

Abstract
Context
The hypothalamic melanocortin 4 receptor (MC4R) pathway serves a critical role in regulating body weight. Loss of function (LoF) mutations in the MC4R pathway, including mutations in the pro-opiomelanocortin (POMC), prohormone convertase 1 (PCSK1), leptin receptor (LEPR), orMC4R genes, have been shown to cause early-onset severe obesity.
Methods
Through a comprehensive epidemiological analysis of known and predicted LoF variants in thePOMC, PCSK1, andLEPR genes, we sought to estimate the number of US individuals with biallelic MC4R pathway LoF variants.
Results
We predict ~650α-melanocyte-stimulating hormone (MSH)/POMC, 8500PCSK1, and 3600LEPR homozygous and compound heterozygous individuals in the United States, cumulatively enumerating >12,800 MC4R pathway–deficient obese patients. Few of these variants have been genetically diagnosed to date. These estimates increase when we include a small subset of less rare variants:β-MSH/POMC,PCSK1 N221D, and aPCSK1 LoF variant (T640A). To further define the MC4R pathway and its potential impact on obesity, we tested associations between body mass index (BMI) and LoF mutation burden in thePOMC, PCSK1, andLEPR genes in various populations. We show that the cumulative allele burden in individuals with two or more LoF alleles in one or more genes in the MC4R pathway are predisposed to a higher BMI than noncarriers or heterozygous LoF carriers with a defect in only one gene.
Conclusions
Our analysis represents a genetically rationalized study of the hypothalamic MC4R pathway aimed at genetic patient stratification to determine which obese subpopulations should be studied to elucidate MC4R agonist (e.g., setmelanotide) treatment responsiveness.
This is a genetic epidemiology study of melanocortin 4 receptor (MC4R) pathway dysfunction in obesity aimed at genetic patient stratification for clinical study of MC4R agonist treatment responsiveness.
The hypothalamic melanocortin 4 receptor (MC4R) pathway plays a critical role in controlling food intake and energy expenditure through brain periphery signaling networks. Genetic case studies of several monogenic forms of early-onset obesity have revealed the significance of loss of function (LoF) mutations within the MC4R pathway, including leptin, the leptin receptor (LEPR) (1), pro-opiomelanocortin (POMC) (2), prohormone convertase 1 (PCSK1), and theMC4R genes (3–6) (Fig. 1), with clinical examples described in the literature (7–12). Leptin, a hormone released from white adipose tissue, regulates appetite and energy expenditure in part through engagement of its cognate receptor, LEPR, expressed on arcuate nucleus POMC neurons (13). POMC is processed into multiple neuropeptides by enzymes including the prohormone convertase PCSK1. The POMC-derived neuropeptides,α- andβ-melanocyte-stimulating hormone (MSH), activate MC4Rs on second-order neurons to induce satiety and increase energy utilization, thereby promoting weight loss (14). Inactivating mutations in these key MC4R pathway genes result in early-onset obesity, providing clinical validation of the MC4R pathway as a therapeutic obesity target. Preclinical (15) and clinical studies (1–3) indicate that the MC4R agonist setmelanotide (also known as RM-493 or BIM-22493) is effective at promoting weight loss in patients with diverse MC4R pathway deficits. Setmelanotide phase II studies have demonstrated a more dramatic clinical benefit in patients with POMC (1) or LEPR deficiency (2) when compared with setmelanotide treatment in general obesity or MC4R haploinsufficiency obesity (3). Given our lack of understanding of the genetic factors that predispose to severe obesity, we initiated a genetic epidemiological analysis of MC4R pathway defects (inPOMC, PCSK1, orLEPR genes) to determine their contribution to severe obesity.
Figure 1.

Schematic outline of the hypothalamic MC4R pathway with POMC, PCSK1, LEPR, Leptin (LEP), and the MC4R as critical mediators of appetite, energy expenditure, and body weight. Red crosses on each gene indicate that genetic evidence is available linking a genetic deficiency in each gene with severe early-onset childhood obesity and hyperphagia.
In addition to deepening our understanding of the genetics underlying obesity, such efforts could identify biologically rationalized subpopulations of individuals who may benefit from a precision medicine strategy that seeks to pharmacologically compensate for genetic MC4R pathway deficits. Genetic defects in the MC4R pathway leading to setmelanotide-responsive obesity [LEPR, POMC, and MC4R (1–3)] are predicted to includePCSK1 deficiency (16,17),Magel2 deficiency (15), and, based on our clinical data, Bardet Biedl syndrome (BBS) (18). In this study, initiated in late 2012, we performed an epidemiological analysis of MC4R pathway genetic variants in theLEPR, PCSK1, andPOMC genes, identified in multiple biobanks, to build a real-world understanding of the prevalence and impact of MC4R pathway defects on the obese patient population. Identification and characterization of LoF variants in this pathway may increase our understanding of the control of energy balance and may predict a subset of patients responsive to MC4R agonist therapy.
Materials and Methods
Compilation of LoF variants forPOMC,PCSK1, andLEPR
Obesity LoF variants were curated via personal communication, a PubMed search for association and functional studies, and searches of databases including the Human Gene Mutation Database (19) and ClinVar (20). An additional search was done for any potential LoF variants not reported in the literature but seen in one of our cohorts. We used SnpEff version 4.0b (21) to define nonsense, frameshift, and splice site variants, and WuXi NextCODE DeepCODE (likelihood scores >0.90) to define likely functional missense variants as described inSupplemental Materials and Methods. These variants were classified into two groups based on the nature of the supporting evidence for LoF (Table 1). Group 1 includes variants experimentally validated as LoFs in the literature and any additional variants encoding nonsense, frameshift indel, or splicing altering variants that, based on previous published protein functional studies, can be confidently predicted to be LoF. Group 2 includes predicted LoF missense variants with a high functional impact based on a Deep Artificial Neural Network, DeepCODE algorithm (Supplemental Materials and Methods).
Table 1.
Grouping of MC4R Pathway Variants
| Category | Gene Source | POMC | PSCK1 | LEPR | Total |
|---|---|---|---|---|---|
| Group 1 | Literature | 30 | 31 | 22 | 83 |
| Novel (nonsense, splice site, frameshift) | 20 | 24 | 39 | 83 | |
| Group 2 | Computationally predicted high-impact missense | 85 | 176 | 160 | 421 |
| Total | 135 | 231 | 221 | 587 |
Key criteria for defining LoF group 1 variants
The criteria by which all nucleotide sequenced DNA will be interpreted and will be considered for LoF status include the following:
Being positive for a mutation (nonsense, insertions, deletions) that prematurely terminates the protein product at or before the last documented deleterious mutation of the gene (precedent for this criterion is in the BRACAnalysis CDxTM genetic testing label) or is shown or predicted, based on sound scientific principles, to lead to an LoF.
-
Variants that:
○ lead to the production of a protein with predicted changes to the amino acid sequence which, based on sound scientific principles, includes informative nonsense mutations, informative missense mutations, or informative frameshift mutations AND
○ exhibit clinical symptoms, either as published data or based on unpublished patient data, such as family pedigree data predicting high body mass index (BMI) segregation with the LoF variants.
