

Abstract
The urea functionality is inherent to numerous bioactive compounds, including a variety of clinically approved therapies. Urea containing compounds are increasingly used in medicinal chemistry and drug design in order to establish key drug-target interactions and fine-tune crucial drug-like properties. In this perspective, we highlight physicochemical and conformational properties of urea derivatives. We provide outlines of traditional reagents and chemical procedures for the preparation of ureas. Also, we discuss newly developed methodologies mainly aimed at overcoming safety issues associated with traditional synthesis. Finally, we provide a broad overview of urea-based medicinally relevant compounds, ranging from approved drugs to recent medicinal chemistry developments.
Graphical Abstract

1. INTRODUCTION
The synthesis of urea by the German chemist Fredrich Wöhler in 1828 marked the beginning of organic chemistry.1,2 Since then, rapid evolution of organic chemistry and further developments in synthesis enabled medicinal chemistry and drug discovery in the latter half of the 20th century.3,4 Urea and its derivatives (1 and 2, Figure 1) have a central role in drug development and medicinal chemistry due to the capability of the urea functionality to form multiple stable hydrogen bonds with protein and receptor targets. Such drug-target interactions are responsible for specific biological activity, drug actions, and drug properties. It is not surprising that a large number of urea derivatives are utilized in a broad range of medicinal applications. In particular, a urea functionality is incorporated to modulate drug potency and selectivity and improve drug properties in the development of anticancer, antibacterial, anticonvulsive, anti-HIV, antidiabetic agents, and other medicinal compounds.5,6 With the advances of protein structures and identification of new disease targets, there is a growing interest in urea-based derivatives for drug design and development.7,8
Figure 1.
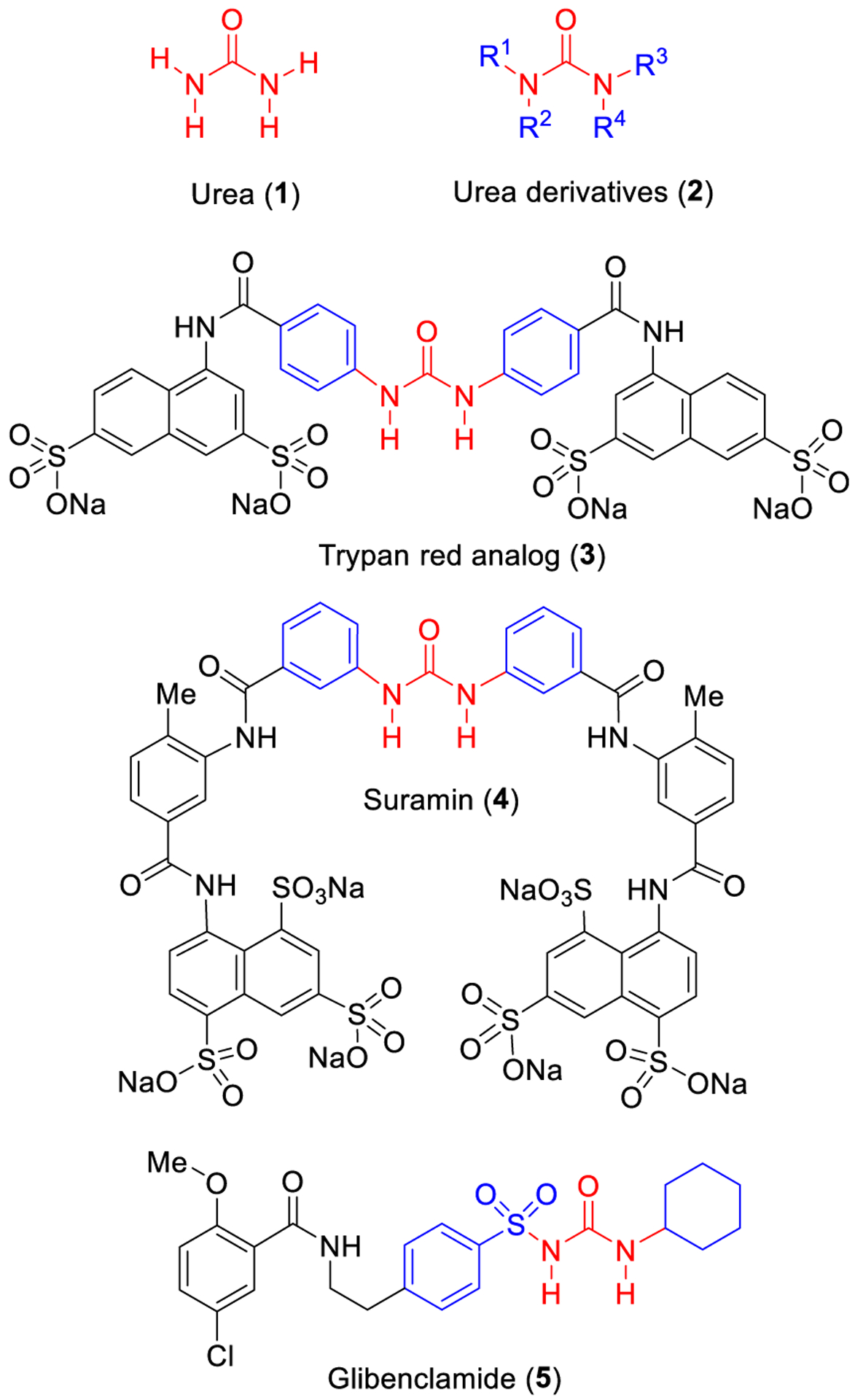
Structures of urea and selected urea derivatives.
One of the early examples of urea derivatives as a medicinal agent is the development of compound 3 by Bayer’s laboratories in Germany. Urea derivative 3 is a colorless derivative of trypan red and has shown potent antitrypanosomal activity. Further optimization of urea compound 3 led to the discovery of suramin (4) with potent antitrypanosomal properties. Suramin is used as an effective therapy during the early stage of sleeping sickness in humans that is caused by the protozoan parasites T. Gambiense and T. Rhodesiense.9,10 Urea-derived Glibenclamide (5), also known as glyburide, is a potent antidiabetic drug that prolongs the hypoglycemic effect. It was used to treat patients with type II diabetes.11 Today, there are many urea containing compounds that are FDA approved drugs for a variety of human diseases.12–15 We will outline these approved therapies in the next section.
Beyond their presence in approved drugs, oligoureas play a pivotal role as starting motifs for the generation of artificial β-sheets16,17 and peptidomimetics.18,19 Urea derivatives are also widely employed as linkers for the development of antibody-drug conjugates as well as in combinatorial chemistry building blocks.20,21 Due to the importance of urea derivatives in the syntheses of medicinal agents and other applications in material science and organocatalytic reactions, a variety of methods have been developed for their syntheses.22,23 Traditional syntheses of urea-containing compounds involve the reaction of amines with phosgene, carbon monoxide, or isocyanates. However, these synthetic procedures have relevant safety and environmental issues due to the use of toxic agents. Alternative routes involving reactions of amines with less toxic agents such as ethylene carbonate or diethyl carbonate have been developed. Also, many other direct routes to urea derivatives from amines and CO2 in the presence of numerous catalysts were investigated. A number of ionic liquids were also examined as suitable solvents for the syntheses of urea derivatives.24
In recent years, urea containing compounds have received much attention due to their growing application in drug design and medicinal chemistry.25,26 In the present perspective, we plan to outline physicochemical properties of urea derivatives, highlighting the key role of the urea functionality in the drug-target interaction. We outline the chemical methodologies for the synthesis of urea derivatives. We cover the most recent methodologies that highlight the progress in terms of both process safety and efficiency. Finally, we provide a broad overview of the relevance of the urea functionality and urea derivatives in modern drug discovery and medicinal chemistry.
2. CONFORMATION OF UREA DERIVATIVES
The urea functionality shows a certain degree of conformational restriction due to the presence and delocalization of nonbonded electrons on nitrogens into the adjacent carbonyl group. Accordingly, three resonance structures can be drawn for ureas (namely A, B, and C; Figure 2).27 Data on the X-ray structure of N,N′-diphenyl-N,N′-diethylurea revealed that the two urea nitrogens adopt a geometry between trigonal and tetrahedral. The amide groups displayed a nonplanar distortion of approximately 30° with amide C-N bond lengths of 1.37 Å.27
Figure 2.
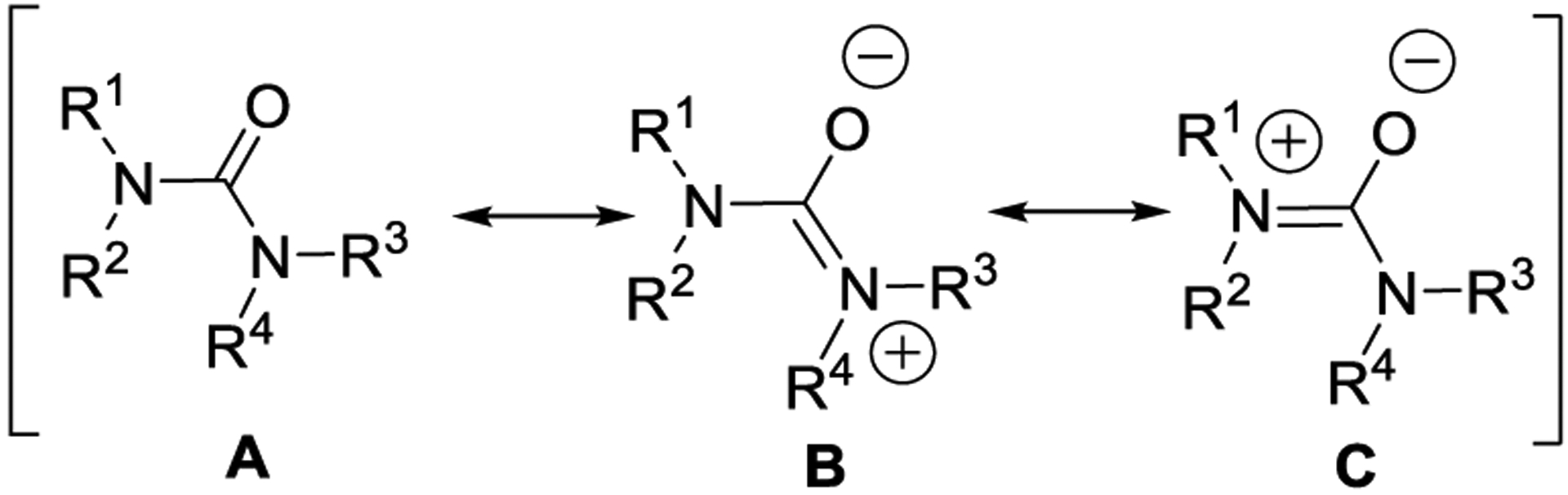
Possible resonance structures for the urea moiety.
Substitution on the nitrogen atoms of the urea moiety plays a key role on the conformational preferences of urea derivatives.28 As shown in Figure 3, three conformations (6a-c) are possible for N,N′-diphenylureas. In solution and in solid state, they are generally characterized by a trans,trans conformation.29,30 Interestingly, sequential introduction of N-methyl group(s) to the free NHs of N,N′-diphenylurea prompted the shift from trans,trans (7) to cis,cis (8a) conformation. Thus, N,N′-diaryl-N,N′-dimethylurea exhibits intrinsic preference for a cis,cis conformation (8a) over trans,cis conformation (8b), characterized by the two aromatic portions located in a face-to-face arrangement, thus allowing π-π stacking interactions.28
Figure 3.
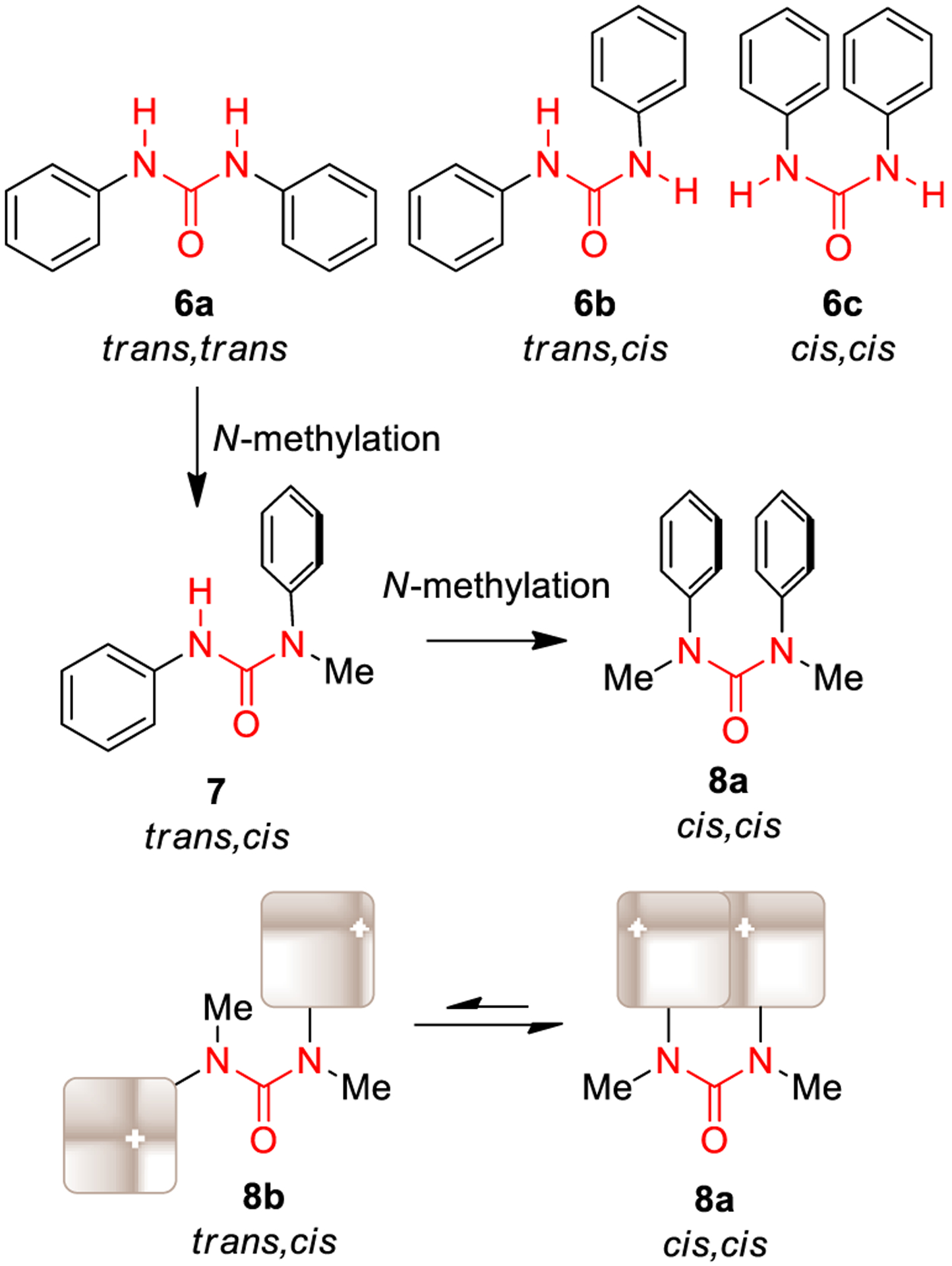
Conformations of N,N′-diphenyl urea, N-methyl-N,N′-diphenyl urea, and N,N′-dimethyl-N,N′-diphenyl urea.
Extensive NMR studies and ultraviolet spectra analysis in solution on several substituted diphenylureas were performed to obtain insight into the stacking of the aromatic rings. Interestingly, the conformation from the X-ray crystallographic data and the conformations adopted in solution are in good agreement. These studies supported similar hybridization of the nitrogens and relative positions of the aromatic rings.31,32
Theoretical studies suggest that the conformation showing the two aromatic rings with a face to face disposition in a mirror image relationship is disfavored as shown in Figure 4. The X-ray structural studies revealed a staggered relationship where the overlap of the HOMO of one ring with the LUMO of the other is enhanced (Figure 4).31
Figure 4.
HOMO and LUMO overlap in substituted diarylureas.
More recent studies on the conformational preferences of urea derivatives employing a combination of IR, NMR, and computational studies (DFT-D, M06–2X, quantum calculations, molecular dynamics) highlighted a more dynamic behavior of the N,N′-aryl ureas and N,N′-diaryl-N,N′-dialkyl ureas, although still in favor of the predominant isomer from the solid state studies.33,34 In studies with synthetic retinoids and cytokinins, the stereochemical alteration following N-methylation of aromatic amides was utilized in modulating the biological activity.35 Similarly, stereochemical switching through N-methylation of ureas from a transoid structure to the preferred bis-cis-N,N′-dimethyl-N,N′-diphenylureas has been exploited. This strategy is particularly attractive in molecular design due to ease of introduction of the N-methylurea substructure in organic molecules. As shown in Figure 5, the N,N′-dimethyl-N,N′-dinaphthylurea (9) and N-methyl derivative of poly-(phenyleneureido)benzene (10) formed nice π-stacked aromatic arrays as observed based upon NMR and X-ray crystallographic studies.36 This concept has been successfully applied to the construction of conformationally defined oligomers (foldamers) and dynamic helical structures as biomimetics.37
Figure 5.
