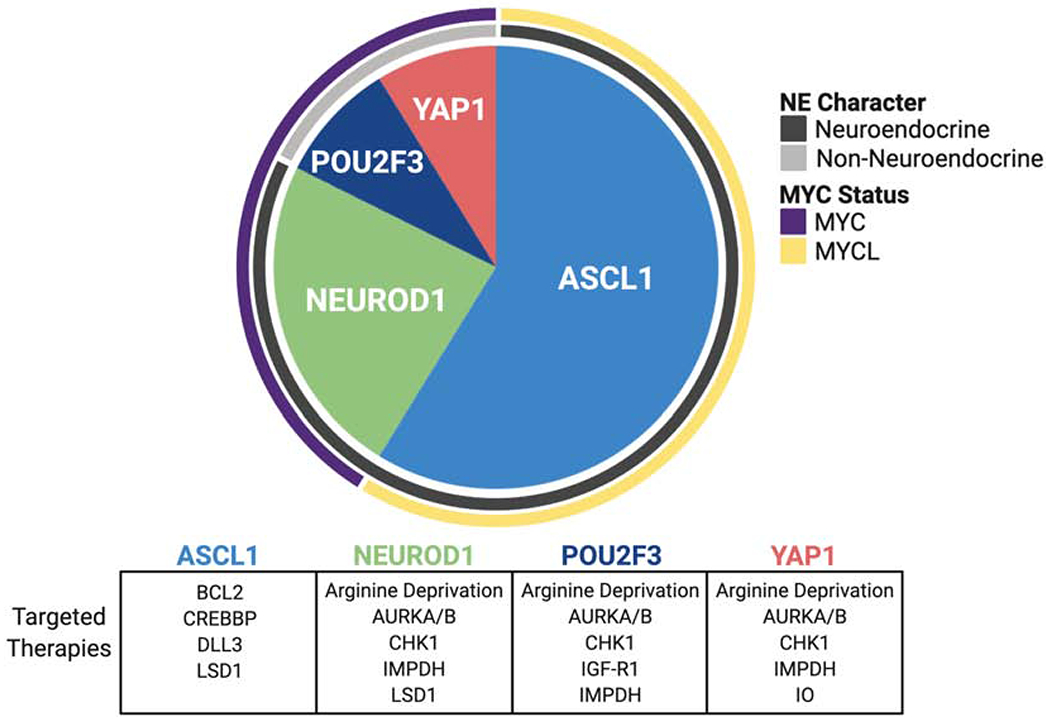
These subtypes may exhibit distinct targetable vulnerabilities, which are represented in the table beneath the pie chart. Proportions of each subtype are as follows: ASCL1 (0.70 95% CI [0.60, 0.79]), NEUROD1 (0.11 95% CI [0.06, 0.20]), YAP1 (0.02 95% CI [0.01, 0.09]), POU2F3 (0.16 95% CI [0.10, 0.26]). ASCL1, achaete-scute homologue 1; AURKA/B, Aurora kinase A/B; BCL2, B-cell lymphoma 2; CREBBP, CREB binding protein; CHK1, checkpoint kinase 1; DLL3, delta-like ligand 3; IMPDH, inosine-5’ monophosphate dehydrogenase; IGF-R1, insulin-like growth factor 1 receptor; IO, immuno-oncology; LSD1, lysine-specific histone demethylase 1; NE, neuroendocrine; NEUROD1, neurogenic differentiation factor 1; POU2F3, POU class 2 homeobox 3; YAP1, yes-associated protein 1.
CELL OF ORIGIN, TUMOR INITIATION, AND LINEAGE-RELATED PATHWAYS
Mouse model studies have suggested that a predominant cell of origin for SCLC is the neuroendocrine (NE) cell [23, 24], which according to our current understanding is also the proposed cell of origin of pulmonary carcinoids and LCNECs. However, interpretation of these studies is complicated by technical issues related to inhalation of viruses using cell type-specific promoters. In order to further characterize the function of NE cells in the normal airway epithelium, lineage tracing approaches have been employed in animal models to track the fate of NE cells following airway injury [25]. This work showed that only a subset of the NE cell population proliferates following lung injury. Interestingly, this subset of cells appears to be the same population that proliferates after sequential injuries, suggesting that these cells possess a unique capacity for self-renewal. This rare population of NE cells has been termed NEstem and is characterized by Notch2 expression. Consistent with the loss of RB1 and TP53 in human tumors, loss of both tumor suppressors in the NE population in mice led to constitutive activation of the self-renewing NE cell population. In accordance with frequent loss of function alterations in the NOTCH pathway in human SCLC, pharmacological Notch inhibition in the mouse blocked NE cell reprogramming and clonal expansion [25]. Loss of function of RB1, TP53 and NOTCH presumably lock NE progenitors into a self-renewal program and thereby contribute to transformation.
While NE cells have been implicated as a major cell of origin, recent studies from animal models suggest that they may not be the only cells eligible for transformation to SCLC. A recent study on the molecular profiles of NFIB-amplified mouse tumors proposed the existence of alternative, as yet unidentified, cell(s) of origin that impact metastatic trajectories [26]. Another recent study on human SCLC cell lines discovered a subset of SCLCs lacking expression of neuroendocrine markers and harboring a tuft cell signature [21]. Tuft cells, also known as brush cells, are marked by expression of Pou2f3, Trmp5 and Ascl2. These cells have been identified in the lung by single cell transcriptome sequencing [27, 28]. Using a transcription factor-focused CRISPR knockout screen, it was shown that SCLC cell lines expressing tuft cell markers are exquisitely dependent on the transcription factor POU2F3, a lineage specific marker and master regulator of tuft cell fate. Chromatin profiling of human POU2F3-positive SCLC cell lines shows that POU2F3 is responsible for maintaining a tuft cell enhancer landscape. This indicates that the tuft cell may be a potential cell of origin for SCLC, or alternatively, a different cell of origin may have the capacity to transdifferentiate to a state resembling the tuft cell. Established SCLC cell lines with a tuft cell program are particularly vulnerable to IGF1R inhibitors [21], highlighting the possibility of subtype-specific therapies for SCLC. A randomized phase 2 study of cisplatin and etoposide alone or in combination with either vismodigib, a Hedgehog inhibitor, or cixutumumab, an IGF1R-directed monoclonal antibody, was negative [29]. However, this trial was conducted without biomarker selection and the number of patients with POU2F3-positive tumors in each arm would likely be few.
As molecular subtypes of SCLC are being increasingly appreciated, there is a need to define the underlying epigenetic states of each subtype. Chromatin immunoprecipitation (ChIP) for acetylated H3K27 to examine the SCLC subtype-specific super-enhancer landscape revealed distinct super-enhancer profiles between human SCLC-A, SCLC-N and SCLC-P [18, 20] (J.E.J. et al., and C.L.C., et al, unpublished data). Further examination of the SCLC-A subtype by IP-mass spectrometry approaches identified three putative ASCL1-interacting partners, including two transcription factors: NKX2-1 and PROX1, and a nuclear import protein, KPNB1. Strong overlap was found between the genomic targets of ASCL1, NKX2-1, and PROX1, delineating a putative transcription factor network. Further studies of KPNB1 in human SCLC cells showed that inhibition of KPNB1 led to decreased viability and colony formation of ASCL1+ cells (J.E.J. et al., unpublished data). These findings suggest that the potential exists for developing subtype-specific therapies for SCLC. Other unbiased approaches are being used to discriminate SCLC subtypes. A recent study investigated SCLC subtypes from a gene regulatory network perspective seeking to account for genetic, epigenetic, and intrinsic stochastic noise within gene expression data [30]. Theoretical modeling with Boolean logic predicted four subtypes amongst SCLC (named NE, NE-v1, NE-v2 and Non-NE) with a high degree of overlap with other proposed subtypes [6]. For example, NE resembles the SCLC-A, NE-v1 resembles the SCLC-N, and the Non-NE resembles the SCLC-Y subtypes. Interestingly, the NE-v2 subtype appeared distinct from the NE cluster, although both express characteristic markers of SCLC-A, suggesting further heterogeneity among the SCLC-A cells with predicted differences in morphology, gene expression and drug response. The NE-v2 state was present in classic (Rb1/Trp53/Rbl2, RPR2) GEMMs, but largely absent from variant (Rb1/Trp53/Myc, RPM) GEMMs, consistent with higher expression of ASCL1 and other canonical neuroendocrine genes in the RPR2 model. Variant MYC-driven GEMMs, in contrast, were enriched for NE-v1 and Non-NE phenotypes. Importantly, these subtypes are postulated to be dynamic states of transition, likely with different thresholds for change [30]. Indeed, in silico predictions of master regulators and destabilizes of these states highlight potential mechanisms of tumor plasticity that will require functional studies to validate.
TUMOR PROGRESSION, INTRATUMORAL HETEROGENEITY, AND METASTASIS
New advances in CRISPR-Cas9 genome engineering technology and mouse genomics have provided a platform with which to functionally interrogate driver genes and provide insight into the mechanisms of SCLC tumor progression. SCLC frequently harbors genomic alterations in genes encoding the histone acetyl transferases CREBBP and EP300 with a significant enrichment of hotspot mutations in the histone acetyltransferase domain. Early functional studies in cell lines pointed to a tumor suppressive function of CREBBP and EP300 in SCLC [14]. Recent CRISPR-based approaches to alter genes in early RPR2 mouse tumor cells further demonstrated a tumor suppressive role for Crebbp in SCLC [31]. In an autochthonous mouse model for SCLC, inactivation of Crebbp (Rb1fl/fl;Trp53fl/fl;Crebbpfl/fl) accelerated tumor formation. Loss of Crebbp led to reduced levels of histone acetylation, which impacted the transcription of cellular adhesion genes [31]. Using a similar approach, the authors tested the oncogenic potential of other mutations. TP73 is altered by mutation or genomic rearrangement in ~13% of human SCLC [13] and this is predicted to lead to loss of TP73 function or expression of dominant negative TP73 isoforms. Targeting Trp73 with sgRNAs directed at its transactivation domain accelerated tumor growth consistent with a tumor suppressive role for TP73 [32]. Ectopic expression of Fgfr1 promoted tumor growth while conversely Fgfr1 loss inhibited tumor growth, suggesting an oncogenic role for FGFR1, consistent with its amplification in ~6% of SCLC [13, 32, 33]. Interestingly, this approach validated unpublished findings that Fgfr1 alterations impact anatomical tumor location in GEMMs (A.B. et al., unpublished data), suggesting a potential cell-type specific impact of FGFR1 in SCLC.
