

Abstract
Nitric oxide (NO) and hydrogen sulfide (H2S) are two gasotransmitters that are produced in the human body and have a key role in many of the physiological activities of the various organ systems. Decreased NO bioavailability and deficiency of H2S are involved in the pathophysiology of type 2 diabetes and its complications. Restoration of NO levels have favorable metabolic effects in diabetes. The role of H2S in pathophysiology of diabetes is however controversial; H2S production is decreased during development of obesity, diabetes, and 8 its complications, suggesting the potential therapeutic effects of H2S. On the other hand, 9 increased H2S levels disturb the pancreatic β-cell function and decrease insulin secretion. In addition, there appear to be important interactions between NO and H2S at the levels of both biosynthesis and signaling pathways, yet clear an insight into this relationship is lacking. H2S potentiates the effects of NO in the cardiovascular system as well as NO release from its storage pools. Likewise, NO increases the activity and the expression of H2S-generating enzymes. Inhibition of NO production leads to elimination/attenuation of the cardioprotective effects of H2S. Regarding the increasing interest in the therapeutic applications of NO or H2S-releasing molecules in a variety of diseases, particularly in the cardiovascular disorders, much is to be learned about their function in glucose/insulin metabolism, especially in diabetes. The aim of this 18 review is to provide a better understanding of the individual and the interactive roles of NO and 19 H2S in carbohydrate metabolism.
Keywords: Nitric oxide, hydrogen sulfide, diabetes, carbohydrate metabolism
Graphical Abstract
1. Introduction
Nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S) collectively are known as gasotransmitters [1]. Although all three are considered to be toxic gases, they are nevertheless synthesized in the human body and play a key role in many of the physiological activities of the various organ systems [1–7]. Although the origins of NO go back to the late eighteenth century, its biological role was recognized in 1980 [8] and its vasorelaxatory effects were established a few years later in 1987 [9]. In comparison, H2S is relatively a new player on the scene and the gradual discovery of H2S-producing enzymes has shed more light on its physiological functions 15 and cellular signaling.
Decreased NO bioavailability and deficiency of H2S are considered to be involved in pathophysiology of many disease such as type 2 diabetes [10–12]. Restoration of NO levels has been associated with many favorable metabolic effects in type 2 diabetes [10, 13, 14]. The role of H2S in pathophysiology of diabetes is however controversial, as both inhibition and stimulation of the H2S system have been suggested to be potential therapeutic approaches [15, 16].
There are many similarities between the biological characteristics of NO and H2S in terms of their biosynthesis, biological targets, effects, metabolism, and elimination. In addition, these molecules regulate many physiological functions with some cross talk between the enzymes that are involved in their generation and also the pathways that these two gasotransmitters affect (reviewed in [17, 18]). For example, both NO and H2S are well known antioxidants and recently it was demonstrated that H2S at low doses potentiates the anti-oxidative effects of NO in diabetic rats [19]. Cross talk between H2S and NO was initially reported in 1997 by Hosoki et al. who showed that H2S at a concentration that did not produce any appreciable vascular relaxation, it potentiated the vasorelaxatory effects of NO [20]. While the individual physiological functions of H2S and NO as well as their potential relationship in many organ systems are extensively studied, our understanding about their potential roles in regulation of carbohydrate metabolism in particular the role of H2S is woefully incomplete. This review discusses the effects of NO and 10 H2S and also their interactions in carbohydrate metabolism.
2. Nitric oxide synthesis
NO is produced in all tissues [21–24] by NO synthase (NOS)-dependent and independent pathways (Figure 1) [25, 26]. In mice, total NO formation has been reported to be about 0.2 mmol/kg/day of which approximately 70% is derived from endothelial NOS (eNOS, NOS-3) [10]. In Wistar rats and humans, the rate of NO production is about 0.33–0.85μmol/kg/h and 0.9 μmol/kg/h, respectively [27–30].
Figure 1. Hydrogen sulfide and nitric oxide biosynthetic pathways.

Hydrogen sulfide (H2S) and nitric oxide (NO) are produced by enzymatic and non-enzymatic pathways. Non-enzymatic production of H2S is mediated through reducing elemental sulfur or organic polysulfides. Enzymatic production of H2S is mediated by cystathionine γ-lyase (CSE), cystathionine-beta synthase (CBS), and 3-mercaptopyruvate sulfuretransferase (3-MST). NO is produced by nitrate/nitrite pathway which can be enzymatic or non-enzymatic. NO is also produced from L8 arginine by neuronal NO synthase (nNOS), inducible NO synthase (iNOS), and endothelial NO 9 synthase (eNOS).
2.1. NOS-dependent NO synthesis
In NOS-dependent pathway, NO is produced from L-arginine (the L-arginine-NO pathway) by the three isoforms of NOS namely, neuronal (nNOS/NOS-1), inducible (iNOS/NOS-2), and eNOS/NOS-3 which are heme-containing dioxygenases enzymes [31–34]. These isoforms are active as homodimers [35–38] and proper dimerization is critical for their activity [39]; L-arginine, tetrahydrobiopterin (BH4), and heme are essential for stabilizing the active dimeric form of all NOS isoforms [38]. NOS monomers cannot catalyze NO formation and have a limited capacity to produce superoxide anions [37]. These isoforms vary in amino acid sequence, cellular location, function, and post-translational modifications [35, 40]. eNOS which produces relatively low quantities of NO, is mostly expressed in the vascular endothelium, but has also been found in epithelial cells, neurons, and cardiomyocytes as well as in hepatocytes and adipocytes [41, 42]. nNOS is expressed to highest relative abundance in neurons, skeletal muscle, and epithelial cells [43, 44]. eNOS and nNOS are firmly regulated through phosphorylation, compartmentalization in caveolae, calcium/calmodulin, and interact with plasma membrane ionotropic receptors [45]. iNOS is primarily identified in macrophages but its expression can be stimulated in virtually any cells or tissues and can produce large amounts of NO for long periods of time provided the necessary substrate is available [45, 46]. Some studies have also suggested a mitochondria-localized NOS isoform [47]; however, the specific contribution of this isoform remains unclear.
All isoforms of NOS bind calmodulin as a prosthetic group; increased in intracellular Ca2+ levels are necessary for binding of calmodulin to eNOS and nNOS (half-maximal activity between 200–400 nM) while due to a different amino acid structure of the calmodulin-binding site in iNOS, in this isoform calmodulin is constitutively active at extremely low intracellular Ca2+ concentrations (<40 nM) [35, 37]. Overproduction of NO by iNOS and also exogenous NO could lead to inhibition of eNOS and nNOS [32, 48]. Of note, the NO concentration necessary for the inhibition of eNOS and nNOS is considerably lower than that required for iNOS inhibition [48]. NO could be protective or toxic depending on its concentration, source, location, and environment [49].
For NO production, NOS isoforms catalyze oxidation of L-arginine to NO and L-citrulline [23, 50, 51]. Reduced nicotinamide-adenine-dinucleotide phosphate (NADPH) and molecular oxygen are co-substrates [36, 52] and flavin mononucleotide, flavin adenine dinucleotide, and BH4 are cofactors of NOS [36, 52], of which, BH4 is critical and rate-limiting [23]. Exposure to oxidative stress such as seen in diabetes, results in the conversion of BH4 to 7,8- dihydrobiopterin (BH2) and thus leads to a dysfunctional eNOS, as BH2 is inactive as a cofactor and competes with BH4 for BH4 binding [53]. Anti-oxidants like vitamin C and folate increase BH4 bioavailability and could affect NO formation [23].
2.2. NOS-independent NO synthesis
NOS-independent NO production from nitrate and nitrite was initially reported in 1994 in the stomach following protonation of swallowed salivary nitrite [54, 55]. Decreased NOS-derived NO production in tissues makes the nitrate-nitrite-NO pathway important [22]. Oxidation of NOS-derived NO as well as direct exposure through the diet are two major sources of nitrate in mammals [56]. Total body pool of nitrate is about 0.53 and 0.47 mM in men and women, respectively [57]; the relative contribution of the dietary source and NOS-derived NO to the total body pool of nitrate varies, but with a moderately high intake of green leafy vegetables (~200 g per day), the dietary source clearly dominates [58].
About 25% (20–28%) of circulating nitrate (from diet or endogenous NO) is actively taken up by the salivary glands and concentrated by a factor of 10 in about 5 hours [59–63]; nitrate is reduced to the more reactive nitrite anion (NO2−) by the oral commensal bacteria with potent nitrate reductase enzymes [64]. For this reason, the salivary nitrate and nitrite concentrations are normally 10–100 and 1000-fold higher than their plasma levels, respectively [65, 66]. After oral loading, nitrate/nitrite is rapidly absorbed in the duodenum and jejunum [10, 67]. In the stomach, part of this nitrite is reduced to NO but most of it is absorbed to the circulation [10, 60, 68]. Nitrite reduction to NO in blood and tissues could be enzymatic or non-enzymatic [64, 69–71], which is generally enhanced during hypoxic, ischemic, and acidic conditions [32, 46, 50, 64, 68–71].
3. Nitric oxide signaling pathways
Actions of NO could be cyclic guanosine monophosphate (cGMP)-dependent and/or cGMP independent (mostly reactive nitrogen species-mediated) [72]. cGMP-dependent signaling pathway is the most important physiologic signaling pathway activated by NO [26, 65, 73–75]. In this pathway, only low concentrations of NO (5–10 nM) are required to activate guanylyl cyclase (GC) [36, 74, 76], which converts guanosine triphosphate to cGMP [36]. GC has two isoforms: soluble (sGC is cytosolic) and membrane (particulate), of which, sGC is the receptor for NO [36]. sGC is a heterodimer of α and β subunits which contains a ferrous heme prosthetic group on histidine 105 residue of the β subunit [36]. When NO binds to the ferrous heme iron, it causes disruption of histidine 105 and inhibition of the catalytic activity of sGC by the heme is overcomed. The catalytic domain near the C-terminal of the α and β subunits is then activated resulting in an increase in Vmax and a decrease in Km of the enzyme [36]. Elevated cGMP levels activate protein kinase G (PKG), which is a serine/threonine kinase and exists as a homodimer [35]. PKG has two isoforms: PKGI and PKGII [77]; PKGI is the common isoform involved in NO/cGMP/PKG signaling pathway [78], which exerts its vasodilatory effects in the vascular smooth muscle cells through decreased [Ca2+]i [79] and Ca2+ sensitivity [80].
In addition to NO/cGMP/PKG signaling pathway, nitrosative post-translational modifications such as S-nitrosylation, which is a reversible covalent attachment of NO to cysteine residues of proteins, is a key mechanism for NO signaling [35, 73, 81]. S-nitrosylation activates or inhibits protein function and therefore can be beneficial or detrimental [82, 83]. Despite the presence of cysteine residues on almost all proteins and production of NO by most cells, only some proteins are nitrosylated [84]. Specificity in S-nitrosylation depends on the presence of metal ions (Mg2+ or Ca2+), local pH, and the acid-base motifs [85].
4. Hydrogen sulfide synthesis
H2S is a colorless gas produced by enzymatic and non-enzymatic pathways in the body (Figure 1) [86]. Non-enzymatic production of H2S is responsible for a limited amount of H2S in mammalian cells [87] and is mediated through reducing elemental sulfur or organic polysulfides via glucose-supported and thiol-dependent as well as glutathione-dependent cellular reactions [88–90]. Enzymatic production of H2S is organ-specific and has been attributed to two pyridoxal 5′-phosphate (PLP) dependent enzymes, cystathionine β synthase (CBS, EC 4.2.1.22) and cystathionine γ lyase (CSE, EC 4.4.1.1), as well as a non-PLP dependent enzyme, 3-mercaptopyruvate sulfurtransferase (3-MST, EC 2.8.1.2) along with cysteine aminotransferase (CAT, EC 2.6.1.3) [2, 86, 91]. CBS and CSE are heme-proteins being in the cytosol, while 3-MST is a zinc-dependent protein, localized in the cytosol and in particular in the mitochondrial matrix, with an optimal pH of ~8 [86, 92].
L-cysteine, which is the major source of H2S production, is desulfhydrated during the transsulfuration pathway by all H2S-producing enzymes [2, 91]. H2S can also be produced from homocysteine; in normal conditions, about 70% of H2S is produced from L-cysteine and 30% from homocysteine [93]. An additional biosynthetic pathway for H2S production has also been reported from D-cysteine involving 3-MST and D-amino acid oxidase [94]. This pathway operates predominantly in the cerebellum and the kidneys. In the cerebellum it protects the cerebellar neurons from oxidative stress and in the kidneys, it attenuates ischemia-reperfusion injury more effectively than L-cysteine [94].
Tissue production of H2S and in particular the liver contributes to plasma level of H2S [95]. Using a polarographic H2S sensor, rate of H2S production in rat liver, brain, aorta, and heart is 12.3 ± 4.6, 10.6 ± 3.2, 5.8 ± 1.7, and 1.1 ± 0.3 pmol/s/mg protein, respectively [96]. Concentration of H2S in the plasma and tissue is under 1 μM, however using the methylene blue method and S2− ion selective electrodes, which are the most commonly employed methods, artificially inflated H2S concentrations and values from 20 to 300 μM are still being reported as “physiological” [97]. H2S at high concentrations (> 700 ppm or ~ 20,000 μM) is lethal to both 10 humans and animals [98].
H2S is a weak acid and equilibrates with hydrosulfide anions (HS−, pKa 7.04 and S2−, pKa 11.96) in aqueous solution; with a pKa1 value of 6.76 and pKa2 >12 at 37 °C [17, 99], 18.5% of H2S remains undissociated at physiologic pH=7.40 [99]. H2S dissolves well in lipids and its solubility is 5-fold higher than in water and therefore can freely penetrate cells [99]; while HS− does not pass. Much of the inactivation of H2S occurs through mitochondrial oxidation during three consecutive reactions; two membrane-bound sulfide: quinone oxidoreductases and a sulfur dioxygenase are involved in oxidation of sulfide to thiosulfate [100].
5. H2S signaling pathways
It is believed that most of H2S signaling is done through S-sulfhydration of target proteins, a reaction which transfers a sulfhydryl group (-SH) to a cysteine residue of a protein to form a hydropersulfid moiety (-SSH) [101, 102] or persulfide group [103]. S-sulfhydration or more correctly sulfuration of a protein modifies its functions, stability, and localization within the cells [104]. S-sulfhydration also contributes to modification of inflammation, endoplasmic reticulum (ER) stress signaling and vascular tone [104]. Regulation of ion channels via S-sulfhydration has been reported in several studies; S-sulfhydration of adenosine triphosphate (ATP)-dependent K+ channels (KATP) activates it by decreasing ATP binding and increasing phosphatidylinositol 4, 5-bisphosphate (PIP2) binding; S-sulfhydration also activates small and intermediate calcium-activated potassium channels (SKCa and IKCa) in vascular endothelial cells [95]. In addition, S-sulfhydration of inositol-3-phosphate receptor (IP3R) inhibits Ca2+ release from the ER [103].
6. Role of NO in carbohydrate metabolism
6.1. Insulin secretion and NO
Glucose enters the pancreatic β-cells through a low affinity glucose transporter (GLUT-2). Glucose is phosphorylated by glucokinase and pyruvate is generated through glycolysis in the cytoplasm; pyruvate is then metabolized by pyruvate carboxylase and pyruvate dehydrogenase and passes into the mitochondria where it increases the cytoplasmic ATP/adenosine diphosphate (ADP) ratio; increased ATP/ADP ratio causes closure of KATP channels [105, 106]. In the β-cells, KATP channels are the primary determinant of the membrane potential, so closure of these channels causes membrane depolarization and the subsequent activation of L-type voltage-dependent Ca2+ channels (VDCC). Elevation of cytosolic free Ca2+ concentrations ([Ca2+]i) is followed by insulin vesicle exocytosis [105], which is mediated by SNAp REceptors (SNARE), located in both vesicle (v-SNAREs) and target (t-SNAREs) membrane. Syntaxin and synaptosome associated protein-25 (SNAP-25) families are known as t-SNARE and v-SNAREs including the vesicle-associated membrane proteins (VAMPs) [107].
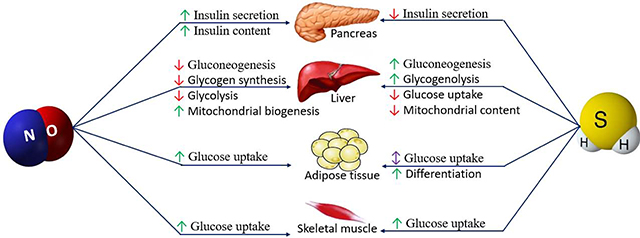
Expression of all three isoforms of NOS has been reported in the pancreatic β-cells [56, 106, 108–110]. The role of NO in insulin secretion is controversial; iNOS-derived NO decreases, while eNOS-derived NO increases insulin secretion [32, 111]. In addition, NO stimulates the activity of the insulin gene promoter, with comparable increases in endogenous insulin mRNA levels in both Min6 β-cells (a pancreatic β-cell line derived from transgenic mouse expressing the large T-antigen of SV40 [112]) and isolated rat islets of Langerhans [113]. Both glucose and insulin increase NO production in β-cells [114]; glucose-induced NO production at physiological concentrations increases insulin secretion, however, higher NO levels inhibit insulin secretion [114]. By contrast, it has been reported that both eNOS- and iNOS-activity are greatly increased by high glucose concentrations (20 mM) in intact islets from freely fed mice; fasting induces islet iNOS activity at both physiological (7 mM) and high (20 mM) glucose concentrations; inhibition of NOS by L-NAME (L-NG-Nitroarginine methyl ester) increases insulin secretion both during freely fed conditions and after fasting [115].
Possible mechanisms for increased insulin secretion by NO include: a) mitochondrial depolarization, which induces calcium release from the mitochondria and therefore increases insulin secretion [32], b) increase in islet blood flow, which supplies oxygen and nutrients to the islets [111], c) increased formation of mitochondrial reactive oxygen species (ROS), which is an obligatory signal for glucose-induced insulin secretion [111], and d) S-nitrosylation of glucokinase (at cysteine-371) or syntaxin 4 (at cysteine-141), which facilitates glucose stimulated insulin secretion (Figure 2) [116]. Besides effects on insulin secretion, NO also inhibits an insulin-degrading enzyme (IDE) [117], a ubiquitously expressed cytosolic protease, by S-nitrosylation [116]. Inhibition of NOS causes glucose intolerance by doubling degradation of the secreted insulin and a 40% drop in β-cell glucose sensitivity in non-diabetic subjects [114, 116], suggesting that NO reduces the burden of β-cells via inhibiting circulating insulin clearance [114]. Interleukin 1 beta (IL-1β) has been shown to be a contributing factor in β-cell dysfunction and decreased insulin secretion [118], nitrite-mediated NO decreased elevated IL-1β levels and increased insulin secretion in diabetic rats [13], indicating the anti-inflammatory effects of NO in increased insulin secretion.
Figure 2. Mechanisms of nitric oxide-stimulated insulin secretion in pancreatic β-cell.

Glucose enters the pancreatic β-cells through glucose transporter type 2 (GLUT-2) (1). Glucose is phosphorylated by glucokinase (2) and increases cytoplasmic adenosine triphosphate (ATP)/adenosine diphosphate (ADP) ratio (3); increased ATP/ADP ratio closes (ATP)15 dependent K+ (KATP) channels (4) and causes membrane depolarization (5) and the subsequent activation of L-type voltage-dependent Ca2+ channels (VDCC) (6). Elevation of cytosolic free Ca2+ concentration is followed by activation of synaptotagmin as a calcium sensor (7) and then exocytosis of insulin granules into the circulation (8).