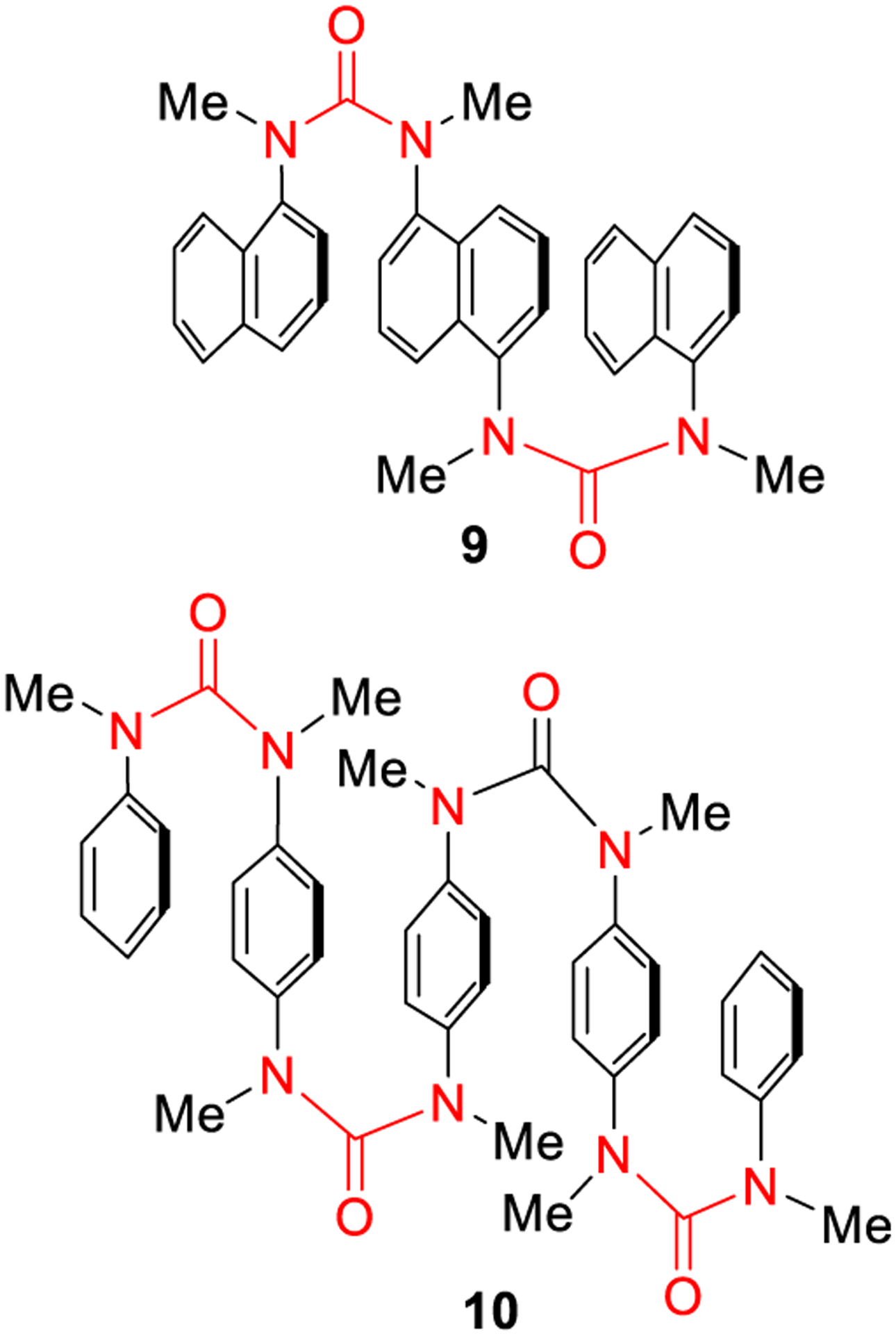
Formation of π-stacked aromatic arrays using substituted diarylureas.
Employing N,N′-diaryl ureas to relay stereochemical information over long distances, thus achieving stereochemical control over conformation, is another important feature of this functional group. As shown in Figure 6, reduction of N,N′-diaryl urea 11 containing a chiral sulfinyl group proceeded with excellent diastereoselectivity, providing alcohol 13 as the major diastereomer (dr 95:5), even though the chiral sulfinyl group is localized many bond lengths away. Presumably, the observed remote diastereoselectivity is due to the defined conformation of the chiral N,N′-diaryl urea 12 which resulted in the preferential nucleophilic attack from the Re-face of the carbonyl center affording alcohol 13.38
Figure 6.
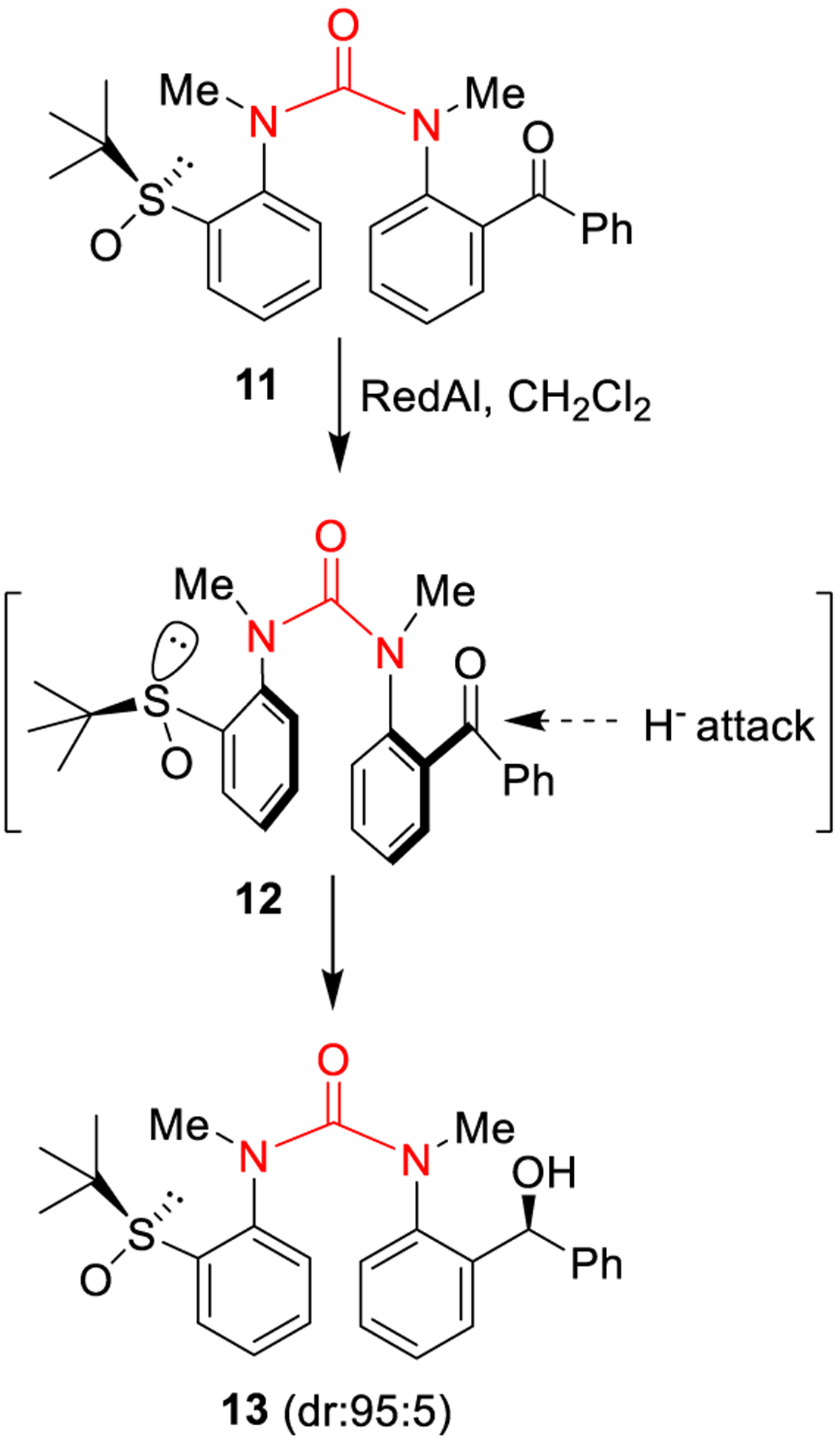
Stereoselective reduction of acylurea 11 containing a chiral sulfinyl group.
Oligomeric aromatic ureas characterized by N,N′-dimethylated urea moieties display a multilayered structure due to the (cis,cis)-urea arrangement. Such derivatives, containing a chiral center on the R2 group, display a dynamic helical architecture (all-S or all-R axis configuration) in solution when the phenyl moieties are connected through urea linkages at the meta positions. These dual dynamic helical and aromatic multilayered properties can be exploited to construct aromatic functional molecules with unique physicochemical properties.39,40
3. PHYSICOCHEMICAL PROPERTIES OF UREAS
The oral bioavailability of a drug depends upon many factors including solubility, dissociation, permeability, first-pass metabolism, and efflux properties. Drug solubility in water is vitally important for drug absorption, bioavailability, and drug administration. The development of appropriate formulations for poorly soluble drugs is a major challenge in drug design and development. The structure of a drug molecule typically contains multiple functional groups which determine the overall hydrophobic or hydrophilic nature of the molecule. This hydrophobic/hydrophilic balance affects the capability of a given drug to cross biological membranes.41
The presence of a urea functionality plays an important role in a drug’s aqueous solubility and permeability due to its dual nature as a hydrogen bond donor and acceptor. For a central nervous system (CNS) acting drug, a moderate level of lipophilicity would allow the drug to cross the blood-brain barrier (BBB) by a passive diffusion process. The hydrogen bonding capability42 in addition to ionization, polar surface area, and flexibility also strongly affects drug transport across the BBB.43,44
Hydrotropic solubilization involves the addition of one solute to promote the solubility of another and is a strategy explored in pharmaceutical formulations. Hydrotropes are mostly aromatic or nonaromatic anions but can also be represented by neutral compounds, such as ureas.45 Hydrotropic solubilization strategy was employed for the solubilization of nifedipine, a poorly soluble antihypertensive drug, by a series of urea analogues in an aqueous environment. The solubilizing effect ranking followed the trend butylurea > ethylurea > methylurea > urea.46 Sometimes, urea containing drugs show poor pharmacokinetic properties due to solubility and permeability issues. Many strategies have been developed to circumvent these issues. They are shown below.
3.1. Modulation of the Hydrogen Bonding Ability of Ureas.
In order to modulate hydrogen bonding capability, electron donating and electron withdrawing functionalities have been introduced on the substituents on the urea nitrogen atoms. The nature of aliphatic moieties placed on the urea nitrogen has been shown to affect self-association properties thus controlling drug solubility in nonpolar solvents.46 A representative example is the N,N′-di(2,6-diisopropylphenyl)urea (DIPPU, 14) which is the only diaryl urea that is soluble in carbon tetrachloride among eight other symmetrical diaryl ureas examined. DIPPU does not form a supramolecular polymer. When the branched alkyl moieties are not too large, the corresponding dialkylureas self-assemble to form supramolecular polymers in solution (Figure 7A).47 However, DIPPU stabilizes the out-trans conformation due to large steric bulk of the alkyl chain on the aromatic rings. Therefore, the formation of dimers occurs through hydrogen bonding of the N-H groups in the out conformation as shown (Figure 7B).
Figure 7.
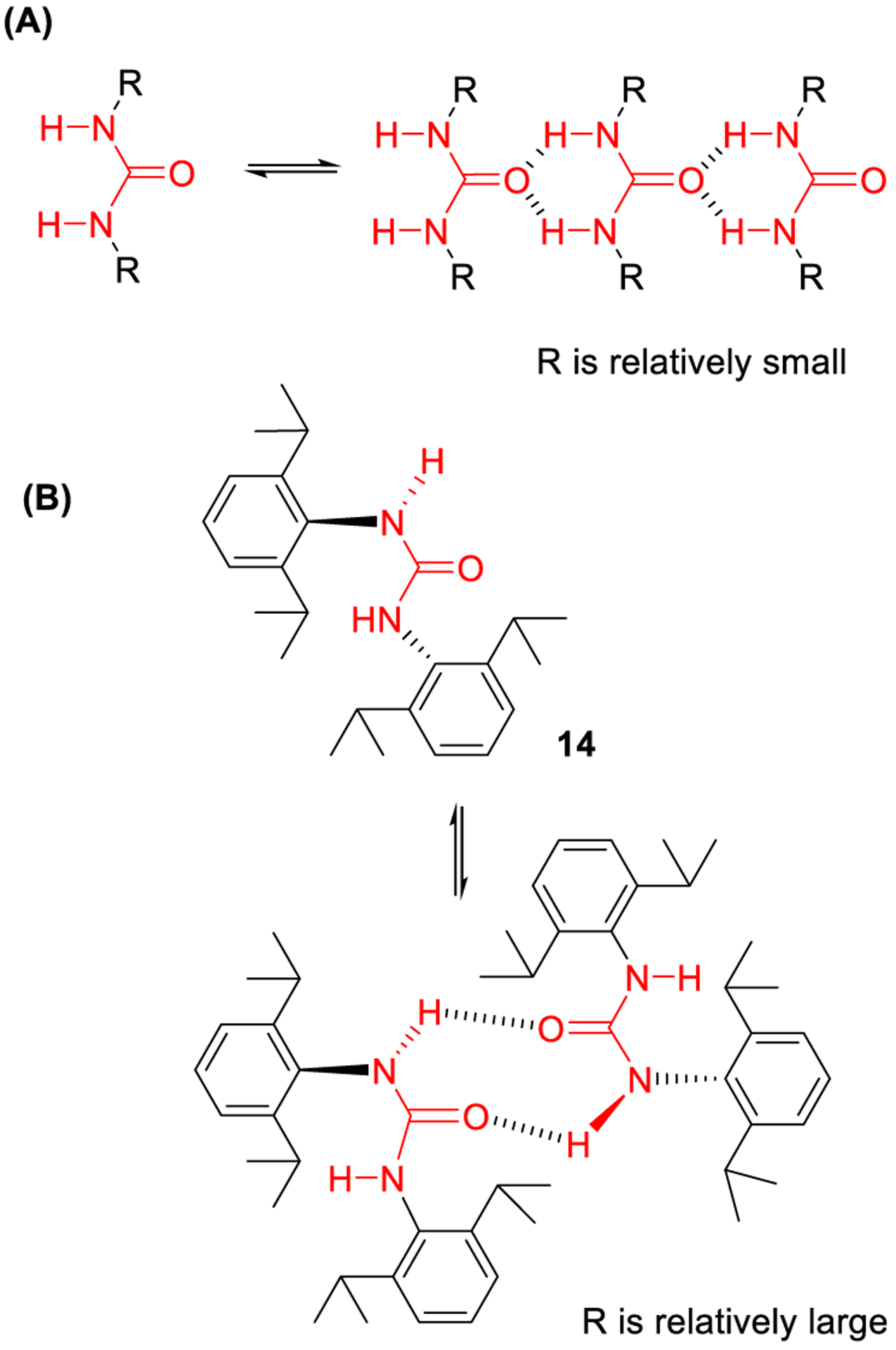
(A) Association pattern of N,N′-disubstituted ureas; (B) proposed association pattern for DIPPU (14).
3.2. Engagement of the Urea Moiety in the Formation of Intramolecular Hydrogen Bonding.
This strategy can be exploited to increase permeability and solubility with the formation of a transient or pseudoring structure. This can be achieved by engagement of a urea functionality in a novel monocyclic template that mimics a bicyclic structure.48 As shown in Figure 8, based on the structure of a bicyclic kinase inhibitor (15), a series of pyrimidin-4-ylureas (16) were designed. This monocyclic structural template could function as suitable bioisostere due to its tendency to form resonance-assisted intramolecular hydrogen bonds.49 The feasibility of this approach was demonstrated since this compound class exhibits multikinase inhibitory activity. A cocrystal structure revealed the hypothetical binding mode. Furthermore, NMR and theoretical studies substantiated the existence of an intramolecular hydrogen bond and its existence in water medium.50
Figure 8.
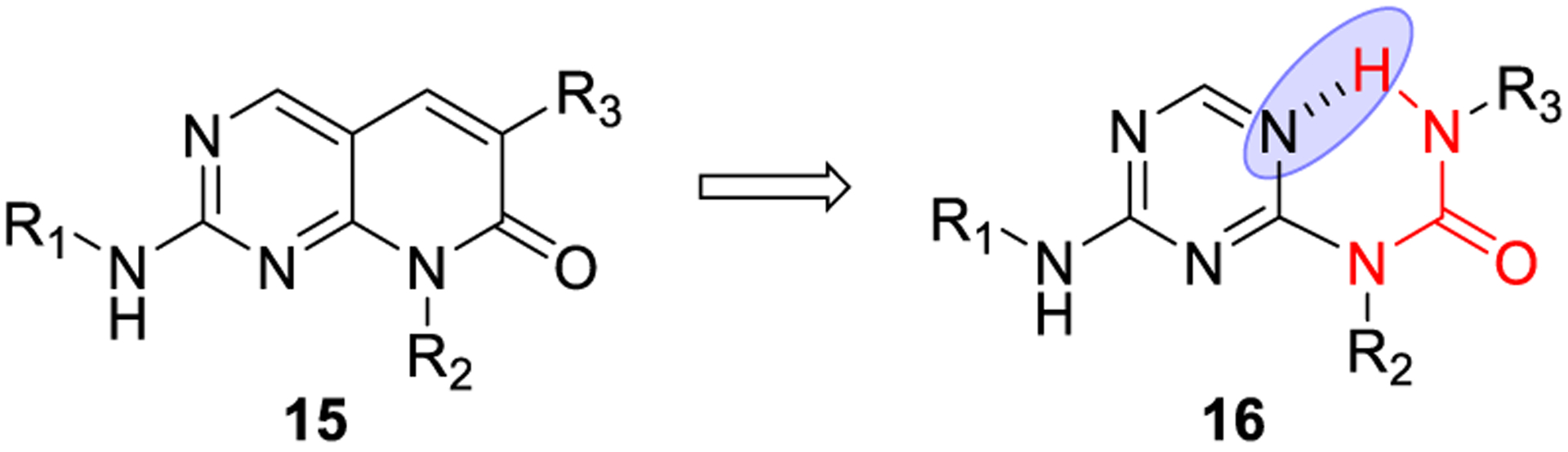
Construction of pseudoring systems exploiting intra-molecular hydrogen bonding.
3.3. Disruption of the Planarity of Urea Derivatives.
In this approach, water solubility is enhanced by disrupting the molecular planarity of the solutes in order to reduce their crystal packing energy.51 The strategy involves the breaking of the urea symmetry by introducing a substituent on one of the urea nitrogens which disrupts planarity.52–54 A practical example is shown in Figure 9. N-Methyl-N-1-naphthyl urea (18) acts as a disruptor of cytokine-mediated STAT1 signaling in β-cells. Incorporation of a methyl group on the urea nitrogen of 17 provided N-methyl-N-1-naphthyl urea (18). The presence of a methyl group disrupts the existence of planar conformations, due to steric clash between hydrogens at C-2 or C-8 positions of the quinoline ring and the appended N-methyl group. Compound 18 showed a 110-fold solubility increase over compound 17. This data also well correlated with a decreased melting point for compound 18 compared to compound 17 (~145 °C vs ~171 °C, respectively).53
Figure 9.