-
Variants that:
○ lead to the production of a protein with predicted changes to the amino acid sequence which, based on sound scientific principles, includes informative nonsense mutations, informative missense mutations, or informative frameshift mutations, AND
○ with epidemiological data that show an association of the POMC obesity deficiency variant with obesity based on published data or based on new studies involving epidemiology data, such as data that predict high BMI association with the LoF variants.
-
Variants that:
○ lead to the production of a variant messenger RNA (mRNA) or protein with predicted changes to the amino acid sequence which, based on sound scientific principles, include informative nonsense mutations, informative missense mutations, or informative frameshift mutations AND
-
○ evidence that the variant shows impaired expression or functionality, which can include:
▪ expression of the mutant protein or the relevant ligand in a cell-based system providing evidence for LoF in a cell-based assay; LoF can be shown in tests where the variant ligand is compared with a positive control, revealing an impaired ability to activate the MC4R as either a full or partial agonist.
▪ impaired ability to detect the mutated variant ligand protein at concentrations equivalent to that of the wild-type (WT) ligand.
▪ inability to detect the enzymatic activity, at or near WT levels.
▪ reduced mRNA quantity or functionality or inability to detect the mRNA at or near WT levels.
▪ novel nucleotide sequence changes that lead to previously documented LoF amino acid changes.
Variants where expression of the variant ligand or enzyme replaces the WT ligand or enzyme in anin vivo/ex vivo preclinical model and shows LoF or reveals association with a metabolic phenotype, providing evidence for obesity as defined by significantly increased fat mass, when compared with the WT control gene.
Genetic variants for which available evidence indicates a strong likelihood, but not definitive proof, that the mutation is deleterious (i.e., LoF based on computational modeling).
Genetic variants for which a strong, clinically important response to an MC4R agonist (e.g., setmelanotide) is available.
Criteria for defining LoF group 2 variants (variants with minor allele frequency <0.1%)
Available evidence indicates a strong likelihood, but not definitive proof, that the mutation is deleterious based on computational modeling with a DeepCODE score >0.9.
The DeepCODE score had a >70% positive predictive value, based on our MC4R pathway data training set. We limited the missense variants to allele frequency about <0.1% [in Genome Aggregation Database (gnomAD)] for nonpublished variants, because variants with a higher frequency probably would have been reported in previous studies if they had functional relevance.
Although missense variants inPOMC, PCSK1, andLEPR with computationally predicted high functional impact do not necessarily lead to loss or diminished protein functions, because of the lack of known gain-of-function variants, we reasoned that variants with the highest predicted functional impact exhibit likely LoF.
LoF variants as defined are scored according to the American College of Medical Genetics and Genomics guidelines to conform to the definitions ofpathogenic, likely pathogenic, or variants of uncertain significance. In this effort, the rule-based approach just defined clarifies criteria that we will research further to support variant classification.
Prevalence estimate of LoF variant carriers
We calculated the frequencies of carriers and prevalence of homozygotes and compound heterozygotes in each gene by using the nucleotide sequence–based gnomAD data (available athttp://gnomad.broadinstitute.org/, downloaded in November 2016). For each gene, we estimated the number of individuals with homozygous or compound heterozygous genotypes from the allele frequencies by using Hardy-Weinberg proportions. We assumed no negative selection of LoF variants. We also assumed random mating between obese and nonobese populations and between different races and ethnicities; however, nonrandom mating in these populations may increase the LoF frequencies within the obese population or in some races or ethnicities. Furthermore, because most of the variants are rare and in close physical proximity, we assume the mutations occurred on separate haplotypes and that a recombination event has not yet occurred between them. The CIs for each individual genotype were approximated with the derived variance (22). For prevalence estimates, we used a US population size of 300 million, and prevalence was estimated per 100,000 individuals within each group.
Association analysis
Association of variants with obesity-related traits was done by regressing the genotypes of interest on either BMI or obesity, with age, age squared, sex, and six genetic ancestry measures as covariates. The BMI effect size (β) was computed with linear regression. The obesity ORs were computed by dichotomizing to severe obese cases (BMI >40 kg/m2) and normal controls (BMI <25 kg/m2). Logistic regression was used to analyze the common variants, and Firth regression was used for rare variants. For burden tests, individuals with the relevant genotypes were grouped into a single test variable in the regression model and were compared with those with the reference (normal) allele (noncarriers or WT) for all variants tested. Meta-analyses of the different datasets and their subpopulations were performed on the individual study effect size estimates via a random effects model in the R metafor package (http://www.metafor-project.org/) to obtain an overall effect size estimate andP value.
Results
Known and predicted LoF genetic variants inPOMC,PCSK1, andLEPR
Using literature search criteria that covered genetic studies on severe early-onset MC4R pathway deficiency; obesity syndrome association studies of common variants with obesity; molecular functional studies; and predictive algorithms on genetic variants, we assembled a list of potential LoF genetic variants in thePOMC,PCSK1, andLEPR genes (see Materials and Methods for a description of our rule-based approach to define LoF). All published variants were systematically evaluated based on a combination of factors including functional studies, identification in patients, prediction to be nonsense, categorization as frameshift or splice site mutation, categorization as likely impactful missense variants, and association with obesity, or segregation analysis, to create a list of 83 credible LoF variants (Table 1;Supplemental Tables 1–3). We also queried publicly available genetic datasets including gnomAD and the UK10K and UK Biobank, along with Mt Sinai Hospital (MSH) internal data (Supplemental Table 4) for potentially novel LoF variants and found an additional 83 nonsense, frameshift, and splice site variants and 421 likely LoF missense variants identified through computational methods (Supplemental Tables 5 and 6;Supplemental Fig. 1). In total, our analyses included 587 known and predicted LoF variants, including 135, 231, and 221 variants inPOMC,PCSK1, andLEPR, respectively (Table 1;Supplemental Tables 1–3). These variants were classified into two groups as described in Materials and Methods (Table 1). The locations of the group 1 and group 2 variants with respect to protein functional domains are outlined inSupplemental Fig. 2. All the group 1 variants exceptPCSK1 N221D andPOMCβ-MSH R236G are rare variants with minor allele frequency (MAF) <0.1%.
Prevalence estimation of LoF variants based on gnomAD analysis
Based on our curated list of group 1 and 2 LoF variants, we next estimated the prevalence of homozygotes and compound heterozygotes in the United States. These individuals represent the subpopulation predicted to be particularly responsive to setmelanotide. Allele frequencies in gnomAD (a database of ~140,000 sequenced individuals of varying ethnicities) were used to compute cumulative allele frequency and to estimate prevalence of homozygotes and compound heterozygotes for each gene (see Materials and Methods for details and assumptions). The gnomAD database has an overall race and ethnicity distribution that is very similar to US census results (Supplemental Fig. 3;Supplemental Table 7). However, heterogeneity within racial and ethnic groups has not been studied, which is probably important for participants of Hispanic ethnicity. Based on a US population of ~300 million, the estimated numbers of homozygotes and compound heterozygotes are 656 individuals (95% CI, 579 to 733) forα-MSH/POMC, 8546 (95% CI, 7368 to 9725) forPCSK1, and 3638 (95% CI, 3308 to 3967) forLEPR. In total, we predict just over 12,800 individuals in the United States who may have a genetically deficient (homozygous or compound heterozygous) MC4R pathway (Fig. 2). This predicted patient population remains largely undiagnosed, because genetic testing is rarely performed in obese patients. Based on available preclinical and clinical data and the LoF status for each of the variants, we anticipate this patient population to respond to MC4R agonist treatment.