MYC family transcription factors (MYC, MYCL and MYCN) are frequently amplified in SCLC tumors. Intriguingly, genomic alterations of these MYC paralogs occur in a mutually exclusive manner in SCLC [34]. Using a MYC-driven GEMM and a pharmacological vulnerability screen in SCLC cell lines, oncogenic activation of MYC was found to pose a specific susceptibility to Aurora kinase inhibition [35, 36]. A recent study employed CRISPR-activation approaches in mouse cell lines with Rb1/Trp53 loss to study the effects of MYC paralogs (C, L and N-MYC) [37]. Myc-activated cells recapitulated previously discovered MYC-dependent drug sensitivities including to Aurora A inhibitors [35, 36, 38]. Mechanistically, MYC was found to epigenetically repress BCL2 transcription through its interactions with DNMT3a and MIZ1. This interaction results in the methylation of the BCL2 promoter with a subsequent decrease in BCL2 protein expression, consistent with observations that the BCL2 promoter is silenced by DNA methylation in the variant SCLC subtype [39]. MYC activation also increases DNA damage signaling, apoptosis, and is associated with increased BH3-apoptotic priming and MCL1 dependency, providing a rationale for MYC-driven SCLC sensitivity to DNA damage checkpoints like CHK1 inhibition [37]. Indeed, combining MYC-dependencies (CHK1 + AURKA inhibition) suppressed tumor growth and enhanced survival of MYC-driven GEMMs compared to the standard of care chemotherapy [37]. These studies suggest the ability to exploit MYC-specific therapeutic vulnerabilities in SCLC.
On the basis that RB1 is almost universally lost in SCLC, two recent studies discovered a dependency of RB1-deficient SCLC cells on Aurora kinases [40, 41]. Specifically, a CRISPR-based synthetic lethal screen determined that RB1 loss is synthetic lethal with Aurora B kinase inhibition in SCLC cell lines [40, 41]. RB1-mutant SCLC cells are sensitive to AZD2811, an AURKB inhibitor, similar to what has been shown in MYC-overexpressing tumors [35, 36, 38, 42]. Together, this suggests that both RB1 loss and MYC-overexpression may be required for the sensitivity of SCLC to Aurora kinase inhibitors. A Phase 1 clinical trial is ongoing for AZD2811 as a monotherapy in patients with relapsed or refractory SCLC. It will be important to determine whether responses in the AZD2811 trial correlate with MYC status as was observed in earlier clinical trials treating SCLC patients with combination Alisertib and paclitaxel [43]. Together, these studies highlight the importance of collecting biomarker information during clinical trials to identify relevant patient populations for larger definitive studies.
In addition to MYC, NOTCH signaling has been implicated as a major pathway contributing to transcriptional heterogeneity in SCLC [44]. While Notch can be tumor suppressive and exhibits frequent loss of function alterations in SCLC [13], it can also be oncogenic. Indeed, NOTCH activation is found in a subset of mouse and human tumors. NOTCH-high tumors express REST, a transcriptional repressor that can suppress neuroendocrine gene expression, thereby contributing to a non-neuroendocrine cell fate. MYC-high SCLC is also associated with a non-neuroendocrine fate, and MYC can drive a non-neuroendocrine phenotype in GEM models [13, 35, 45]. Consistently, in a large panel of human SCLC cell lines, MYC, NOTCH and REST (as well as HIPPO and TGFB pathways) are highly associated with non-neuroendocrine SCLC fate [45]. In contrast, neuroendocrine-high cell lines express lower levels of those genes and related pathways (i.e. MYC, NOTCH, REST) and higher levels of ASCL1 and NKX2-1. Understanding the functional relevance of these pathways and their relationships to one another constitutes a major unmet need in SCLC since these pathways may be therapeutically relevant.
SCLC is a highly metastatic tumor type, which is a major cause of morbidity. Recent work has shed light on mechanisms of metastasis using GEM models. Data from multiple groups demonstrate that the transcription factor NFIB promotes metastases in SCLC [46–48]. NFIB is important for lung and brain development and Nfib is genomically amplified in classic SCLC tumors from GEM models [49], where it has been implicated as an ASCL1 target gene [18]. NFIB is also highly expressed in ASCL1-low MYC-driven variant tumors from GEM models where it is not amplified but appears to be a MYC target gene [36]. Overexpression of Nfib in classic GEMMs accelerates tumor formation and metastases, and this is due to NFIB’s ability to open chromatin and promote a pro-metastatic neuronal gene expression program [46–48]. A recent study suggests that metastastic tumor cells are more neuronal than neuroendocrine and exhibit protrusions that resemble axons; knockdown of genes implicated in protrusion-formation inhibited metastatic capacity, suggesting a potential new avenue for blocking metastases [50]. NEUROD1 has also been implicated in SCLC migration and is associated with neuronal gene expression programs [18, 51, 52], but the relationship between NFIB, NEUROD1 and neuronal behavior, if there is one, is currently not well understood. More recently, it was shown that the cell of origin impacts the metastatic trajectory of tumors in GEMMs and therefore, different mechanisms of metastases may occur in SCLC [26]. Further studies are needed to determine whether these metastatic programs could be pharmacologically inhibited for patient benefit.
In order to further delineate heterogeneous phenotypes in SCLC, new technologies are being explored including single cell transcriptome sequencing and mass cytometry (CyTOF). CyTOF is a mass cytometry approach coupling antibodies to rare earth metals, similar to the fluorophores of traditional flow cytometry. CyTOF allows for increased multiplexing capacity and sensitivity per cell. CyTOF is being employed to investigate intra- and intertumoral heterogeneity among PDX samples, to identify novel populations of SCLC, and to probe mechanisms of chemotherapy resistance (J.L. et al., unpublished data). These unbiased approaches to investigate tumor cell populations will undoubtedly increase our understanding of tumor heterogeneity, plasticity, and treatment resistance mechanisms.
LARGE SCALE MOLECULAR PROFILING
Large-scale profiling studies have provided valuable insight into the epi-genomic, genomic, proteomic, and metabolomic landscape of SCLC tumors [14, 15, 18, 20, 39, 53–55]. Quantification of intra-tumoral heterogeneity using genome sequencing data of SCLC tumors showed that sub-clonal diversity was three-fold lower in SCLC than in lung adenocarcinomas [13]. This disparity may point to pronounced differences in the evolution and progression of SCLC and lung adenocarcinomas. This view is further challenged by the evolutionary dynamics of lung adenocarcinomas with mutations in the Epidermal Growth Factor Receptor (EGFR) gene that undergo histological trans-differentiation to SCLC through a poorly understood mechanism of lineage plasticity. SCLC tumors may originate from multiple cell types, whereas lung adenocarcinomas have been reported to mainly develop from type II alveolar cells [56, 57]. Despite their reportedly distinct cells of origin, EGFR--mutant lung adenocarcinomas have been shown to transform to SCLC following treatment with tyrosine kinase inhibitors [58–60]. The molecular mechanisms of this phenomenon have been strongly associated with the loss of RB1 [61]. A recent study on serial sampling and longitudinal sequencing of four trans-differentiated EGFR-mutant lung adenocarcinomas provided further insight into the genomic and evolutionary dynamics of transformed SCLC tumors [62]. The genomic profiles of these cases revealed a branched evolution in which all SCLC-transformed cases harbored early clonal EGFR mutations and early clonal dual inactivation of TP53 and RB1. As part of the evolutionary early branching, acquired EGFR resistance mutations and other driver gene alterations (e.g. MYC, PIK3CA, PTEN, ARID1A) were found to be late events and branch-specific. Private tumor-specific late branching events were enriched for APOBEC-related mutational signatures [62]. This observation is in line with previous studies on the evolution of lung adenocarcinomas that revealed reduced rates of smoking-related C>A transversions in the branches and increased APOBEC-related sub-clonal mutations [57, 63–65]. In light of the evolutionary dynamics of lung adenocarcinomas, further studies on serial SCLC tumor samples are required to obtain more insight on genomic changes throughout tumor progression and therapy resistance.
In a recent study, whole exome sequencing of 30 chemo-resistant SCLC tumors was performed, and for 12 of these cases the comparative analysis of matched treatment-naive tumors provided information on potential mechanisms of resistance to chemotherapy [66]. The genomic profile of chemo-resistant tumors mirrored the alterations identified in treatment-naïve SCLC. Tumors from chemo-refractory SCLC patients were reported with recurrent somatic alterations and with transcriptional up-regulation of WNT pathway genes. WNT activation was enriched in patient tumors that specifically harbored low levels of ASCL1 expression. Additionally, some patient cases were found with alterations in mismatch repair genes and amplification of ABCC1, which in earlier studies was identified in chemo-resistant SCLC cell lines [66]. These analyses warrant further studies on WNT pathway genes and their role in distinct genomic and transcriptional subgroups of SCLC.