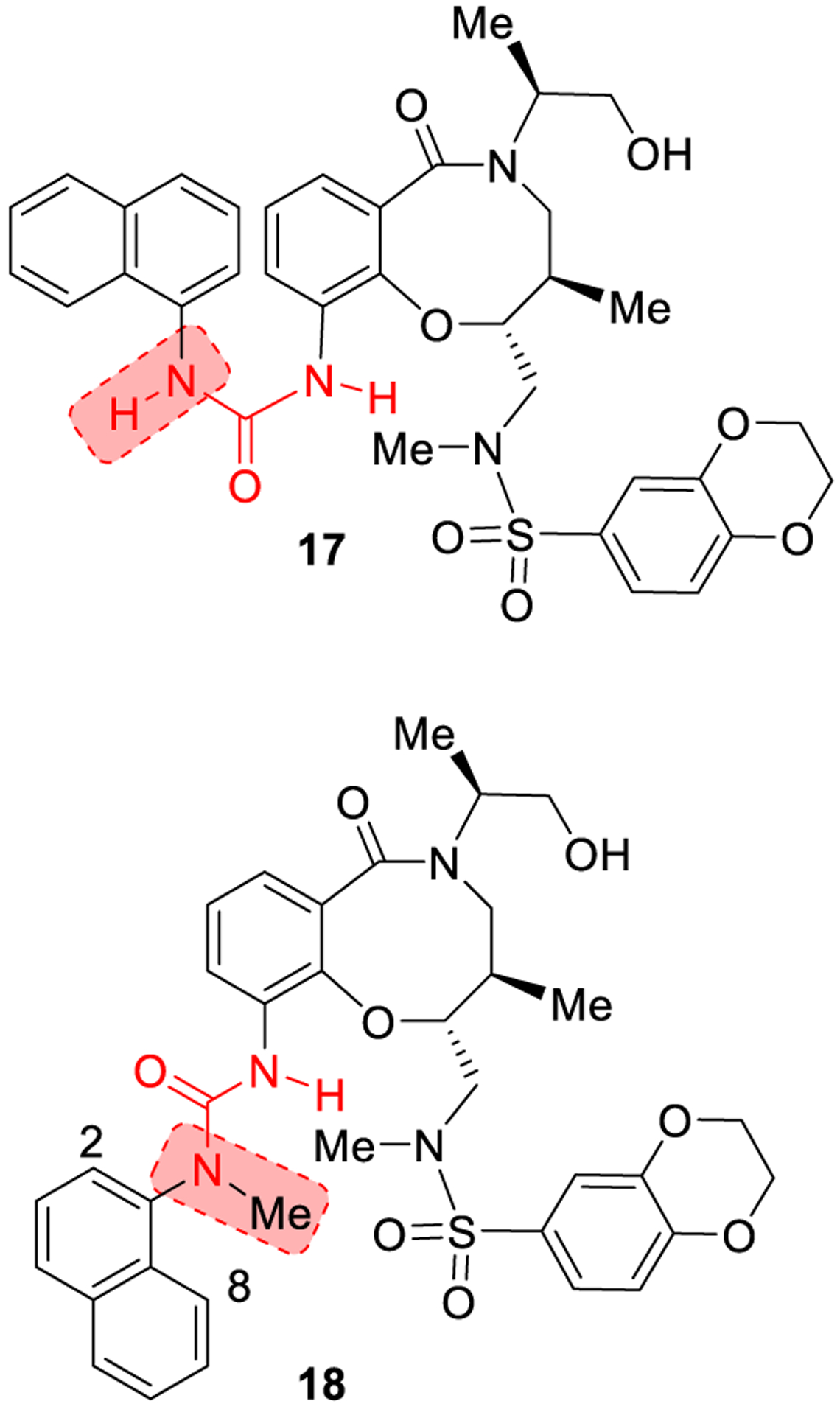
Disruption of the planarity of urea derivatives through urea alkylation.
Another strategy to disrupt planarity is represented by the insertion of substituents at the ortho position of the N-aryl group of arylureas. Thus, the presence of two halogen atoms at the ortho position of the urea functionality could promote the formation of intermolecular hydrogen bonds due to a better conformational preorganization of the monomer. FT-IR and ab initio calculations revealed that an intramolecular hydrogen bond is formed when phenylureas are functionalized with chlorine or bromine atoms at the ortho positions. This halogen effect is at least in part due to the significant influence of the substituents on the dihedral angle (φ) between the urea functionality and the aromatic groups. As shown in Figure 10, in the absence of substituents in compound 19, the most stable conformation for the compound is coplanar (φ = 0°), while in the presence of chlorine or bromine substituents (compounds 20 and 21), the energy surface is completely flat with φ values ranging from 60° to 120°.55
Figure 10.
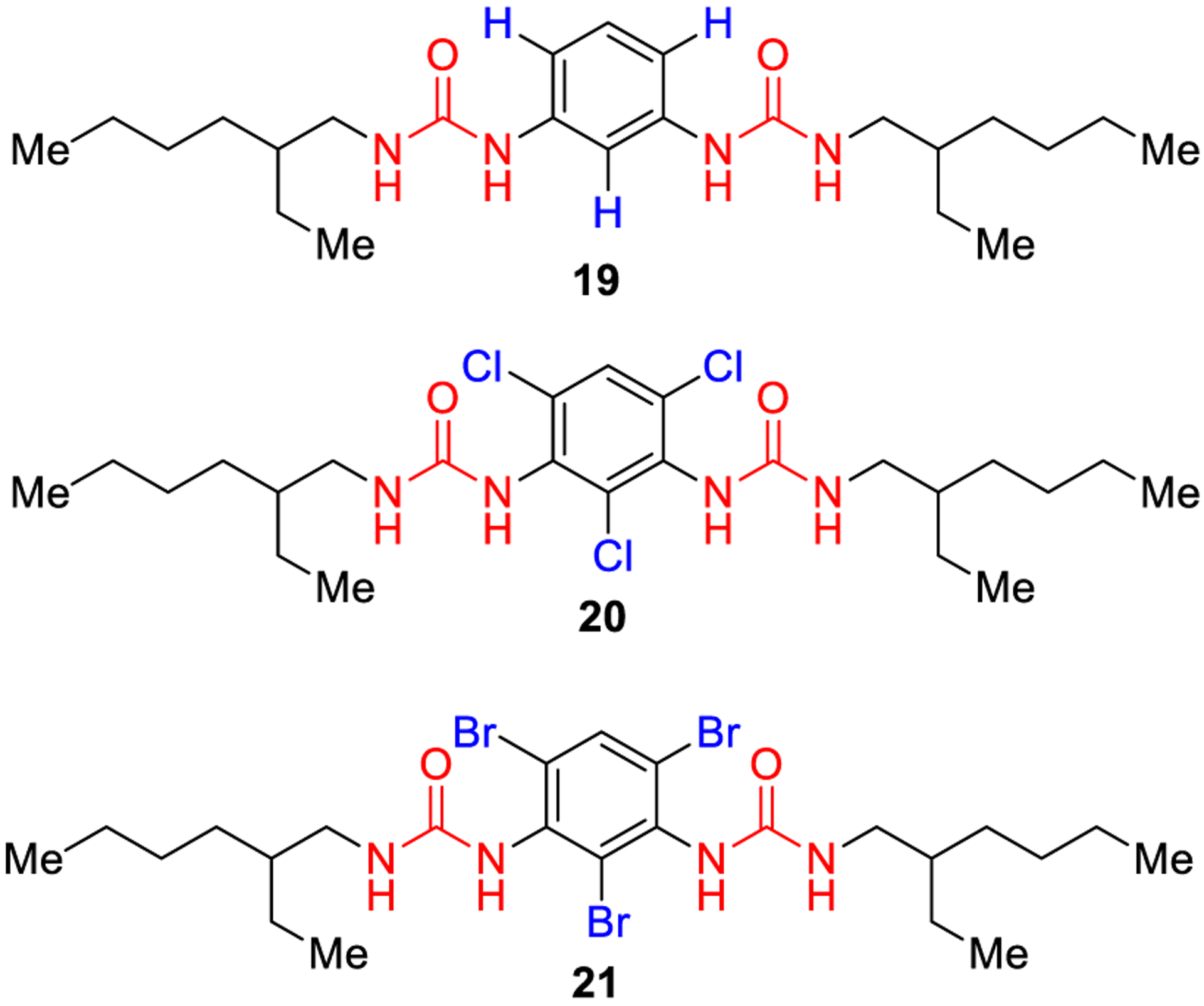
Disruption of urea planarity by introduction of orthosubstituents at the aryl urea system.
3.4. Stacking Interactions of Ureas with Aromatic Rings.
Although the hydrogen bonding ability of the urea moiety has been long known and studied, this functionality can also be involved in other types of important interactions, especially those involving urea functionality and aromatic side chains in proteins.56,57 Several crystal structures of proteins bound to urea/urea derivatives have been identified to exhibit π-stacking-like favorable interactions. Moreover, perpendicular orientation of urea with respect to aromatic groups leads to NH-π type interactions. Such a complex hydrophilic/hydrophobic combination of interactions involving the urea moiety could likely be exploited in the future design of ligand molecules for drug discovery purposes.58
Several protein structures in the Protein Data Bank contain urea and urea derivatives as ligands or cosolvents. Quite interestingly, 38% of the structures containing urea and 25% of those containing urea derivatives were found to fall within the geometric requirements for a stacking arrangement with aromatic rings (e.g., PDB ID: 3IPU and 4EV9).59,60 The relevance of stacking interactions between urea and aromatic side chains in urea transporters (UTs) has been modeled by Priyakumar and co-workers.61 UTs are trimeric transmembrane proteins with a parallel arrangement of aromatic rings in the pore which allows stacking of urea and urea homologues (e.g., formamide, acetamide, and dimethylurea). This results in lowering of the energy barrier for urea based solute to transport.62,63
The aromatic ring stacking interactions play an important role in urea-assisted protein denaturation. Using Trp-cage mini-protein, Priyakumar and co-workers61 investigated distortion of the protein hydrophobic core in the presence of urea and showed that aqueous urea optimally solvates aromatic groups in the protein. Furthermore, studies suggested the presence of stacking and NH-π interactions involving aromatic groups and urea. In particular, three kinds of interactions were shown between the Trp6 side-chain and urea: (i) stacking, where the urea moiety and indole ring were parallel to each other, (ii) NH-π interaction, where one of the NH bonds of the urea was perpendicular to the indole ring; (iii) typical hydrogen-bonding interaction between the urea oxygen atom and the nitrogen atom of the Trp side chain. Stacking and NH-π interactions with urea are responsible for stabilizing Trp6 in the unfolded state (Figure 11, PDB ID: 1L2Y).61
Figure 11.
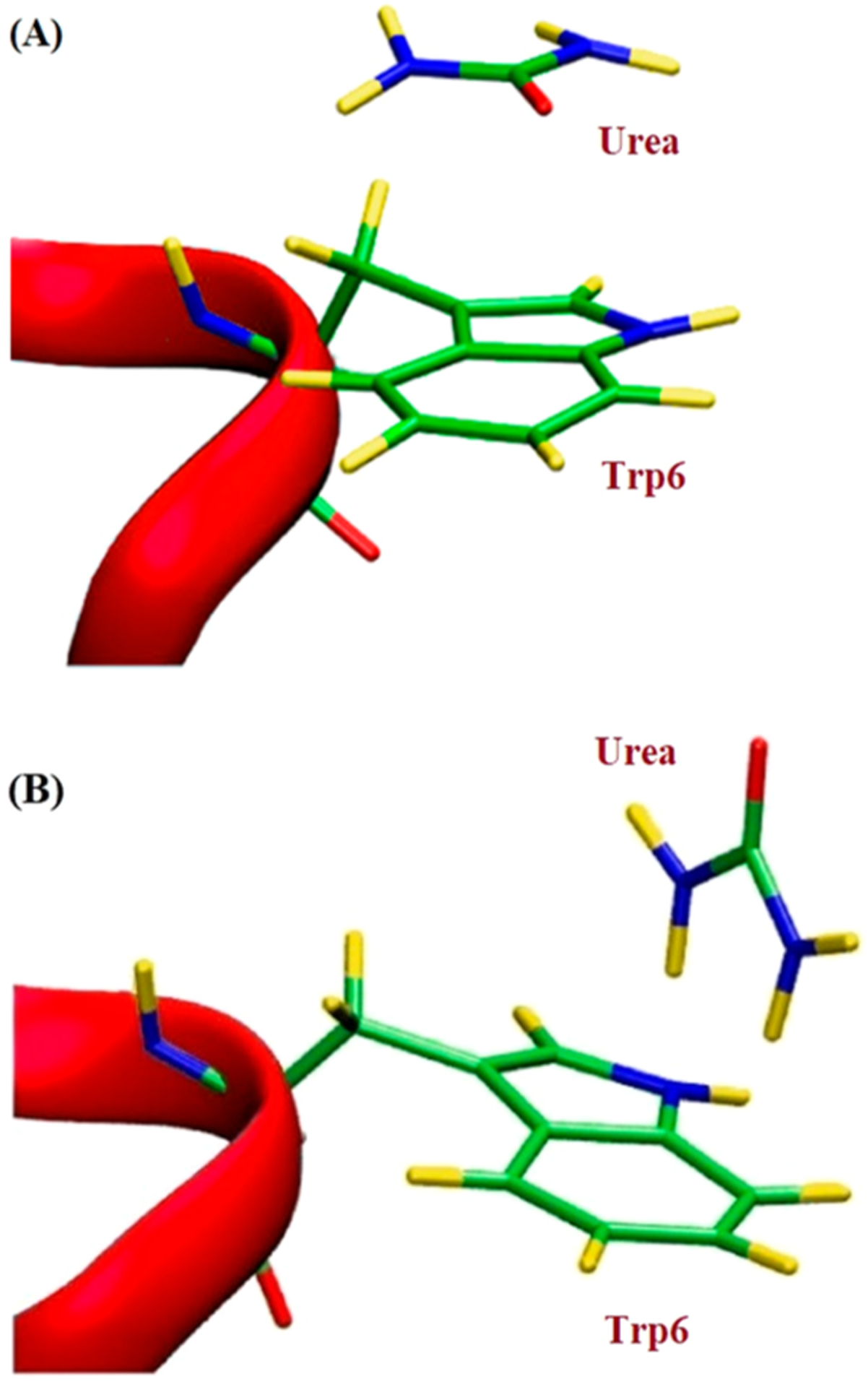
(A) Representation of the stacking interaction between Trp6 and urea; (B) NH-π interaction between the tryptophan 6 side-chain and urea. Part of the Trp-cage miniprotein is shown in magenta; carbon atoms, nitrogen, and hydrogen atoms are shown in green, blue, and yellow, respectively. The figure is modified based upon published structures.61
Urea-assisted RNA unfolding is another phenomenon which can be explained by the engagement of an extrahelical state via favorable π-π, NH-π, and hydrogen bonding interactions with the urea moiety. Both urea and water can establish NH-π and hydrogen bonding interactions with nucleic acid structures. However, stacking interactions are only possible with urea, and this explains its strong denaturing actions. These effects also rely upon the base composition and nature of nucleic acids.64,65
4. IMPORTANCE OF UREA FUNCTIONALITY IN DRUG-RECEPTOR INTERACTION
The bioactivity of drugs is governed by molecular recognition through drug and target protein interactions. Among the multiple forces that are involved in protein-ligand interactions, hydrogen bonds play a key role due to their ability to stabilize the drug-receptor interaction.44,66 The donor-acceptor hydrogen bonding capability of urea derivatives is one of the most important elements of their molecular recognition and bioactivity. A few examples describing key interactions involving urea functionality in biologically active compounds are presented here.
A series of potent HIV protease inhibitors have been developed featuring a urea substructure. In 1993, Getman and co-workers reported a novel class of HIV-1 protease inhibitors incorporating the (hydroxyethyl)urea isostere. Compound 22 (SC-52151) exhibited good inhibitory activity on HIV-1 protease (IC50 = 6.3 nM) and excellent selectivity over other aspartyl proteases, such as renin and cathepsin D. As shown in Figure 12, X-ray structural studies of a related inhibitor 23 containing a n-butyl chain complexed with HIV-1 protease revealed that the inhibitor binds in an extended conformation with the (R)-hydroxyl group positioned symmetrically between the two catalytic aspartates of the enzyme. The urea carbonyl group appears to be involved in water-mediated hydrogen bonding with the Ile50 and Ile50′ residues. The P2′ n-butyl substituent of the urea derivative is occupying the S1′-binding site, and the P1′ isobutyl group has occupied the S2′ subsite.67
Figure 12.
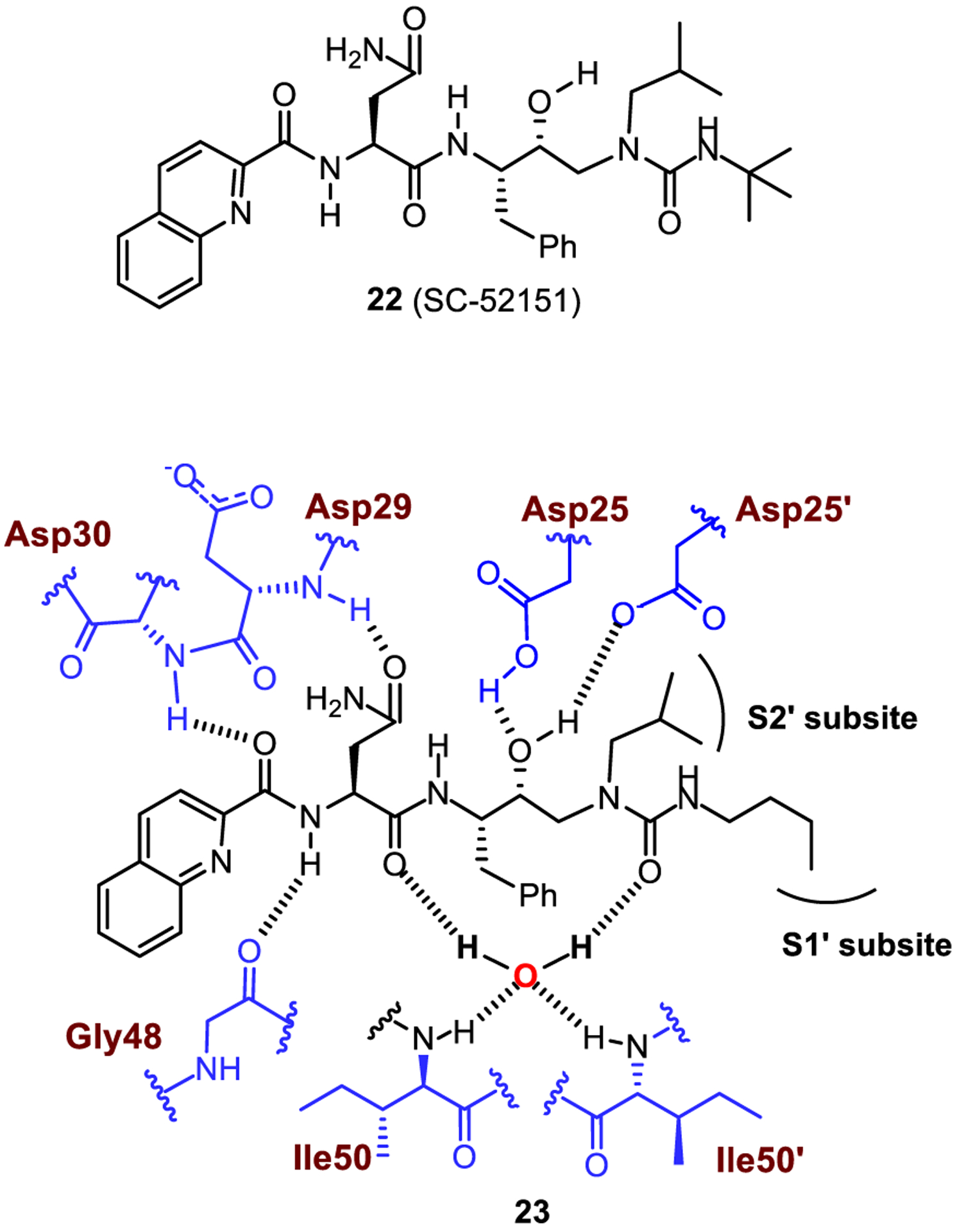
Structure of HIV-1 protease inhibitor 23 and highlight of the interaction with protease. Hydrogen bonding interactions are shown by blue dotted lines.