Figure 2.

Prevalence estimations of group 1 and 2 predicted patient populations in the United States. Based on a US population of ~300 million, the estimated numbers of homozygotes and compound heterozygotes (error bars represent 95% CIs).
These estimates were restricted to rare variants. There are a small number of additional predicted LoF,β−MSHPOMC, andPCSK1 alleles with frequencies >0.1%, but the impact of these variants on BMI is less clear. When we include these less rare alleles and combineα-MSH withβ−MSH/POMC, 9914 (95% CI, 8474 to 11,354) homozygous or compound heterozygous individuals are predicted in the United States. ForPCSK1, the N221D variant has evidence of association with obesity in epidemiological studies (23–28). Although functional studies have shown a modest, 10% reduction in PCSK1 activity in N221D mutants compared with WT (29), it is possible thatPCSK1 N221D may confer some level of dominant negativity on the remaining WT PCSK1 protein, thereby potentially increasing its impact (30). When thePCSK1 N221D allele is included in prevalence estimates, 564,529 homozygous or compound heterozygous individuals (95% CI, 493,338 to 635,720) are predicted in the United States. For theseβ-MSH/POMC andPCSK1 deficiencies, evidence that the mutations predispose to severe obesity and may confer MC4R agonist sensitivity remains to be established. Therefore, our conservative estimate of MC4R pathway deficiency, which excludes these less rare variants, assumes about 12,800 obese patients in the United States.
To determine whether proportional prevalence estimates vary by race or ethnicity, we calculated the cumulative allele frequency and genotype prevalence within each race or ethnic group (as defined in gnomAD), assuming preferential mating within the ethnicity (see Materials and Methods,Fig. 3, andSupplemental Fig. 4). We found substantial variability of predicted allele prevalence for the three MC4R pathway genes across race and ethnicity (Fig. 3), suggesting that the identification of treatment target populations should consider ancestral background. Applying roughly estimated US racial or ethnic proportions from the 2010 to 2014 censuses to the gnomAD ancestry groups, we next estimated the cumulative US prevalence but allowed for violations of the random mating assumption by racial or ethnic group [“All (NRM)” inFig. 3E], and we compared this result to the original random mating estimates (“All” inFig. 3E). Overall, violations of the random mating assumption do not greatly affect the overall US prevalence estimates (a ≤25% increase).
Figure 3.
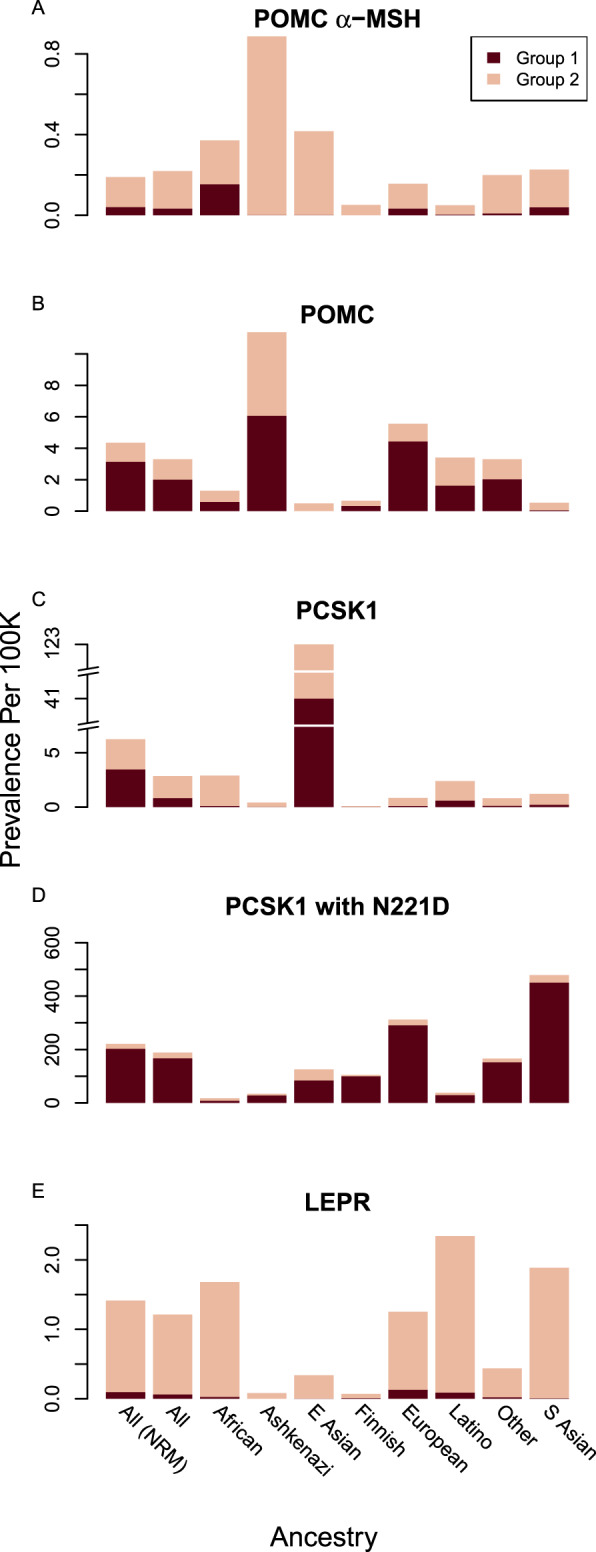
Stacked barplots for prevalence estimates of homozygous and compound heterozygous carriers of group 1 and 2 variants, listed separately, in (A)POMCα-MSH, (B)POMC, (C)PCSK1 without N221D, (D)PCSK1 with N221D, and (E)LEPR, based on the gnomAD database, which includes estimated ancestry data on a number of US races and ethnicities. Prevalence is reported as the number of homozygous and compound heterozygous LoF genotyped individuals per 100,000 individuals within each group. The “All” group is based on the overall gnomAD allele frequencies, and the “All (NRM)” group is based on nonrandom mating within ethnicities given the estimated percentage of individuals within each ethnic group in the United States. E, East; S, South.
Allele burden of LoF variants is associated with BMI and obesity in the UK Biobank
Beyond the known severe childhood obesity conditions defined by homozygous or compound heterozygous group 1 mutations inPOMC, PCSK1, orLEPR, individuals carrying multiple LoF alleles across these three genes may also exhibit an increased risk for obesity because of a cumulative burden of genetic deficiency on the MC4R pathway. We hypothesize that if this cumulative genetic deficiency burden indeed affects the risk of obesity, it may also predict a clinical response to MC4R agonist treatment.
We therefore analyzed the 120,000 British individuals from the UK Biobank (31) to investigate whether allele burden of LoF variants within the three genes was associated with BMI. We first tested whether any individual variants in groups 1 and 2 were associated with BMI. We assume that because of the low frequency of these LoF variants and because the UK Biobank genetic data are genotyped, none of these variants were found to be statistically significant at theα = 0.05 level, except for the well-studiedPCSK1 N221D variant [additive effectβ = 0.14, SE = 0.04,P = 0.002, with genotypic effects presented inSupplemental Fig. 5B]. Analysis of additional rare missense variants with MAF <0.5% found a potential LoF variant inPCSK1, T640A (MAF 0.3%,β = 0.68, SE = 0.18,P = 0.00017;Supplemental Fig. 5B), which was significantly associated with BMI. Because the effect sizes for N221D and the newly identified T640A variant were consistent across other populations and the overall effect was statistically significant (Supplemental Figs. 5 and 6), we add T640A to group 1, and both variants are also included in subsequently described allele burden tests. Including T640A in thePCSK1 prevalence estimate adds >33,000 individuals, mainly N221D/T640A compound heterozygotes.