Nitric oxide (NO) causes mitochondrial depolarization, which induces calcium release from mitochondria. NO also facilitates glucose-stimulated insulin secretion by S-nitrosylation of glucokinase or syntaxin 4.
6.2. Insulin signaling and NO
Insulin activates the insulin receptor (IR), which belongs to a tyrosine kinase family of transmembrane signaling proteins containing two extracellular α-subunits and two transmembrane β-subunits [118, 119]. When insulin binds to the α-subunits, this results results in tyrosine autophosphorylation of the β subunits [119]. Transphosphorylation among β-subunits leads to further kinase activity and recruitment and phosphorylation of receptor substrates including insulin receptor substrates (IRSs), SH2 (Src-homology2)-containing proteins (Shc), and Grb2 (Growth factor receptor binding protein 2)-associated binder (GAB) [119]. Phosphorylated IRSs provide docking sites for intracellular molecules containing SH2 domains including type 1A phosphatydylinositol (3, 4, 5)-triphosphate kinase (PI3K), which phosphorylates PIP2 to generate PI (3, 4, 5)-triphosphate (PIP3) [120]. PIP3 is an allosteric regulator of phosphoinositide-dependent kinase (PDK), which in turn leads to phosphorylation and activation of Akt (PKB), a serine/threonine protein kinase [120, 121]. Mitogen-activated protein kinase (MAPK) pathway and PI3K-Akt pathway are two major signaling pathways for insulin actions [122]. Most of insulin’s metabolic actions are mediated through the PI3K-Akt 19 pathway [119].
In endothelial cells, insulin increases eNOS activity through the PI3K-Akt pathway (PI3K-Akt-eNOS pathway) [37, 38, 121, 123] and provides an important step in regulating eNOS activity and glucose uptake [39]. Insulin by enhancing the synthesis and therefore availability of NADPH and BH4 in endothelial cells increases NO production [38]. In the PI3K-Akt-eNOS pathway, eNOS is activated by phosphorylation at serine 1177 [39, 123], which is not phosphorylated in the resting endothelial cells [37].
6.3. NO and glucose metabolism in skeletal muscle
nNOS is the main isoform of NOS in skeletal muscle and is called nNOSμ [43, 44]; eNOS is expressed at low levels and is mainly associated with the vascular endothelium [44]; although there is essentially no expression of iNOS in healthy skeletal muscle [43], it can however be induced in response to inflammatory cytokines [124]. All isoforms may transcriptionally be regulated by hypoxia; expressions of both vascular and skeletal muscle eNOS are increased by chronic exercise [125, 126]; eNOS in blood vessels is also increased by sheer stress [127]. Expression of nNOS is upregulated following muscle activity [125], crush injury [128], and 11 ageing [129], while it is downregulated following denervation [130].
NOS activity is regulated developmentally; total activity in diaphragm homogenates has been reported to be 40–45 pmol/min/mg protein from embryonic day 18 to postnatal day 1; the activity is decreased to ~25 pmol/min/mg protein by day 7 and then to the adult rate by day 30 [131]. Rat diaphragm produces ~3–5 pmol NO/min/mg muscle during passive incubation and is increased approximately six-fold in actively contracting muscle in a tissue bath [132, 133]. Resting cordofemoralis and quadratus femoralis muscles produces ~1 pmol/min/mg muscle and this is increased two-three-fold by electrical stimulation in vitro [134]. Resting mouse diaphragm and soleus muscles produce ~13 pmol NO/min/mg and there is no difference between muscle types or between wild-type and eNOS-deficient mice [135].
Effect of NO on glucose uptake was first recognized when NOS inhibition was shown to attenuate 2-deoxyglucose uptake by rat limb muscle [134] both under basal conditions and during repetitive contractions in vitro [125]. These studies further demonstrated that exogenous NO was associated with an increase in 2-deoxyglucose uptake which was additive with the increase in uptake stimulated by insulin. Local infusion of the NOS inhibitor, NG-monomethyl-L-arginine (L-NMMA), into the femoral artery attenuates elevated skeletal muscle glucose uptake during moderate cycling exercise in healthy and type 2 diabetes participants and this effect occurs independently of blood flow [136]. In addition, NO stimulates rate of lactate release and glucose oxidation in isolated rat skeletal muscle [137], which is responsible for approximately 80% of insulin-stimulated glucose uptake [138].
The mechanism(s) by which NO stimulate glucose metabolism in skeletal muscle are not well characterized, however it has been demonstrated that NO decreases insulin resistance by increasing mRNA expression, protein levels, and translocation of GLUT4 in skeletal muscle of type 2 diabetic rats [13, 14, 121]. sGC/cGMP/PKG pathway as well as various post-translational protein modifications are involved in the NO-mediated intramuscular GLUT4 translocation [137]. In resting muscle, using various NO donors, the cGMP analogue (8-bromo-cGMP), and phosphodiesterase 5 (PDE5) inhibitor (zaprinast), which prevents degradation of cGMP, increases cGMP level with a parallel increase in muscle glucose uptake [137, 139], whereas inhibiting sGC, decreases cGMP level and NO-induced glucose uptake [137]. In contrast to basal muscle glucose uptake, inhibition of sGC and PKG during ex vivo contraction have no effect on muscle glucose uptake [140], indicating that a cGMP/PKG-independent mechanism may be involved in NO-induced glucose uptake during contraction.
6.4. NO and glucose metabolism in adipose tissue
The expression of iNOS and eNOS have been reported in white adipose tissue [41]. eNOS is mostly membrane bound, while iNOS is found in the cytoplasm of adipocytes and macrophages. Although protein expression of nNOS has been reported in the cytoplasm and the mitochondria of adipocytes [141, 142], its expression does not appear to be present in significant amounts [143]. In addition, nitrate-mediated NO production is increased in response to hypoxia in rats and primary adipocytes [69].
In adipose tissue, NO stimulates insulin-dependent [144] and insulin-independent [41, 142] uptake and oxidation of glucose. NO-released from S-Nitrosoglutathione and S-Nitroso-N acetylpenicillamine (GSNO and SNAP) at low doses (<1 mM), increases while at high doses (10 and 20 mM) inhibits insulin-stimulated glucose uptake in isolated adipocytes of normoglycemic and streptozotocin (STZ)-induced diabetic rats [145]. Increased NO production also up-regulates brown adipocyte-associated genes [69], which can be a physiological adaptation of adipocytes undergoing hypertrophy in obesity.
In adipose tissue, the reaction between eNOS-mediated NO and sGC occurs at nM concentrations of NO [146, 147]. In addition, nitrite-mediated NO production by the nitrite reductase activity of xanthine oxidase (XO) also acts through the sGC/cGMP-signaling pathway [69]. NO scavenger (CPTIO) and GC inhibitor (LY83583) reduce SNP-stimulated glucose uptake to the basal level, suggesting that SNP-stimulated glucose uptake is mediated by NO and GC [41]. sGC/cGMP-dependent stimulation of AMP-activated protein kinase (AMPK) increases gene expression and PKG-dependent AMPK phosphorylation and therefor increases glucose uptake in adipose tissue [142]. The anti-inflammatory effects of NO may have an important role in decreasing insulin resistance, since in adipose tissue of diabetic rats; nitrite-derived NO in a dose-dependent manner reduced both mRNA levels of TNF-α and adipocyte size [149].
6.5. NO and glucose metabolism in hepatocytes
Glucose uptake by the liver is not affected by insulin directly and is accomplished passively via glucose transporters of which GLUT2 is the main hepatic glucose transporter [148]. The physiological control of glucose uptake across the plasma membrane of a hepatocyte depends on the intracellular glucose phosphorylation/dephosphorylation balance; however, insulin by stimulating the activity of glucokinase, it indirectly promotes hepatic glucose uptake (reviewed in [149]). Within the cells, hexokinase isoenzymes phosphorylate free glucose to glucose 6-phosphate which may follow three metabolic pathways: a) isomerization into glucose 1-phosphate, that is transformed into UDP–glucose (precursor of glycogen), UDP–glucuronate and UDP–galactose; b) isomerization into fructose 6-phosphate, which may either start the hexosamine pathway by combining with glutamine or continue into the glycolytic pathway to form pyruvate and then acetyl-CoA; c) oxidation into gluconolactone and start the pentose phosphate pathway [150].
In the liver, iNOS is expressed primarily in the cytoplasm of periportal hepatocytes and eNOS is expressed in hepatocytes, hepatic sinusoidal, endothelium of hepatic arteries, terminal hepatic venules, and epithelium of biliary ducts [42]. In the liver, NO induces mitochondrial biogenesis through an increase in cGMP levels and activation of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) [151], decreases gluconeogenesis through decreasing mRNA expression of phosphoenolpyruvate carboxykinase (PEPCK) [53], inhibits glycogen synthesis by decreasing the activity of glycogen synthase [152], and reduces glycolysis. Besides hepatic glucose metabolism, NO is also involved in the regulation of lipid metabolism at the level of lipogenesis and lipolysis. Whether NO stimulates or inhibits lipid oxidation or synthesis appears to be dependent on the isoforms on NOS stimulated, tissue site and the intracellular redox state [142].
BH4 suppresses gluconeogenesis and increases AMPKα phosphorylation in hepatocytes from wild-type mice but not in hepatocytes isolated from eNOS−/− mice or in the presence of NOS inhibitors, suggesting that eNOS acts upstream of AMPK activation in suppression of hepatic gluconeogenesis by BH4 [53]. AMPK is a central regulator of glucose metabolism as liver-specific AMPK−/− mice exhibit hyperglycemia, glucose intolerance, and increased hepatic glucose production [153]. BH4 is mainly produced in the liver [154] and this is impaired by oxidative stress, such as seen in liver cirrhosis and diabetes [155, 156]. Furthermore, nitrate/nitrite-derived NO restores decreased phospho-AMPK (p-AMPK)/AMPK ratios in the liver of high fat feeding mice and this effect is abolished in the presence of sGC inhibitors [157]. NO decreases glucose production from lactate and also by inhibiting the conversion of glycogen synthase b into synthase a, it inhibits glycogen synthesis as observed in isolated rat hepatocytes [152].
6.6. NO and insulin resistance
Insulin resistance can be defined as the reduced metabolic actions of insulin in target tissues namely liver, skeletal muscle, and adipose tissue [158]. Insulin responsiveness is defined as the maximal effect of insulin (Vmax or concentration of insulin exerting maximal biological response), and insulin sensitivity is defined as the insulin concentration that is required for 50% of its maximal response (EC50/ED50) [158]. Defects in insulin receptor decreases insulin sensitivity while defects that are post-receptor, reduce its responsiveness [118].
Insulin resistance is associated with decreased NO bioavailability as eNOS−/− animals show a number of features of insulin resistance; in addition, polymorphisms in the eNOS gene are associated with insulin resistance susceptibility and metabolic syndrome in humans; inhibition of iNOS prevents while inhibition of eNOS and nNOS promotes insulin resistance [159, 160]. It is interesting to note that absence of eNOS promotes insulin resistance in both skeletal muscle and liver, while absence of nNOS impairs insulin sensitivity only in the liver of eNOS/nNOS doubleknockout mice [160]. Deletion of all isoforms of NOS (eNOS/nNOS/iNOS triple knockout) in mice, causes visceral obesity, hypertension, hypertriglyceridemia, and impaired glucose tolerance [161]. Insulin resistance caused by eNOS dysfunction is thought to be induced by endothelial dysfunction, which decreases skeletal muscle blood flow and glucose uptake [162]. eNOS−/− mice have lower glucose transport in skeletal muscle, indicating that eNOS also regulates glucose uptake in skeletal muscle [162].
Absence of iNOS improves glucose tolerance, normalizes insulin sensitivity, and prevents disorders in PI3K/Akt signaling in high fat-fed mice [163]. Increased iNOS expression is associated with increased S-nitrosation of the insulin receptor, IRS-1, and Akt in skeletal muscle of obese mice, suggesting that S-nitrosation of proteins in insulin signaling pathway is responsible for iNOS-induced insulin resistance [164]. Furthermore, free fatty acid (FFA) induced loss of pancreatic β-cells is due to NO overproduction, which leads to interleukin 1 beta (IL-1β)-mediated β-cell dysfunction and death [108]. Selective overexpression of iNOS in liver causes hepatic insulin resistance, hyperglycemia and hyperinsulinemia [165]; the iNOS-specific inhibitor (L-NIL), reverses hyperglycemia, hyperinsulinemia, and insulin resistance in ob/ob mice [166].
These studies suggest that NOS isoforms play a central role in the regulation of glucose metabolism and insulin resistance and represent several therapeutic targets for management of type 2 diabetes.
6.7. Obesity and diabetes: The NO connection
NO bioavailability has been shown to be decreased in obesity and type 2 diabetes in animal models of obesity and diabetes as well as in obese and diabetic humans [32, 38, 121, 167]. Diminished NO bioavailability is an independent predictor of type 2 diabetes [116], hypertension [168], and atherosclerosis [168]. The role of glucose levels in NO production is controversial; it has been reported that NOS activity and subsequently NO production gradually increases due to an elevation in glucose concentrations within the pancreatic islets [169] and cultured human aortic endothelial cells (HAECs) [170]. In contrast, other groups have demonstrated that hyperglycemia contributes to endothelial dysfunction and leads to a decrease in NO bioavailability by inhibiting basal levels of eNOS expression/activity or increased NO quenching (increased NO oxidation) [32, 38, 39]. Moreover, it has been shown that uncoupling of NOS, led to decreased availability/transport of L-arginine, and an increased in arginase activity resulted in reduced NO production [57, 171, 172].
Diminished expression of NOS isoforms in particular, expression and activity of eNOS are found in both adipose tissue and skeletal muscle of obese humans and rodents [173–175]. Increased mRNA expression of caveolin-1 in adipose tissues is another mechanism for reduced eNOS-derived NO in obesity and type 2 diabetes [176]. Caveolae are a specialized type of lipid raft that appear in the plasma membrane, especially in adipocytes [176]. Caveolin-1, an essential protein for caveolae formation [177], directly binds to eNOS [168] and inhibits its activity [37] resulting in decreased NO production [168, 177]. Obesity and high-fat diets (HFD) by impairing eNOS phosphorylation at serine 1177, reduce eNOS activity [39, 46]. This site of phosphorylation is critical for NO production and can be regulated by Akt [178], which is activated by insulin.
The reason for decreased eNOS phosphorylation in obesity and diabetes could be due to FFA-induced insulin resistance [179]. Elevated FFA which is observed in obesity and type 2 diabetes, by stimulation of Toll-like receptor 4 (TLR4) or TLR2 and NFκB decreases PI3K-Akt-mediated phosphorylation of eNOS at serine 1177 and therefore reduces eNOS activity [32]. Increases in tumor necrosis factor-α (TNF-α) that are seen in obesity and diabetes, by decreasing the stability of eNOS mRNA [180] result in downregulation of its expression and abundance [175, 181]. Increased TNF-α acutely increases eNOS activity most likely through activation of PI3K-Akt and sphingomyelinase/sphingosine-1-phosphate pathways; high levels of NO however through a negative feedback loop, leads to downregulation of eNOS [32]. Elevated ROS levels in obesity and type 2 diabetes [36, 37, 182] also cause PI3K-Akt-eNOS pathway inhibition, which also 6 decreases NO bioavailability [39].
Expression of iNOS is increased in pancreatic β-cells [108], skeletal muscle [183], liver [166], and adipose tissue [184] in obesity and diabetes. Elevated TNF-α levels also increase iNOS in adipocytes which downregulates uncoupling protein 2 (UCP-2) [185] and decreases energy expenditure in adipose tissue.
In addition to decreases in eNOS-mediated NO production, uncoupling of NOS and NO quenching contribute to decreased NO bioavailability in obesity and diabetes. Obesity and diabetes are associated with decreased BH4 and increased BH2 levels; indeed, BH4 to BH2 ratio is very critical in preventing glucose-induced eNOS uncoupling [186]. Uncoupling of nNOS has also been reported in penile arteries of obese rats and leads to nitrergic dysfunction, which is corrected by elevating BH4 levels [187].
Increased oxidative stress and inflammation are the major causes of increased NO quenching in obesity and diabetes. Uncoupled eNOS produces superoxide anion instead of NO [23, 32], which rapidly combines with NO to produce peroxynitrite [36, 39]. On the other hand, excessive iNOS-derived NO increases the formation of peroxynitrite, which enhances eNOS uncoupling [32]. Elevated peroxynitrite levels in obesity and diabetes leads to oxidation, nitration, and S-nitrosylation of proteins, lipids, and DNA [37, 116]. Obesity and diabetes are associated with chronic inflammation [72, 188]. Inflammatory cytokines by decreasing the stability of eNOS mRNA reduce its expression [39]. Moreover, inflammatory cytokines upregulate cationic amino acid transporter-2 and downregulate cationic amino acid transporter-1, the arginine transporters that enhance L-arginine availability for iNOS and decrease it for eNOS [23].
6.8. NO and diabetes complications
NO exerts several protective roles in the prevention and treatment of diabetes complications [189, 190]; NO has cardioprotective effect in ischemia-reperfusion injury [190] and it improves endothelial dysfunction, cardiomyopathy, and nephropathy in animal models of diabetes [189–191].
6.8.1. NO and diabetic cardiovascular complications
It is well known that cardiovascular disease is a common complication of diabetes that can lead to a significant number of mortalities in diabetic patients; diabetic men and women being two times and five times more likely to suffer from congestive heart failure than non-diabetic individuals, respectively [192]. High glucose levels in diabetes cause multiple biochemical modifications in endothelial cells and myocytes; glucose enters the cells by GLUT-1, whose activity is insulin independent and is predominantly regulated by extracellular glucose concentrations [193][194]. Therefore, endothelial cells are more sensitive to hyperglycemia induced injury than other cell types. During diabetes, hyperglycemia causes activation of NAD(P)H oxidase (NOX), which by using NADPH converts oxygen into superoxide anions, which in turn reacts with NO and forms peroxynitrite (ONOO-); peroxynitrite then reacts with BH4, and that this loss of BH4 leads eNOS uncoupling in endothelial cells [192].
Blocking NO production with L-NAME in diabetic rats increase mean arterial pressure (MAP); suggesting that in the onset of diabetes, NO is important in prevention of hypertension, most likely through actions to maintain glomerular filtration and suppress renin secretion [189]. Nitrate-mediated NO in STZ-nicotinamide-induced diabetic rats was shown to provide cardioprotection against ischemia-reperfusion injury through regulating eNOS and iNOS expression and inhibiting lipid peroxidation in an ex vivo heart preparation [190]. Nitrite4 mediated NO in db/db mice subjected to permanent unilateral femoral artery ligation, restored ischemic hind limb blood flow in a vascular endothelial growth factor (VEGF)-dependent and NO-mediated manner [195]. L-arginine treatment improved hypertension and vascular responsiveness in STZ-induced diabetic rats [191]. SNP infusions in diabetic rats decreased MAP and increased vascular conductance (flow/MAP) in a dose-dependent manner [196]. These findings indicate that impaired metabolic pathways in diabetes by decreasing NO synthesis and bioavailability lead to the impaired of endothelium-dependent vasodilatation, which could be improved by exogenous NO donors.
6.8.2. NO and renal complications of diabetes
Approximately 30% of patients with type 2 diabetes develop nephropathy; thus, it appears that hyperglycemia is necessary but not sufficient to result renal failure [197]. In the kidney, NO controls glomerular and renal hemodynamics and promotes natriuresis and diuresis [198], along with renal adaptation to dietary salt intake [199]. nNOS is mostly expressed in macula densa [200] and in small degree in specialized neurons within renal arteries of the hilus, arcuate and interlobular arteries [201]. eNOS is mostly found in renal vascular endothelium, although tubular expressions of eNOS have also been reported [201]. Although iNOS is weakly expressed in the kidney, its expression is dramatically increased by pro-inflammatory stimuli such as ischemia– reperfusion and lipopolysaccharide [202, 203]. Expression and activity of nNOS [204], iNOS [205], and eNOS [205] as well as NO metabolites [204, 206, 207] were found to be decreased in the kidney of diabetic animals.