HIV protease inhibitors featuring a cyclic urea substructure have also been designed and developed into a number of preclinical candidates. One of the specific design objectives was to replace the structural water molecule found in cocrystal structures of HIV protease and linear inhibitors using the urea functionality. The resulting inhibitors maximized interactions in the protease active site and maintained good activity against mutant proteases.68 Compound 24 (DMP450) and a number of other urea-based inhibitors exhibited broad spectrum antiviral activity against multidrug-resistant HIV-1 variants.69 The cocrystal structure (PDB id: 1DMP) of DMP450-bound HIV-1 protease revealed that the urea functionality nicely mimicked the flap ligand-bridging water molecule (Figure 13). Also, the conformationally locked cyclic core allowed optimal interaction and accommodation of ligands within the S1, S1′, S2, and S2′ subpockets of the HIV protease active site.68
Figure 13.
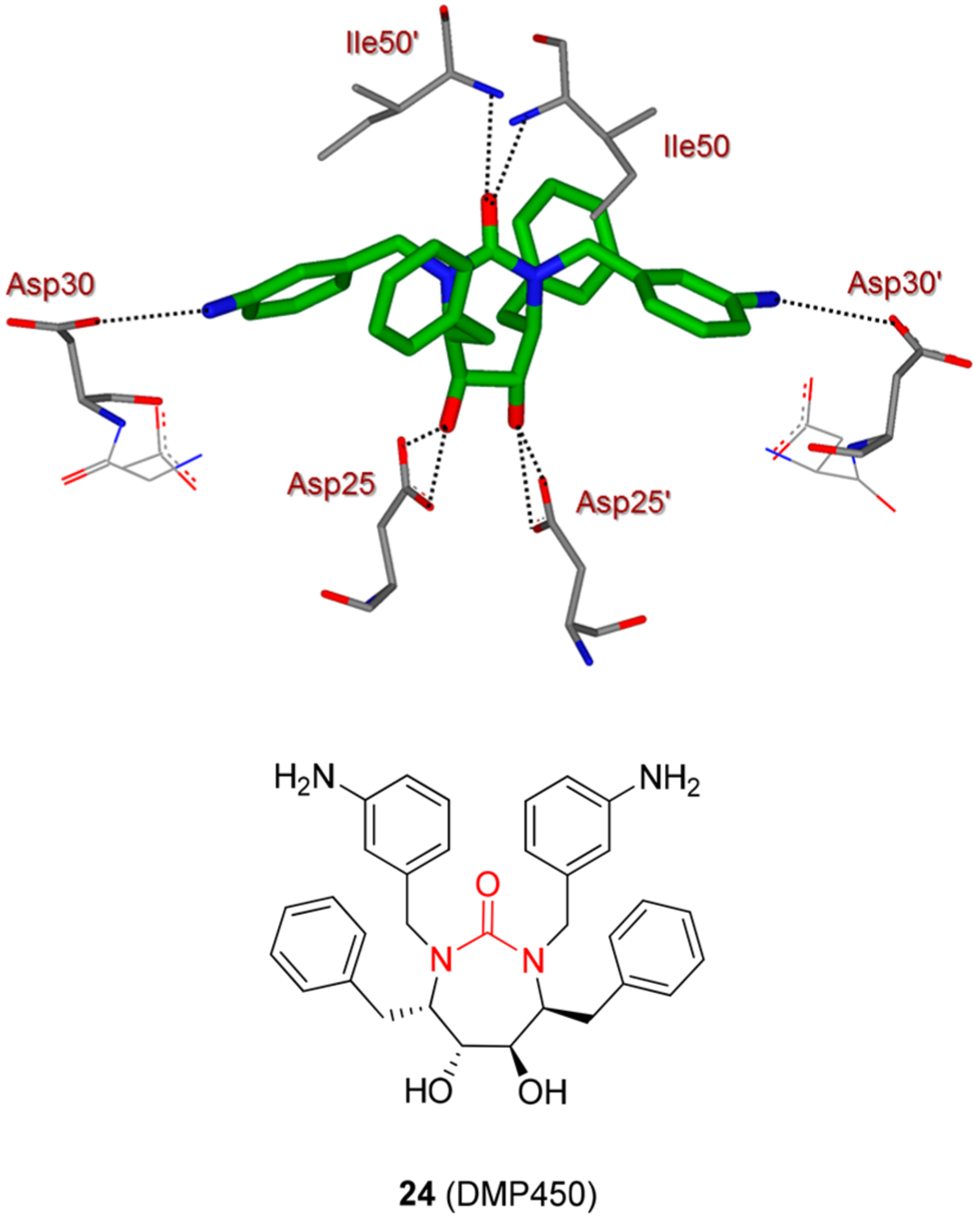
X-ray structure of urea derivative 24 and HIV-1 protease complex (pdb code: 1DMP). Hydrogen bonding interactions are shown by black dotted lines.
The urea moiety also plays a prominent role in molecular recognition of sorafenib (25) and lenvatinib (26) by the protein kinase active site (Figures 14 and 15). The X-ray crystal structures of vascular endothelial growth factor receptor 2 (VEGFR2) in complex with either lenvatinib or sorafenib were determined. These protein-ligand complexes with lenvatinib and sorafenib highlighted the importance of the urea functionality. As shown, the urea functionality is involved in critical hydrogen bonding with main-chain atoms of Asp1046 and the side-chain atoms of Glu885 (Figures 14 and 15).70 The quinoline moiety of lenvatinib and the pyridine core of sorafenib
Figure 14.
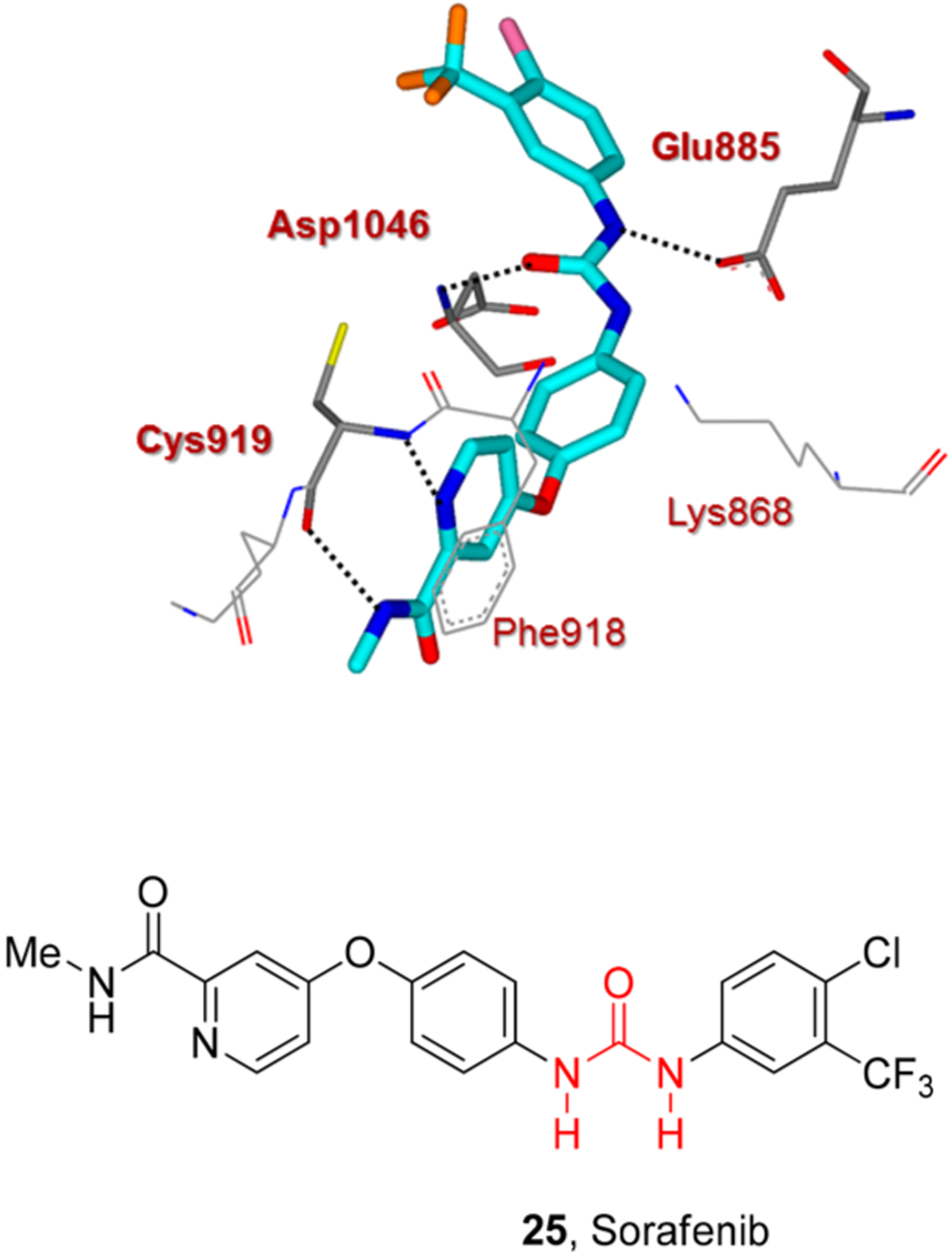
X-ray structure of sorafenib (25) and VEGFR2 complex (pdb code: 4ASD). Hydrogen bonding interactions are shown by black dotted lines.
Figure 15.
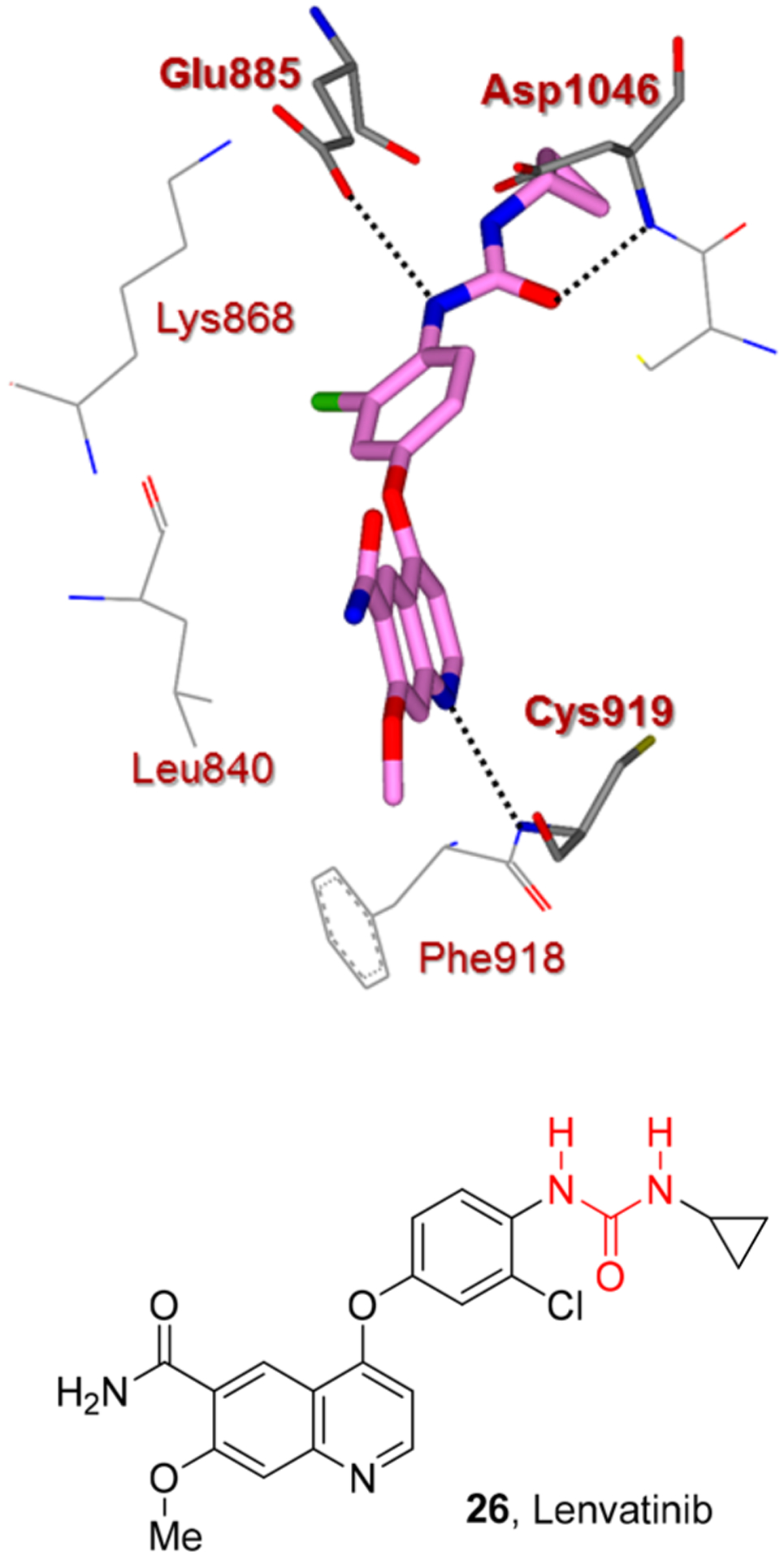
X-ray structure of lenvatinib (26) and VEGFR2 complex (pdb code: 3WZD). Hydrogen bonding interactions are shown by black dotted lines.
Urea derivative 27 is a potent Map kinase inhibitor that has undergone human clinical trials for the treatment of inflammatory diseases. Interestingly, the N-pyrazole-N-aryl urea occupies the binding domain on p38 that is exposed when the conserved binding loop containing Asp168, Phe169, and Gly170 adopts a DFG-out conformation (Figure 16). The X-ray structural studies revealed that both urea NHs form strong hydrogen bonds with the Glu71 side chain carboxylic acid while the urea carbonyl forms a strong hydrogen bond with the Asp168 backbone amide NH. The naphthalene moiety binds in the kinase specificity pocket. The ethoxymorpholine chain adopts a Gauche conformation and orients the morpholine oxygen to form a strong hydrogen bond with the backbone NH of Met109 in the ATP binding region of p38.6
Figure 16.
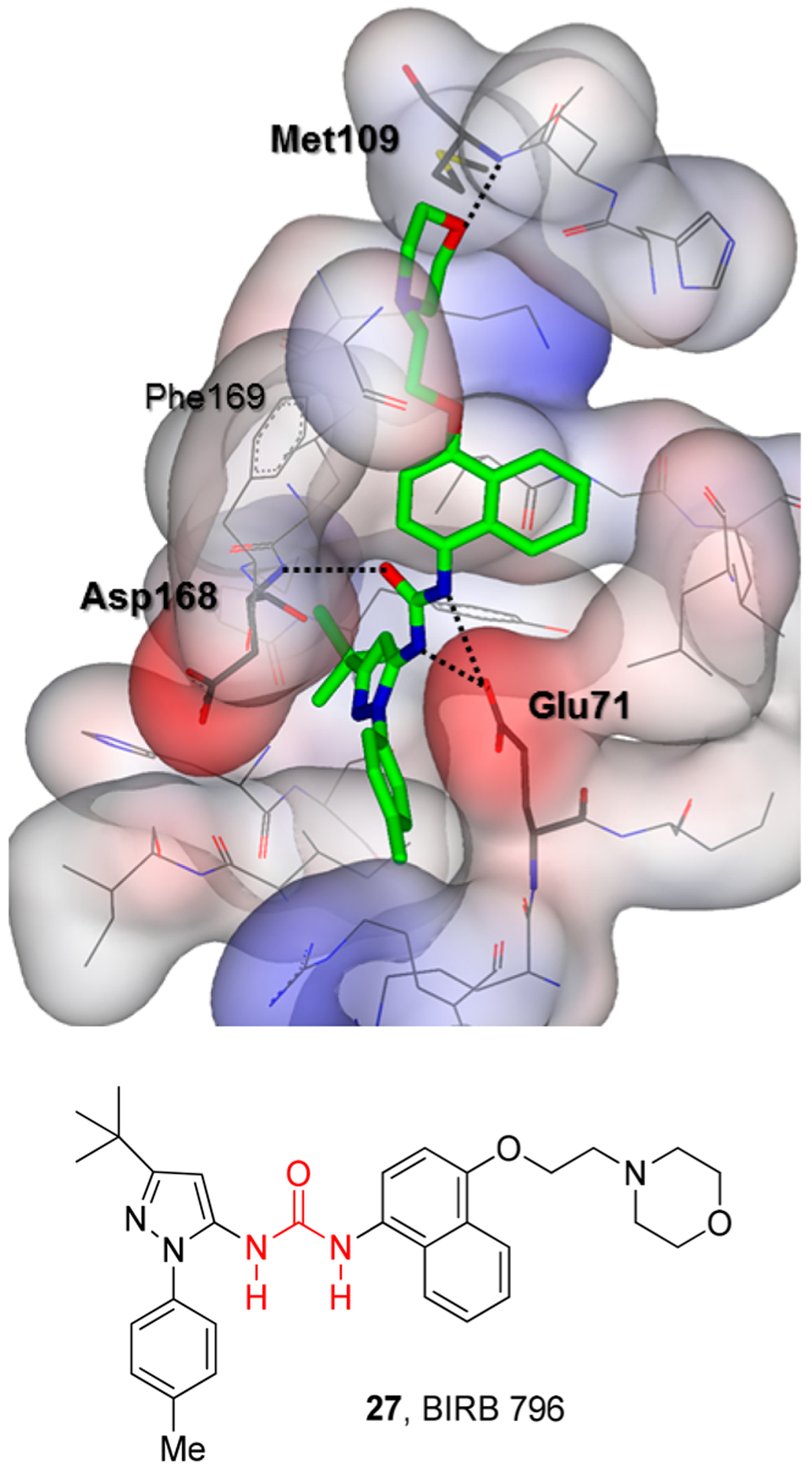
X-ray structure of MAP kinase inhibitor BIRB 796 (27) with human p38 MAP kinase (PDB code: 1KV2). Hydrogen bonding interactions are shown by black dotted lines.