We calculated the genetic burden across the populations for group 1 and 2 alleles, includingPCSK1 N221D andPCSK1 T640A. We first examined those who are heterozygous for only one of the LoF variants and noted an association with higher BMI (β = 0.35, SE = 0.14,P = 0.012) when compared with individuals who did not carry these alleles (Fig. 4A). We then evaluated those who carry two or more variants (have two or more alleles) and observed an association with higher levels of BMI (β = 2.79, SE = 1.16,P = 0.016) compared with those that do not carry these alleles (Fig. 4B). To confirm that this result was not driven primarily byPCSK1 N221D because of its high allele frequency, and to evaluate the significance of the contribution from other group 1 or 2 variants on top of a heterozygous N221D background, we compared individuals who were heterozygous for N221D and at the same time heterozygous for another group 1 or 2 allele to those who were N221D heterozygotes without any additional variants. The additional allele on the N221D heterozygote background results in an association with a higher BMI of 0.57 in this population (P = 0.058, SE = 0.30,Fig. 4C). Finally, we examined individuals heterozygous for LoF variants in at least two of the three genes (defined as a composite genotype). The number of individuals in this group was small (n = 234), and most of them carried one N221D allele. However, we still observed a strong trend toward an association with higher BMI (β = 0.52, SE = 0.32,P = 0.098) when compared with noncarrier or WT individuals (Fig. 4D). We also saw a significant increase in the proportion of individuals with two or more group 1 or group 2 alleles in two or more genes in a single individual as we compared different BMI categories represented in the UK Biobank (Fig. 5;P = 0.0008). Overall, the magnitude of association betweenPOMC, PCSK1, orLEPR variants and BMI was small. This finding may be related to the lack of availability of sequencing data for these analyses, which may result in missing information on potentially high-effect variants that could affect BMI. Additional studies are needed to carefully define the magnitude of effects of carrying two or more LoF alleles in two or more genes. Nevertheless, these results support an association between a burden of LoF variants in the MC4R pathway and higher levels of BMI, in comparison with noncarriers of these LoF variants. Based on these findings we are now investigating whether the MC4R pathway LoF allele burden indeed predisposes patients to severe obesity (additional results from analyses on rare variants and allele burdens are presented inSupplemental Results,Supplemental Tables 8–10, andSupplemental Figs. 7–9).
Figure 4.

Forest plots for (A) effects of carrying one or more group 1 plus group 2 variants vs WT for all group 1 plus group 2 variants; (B) effects of carrying two or more group 1 plus group 2 variants vs WT for all group 1 plus group 2 variants; (C) effects of carrying only one copy ofPCSK1 N221D plus another group 1 or group 2 variant vs N221D heterozygotes; and (D) effects of carrying two group 1 plus group 2 variants from different genes vs WT for all group 1 plus group 2 variants. The forest plots represent the estimated effect sizes and CIs for each dataset and subgroup. Groups that did not have any individuals with the tested variants are not represented in the plots. Each dataset is assigned a weight based on the inverse of the variance in the estimate, and thus larger or more homogenous datasets tend to have larger weights. The overall effect size andP value are a weighted average of these studies. A test of heterogeneity was also performed to determine whether the different studies had significantly different estimated effects (theI2 statistic describes the percentage of variation across studies that is due to heterogeneity rather than chance). Beta reflects the BMI shift. AA, black or African American from the United States; AFR, black or African American not from the United States with high African ancestry; BB, MSH Biobank; BM, MSH BioMe Biobank; EA, white of European descent; EA_AJ, white of Jewish descent; HA_DOM, Hispanic from the Dominican Republic; HA_LAT, Hispanic from Central or South America not identifying as white; HA_PUR, Hispanic from Puerto Rico; UKBB, UK Biobank; UKBB_BB, UK Biobank array; UKBB_BL, BiLEVE array.
Figure 5.

The proportion of individuals in the UK Biobank carrying two or more group 1 and 2 variants (plusPCSK1 N221D and T640A) across the three genes for BMI category (comparison of BMI <20, 20–30, 30–40, and ≥40 kg/m2). For groups 1 and 2, the carrier frequency tends to increase as BMI increases, and there is a significant difference in the carrier frequencies between the BMI <20 and BMI >40 groups (error bars represent 95% CIs). Analyzing group 1 data only did not reveal a significant difference between the groups, because of the low numbers or absence of group 1 homozygotes, heterozygotes, or composites in the BMI <20 kg/m2 and BMI >40 kg/m2 groups. The total number of noncarriers (top) and carriers (bottom) for each BMI category are shown in the columns.
We also examined many common variants in these three genes that have been heavily studied in published genetic association studies for BMI and other anthropometric traits (seeSupplemental Results for a description of the data andSupplemental Table 11). Our data on common variants in the MC4R pathway based on a polygenic risk score (Supplemental Results,Supplemental Table 10, andSupplemental Figs. 10–11) indicate that it is likely that overall allele burden in the MC4R pathway when common alleles are superimposed on the monogenic impactful group 1 or group 2 alleles contributes to increased BMI. This effect is analogous to the substantial effect of the polygenic risk score of 94 common single nucleotide polymorphisms on the risk of breast and ovarian cancer in BRCA1 and BRCA2 rare LoF carriers (32). It will be worthwhile evaluating to what extent these common alleles contribute to severe obesity when in combination with group 1 or group 2 alleles and predict sensitivity to MC4R pathway supplementation therapy, to mitigate severe obesity and its comorbidities.
LoF carriers in Mount Sinai BioMe Biobank
To assess the ease of identifying potential patients with MC4R pathway dysfunction, we evaluated the MSH Biobank for individuals potentially eligible (based on severe obesity) and responsive (based on genotype) for setmelanotide to facilitate a future phase II proof-of-concept clinical trial. Of ~16,000 individuals, 1273 met the BMI ≥40 kg/m2 requirement, of which 89 adults and 3 children or adolescents (>97th percentile for BMI) were carriers of one or more group 1 or 2 alleles, with a total of 15 distinct alleles detected. We identified threePCSK1 N221D homozygotes and 84 heterozygotes, including fourβ-MSH R236G carriers with BMI >40 kg/m2. A single composite genotype (an affected allele in more than one gene, thereby providing a potential MC4R pathway allele burden) was identified in a severely obese 5-year-oldLEPR S274*,PCSK1 N221D carrier who shows other symptoms related to rare forms ofPCSK1-deficiency childhood obesity, including repeated instances of gastroenteritis that began at 17 months of age (33). Non–PCSK1 N221D allele carrier composites were not identified in this cohort.