Sodium nitrite, L-arginine, and daidzein (caveolin inhibitor) administration in HFD-STZ-induced diabetic rats have been shown to reduce blood urea nitrogen (BUN), serum creatinine, proteinuria, urinary output, kidney weight/ body weight, and renal cortical collagen content [207]; however, treatment with L-NAME, decreased the L-arginine-and daidzein-induced ameliorative effects in diabetic nephropathy [207]. In addition, nitrite treatment improved some parameters of glomerular injury, including urinary protein and albumin excretion, histopathological glomerular hypertrophy, and mesangial matrix accumulation in STZ-induced diabetic rats [206]. Improvement in renal function following L-arginine treatment and deterioration following NOS inhibitor (Nω-Nitro-L-arginine) have been reported in the ischemic acute renal failure of diabetic rats [208]. L-arginine prevents reduction in protein and mRNA expression of aquaporin-2, a water and sodium transporter in the renal outer medulla of diabetic rats [209].
7. Role of H2S in carbohydrate metabolism
The potential role of H2S in carbohydrate metabolism was first reported in 1990 when Hayden and colleagues demonstrated that H2S inhalation (2.2 mM) increased circulating glucose levels in postpartum rats [210]. Later on, a growing body of evidence showed that H2S is generated in pancreatic β-cells as well as in insulin target tissues including the liver, adipose tissue and skeletal muscles where it may control insulin secretion and insulin resistance.
7.1. H2S and the pancreas
Expressions of CSE [211–213], CBS [212–214], and 3-MST [215] have been documented in the pancreas; further analyses in the islets showed that expression of CSE, but not CBS, was dramatically increased following glucose stimulation, which suggests that CSE may act as an inducible H2S-generating enzyme, while CBS is constitutive [216]. CSE appears to be the major H2S-synthesisng enzyme in the pancreas, as most of the H2S produced from cultured insulinoma INS-1E cells was abolished after inhibition of CSE by propargylglycine (PPG) [211], and also following partially knockdown of the CSE gene, using an CSE-siRNA technique [211]. H2S production rate at basal glucose concentration is about 12 nmole/g/min in INS-1E cells [211], about 30 nmole/g/30 min in fresh rat pancreas [212], and 8 nmole/g/min in isolated rat islets [213].
H2S protects pancreatic β-cells against apoptosis induced oxidative stress or glucotoxicity in mice treated with glucose (5 and 20 mM); in this study, both low and high-glucose concentrations increased CSE expression but CBS expression remained relatively constant, suggesting that CSE-mediated H2S may act as an “intrinsic brake” against glucose-induced apoptotic death in β-cells [216]. Exogenous H2S has also been shown to protect β-cells from apoptosis induced by hydrogen peroxide, fatty acids, and cytokines; and through phosphorylation and activation of Akt signaling, promote cell proliferation and survival [217]. H2S by reducing elevated thioredoxin binding protein-2 (TBP-2) expression in isolated islets of HFD-fed CSE knockout (CSE-KO) mice, protected β-cells from glucotoxicity-induced apoptosis [218]; considering the role of TBP-2 in β-cell apoptosis, it can be speculated that H2S may protect β-cells from glucotoxicity-induced apoptosis by suppressing TBP-2 expression levels.
Conversely, exogenous H2S or overexpression of CSE in INS-1E cells, reduced cell viability and induced apoptosis; in this study, H2S increased expression of ER stress indices such as binding of immunoglobulin protein (BiP), CCAAT/enhancer-binding protein homologous protein (CHOP), and sterol regulatory element-binding transcription factor 1 (SREBF1) [219], which play a key role in pancreatic β-cell apoptosis and development of diabetes [220]. These contradictory effects of H2S on β-cell apoptosis may be due to the different types of insulin secreting cells used in different studies and their sensitivity to H2S [218], differences between exogenous and endogenous H2S [221], and differences in the dose and duration of exposure [222].
7.2. Insulin secretion and H2S
H2S can influence insulin secretion [211, 214, 223] and modulate circulating glucose levels [224]. Inhibitory effects of sodium hydrosulfide (NaSH, 10 μM-1 mM) and L-cysteine (0.1–10 7 mM) on glucose (10 mM)-induced insulin secretion have been observed in both isolated mouse islets and pancreatic beta cell line, MIN6 cells, an effect that was not observed at a low glucose concentration (3 mM) [214]. In addition, NaSH treatment (100 μM) inhibited insulin secretion by about 70% from HIT-T15 cells [225]. Overexpression of CSE in INS-1E cells virtually abolished high glucose (16 mM)-stimulated insulin secretion; however, basal insulin secretion was not altered [211]. NaSH at concentrations of 100 and 300 μM decreased glucose-stimulated insulin secretion by about 26% and 45%, respectively from isolated mouse β-cells; moreover, glucose induced insulin release in CSE-KO mice was three times higher than wild-type mice [223]. Indeed, increased extracellular glucose levels have been shown to decrease intracellular H2S production followed by increased insulin secretion [211]. With an increase in glucose level from 5 mM to 16 mM, the insulin secretion became 3-fold greater in INS-1E cells [211]. Taken together, these studies showan inhibitory effect of H2S on glucose stimulated insulin secretion.
One of the mechanisms through which H2S inhibits insulin secretion is through opening of KATP channels, as the inhibitory effects of NaSH and L-cysteine on insulin secretion were reproduced after using tolbutamide (a KATP blocker), α-ketoisocaproate (a mitochondrial fuel), and high K+ condition (30 mmol/L) [214]. Similar conclusions were drawn by an independent group of investigators, who showed that endogenous H2S levels may be a switch for turning KATP channels on/off at different glucose concentrations in INS-1E cells [211]. Interaction of H2S with KATP channels seems to be mediated through functional manipulation, probably by decreasing selective cysteine residues of KATP channel protein, independent of cytosolic second messengers [211]. It has been suggested that S-sulfhydration of KATP channels is an underlying mechanism by which H2S could influence insulin secretion (Figure 3) [222].
Figure 3. Mechanisms of hydrogen sulfide-inhibited insulin secretion from pancreatic β-cell.

Hydrogen sulfide (H2S) inhibits insulin secretion by opening of KATP channels via Ssulfhydration. Opening of KATP channels causes membrane hyperpolarization and therefore closing of VDCC. H2S also inhibits VDCC directly via S-sulfhydration. H2S inhibits glucose-induced mitochondrial membrane hyperpolarization and ATP production. G6P, Glucose 6-phosphate; SNAP 23/25, Synaptosome Associated Protein 23/25; VAMP, Vesicle Associated Membrane Protein.
In a study by Kaneko et al., L-cysteine and NaSH inhibited glucose-induced [Ca2+]i oscillation without obvious changes in the mean [Ca2+]i value in mouse pancreatic β-cells [214]. L-cysteine and NaSH also inhibited Ca2+-stimulated insulin secretion from permeabilized islets (treated with 9 streptolysin-O; SLO); interestingly, NaSH and L-cysteine also inhibited insulin release induced by the co-presence of guanosine 5′−0–3-thiotriphosphate (GTPγS) and Ca2+ from SLO-treated islets, under this condition, [Ca2+]i is not altered by either Ca2+ influx or mobilization, because of its chelation by ethylene glycol-bis (β-aminoethyl ether)-N,N,N’,N’-tetra acetic acid (EGTA) [214], indicating that the inhibitory effect of H2S on insulin release at least in part is independent of [Ca2+]i. This study also revealed that modulation of glucose metabolism in the pancreatic β-cells may be another mechanism which H2S inhibits insulin secretion, as NaSH and L-cysteine inhibited glucose-induced mitochondrial membrane hyperpolarization and ATP production [214].
Inhibition of L-type VDCC is another mechanism through which H2S inhibits insulin secretion in β-cells. Tang and colleagues demonstrated that NaSH and ACS67, another H2S donor, decreased insulin secretion from pancreatic β-cell as well as L-type VDCC current density by 45% and 18%, respectively [223]. Baseline VDCC current was higher in β-cells of CSE-KO mice and PPG increased baseline Ca2+ current in β-cells of wild-type mice; in addition, Bay K-8644, the specific agonist of VDCC, increased glucose-induced insulin secretion, an effect which was abolished by NaSH [223].
Despite the reported inhibitory effects of H2S on insulin secretion from the pancreatic β-cells, lower plasma H2S levels in type 2 diabetic patients may be a compensatory response [222], and contribute to the development of hyper-insulinemia to maintain normal glucose concentrations [222].
By contrast to these studies, the stimulatory effect of H2S on insulin secretion has been reported by Takahashi et al., who showed that inhibition of CBS by β-cyano-L-alanine, reduces cysteine hydropersulfide (Cys-SSH) levels, and 2-methylthio modification, as well as decreases in glucose-induced insulin release in two different β-cell lines were abolished by Cys-SSH precursor, Cys-S2-Cys, but not by NaSH [226]. Silencing of CBS or CSE by the respective siRNAs resulted in a decrease in 2-methylthio modification in HeLa cells concomitantly with reducing intracellular cysteine persulfide (Cys-SSH), one of the possible products of CBS and CSE-catalyzed reactions [226].
7.3. Glucose metabolism in skeletal muscle and H2S
Both CSE and CBS are expressed in human skeletal muscle cells [227]; although all H2S producing enzymes (CBS, CSE, 3-MST) are expressed in rat skeletal muscle, these enzymes surprisingly are absent in mice skeletal muscle [228]. The rate of H2S production in the skeletal muscles of Sprague-Dawley rats has been found to be 0.17 nmol/min/mg with an H2S content of 2.06 nmol/mg [228].
There is limited evidence demonstrating the potential role of H2S on carbohydrate metabolism in skeletal muscle. CSE gene knockdown resulted in a decrease in glucose uptake by cultured rat L6 myotubes and NaSH treatment at 25, 50, and 100 μM for 24h potentiated insulin-induced glucose uptake by 1.54, 1.72 and 2.06-fold, respectively, an effect which was mediated through increased phosphorylation of insulin receptors (IRs), PI3K, and Akt [229]. Moreover, NaSH (30 μmol/kg/day) increased phosphorylation of PI3K and Akt in skeletal muscles of Goto-Kakizaki rats, an experimental model of type 2 diabetes [229]. Inhalation of 5 mg/L H2S inhibited aerobic metabolism, resulting in a significant accumulation of circulating lactate during exercise in healthy men [230]. This metabolic shift was probably due to a decrease in citrate synthase levels, which is a rate limiting enzyme in the tricarboxylic acid (TCA) cycle in skeletal muscle [230].
H2S-mediated activation of Wnt/β-catenin signaling pathway [231] may partly elucidate the H2S effect on insulin sensitivity in skeletal muscle cells. Activation of skeletal muscle Wnt/β-catenin signaling increases insulin sensitivity through decreasing intra-myocellular lipid deposition and down-regulation of SREBP-1c, inhibiting MAPK pathway, and also trough increasing activation of the Akt/PKB and AMPK pathways [232].
7.4. Glucose metabolism in adipose tissue and H2S
H2S is produced in epidydimal, perirenal, and brown adipose tissue with the rate of 4.76, 2.93, and 4.65 nmol/min/protein, respectively. In addition, H2S-producing rates in cultured adipocytes and preadipocytes were 2.89 and 2.17 nmol/min/mg protein, respectively [233]. Although both CBS and CSE are expressed in adipose tissue, CSE appears to be the main H2S-producing enzyme [233], as CSE inhibitors decreased H2S production by more than 80% in adipose tissue.
Aging is associated with upregulation of CSE expression and H2S production in adipocytes while hyperglycemia through increases in ROS down-regulates CSE expression [233]. Moreover, CSE expression was higher in adipose tissue macrophages of obese animals; however, endogenous H2S level was lower, which indicates reduced H2S bioavailability in obesity [234]. Decreased H2S bioavailability in obesity activates store-operated Ca2+ entry pathway in adipose tissue macrophages and increases cytokine production [234]. CSE protein expression has been shown to be increased in lipopolysaccharides (LPS)-induced inflammation in a mouse macrophage cell line (RAW264.7 cells), whereas H2S level is decreased due to enhanced cellular demand and/or consumption of H2S [234]. Indeed, decreased H2S bioavailability following hyperglycemia and 5 obesity plays a key role in the development of metabolic syndrome [235].
H2S plays a regulatory role in adipogenesis [236] and adipose tissue metabolism [235]. Adipogenesis and adipose tissue maturation are promoted by endogenous and exogenous H2S [236]. Differentiation of 3T3L1 cells is associated with upregulation of all H2S-producing enzymes (CBS, CSE, 3-MST) and incubation of preadipocytes with GYY4137 (a slow H2S releasing compound) or NaSH, increase adipocyte differentiation factors, such as proliferator11 activated receptor γ (PPARγ) and CCAAT/enhancer binding protein α (CEBPα) [236].
The role of H2S on glucose uptake in adipose tissue and therefore development of insulin resistance is controversial as both inhibitory and stimulatory effects of H2S have been reported [233, 237–239]. H2S (10–1000 μM) as well as L-cysteine decreased basal and insulin-stimulated glucose uptake in the mature adipocytes, an effect which was reversed by CSE inhibitors (PPG or BCA) [233]. CSE expression and H2S production increased in adipose tissue of high fructose fed rats, which is a commonly used model of insulin resistance and hyperlipidemia [233]. Increased adipose tissue CSE-H2S system in insulin-resistant rats correlated with impaired insulin-induced glucose uptake [233]. In this study, H2S-inhibited glucose uptake of adipocytes was mediated through PI3K but not KATP channel pathway, which may inhibit GLUT4 translocation [233]. These findings suggest that adipose-released H2S may contribute to the pathogenesis of insulin resistance and diabetes [233]. In addition, TNF-α inhibits insulin-stimulated glucose uptake of 3T3-L1 adipocytes, this effect is accompanied by an increase in CSE activity, expression, and H2S generation [240]. A CSE inhibitor attenuated the detrimental effect of TNF-α on insulin-stimulated glucose uptake of 3T3-L1 adipocytes; however, a CBS inhibitor had no effect [240]. The authors accordingly suggested that the detrimental effects of TNF-α on insulin sensitivity may be partially mediated by H2S and modulation of CSE-H2S 5 system can be a potential therapeutic approach for insulin resistance [240].
By contrast, Manna et al. examined the effects of L-cysteine (100, 500, and 1000 μM) and Na2S (10 and 100 μM) on phosphatase and tensin homolog (PTEN), PI3K, PIP3, phospho-AKT and glucose uptake in high glucose (25 mM)-treated 3T3L1 adipocyte cells. H2S decreased PTEN (a negative regulator of glucose utilization) and increased PI3K and PIP3 levels, as well as restored phosphorylation of IRS1, phospho-AKT, and GLUT4 translocation in adipocytes, and eventually increased glucose utilization [237]. The protective role of L-cysteine was blocked by PAG, in addition, H2S and L-cysteine had no effect on glucose utilization by cells cultured at normoglycemic conditions [237]. The same group also reported that hyperglycemia decreases CSE expression and H2S production by 3T3-L1 cells and suggested that hyperglycemia-induced insulin resistance at least in part is mediated through downregulation of the CSE-H2S system [16].
Similarly, Cai et al. demonstrated that H2S gas and GYY4137 stimulate glucose uptake by 3T3-L1 adipocytes which was associated with persulfidation of PPAR-γ at Cys139 [238]. H2S-stimulated glucose uptake was abolished when Cys139 was replaced by serine producing a mutant PPAR-γ; this mutant could not be persulfidated, indicating that PPARγ C139 site is a major sulhydration site [238]. In addition, CSE‒H2S inhibited adipose-tissue PDE activity whereas it increased PPARγ activity and adipocyte numbers in mice fed a HFD; H2S induced PPARγ sulfhydration and reduced insulin resistance but did not continuously increase obesity [238], which may explain why some obese patients (CSE‒H2S system not downregulated) do not have diabetes [238]. H2S also inhibited lipolysis through the protein kinase A (PKA)perilipin/hormone-sensitive lipase pathway, which promotes and sensitizes insulin response in adipocytes [239].
Vitamin D has a favorable effect on insulin sensitivity and it is considered to be of potential value in management of type 2 diabetes. It has been shown that, 1, 25-dihydroxycholecalciferol (1,25-(OH)2-D3), the active metabolite of vitamin D3, up-regulates CSE expression and H2S production in 3T3-L1 adipocytes cultured in high glucose medium [16]. In addition, 1,25-(OH)2 D3 through increased IRS-1 phosphorylation, PI3K activity and Akt phosphorylation, increased insulin-stimulated glucose uptake, these effects were abolished by CSE inhibitor or CSE gene knockdown [16].
NaSH (25 and 50 μM) promoted insulin-stimulated glucose uptake in 3T3-L1 adipocytes; H2S increased phosphorylation of the insulin receptor β-subunit, PI3K activity and Akt phosphorylation in cells cultured at both normo- or hyperglycemic conditions [229]. NaSH also increased insulin receptor tyrosine phosphorylation and its kinase activity in a cell free system, indicating that insulin receptor may be the direct target for the stimulatory effects of H2S [229].
7.5. Effects of H2S on hepatic glucose metabolism
All three H2S-producing enzymes are expressed in the liver but H2S production seems to be primarily catalyzed by CSE [241, 242]. H2S plays an important role in glucose metabolism and insulin signaling in hepatocytes; H2S stimulates hepatic glucose production and activates gluconeogenesis and glycogenolysis [243]. CSE protein was shown to be downregulated upon starvation in murine liver extracts [244], and fasting-induced downregulation of CSE can prevent hepatic glucose production and release into the circulation. Hepatic CBS expression is up-regulated in both prediabetic insulin-resistance and frank diabetic stages of Zucker diabetic fatty (ZDF) rats [245].
NaSH (100 μM) decreased glucose uptake and glycogen content in HepG2 cells; in addition, primary hepatocytes from CSE-KO mice showed a 2-fold increase in glucose consumption rate [246]. CSE-KO mice had higher glucose consumption and glycogen content in their liver tissues; however, lower glucose was produced by hepatocytes via gluconeogenesis and glycogenolysis pathways in these mice [246]. Overexpression of CSE in HepG2 cells, increased endogenous H2S production and decreased glycogen content [246]. Decreased AMPK activation and suppression of glucokinase activity were responsible for H2S-downregulated glucose uptake and glycogen storage; furthermore, H2S-increased glucose production was mediated by inhibition of AKT signaling which is followed by activation of PEPCK [246]. H2S-increased PEPCK activity has also been reported to be induced by increased glucocorticoid receptor activity and decreased AMPK phosphorylation [221]. Similarly, activity of glucose 6-phosphatase and fructose-1,6-bisphosphatase, the rate-limiting gluconeogenic enzymes, is promoted by H2S through S- sulfhydration [247]. H2S also increases the expression of theses enzymes indirectly through S- sulfhydration of the peroxisome proliferator-activated receptor gamma coactivator 1-ɑ (PGC-1ɑ) [221]. H2S-induced glucose production is also mediated by S-sulfhydration and increased activity of pyruvate carboxylase, a key enzyme providing fuel for gluconeogenesis [248]. Taken together, these studies suggest that H2S may play a pivotal role in hepatic insulin resistance and is further involved in the pathogenesis of type 2 diabetes.