SCLC tumors with low expression of ASCL1 generally harbor high expression levels of the MYC transcription factor. In addition to their distinct expression patterns, MYC-dependent SCLC tumors demonstrate therapeutic vulnerabilities to Aurora kinase inhibitors [35, 36]. Metabolomics of human SCLC cell lines and mouse-derived SCLC tumors revealed distinct metabolic profiles of MYC-driven tumors. Metabolite set enrichment analyses pointed to an inosine monophosphate dehydrogenase (IMPDH) dependency in ASCL1low cells [54]. The enzymatic activities of IMPDH1/2 allow for de novo guanosine nucleotide synthesis, and ASCL1low SCLC cells depend on this mechanism for cell survival. IMPDH1/2 inhibitors suppressed cell growth in xenograft models of ASCL1low SCLC cell lines, and GEMMs of ASCL1low/MYChigh tumors were sensitive to the combined inhibition of IMPDH1/2 with chemotherapy [54]. In addition to guanosine metabolism, recent studies also revealed subtype-specific dependencies of SCLC on arginine [55]. Arginine has pleiotropic cellular functions in nitric oxide signaling, polyamine biosynthesis and mTOR activation. Pharmacological interventions revealed that MYC-driven SCLC tumor cells are dependent on arginine for mTOR pathway activity and polyamine biosynthesis. Pharmacological depletion of arginine was shown to promote survival of mice bearing MYC-driven SCLC tumors in GEMMs, human cell line xenografts, and PDX models [55]. Arginine deprivation agents such as pegylated arginine deiminase (ADI-PEG20) and pegylated human arginase have been in clinical trials for SCLC, and these data suggest that biomarker analysis may be critical to interpretation of these findings [67].
PLATFORMS FOR DISCOVERY
In contrast to the large-scale genome sequencing efforts of primary lung adenocarcinoma that proved to be a fertile ground for drug target discovery, genomic surveys of SCLC tumors have produced few directly-targetable alterations [13–15]. A growing theme among recent discoveries has been the importance of neuroendocrine transcriptional networks, epigenetic modifiers, metabolism, and global transcriptional addiction in the maintenance of SCLC tumors. This reality has forced a strategic shift in therapeutic discovery efforts away from target nomination from the set of recurrent genetic alterations in spontaneous human tumors and toward pharmacologic targeting of candidate pathways, targeted gene disruption, synthetic lethal screens, and chemistry-first screening as platforms for target identification. The field is increasingly gravitating toward approaches that combine multi-omic characterization, diverse chemical libraries that include natural products, and focused gene disruption of defined target classes, such as epigenetic modifiers and kinases using CRISPR-Cas9 genome engineering technology in cell lines and mouse models as a means to discover new SCLC biology.
Landmark studies to identify the key drivers of drug response in cancer cell lines uncovered several important chemical:genetic or chemical:epigenetic interactions that have subsequently been validated in patients [68]. These initial studies focused on over 1,000 unique cell lines across cancer types and a relatively small number of drugs with known antineoplastic activity in order to power the discovery of molecular predictors of response. A study focusing on SCLC cell lines screened 526 compounds in 63 SCLC cell lines and included gene and miRNA expression measurement; this study constitutes one of the largest public chemical screens in SCLC cell lines [69]. Recent efforts in lung cancer have focused on screening larger and more diverse sets of compounds in order to discover “therapeutic triads” of novel targets, compounds, and response biomarkers. In NSCLC, >100 cell lines were recently screened with a 200,000 compound diversity-oriented chemical library as part of the precision oncology probe set, lung project (POPS-Lung) [70]. This effort has been expanded to include 30 SCLC cell lines to identify compounds with selectivity for SCLC over NSCLC, increasing the total chemical space explored by two orders of magnitude over the prior benchmark study (J.M. et al., unpublished data). Natural product screening for new classes of biologically active compounds and identification of their relevant targets through forward genetic approaches may further augment screens of synthetic compound libraries.
CRISPR-Cas9 gene disruption screens have revolutionized forward genetics in human cell lines. This powerful technology has been recently employed to uncover transcription factor dependencies in SCLC cell lines, as discussed above [21]. Dropout screens targeting developmental pathways or genes required for stem cell maintenance, kinases, or other classes of proteins may uncover previously unappreciated subtype-specific gene dependencies. Screens that target genes in tandem or that combine chemical perturbation with gene disruption constitute a powerful technology for interrogating genetic or chemical:genetic interactions. One recent study identified a role for MAST1 kinase in driving cisplatin resistance through impingement on MAP kinase signaling through a kinome-wide RNAi screen in combination with cisplatin [71]. Screens of this nature constitute a particularly powerful discovery engine.
Discovery platforms by their nature generate large scale datasets that require specialized expertise to curate and mine for biological signal. Pharmacogenomic data portals are a useful resource to make these datasets readily accessible to a broad audience. Recently, CellMinerCDB has been established as a web-based data portal (https://discover.nci.nih.gov/cellminercdb/) that combines drug response and multi-omics data from multiple cell line datasets (the NCI-60, NCI-SCLC, Sanger/MGH GDSC, and Broad CCLE/CTRP) and additionally provides integrative and exploratory tools to mine these data for potential molecular predictors of response [72]. Beyond studying a large panel of cell lines derived from various cancer types, a SCLC-specific data portal will be launched (SCLC-CellMinerCDB) providing analysis tools for drug screening data from 118 SCLC cell lines from four drug screen portals (NCI-DTP, CCLE, GDSC, CTRP) and the UT Southwestern data. SCLC-CellMinerCDB will allow cross database analysis for whole genome, somatic alterations (mutations and copy number alterations), DNA methylome, gene expression and proteomic data. This integrative approach will allow for cross-comparisons of SCLC and other cancer cell lines, and for pharmacogenomic data exploration to link drug treatment response to genomic and molecular characteristics.
These discovery efforts ultimately demand clean genetic models for mechanistic validation. The Rb1/Trp53 GEMM developed in the laboratory of Anton Berns, which gives rise to lung tumors that closely resemble human SCLC-A, continues to be a widely used model for mechanistic validation of potentially cooperating genetic events, although it has a long latency of 10-12 months [73]. This model has proven to be useful in ranking therapeutic liabilities that may arise from the underlying biology of a given genetic lesion. An ever-expanding number of triple- and quadruple-transgenic GEMMs based on the original double-transgenic model have expedited tumor latency and are helping to unravel the early steps of SCLC carcinogenesis and validate new therapeutic approaches.
GEMMs have many advantages; however, in some instances, certain hypotheses cannot be tested in mice due to a lack of direct homology between mouse and human. Pulmonary neuroendocrine cells (PNECs) are considered by many to be a likely cell of origin for SCLC. Since these relatively rare cells constitute <1% of an adult human lung and there are no effective in vitro culture systems, primary PNECs have not been studied directly. The generation of PNECs and SCLC-like tumors from human embryonic stem cells through chemical and genetic recapitulation of the key events in SCLC carcinogenesis holds promise as a clean in vitro genetic model for the initiation of human SCLC [74]. In this system, NOTCH inhibition and RNAi knockdown of TP53 and RB1 in lung progenitor cells results in a massive expansion of PNECs to constitute 10–30% of the culture population.
Together, complementary mouse and in vitro human model systems enable the discovery and preclinical validation of disease-relevant molecular alterations identified in spontaneous human cancers and their relation to therapeutic responses. A deeper understanding of the mechanistic basis of drug sensitivity is likely to be a source of candidate predictive biomarkers of response.
BIOMARKERS
Liquid biopsies for molecular profiling in SCLC patients
Serial analysis of tumor biopsies beyond the initial diagnostic specimens are not routinely performed in SCLC patients, which limits molecular studies and biomarker assessments of treatment-induced changes in this cancer type. The analysis of blood-based tumor components such as circulating tumor cells (CTCs), circulating tumor DNA (ctDNA) and tumor-derived extracellular vesicles (EV) could provide alternative opportunities to monitor the molecular phenotype of a patient’s tumor throughout disease, and to assess biomarkers of treatment response and tumor progression.
In comparison to other lung cancers, SCLC patients have the greatest number of CTCs [75]. The number of CTCs has been demonstrated to serve as a prognostic marker for clinical response to therapy [76]. Furthermore, isolated CTCs from SCLC patients can be utilized to generate CTC-derived xenograft models (CDX), which allow for the enrichment and subsequent analysis of the tumor material [77]. The generation of serial CDX proved feasible at multiple time points during treatment. The established tumor models were shown to faithfully reflect the matched patient tumor, and to recapitulate the mutational landscape and transcriptional diversity found in SCLC tumors [77, 78]. Furthermore, pharmacological drug screening using CDXs mirrors the therapeutic responses of the donor patient [77, 78]. The establishment of CDX requires significant resources and time, which limits the chances for a donor patient to benefit from pre-clinical interrogations of their own tumor. New approaches and technologies are focusing on the direct study of isolated CTCs, which provide prospects for ex vivo expansion and short-term culture for gene manipulation and preclinical drug screening [79]. Thus, CTCs and CDXs hold promise as additional tools to gain mechanistic insights into treatment sensitivity and resistance.
CTC-derived nucleic acids or ctDNA similarly allow for longitudinal disease monitoring of SCLC patients [80, 81]. Customized targeted sequencing panels aid in assessing and tracking tumor-related mutations and copy number changes. SCLC-associated genomic alterations were successfully identified in up to 85% of plasma samples as recently reported by genomic profiling studies using ctDNA [80]. Tracking such genomic alterations in the ctDNA of SCLC patients also served as an indicator of disease relapse [80]. Furthermore, comparative analyses of CTC-derived nucleic acids from chemosensitive and chemo-refractory SCLC patients revealed distinct copy number profiles, which may serve as biomarkers for patient stratification [81].
The isolation and characterization of extracellular vesicles (EVs) such as exosomes may constitute an alternative source for multiple analytes in liquid biopsies. EVs contain cytosolic proteins, lipids, and nucleic acids and are released from all cells including cancer cells [82]. Tumor-related EVs can be found in cancer patients and it is critical to distinguish those from EVs of normal cells by tracking tumor-specific markers. To this end, a recent study demonstrated that Glypican-1 positive exosomes can serve as a diagnostic marker for pancreatic cancer [83]. The signal of EV-associated Glypican-1 RNA was shown to be further amplified with the use of molecular beacons and lipoplex nanoparticles [84]. SCLC tumors harbor several tumor-specific expression markers [6], including DLL3 [85]. The technological advances in EV capture and analyses could be resourceful for studying SCLC tumor markers and could aid in diagnosis and in monitoring disease progression.