Discussion
Our aim has been to initiate an analysis of the possible extent of the obese US patient population that is associated with diverse forms of MC4R pathway impairments and to unravel the importance of allele burden in this pathway for obesity. These studies are aimed at the genetic characterization of a patient population with MC4R pathway deficiency, predicted to be eligible for setmelanotide treatment. Because of the enormous defects in appetite regulation in these patients, bariatric surgery is unlikely to have clinical benefit, further underscoring the importance of pharmacotherapy with an MC4R agonist and genetic screening to determine eligibility for surgery. Through comprehensive epidemiological analysis of the MC4R pathway genesPOMC,PCSK1, andLEPR, we estimate that there are ~12,800 MC4R pathway–defective obese patients in the United States. The alleles that make up the repertoire of LoF variants in these three genes are based on a select set of LoF rules (seeSupplemental Materials and Methods) that define variant function. The estimate of the US-based MC4R pathway–deficient population is based on random mating in the population, assuming Hardy-Weinberg equilibrium. Because we noted significant allele prevalence variability between racial and ethnic groups, where random mating assumptions in the United States may not hold, the prevalence of biallelic LoF obesity could be higher. When we assumed selected mating preferences within racial and ethnic groups, overall prevalence estimates increased moderately, by ~25%. Furthermore, it is known that biallelic LoF alleles result in early mortality, and therefore negative selection may have reduced our prevalence estimates in these adult populations. We therefore believe that the estimate of ~12,800α-MSHPOMC, PCSK1, andLEPR biallelic rare allele (prevalence <0.1%) deficiencies represents a conservative assumption of the prevalence of such patients in the United States. The number of potential patients with LoF increases if composite group 1 or group 2 genotypes contribute to severe obesity and if some of the more abundant group 1 and group 2 LoF alleles not considered in the prediction of ~12,800 patients (such asPOMCβ-MSH andPCSK1 N221D, T640A) may predispose to severe obesity in some patients. Screening for these genetic risk factors is not part of routine medical practice, even though the pediatric endocrinology guidelines (11) now favor genetic screening for obesity. Newborn screening to detect MC4R pathway deficiencies can be predicted to improve patient identification and patient care. Based on previously published work and our analysis we believe that a discussion is warranted, aimed at defining an obesity gene panel for a severe obesity screening initiative, where early detection may lead to early intervention.
The selective MC4R agonist setmelanotide (34), uniquely within this class of compounds, promotes significant and persistent weight loss via appetite suppression (2) and increased energy utilization (35), without evidence of adverse cardiovascular effects. Preliminary support for lack of effects of setmelanotide on cardiovascular parameters was initially obtained in a preclinical obese primate model (36) and subsequently in clinical trials, including in patients with general obesity (37), MC4R haploinsufficiency (3), POMC (2), and LEPR deficiency (1). Weight loss from setmelanotide treatment in the general obese andMC4R heterozygous populations was noted at ~0.9 kg/week and ~0.6 kg/week (3), respectively. Setmelanotide treatment of two adult patients with genetic POMC deficiency not only completely normalized hyperphagia, leading to rapid weight loss, on the order 2.5 kg/week (primarily through fat mass reduction), but also improved hyperleptinemia and hyperinsulinemia (2). Furthermore, one patient had significant reductions in blood pressure and heart rate, and neither patient had any serious side effects (2). We aim to incorporate genetic MC4R pathway deficiency in the proposed efficacy evaluations of setmelanotide applied to specific MC4R pathway–impaired patient populations, exemplified in this study.
We provide evidence that allele burden in the MC4R pathway can contribute to obesity. Interestingly, our allele burden analysis provides evidence of an impact on BMI, even though we have thus far tested only variants in thePOMC,PCSK1, andLEPR genes. We anticipate that many other genes will play a role in MC4R pathway–mediated obesity. For example, mutations in the protein complex of the basal body of primary cilia, as characterized by BBS, predispose to obesity and are believed to impair LEPR and MC4R signaling. This assertion is further supported by the finding that several BBS knockout rodents retain sensitivity to the melanocortin agonist melanotan II (38). Indeed, we have recently shown that patients with BBS are sensitive to setmelanotide treatment (18). Furthermore, there is evidence of allelic burden in BBS where a specific composite genotype in the Newfoundland population with BBS is proposed to contribute to BBS allele burden and obesity (39). Similarly, mutations in the5HT2C gene lead to obesity in rodents while remaining responsive to melanotan II, befitting its positioning as an afferent regulator of POMC neuron function (40). We anticipate that monogenic forms of MC4R pathway obesity, in addition to the genes listed earlier, may include carboxypeptidase E deficiency (41), Alström syndrome (42), Smith-Magenis syndrome (43), Magel2 deficiency as in Schaaf-Yang syndrome, Prader-Willi syndrome (15), and potentially hyperphagia and obesity in fragile X syndrome (44). We are in the process of defining which of these genetic deficiencies are involved in regulation of the MC4R pathway so we can develop clinical trials to assess the utility of setmelanotide therapy in each of these patient populations. We also plan to enroll severely obese patients with a burden of different MC4R pathway alleles to determine setmelanotide responsiveness for weight loss. Interestingly, our data indicate that some heterozygous LoF alleles, and some common alleles in these genes, may contribute to the prevalence of obesity. As demonstrated, a polygenic risk score that aggregated common and group 1 and group 2 alleles was associated with both BMI and obesity (Supplemental Results;Supplemental Table 10). However, based on published data for these common alleles, we predict that in the absence of an LoF group 1 or group 2 allele, these common alleles cannot significantly contribute to severe obesity. We conclude that overall MC4R pathway allele burden may outline a risk for increased BMI and severe obesity, where a core of impactful LoF alleles appears essential for MC4R pathway dysfunction. Establishing the response to setmelanotide in the severely obese patient population for these diverse classes of MC4R pathway dysfunctions is a complex genetic and clinical challenge. Because the rank order of treatment sensitivity to setmelanotide or other MC4R agonists, in the analysis of selected monogenic defects in the MC4R pathway, correlates well when compared between rodent and human physiological responses (1–3,15), we have excellent preclinical and clinical models at our disposal to further unravel MC4R pathway genetic contributors to obesity.
Our study has several limitations. First, the prediction of LoF allele status is based on several assumptions, all of which may have affected our results. Whenever possible, we applied conservative metrics to define allele function. These metrics are summarized in Materials and Methods, where we outline a set of rules that define whether a variant can be considered LoF. However, group 2 variants lack experimental data from biochemical or pharmacological studies, and additional research comparing diverse alleles in standardized cell based assays is needed to shed light on a variant’s relative LoF status. Second, the detection of indels is compromised by false positives and false negatives, and thus we may be missing important frameshift or copy number variants. Here, common variants associated with BMI might be of use for tagging haplotypes or rare indels that are functional but difficult to detect, with the caveat that these are predicted to be population specific and may not carry across race or ethnicity because of differences in linkage disequilibrium structure. Third, we were underpowered to detect associations between rare variants and BMI. For example, although we identified severalβ-MSH variants, including R236G (a weak partial MC4R agonist), Y221C, and E206X, an association with obesity could not be confirmed in the UK Biobank. Some POMCβ-MSH alleles have been correlated with obesity in previous studies, including two studies in humans and a study in obese Labrador dogs (45–47). If these variants are indeed LoF, with a causative role in severe obesity, our lack of detection may reflect the limited sample size or a less obese UK Biobank population (Supplemental Fig. 12). Considering the biochemical evidence supporting reduced functionality of the R236G variant (48), we are testing in a clinical trial whether the R236G allele carriers respond uniquely to setmelanotide to assess its functional relevance in the MC4R pathway. Fourth, there could be larger environmental effects in a population, more diverse genetic effects, or different genetic effects because most variant discovery occurred in European populations. Fifth, we could be missing synonymous variants that are cryptic splice sites. Sixth, our data indicate that it is critical to pay close attention to the race and ethnicity of the population under study. Enrichment for unique alleles will probably occur in selected isolated populations, where specific alleles can be found at unusually high prevalence (49). This consideration, in conjunction with the identification of a cumulative or synergistic genetic burden for obesity, highlights the complexity of the planned clinical studies aimed at unraveling the MC4R pathway, where we anticipate that evaluation of numerous subgroups with specific genotypes will be necessary to determine the validity of MC4R pathway deficiency and drug treatment responsiveness. Finally, the UK Biobank does not have sequencing data, and therefore many rare and interesting alleles will be missing, making the genetic burden test an underrepresentation of the actual allele burden effect.