Mitochondrial dysfunction is associated with the pathogenesis of insulin resistance. CSE-KO hepatocytes were shown to have less mitochondrial content and DNA which were restored by NaSH [249]; CSE-KO hepatocytes exhibited lower levels of transcription factors involved in mitochondrial biogenesis including nuclear respiratory factors-1 and −2 (NRF-1, NRF-2), PGC-1α, PGC-1β, and PGC-related protein (PPRC) [249]. NaSH treatment (30 μM) upregulated PPRC by S-sulfhydration, yet downregulated PGC-1β protein levels [249]. Knockdown of either PGC-1α or PPRC reduced NaSH-induced mitochondrial biogenesis in hepatocytes, while knockdown of both genes completely abolished effects of NaSH on mitochondrial biogenesis [249].
7.6. Pathophysiology of diabetes: Role of H2S
H2S deficiency is related to the pathophysiology of diabetes; however, the potential role of H2S in diabetes seems to be complex. Contribution of H2S in the onset and progression of diabetes has been reported in several studies. Expressions of CSE and CBS mRNAs, as well as endogenous H2S production are higher in both the liver and pancreas of STZ-induced diabetic rats [212]. Similarly, activities of the hepatic CBS and CSE increased in STZ-induced diabetic rats, this effect was normalized by insulin treatment [250]. Pancreatic CSE expression and H2S production are higher in ZDF rats than in Zucker fatty and Zucker lean rats [213]; PPG treatment of ZDF rats resulted in an increased serum insulin concentration and decreased hyperglycemia [213]. We recently showed that NaSH at high doses (1.6–5.6 mg/kg) aggravated carbohydrate metabolism while at low doses (0.28 and 0.56 mg/kg) it had no effect [251]. A positive 18 correlation has been found between H2S concentration and disrupted insulin secretion in pancreatic β-cells [252], suggesting that inhibition of pancreatic H2S may be a new therapeutic approach for the management of diabetes [213, 252]. However, it has been suggested that hyperglycemia-induced pancreatic CSE overexpression in the early stages of diabetes, may have as a protective mechanism, as H2S neutralizes oxidative/nitrosative stress [15].
Conversely, endogenous H2S production and plasma H2S levels decreased in both non-obese diabetic mice [253], STZ-treated diabetic rats [12, 251] and patients with type 2 diabetes [11, 12] which was parallel with the severity of diabetes [11, 253], in particular in those with a history of cardiovascular disease [11]. Moreover, metformin administration in diabetic rats was associated with elevated H2S levels in the kidney, heart, liver, and brain [254]. Hyperglycemia causes H2S deficiency in endothelial cells and administration of H2S could be a potential therapeutic approach in hyperglycemia [255]. Indeed, high glucose levels have been shown to inhibit H2S production via specificity protein 1 (SP1) and p38 MAPK phosphorylation in INS-1E cells and freshly isolated rat pancreatic islets [256]. In addition, hyperglycemia and increased ROS levels decrease CSE expression [257] and increase H2S consumption [255], leading to lower H2S levels in diabetes.
To summarize, whether H2S has a beneficial or deleterious effects on glucose metabolism in type 2 diabetes is inconclusive and further studies are needed to clear the potential role of H2S manipulation in treatment of diabetes. Regarding the association between diabetes and H2S deficiency [12, 258], H2S modulation may have potential therapeutic effects in diabetes.
7.7. Diabetes complications and H2S
H2S plays multiple protective roles in the prevention and improvement of complications associated with diabetes [259]. Favorable effects of H2S have been reported in endothelial dysfunction, retinopathy, cardiomyopathy, and nephropathy [255, 260–262].
7.7.1. Diabetic cardiovascular complications and H2S
Increasing evidence indicates that H2S has multiple beneficial roles in diabetic cardiovascular complications. H2S attenuates the development of diabetic cardiomyopathy as exemplified by Barr and colleagues who examined the role of H2S in the pathogenesis of diabetic cardiomyopathy in mice that were fed a HFD [263]. These mice had reduced circulating and cardiac H2S levels, hallmark features of type-2 diabetes, and also marked cardiac dysfunction. H2S treatment restored sulfide levels and attenuated HFD-induced cardiac dysfunction; the protective effects of H2S were associated with the activation of adiponectin-AMPK signaling and suppression of HFD-induced ER stress [263]. Adiponectin plays an important role in maintaining cardiovascular health and there is a correlation between decreased adiponectin levels and increase cardiovascular risk [264]. Adiponectin delivers much of its metabolic-regulatory effects through the induction of AMPK [265]. AMPK stimulates glucose transport by increasing the expression of GLUT4 and also modulates cardiac fatty acid oxidation and subsequent lipid accumulation through the phosphorylation and inhibition of acetyl-coenzyme A carboxylase [266].
NaSH administration was found to improve ventricular function and attenuate cardiac hypertrophy and myocardial fibrosis in STZ-induced diabetic rats [262]; NaSH also reduced hyperglycemia-induce inflammation, oxidative stress and apoptosis in the cardiac tissue; favorable effects of NaSH were associated with increased activation of Nrf2 and protein expression of its downstream targets, increased activation of PI3K/Akt pathway and also blockade of c-Jun N-terminal kinase (JNK) and p38 MAPK pathways [262]. Similarly, using diabetic mice, Ye et al. demonstrated that the protective effects of NaSH on cardiac structure and function as well as reduction in apoptosis are associated with increased FoxO1 phosphorylation and the prevention of FoxO1 nuclear translocation in the injured tissue [267]. FoxO1 is critical for cellular processes that are involved in reducing oxidative stress, cell resistance to apoptosis, cell survival, energy metabolism, and cell death [268]. Furthermore, favorable effects of H2S in diabetic cardiomyopathy were attributed to considerable reductions in the up-regulated matrix metalloproteinase 2 and transforming growth factor (TGF)-β1 in the hearts of diabetic animals [269].
Xie et al. showed that H2S reduces aortic atherosclerotic plaque formation with reductions in superoxide generation and the adhesion molecules in STZ-induced LDLr(−/−) mice [270], a protective effect attributed to inhibition of oxidative stress via kelch ECH associating protein 1 (Keap1) sulfhydrylation at Cys151 to activate Nrf2 signaling [270]. H2S also inhibits development of atherosclerosis via blocking diabetes-induced oxidative and inflammatory stress in endothelial cells, decreases in ROS levels and prevents foam cell formation by macrophages [271]. H2S supplementation in diabetic rats normalized plasma H2S levels and improved the endothelium-dependent relaxant responses of the thoracic aorta ex vivo, without affecting the degree of hyperglycemia [255].
7.7.2. Renal complications of diabetes and H2S
Yamamoto and colleagues examined the role of NaSH in diabetic nephropathy, using pancreatic β-cell specific calmodulin-overexpressing transgenic mice as a model of diabetes [272]. In this study, diabetic mice had higher BUN and albuminuria as well as decreased CSE expression in the kidneys, however CBS expression was not altered. Renal peritubular capillary (PTC) blood flow velocity, diameter and blood flow were decreased in the kidneys and the hematocrit was on the low side. NaSH treatment dilated PTC diameter and increased blood flow in diabetic mice but had no effect on PTC blood flow velocity [272]. These findings suggest that the CSE-H2S system in the proximal tubules may regulate the interstitial microcirculation and that H2S releasing compounds may offer a useful strategy for the treatment of diabetic nephropathy.
NaSH improved renal tissue fibrosis in STZ-treated diabetic rats by inhibition of autophagy, up regulation of superoxide dismutase, and down-regulation of serine/threonine kinase, TGF-β1 and NF-κB protein, as key mediators of diabetic nephropathy [273]. Similarly, NaSH administration to diabetic rats attenuated high BUN levels, and reduced renal collagen and TGF-β1 expression, without affecting hyperglycemia levels [274]. NaSH also improved renal function and decreased mesangial matrix deposition, glomerular basement membrane thickening, and renal interstitial fibrosis in diabetic rats [260]; furthermore, NaSH by activation of the Nrf2 antioxidant pathway and inhibiting NF-κB signaling decreased high glucose-induced oxidative stress [260]. H2S inhibited the renin-angiotensin system in the diabetic kidney and attenuated high glucose induced mesangial cell proliferation by suppression of the MAPK signaling pathway [260].
8. NO and H2S interactions in carbohydrate metabolism
In the past few years, much attention has been given to the interactions between NO and H2S. There is a growing body of evidence showing that these two gasotransmitters interact with each other at the levels of both biosynthesis and biological responses. Although the individual roles of these two gasotransmitters in both physiological and pathophysiological function are well appreciated, consequences of their interactions need further research. Understanding the interactions between these two molecules will prove fruitful in managing several pathophysiological conditions.
8.1. Biosynthesis of H2S and NO and their interaction
We recently examined the combination effects of sodium nitrite and NaSH on carbohydrate metabolism in type 2 diabetic rats [19]. In this study, nitrite supplementation increased serum total sulfide levels and NaSH administration increased serum NOx (nitrate+nitrite), however, the combined treatments further increased NOx but not sulfide levels. In addition, nitrite supplementation increased mRNA expression levels of CSE and CBS in the soleus muscle and CBS in the liver and epididymal adipose tissue of diabetic rats [19]. NaSH administration was also shown to decrease total NOS and iNOS activities as well as NO content in diabetic rats [275].
Effects of H2S on activity and expression of NOS enzymes are controversial. H2S was shown to increase eNOS activity [17, 101, 276], have no effect on iNOS and nNOS activities [99], or inhibit all NOS isoforms [4]. These controversies may at least in part be due to the time of NOS activity measurement after H2S administration, as the stimulatory effects of H2S on eNOS activity are transient [277]. Moreover, low concentrations of L-cysteine (0.1–3 mM and 0.01–1 mM, respectively) promote the activities of nNOS and iNOS but not that of eNOS, and high concentrations inhibit the activity of all NOS enzymes (10 mM for nNOS and 3–10 mM for iNOS or eNOS) [4]. This effect of L-cysteine is considered to be independent of its property as a reductant and zinc-chelator, because DTT (threo-1, 4-dimercapto-2, 3-butanediol), a reducing agent and also NaSH that possess similar properties did not mimic the facilitating effects of L-cysteine at low concentrations [4], thus the underlying mechanisms for these paradoxical effects of L-cysteine on the NOS activity have yet to be clarified.
The stimulatory effects of H2S on eNOS activity is partly due to an elevated [Ca2+]i levels [1, 17, 95, 101, 278–280], resulting in phosphorylation of eNOS at S1177 [103, 280, 281], and inhibition of eNOS S-nitrosylation [85]. Furthermore, H2S prevents eNOS degradation [281] and stabilizes the dimeric active form of eNOS [103] through sulfhydration of Cys443 [17, 85]. H2S-increased [Ca2+]i is mediated through increasing IP3-dependent intracellular Ca2+ mobilization, activating KATP channels, and favoring the reverse mode of Na+-Ca2+ exchanger [1, 17, 101]. Inhibitory effect of NaSH on eNOS is concentration dependent with an IC50 of 170 μM [4]; which is due to the inhibition of BH4 function [282] and decreasing the phospho-eNOS (serine 1177) [283].
The interaction between NO and H2S is also influences the diabetic cardiomyopathy; Yang and colleagues showed that iNOS expression in STZ-induced diabetic rats is positively correlated with the severity of myocardial injury [275]. In this study, the cardioprotective effects of H2S (56 μmol/kg/day NaSH) were related to the decrease in activity and expression of iNOS; inhibition of H2S exacerbated myocardial injury [275].
Effect of NO on H2S synthesis is also not straightforward. It has been reported that NO increases the activity and expression of CSE [93, 102, 282, 284, 285], has no effect on H2S-producing enzymes [286], or even directly inhibits CSE activity in vitro with an IC50 of about 100 nM [95]. NO-increased H2S synthesis is mediated by cGMP as it is reduced by inhibition of sGC [287]. CSE also is a target of S-nitrosylation at its multiple reactive cysteine residues [288]. Heme-containing proteins are targets of NO, thus, CBS activity might be affected by NO [289].
8.2. Interaction between NO and H2S in biological responses
The interaction between thiol molecules and NO is the bases of NO-induced S-nitrosylation. As H2S is a thiol molecule, it is possible that H2S interacts with NO to form nitrothiols (RSNO). Incubation of NaSH with SNP in vitro resulted in a time dependent release of nitrite, indicating formation of nitrosothiol [290]. In addition, incubation of liver homogenates from LPS-treated rats with NaSH or L-cysteine and pyridoxal phosphate to increase endogenous production of H2S, increased nitrite formation as an outcome of the interaction between exogenous NaSH and endogenous NO [290]. In addition to nitrosothiol, interaction of NO and H2S and their respective metabolites can generate different species that have significantly different physiological functions as compared to either NO and/or H2S [291–294]. These products include nitrosopersulfide (SSNO−) [294–298], thionitrous acid (HSNO) [293, 295, 297, 299], and nitroxyl (HNO) [294, 296, 297].
In our previous study, NaSH at a low dose, which had no effect on carbohydrate metabolism, potentiated the favorable metabolic effects of sodium nitrite in type 2 diabetic rats [19]. These favorable effects were associated with improvement of liver function, reduced oxidative stress, and increased mRNA expression and protein levels of GLUT4 in insulin-sensitive tissues [19]. H2S stimulates NO release from nitrite via increasing the activity of XO [300] and also enhances production of sulfinyl nitrite (an NO donor) [301], suggesting that nitrite in the presence of H2S becomes more biologically active. In addition, H2S maintains sGC in an NO-activatable form [103] and increases cGMP levels by inhibiting PDE5 [17].
The interaction between NO and H2S has also been reported in diabetes-induced nephropathy; NaSH administration decreased fasting blood glucose, insulin, lipid profile, urea and creatinine, as well as insulin resistance in HFD-STZ-induced type 2 diabetic rats [302]. NaSH also decreased TNF-α, NF-κB, TGF-β, caspase 3, malondialdehyde (MDA), and H2O2 production, whereas it increased catalase and superoxide dismutase activities in renal tissue of diabetic rats. However, chronic administration of L-NAME in combination with NaSH diminished the favorable effects of NaSH on diabetes-induced nephropathy, serum insulin, urea, and creatinine, as well as tissue levels of TNF-α, NF-κB, TGF-β, caspase 3, MDA, and H2O2 production, and activity of antioxidant enzymes [302]. Collectively, these data strongly suggest that NO has a significant role in the protective effects of H2S in diabetes-induced nephropathy.
Expression of NOX4, the major source of ROS in the kidney, is increased in diabetes. Genetic deletion or chemical inhibitors of NOX4 ameliorate kidney injury that is diabetes induced [303]. Reduced activity of AMPK is also associated with high glucose-induced increases in NOX4 expression; AMPK activation prevents the deleterious effects of glucose in the kidney via inhibition of high glucose-induced NOX4 protein expression and subsequent ROS generation [304]. NaSH inhibited high glucose-induced NOX4 expression via activation of AMPK, an effect which was reversed by L-NAME, suggesting a role for NO in mediating the H2S effect; in this study, NaSH increased mRNA and protein expression of iNOS but not that of eNOS in the mouse kidney proximal tubular epithelial cells [305]. In addition, compound C, a selective AMPK inhibitor, blocked the NaSH-induced increases in iNOS expression [305]. These data demonstrate that H2S stimulates iNOS expression in an AMPK-dependent manner to inhibit the high glucose-induced increase in NOX4.
9. Conclusion and perspectives
All H2S-producing enzymes are found in the pancreas and insulin sensitive tissues; however, CSE seems to be the major enzyme in these tissues. All NO-producing enzymes are also expressed in the pancreas and insulin sensitive tissues; although nNOS expression does not appear to be present in any significant amounts in the adipose tissue, it is the main isoform of NOS in skeletal muscle. Both NO and H2S have roles in pathophysiology of diabetes and regulation of blood glucose levels. Deficiencies in H2S and NO systems have been reported in diabetic animal models as well as in human studies. The role of H2S in carbohydrate metabolism is however controversial, H2S causes inhibition of insulin secretion by activation of KATP channels and inhibition of VDCC in pancreatic β-cells. In addition, H2S through inhibition of GLUT4, inhibits insulin-stimulated glucose uptake in insulin sensitive tissues. By contrast, H2S protects β-cells against glucotoxicity-induced apoptosis and up-regulates insulin signaling pathways essential for glucose utilization, suggesting a potential therapeutic approach in diabetes. Regarding H2S administration in animal models, its multiple functions against diabetes-induced cardiovascular diseases, nephropathy, and other complications have been documented.
In addition, iNOS-derived NO decreases while eNOS-derived NO increases insulin secretion. eNOS−/− mice have lower glucose transport in skeletal muscle, indicating regulation of glucose uptake by NO. Regarding effects of these gasotranmitters on carbohydrate metabolism, translational work in this area requires a deep understanding of the biology and pharmacology of H2S and NO, as well as an ability to integrate this scientific knowledge with the principles of drug development. In addition, it should be noted that in compare to NO, H2S is the youngest member of the gasotransmitter family and certainly needs further work to fully elucidate its effects on carbohydrate metabolism. Deficiencies in both H2S and NO contribute to the development of diabetes, and in this context, combination treatment modalities may prove fruitful in managing diabetes. Another area that warrants attention is the possible roles of these gasotransmitters in the diabetes-cancer-axis; since undesirable changes in the H2S and NO systems contribute to the pathophysiological conditions of both, this topic needs further research.
Acknowledgments
KK, supported in part by the National Institutes of Health [R24 DA018055; R01GM123508] and the Professional Staff Congress-City University of New York (PSC-CUNY) [TRADB-49-271].