Another example of the key role of urea functionality in drug-target interactions is shown in urea-derived soluble epoxide hydrolase (sEH) inhibitors. The sEH is involved in the metabolism of endogenous mediators (e.g., epoxides of linoleic acid, arachidonic acid, and other lipids).71 The catalytic mechanism of sEH as shown in Figure 17A, involves a SN2-type reaction in which the epoxide is first activated by forming hydrogen bonds with Tyr381 and/or Tyr465 residues. Subsequent nucleophilic attack by Asp333 forms the acylenzyme intermediate which is attacked by a water molecule activated by His523. The resulting tetrahedral intermediate finally collapses to provide the diol product (Figure 17A). 1,3-Disubstituted ureas have become interesting structural templates for sEH inhibition.72–75 The X-ray structural studies with the urea-derived inhibitor 4-(3-cyclohexylureido)-ethanoic acid (28) demonstrated that the urea moiety is able to mimic the transition state of the sEH-catalyzed epoxide ring opening, establishing strong hydrogen bonding interactions with Asp333 and Tyr381 residues (Figure 17B).76–78
Figure 17.
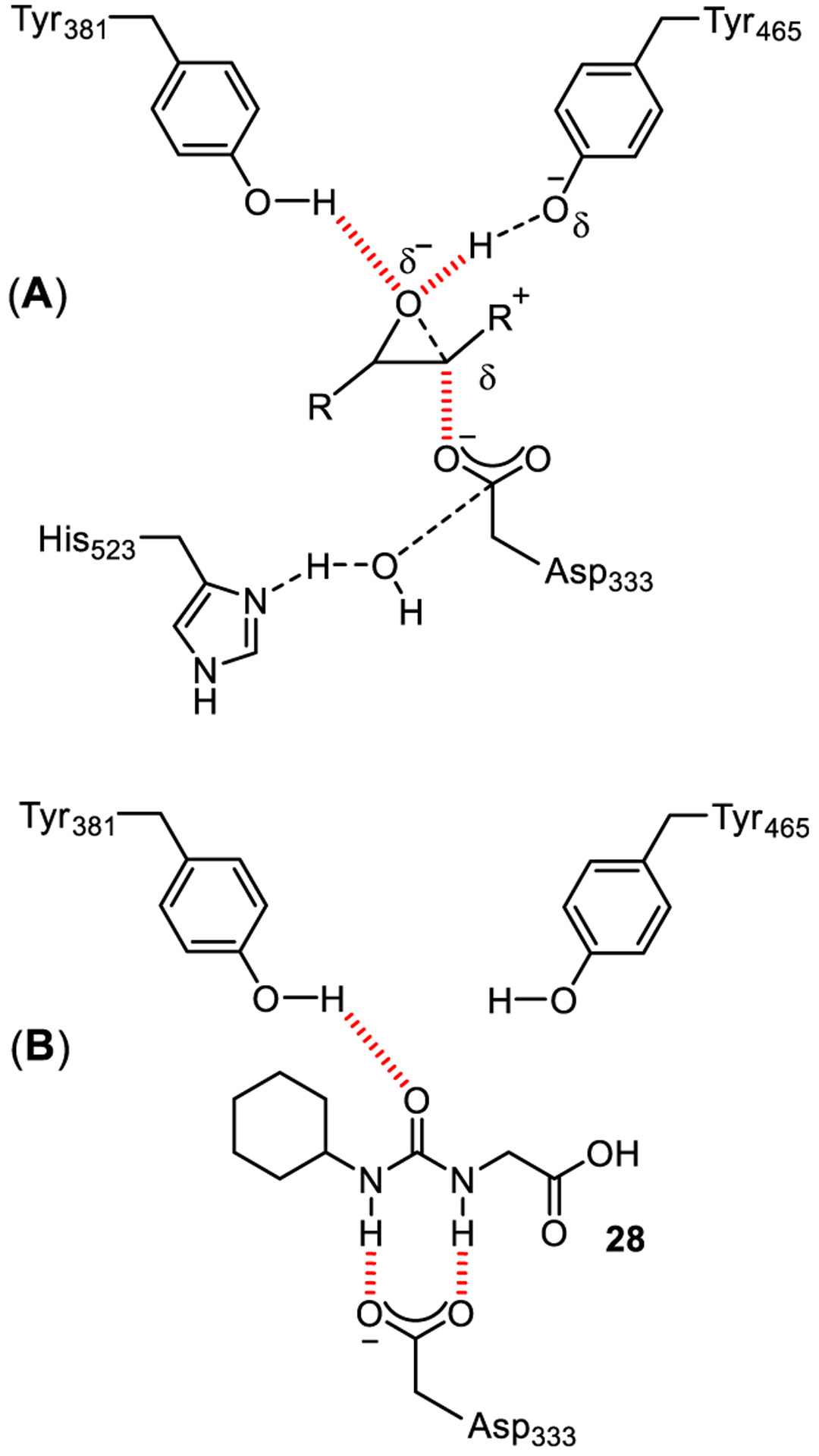
(A) Transition state for sEH-catalyzed oxirane ring opening; (B) Interaction of inhibitor 28 in the enzyme active site.
PF-04457845 (29) is a highly potent inhibitor of fatty acid amide hydrolase (FAAH).79 FAAH is a serine hydrolase responsible for the metabolism of signaling lipids, such as the endocannabinoid anandamide.80 FAAH is considered a potential therapeutic target for the treatment of pain and nervous system disorders. Thus, small-molecule FAAH inhibitors would exert their beneficial effects without the potential drawbacks associated with direct stimulation of cannabinoid receptors.81,82
Compound 29 contains a benzylidenepiperidine pyridazine urea. The X-ray and kinetic studies demonstrated that these urea-containing derivatives are able to covalently and irreversibly inhibit FAAH through involvement of the Ser241-Ser217-Lys142 catalytic triad as shown in Figure 18. The urea functionality interacts with the catalytic Ser241 nucleophile, thus generating a tetrahedral intermediate (30) and sub-sequently inducing carbamoylation (inactivation) of the FAAH enzyme through formation of carbamate 31.83,84 Recently, PF-04457845 has been shown to reduce cannabis withdrawal symptoms and cannabis use, in a phase IIa clinical trial.85
Figure 18.
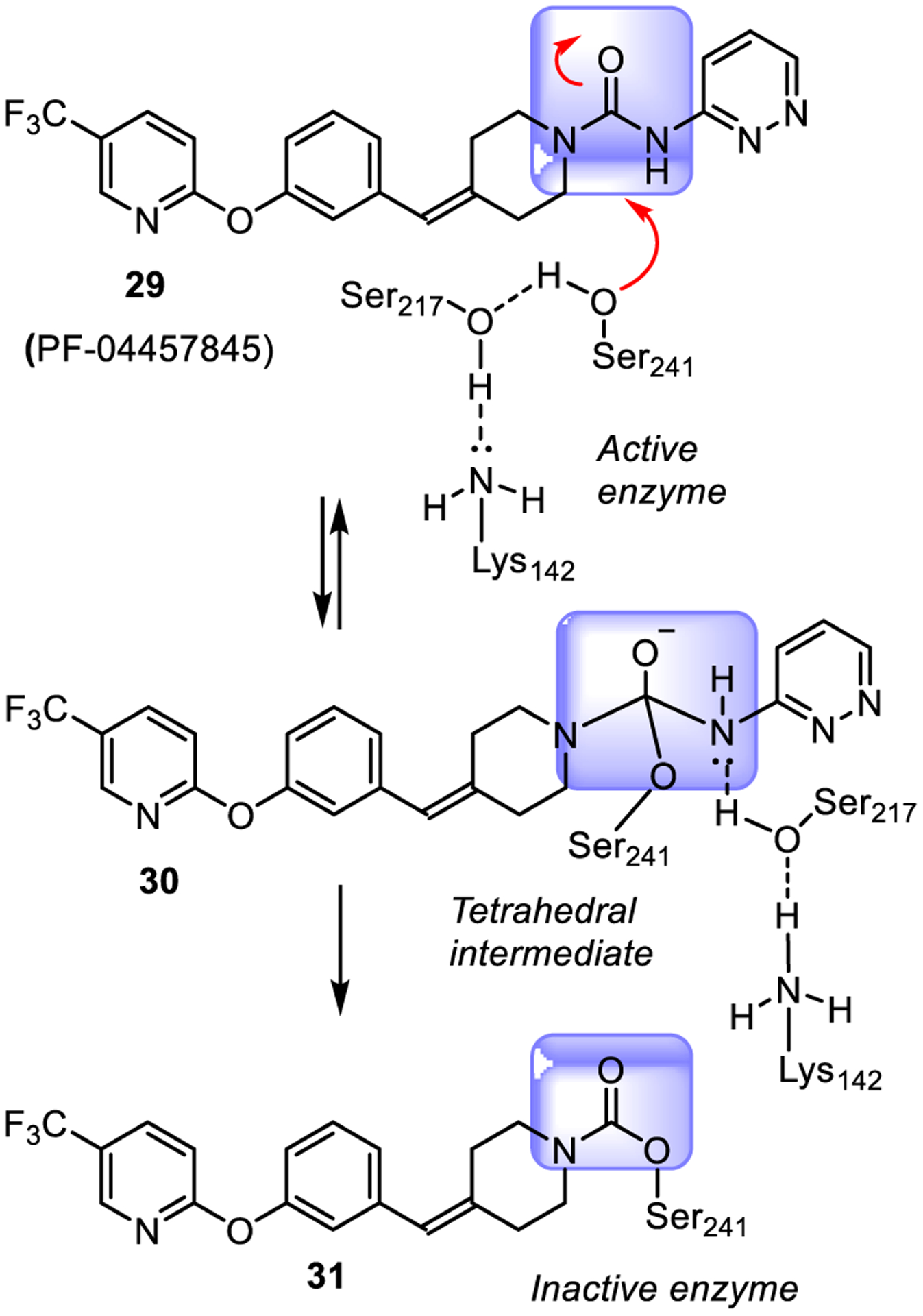
Mechanism of FAAH inhibition by urea derivative PF-04457845 (29).
5. UREA CONTAINING FDA APPROVED DRUGS
Urea substructures are characteristic of several FDA approved drugs as shown in Figure 1 and Figure 19. Many urea containing compounds are also undergoing clinical development which will be discussed later. In this section, approved urea derivatives for therapeutic applications are highlighted.
Figure 19.
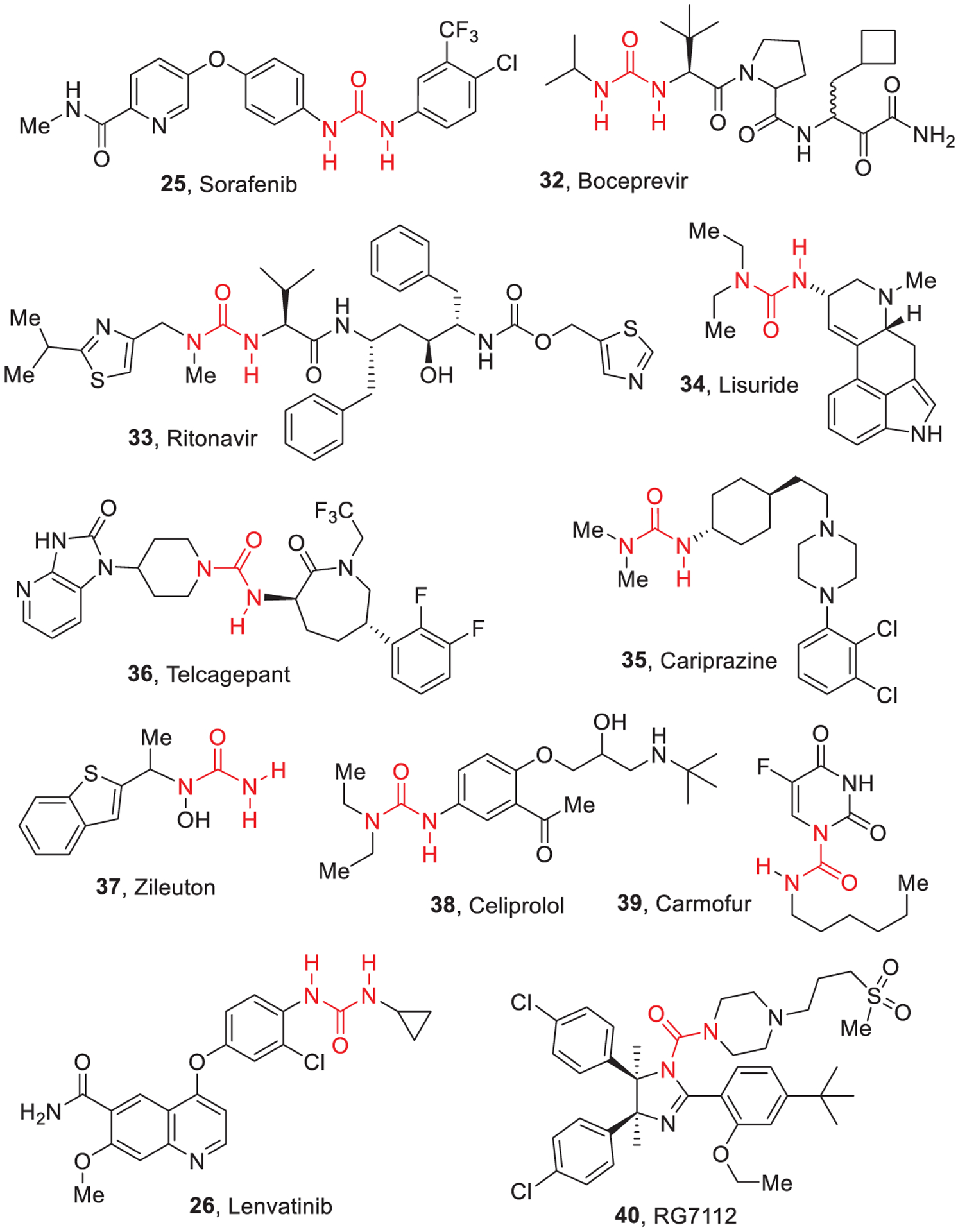
Structures of urea containing FDA approved drugs.
5.1. Sorafenib.
Sorafenib (25, Nexavar, Bayer and Onyx Pharmaceuticals, Figure 19) is a diaryl urea multikinase inhibitor acting on c-RAF, B-RAF, c-KIT, FLT3, platelet derived growth factor receptor (PDGFR) α and β, and vascular endothelial growth factor receptor (VEGFR) 1, 2, and 3, among others.12 It was approved in 2007 for the therapy of hepatocellular carcinoma and advanced renal cell carcinoma and is under evaluation for acute myeloid leukemia (AML).13,86 Sorafenib is converted by UDP-glucuronosyltransferase 1A9 (UGT1A9) to its glucuronide and by CYP3A4 to its active metabolite sorafenib N-oxide.87,88
5.2. Boceprevir.
Boceprevir (32, Victrelis, Merck, Figure 19) is a urea containing protease inhibitor useful against hepatitis C virus (HCV). Boceprevir reversibly binds the active site of nonstructural protein 3 (NS3) and displays high potency in the replicon system alone or combined with interferon α−2b and ribavirin.14 Boceprevir is a diastereomeric mixture of two compounds differing in the stereochemical configuration at the cyclobutylmethyl functionality appended to its ketoamide end. In January 2015, Merck announced market withdrawing for boceprevir due to the striking superiority of newer agents (e.g., ledipasvir/sofosbuvir). Oxidative metabolites of boceprevir are formed upon interaction with CYP3A4 and CYP3A5, while the keto-reduced metabolites mostly form upon transformations mediated by aldo-keto reductases AKR1C2 and AKR1C3. Since boceprevir metabolism encompasses two diverse enzymatic pathways, the drug undergoes drug-drug interaction to a lesser extent.89
5.3. Ritonavir.
Ritonavir (33, Norvir, ABT-538, A-84538, AbbVie, Inc., Figure 19) features a thiazolyl methyl urea substructure. In 1996, it was approved as inhibitor of the HIV protease.90 Ritonavir displayed an EC50 of 0.025 μM, plasma half-life of 1.2 h, and bioavailability of 78%. Ritonavir showed an excellent pharmacokinetic profile due to the higher stability of the thiazole moieties toward oxidative metabolism. Ritonavir inhibits CYP3A4 irreversibly binding to the heme iron via the thiazole nitrogen91 thus increasing plasma concentrations of other CYP3A4 substrate anti-HIV drugs, improving their efficacy.