TARGETED THERAPIES
DNA Damage Response Inhibition
In comparison to lung adenocarcinoma, SCLC tumors exhibit high expression levels of DNA damage response (DDR) proteins [53]. Many DDR proteins, such as PARP, ATR, CHK1, and WEE1, have small molecule inhibitors in various stages of development (summarized in Table 1). Among the DDR proteins, PARP is perhaps the most actively studied drug target as a single agent as well as in combination with other therapies.
Table 1.
Recent and ongoing clinical trials
| Agent | Target | Trial Phase | Clinical Trial ID(s) |
|---|---|---|---|
| Navitoclax (ABT-263) | BCL-2, BCL-xL, BCL-W | I, II | NCT03366103 |
| APG-1252 | BCL-2, BCL-xL, BCL-W | I | NCT03080311, NCT03387332 |
| ABBV-075 | BET | I | NCT02391480 |
| Lurbinectedin (PM01183) | CG-rich promoter sequences | III | NCT02566993 |
| Prexasertib (SRA737) | CHK1 | II | NCT02735980 |
| Rova-T | DLL3 | III | NCT03543358 |
| AMG 757 | DLL3 | I | NCT03319940 |
| AMG 119 | DLL3 | I | NCT03392064 |
| BMS-986012 | FucGM1 | I,II | NCT02247349, NCT02815592 |
| Vistusertib | mTORC1/2 | I,II | NCT03366103 |
| Olaparib | PARP | I,II | NCT02446704, NCT03009682, NCT02769962, NCT03532880, NCT03923270, NCT02511795 |
| Talazoparib | PARP | II | NCT03672773 |
| Veliparib | PARP | II | NCT03227016 |
| Rucaparib | PARP | II | NCT03958045 |
| Niraparib | PARP | II | NCT03830918 |
| II | NCT03516084 | ||
The mechanism of action of PARP inhibitors is two-fold: to inhibit the formation of poly-ADP ribose (PAR) at single-strand breaks (SSB) and to prevent the release of PARP complexes from DNA single-strand breaks. Replication forks stall when they encounter PARP trapped on DNA, which can ultimately result in double-strand breaks (DSB). It is now recognized that PARP inhibitors differ in their PARP trapping potency, a crucial factor for cytotoxicity of these agents [86]. In addition to their action as single agents, PARP inhibitors can synergize with other agents that induce single-stranded breaks such as the alkylating agent temozolomide (TMZ) [86–88]. SLFN11 has been tested as a biomarker of PARP inhibitor sensitivity in SCLC in preclinical models and as an exploratory clinical correlate [87, 89].
The efficacy of PARP inhibitors has been tested in various settings in SCLC patients [43, 89–91]. While there is reproducible evidence of efficacy, the magnitude of benefit appears modest in the absence of predictive biomarkers to assist in patient selection. Emerging data from the preclinical and clinical arena are coalescing to inform the best strategies to successfully incorporate this class of agents into SCLC treatment. A recent clinical study of the PARP inhibitor, olaparib, in combination with TMZ in SCLC patients included a co-clinical research program to establish PDX models and further interrogate patient-specific treatment responses [91]. Longitudinal SCLC xenograft models established from tumor material from this trial recapitulated the clinical responses of the patients to chemotherapy and to the combination treatment of TMZ with PARPi [78]. This co-clinical framework provides a valuable platform for in-depth functional studies of treatment responses and biomarker discovery.
Synergy between DDR inhibitors and ionizing radiation therapy has been demonstrated in various tumor types [92–95]. The addition of the PARPi talazoparib to escalating doses of ionizing radiation (IR) showed potentiation of the IR effect in a panel of SCLC cell lines using both short-term cytotoxicity and long-term clonogenic assays [96]. Moreover, IR potentiation correlates with PARP trapping ability of PARP inhibitors with a stronger effect recorded with talazoparib compared to veliparib, which has been shown to have the weakest PARP trapping effect. Similar potentiation effects were noted in in vivo models using SCLC PDX representative of chemo-naïve and chemo-resistant disease. An investigator-initiated clinical trial is now testing the combination of olaparib and low-dose fractionated thoracic radiation (10×3 Gy over 2 weeks) ( NCT03532880).
G2/M Mitotic Checkpoint Inhibition
The DNA damage checkpoint represents an attractive target in SCLC, because of the aberrant expression of various DNA damage response genes and intrinsic cellular vulnerabilities [37, 53, 97]. In addition to Aurora kinases and WEE1, PLK1 is a node in the G2/M mitotic checkpoint that plays a major role in driving centrosome disjunction and separation. It is frequently over-expressed in human cancers including lung cancer and its inhibition leads to a characteristic Polo arrest phenotype leading to cell cycle arrest in mitosis due to monopolar spindles and apoptosis [98, 99]. Inhibitors of PLK1 previously evaluated in preclinical and clinical settings include SBE, onvansertib (NMS-P937), Ro3280, MLN0905, HMN-214, GSK461364, Rigosertib (ON-01910), Volasertib (BI6727) and BI2536 [100]. Agnostic low-throughput screening showed exquisite sensitivity of SCLC cell lines and in vivo models to different PLK1 inhibitors, in particular, onvansertib and volasertib (T.O., et al., unpublished). In addition, TP53 gene mutation and MYC expression appeared to predict for sensitivity to this class of agents [37, 42], consistent with observations in other tumor types. The combination of PARP inhibitors and other targeted agents that act through G2 checkpoint blockade such as ATR, CHK1 and WEE1 inhibitors has been the focus of preclinical testing [97, 101–103].
Genomic instability characterized by high tumor mutational burden (TMB) and the presence of microsatellite instability or BRCA mutations have been associated with sensitivity to immunotherapeutic agents targeting the PD-1 signaling axis. PARP inhibition was shown to induce an immune response and increase the efficacy of PD-1 targeted immunotherapy agents through a mechanism that implicates T-cell activation through the stimulator of interferon genes (STING) pathway [104]. Furthermore, the combination of PARP inhibitor olaparib and anti-PD1 therapy was synergistic in a syngeneic transplantable GEMM (RPR2) of SCLC with increased cytotoxic T cell infiltration in SCLC tumors treated with the combination compared to either agent alone [105]. A similar observation was made with the combination of a CHK1 inhibitor (prexasertib or SRA737) and anti-PD1 antibodies in a classic SCLC GEMM [106]. Both PARP and CHK1 inhibitors were shown to induce increased expression of PDL1, IFNβ, CXCL10, and CCL5. Also, the synergistic interaction of PARP and CHK1 inhibitors with PD-1 targeted agents was shown to be strongly dependent on the activation of the cGAS–STING pathway [105, 106]. These preclinical data support the clinical evaluation of CHK1 and PARP inhibitors as rational combination partners for immunotherapy in SCLC [107].
Apoptosis
BCL2, a key regulator of the intrinsic apoptotic pathway, is over-expressed in a subset of SCLC. Inhibitors of this pathway have been evaluated in patients with recurrent SCLC but the promise for clinical use has been limited because of modest efficacy along with high rates of hematologic toxicity [108, 109]. ABT-263 (navitoclax) is a sub-nanomolar inhibitor of BCL2, BCL2L1 (BCL-XL), and BCL2L2 (BCL-W). Navitoclax has shown efficacy in preclinical models of SCLC [110]. Preclinical data using cell line and xenograft models demonstrated synergy with concurrent inhibition of the PI3K/mTOR and BCL2 pathways [111–113]. A phase I/II study is currently evaluating the safety of this combination using navitoclax and vistusertib, a dual TORC1/2 kinase inhibitor, in relapsed SCLC patients ( NCT03366103). In addition, APG-1252, which also inhibits BCL2, BCL2L1 and BCL2L2, is also being tested as a single agent in relapsed SCLC ( NCT03387332, NCT03080311). Preclinical studies suggest that BCL2 inhibitors may be most relevant to classic SCLC-A subsets [37], suggesting biomarker information will be important in these clinical trials. As mentioned earlier, MYC can repress BCL2 expression, consistent with functional studies showing that MYC-driven SCLC is more reliant on MCL1 than BCL2 [37]. These studies also suggested that high BCL2 expression in the SCLC-A subset may be responsible for the relative resistance of these cells to Aurora kinase inhibition. Future studies are warranted to determine the utility of BCL2 family targets and to identify novel combination strategies that can sensitize SCLC to apoptosis.
Lurbinectedin (PM01183)
Lurbinectedin (PM01183) is a synthetic analog of trabectedin (Yondelis®, ET-743), and belongs to the natural marine-based tetrahydroisoquinoline family of antitumor agents. It is a selective inhibitor of active transcription and binds to CG-rich sequences within the promoter region of select genes, leading to irreversible stalling and degradation of elongating RNA polymerase II on the DNA template, generation of single- and double-strand DNA breaks, and subsequent cell death [114, 115]. In preclinical models, lurbinectedin can also reduce type 2 tumor-associated macrophages and modulate the inflammatory tumor microenvironment [116, 117]. A phase I trial of doxorubicin (50 mg/m2) and lurbinectedin (4 mg) as second line therapy in 48 patients with relapsed SCLC reported a response rate of 37-67% with myelosuppression as the main toxicity [118]. As a single agent, lurbinectedin also showed impressive efficacy in relapsed SCLC, particularly in patients with platinum-sensitive relapse with a response rate of 44% and median overall survival of 15.8 months [119]. The ATLANTIS study is a phase III trial that randomized patients with relapsed SCLC to receive doxorubicin plus lurbinectedin on the experimental arm and topotecan or a three-drug regimen (cyclophosphamide, doxorubicin, vincristine) on the control arm ( NCT02566993). The study has completed accrual and final results are awaited to provide a basis for potential regulatory approval.