Abbreviations
- 3-MST
3-Mercaptopyruvate sulfurtransferase
- AMPK
AMP-activated protein kinase
- ATP
Adenosine triphosphate
- BH2
7,8- Dihydrobiopterin
- BH4
Tetrahydrobiopterin
- CAT
Cysteine aminotransferase
- CBS
Cystathionine β synthase
- cGMP
Cyclic guanosine monophosphate
- CSE
Cystathionine γ lyase
- EGTA
Ethylene glycol-bis (β-aminoethyl ether)-N,N,N’,N’-tetra acetic acid
- eNOS
Endothelial nitric oxide synthase
- ER
Endoplasmic reticulum
- FFA
Free fatty acid
- G6P
Glucose 6-phosphate
- GSNO
S-Nitrosoglutathione
- H2S
Hydrogen sulfide
- HFD
High-fat diet
- IL-1β
Interleukin 1 beta
- iNOS
Inducible nitric oxide synthase
- IP3R
Inositol-3-phosphate receptor
- KATP
K+ channels
- L-NAME
L-NG-Nitroarginine methyl ester
- L-NMMA
NG-monomethyl-L-arginine
- MAPK
Mitogen-activated protein kinase
- NADPH
Nicotinamide-adenine-dinucleotide phosphate
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- nNOS
Neuronal nitric oxide synthase
- NO
Nitric oxide
- NO2−
Nitrite
- NO3−
Nitrate
- NOS
Nitric oxide synthase
- Nrf2
Nuclear factor-like 2
- PDE5
Phosphodiesterase 5
- PEPCK
Phosphoenolpyruvate carboxykinase
- PIP2
Phosphatidylinositol 4, 5-bisphosphate
- PIP3
Phosphatidylinositol (3, 4, 5)-triphosphate
- PKG
Protein kinase G
- PLP
Pyridoxal-5′-phosphate
- PPG
Propargylglycine
- ROS
Reactive oxygen species
- sGC
Soluble guanylyl cyclase
- SH
Sulfhydryl group
- SNAP
S-Nitroso-N-acetylpenicillamine
- SNAP23/25
Synaptosome associated protein 23/25
- SNP
Sodium nitroprusside
- SREBF1
Sterol regulatory element-binding transcription factor 1
- SSH
Hydropersulfid moiety
- STZ
Streptozotocin
- TLR4
Toll-like receptor 4
- TNF-α
Tumor necrosis factor-α
- UCP-2
Uncoupling protein 2
- VAMP
Vesicle associated membrane protein
- VDCC
L-type voltage-dependent Ca2+ channels
- XO
Xanthine oxidase
Footnotes
Conflicts of interest
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Moccia F, Bertoni G, Pla AF, Dragoni S, Pupo E, Merlino A, Mancardi D, Munaron L, Tanzi F, Hydrogen sulfide regulates intracellular Ca2+ concentration in endothelial cells from excised rat aorta, Current pharmaceutical biotechnology 12(9) (2011) 1416–26. [DOI] [PubMed] [Google Scholar]
- [2].Kamoun P, Endogenous production of hydrogen sulfide in mammals, Amino Acids 26(3) (2004) 243–54. [DOI] [PubMed] [Google Scholar]
- [3].Martelli A, Testai L, Breschi MC, Lawson K, McKay NG, Miceli F, Taglialatela M, Calderone V, Vasorelaxation by hydrogen sulphide involves activation of Kv7 potassium channels, Pharmacological research 70(1) (2013) 27–34. [DOI] [PubMed] [Google Scholar]
- [4].Kubo S, Kurokawa Y, Doe I, Masuko T, Sekiguchi F, Kawabata A, Hydrogen sulfide inhibits activity of three isoforms of recombinant nitric oxide synthase, Toxicology 241(1–2) (2007) 92–7. [DOI] [PubMed] [Google Scholar]
- [5].Greaney JL, Kutz JL, Shank SW, Jandu S, Santhanam L, Alexander LM, Impaired Hydrogen Sulfide-Mediated Vasodilation Contributes to Microvascular Endothelial Dysfunction in Hypertensive Adults, Hypertension 69(5) (2017) 902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gheibi S, Aboutaleb N, Khaksari M, Kalalian-Moghaddam H, Vakili A, Asadi Y, Mehrjerdi FZ, Gheibi A, Hydrogen sulfide protects the brain against ischemic reperfusion injury in a transient model of focal cerebral ischemia, J Mol Neurosci 54(2) (2014) 264–70. [DOI] [PubMed] [Google Scholar]
- [7].Gheibi S, Jeddi S, Kashfi K, Ghasemi A, Regulation of vascular tone homeostasis by NO and H2S: Implications in hypertension, Biochemical pharmacology 149 (2018) 42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Furchgott RF, Zawadzki JV, The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine, Nature 288(5789) (1980) 373–376. [DOI] [PubMed] [Google Scholar]
- [9].Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G, Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide, Proc Natl Acad Sci U S A 84(24) (1987) 9265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Carlstrom M, Larsen FJ, Nystrom T, Hezel M, Borniquel S, Weitzberg E, Lundberg JO, Dietary inorganic nitrate reverses features of metabolic syndrome in endothelial nitric oxide synthase-deficient mice, Proc Natl Acad Sci USA 107(41) (2010) 17716–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Suzuki K, Sagara M, Aoki C, Tanaka S, Aso Y, Clinical Implication of Plasma Hydrogen Sulfide Levels in Japanese Patients with Type 2 Diabetes, Internal medicine (Tokyo, Japan) 56(1) (2017) 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jain SK, Bull R, Rains JL, Bass PF, Levine SN, Reddy S, McVie R, Bocchini JA, Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation?, Antioxidants & redox signaling 12(11) (2010) 1333–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gheibi S, Bakhtiarzadeh F, Jeddi S, Farrokhfall K, Zardooz H, Ghasemi A, Nitrite increases 34 glucose-stimulated insulin secretion and islet insulin content in obese type 2 diabetic male rats, Nitric oxide : biology and chemistry 64 (2017) 39–51. [DOI] [PubMed] [Google Scholar]
- [14].Gheibi S, Jeddi S, Carlström M, Gholami H, Ghasemi A, Effects of long-term nitrate 37 supplementation on carbohydrate metabolism, lipid profiles, oxidative stress, and inflammation in male obese type 2 diabetic rats, Nitric oxide : biology and chemistry 75 (2018) 27–41. [DOI] [PubMed] [Google Scholar]
- [15].Szabo C, Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications, Antioxidants & redox signaling 17(1) (2012) 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Manna P, Jain SK, Vitamin D up-regulates glucose transporter 4 (GLUT4) translocation and glucose utilization mediated by cystathionine-gamma-lyase (CSE) activation and H2S formation in 3T3L1 adipocytes, J Biol Chem 287(50) (2012) 42324–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Szabo C, Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: mechanisms and implications, American journal of physiology. Cell physiology 312(1) (2017) C3–C15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Szabo C, Gaseotransmitters: new frontiers for translational science, Science translational medicine 2(59) (2010) 59ps54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gheibi S, Jeddi S, Carlstrom M, Kashfi K, Ghasemi A, Hydrogen sulfide potentiates the favorable metabolic effects of inorganic nitrite in type 2 diabetic rats, Nitric oxide : biology and chemistry 92 (2019) 60–72. [DOI] [PubMed] [Google Scholar]
- [20].Hosoki R, Matsuki N, Kimura H, The possible role of hydrogen sulfide as an endogenous smooth 7 muscle relaxant in synergy with nitric oxide, Biochemical and biophysical research communications 237(3) (1997) 527–31. [DOI] [PubMed] [Google Scholar]
- [21].Higashino H, Tabuchi M, Yamagata S, Kurita T, Miya H, Mukai H, Miya Y, Serum nitric oxide metabolite levels in groups of patients with various diseases in comparison of healthy control subjects Journal of Medical Sciences 10(1) (2010) 1–11. [Google Scholar]
- [22].Ghasemi A, Zahedi Asl S, Is nitric oxide a hormone?, Iranian Biomedical Journal 15 (2011) 59–65. [PMC free article] [PubMed] [Google Scholar]
- [23].Luiking YC, Ten Have GA, Wolfe RR, Deutz NE, Arginine de novo and nitric oxide production in disease states, Am J Physiol Endocrinol Metab 303(10) (2012) E1177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tan B, Li X, Yin Y, Wu Z, Liu C, Tekwe CD, Wu G, Regulatory roles for L-arginine in reducing white adipose tissue, Front Biosci (Landmark Ed) 17 (2012) 2237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ghasemi A, Jeddi S, Anti-obesity and anti-diabetic effects of nitrate and nitrite, Nitric oxide : biology and chemistry 70 (2017) 9–24. [DOI] [PubMed] [Google Scholar]
- [26].Aronstam RS, Martin DC, Dennison RL, Cooley HG, S-nitrosylation of m2 muscarinic receptor thiols disrupts receptor-G-protein coupling, Ann N Y Acad Sci 757 (1995) 215–7. [DOI] [PubMed] [Google Scholar]
- [27].Sakinis A, Wennmalm A, Estimation of total rate of formation of nitric oxide in the rat, The Biochemical Journal 330 ( Pt 1) (1998) 527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wickman A, Klintland N, Gan LM, Sakinis A, Soderling AS, Bergstrom G, Caidahl K, A technique to estimate the rate of whole body nitric oxide formation in conscious mice, Nitric oxide : biology and chemistry 9(2) (2003) 77–85. [DOI] [PubMed] [Google Scholar]
- [29].Wagner DA, Schultz DS, Deen WM, Young VR, Tannenbaum SR, Metabolic fate of an oral dose of 15N-labeled nitrate in humans: effect of diet supplementation with ascorbic acid, Cancer Res 43(4) (1983) 1921–5. [PubMed] [Google Scholar]
- [30].Kelm M, Nitric oxide metabolism and breakdown, Biochim Biophys Acta 1411(2–3) (1999) 273–89. [DOI] [PubMed] [Google Scholar]
- [31].Knowles RG, Moncada S, Nitric oxide synthases in mammals, Biochem J 298 ( Pt 2) (1994) 249–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sansbury BE, Hill BG, Regulation of obesity and insulin resistance by nitric oxide, Free Radic Biol Med 73 (2014) 383–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].R.O.r. Cannon, Role of nitric oxide in cardiovascular disease: focus on the endothelium, Clin Chem 44(8 Pt 2) (1998) 1809–19. [PubMed] [Google Scholar]
- [34].Yoon Y, Song J, Hong SH, Kim JQ, Plasma nitric oxide concentrations and nitric oxide synthase gene polymorphisms in coronary artery disease, Clin Chem 46(10) (2000) 1626–30. [PubMed] [Google Scholar]
- [35].Berridge MJ, Cell signalling biology, Portland Press; 2014. [Google Scholar]
- [36].Murad F, Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development, N Engl J Med 355(19) (2006) 2003–11. [DOI] [PubMed] [Google Scholar]
- [37].Forstermann U, Sessa WC, Nitric oxide synthases: regulation and function, Eur Heart J 33(7) (2012) 829–37, 837a–837d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wu G, Meininger CJ, Nitric oxide and vascular insulin resistance, Biofactors 35(1) (2009) 21–7. [DOI] [PubMed] [Google Scholar]
- [39].Kobayashi J, Nitric oxide and insulin resistance, Immunoendocrinology 2(1) (2015) 10–14800/ie.657. [Google Scholar]
- [40].Santolini J, Meade AL, Stuehr DJ, Differences in three kinetic parameters underpin the unique catalytic profiles of nitric-oxide synthases I, II, and III, The Journal of Biological Chemistry 276(52) (2001) 48887–98. [DOI] [PubMed] [Google Scholar]
- [41].Tanaka T, Nakatani K, Morioka K, Urakawa H, Maruyama N, Kitagawa N, Katsuki A, Araki-Sasaki R, Hori Y, Gabazza EC, Yano Y, Wada H, Nobori T, Sumida Y, Adachi Y, Nitric oxide stimulates glucose transport through insulin-independent GLUT4 translocation in 3T3-L1 adipocytes, Eur J Endocrinol 149(1) (2003) 61–7. [DOI] [PubMed] [Google Scholar]
- [42].McNaughton L, Puttagunta L, Martinez-Cuesta MA, Kneteman N, Mayers I, Moqbel R, Hamid Q, Radomski MW, Distribution of nitric oxide synthase in normal and cirrhotic human liver, Proc Natl Acad Sci U S A 99(26) (2002) 17161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].McConell GK, Bradley SJ, Stephens TJ, Canny BJ, Kingwell BA, Lee-Young RS, Skeletal muscle nNOS mu protein content is increased by exercise training in humans, American journal of physiology. Regulatory, integrative and comparative physiology 293(2) (2007) R821–8. [DOI] [PubMed] [Google Scholar]
- [44].Frandsen U, Lopez-Figueroa M, Hellsten Y, Localization of nitric oxide synthase in human skeletal muscle, Biochemical and biophysical research communications 227(1) (1996) 88–93. [DOI] [PubMed] [Google Scholar]
- [45].Lee J, Ryu H, Ferrante RJ, Morris SM Jr., Ratan RR, Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox, Proc Natl Acad Sci USA 100(8) (2003) 4843–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Luiking YC, Engelen MP, Deutz NE, Regulation of nitric oxide production in health and disease, Curr Opin Clin Nutr Metab Care 13(1) (2010) 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Finocchietto PV, Franco MC, Holod S, Gonzalez AS, Converso DP, Antico Arciuch VG, Serra MP, Poderoso JJ, Carreras MC, Mitochondrial nitric oxide synthase: a masterpiece of metabolic adaptation, cell growth, transformation, and death, Exp Biol Med (Maywood) 234(9) (2009) 1020–8. [DOI] [PubMed] [Google Scholar]
- [48].Schwartz D, Mendonca M, Schwartz I, Xia Y, Satriano J, Wilson CB, Blantz RC, Inhibition of constitutive nitric oxide synthase (NOS) by nitric oxide generated by inducible NOS after lipopolysaccharide administration provokes renal dysfunction in rats, The Journal of clinical investigation 100(2) (1997) 439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jung KH, Chu K, Ko SY, Lee ST, Sinn DI, Park DK, Kim JM, Song EC, Kim M, Roh JK, Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury, Stroke; a journal of cerebral circulation 37(11) (2006) 2744–50. [DOI] [PubMed] [Google Scholar]
- [50].Lundberg JO, Weitzberg E, NO-synthase independent NO generation in mammals, Biochemical and biophysical research communications 396(1) (2010) 39–45. [DOI] [PubMed] [Google Scholar]
- [51].Caldwell RB, Toque HA, Narayanan SP, Caldwell RW, Arginase: an old enzyme with new tricks, Trends Pharmacol Sci 36(6) (2015) 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Marletta MA, Nitric oxide synthase structure and mechanism, The Journal of Biological Chemistry 268(17) (1993) 12231–4. [PubMed] [Google Scholar]
- [53].Abudukadier A, Fujita Y, Obara A, Ohashi A, Fukushima T, Sato Y, Ogura M, Nakamura Y, Fujimoto S, Hosokawa M, Hasegawa H, Inagaki N, Tetrahydrobiopterin has a glucose-lowering effect by suppressing hepatic gluconeogenesis in an endothelial nitric oxide synthase-dependent manner in diabetic mice, Diabetes 62(9) (2013) 3033–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Benjamin N, O’Driscoll F, Dougall H, Duncan C, Smith L, Golden M, McKenzie H, Stomach NO synthesis, Nature 368(6471) (1994) 502. [DOI] [PubMed] [Google Scholar]
- [55].Lundberg JO, Weitzberg E, Lundberg JM, Alving K, Intragastric nitric oxide production in humans: measurements in expelled air, Gut 35(11) (1994) 1543–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lundberg JO, Weitzberg E, Biology of nitrogen oxides in the gastrointestinal tract, Gut 62(4) (2013) 616–29. [DOI] [PubMed] [Google Scholar]
- [57].Erzurum SC, Ghosh S, Janocha AJ, Xu W, Bauer S, Bryan NS, Tejero J, Hemann C, Hille R, Stuehr DJ, Feelisch M, Beall CM, Higher blood flow and circulating NO products offset high-altitude hypoxia among Tibetans, Proc Natl Acad Sci USA 104(45) (2007) 17593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lundberg JO, Carlstrom M, Weitzberg E, Metabolic Effects of Dietary Nitrate in Health and Disease, Cell metabolism 28(1) (2018) 9–22. [DOI] [PubMed] [Google Scholar]
- [59].Bartholomew B, Hill MJ, The pharmacology of dietary nitrate and the origin of urinary nitrate, Food Chem Toxicol 22(10) (1984) 789–95. [DOI] [PubMed] [Google Scholar]
- [60].Weitzberg E, Lundberg JO, Nonenzymatic nitric oxide production in humans, Nitric oxide : biology and chemistry 2(1) (1998) 1–7. [DOI] [PubMed] [Google Scholar]
- [61].Gladwin MT, Haldane, hot dogs, halitosis, and hypoxic vasodilation: the emerging biology of the nitrite anion, The Journal of clinical investigation 113(1) (2004) 19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].van Velzen AG, Sips AJ, Schothorst RC, Lambers AC, Meulenbelt J, The oral bioavailability of nitrate from nitrate-rich vegetables in humans, Toxicol Lett 181(3) (2008) 177–81. [DOI] [PubMed] [Google Scholar]
- [63].Pannala AS, Mani AR, Spencer JP, Skinner V, Bruckdorfer KR, Moore KP, Rice-Evans CA, The effect of dietary nitrate on salivary, plasma, and urinary nitrate metabolism in humans, Free Radic Biol Med 34(5) (2003) 576–84. [DOI] [PubMed] [Google Scholar]
- [64].Lundberg JO, Weitzberg E, Gladwin MT, The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics, Nature reviews. Drug discovery 7(2) (2008) 156–67. [DOI] [PubMed] [Google Scholar]
- [65].Omar SA, Webb AJ, Lundberg JO, Weitzberg E, Therapeutic effects of inorganic nitrate and nitrite in cardiovascular and metabolic diseases, J Intern Med 279(4) (2016) 315–336. [DOI] [PubMed] [Google Scholar]
- [66].Rammos C, Luedike P, Hendgen-Cotta U, Rassaf T, Potential of dietary nitrate in angiogenesis, World J Cardiol 7(10) (2015) 652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kevil CG, Kolluru GK, Pattillo CB, Giordano T, Inorganic nitrite therapy: historical perspective and future directions, Free Radic Biol Med 51(3) (2011) 576–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Dauncey S, Can Dietary Nitrate Supplements Improve Tolerance to Hypoxia?, Journal of the Intensive Care Society 13(3) (2012) 198–204. [Google Scholar]
- [69].Roberts LD, Ashmore T, Kotwica AO, Murfitt SA, Fernandez BO, Feelisch M, Murray AJ, Griffin JL, Inorganic nitrate promotes the browning of white adipose tissue through the nitrate-nitrite-nitric oxide pathway, Diabetes 64(2) (2015) 471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lundberg JO, Weitzberg E, The biological role of nitrate and nitrite: the times they are a-changin’, Nitric oxide : biology and chemistry 22(2) (2010) 61–3. [DOI] [PubMed] [Google Scholar]
- [71].Parthasarathy DK, Bryan NS, Sodium nitrite: the “cure” for nitric oxide insufficiency, Meat Sci 92(3) (2012) 274–9. [DOI] [PubMed] [Google Scholar]
- [72].Cordes CM, Bennett RG, Siford GL, Hamel FG, Nitric oxide inhibits insulin-degrading enzyme activity and function through S-nitrosylation, Biochemical pharmacology 77(6) (2009) 1064–73. [DOI] [PubMed] [Google Scholar]
- [73].Huynh NN, Chin-Dusting J, Amino acids, arginase and nitric oxide in vascular health, Clinical and experimental pharmacology & physiology 33(1–2) (2006) 1–8. [DOI] [PubMed] [Google Scholar]
- [74].Pacher P, Beckman JS, Liaudet L, Nitric oxide and peroxynitrite in health and disease, 34 Physiological reviews 87(1) (2007) 315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Modis K, Panopoulos P, Asimakopoulou A, Gero D, Sharina I, Martin E, Szabo C, Hydrogen sulfide and nitric oxide are mutually dependent in the 37 regulation of angiogenesis and endothelium-dependent vasorelaxation, Proc Natl Acad Sci U S A 109(23) (2012) 9161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cooper CE, Brown GC, The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance, Journal of bioenergetics and biomembranes 40(5) (2008) 533–9. [DOI] [PubMed] [Google Scholar]