5.4. Lisuride.
Lisuride (34, Dopergin, Proclacam, or Revanil, Figure 19) is a urea-based ergot-related dopamine agonist used for the therapy of Parkinson’s disease (PD). Lisuride also binds the serotonin 5-HT1A, 5-HT2A/2C, and histamine H1 receptors.92 Studies aimed at examining the metabolism of 14C-lisuride hydrogen maleate in humans and rhesus monkeys demonstrated that 2-keto-3-hydroxy-lisuride was the main metabolic derivative. Parallel metabolic transformations of lisuride were identified, including hydroxylation of the phenyl ring, oxidative N-deethylation, monooxygenation at C2 and C9, and oxidation of C2/C3 and C9/C10 double bonds.93
5.5. Cariprazine.
Cariprazine (35, Vraylar in the United States and Reagila in Europe, Gedeon Richter and Actavis, Figure 19) is a urea-containing dopamine D3/D2 receptor partial agonist, acting as atypical antipsychotic for treating schizophrenia and bipolar disorders.94,95 Cariprazine received FDA approval on September 2015. It is highly metabolized by cytochrome CYP3A4 with the formation of active metabolites and, to a limited extent, by CYP2D6.96 Cariprazine displays a mean half-life of 2–5 days (1.5–12.5 mg dose). It generates two clinically relevant metabolites by modification of its urea moiety: desmethyl-cariprazine and didesmethyl-cariprazine, this latter displaying an extended half-life with respect to cariprazine.97
5.6. Telcagepant.
Telcagepant (36, code name MK-0974, Figure 19) was a urea containing oral calcitonin gene-related peptide (CGRP) receptor antagonist launched by Merck as an investigational drug for the treatment and prevention of migraine; it was the first orally available drug in this class.98 A Phase IIa clinical trial evaluating telcagepant for the prophylaxis of episodic migraine was halted on March 26, 2009 after the observation of a significant increase of serum transaminase levels upon treatment.99 MK-0974 undergoes significant oxidative metabolism (CYP3A) in monkey intestinal microsomes. The drug is mainly metabolized as pyridine N-oxide, and its metabolism is slower in rat than in monkey intestinal microsomes.100
5.7. Zileuton.
Zileuton (37, Zyflo, Cornerstone Therapeutics Inc., Figure 19) is a benzothiophene N-hydroxyurea. It is an inhibitor of 5-lipoxygenase (5-LOX), and it alleviates allergic and inflammatory states by suppressing leukotriene biosyn-thesis.101,102 Zileuton inhibits 5-LOX by coordinating the iron ion in the active site, and it also displays weak reducing properties. Zileuton and its N-dehydroxylated metabolite are oxidatively metabolized by CYP450 isoenzymes 1A2, 2C9, and 3A4. Interactions of zileuton with other drugs are related to inhibition of CYP1A2.103
5.8. Celiprolol.
Celiprolol (38, brand names Cardem, Selectol, Celipres, Celipro, Celol, Cordiax, Dilanorm, Acer Therapeutics, Figure 19) is a β-blocker characterized by a unique pharmacologic profile: it behaves as a β1-andrenoceptor antagonist with partial β2 agonist activity. Based on these properties, it can be defined as a selective adrenoreceptor modulator with antihypertensive and antianginal properties.104 Recently, the unique properties of celiprolol prompted investigation into its use for the treatment of a rare connective tissue disorder, namely Ehlers-Danlos syndrome.105 Celiprolol is minimally metabolized, with only a very low percentage of a dose being excreted.106
5.9. Carmofur.
Carmofur (39, HCFU, 1-hexylcarbamoyl-5-fluorouracil, Figure 19) is a pyrimidine analogue used as an antineoplastic agent, and it is an orally available lipophilic-masked derivative of 5-fluorouracil. Its carbamoyl moiety is cleaved in vivo to release 5-FU. Carmofur has been employed for the therapy of colorectal cancer,107 although causing delayed leukoencephalopathy.108 Significant side effects and no survival advantage in patients with stage II hepatocellular carcinoma led to clinical trial discontinuation.109
5.10. Lenvatinib.
Lenvatinib (26, Figure 19) is a multi-kinase urea-based inhibitor targeting VEGF receptors 1–3, fibroblast growth factor receptors 1–4, PDGFR α, and RET and KIT proto-oncogenes.110–112 Lenvatinib is approved for treating radioiodine-refractory differentiated thyroid cancer, and combined with Everolimus, it is used to treat advanced renal cell carcinoma.113 Moreover, lenvatinib is under study for the treatment of hepatocellular carcinoma. The main metabolic products isolated from human liver microsomes derive from demethylation, decyclopropylation, O-dearylation, and N-oxidation.114 CYP-related metabolism was reported in human, monkeys, dogs, and rats, while non-CYP-mediated metabolism, mainly ascribable to the action of aldehyde oxidase, has been found in monkeys and humans.115
5.11. RG7112.
RG7112 (40, Figure 19) is a urea-containing small-molecule acting as an MDM2 antagonist. MDM2 downregulates the tumor suppressor p53.116 RG7112 underwent a phase I clinical trial in patients with hematologic malignancies. The main goal of the study was the estimation of the posology and safety profile. The secondary goals involved the assessment of pharmacokinetics, pharmacodynamics, and preliminary clinical efficacy.117 RG7112 is mainly metabolized by CYP3A4/5 and also behaves as a moderate inhibitor of CYP3A4/5 and P-glycoprotein (P-gp).118
6. PROCEDURES FOR THE SYNTHESIS OF UREA DERIVATIVES
The urea functionality is present in drug molecules, agro-chemicals, resins, and dyes. For this reason, a variety of synthetic methodologies for the preparation of urea derivatives have been developed (Scheme 1). The classical approach for the preparation of urea derivatives involves reagents such as phosgene or its derivative, triphosgene. Urea synthesis typically proceeds through an isocyanate intermediate, which upon reaction with a second amine forms the urea derivatives (Scheme 1). Over the years, a series of environmentally friendly phosgene substitutes were developed in order to overcome the hazardous nature of phosgene. In particular, carbonates, N,N′-carbonyldiimidazole, 1,1′-carbonylbisbenzotriazole, S-methylthiocarbamate, formamides, and chloroformates have been utilized for the synthesis of urea derivatives. Also, Curtius, Lossen, and Hofmann rearrangements have been employed to generate urea derivatives through the isocyanate intermediates.
Scheme 1.
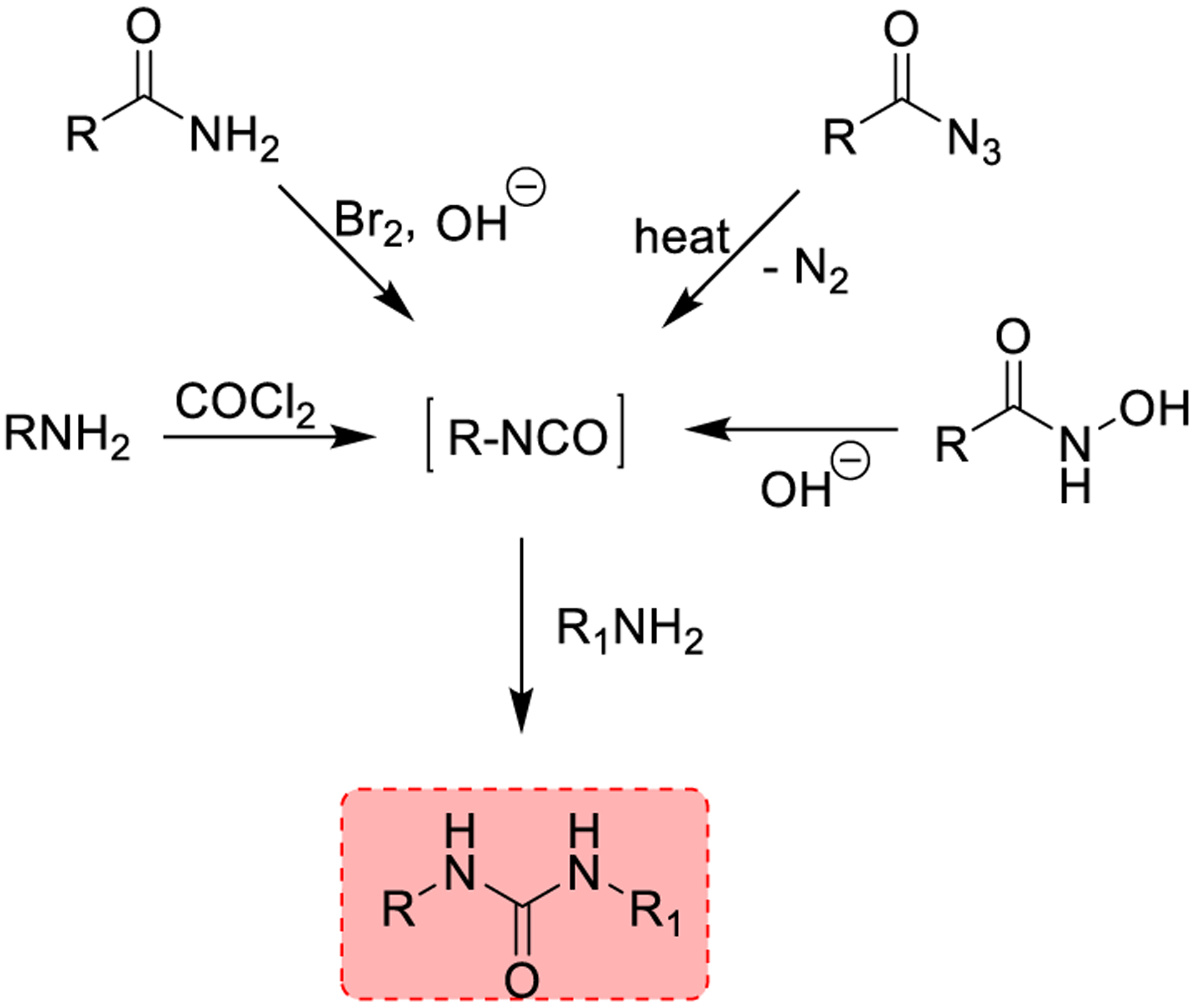
Traditional Methodologies for the Synthesis of Ureas
More recently, carbon monoxide has been utilized as a reliable alternative to phosgene. Accordingly, catalytic oxidative carbonylation employs amines, an oxidant, and carbon monoxide as starting materials to provide ureas. These reactions are appealing in terms of atom economy standards, with the only byproducts being the reduced form of the oxidant. Various transition metal catalysts including Pd, Co, Ni, Ru, Mn, and Au have been employed to afford urea derivatives. However, outcomes varied and often reaction conditions were harsh. As a result, complex mixtures of ureas, oxamides, and formamides were obtained.
6.1. Synthesis of Urea Derivatives Using Phosgene or Phosgene Equivalents.
The reaction of amines with phosgene represents the most traditional methodology for the generation of urea derivatives. This methodology is commonly preferred for the generation of symmetrical ureas, but also unsymmetrical urea derivatives can be prepared efficiently. In general, amines react with phosgene in the presence of a base to provide the desired isocyanate intermediates.119,120 Subsequent reactions of the isocyanates with diverse amine nucleophiles provide N,N-disubstituted or N,N,N′-trisubstituted unsymmetrical urea derivatives. This methodology is widely employed in drug discovery and pharmaceutical research since it represents a convenient method to generate urea derivatives.5 A synthesis of a triazole urea derivative containing a β-lactam ring is shown in Scheme 2. Beta-lactam derivative 41 was reacted with phosgene and 1,2,4-triazole (42) to provide triazole urea 43, a potent and selective monoacylglycerol lipase (MAGL) inhibitor.121
Scheme 2.
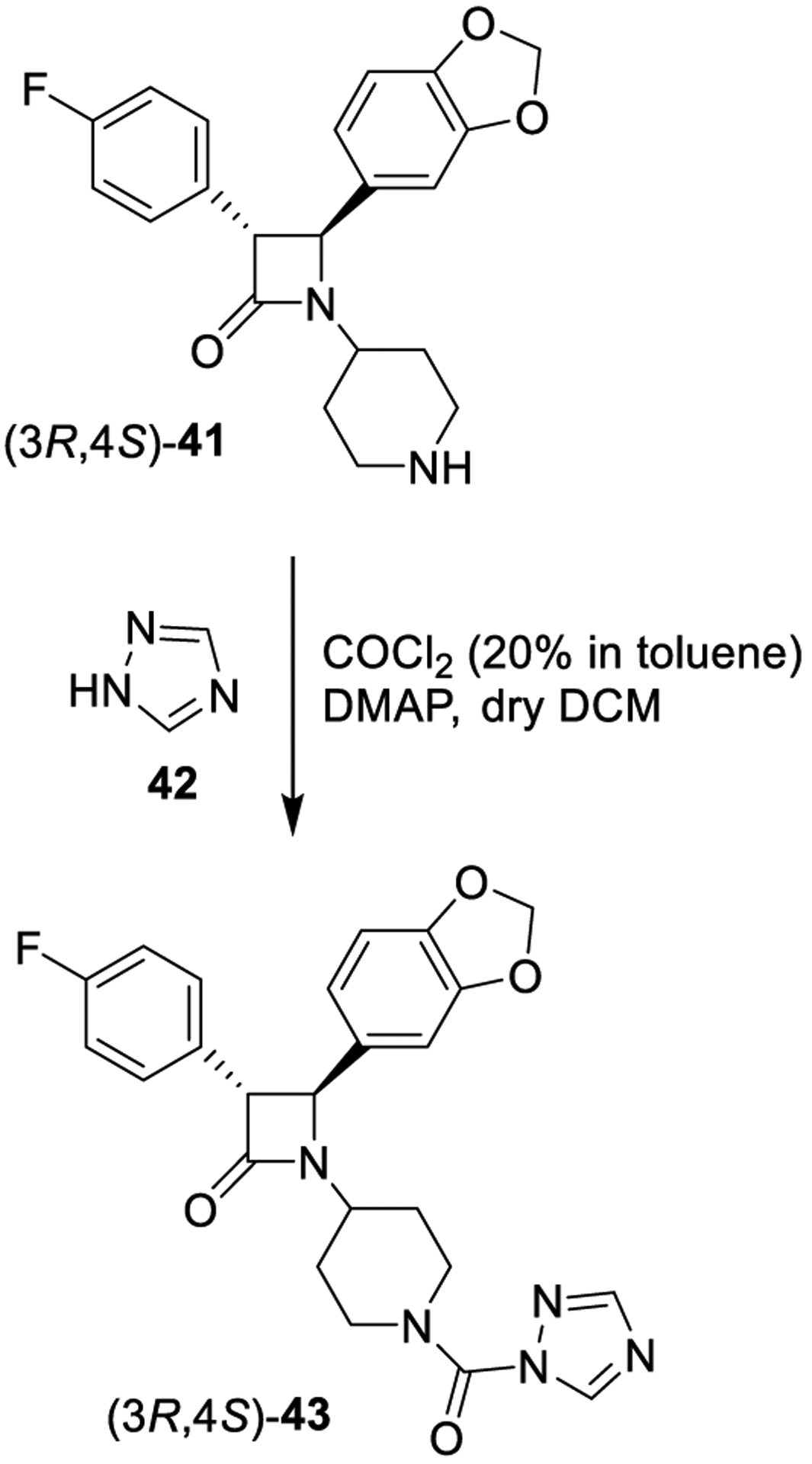
Formation of Urea-Containing MAGL Inhibitor 43 Using Phosgene
Bis(trichloromethyl)carbonate (46, BTC, or triphosgene) is a crystalline and stable solid, allowing safe handling. For this reason, it has been considered a safer replacement to phosgene.5 However, the vapor pressure is sufficiently high to easily result in toxic concentrations, therefore safety concerns are also associated with its prolonged use.122,123 The use of triphosgene in the synthesis of urea derivative 47, a potent antitrypanosomal agent, is shown in Scheme 3.124 Aniline derivative 44 was reacted with BTC in the presence of Et3N. The resulting intermediate was reacted with amine 45 to provide 47.
Scheme 3.
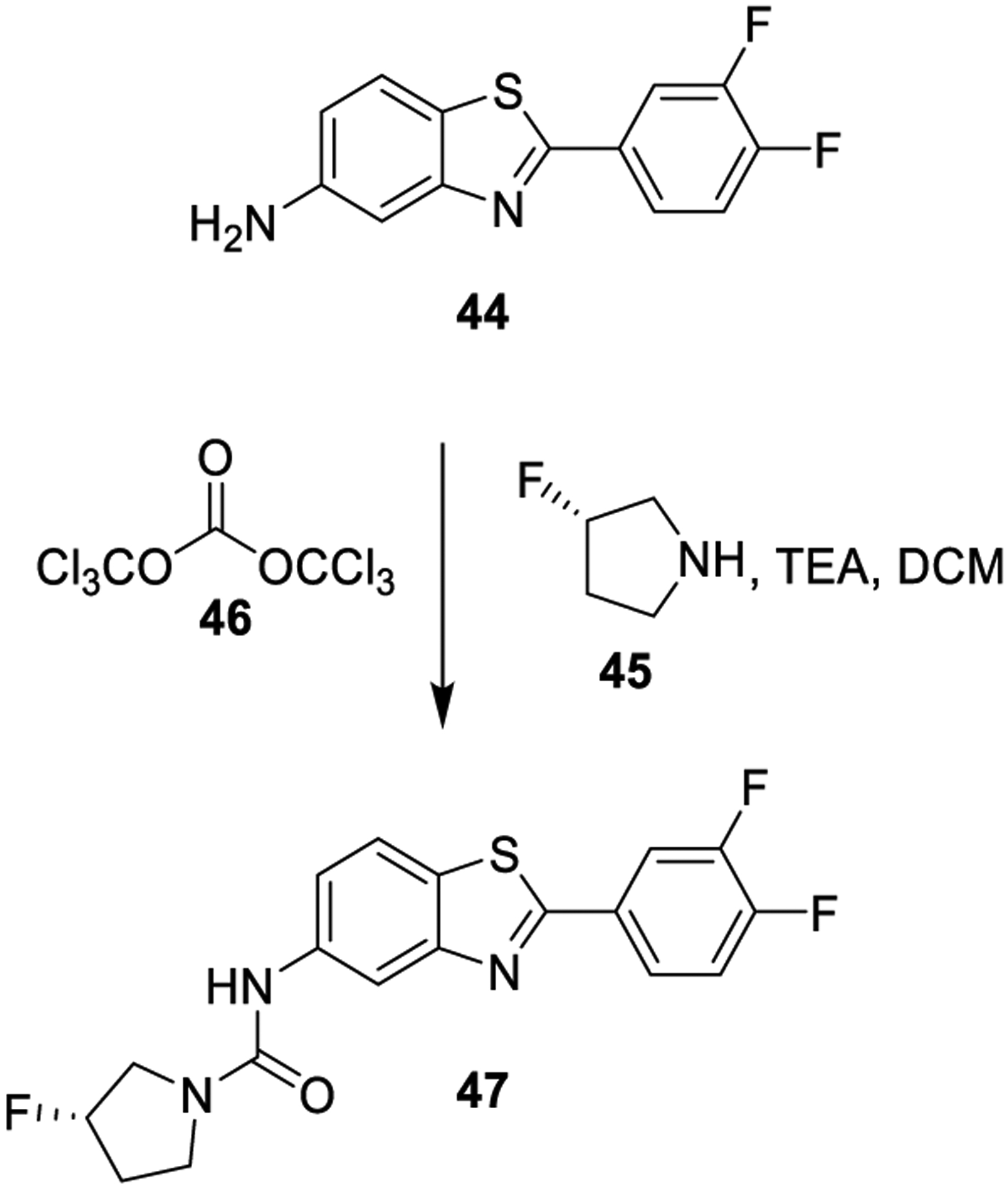
Formation of the Urea Derivative 47 Using Triphosgene
6.2. Synthesis of Urea Derivatives via Safer Phosgene Substitutes.
N,N′-Carbonyldiimidazole (50, CDI) is a widely exploited alternative to phosgene reagent for the preparation of ureas. CDI is a crystalline solid and is commercially available. CDI does not produce chlorine or chlorinated byproducts and is thus a safer and less toxic alternative to the use of BTC and phosgene for the generation of biologically active unsymmetrical ureas.125,126 As shown in Scheme 4, CDI has been used for the synthesis of urea derivative 51, a potent inhibitor of the DCN1-UBE2M interaction, starting from amine 48 and aniline 49.127
Scheme 4.