Delta-like ligand 3 (DLL3)
Delta-like canonical Notch Ligand 3 (DLL3) is a Notch ligand with restricted expression in SCLC and other neuroendocrine tumors and a validated target for therapy. In a phase 1 clinical trial of rovalpituzumab teserine (Rova-T; SC16LD6.5), a DLL3-targeted antibody drug conjugate, objective responses were observed in 16% of 56 patients with recurrent SCLC. This was associated with a median OS of 5.8 months [120]. The follow-up TRINITY study enrolled 339 eligible patients with recurrent DLL3-positive SCLC to receive Rova-T (0.3 mg/kg IV every six weeks for 2 doses) as 3rd line treatment or beyond. Approximately 70% of these patients had tumors with high DLL3 expression (i.e. ≥75% cells DLL3+) using a standard companion immunohistochemistry assay. The confirmed response rate was 18% in all patients and 19.7% in patients with high DLL3. Median OS was identical in both populations at 5.6 months and 5.7 months, respectively. Treatment emergent adverse events attributable to Rova-T occurred in 91% of patients with serious adverse events in 30% of patients. Fatal adverse events were recorded in 10 patients [121]. TAHOE was a randomized phase III trial of Rova-T versus topotecan as second line treatment in recurrent SCLC ( NCT03061812). This study was discontinued due to a shorter survival noted in patients on the experimental arm versus the control arm. MERU was a phase III trial to evaluate Rova-T as a maintenance therapy following frontline platinum-based chemotherapy in patients with extensive stage SCLC ( NCT03033511). This study was terminated due to a lack of survival benefit at the interim analysis. Based on these two negative studies, research and development of Rova-T has been discontinued. Rova-T dosing was limited by a toxicity profile that has been attributed to its pyrrolobenzodiazepine (PBD) warhead, including persistent pleural and pericardial effusions, peripheral edema, and in some cases anasarca. These toxicities are believed to be an off-target liability of the ADC, and therefore DLL3 remains an intriguing target for multiple alternative therapeutic strategies including bispecific T cell engagers (BiTEs) (AMG 757) and a chimeric antigen receptor T cell (CAR-T) therapy (AMG 119) [121].
AMG 757 is a bispecific T cell engager (BiTE) antibody construct that binds to DLL3 on the cancer cell surface and the CD3 on cytotoxic lymphocytes as a method to directly recruit immune effector cells into the tumor microenvironment. Binding brings the T-cell and cancer cell into close proximity leading to T-cell receptor independent activation [122, 123]. In preclinical models, AMG 757 demonstrated strong potency against SCLC cell lines with varying levels of DLL3 receptor density. It also achieved effective antitumor effects in orthotopic models of SCLC. Finally, AMG 757 showed good tolerability and dose proportional exposure with an extended half-life in toxicology studies in cynomolgus monkeys [124]. The agent is currently in human clinical testing in a first-in-human phase I clinical trial designed to evaluate its safety, tolerability, and pharmacokinetics ( NCT03319940). Two populations of SCLC patients will be enrolled including patients with relapsed/refractory SCLC (part A) and as maintenance in patients with ongoing clinical benefit following frontline platinum-based chemotherapy (part B).
From a diagnostic perspective, the use of 89Zr-SC16, a PET radiotracer, is under development for in vivo imaging and as a companion diagnostic to optimize the selection of patients for treatment with DLL3-directed pharmacophores. 89Zr-labeled SC16 antibody successfully delineated normal tissue from subcutaneous and orthotopic SCLC PDX models [125]. Radiotracer accumulation in tumors was directly correlated with the degree of DLL3 expression and also correlated with response to SC16LD6.5 (Rova-T) therapy in SCLC PDX models [125]. Ongoing attempts to further improve the imaging characteristics of the 89Zr-labeled-SC16 radiotracer include evaluation of different site-selective linker chemistries with improved in vivo stability [126].
Epigenetic inhibitors
Bromodomain and Extra-Terminal motif protein (BET) proteins recognize acetylated histones and recruit proteins to promoters and enhancers, especially super-enhancers [127–129]. A number of small molecule BET inhibitors are currently in clinical development [130]. This class of agents inhibits the expression of cancer-related target genes including MYC, MYCN, IL7R, FOSL1, AR, ER, BCL2, BCL6, PAX5, CDK4, and CDK6. Limited efficacy of BET inhibitors was observed as a single agent in preclinical models of SCLC; however, activity was more promising when combined with BCL2 inhibitors or cytotoxic chemotherapy [131–133]. A study of the BET inhibitor ABBV-075 in combination with the BCL2 inhibitor venetoclax in patients with cancer, including SCLC, recently completed accrual ( NCT02391480).
The polycomb repressive complex 2 (PRC2) maintains epigenetic gene silencing during normal development and tissue differentiation by methylating histone H3 at lysine 27 (K27), an inhibitory chromatin mark [134]. The methyltransferase subunit of PRC2 can be either enhancer of zeste homolog 1 or 2 (EZH1/2), which use the cofactor S-adenosyl methionine (SAM) as a methyl donor for H3K27 mono-, di- and tri-methylation reactions. EZH2 is known to be overexpressed or mutated in multiple cancers [135]. While EZH2 is not frequently mutated in SCLC, it can be overexpressed at the mRNA and protein levels relative to other cancer types [39, 53]. Small molecule SAM mimetics that selectively inhibit EZH1/2 to different degrees are in active development; however, they have thus far demonstrated limited single agent activity in preclinical models of SCLC. One study found that EZH1/2 inhibitors could reverse an epigenetic mechanism of acquired chemoresistance caused by epigenetic silencing of SLFN11 [136]. In this context, inhibition of EZH1/2 could rescue SLFN11 expression and synergize with a variety of DNA damaging agents in vitro and in vivo. These results supported the initiation of a phase I/II trial evaluating the safety and tolerability of valemetostat (DS-3201b), a potent dual EZH1/2 inhibitor, in combination with irinotecan in patients with recurrent SCLC ( NCT03879798).
IMMUNOTHERAPY
After decades without a change in the standard of care, 2018 produced a new landmark in the treatment of SCLC, with the US Food and Drug Administration (FDA) approval of atezolizumab (anti-PDL1) in combination with first-line platinum doublet chemotherapy for extensive stage disease [5]. This approval established immune checkpoint blockade as a new treatment for SCLC, as it has been for several other lung cancer subtypes. IMPOWER 133 was a randomized phase III study of carboplatin and etoposide with or without atezolizumab. The addition of atezolizumab resulted in a statistically significant improvement in progression-free survival (PFS) (HR 0.77; 95% CI 0.62-0.96; p=0.02) and overall survival (OS) (HR 0.70; 95% CI 0.54-0.91; p=0.007). No significant difference in objective response rate was observed. In an exploratory analysis, blood-based tumor mutational burden was not predictive of benefit from the addition of atezolizumab to chemotherapy. Despite the milestone significance of this advance, the benefits of atezolizumab in this context were limited – the improvements in median PFS and OS were approximately 1 and 2 months, respectively, with just over half of patients treated with the triplet regimen alive at the 1-year mark [5].
Initial results from the phase III CASPIAN study, similarly first-line carboplatin-etoposide or cisplatin-etoposide with or without durvalumab, were recently presented [119]. This study demonstrated very similar improvements in both PFS (HR 0.78; 95% CI 0.65-0.94) and OS (HR 0.73; 95% CI 0.59-0.91) to those seen in IMPOWER 133, using a different PD-L1 inhibitor. Objective response rate was modestly higher with durvalumab (79% vs. 70 %; odds ratio 1.64, 95% CI 1.11-2.44). Results of the third arm of the CASPIAN study, adding the CTLA-4 inhibitor tremelimumab to platinum, etoposide, and durvalumab, have not yet been reported.
Excitement about the application of immune checkpoint blockade in unselected patients with SCLC was further tempered by negative results in other studies, in which the activity of immune checkpoint inhibitors alone was tested. Notably the CheckMate 331 study, a randomized phase III study of nivolumab (anti-PD1) vs. standard of care topotecan in the second-line (recurrent metastatic) setting demonstrated no statistically significant improvement in progression-free or overall survival [137]. Post hoc subset analysis suggested a benefit from nivolumab in patients defined as chemoresistant based on duration of response to first line platinum-based chemotherapy (HR 0.71; 95% CI 0.54-0.94), a population in particular need of better therapeutic options, but this result would require confirmation in a dedicated prospective study.
Putting the results of IMPOWER 133 and CASPIAN into context with other studies of immune checkpoint blockade in SCLC, it is important to emphasize that the modest improvements in median PFS and OS do not tell the full story. Notably, across several studies a small subset of SCLC patients appear to benefit substantially from treatment with immune checkpoint blockade, with durable responses observed in patients treated with either single agent PD-(L)1 therapy or combined PD-(L)1 plus CTLA4 blockade [138–141]. The CheckMate 032 study was a randomized study of nivolumab and nivolumab plus ipilimumab (anti-CTLA-4). In late 2018, nivolumab was granted accelerated FDA approval for third-line treatment of metastatic SCLC based primarily on response data from a subset of patients treated on the nivolumab arm of this study. Treatment with nivolumab in the third line setting was associated with a response rate of only 12% – but notably these responses were durable for at least 6 months in 77%, and at least 12 months in 62% of cases.
Together these observations suggest two directions of active investigation in SCLC immunotherapy: first, to further define predictive biomarkers identifying SCLC patients for whom immune checkpoint blockade might offer a durable benefit, and second, to build on the initial success of chemoimmunotherapy through new combinations that might impact a broader fraction of patients. Both strategies are being actively pursued.