- [77].Mitschke MM, Hoffmann LS, Gnad T, Scholz D, Kruithoff K, Mayer P, Haas B, Sassmann A, Pfeifer A, Kilic A, Increased cGMP promotes healthy expansion and browning of white adipose tissue, FASEB Journal 27(4) (2013) 1621–30. [DOI] [PubMed] [Google Scholar]
- [78].Francis SH, Busch JL, Corbin JD, cGMP-Dependent Protein Kinases and cGMP 46 Phosphodiesterases in Nitric Oxide and cGMP Action, Pharmacological Reviews 62(3) (2010) 525–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Gewaltig MT, Kojda G, Vasoprotection by nitric oxide: mechanisms and therapeutic potential, Cardiovascular research 55(2) (2002) 250–60. [DOI] [PubMed] [Google Scholar]
- [80].Bastin G, Heximer SP, Intracellular regulation of heterotrimeric G-protein signaling modulates vascular smooth muscle cell contraction, Archives of biochemistry and biophysics 510(2) (2011) 182–9. [DOI] [PubMed] [Google Scholar]
- [81].Crawford NM, Guo FQ, New insights into nitric oxide metabolism and regulatory functions, Trends Plant Sci 10(4) (2005) 195–200. [DOI] [PubMed] [Google Scholar]
- [82].Wiseman DA, Thurmond DC, The good and bad effects of cysteine S-nitrosylation and tyrosine nitration upon insulin exocytosis: a balancing act, Curr Diabetes Rev 8(4) (2012) 303–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zheng H, Wu J, Jin Z, Yan LJ, Protein Modifications as Manifestations of Hyperglycemic 8 Glucotoxicity in Diabetes and Its Complications, Biochemistry insights 9 (2016) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Mannick JB, Schonhoff CM, Nitrosylation: the next phosphorylation?, Archives of biochemistry and biophysics 408(1) (2002) 1–6. [DOI] [PubMed] [Google Scholar]
- [85].Altaany Z, Ju Y, Yang G, Wang R, The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide, Science signaling 7(342) (2014) ra87. [DOI] [PubMed] [Google Scholar]
- [86].Kolluru GK, Shen X, Bir SC, Kevil CG, Hydrogen sulfide chemical biology: pathophysiological roles and detection, Nitric oxide : biology and chemistry 35 (2013) 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Singh SB, Lin HC, Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract, Microorganisms 3(4) (2015) 866–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Pan LL, Liu XH, Gong QH, Yang HB, Zhu YZ, Role of cystathionine gamma-lyase/hydrogen sulfide pathway in cardiovascular disease: a novel therapeutic strategy?, Antioxidants & redox signaling 17(1) (2012) 106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Moore PK, Whiteman M, Chemistry, Biochemistry and Pharmacology of Hydrogen Sulfide, Springer International Publishing; 2015. [Google Scholar]
- [90].Benavides GA, Squadrito GL, Mills RW, Patel HD, Isbell TS, Patel RP, Darley-Usmar VM, Doeller JE, Kraus DW, Hydrogen sulfide mediates the vasoactivity of garlic, Proc Natl Acad Sci U S A 104(46) (2007) 17977–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Predmore BL, Lefer DJ, Gojon G, Hydrogen sulfide in biochemistry and medicine, Antioxidants & redox signaling 17(1) (2012) 119–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Nagahara N, Ito T, Kitamura H, Nishino T, Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis, Histochemistry and cell biology 110(3) (1998) 243–50. [DOI] [PubMed] [Google Scholar]
- [93].Kimura H, Hydrogen sulfide: its production, release and functions, Amino Acids 41(1) (2011) 113–21. [DOI] [PubMed] [Google Scholar]
- [94].Shibuya N, Kimura H, Production of hydrogen sulfide from d-cysteine and its therapeutic potential, Frontiers in endocrinology 4 (2013) 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE, Snyder SH, Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels, Circulation research 109(11) (2011) 1259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Doeller JE, Isbell TS, Benavides G, Koenitzer J, Patel H, Patel RP, Lancaster JR Jr., Darley Usmar VM, Kraus DW, Polarographic measurement of hydrogen sulfide production and consumption by mammalian tissues, Analytical biochemistry 341(1) (2005) 40–51. [DOI] [PubMed] [Google Scholar]
- [97].Kashfi K, Olson KR, Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide releasing chimeras, Biochemical pharmacology 85(5) (2013) 689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Khan AA, Schuler MM, Prior MG, Yong S, Coppock RW, Florence LZ, Lillie LE, Effects of hydrogen sulfide exposure on lung mitochondrial respiratory chain enzymes in rats, Toxicology and applied pharmacology 103(3) (1990) 482–90. [DOI] [PubMed] [Google Scholar]
- [99].Liu YH, Yan CD, Bian JS, Hydrogen sulfide: a novel signaling molecule in the vascular system, Journal of cardiovascular pharmacology 58(6) (2011) 560–9. [DOI] [PubMed] [Google Scholar]
- [100].Hildebrandt TM, Grieshaber MK, Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria, The FEBS journal 275(13) (2008) 3352–61. [DOI] [PubMed] [Google Scholar]
- [101].Wang R, Signaling pathways for the vascular effects of hydrogen sulfide, Current opinion in nephrology and hypertension 20(2) (2011) 107–12. [DOI] [PubMed] [Google Scholar]
- [102].Peers C, Bauer CC, Boyle JP, Scragg JL, Dallas ML, Modulation of ion channels by hydrogen sulfide, Antioxidants & redox signaling 17(1) (2012) 95–105. [DOI] [PubMed] [Google Scholar]
- [103].Kanagy NL, Szabo C, Papapetropoulos A, Vascular Biology of Hydrogen Sulfide, American journal of physiology. Cell physiology (2017) ajpcell 00329 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Paul BD, Snyder SH, H2S signalling through protein sulfhydration and beyond, Nat Rev Mol Cell Biol 13(8) (2012) 499–507. [DOI] [PubMed] [Google Scholar]
- [105].Henquin JC, Triggering and amplifying pathways of regulation of insulin secretion by glucose, Diabetes 49(11) (2000) 1751–60. [DOI] [PubMed] [Google Scholar]
- [106].Novelli M, Pocai A, Lajoix AD, Beffy P, Bezzi D, Marchetti P, Gross R, Masiello P, Alteration of beta-cell constitutive NO synthase activity is involved in the abnormal insulin response to arginine in a new rat model of type 2 diabetes, Mol Cell Endocrinol 219(1–2) (2004) 77–82. [DOI] [PubMed] [Google Scholar]
- [107].Han J, Pluhackova K, Böckmann RA, The Multifaceted Role of SNARE Proteins in Membrane Fusion, Frontiers in physiology 8 (2017) 5–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Shimabukuro M, Ohneda M, Lee Y, Unger RH, Role of nitric oxide in obesity-induced beta cell disease, The Journal of clinical investigation 100(2) (1997) 290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Lajoix AD, Reggio H, Chardes T, Peraldi-Roux S, Tribillac F, Roye M, Dietz S, Broca C, Manteghetti M, Ribes G, Wollheim CB, Gross R, A neuronal isoform of nitric oxide synthase expressed in pancreatic beta-cells controls insulin secretion, Diabetes 50(6) (2001) 1311–23. [DOI] [PubMed] [Google Scholar]
- [110].Broniowska KA, Oleson BJ, Corbett JA, Beta-Cell responses to nitric oxide, Vitamins and Hormones 95 (2014) 299–322. [DOI] [PubMed] [Google Scholar]
- [111].Nystrom T, Ortsater H, Huang Z, Zhang F, Larsen FJ, Weitzberg E, Lundberg JO, Sjoholm A, Inorganic nitrite stimulates pancreatic islet blood flow and insulin secretion, Free Radic Biol Med 53(5) (2012) 1017–23. [DOI] [PubMed] [Google Scholar]
- [112].Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, Kikuchi M, Yazaki Y, Miyazaki JI, Oka Y, Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose30 stimulated insulin secretion similar to those of normal islets, Diabetologia 36(11) (1993) 1139–45. [DOI] [PubMed] [Google Scholar]
- [113].Campbell SC, Richardson H, Ferris WF, Butler CS, Macfarlane WM, Nitric oxide stimulates insulin gene transcription in pancreatic beta-cells, Biochemical and biophysical research communications 353(4) (2007) 1011–6. [DOI] [PubMed] [Google Scholar]
- [114].Natali A, Ribeiro R, Baldi S, Tulipani A, Rossi M, Venturi E, Mari A, Macedo MP, Ferrannini E, Systemic inhibition of nitric oxide synthesis in non-diabetic individuals produces a significant deterioration in glucose tolerance by increasing insulin clearance and inhibiting insulin secretion, Diabetologia 56(5) (2013) 1183–91. [DOI] [PubMed] [Google Scholar]
- [115].Eckersten D, Henningsson R, Nitric oxide (NO) — Production and regulation of insulin secretion in islets of freely fed and fasted mice, Regulatory Peptides 174(1) (2012) 32–37. [DOI] [PubMed] [Google Scholar]
- [116].Kruszelnicka O, Nitric oxide vs insulin secretion, action and clearance, Diabetologia 57(1) (2014) 257–8. [DOI] [PubMed] [Google Scholar]
- [117].Cordes CM, Bennett RG, Siford GL, Hamel FG, Nitric oxide inhibits insulin-degrading enzyme activity and function through S-nitrosylation, Biochemical pharmacology 77(6) (2009) 1064–73. [DOI] [PubMed] [Google Scholar]
- [118].Krentz A, Insulin resistance: a clinical handbook, John Wiley & Sons; 2002. [Google Scholar]
- [119].Boucher J, Kleinridders A, Kahn CR, Insulin receptor signaling in normal and insulin-resistant states, Cold Spring Harb Perspect Biol 6(1) (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Saltiel AR, Pessin JE, Insulin signaling pathways in time and space, Trends Cell Biol 12(2) (2002) 65–71. [DOI] [PubMed] [Google Scholar]
- [121].Jiang H, Torregrossa AC, Potts A, Pierini D, Aranke M, Garg HK, Bryan NS, Dietary nitrite improves insulin signaling through GLUT4 translocation, Free Radic Biol Med 67 (2014) 51–7. [DOI] [PubMed] [Google Scholar]
- [122].Ishii M, Shimizu S, Nagai T, Shiota K, Kiuchi Y, Yamamoto T, Stimulation of tetrahydrobiopterin synthesis induced by insulin: possible involvement of phosphatidylinositol 3-kinase, Int J Biochem Cell Biol 33(1) (2001) 65–73. [DOI] [PubMed] [Google Scholar]
- [123].Yu Q, Gao F, Ma XL, Insulin says NO to cardiovascular disease, Cardiovascular research 89(3) 7 (2011) 516–24. [DOI] [PubMed] [Google Scholar]
- [124].Williams G, Brown T, Becker L, Prager M, Giroir BP, Cytokine-induced expression of nitric oxide synthase in C2C12 skeletal muscle myocytes, The American journal of physiology 267(4 Pt 2) (1994) R1020–5. [DOI] [PubMed] [Google Scholar]
- [125].Balon TW, Nadler JL, Evidence that nitric oxide increases glucose transport in skeletal muscle, Journal of applied physiology (Bethesda, Md. : 1985) 82(1) (1997) 359–63. [DOI] [PubMed] [Google Scholar]
- [126].Tidball JG, Lavergne E, Lau KS, Spencer MJ, Stull JT, Wehling M, Mechanical loading regulates NOS expression and activity in developing and adult skeletal muscle, The American journal of physiology 275(1) (1998) C260–6. [DOI] [PubMed] [Google Scholar]
- [127].Stamler JS, Meissner G, Physiology of nitric oxide in skeletal muscle, Physiological reviews 81(1) (2001) 209–237. [DOI] [PubMed] [Google Scholar]
- [128].Rubinstein I, Abassi Z, Coleman R, Milman F, Winaver J, Better OS, Involvement of nitric oxide system in experimental muscle crush injury, The Journal of clinical investigation 101(6) (1998) 1325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Capanni C, Squarzoni S, Petrini S, Villanova M, Muscari C, Maraldi NM, Guarnieri C, Caldarera CM, Increase of neuronal nitric oxide synthase in rat skeletal muscle during ageing, Biochemical and biophysical research communications 245(1) (1998) 216–9. [DOI] [PubMed] [Google Scholar]
- [130].Tews DS, Goebel HH, Schneider I, Gunkel A, Stennert E, Neiss WF, Expression of different isoforms of nitric oxide synthase in experimentally denervated and reinnervated skeletal muscle, Journal of neuropathology and experimental neurology 56(12) (1997) 1283–9. [DOI] [PubMed] [Google Scholar]
- [131].El Dwairi Q, Guo Y, Comtois A, Zhu E, Greenwood MT, Bredt DS, Hussain SN, Ontogenesis of nitric oxide synthases in the ventilatory muscles, American journal of respiratory cell and molecular biology 18(6) (1998) 844–52. [DOI] [PubMed] [Google Scholar]
- [132].Kobzik L, Reid MB, Bredt DS, Stamler JS, Nitric oxide in skeletal muscle, Nature 372(6506) (1994) 546–8. [DOI] [PubMed] [Google Scholar]
- [133].Reid MB, Kobzik L, Bredt DS, Stamler JS, Nitric oxide modulates excitation-contraction coupling in the diaphragm, Comparative biochemistry and physiology. Part A, Molecular & integrative physiology 119(1) (1998) 211–8. [DOI] [PubMed] [Google Scholar]
- [134].Balon TW, Nadler JL, Nitric oxide release is present from incubated skeletal muscle 35 preparations, Journal of applied physiology (Bethesda, Md. : 1985) 77(6) (1994) 2519–21. [DOI] [PubMed] [Google Scholar]
- [135].Hirschfield W, Moody MR, O’Brien WE, Gregg AR, Bryan RM Jr., Reid MB, Nitric oxide release and contractile properties of skeletal muscles from mice deficient in type III NOS, American journal of physiology. Regulatory, integrative and comparative physiology 278(1) (2000) R95–r100. [DOI] [PubMed] [Google Scholar]
- [136].Kingwell BA, Formosa M, Muhlmann M, Bradley SJ, McConell GK, Nitric oxide synthase inhibition reduces glucose uptake during exercise in individuals with type 2 diabetes more than in control subjects, Diabetes 51(8) (2002) 2572–80. [DOI] [PubMed] [Google Scholar]
- [137].Young ME, Radda GK, Leighton B, Nitric oxide stimulates glucose transport and metabolism in rat skeletal muscle in vitro, Biochem J 322 ( Pt 1) (1997) 223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Eghbalzadeh K, Brixius K, Bloch W, Brinkmann C, Skeletal muscle nitric oxide (NO) synthases and NO-signaling in “diabesity”--what about the relevance of exercise training interventions?, Nitric oxide : biology and chemistry 37 (2014) 28–40. [DOI] [PubMed] [Google Scholar]
- [139].Young ME, Leighton B, Evidence for altered sensitivity of the nitric oxide/cGMP signalling cascade in insulin-resistant skeletal muscle, Biochem J 329 ( Pt 1) (1998) 73–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Merry TL, Lynch GS, McConell GK, Downstream mechanisms of nitric oxide-mediated skeletal muscle glucose uptake during contraction, American journal of physiology. Regulatory, integrative and comparative physiology 299(6) (2010) R1656–65. [DOI] [PubMed] [Google Scholar]
- [141].Fu WJ, Haynes TE, Kohli R, Hu J, Shi W, Spencer TE, Carroll RJ, Meininger CJ, Wu G, Dietary L-arginine supplementation reduces fat mass in Zucker diabetic fatty rats, J Nutr 135(4) (2005) 714–21. [DOI] [PubMed] [Google Scholar]
- [142].Jobgen WS, Fried SK, Fu WJ, Meininger CJ, Wu G, Regulatory role for the arginine-nitric oxide pathway in metabolism of energy substrates, J Nutr Biochem 17(9) (2006) 571–88. [DOI] [PubMed] [Google Scholar]
- [143].Engeli S, Janke J, Gorzelniak K, Bohnke J, Ghose N, Lindschau C, Luft FC, Sharma AM, Regulation of the nitric oxide system in human adipose tissue, J Lipid Res 45(9) (2004) 1640–8. [DOI] [PubMed] [Google Scholar]
- [144].Roy D, Perreault M, Marette A, Insulin stimulation of glucose uptake in skeletal muscles and 11 adipose tissues in vivo is NO dependent, The American journal of physiology 274(4) (1998) E692–9. [DOI] [PubMed] [Google Scholar]
- [145].McGrowder D, Ragoobirsingh D, Brown P, Modulation of glucose uptake in adipose tissue by nitric oxide-generating compounds, J Biosci 31(3) (2006) 347–54. [DOI] [PubMed] [Google Scholar]
- [146].Bellamy TC, Garthwaite J, Sub-second kinetics of the nitric oxide receptor, soluble guanylyl cyclase, in intact cerebellar cells, J Biol Chem 276(6) (2001) 4287–92. [DOI] [PubMed] [Google Scholar]
- [147].Rodríguez-Juárez F, Aguirre E, Cadenas S, Relative sensitivity of soluble guanylate cyclase and mitochondrial respiration to endogenous nitric oxide at physiological oxygen concentration, The Biochemical journal 405(2) (2007) 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Adkins A, Basu R, Persson M, Dicke B, Shah P, Vella A, Schwenk WF, Rizza R, Higher insulin concentrations are required to suppress gluconeogenesis than glycogenolysis in nondiabetic humans, Diabetes 52(9) (2003) 2213–20. [DOI] [PubMed] [Google Scholar]
- [149].Leclercq IA, Da Silva Morais A, Schroyen B, Van Hul N, Geerts A, Insulin resistance in hepatocytes and sinusoidal liver cells: Mechanisms and consequences, Journal of Hepatology 47(1) (2007) 142–156. [DOI] [PubMed] [Google Scholar]
- [150].Adeva-Andany MM, Pérez-Felpete N, Fernández-Fernández C, Donapetry-García C, Pazos García C, Liver glucose metabolism in humans, Biosci Rep 36(6) (2016) e00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Kim S-K, Joe Y, Zheng M, Kim HJ, Yu J-K, Cho GJ, Chang KC, Kim HK, Han J, Ryter SW, Chung HT, Resveratrol induces hepatic mitochondrial biogenesis through the sequential activation of nitric oxide and carbon monoxide production, Antioxidants & redox signaling 20(16) (2014) 2589–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Sprangers F, Sauerwein HP, Romijn JA, van Woerkom GM, Meijer AJ, Nitric oxide inhibits glycogen synthesis in isolated rat hepatocytes, Biochem J 330 ( Pt 2) (1998) 1045–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Andreelli F, Foretz M, Knauf C, Cani PD, Perrin C, Iglesias MA, Pillot B, Bado A, Tronche F, Mithieux G, Vaulont S, Burcelin R, Viollet B, Liver adenosine monophosphate-activated kinase-alpha2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin 35 but not insulin, Endocrinology 147(5) (2006) 2432–41. [DOI] [PubMed] [Google Scholar]
- [154].Hoshiga M, Hatakeyama K, Watanabe M, Shimada M, Kagamiyama H, Autoradiographic distribution of [14C]tetrahydrobiopterin and its developmental change in mice, J Pharmacol Exp Ther 267(2) (1993) 971–8. [PubMed] [Google Scholar]
- [155].Elrod JW, Duranski MR, Langston W, Greer JJ, Tao L, Dugas TR, Kevil CG, Champion HC, Lefer DJ, eNOS gene therapy exacerbates hepatic ischemia-reperfusion injury in diabetes: a role for eNOS uncoupling, Circulation research 99(1) (2006) 78–85. [DOI] [PubMed] [Google Scholar]
- [156].Matei V, Rodriguez-Vilarrupla A, Deulofeu R, Garcia-Caldero H, Fernandez M, Bosch J, Garcia-Pagan JC, Three-day tetrahydrobiopterin therapy increases in vivo hepatic NOS activity and reduces portal pressure in CCl4 cirrhotic rats, J Hepatol 49(2) (2008) 192–7. [DOI] [PubMed] [Google Scholar]