We initiated a broad array of collaborative efforts with existing biobank and electronic medical record datasets, aimed at evaluating the genetic and clinical etiology relationships in the MC4R pathway. We anticipate that genes of interest will continue to be identified in this pathway. For example, it has been recently reported that LoF mutations in ADCY3 colocalizing with MC4R at the primary cilia of hypothalamic neurons are causal for monogenic severe obesity, highlighting the importance of ADCY3 as a therapeutic target for obesity (50–53). We aim to use a similar approach as described in this study for these genes, to identify relevant variants, estimate prevalence, and determine the cumulative impact on BMI, based on the notion that LoF allele burden can drive severe obesity. In addition, focused nucleotide sequencing efforts are being initiated for newly recruited severely obese patients, where we will expand the gene panel to include ~100 candidate MC4R pathway genes and compare them with available nucleotide sequencing data from normal-weight controls. We anticipate that these efforts will shed light on the genetic factors that contribute to MC4R pathway deficiency and severe obesity.
In summary, we conservatively estimate ~12,800 patients with biallelic MC4R pathway LoF in the United States. Our rule-based approach to identify LoF variants and stratify MC4R pathway–deficient patients provides a controlled route to the continued discovery and expansion of MC4R pathway variants. Our data indicate that MC4R pathway deficiency in the three canonical genes studied here contributes to obesity and can lead to a cumulative burden of LoF alleles, thus explaining important genetic aspects of severe obesity. Finally, we aim to test whether obese populations with diverse MC4R pathway genetic deficits may benefit from setmelanotide treatment of body weight control.
Supplementary Material
Acknowledgments
We thank Cathy Folster, Ally Leonard, Michael Feloni, Genie Meca, Scott Baver, Monica Fay, and Ida Hatoum Moeller for critical feedback and for expert clinical and administrative support.
Financial Support: This study was supported by Icahn School Medicine at Mount Sinai, Sema 4, and Rhythm Pharmaceuticals.
Disclosure Summary: K.L.A., E.E.S., S.D.L., and R.C. are employees of Sema4, a Mount Sinai venture. A.S.G., S.R., F.F., K.G., and L.H.T.V.d.P. are employees of Rhythm Pharmaceuticals. S.L., J.A.W., P.Y., L.D., T.W.C., and J.R.G. are employees of WuXi NextCode. S.C. is an employee of EpidStat. B.S.G. and K.E.N. have nothing to disclose.
Glossary
- Abbreviations
BBS, Bardet Biedl syndrome
- BMI
body mass index
- gnomAD
Genome Aggregation Database
- LEPR
leptin receptor
- LoF
loss of function
- MAF
minor allele frequency
- MC4R
melanocortin 4 receptor
- mRNA
messenger RNA
- MSH
melanocyte-stimulating hormone
- PCSK1
prohormone convertase 1
- POMC
pro-opiomelanocortin
- WT
wild-type.
References
- 1. Kühnen P,Clément K,Poitou-Bernert C,Fiedorek F,Van Der Ploeg LH,Connors H,Gottesdiener K,Farooqi S,Wiegand S,Grüters A,Krude H Proof of concept for treatment of a second rare genetic disorder of the leptin-melanocortin pathway: successful therapy of extreme obesity in a leptin-receptor (LepR) deficient patient with setmelanotide. Available at:www.rhythmtx.com/wp-content/uploads/2017/03/2017-11-ObesityWeek-Rhythm-Kuhnen.pdf. Accessed 16 May 2018.
- 2. Kühnen P,Clément K,Wiegand S,Blankenstein O,Gottesdiener K,Martini LL,Mai K,Blume-Peytavi U,Grüters A,Krude H. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist.N Engl J Med.2016;375(3):240–246. [DOI] [PubMed] [Google Scholar]
- 3. Collet T-H,Dubern B,Mokrosinski J,Connors H,Keogh JM,Mendes de Oliveira E,Henning E,Poitou-Bernert C,Oppert J-M,Tounian P,Marchelli F,Alili R,Le Beyec J,Pépin D,Lacorte J-M,Gottesdiener A,Bounds R,Sharma S,Folster C,Henderson B,O’Rahilly S,Stoner E,Gottesdiener K,Panaro BL,Cone RD,Clément K,Farooqi IS,Van der Ploeg LHT. Evaluation of a melanocortin-4 receptor (MC4R) agonist (setmelanotide) in MC4R deficiency.Mol Metab.2017;6(10):1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Farooqi IS,O’Rahilly S. Mutations in ligands and receptors of the leptin-melanocortin pathway that lead to obesity.Nat Clin Pract Endocrinol Metab.2008;4(10):569–577. [DOI] [PubMed] [Google Scholar]
- 5. Walley AJ,Asher JE,Froguel P. The genetic contribution to non-syndromic human obesity.Nat Rev Genet.2009;10(7):431–442. [DOI] [PubMed] [Google Scholar]
- 6. Yeo GS,Heisler LK. Unraveling the brain regulation of appetite: lessons from genetics.Nat Neurosci.2012;15(10):1343–1349. [DOI] [PubMed] [Google Scholar]
- 7. Farooqi IS,Keogh JM,Yeo GS,Lank EJ,Cheetham T,O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene.N Engl J Med.2003;348(12):1085–1095. [DOI] [PubMed] [Google Scholar]
- 8. Farooqi IS,Matarese G,Lord GM,Keogh JM,Lawrence E,Agwu C,Sanna V,Jebb SA,Perna F,Fontana S,Lechler RI,DePaoli AM,O’Rahilly S. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency.J Clin Invest.2002;110(8):1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jackson RS,Creemers JW,Ohagi S,Raffin-Sanson ML,Sanders L,Montague CT,Hutton JC,O’Rahilly S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene.Nat Genet.1997;16(3):303–306. [DOI] [PubMed] [Google Scholar]
- 10. Krude H,Biebermann H,Luck W,Horn R,Brabant G,Grüters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans.Nat Genet.1998;19(2):155–157. [DOI] [PubMed] [Google Scholar]
- 11. Styne DM,Arslanian SA,Connor EL,Farooqi IS,Murad MH,Silverstein JH,Yanovski JA. Pediatric obesity-assessment, treatment, and prevention: an Endocrine Society clinical practice guideline.J Clin Endocrinol Metab.2017;102(3):709–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wabitsch M,Funcke JB,Lennerz B,Kuhnle-Krahl U,Lahr G,Debatin KM,Vatter P,Gierschik P,Moepps B,Fischer-Posovszky P. Biologically inactive leptin and early-onset extreme obesity.N Engl J Med.2015;372(1):48–54. [DOI] [PubMed] [Google Scholar]