Biomarkers of immune response
High TMB is increasingly recognized as an important determinant of the likelihood of response to immune checkpoint blockade across disease types [142]. Given that SCLC is predominantly a disease associated with the chemical mutagenic effects of substantial tobacco exposure, it was not clear whether any predictive TMB threshold could be established in a patient population typified by consistently high clonal mutational load. However, recent exploratory analyses from the CheckMate 032 study showed that TMB could serve as a predictive biomarker in SCLC [140]. Patients with tumors in the highest tertile of TMB appeared more likely to benefit from either nivolumab or the combination of ipilimumab and nivolumab, as measured by progression-free and overall survival. These differences were most striking in the combination arm, with 1-year survival of 62.4% in the highest TMB tertile vs. 19.6% and 23.4% survival in the middle and lowest tertile, respectively.
PD-L1 expression is also a predictive biomarker for immunotherapy across several solid tumors. However, the frequency and intensity of PD-L1 staining in SCLC tumor cells is quite low in comparison to NSCLCs and other solid tumors [143]. Initial data from the CheckMate 032 study suggested no correlation between PD-L1 expression and clinical benefit [138]. However, in the Keynote-028 and Keynote-158 studies of pembrolizumab (anti-PD1), PD-L1 expression, especially combined expression on tumor and immune stromal cells, was associated with improved response to pembrolizumab (35.7% vs. 6%) [139, 141].
Novel immunotherapy combinations and targets
Beyond PD-(L)1 and CTLA-4, a number of other combinatorial approaches to augment the efficacy of immune checkpoint blockade are under active exploration. One approach showing remarkable preclinical promise are combinations of agents targeting DNA damage repair pathways and cell cycle regulators, including PARP1 and CHK1/2 [105]. CDK7 is a central regulator of cell cycle progression, controlling the activity of multiple other CDK complexes involved in both G1-S and G2-M transitions. The recognition of CDK7 as a vulnerability that could be selectively targeted with an initial inhibitor, THZ1, was reported in 2015 [20]. A novel and more selective inhibitor of CDK7, YKL-5-124, has recently been characterized [144]. YKL-5-124 is being extensively evaluated in preclinical models of SCLC, both as a single agent and in combination with PD-1 blockade (H.Z.; C.L.C. et al., unpublished data).
Ganglioside fucosyl-GM1 (FucGM1) is a tumor-associated antigen with restricted expression in SCLC that is absent in most normal tissues [145]. BMS-986012 is a non-fucosylated, fully human IgG1 antibody that binds specifically to FucGM1. It showed strong antitumor efficacy against SCLC cell lines and xenograft models when used as a single agent and in combination with chemotherapy and immunomodulatory agents [145]. The safety of BMS-986012 in combination with a platinum doublet was tested in SCLC patients ( NCT02949895, NCT02815592). In addition, BMS-986012 was combined with nivolumab ( NCT02247349) in relapsed SCLC patients and was safe and tolerable [146]. A promising response rate > 20% was recorded in this study and future development of the regimen is awaited.
A final immunologic strategy under rigorous interrogation by multiple SCLC investigators involves the harnessing of key components of the innate immune system including macrophages and natural killer (NK) cells. Macrophages are found in SCLC stroma, and many SCLCs express CD47, the “don’t-eat-me” signal that inhibits macrophage phagocytosis. In preclinical GEMM and PDX SCLC models, CD47 blockade can induce tumor responses in vivo [147]. SCLC can lose expression of MHC class I [148], which may in part explain why this tumor type seems less responsive to PD-1 blockade, directed primarily at activating cytolytic T cell responses. Loss of MHC class I on the cell surface may make SCLC susceptible to NK cell activating therapies, an area of active investigation. A recent study demonstrated that loss of MHC class I may be driven by PRC2 and suggests the tantalizing possibility that resistance to immunotherapy may arise in SCLC via an epigenetic mechanism with the potential for pharmacologic intervention [149].
CONCLUSION
Despite recent advances in SCLC research, many key knowledge gaps remain in our understanding of the underlying pathobiology that drives this disease and how the therapeutic liabilities that it causes can be exploited in the clinic. We have identified eight thought-provoking questions that are of significant interest to the field:
Which therapeutic liabilities and treatment outcomes are associated with different molecular subtypes of SCLC (SCLC-A, SCLC-N, SCLC-Y, SCLC-P)?
To what extent will molecular subtypes of SCLC be used in the diagnosis of SCLC? What are the best biomarkers for molecular subtype discrimination?
What are the potential cells of origin of SCLC and what is the impact on the natural history of the disease? Are different SCLC characteristics driven by genetics, cell of origin, lineage plasticity, or some combination of factors?
What roles do NOTCH and WNT signaling play in intratumoral heterogeneity and the development of acquired resistance? What are the functional roles of different cell populations to the extent that they can be defined?
What are the underlying mechanisms of lineage plasticity? Can this be understood and targeted?
How can metabolic liabilities and nutrient sensing pathways be exploited?
What are the most effective ways to measure different circulating analytes with respect to early detection, treatment response, relapse, prognosis, etc.?
How can we improve immunotherapy outcomes? Can we identify predictive biomarkers?
ACKNOWLEDGEMENTS
The Third Biennial IASLC SCLC meeting and this manuscript are dedicated to the memory of an inspirational friend, mentor, and colleague, Dr. Adi Gazdar, whose pioneering work has greatly impacted the discoveries described herein. The NCI Small Cell Research Consortium Coordinating Center (U24 CA213274) and the International Association for the Study of Lung Cancer supported the meeting on which this review is based. The authors were supported by NIH R01 CA213448 (JTP and CMR), U01 CA213359 (JTP), U01 CA231844 (TGO and RG), U24 CA213274 (CMR and TGO), and R01 CA207295 (LAB). The authors would also like to thank Dr. Peter Ujhazy, Dr. Eva Szabo, and Dr. Suzanne Forry from the NCI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Howlader N, et al. , SEER Cancer Statistics Review, 1975-2016, National Cancer Institute; 2019. [Google Scholar]
- 2.Hanahan D and Weinberg RA, Hallmarks of cancer: the next generation. Cell, 2011. 144(5): p. 646–74. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D and Weinberg RA, The hallmarks of cancer. Cell, 2000. 100(1): p. 57–70. [DOI] [PubMed] [Google Scholar]
- 4.Bunn PA Jr., et al. , Small Cell Lung Cancer: Can Recent Advances in Biology and Molecular Biology Be Translated into Improved Outcomes? J Thorac Oncol, 2016. 11(4): p. 453–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horn L, et al. , First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med, 2018. 379(23): p. 2220–2229. [DOI] [PubMed] [Google Scholar]
- 6.Rudin CM, et al. , Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer, 2019. 19(5): p. 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Travis WD, et al. , The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J Thorac Oncol, 2015. 10(9): p. 1243–1260. [DOI] [PubMed] [Google Scholar]
- 8.Nicholson SA, et al. , Small cell lung carcinoma (SCLC): a clinicopathologic study of 100 cases with surgical specimens. Am J Surg Pathol, 2002. 26(9): p. 1184–97. [DOI] [PubMed] [Google Scholar]
- 9.Rekhtman N, et al. , Stage IV lung carcinoids: spectrum and evolution of proliferation rate, focusing on variants with elevated proliferation indices. Mod Pathol, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eguchi T, et al. , Lobectomy Is Associated with Better Outcomes than Sublobar Resection in Spread through Air Spaces (STAS)-Positive T1 Lung Adenocarcinoma: A Propensity Score-Matched Analysis. J Thorac Oncol, 2019. 14(1): p. 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu S, et al. , Spread through Air Spaces (STAS) Is an Independent Predictor of Recurrence and Lung Cancer-Specific Death in Squamous Cell Carcinoma. J Thorac Oncol, 2017. 12(2): p. 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aly RG, et al. , Spread Through Air Spaces (STAS) Is Prognostic in Atypical Carcinoid, Large Cell Neuroendocrine Carcinoma, and Small Cell Carcinoma of the Lung. J Thorac Oncol, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.George J, et al. , Comprehensive genomic profiles of small cell lung cancer. Nature, 2015. 524(7563): p. 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peifer M, et al. , Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet, 2012. 44(10): p. 1104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudin CM, et al. , Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet, 2012. 44(10): p. 1111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borges M, et al. , An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature, 1997. 386(6627): p. 852–5. [DOI] [PubMed] [Google Scholar]
- 17.Neptune ER, et al. , Targeted disruption of NeuroD, a proneural basic helix-loop-helix factor, impairs distal lung formation and neuroendocrine morphology in the neonatal lung. J Biol Chem, 2008. 283(30): p. 21160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borromeo MD, et al. , ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep, 2016. 16(5): p. 1259–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaffer BE, et al. , Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res, 2010. 70(10): p. 3877–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christensen CL, et al. , Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell, 2014. 26(6): p. 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang YH, et al. , POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev, 2018. 32(13–14): p. 915–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McColl K, et al. , Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget, 2017. 8(43): p. 73745–73756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutherland KD, et al. , Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell, 2011. 19(6): p. 754–64. [DOI] [PubMed] [Google Scholar]