- [157].Cordero-Herrera I, Kozyra M, Zhuge Z, McCann Haworth S, Moretti C, Peleli M, Caldeira Dias M, Jahandideh A, Huirong H, Cruz JC, Kleschyov AL, Montenegro MF, Ingelman-Sundberg M, Weitzberg E, Lundberg JO, Carlstrom M, AMP-activated protein kinase activation and NADPH oxidase inhibition by inorganic nitrate and nitrite prevent liver steatosis, Proc Natl Acad Sci U S A 116(1) (2019) 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Muniyappa R, Lee S, Chen H, Quon MJ, Current approaches for assessing insulin sensitivity and resistance in vivo: advantages, limitations, and appropriate usage, American Journal of Physiology: Endocrinology and Metabolism 294(1) (2008) E15–26. [DOI] [PubMed] [Google Scholar]
- [159].Joost HG, Tschop MH, NO to obesity: does nitric oxide regulate fat oxidation and insulin sensitivity?, Endocrinology 148(10) (2007) 4545–7. [DOI] [PubMed] [Google Scholar]
- [160].Shankar RR, Wu Y, Shen HQ, Zhu JS, Baron AD, Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance, Diabetes 49(5) (2000) 684–7. [DOI] [PubMed] [Google Scholar]
- [161].Nakata S, Tsutsui M, Shimokawa H, Suda O, Morishita T, Shibata K, Yatera Y, Sabanai K, Tanimoto A, Nagasaki M, Tasaki H, Sasaguri Y, Nakashima Y, Otsuji Y, Yanagihara N, Spontaneous myocardial infarction in mice lacking all nitric oxide synthase isoforms, Circulation 117(17) (2008) 2211–23. [DOI] [PubMed] [Google Scholar]
- [162].Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B, Scherrer U, Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase, Circulation 104(3) (2001) 342–5. [DOI] [PubMed] [Google Scholar]
- [163].Perreault M, Marette A, Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle, Nature medicine 7(10) (2001) 1138–43. [DOI] [PubMed] [Google Scholar]
- [164].Carvalho-Filho MA, Ueno M, Hirabara SM, Seabra AB, Carvalheira JB, de Oliveira MG, Velloso LA, Curi R, Saad MJ, S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance, Diabetes 54(4) (2005) 959–67. [DOI] [PubMed] [Google Scholar]
- [165].Shinozaki S, Choi CS, Shimizu N, Yamada M, Kim M, Zhang T, Shiota G, Dong HH, Kim YB, Kaneki M, Liver-specific inducible nitric-oxide synthase expression is sufficient to cause hepatic insulin resistance and mild hyperglycemia in mice, J Biol Chem 286(40) (2011) 34959–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Fujimoto M, Shimizu N, Kunii K, Martyn JA, Ueki K, Kaneki M, A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice, Diabetes 54(5) (2005) 1340–8. [DOI] [PubMed] [Google Scholar]
- [167].Bakhtiarzadeh F, Siavoshi F, Gheibi S, Kashfi K, Samadi R, Jeddi S, Ghasemi A, Effects of long term oral nitrate administration on adiposity in normal adult female rats, Life Sci 210 (2018) 76–85. [DOI] [PubMed] [Google Scholar]
- [168].Trane AE, Pavlov D, Sharma A, Saqib U, Lau K, van Petegem F, Minshall RD, Roman LJ, Bernatchez PN, Deciphering the binding of caveolin-1 to client protein endothelial nitric-oxide synthase (eNOS): scaffolding subdomain identification, interaction modeling, and biological significance, The Journal of Biological Chemistry 289(19) (2014) 13273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Henningsson R, Salehi A, Lundquist I, Role of nitric oxide synthase isoforms in glucose-stimulated insulin release, Am J Physiol Cell Physiol 283(1) (2002) C296–304. [DOI] [PubMed] [Google Scholar]
- [170].Cosentino F, Hishikawa K, Katusic Zvonimir S, Lüscher Thomas F, High Glucose Increases Nitric Oxide Synthase Expression and Superoxide Anion Generation in Human Aortic Endothelial Cells, Circulation 96(1) (1997) 25–28. [DOI] [PubMed] [Google Scholar]
- [171].Rajapakse NW, Nanayakkara S, Kaye DM, Pathogenesis and treatment of the cardiorenal syndrome: Implications of L-arginine-nitric oxide pathway impairment, Pharmacol Ther 154 (2015) 1–12. [DOI] [PubMed] [Google Scholar]
- [172].Tanrikulu-Kucuk S, Kocak H, Oner-Iyidogan Y, Seyithanoglu M, Topparmak E, Kayan-Tapan T, Serum fetuin-A and arginase-1 in human obesity model: Is there any interaction between inflammatory status and arginine metabolism?, Scand J Clin Lab Invest 75(4) (2015) 301–7. [DOI] [PubMed] [Google Scholar]
- [173].Georgescu A, Popov D, Constantin A, Nemecz M, Alexandru N, Cochior D, Tudor A, Dysfunction of human subcutaneous fat arterioles in obesity alone or obesity associated with Type 2 diabetes, Clin Sci (Lond) 120(10) (2011) 463–72. [DOI] [PubMed] [Google Scholar]
- [174].Kraus RM, Houmard JA, Kraus WE, Tanner CJ, Pierce JR, Choi MD, Hickner RC, Obesity, insulin resistance, and skeletal muscle nitric oxide synthase, Journal of applied physiology (Bethesda, Md. : 1985) 113(5) (2012) 758–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L, Cantoni O, Clementi E, Moncada S, Carruba MO, Nisoli E, TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents, The Journal of clinical investigation 116(10) 7 (2006) 2791–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Catalan V, Gomez-Ambrosi J, Rodriguez A, Silva C, Rotellar F, Gil MJ, Cienfuegos JA, Salvador J, Fruhbeck G, Expression of caveolin-1 in human adipose tissue is upregulated in obesity and obesity-associated type 2 diabetes mellitus and related to inflammation, Clin Endocrinol (Oxf) 68(2) 11 (2008) 213–9. [DOI] [PubMed] [Google Scholar]
- [177].Michel JB, Feron O, Sacks D, Michel T, Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin, The Journal of Biological Chemistry 272(25) (1997) 15583–6. [DOI] [PubMed] [Google Scholar]
- [178].Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM, Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation, Nature 399(6736) (1999) 601–5. [DOI] [PubMed] [Google Scholar]
- [179].Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A, Schwartz MW, Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance, Arterioscler Thromb Vasc Biol 28(11) (2008) 1982–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Alonso J, Sanchez de Miguel L, Monton M, Casado S, Lopez-Farre A, Endothelial cytosolic proteins bind to the 3’ untranslated region of endothelial nitric oxide synthase mRNA: regulation by tumor necrosis factor alpha, Mol Cell Biol 17(10) (1997) 5719–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Anderson HD, Rahmutula D, Gardner DG, Tumor necrosis factor-alpha inhibits endothelial nitric-oxide synthase gene promoter activity in bovine aortic endothelial cells, J Biol Chem 279(2) (2004) 963–9. [DOI] [PubMed] [Google Scholar]
- [182].Mueller CF, Laude K, McNally JS, Harrison DG, ATVB in focus: redox mechanisms in blood vessels, Arteriosclerosis, Thrombosis, and Vascular Biology 25(2) (2005) 274–8. [DOI] [PubMed] [Google Scholar]
- [183].Sugita H, Fujimoto M, Yasukawa T, Shimizu N, Sugita M, Yasuhara S, Martyn JA, Kaneki M, Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells, J Biol Chem 280(14) (2005) 14203–11. [DOI] [PubMed] [Google Scholar]
- [184].Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR, Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity, Diabetes 56(1) (2007) 16–23. [DOI] [PubMed] [Google Scholar]
- [185].Merial C, Bouloumie A, Trocheris V, Lafontan M, Galitzky J, Nitric oxide-dependent 33 downregulation of adipocyte UCP-2 expression by tumor necrosis factor-alpha, American journal of physiology. Cell physiology 279(4) (2000) C1100–6. [DOI] [PubMed] [Google Scholar]
- [186].Crabtree MJ, Smith CL, Lam G, Goligorsky MS, Gross SS, Ratio of 5,6,7,8-tetrahydrobiopterin to 7,8-dihydrobiopterin in endothelial cells determines glucose-elicited changes in NO vs. superoxide production by eNOS, Am J Physiol Heart Circ Physiol 294(4) (2008) H1530–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Sanchez A, Contreras C, Martinez MP, Climent B, Benedito S, Garcia-Sacristan A, Hernandez M, Prieto D, Role of neural NO synthase (nNOS) uncoupling in the dysfunctional nitrergic vasorelaxation of penile arteries from insulin-resistant obese Zucker rats, PloS one 7(4) (2012) e36027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Kashfi K, Rosen C, Aslan M, Obesity, Type-2 Diabetes and Cancer: Mechanistic insights, Critical Reviews in Oncogenesis 24(3) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Fitzgerald SM, Brands MW, Nitric oxide may be required to prevent hypertension at the onset of diabetes, American Journal of Physiology-Endocrinology and Metabolism 279(4) (2000) E762–E768. [DOI] [PubMed] [Google Scholar]
- [190].Jeddi S, Khalifi S, Ghanbari M, Bageripour F, Ghasemi A, Effects of Nitrate Intake on Myocardial Ischemia-Reperfusion Injury in Diabetic Rats, Arq Bras Cardiol 107(4) (2016) 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Ozcelikay AT, Tay A, Guner S, Tasyaran V, Yildizoglu-Ari N, Dincer UD, Altan VM, Reversal effects of L-arginine treatment on blood pressure and vascular responsiveness of streptozotocin-diabetic rats, Pharmacological research 41(2) (2000) 201–9. [DOI] [PubMed] [Google Scholar]
- [192].Knapp M, Tu X, Wu R, Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy, Acta Pharmacologica Sinica 40(1) (2019) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Mandarino LJ, Finlayson J, Hassell JR, High glucose downregulates glucose transport activity in retinal capillary pericytes but not endothelial cells, Invest Ophthalmol Vis Sci 35(3) (1994) 964–72. [PubMed] [Google Scholar]
- [194].Kaiser N, Sasson S, Feener EP, Boukobza-Vardi N, Higashi S, Moller DE, Davidheiser S, Przybylski RJ, King GL, Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells, Diabetes 42(1) (1993) 80–9. [DOI] [PubMed] [Google Scholar]
- [195].Bir SC, Pattillo CB, Pardue S, Kolluru GK, Shen X, Giordano T, Kevil CG, Nitrite anion therapy protects against chronic ischemic tissue injury in db/db diabetic mice in a NO/VEGF-dependent manner, Diabetes 63(1) (2014) 270–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Martinez-Nieves B, Dunbar JC, Vascular dilatatory responses to sodium nitroprusside (SNP) and alpha-adrenergic antagonism in female and male normal and diabetic rats, Proc Soc Exp Biol Med 222(1) (1999) 90–8. [DOI] [PubMed] [Google Scholar]
- [197].Dellamea BS, Leitao CB, Friedman R, Canani LH, Nitric oxide system and diabetic nephropathy, Diabetol Metab Syndr 6(1) (2014) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Lahera V, Salom MG, Miranda-Guardiola F, Moncada S, Romero JC, Effects of NG-nitro-L20 arginine methyl ester on renal function and blood pressure, The American journal of physiology 261(6 Pt 21 2) (1991) F1033–7. [DOI] [PubMed] [Google Scholar]
- [199].Shultz PJ, Tolins JP, Adaptation to increased dietary salt intake in the rat. Role of endogenous nitric oxide, The Journal of clinical investigation 91(2) (1993) 642–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Mount PF, Fraser SA, Watanabe Y, Lane N, Katsis F, Chen ZP, Kemp BE, Power DA, Phosphorylation of neuronal and endothelial nitric oxide synthase in the kidney with high and low salt diets, Nephron Physiol 102(2) (2006) p36–50. [DOI] [PubMed] [Google Scholar]
- [201].Bachmann S, Bosse HM, Mundel P, Topography of nitric oxide synthesis by localizing 28 constitutive NO synthases in mammalian kidney, The American journal of physiology 268(5 Pt 2) (1995) F885–98. [DOI] [PubMed] [Google Scholar]
- [202].Kosaka H, Yoneyama H, Zhang L, Fujii S, Yamamoto A, Igarashi J, Induction of LOX-1 and iNOS expressions by ischemia-reperfusion of rat kidney and the opposing effect of L-arginine, Faseb j 17(6) (2003) 636–43. [DOI] [PubMed] [Google Scholar]
- [203].Zhang C, Walker LM, Mayeux PR, Role of nitric oxide in lipopolysaccharide-induced oxidant stress in the rat kidney, Biochemical pharmacology 59(2) (2000) 203–9. [DOI] [PubMed] [Google Scholar]
- [204].Keynan S, Hirshberg B, Levin-Iaina N, Wexler ID, Dahan R, Reinhartz E, Ovadia H, Wollman Y, Chernihovskey T, Iaina A, Raz I, Renal nitric oxide production during the early phase of experimental diabetes mellitus, Kidney International 58(2) (2000) 740–747. [DOI] [PubMed] [Google Scholar]
- [205].Prabhakar S, Starnes J, Shi S, Lonis B, Tran R, Diabetic Nephropathy Is Associated with Oxidative Stress and Decreased Renal Nitric Oxide Production, Journal of the American Society of Nephrology 18(11) (2007) 2945. [DOI] [PubMed] [Google Scholar]
- [206].Ohtake K, Ishiyama Y, Uchida H, Muraki E, Kobayashi J, Dietary nitrite inhibits early glomerular injury in streptozotocin-induced diabetic nephropathy in rats, Nitric oxide : biology and chemistry 17(2) (2007) 75–81. [DOI] [PubMed] [Google Scholar]
- [207].Arya A, Yadav HN, Sharma PL, Involvement of vascular endothelial nitric oxide synthase in development of experimental diabetic nephropathy in rats, Mol Cell Biochem 354(1–2) (2011) 57–66. [DOI] [PubMed] [Google Scholar]
- [208].Goor Y, Peer G, Iaina A, Blum M, Wollman Y, Chernihovsky T, Silverberg D, Cabili S, Nitric oxide in ischaemic acute renal failure of streptozotocin diabetic rats, Diabetologia 39(9) (1996) 1036–40. [DOI] [PubMed] [Google Scholar]
- [209].Ortiz MC, Albertoni Borghese MF, Balonga SE, Lavagna A, Filipuzzi AL, Elesgaray R, Costa MA, Majowicz MP, Renal response to L-arginine in diabetic rats. A possible link between nitric oxide system and aquaporin-2, PloS one 9(8) (2014) e104923–e104923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Hayden LJ, Goeden H, Roth SH, Exposure to low levels of hydrogen sulfide elevates circulating glucose in maternal rats, Journal of toxicology and environmental health 31(1) (1990) 45–52. [DOI] [PubMed] [Google Scholar]
- [211].Yang W, Yang G, Jia X, Wu L, Wang R, Activation of KATP channels by H2S in rat insulin-secreting cells and the underlying mechanisms, The Journal of physiology 569(Pt 2) (2005) 519–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Yusuf M, Kwong Huat BT, Hsu A, Whiteman M, Bhatia M, Moore PK, Streptozotocin-induced diabetes in the rat is associated with enhanced tissue hydrogen sulfide biosynthesis, Biochemical and biophysical research communications 333(4) (2005) 1146–52. [DOI] [PubMed] [Google Scholar]
- [213].Wu L, Yang W, Jia X, Yang G, Duridanova D, Cao K, Wang R, Pancreatic islet overproduction of H2S and suppressed insulin release in Zucker diabetic rats, Laboratory investigation; a journal of technical methods and pathology 89(1) (2009) 59–67. [DOI] [PubMed] [Google Scholar]
- [214].Kaneko Y, Kimura Y, Kimura H, Niki I, L-cysteine inhibits insulin release from the pancreatic beta cell: possible involvement of metabolic production of hydrogen sulfide, a novel gasotransmitter, Diabetes 55(5) (2006) 1391–7. [DOI] [PubMed] [Google Scholar]
- [215].Tomita M, Nagahara N, Ito T, Expression of 3-Mercaptopyruvate Sulfurtransferase in the Mouse, Molecules (Basel, Switzerland) 21(12) (2016) 1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Kaneko Y, Kimura T, Taniguchi S, Souma M, Kojima Y, Kimura Y, Kimura H, Niki I, Glucose induced production of hydrogen sulfide may protect the pancreatic beta-cells from apoptotic cell death by high glucose, FEBS Lett 583(2) (2009) 377–82. [DOI] [PubMed] [Google Scholar]
- [217].Taniguchi S, Kang L, Kimura T, Niki I, Hydrogen sulphide protects mouse pancreatic β-cells from cell death induced by oxidative stress, but not by endoplasmic reticulum stress, British journal of pharmacology 162(5) (2011) 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Okamoto M, Yamaoka M, Takei M, Ando T, Taniguchi S, Ishii I, Tohya K, Ishizaki T, Niki I, Kimura T, Endogenous hydrogen sulfide protects pancreatic beta-cells from a high-fat diet-induced glucotoxicity and prevents the development of type 2 diabetes, Biochemical and biophysical research communications 442(3–4) (2013) 227–33. [DOI] [PubMed] [Google Scholar]
- [219].Yang G, Yang W, Wu L, Wang R, H2S, Endoplasmic Reticulum Stress, and Apoptosis of Insulin secreting Beta Cells, Journal of Biological Chemistry 282(22) (2007) 16567–16576. [DOI] [PubMed] [Google Scholar]
- [220].Oyadomari S, Araki E, Mori M, Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells, Apoptosis : an international journal on programmed cell death 7(4) (2002) 335–45. [DOI] [PubMed] [Google Scholar]
- [221].Untereiner A, Wu L, Hydrogen sulfide and glucose homeostasis - a tale of sweet & the stink, Antioxidants & redox signaling (2017). [DOI] [PubMed] [Google Scholar]
- [222].Carter RN, Morton NM, Cysteine and hydrogen sulphide in the regulation of metabolism: insights from genetics and pharmacology, The Journal of pathology 238(2) (2016) 321–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Tang G, Zhang L, Yang G, Wu L, Wang R, Hydrogen sulfide-induced inhibition of L-type Ca2+ channels and insulin secretion in mouse pancreatic beta cells, Diabetologia 56(3) (2013) 533–41. [DOI] [PubMed] [Google Scholar]
- [224].Patel MA, Shah GB, Possible Role of Hydrogen Sulfide in Insulin Secretion and in Development of Insulin Resistance, Journal of Young Pharmacists : JYP 2(2) (2010) 148–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Ali MY, Whiteman M, Low CM, Moore PK, Hydrogen sulphide reduces insulin secretion from HIT-T15 cells by a KATP channel-dependent pathway, J Endocrinol 195(1) (2007) 105–12. [DOI] [PubMed] [Google Scholar]
- [226].Takahashi N, Wei FY, Watanabe S, Hirayama M, Ohuchi Y, Fujimura A, Kaitsuka T, Ishii I, Sawa T, Nakayama H, Akaike T, Tomizawa K, Reactive sulfur species regulate tRNA methylthiolation and contribute to insulin secretion, Nucleic Acids Res 45(1) (2017) 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Chen NC, Yang F, Capecci LM, Gu Z, Schafer AI, Durante W, Yang X-F, Wang H, Regulation of homocysteine metabolism and methylation in human and mouse tissues, The FASEB Journal 24(8) (2010) 2804–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Du JT, Li W, Yang JY, Tang CS, Li Q, Jin HF, Hydrogen sulfide is endogenously generated in rat skeletal muscle and exerts a protective effect against oxidative stress, Chinese medical journal 126(5) (2013) 930–6. [PubMed] [Google Scholar]
- [229].Xue R, Hao DD, Sun JP, Li WW, Zhao MM, Li XH, Chen Y, Zhu JH, Ding YJ, Liu J, Zhu YC, Hydrogen sulfide treatment promotes glucose uptake by increasing insulin receptor sensitivity and ameliorates kidney lesions in type 2 diabetes, Antioxidants & redox signaling 19(1) (2013) 5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Bhambhani Y, Burnham R, Snydmiller G, MacLean I, Martin T, Effects of 5 ppm hydrogen sulfide 8 inhalation on biochemical properties of skeletal muscle in exercising men and women, Am Ind Hyg Assoc J 57(5) (1996) 464–8. [DOI] [PubMed] [Google Scholar]
- [231].Matei IV, Ii H, Yaegaki K, Hydrogen sulfide enhances pancreatic beta-cell differentiation from human tooth under normal and glucotoxic conditions, Regen Med 12(2) (2017) 125–141. [DOI] [PubMed] [Google Scholar]