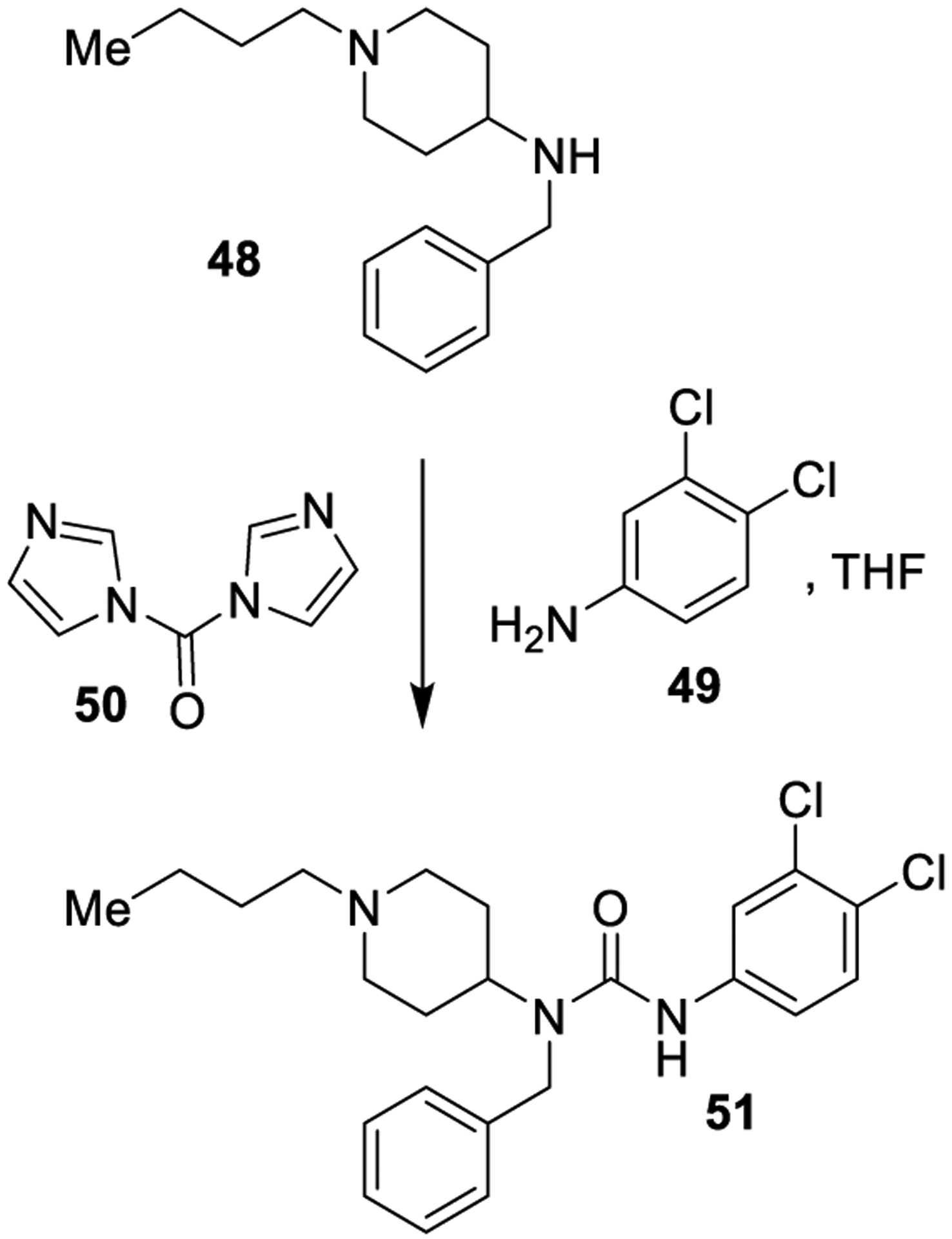
Formation of Urea Derivative 51 Using CDI (50)
1,l′-Carbonylbisbenzotriazole (CBT, 53) was proposed as a safer and milder alternative to phosgene for the preparation of unsymmetrical urea derivatives. As an example, compound 53 reacts with secondary amine 52 to provide the corresponding carbamoyl benzotriazole derivative 54. Intermediate 54 is subsequently treated with a different secondary amine (55), to smoothly provide tetrasubstituted unsymmetrical urea derivative 56 (Scheme 5). Steric hindrance of the substituents placed on the secondary amine is the main factor influencing reaction outcome. For cyclic and aliphatic amine derivatives, the reaction proceeds at room temperature and provides good yields, while it may require higher temperatures or reflux condition for aromatic amines.128
Scheme 5.
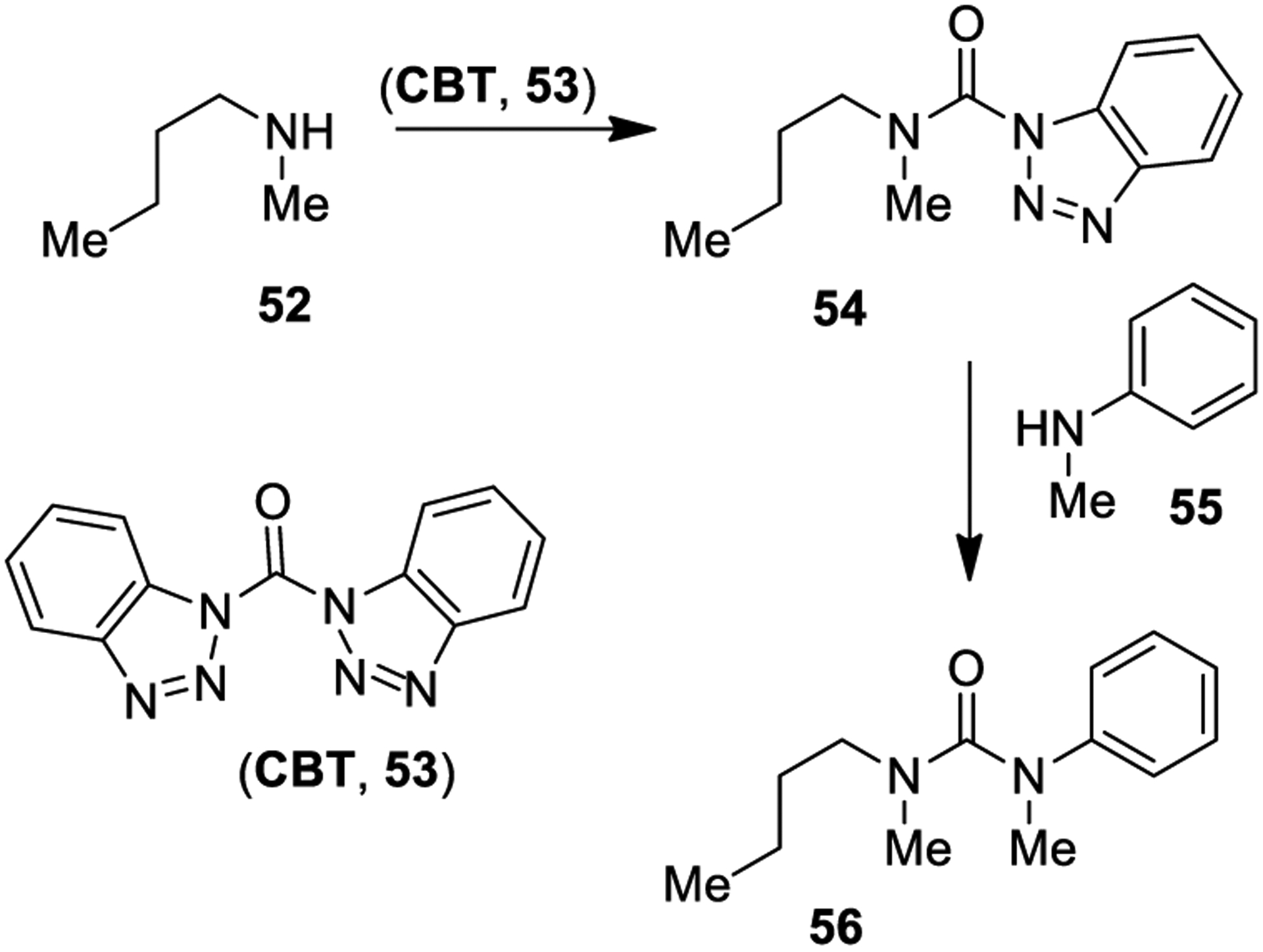
Formation of Urea Derivatives Using 1,l′-Carbonylbisbenzotriazole (53)
The aminolysis of a urethane derivative is a well-known procedure for the synthesis of ureas. Urethanes are usually generated from carbonates or chloroformates.129,130 Urethanes can also be prepared by reacting amine derivatives with carbon dioxide or by utilizing other safer and more recent methodologies.131 Urethanes react with amines with varying efficacy, depending upon the nature of the leaving alkoxide group.5,129,132
As an example, addition of one equivalent of the indoline 58 to phenylcarbamate 57 in the presence of triethylamine followed by heating to reflux resulted in almost quantitative conversion to the urea 59 (Scheme 6). The product was isolated in 78% yield and >99% purity through the addition of water and collection by filtration.129
Scheme 6.
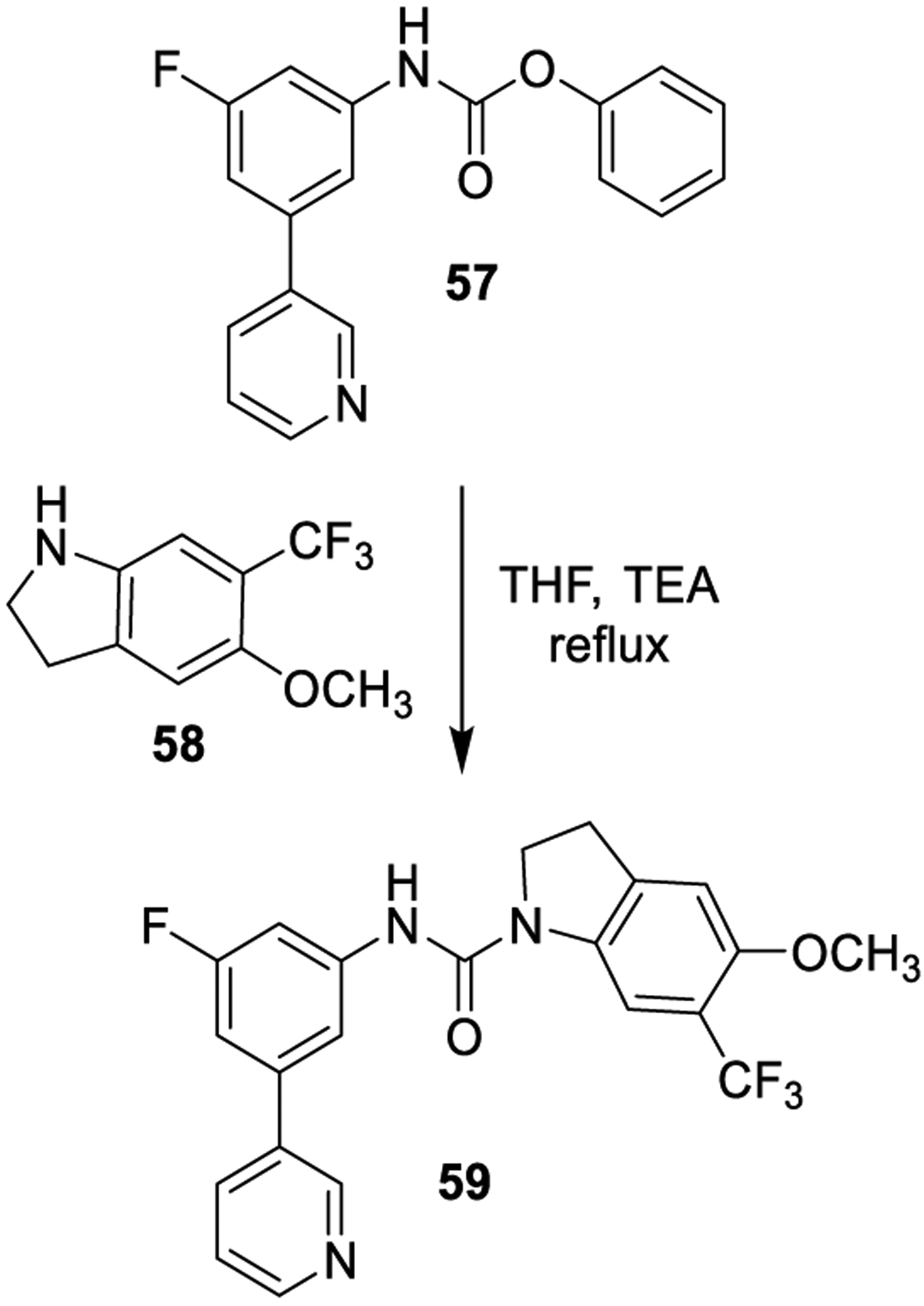
Formation of Urea Derivatives through Aminolysis of Carbamates
For the effective synthesis of unsymmetrical tetrasubstituted ureas, Batey and co-workers formed imidazolium salts and developed effective carbamoyl transfer reagents.133 As shown in Scheme 7, carbamoylimidazolium salts are readily synthesized by reaction of secondary amines with CDI and successive alkylation with iodomethane to form salt 61.131 Addition of primary or secondary amines to these salts in the presence of triethylamine efficiently provided disubstituted or tetrasubstituted ureas, respectively. The resulting byproducts (namely N-methylimidazole and triethylamine hydrochloride salt) could be washed away by treatment of the organic layer with a dilute acid. The protocol was not successful for poorly nucleophilic aromatic amines. However, their conversion to anilide anions, upon treatment with KHMDS or n-BuLi led to more reactive nucleophiles. Reaction of these anions with imidazolium salts smoothly provided the corresponding ureas in good yields.134
Scheme 7.
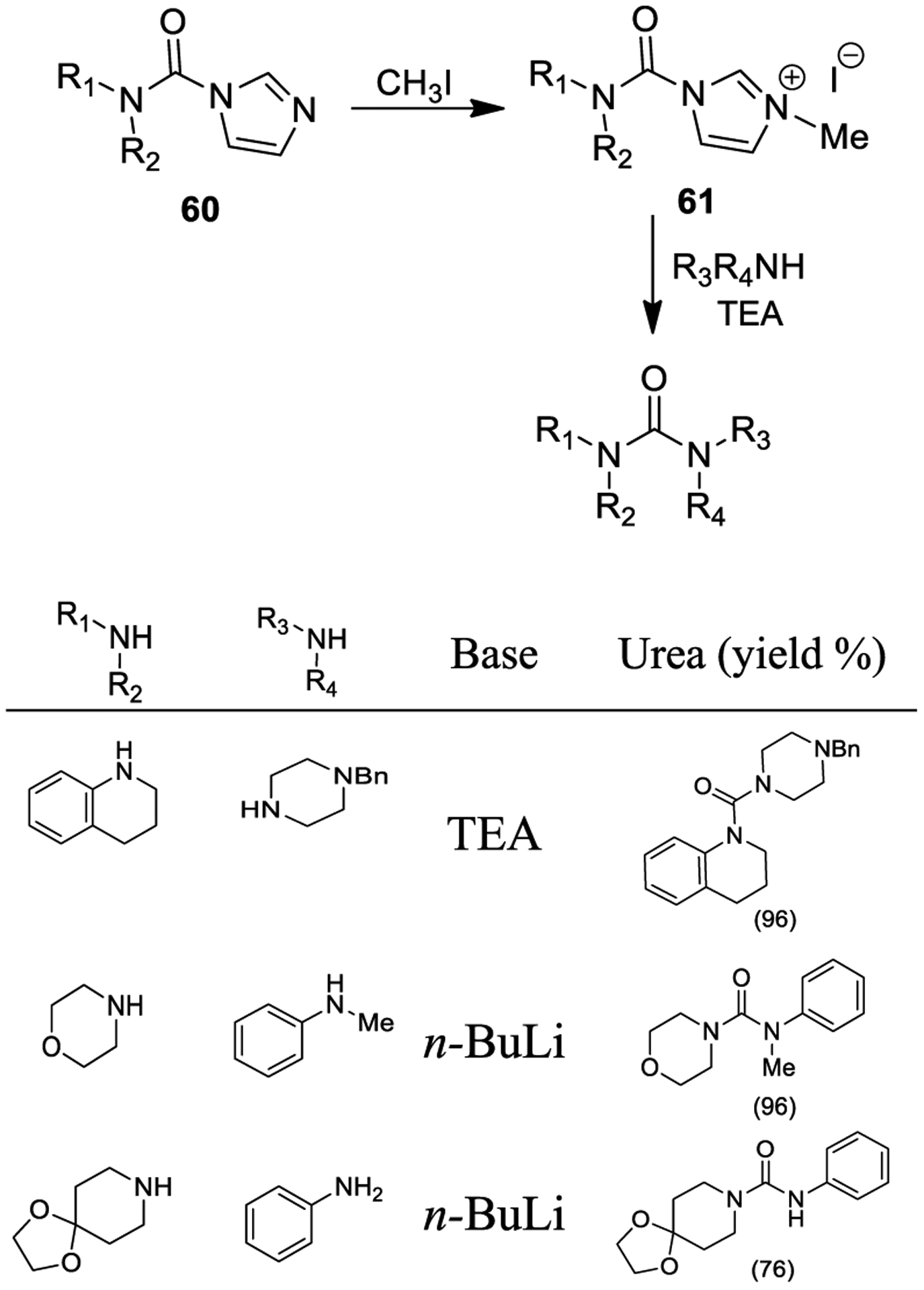
Synthesis of Urea Derivatives Using Carbamoylimidazolium Salts
Artuso and co-workers validated S,S-dimethyl dithiocarbonate (DMDTC, 63) as a valuable replacement of phosgene for the in-water carbonylation of amines for the convenient generation of mono-, di-, and trisubstituted urea derivatives (Scheme 8). Symmetrical disubstituted ureas were prepared using a 1:2 molar ratio of DMDTC and amine, respectively, at 60 °C. Unsymmetrical urea derivative 65 was synthesized in two steps involving: (i) formation of the S-methyl N-alkylthiocarbamate derivative 64 (room temperature); and (ii) reaction of this intermediate with butylamine (50–70 °C). This protocol provided urea derivatives in excellent yields and purity. The protocol allowed for easy and straightforward isolation of reaction products.135
Scheme 8.