- 24.Park KS, et al. , Characterization of the cell of origin for small cell lung cancer. Cell Cycle, 2011. 10(16): p. 2806–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ouadah Y, et al. , Rare Pulmonary Neuroendocrine Cells Are Stem Cells Regulated by Rb, p53, and Notch. Cell, 2019. 179(2): p. 403–416.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang D, et al. , Intertumoral Heterogeneity in SCLC Is Influenced by the Cell Type of Origin. Cancer Discov, 2018. 8(10): p. 1316–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montoro DT, et al. , A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature, 2018. 560(7718): p. 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plasschaert LW, et al. , A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature, 2018. 560(7718): p. 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belani CP, et al. , Vismodegib or cixutumumab in combination with standard chemotherapy for patients with extensive-stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E1508). Cancer, 2016. 122(15): p. 2371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wooten DJ, et al. , Systems-level network modeling of Small Cell Lung Cancer subtypes identifies master regulators and destabilizers. bioRxiv, 2019: p. 506402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jia D, et al. , Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov, 2018. 8(11): p. 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim K-B, et al. , Oncogenic role of FGFR1 and vulnerability of RBL2-FGFR1 axis in small cell lung cancer development. bioRxiv, 2019: p. 796607. [Google Scholar]
- 33.Schultheis AM, et al. , Fibroblast growth factor receptor 1 (FGFR1) amplification is a potential therapeutic target in small-cell lung cancer. Mod Pathol, 2014. 27(2): p. 214–21. [DOI] [PubMed] [Google Scholar]
- 34.Bragelmann J, et al. , Family matters: How MYC family oncogenes impact small cell lung cancer. Cell Cycle, 2017. 16(16): p. 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sos ML, et al. , A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci U S A, 2012. 109(42): p. 17034–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mollaoglu G, et al. , MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell, 2017. 31(2): p. 270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dammert MA, et al. , MYC paralog-dependent apoptotic priming orchestrates spectrum of vulnerabilities in small cell lung cancer. Nature Communications, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Helfrich BA, et al. , Barasertib (AZD1152), a Small Molecule Aurora B Inhibitor, Inhibits the Growth of SCLC Cell Lines In Vitro and In Vivo. Mol Cancer Ther, 2016. 15(10): p. 2314–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poirier JT, et al. , DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene, 2015. 34(48): p. 5869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oser MG, et al. , Cells Lacking the RB1 Tumor Suppressor Gene Are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov, 2019. 9(2): p. 230–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong X, et al. , Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov, 2019. 9(2): p. 248–263. [DOI] [PubMed] [Google Scholar]
- 42.Cardnell RJ, et al. , Protein expression of TTF1 and cMYC define distinct molecular subgroups of small cell lung cancer with unique vulnerabilities to aurora kinase inhibition, DLL3 targeting, and other targeted therapies. Oncotarget, 2017. 8(43): p. 73419–73432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Owonikoko TK, et al. , Randomized Phase II Trial of Cisplatin and Etoposide in Combination With Veliparib or Placebo for Extensive-Stage Small-Cell Lung Cancer: ECOG-ACRIN 2511 Study. J Clin Oncol, 2019. 37(3): p. 222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim JS, et al. , Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature, 2017. 545(7654): p. 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang W, et al. , Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl Lung Cancer Res, 2018. 7(1): p. 32–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Denny SK, et al. , Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell, 2016. 166(2): p. 328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Semenova EA, et al. , Transcription Factor NFIB Is a Driver of Small Cell Lung Cancer Progression in Mice and Marks Metastatic Disease in Patients. Cell Rep, 2016. 16(3): p. 631–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu N, et al. , NFIB overexpression cooperates with Rb/p53 deletion to promote small cell lung cancer. Oncotarget, 2016. 7(36): p. 57514–57524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dooley AL, et al. , Nuclear factor I/B is an oncogene in small cell lung cancer. Genes Dev, 2011. 25(14): p. 1470–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang D, et al. , Axon-like protrusions promote small cell lung cancer migration and metastasis. bioRxiv, 2019: p. 726026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Osborne JK, et al. , NeuroDl mediates nicotine-induced migration and invasion via regulation of the nicotinic acetylcholine receptor subunits in a subset of neural and neuroendocrine carcinomas. Mol Biol Cell, 2014. 25(11): p. 1782–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Osborne JK, et al. , NeuroD1 regulates survival and migration of neuroendocrine lung carcinomas via signaling molecules TrkB and NCAM. Proc Natl Acad Sci U S A, 2013. 110(16): p. 6524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Byers LA, et al. , Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov, 2012. 2(9): p. 798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang F, et al. , Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell Metab, 2018. 28(3): p. 369–382.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chalishazar MD, et al. , MYC-Driven Small-Cell Lung Cancer is Metabolically Distinct and Vulnerable to Arginine Depletion. Clin Cancer Res, 2019. 25(16): p. 5107–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Desai TJ, Brownfield DG, and Krasnow MA, Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature, 2014. 507(7491): p. 190–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swanton C and Govindan R, Clinical Implications of Genomic Discoveries in Lung Cancer. N Engl J Med, 2016. 374(19): p. 1864–73. [DOI] [PubMed] [Google Scholar]
- 58.Morinaga R, et al. , Sequential occurrence of non-small cell and small cell lung cancer with the same EGFR mutation. Lung Cancer, 2007. 58(3): p. 411–3. [DOI] [PubMed] [Google Scholar]
- 59.Zakowski MF, Ladanyi M, and Kris MG, EGFR mutations in small-cell lung cancers in patients who have never smoked. N Engl J Med, 2006. 355(2): p. 213–5. [DOI] [PubMed] [Google Scholar]
- 60.Sequist LV, et al. , Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med, 2011. 3(75): p. 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Niederst MJ, et al. , RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun, 2015. 6: p. 6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee JK, et al. , Clonal History and Genetic Predictors of Transformation Into Small-Cell Carcinomas From Lung Adenocarcinomas. J Clin Oncol, 2017. 35(26): p. 3065–3074. [DOI] [PubMed] [Google Scholar]
- 63.de Bruin EC, et al. , Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science, 2014. 346(6206): p. 251–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang J, et al. , Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science, 2014. 346(6206): p. 256–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jamal-Hanjani M, et al. , Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med, 2017376(22): p. 2109–2121. [DOI] [PubMed] [Google Scholar]
- 66.Wagner AH, et al. , Recurrent WNT pathway alterations are frequent in relapsed small cell lung cancer. Nat Commun, 2018. 9(1): p. 3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Keshet R, et al. , Rewiring urea cycle metabolism in cancer to support anabolism. Nat Rev Cancer, 2018. 18(10): p. 634–645. [DOI] [PubMed] [Google Scholar]
- 68.Barretina J, et al. , The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 2012. 483(7391): p. 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Polley E, et al. , Small Cell Lung Cancer Screen of Oncology Drugs, Investigational Agents, and Gene and microRNA Expression. J Natl Cancer Inst, 2016. 108(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McMillan EA, et al. , Chemistry-First Approach for Nomination of Personalized Treatment in Lung Cancer. Cell, 2018. 173(4): p. 864–878.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jin L, et al. , MAST1 Drives Cisplatin Resistance in Human Cancers by Rewiring cRaf-Independent MEK Activation. Cancer Cell, 2018. 34(2): p. 315–330.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rajapakse VN, et al. , CellMinerCDB for Integrative Cross-Database Genomics and Pharmacogenomics Analyses of Cancer Cell Lines. iScience, 2018. 10: p. 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meuwissen R, et al. , Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell, 2003. 4(3): p. 181–9. [DOI] [PubMed] [Google Scholar]
- 74.Chen HJ, et al. , Generation of pulmonary neuroendocrine cells and SCLC-like tumors from human embryonic stem cells. J Exp Med, 2019. 216(3): p. 674–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hou JM, et al. , Evaluation of circulating tumor cells and serological cell death biomarkers in small cell lung cancer patients undergoing chemotherapy. Am J Pathol, 2009. 175(2): p. 808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hou JM, et al. , Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J Clin Oncol, 2012. 30(5): p. 525–32. [DOI] [PubMed] [Google Scholar]
- 77.Hodgkinson CL, et al. , Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med, 2014. 20(8): p. 897–903. [DOI] [PubMed] [Google Scholar]
- 78.Drapkin BJ, et al. , Genomic and Functional Fidelity of Small Cell Lung Cancer Patient-Derived Xenografts. Cancer Discov, 2018. 8(5): p. 600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lallo A, et al. , Ex vivo culture of cells derived from circulating tumour cell xenograft to support small cell lung cancer research and experimental therapeutics. Br J Pharmacol, 2019. 176(3): p. 436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Almodovar K, et al. , Longitudinal Cell-Free DNA Analysis in Patients with Small Cell Lung Cancer Reveals Dynamic Insights into Treatment Efficacy and Disease Relapse. J Thorac Oncol, 2018. 13(1): p. 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carter L, et al. , Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med, 2017. 23(1): p. 114–119. [DOI] [PubMed] [Google Scholar]
- 82.Halvaei S, et al. , Exosomes in Cancer Liquid Biopsy: A Focus on Breast Cancer. Mol Ther Nucleic Acids, 2018. 10: p. 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Melo SA, et al. , Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature, 2015. 523(7559): p. 177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hu J, et al. , A signal-amplifiable biochip quantifies extracellular vesicle-associated RNAs for early cancer detection. Nat Commun, 2017. 8(1): p. 1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saunders LR, et al. , A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med, 2015. 7(302): p. 302ra136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Murai J, et al. , Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res, 2012. 72(21): p. 5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lok BH, et al. , PART Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin Cancer Res, 2017. 23(2): p. 523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Palma JP, et al. , ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res, 2009. 15(23): p. 7277–90. [DOI] [PubMed] [Google Scholar]