- [232].Abiola M, Favier M, Christodoulou-Vafeiadou E, Pichard AL, Martelly I, Guillet-Deniau I, Activation of Wnt/beta-catenin signaling increases insulin sensitivity through a reciprocal regulation of Wnt10b and SREBP-1c in skeletal muscle cells, PloS one 4(12) (2009) e8509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Feng X, Chen Y, Zhao J, Tang C, Jiang Z, Geng B, Hydrogen sulfide from adipose tissue is a novel insulin resistance regulator, Biochemical and biophysical research communications 380(1) (2009) 153–9. [DOI] [PubMed] [Google Scholar]
- [234].Velmurugan GV, Huang H, Sun H, Candela J, Jaiswal MK, Beaman KD, Yamashita M, Prakriya M, White C, Depletion of H2S during obesity enhances store-operated Ca2+ entry in adipose tissue macrophages to increase cytokine production, Science signaling 8(407) (2015) ra128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Beltowski J, Jamroz-Wisniewska A, Hydrogen Sulfide in the Adipose Tissue-Physiology, Pathology and a Target for Pharmacotherapy, Molecules (Basel, Switzerland) 22(1) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Tsai CY, Peh MT, Feng W, Dymock BW, Moore PK, Hydrogen sulfide promotes adipogenesis in 3T3L1 cells, PloS one 10(3) (2015) e0119511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Manna P, Jain SK, Hydrogen sulfide and L-cysteine increase phosphatidylinositol 3,4,525 trisphosphate (PIP3) and glucose utilization by inhibiting phosphatase and tensin homolog (PTEN) 26 protein and activating phosphoinositide 3-kinase (PI3K)/serine/threonine protein kinase (AKT)/protein 27 kinase Czeta/lambda (PKCzeta/lambda) in 3T3l1 adipocytes, J Biol Chem 286(46) (2011) 39848–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Cai J, Shi X, Wang H, Fan J, Feng Y, Lin X, Yang J, Cui Q, Tang C, Xu G, Cystathionine γ lyase–hydrogen sulfide increases peroxisome proliferator-activated receptor γ activity by sulfhydration at C139 site thereby promoting glucose uptake and lipid storage in adipocytes, Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1861(5) (2016) 419–429. [DOI] [PubMed] [Google Scholar]
- [239].Geng B, Cai B, Liao F, Zheng Y, Zeng Q, Fan X, Gong Y, Yang J, Cui QH, Tang C, Xu GH, Increase or decrease hydrogen sulfide exert opposite lipolysis, but reduce global insulin resistance in high fatty diet induced obese mice, PloS one 8(9) (2013) e73892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Huang CY, Yao WF, Wu W.g., Lu YL, Wan H, Wang W, Endogenous CSE/H2S system mediates TNF-α-induced insulin resistance in 3T3-L1 adipocytes, Cell biochemistry and function 31(6) (2013) 468–475. [DOI] [PubMed] [Google Scholar]
- [241].Kabil O, Vitvitsky V, Xie P, Banerjee R, The quantitative significance of the transsulfuration 39 enzymes for H2S production in murine tissues, Antioxidants & redox signaling 15(2) (2011) 363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Modis K, Coletta C, Erdelyi K, Papapetropoulos A, Szabo C, Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics, Faseb j 27(2) (2013) 601–11. [DOI] [PubMed] [Google Scholar]
- [243].Mani S, Cao W, Wu L, Wang R, Hydrogen sulfide and the liver, Nitric oxide : biology and chemistry 41 (2014) 62–71. [DOI] [PubMed] [Google Scholar]
- [244].Untereiner AA, Wang R, Ju Y, Wu L, Decreased Gluconeogenesis in the Absence of Cystathionine Gamma-Lyase and the Underlying Mechanisms, Antioxidants & redox signaling 24(3) (2016) 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Wijekoon EP, Hall B, Ratnam S, Brosnan ME, Zeisel SH, Brosnan JT, Homocysteine Metabolism in ZDF (Type 2) Diabetic Rats, Diabetes 54(11) (2005) 3245–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Zhang L, Yang G, Untereiner A, Ju Y, Wu L, Wang R, Hydrogen sulfide impairs glucose utilization and increases gluconeogenesis in hepatocytes, Endocrinology 154(1) (2013) 114–26. [DOI] [PubMed] [Google Scholar]
- [247].Yang G, Protein S-sulfhydration as a major sources of H2S bioactivity, Receptors & Clinical Investigation 1(4) (2014). [Google Scholar]
- [248].Ju Y, Untereiner A, Wu L, Yang G, H 2 S-induced S-sulfhydration of pyruvate carboxylase contributes to gluconeogenesis in liver cells, Biochimica et Biophysica Acta (BBA)-General Subjects 1850(11) (2015) 2293–2303. [DOI] [PubMed] [Google Scholar]
- [249].Untereiner AA, Fu M, Modis K, Wang R, Ju Y, Wu L, Stimulatory effect of CSE-generated H2S on hepatic mitochondrial biogenesis and the underlying mechanisms, Nitric oxide : biology and chemistry 58 (2016) 67–76. [DOI] [PubMed] [Google Scholar]
- [250].Jacobs RL, House JD, Brosnan ME, Brosnan JT, Effects of streptozotocin-induced diabetes and of insulin treatment on homocysteine metabolism in the rat, Diabetes 47(12) (1998) 1967–70. [DOI] [PubMed] [Google Scholar]
- [251].Gheibi S, Jeddi S, Kashfi K, Ghasemi A, Effects of Hydrogen Sulfide on Carbohydrate Metabolism in Obese Type 2 Diabetic Rats, Molecules (Basel, Switzerland) 24(1) (2019) 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Desai KM, Chang T, Untereiner A, Wu L, Hydrogen sulfide and the metabolic syndrome, Expert review of clinical pharmacology 4(1) (2011) 63–73. [DOI] [PubMed] [Google Scholar]
- [253].Brancaleone V, Roviezzo F, Vellecco V, De Gruttola L, Bucci M, Cirino G, Biosynthesis of H2S is impaired in non-obese diabetic (NOD) mice, British journal of pharmacology 155(5) (2008) 673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Wilinski B, Wilinski J, Somogyi E, Piotrowska J, Opoka W, Metformin raises hydrogen sulfide tissue concentrations in various mouse organs, Pharmacological reports : PR 65(3) (2013) 737–42. [DOI] [PubMed] [Google Scholar]
- [255].Suzuki K, Olah G, Modis K, Coletta C, Kulp G, Gero D, Szoleczky P, Chang T, Zhou Z, Wu L, Wang R, Papapetropoulos A, Szabo C, Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function, Proc Natl Acad Sci U S A 108(33) (2011) 13829–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Zhang L, Yang G, Tang G, Wu L, Wang R, Rat pancreatic level of cystathionine gamma-lyase is regulated by glucose level via specificity protein 1 (SP1) phosphorylation, Diabetologia 54(10) (2011) 2615–25. [DOI] [PubMed] [Google Scholar]
- [257].Manna P, Gungor N, McVie R, Jain SK, Decreased cystathionine-gamma-lyase (CSE) activity in livers of type 1 diabetic rats and peripheral blood mononuclear cells (PBMC) of type 1 diabetic patients, J Biol Chem 289(17) (2014) 11767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Whiteman M, Gooding KM, Whatmore JL, Ball CI, Mawson D, Skinner K, Tooke JE, Shore AC, Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide, Diabetologia 53(8) (2010) 1722–6. [DOI] [PubMed] [Google Scholar]
- [259].Wallace JL, Wang R, Hydrogen sulfide-based therapeutics: exploiting a unique but ubiquitous gasotransmitter, Nature reviews. Drug discovery 14(5) (2015) 329–45. [DOI] [PubMed] [Google Scholar]
- [260].Zhou X, Feng Y, Zhan Z, Chen J, Hydrogen Sulfide Alleviates Diabetic Nephropathy in a 39 Streptozotocin-induced Diabetic Rat Model, Journal of Biological Chemistry 289(42) (2014) 28827–28834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Si YF, Wang J, Guan J, Zhou L, Sheng Y, Zhao J, Treatment with hydrogen sulfide alleviates streptozotocin-induced diabetic retinopathy in rats, British journal of pharmacology 169(3) (2013) 619–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Zhou X, An G, Lu X, Hydrogen sulfide attenuates the development of diabetic cardiomyopathy, Clinical Science 128(5) (2015) 325–335. [DOI] [PubMed] [Google Scholar]
- [263].Barr LA, Shimizu Y, Lambert JP, Nicholson CK, Calvert JW, Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress, Nitric oxide : biology and chemistry 46 (2015) 145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Ouchi N, Shibata R, Walsh K, Cardioprotection by adiponectin, Trends Cardiovasc Med 16(5) (2006) 141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T, Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP activated protein kinase, Nature medicine 8(11) (2002) 1288–95. [DOI] [PubMed] [Google Scholar]
- [266].Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, Hardie DG, Macaulay SL, Schertzer JD, Dyck JR, van Denderen BJ, Kemp BE, Steinberg GR, Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin, Nature medicine 19(12) (2013) 1649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Ye P, Gu Y, Zhu YR, Chao YL, Kong XQ, Luo J, Ren XM, Zuo GF, Zhang DM, Chen SL, Exogenous hydrogen sulfide attenuates the development of diabetic cardiomyopathy via the FoxO1 pathway, J Cell Physiol 233(12) (2018) 9786–9798. [DOI] [PubMed] [Google Scholar]
- [268].Kandula V, Kosuru R, Li H, Yan D, Zhu Q, Lian Q, Ge RS, Xia Z, Irwin MG, Forkhead box transcription factor 1: role in the pathogenesis of diabetic cardiomyopathy, Cardiovasc Diabetol 15 (2016) 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].El-Seweidy MM, Sadik NA, Shaker OG, Role of sulfurous mineral water and sodium hydrosulfide as potent inhibitors of fibrosis in the heart of diabetic rats, Archives of biochemistry and biophysics 506(1) (2011) 48–57. [DOI] [PubMed] [Google Scholar]
- [270].Xie L, Gu Y, Wen M, Zhao S, Wang W, Ma Y, Meng G, Han Y, Wang Y, Liu G, Moore PK, Wang X, Wang H, Zhang Z, Yu Y, Ferro A, Huang Z, Ji Y, Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation, Diabetes 65(10) (2016) 3171–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Durante W, Hydrogen Sulfide Therapy in Diabetes-Accelerated Atherosclerosis: A Whiff of Success, Diabetes 65(10) (2016) 2832–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Yamamoto J, Sato W, Kosugi T, Yamamoto T, Kimura T, Taniguchi S, Kojima H, Maruyama S, Imai E, Matsuo S, Yuzawa Y, Niki I, Distribution of hydrogen sulfide (H₂S)-producing enzymes and the roles of the H₂S donor sodium hydrosulfide in diabetic nephropathy, Clin Exp Nephrol 17(1) (2013) 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Li L, Xiao T, Li F, Li Y, Zeng O, Liu M, Liang B, Li Z, Chu C, Yang J, Hydrogen sulfide reduced renal tissue fibrosis by regulating autophagy in diabetic rats, Molecular medicine reports 16(2) (2017) 1715–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Yuan P, Xue H, Zhou L, Qu L, Li C, Wang Z, Ni J, Yu C, Yao T, Huang Y, Wang R, Lu L, Rescue of mesangial cells from high glucose-induced over-proliferation and extracellular matrix secretion by hydrogen sulfide, Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 26(7) (2011) 2119–26. [DOI] [PubMed] [Google Scholar]
- [275].Yang R, Jia Q, Liu XF, Wang YY, Ma SF, Effects of hydrogen sulfide on inducible nitric oxide synthase activity and expression of cardiomyocytes in diabetic rats, Molecular medicine reports 16(4) (2017) 5277–5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Cui J, Zhuang S, Qi S, Li L, Zhou J, Zhang W, Zhao Y, Qi N, Yin Y, Huang L, Hydrogen sulfide facilities production of nitric oxide via the Akt/endothelial nitric oxide synthases signaling pathway to protect human umbilical vein endothelial cells from injury by angiotensin II, Molecular medicine reports (2017) 6255–6261. [DOI] [PubMed] [Google Scholar]
- [277].Predmore BL, Julian D, Cardounel AJ, Hydrogen sulfide increases nitric oxide production from endothelial cells by an akt-dependent mechanism, Frontiers in physiology 2 (2011) 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Bauer CC, Boyle JP, Porter KE, Peers C, Modulation of Ca(2+) signalling in human vascular endothelial cells by hydrogen sulfide, Atherosclerosis 209(2) (2010) 374–80. [DOI] [PubMed] [Google Scholar]
- [279].Potenza DM, Guerra G, Avanzato D, Poletto V, Pareek S, Guido D, Gallanti A, Rosti V, Munaron L, Tanzi F, Moccia F, Hydrogen sulphide triggers VEGF-induced intracellular Ca(2)(+) signals in human endothelial cells but not in their immature progenitors, Cell calcium 56(3) (2014) 225–34. [DOI] [PubMed] [Google Scholar]
- [280].Kida M, Sugiyama T, Yoshimoto T, Ogawa Y, Hydrogen sulfide increases nitric oxide production with calcium-dependent activation of endothelial nitric oxide synthase in endothelial cells, European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences 48(1–2) (2013) 211–5. [DOI] [PubMed] [Google Scholar]
- [281].Lei YP, Liu CT, Sheen LY, Chen HW, Lii CK, Diallyl disulfide and diallyl trisulfide protect endothelial nitric oxide synthase against damage by oxidized low-density lipoprotein, Mol Nutr Food Res 54 Suppl 1 (2010) S42–52. [DOI] [PubMed] [Google Scholar]
- [282].Kubo S, Doe I, Kurokawa Y, Nishikawa H, Kawabata A, Direct inhibition of endothelial nitric oxide synthase by hydrogen sulfide: contribution to dual modulation of vascular tension, Toxicology 232(1–2) (2007) 138–46. [DOI] [PubMed] [Google Scholar]
- [283].Chai Q, Lu T, Wang XL, Lee HC, Hydrogen sulfide impairs shear stress-induced vasodilation in mouse coronary arteries, Pflugers Arch 467(2) (2015) 329–40. [DOI] [PubMed] [Google Scholar]
- [284].Zhao W, Zhang J, Lu Y, Wang R, The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener, The EMBO Journal 20(21) (2001) 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Zuidema MY, Yang Y, Wang M, Kalogeris T, Liu Y, Meininger CJ, Hill MA, Davis MJ, Korthuis RJ, Antecedent hydrogen sulfide elicits an anti-inflammatory phenotype in postischemic murine small intestine: role of BK channels, Am J Physiol Heart Circ Physiol 299(5) (2010) H1554–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Chen P-H, Fu Y-S, Wang Y-M, Yang K-H, Wang DL, Huang B, Hydrogen Sulfide Increases Nitric Oxide Production and Subsequent S-Nitrosylation in Endothelial Cells, The Scientific World Journal 2014 (2014) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Zhao W, Ndisang JF, Wang R, Modulation of endogenous production of H2S in rat tissues, Can J Physiol Pharmacol 81(9) (2003) 848–53. [DOI] [PubMed] [Google Scholar]
- [288].Wang R, Physiological implications of hydrogen sulfide: a whiff exploration that blossomed, Physiological reviews 92(2) (2012) 791–896. [DOI] [PubMed] [Google Scholar]
- [289].Thuillez C, Richard V, Targeting endothelial dysfunction in hypertensive subjects, Journal of human hypertension 19 Suppl 1 (2005) S21–5. [DOI] [PubMed] [Google Scholar]
- [290].Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M, Moore PK, Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide, Biochemical and biophysical research communications 343(1) (2006) 303–10. [DOI] [PubMed] [Google Scholar]
- [291].Lo Faro ML, Fox B, Whatmore JL, Winyard PG, Whiteman M, Hydrogen sulfide and nitric oxide interactions in inflammation, Nitric oxide : biology and chemistry 41(Supplement C) (2014) 38–47. [DOI] [PubMed] [Google Scholar]
- [292].Kolluru GK, Shen X, Kevil CG, A tale of two gases: NO and H2S, foes or friends for life?, Redox Biology 1(1) (2013) 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].King SB, Potential Biological Chemistry of Hydrogen Sulfide (H(2)S) with the Nitrogen Oxides, Free Radic Biol Med 55 (2013) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Nagpure BV, Bian JS, Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System, Oxidative medicine and cellular longevity 2016 (2016) 6904327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Cortese-Krott MM, Kuhnle GGC, Dyson A, Fernandez BO, Grman M, DuMond JF, Barrow MP, McLeod G, Nakagawa H, Ondrias K, Nagy P, King SB, Saavedra JE, Keefer LK, Singer M, Kelm M, Butler AR, Feelisch M, Key bioactive reaction products of the NO/H(2)S interaction are S/Nhybrid species, polysulfides, and nitroxyl, Proc Natl Acad Sci USA 112(34) (2015) E4651–E4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Miyamoto R, Koike S, Takano Y, Shibuya N, Kimura Y, Hanaoka K, Urano Y, Ogasawara Y, Kimura H, Polysulfides (H2Sn) produced from the interaction of hydrogen sulfide (H2S) and nitric oxide (NO) activate TRPA1 channels, Sci Rep 7 (2017) 45995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Filipovic MR, Persulfidation (S-sulfhydration) and H2S, Handb Exp Pharmacol 230 (2015) 29–59. [DOI] [PubMed] [Google Scholar]
- [298].Berenyiova A, Grman M, Mijuskovic A, Stasko A, Misak A, Nagy P, Ondriasova E, Cacanyiova S, Brezova V, Feelisch M, Ondrias K, The reaction products of sulfide and S-nitrosoglutathione are potent vasorelaxants, Nitric oxide : biology and chemistry 46(Supplement C) (2015) 123–130. [DOI] [PubMed] [Google Scholar]
- [299].Filipovic MR, Miljkovic J, Nauser T, Royzen M, Klos K, Shubina T, Koppenol WH, Lippard SJ, Ivanovic-Burmazovic I, Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols, J Am Chem Soc 134(29) (2012) 12016–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Bir SC, Kolluru GK, McCarthy P, Shen X, Pardue S, Pattillo CB, Kevil CG, Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1alpha and vascular endothelial growth factor-dependent angiogenesis, Journal of the American Heart Association 1(5) (2012) e004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Filipovic MR, Miljkovic J, Allgauer A, Chaurio R, Shubina T, Herrmann M, I. Ivanovic12 Burmazovic, Biochemical insight into physiological effects of H(2)S: reaction with peroxynitrite and 13 formation of a new nitric oxide donor, sulfinyl nitrite, Biochem J 441(2) (2012) 609–21. [DOI] [PubMed] [Google Scholar]
- [302].Anter A, Taye A, El-Moselhy M, NOS activity mediates some pathways in the protective effects of H2S in a model of diabetic nephrotpathy, Journal of advanced Biomedical and Pharmaceutical Sciences 16 1(1) (2018) 26–32. [Google Scholar]
- [303].Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HH, Jandeleit-Dahm KA, Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy, J Am Soc Nephrol 25(6) (2014) 1237–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Eid AA, Lee DY, Roman LJ, Khazim K, Gorin Y, Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression, Mol Cell Biol 33(17) (2013) 3439–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Lee HJ, Lee DY, Mariappan MM, Feliers D, Ghosh-Choudhury G, Abboud HE, Gorin Y, Kasinath BS, Hydrogen sulfide inhibits high glucose-induced NADPH oxidase 4 expression and matrix increase by recruiting inducible nitric oxide synthase in kidney proximal tubular epithelial cells, J Biol Chem 292(14) (2017) 5665–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]