- 13. Farooqi IS,O’Rahilly S. 20 years of leptin: human disorders of leptin action.J Endocrinol.2014;223(1):T63–T70. [DOI] [PubMed] [Google Scholar]
- 14. Garfield AS,Shah BP,Burgess CR,Li MM,Li C,Steger JS,Madara JC,Campbell JN,Kroeger D,Scammell TE,Tannous BA,Myers MG Jr,Andermann ML,Krashes MJ,Lowell BB. Dynamic GABAergic afferent modulation of AgRP neurons.Nat Neurosci.2016;19(12):1628–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bischof JM,Van Der Ploeg LH,Colmers WF,Wevrick R. Magel2-null mice are hyper-responsive to setmelanotide, a melanocortin 4 receptor agonist.Br J Pharmacol.2016;173(17):2614–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Low MJ.Neuroendocrinology: new hormone treatment for obesity caused by POMC-deficiency.Nat Rev Endocrinol.2016;12(11):627–628. [DOI] [PubMed] [Google Scholar]
- 17. Müller TD,Tschöp MH,O’Rahilly S. Metabolic precision medicines: curing POMC deficiency.Cell Metab.2016;24(2):194–195. [DOI] [PubMed] [Google Scholar]
- 18. Haws RM,Fletty KL,McIntee TJ,Green C,Pomeroy J,Hylan M,Folster C,Fiedorek FT. Obesity and hyperphagia therapy in Bardet-Biedl syndrome with a melanocortin-4 receptor agonist.ObesityWeek;2017. [Google Scholar]
- 19. Stenson PD,Mort M,Ball EV,Shaw K,Phillips A,Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine.Hum Genet.2014;133(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Landrum MJ,Lee JM,Riley GR,Jang W,Rubinstein WS,Church DM,Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype.Nucleic Acids Res.2014;42(Database issue):D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cingolani P,Platts A,Wang L,Coon M,Nguyen T,Wang L,Land SJ,Lu X,Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome ofDrosophila melanogaster strain w1118; iso-2; iso-3.Fly (Austin).2012;6(2):80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chakraborty R,Srinivasan MR,Daiger SP. Evaluation of standard error and confidence interval of estimated multilocus genotype probabilities, and their implications in DNA forensics.Am J Hum Genet.1993;52(1):60–70. [PMC free article] [PubMed] [Google Scholar]
- 23. Choquet H,Kasberger J,Hamidovic A,Jorgenson E. Contribution of common PCSK1 genetic variants to obesity in 8,359 subjects from multi-ethnic American population.PLoS One.2013;8(2):e57857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kilpeläinen TO,Bingham SA,Khaw KT,Wareham NJ,Loos RJ. Association of variants in the PCSK1 gene with obesity in the EPIC-Norfolk study.Hum Mol Genet.2009;18(18):3496–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Villalobos-Comparán M,Villamil-Ramírez H,Villarreal-Molina T,Larrieta-Carrasco E,León-Mimila P,Romero-Hidalgo S,Jacobo-Albavera L,Liceaga-Fuentes AE,Campos-Pérez FJ,López-Contreras BE,Tusié-Luna T,Del Río-Navarro BE,Aguilar-Salinas CA,Canizales-Quinteros S. PCSK1 rs6232 is associated with childhood and adult class III obesity in the Mexican population.PLoS One.2012;7(6):e39037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Löffler D,Behrendt S,Creemers JW,Klammt J,Aust G,Stanik J,Kiess W,Kovacs P,Körner A. Functional and clinical relevance of novel and knownPCSK1 variants for childhood obesity and glucose metabolism.Mol Metab.2016;6(3):295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nead KT,Li A,Wehner MR,Neupane B,Gustafsson S,Butterworth A,Engert JC,Davis AD,Hegele RA,Miller R,den Hoed M,Khaw KT,Kilpeläinen TO,Wareham N,Edwards TL,Hallmans G,Varga TV,Kardia SL,Smith JA,Zhao W,Faul JD,Weir D,Mi J,Xi B,Quinteros SC,Cooper C,Sayer AA,Jameson K,Grøntved A,Fornage M,Sidney S,Hanis CL,Highland HM,Häring HU,Heni M,Lasky-Su J,Weiss ST,Gerhard GS,Still C,Melka MM,Pausova Z,Paus T,Grant SF,Hakonarson H,Price RA,Wang K,Scherag A,Hebebrand J,Hinney A,Franks PW,Frayling TM,McCarthy MI,Hirschhorn JN,Loos RJ,Ingelsson E,Gerstein HC,Yusuf S,Beyene J,Anand SS,Meyre D;BioBank Japan, AGEN-BMI, GIANT Consortium . Contribution of common non-synonymous variants in PCSK1 to body mass index variation and risk of obesity: a systematic review and meta-analysis with evidence from up to 331 175 individuals.Hum Mol Genet.2015;24(12):3582–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stijnen P,Tuand K,Varga TV,Franks PW,Aertgeerts B,Creemers JW. The association of common variants in PCSK1 with obesity: a HuGE review and meta-analysis.Am J Epidemiol.2014;180(11):1051–1065. [DOI] [PubMed] [Google Scholar]
- 29. Benzinou M,Creemers JW,Choquet H,Lobbens S,Dina C,Durand E,Guerardel A,Boutin P,Jouret B,Heude B,Balkau B,Tichet J,Marre M,Potoczna N,Horber F,Le Stunff C,Czernichow S,Sandbaek A,Lauritzen T,Borch-Johnsen K,Andersen G,Kiess W,Körner A,Kovacs P,Jacobson P,Carlsson LM,Walley AJ,Jørgensen T,Hansen T,Pedersen O,Meyre D,Froguel P. Common nonsynonymous variants in PCSK1 confer risk of obesity.Nat Genet.2008;40(8):943–945. [DOI] [PubMed] [Google Scholar]
- 30. Prabhu Y,Blanco EH,Liu M,Peinado JR,Wheeler MC,Gekakis N,Arvan P,Lindberg I. Defective transport of the obesity mutant PC1/3 N222D contributes to loss of function.Endocrinology.2014;155(7):2391–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sudlow C,Gallacher J,Allen N,Beral V,Burton P,Danesh J,Downey P,Elliott P,Green J,Landray M,Liu B,Matthews P,Ong G,Pell J,Silman A,Young A,Sprosen T,Peakman T,Collins R. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age.PLoS Med.2015;12(3):e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuchenbaecker KB,McGuffog L,Barrowdale D,Lee A,Soucy P,Dennis J,Domchek SM,Robson M,Spurdle AB,Ramus SJ,Mavaddat N,Terry MB,Neuhausen SL,Schmutzler RK,Simard J,Pharoah PDP,Offit K,Couch FJ,Chenevix-Trench G,Easton DF,Antoniou AC. Evaluations of polygenic risk scores for breast and ovarian cancer risk prediction is BRCA1 and BRCA2 mutation carriers.J Natl Cancer Inst.2017;109(7):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martín MG,Lindberg I,Solorzano-Vargas RS,Wang J,Avitzur Y,Bandsma R,Sokollik C,Lawrence S,Pickett LA,Chen Z,Egritas O,Dalgic B,Albornoz V,de Ridder L,Hulst J,Gok F,Aydoğan A,Al-Hussaini A,Gok DE,Yourshaw M,Wu SV,Cortina G,Stanford S,Georgia S. Congenital proprotein convertase 1/3 deficiency causes malabsorptive diarrhea and other endocrinopathies in a pediatric cohort.Gastroenterology.2013;145(1):138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kumar KG,Sutton GM,Dong JZ,Roubert P,Plas P,Halem HA,Culler MD,Yang H,Dixit VD,Butler AA. Analysis of the therapeutic functions of novel melanocortin receptor agonists in MC3R- and MC4R-deficient C57BL/6J mice.Peptides.2009;30(10):1892–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen KY,Muniyappa R,Abel BS,Mullins KP,Staker P,Brychta RJ,Zhao X,Ring M,Psota TL,Cone RD,Panaro BL,Gottesdiener KM,Van der Ploeg LH,Reitman ML,Skarulis MC. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals.J Clin Endocrinol Metab.2015;100(4):1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kievit P,Halem H,Marks DL,Dong JZ,Glavas MM,Sinnayah P,Pranger L,Cowley MA,Grove KL,Culler MD. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques.Diabetes.2013;62(2):490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gottesdiener K,Conners H,Van der Ploeg L,Fiedorek F,Hylan M,Louis W,Lasseter K. Analysis of the synthetic peptide setmelanotide (RM-493), a melanocortin-4 receptor (MC4R) agonist, on cardiovascular parameters in three phase 1b/2a studies.ObesityWeek;2015. [Google Scholar]
- 38. Seo S,Guo DF,Bugge K,Morgan DA,Rahmouni K,Sheffield VC. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling.Hum Mol Genet.2009;18(7):1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beales PL,Badano JL,Ross AJ,Ansley SJ,Hoskins BE,Kirsten B,Mein CA,Froguel P,Scambler PJ,Lewis RA,Lupski JR,Katsanis N. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome.Am J Hum Genet.2003;72(5):1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heisler LK,Cowley MA,Tecott LH,Fan W,Low MJ,Smart JL,Rubinstein M,Tatro JB,Marcus JN,Holstege H,Lee CE,Cone RD,Elmquist JK. Activation of central melanocortin pathways by fenfluramine.Science.2002;297(5581):609–611. [DOI] [PubMed] [Google Scholar]
- 41. Alsters SI,Goldstone AP,Buxton JL,Zekavati A,Sosinsky A,Yiorkas AM,Holder S,Klaber RE,Bridges N,van Haelst MM,le Roux CW,Walley AJ,Walters RG,Mueller M,Blakemore AI. Truncating homozygous mutation of carboxypeptidase E (CPE) in a morbidly obese female with type 2 diabetes mellitus, intellectual disability and hypogonadotrophic hypogonadism.PLoS One.2015;10(6):e0131417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Joy T,Cao H,Black G,Malik R,Charlton-Menys V,Hegele RA,Durrington PN. Alstrom syndrome (OMIM 203800): a case report and literature review.Orphanet J Rare Dis.2007;2(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Edelman EA,Girirajan S,Finucane B,Patel PI,Lupski JR,Smith AC,Elsea SH. Gender, genotype, and phenotype differences in Smith-Magenis syndrome: a meta-analysis of 105 cases.Clin Genet.2007;71(6):540–550. [DOI] [PubMed] [Google Scholar]
- 44. Muzar Z,Lozano R,Kolevzon A,Hagerman RJ. The neurobiology of the Prader-Willi phenotype of fragile X syndrome.Intractable Rare Dis Res.2016;5(4):255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Biebermann H,Castañeda TR,van Landeghem F,von Deimling A,Escher F,Brabant G,Hebebrand J,Hinney A,Tschöp MH,Grüters A,Krude H. A role for beta-melanocyte–stimulating hormone in human body-weight regulation.Cell Metab.2006;3(2):141–146. [DOI] [PubMed] [Google Scholar]
- 46. Lee YS,Challis BG,Thompson DA,Yeo GS,Keogh JM,Madonna ME,Wraight V,Sims M,Vatin V,Meyre D,Shield J,Burren C,Ibrahim Z,Cheetham T,Swift P,Blackwood A,Hung CC,Wareham NJ,Froguel P,Millhauser GL,O’Rahilly S,Farooqi IS. A POMC variant implicates beta-melanocyte–stimulating hormone in the control of human energy balance.Cell Metab.2006;3(2):135–140. [DOI] [PubMed] [Google Scholar]
- 47. Raffan E,Dennis RJ,O’Donovan CJ,Becker JM,Scott RA,Smith SP,Withers DJ,Wood CJ,Conci E,Clements DN,Summers KM,German AJ,Mellersh CS,Arendt ML,Iyemere VP,Withers E,Söder J,Wernersson S,Andersson G,Lindblad-Toh K,Yeo GS,O’Rahilly S. A deletion in the canine POMC gene is associated with weight and appetite in obesity-prone Labrador retriever dogs.Cell Metab.2016;23(5):893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Challis BG,Pritchard LE,Creemers JW,Delplanque J,Keogh JM,Luan J,Wareham NJ,Yeo GS,Bhattacharyya S,Froguel P,White A,Farooqi IS,O’Rahilly S. A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism.Hum Mol Genet.2002;11(17):1997–2004. [DOI] [PubMed] [Google Scholar]
- 49. Novembre J,Johnson T,Bryc K,Kutalik Z,Boyko AR,Auton A,Indap A,King KS,Bergmann S,Nelson MR,Stephens M,Bustamante CD. Genes mirror geography within Europe [published correction appears in Nature. 2008;456:274]Nature.2008;456(7218):98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barroso I.ADCY3, neuronal primary cilia and obesity.Nat Genet.2018;50(2):166–167. [DOI] [PubMed] [Google Scholar]
- 51. Grarup N,Moltke I,Andersen MK,Dalby M,Vitting-Seerup K,Kern T,Mahendran Y,Jørsboe E,Larsen CVL,Dahl-Petersen IK,Gilly A,Suveges D,Dedoussis G,Zeggini E,Pedersen O,Andersson R,Bjerregaard P,Jørgensen ME,Albrechtsen A,Hansen T. Loss-of-function variants in ADCY3 increase risk of obesity and type 2 diabetes.Nat Genet.2018;50(2):172–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Saeed S,Bonnefond A,Tamanini F,Mirza MU,Manzoor J,Janjua QM,Din SM,Gaitan J,Milochau A,Durand E,Vaillant E,Haseeb A,De Graeve F,Rabearivelo I,Sand O,Queniat G,Boutry R,Schott DA,Ayesha H,Ali M,Khan WI,Butt TA,Rinne T,Stumpel C,Abderrahmani A,Lang J,Arslan M,Froguel P. Loss-of-function mutations in ADCY3 cause monogenic severe obesity.Nat Genet.2018;50(2):175–179. [DOI] [PubMed] [Google Scholar]
- 53. Siljee JE,Wang Y,Bernard AA,Ersoy BA,Zhang S,Marley A,Von Zastrow M,Reiter JF,Vaisse C. Subcellular localization of MC4R with ADCY3 at neuronal primary cilia underlies a common pathway for genetic predisposition to obesity.Nat Genet.2018;50(2):180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.