- 89.Pietanza MC, et al. , Randomized, Double-Blind, Phase II Study of Temozolomide in Combination With Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J Clin Oncol, 2018. 36(23): p. 2386–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.de Bono J, et al. , Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov, 2017. 7(6): p. 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Farago AF, et al. , Combination Olaparib and Temozolomide in Relapsed Small Cell Lung Cancer. Cancer Discovery, 2019. 19(9): p. 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dungey FA, Loser DA, and Chalmers AJ, Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-Ribose) polymerase: mechanisms and therapeutic potential. Int J Radiat Oncol Biol Phys, 2008. 72(4): p. 1188–97. [DOI] [PubMed] [Google Scholar]
- 93.Senra JM, et al. , Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol Cancer Ther, 2011. 10(10): p. 1949–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gani C, et al. , In vivo studies of the PARP inhibitor, AZD-2281, in combination with fractionated radiotherapy: An exploration of the therapeutic ratio. Radiother Oncol, 2015. 116(3): p. 486–94. [DOI] [PubMed] [Google Scholar]
- 95.Verhagen CV, et al. , Extent of radiosensitization by the PARP inhibitor olaparib depends on its dose, the radiation dose and the integrity of the homologous recombination pathway of tumor cells. Radiother Oncol, 2015. 116(3): p. 358–65. [DOI] [PubMed] [Google Scholar]
- 96.Laird JH, et al. , Talazoparib Is a Potent Radiosensitizer in Small Cell Lung Cancer Cell Lines and Xenografts. Clin Cancer Res, 2018. 24(20): p. 5143–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Doerr F, et al. , Targeting a non-oncogene addiction to the ATR/CHK1 axis for the treatment of small cell lung cancer. Sci Rep, 2017. 7(1): p. 15511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.de Carcer G, The Mitotic Cancer Target Polo-Like Kinase 1: Oncogene or Tumor Suppressor? Genes (Basel), 2019. 10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu Z, Sun Q, and Wang X, PLK1, A Potential Target for Cancer Therapy. Transl Oncol, 2017. 10(1): p. 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gutteridge RE, et al. , Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol Cancer Ther, 2016. 15(7): p. 1427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Murai J, et al. , Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget, 2016. 7(47): p. 76534–76550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sen T, et al. , CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res, 2017. 77(14): p. 3870–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lallo A, et al. , The Combination of the PARP Inhibitor Olaparib and the WEE1 Inhibitor AZD1775 as a New Therapeutic Option for Small Cell Lung Cancer. Clin Cancer Res, 2018. 24(20): p. 5153–5164. [DOI] [PubMed] [Google Scholar]
- 104.Shen J, et al. , PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res, 2019. 79(2): p. 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sen T, et al. , Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov, 2019. 9(5): p. 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sen T, et al. , Combination treatment of the oral CHK1 inhibitor, SRA737 and low dose gemcitabine, enhances the effect of PD-L1 blockade by modulating the immune microenvironment in small cell lung cancer. J Thorac Oncol, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Thomas A, et al. , Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J Thorac Oncol, 2019. 14(8): p. 1447–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gandhi L, et al. , Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol, 2011. 29(7): p. 909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rudin CM, et al. , Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res, 2012. 18(11): p. 3163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shoemaker AR, et al. , Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res, 2008. 14(11): p. 3268–77. [DOI] [PubMed] [Google Scholar]
- 111.Gardner EE, et al. , Rapamycin rescues ABT-737 efficacy in small cell lung cancer. Cancer research, 2014. 74(10): p. 2846–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Faber AC, et al. , mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer discovery, 2014. 4(1): p. 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Potter DS, et al. , Inhibition of PI3K/BMX Cell Survival Pathway Sensitizes to BH3 Mimetics in SCLC. Mol Cancer Ther, 2016. 15(6): p. 1248–60. [DOI] [PubMed] [Google Scholar]
- 114.Harlow ML, et al. , Trabectedin Inhibits EWS-FLI1 and Evicts SWI/SNF from Chromatin in a Schedule-dependent Manner. Clin Cancer Res, 2019. 25(11): p. 3417–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Santamaria Nunez G, et al. , Lurbinectedin Specifically Triggers the Degradation of Phosphorylated RNA Polymerase II and the Formation of DNA Breaks in Cancer Cells. Mol Cancer Ther, 2016. 15(10): p. 2399–2412. [DOI] [PubMed] [Google Scholar]
- 116.Belgiovine C, et al. , Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br J Cancer, 2017. 117(5): p. 628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cespedes MV, et al. , Lurbinectedin induces depletion of tumor-associated macrophages, an essential component of its in vivo synergism with gemcitabine, in pancreatic adenocarcinoma mouse models. Dis Model Mech, 2016. 9(12): p. 1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Calvo E, et al. , Antitumor activity of lurbinectedin (PM01183) and doxorubicin in relapsed small-cell lung cancer: results from a phase I study. Ann Oncol, 2017. 28(10): p. 2559–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Paz-Ares LG, et al. , Efficacy and safety profile of lurbinectedin in second-line SCLC patients: Results from a phase II single-agent trial. J Clin Oncol, 2019. 37. [Google Scholar]
- 120.Rudin CM, et al. , Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol, 2017. 18(1): p. 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carbone DP, et al. , Efficacy and safety of rovalpituzumab tesirine in patients With DLL3-expressing, ≥ 3rd line small cell lung cancer: Results from the phase 2 TRINITY study. Journal of Clinical Oncology, 2018. 36(15_suppl): p. 8507–8507. [Google Scholar]
- 122.Baeuerle PA, Kufer P, and Bargou R, BiTE: Teaching antibodies to engage T-cells for cancer therapy. Curr Opin Mol Ther, 2009. 11(1): p. 22–30. [PubMed] [Google Scholar]
- 123.Baeuerle PA and Reinhardt C, Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res, 2009. 69(12): p. 4941–4. [DOI] [PubMed] [Google Scholar]
- 124.Giffin MJ, et al. , Abstract 3632: BiTE® antibody constructs for the treatment of SCLC. Cancer Research, 2017. 77(13 Supplement): p. 3632–3632.28446465 [Google Scholar]
- 125.Sharma SK, et al. , Noninvasive Interrogation of DLL3 Expression in Metastatic Small Cell Lung Cancer. Cancer Res, 2017. 77(14): p. 3931–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Adumeau P, Davydova M, and Zeglis BM, Thiol-Reactive Bifunctional Chelators for the Creation of Site-Selectively Modified Radioimmunoconjugates with Improved Stability. Bioconjug Chem, 2018. 29(4): p. 1364–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Falkenberg KJ and Johnstone RW, Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov, 2014. 13(9): p. 673–91. [DOI] [PubMed] [Google Scholar]
- 128.Filippakopoulos P and Knapp S, Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov, 2014. 13(5): p. 337–56. [DOI] [PubMed] [Google Scholar]
- 129.Stathis A and Bertoni F, BET Proteins as Targets for Anticancer Treatment. Cancer Discov, 2018. 8(1): p. 24–36. [DOI] [PubMed] [Google Scholar]
- 130.Doroshow DB, Eder JP, and LoRusso PM, BET inhibitors: a novel epigenetic approach. Ann Oncol, 2017. 28(8): p. 1776–1787. [DOI] [PubMed] [Google Scholar]
- 131.Lam LT, et al. , Vulnerability of Small-Cell Lung Cancer to Apoptosis Induced by the Combination of BET Bromodomain Proteins and BCL2 Inhibitors. Mol Cancer Ther, 2017. 16(8): p. 1511–1520. [DOI] [PubMed] [Google Scholar]
- 132.Wang H, et al. , JQ1 synergizes with the Bcl-2 inhibitor ABT-263 against MYCN-amplified small cell lung cancer. Oncotarget, 2017. 8(49): p. 86312–86324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Teicher BA, et al. , Small cell lung carcinoma cell line screen of etoposide/carboplatin plus a third agent. Cancer Med, 2017. 6(8): p. 1952–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Margueron R and Reinberg D, The Polycomb complex PRC2 and its mark in life. Nature, 2011. 469(7330): p. 343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim KH and Roberts CW, Targeting EZH2 in cancer. Nat Med, 2016. 22(2): p. 128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gardner EE, et al. , Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell, 2017. 31(2): p. 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Reck M, et al. , Efficacy and safety of nivolumab (nivo) monotherapy versus chemotherapy (chemo) in recurrent small cell lung cancer (SCLC): Results from CheckMate 331. Annals of Oncology, 2018. 29. [Google Scholar]
- 138.Antonia SJ, et al. , Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol, 2016. 17(7): p. 883–895. [DOI] [PubMed] [Google Scholar]
- 139.Ott PA, et al. , Pembrolizumab in Patients With Extensive-Stage Small-Cell Lung Cancer: Results From the Phase Ib KEYNOTE-028 Study. J Clin Oncol, 2017. 35(34): p. 3823–3829. [DOI] [PubMed] [Google Scholar]
- 140.Hellmann MD, et al. , Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell, 2018. 33(5): p. 853–861.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chung HC, et al. , Phase 2 study of pembrolizumab in advanced small-cell lung cancer (SCLC): KEYNOTE-158. Journal of Clinical Oncology, 2018. 36(15_suppl): p. 8506–8506. [Google Scholar]
- 142.Goodman AM, et al. , Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther, 2017. 16(11): p. 2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yu H, et al. , PD-L1 Expression by Two Complementary Diagnostic Assays and mRNA In Situ Hybridization in Small Cell Lung Cancer. J Thorac Oncol, 2017. 12(1): p. 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Olson CM, et al. , Development of a Selective CDK7 Covalent Inhibitor Reveals Predominant Cell-Cycle Phenotype. Cell Chem Biol, 2019. 26(6): p. 792–803.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ponath P, et al. , A Novel, Fully Human Anti-fucosyl-GM1 Antibody Demonstrates Potent In Vitro and In Vivo Antitumor Activity in Preclinical Models of Small Cell Lung Cancer. Clin Cancer Res, 2018. 24(20): p. 5178–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chu QS, et al. , Initial Results of BMS-986012, a First-in-Class Fucosyl-GM1 mAb, in Combination With Nivolumab, in Pts With Relapsed/Refractory (rel/ref) Small-Cell Lung Cancer. Annals of Oncology, 2017. 28: p. v539–v542. [Google Scholar]
- 147.Weiskopf K, et al. , CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J Clin Invest, 2016. 126(7): p. 2610–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Doyle A, et al. , Markedly decreased expression of class I histocompatibility antigens, protein, and mRNA in human small-cell lung cancer. J Exp Med, 1985. 161(5): p. 1135–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Burr ML, et al. , An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell, 2019. 36(4): p. 385–401.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]