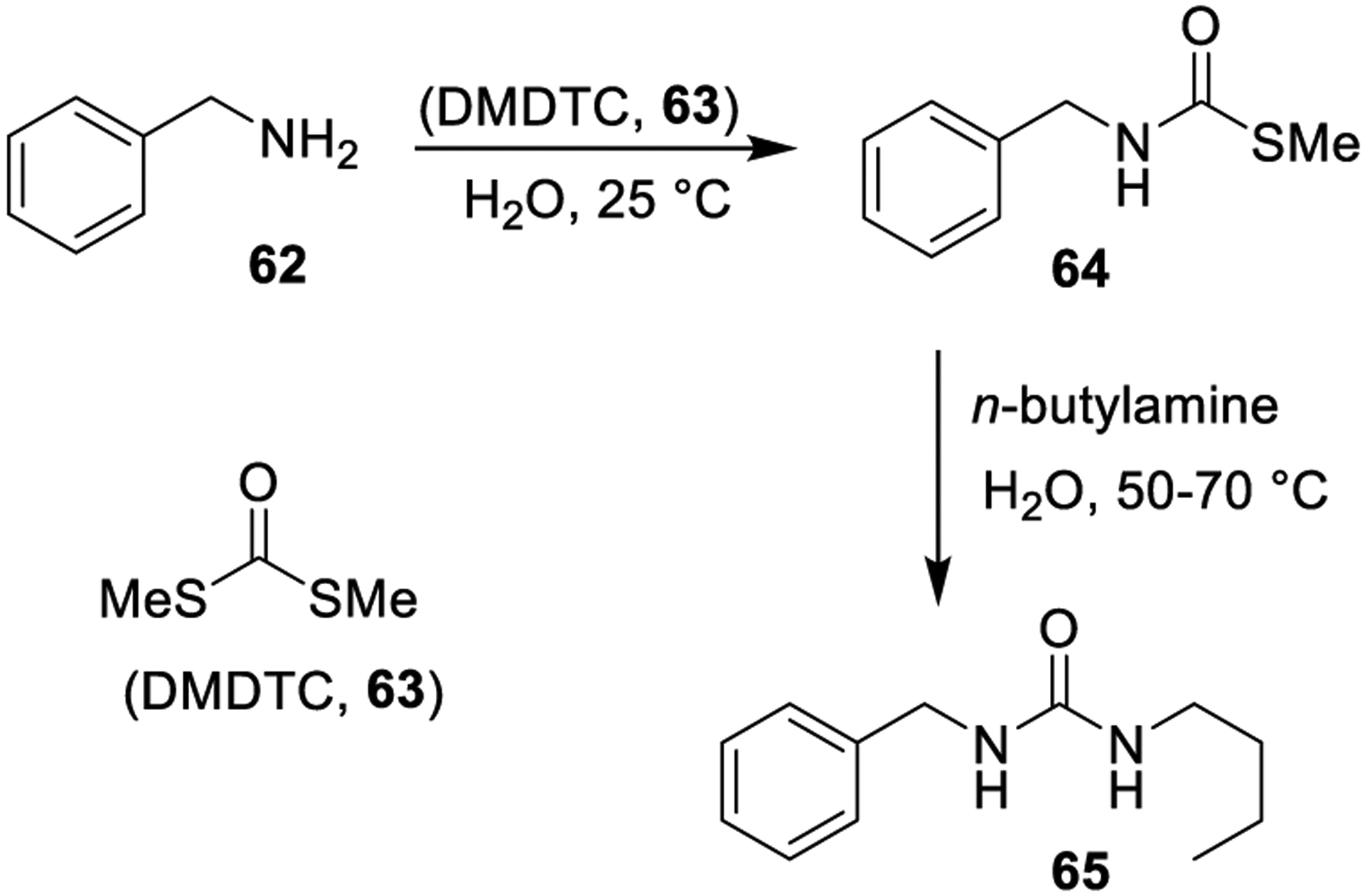
Synthesis of Urea Derivatives Using S,S-Dimethyl Dithiocarbonate (63)
As part of safer phosgene substitutes to be exploited under mild reaction conditions, Yoon and co-workers examined the use of phenyl 4,5-dichloro-6-oxopyridazine-1(6H)-carboxylate (68). Compound 68 was synthesized from 4,5-dichloropyridazin-3(2H)-one (66) and phenyl chloroformate (67) in the presence of triethylamine (Scheme 9). The compound showed stability even at high temperature. Unsymmetrical ureas were synthesized through this protocol in a one pot fashion starting from compound 68. Upon reaction of anilines with 68 to form the corresponding N-aryl carbamates, aliphatic amines were added and refluxed until disappearance of the carbamate to efficiently provide N-aryl-N-alkyl urea derivatives.136
Scheme 9.
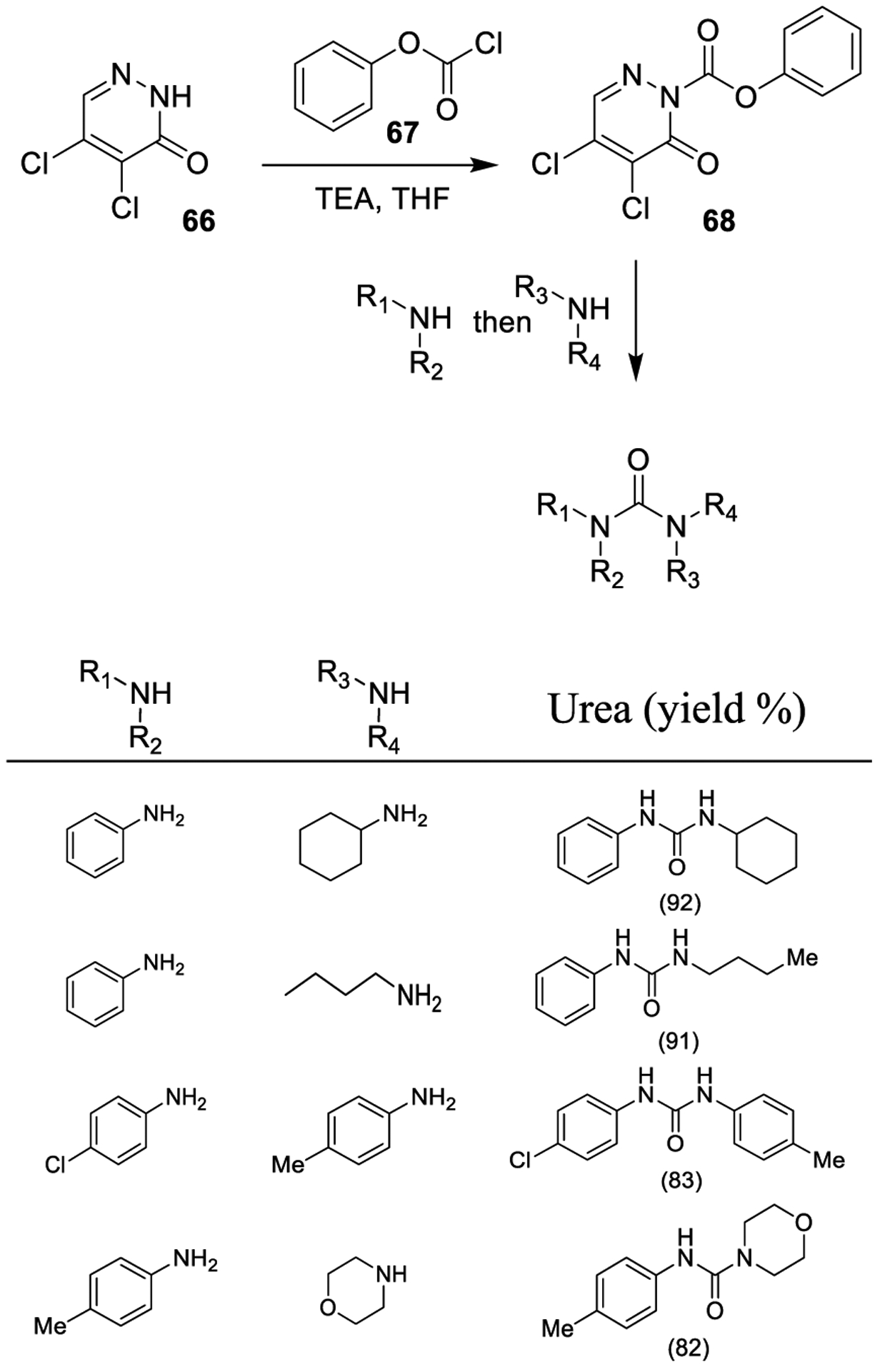
Synthesis of Urea Derivatives Using Phenyl 4,5-Dichloro-6-oxopyridazine-1(6H)-carboxylate (68)
Padiya and co-workers described a useful protocol for synthesis of urea derivatives (Scheme 10) in water. It turns out, CDI reacts with amines in aqueous medium to afford the corresponding N-substituted carbonylimidazolides in good yields. Carbonylimidazolides (such as compound 69) derived from primary amines react in situ with an amine nucleophile (e.g., compound 70) providing the corresponding urea 71. Compounds 72 and 73 are other examples of urea derivatives which can be efficiently prepared through this procedure. Due to product precipitation from the reaction mixture, a high degree of purity was observed.137
Scheme 10.
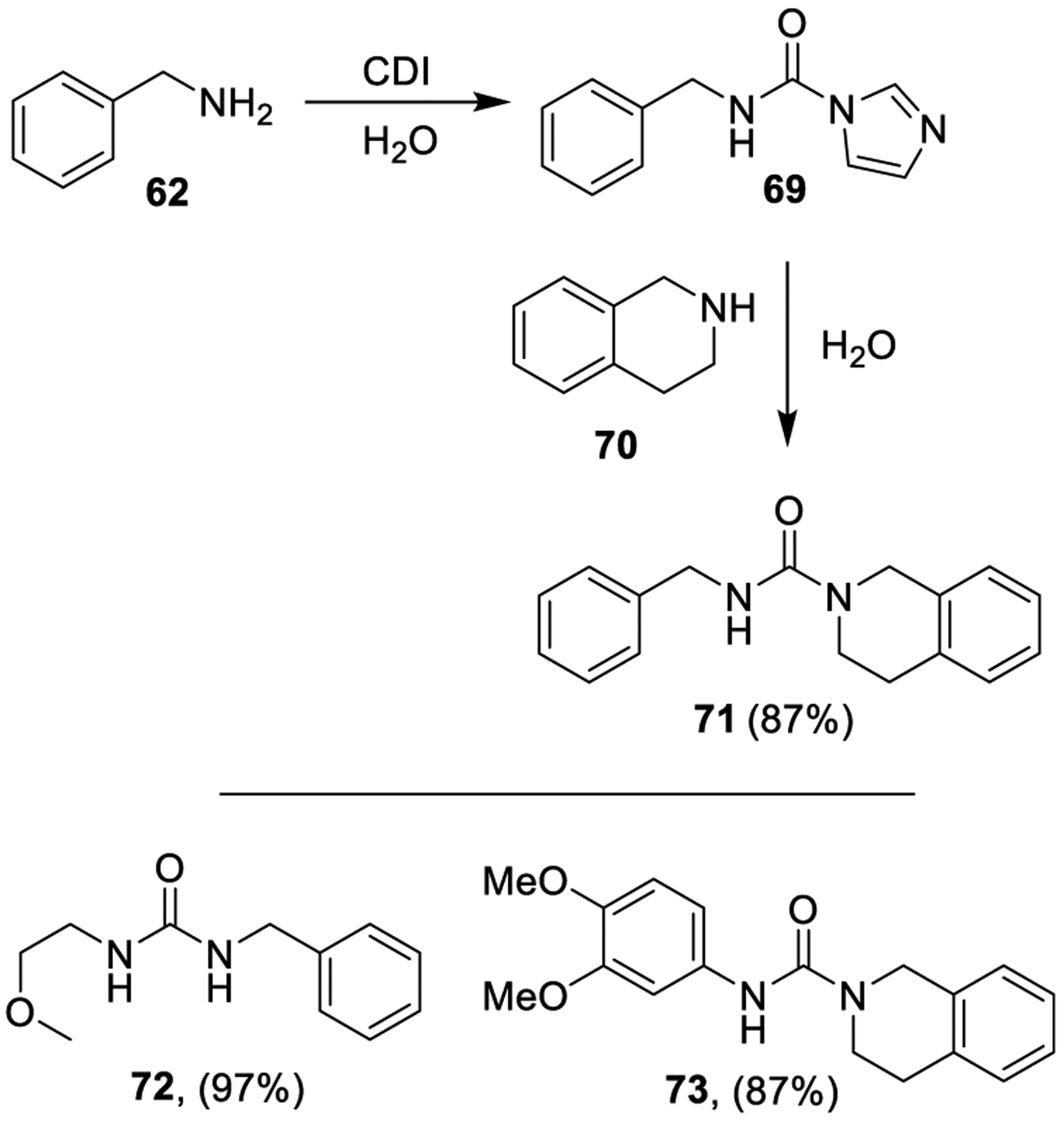
Synthesis of Urea Derivatives 71–73 Using CDI in Water Medium
Recently, Moroz and co-workers investigated bis(2,2,2-trifluoroethyl) carbonate (76) as a convenient alternative to traditional carbonates. Carbonate 76 was prepared by reaction of triphosgene (75) and trifluoroethanol (74). The reaction provided 76 and its trifluoroethylchloroformate counterpart in a nearly 1:3 ratio. Moreover, trifluoroethylchloroformate could provide 76 by further treatment with trifluoroethanol and triethylamine (Scheme 11).
Scheme 11.
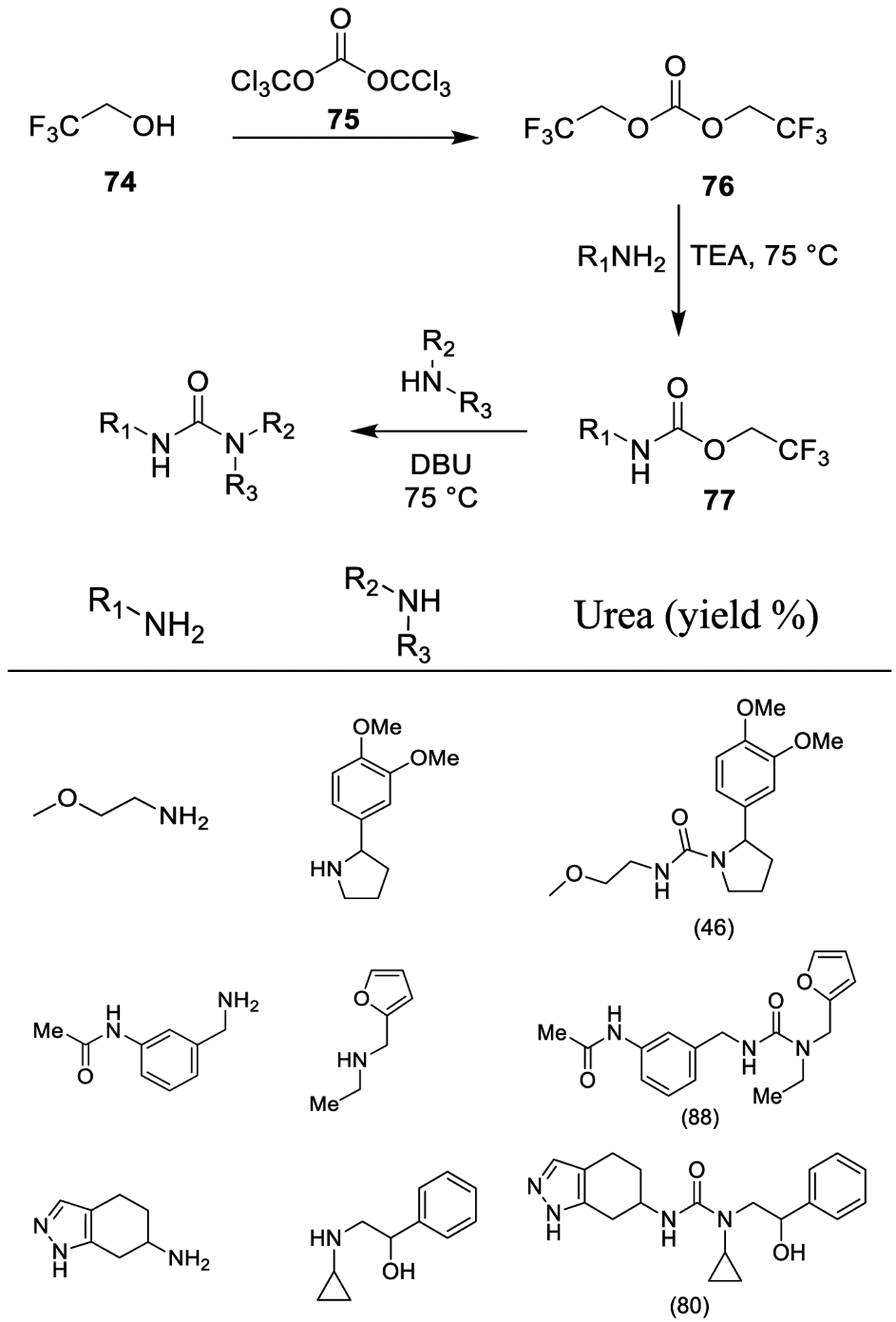
Synthesis of Urea Derivatives Using Bis(2,2,2-trifluoroethyl) Carbonate 76
Trifluoroethyl carbonate 76 smoothly reacts with alkyl amines with high regioselectivity providing trifluoroethyl carbamates (77), rather than symmetrical ureas. Further reaction with amines in the presence of a base provided urea derivatives. This protocol displays a wide scope and utility, since it allowed the straightforward synthesis of ureas in the presence of potentially competing functionalities (e.g., amino, hydroxyl and phenolic groups, pyrazole moieties) (Scheme 11).138
6.3. Urea Synthesis Using Rearrangements.
The Hofmann rearrangement is a common method to access ureas through the formation of their amine precursors. This reaction involves the generation of a primary amine from a carboxamide derivative in the presence of sodium hydroxide and bromine, encompassing the formation of an intermediate isocyanate. The intermediate isocyanate could be converted into urea derivatives by in situ trapping with a suitable amine nucleophile.139 To improve the overall process, a variety of alternative oxidants and different bases arose over years, such as diacetoxyiodobenzene PhI(OAc)2,140 MeOBr,141 N-bromosuccinimide (NBS)-CH3ONa,142 (NBS)-KOH,143 lead(IV) acetate,144 and benzyltrimethylammonium tribromide.145 Hofmann rearrangement has been utilized in the synthesis of 2-pyridylureas as glucokinase activators. As shown in Scheme 12, carboxamide 78 was subjected to Hofmann rearrangement with NBS and potassium hydroxide to provide aniline 79. It was subsequently converted to urea derivative 80, precursor of the target compound 81.146
Scheme 12.
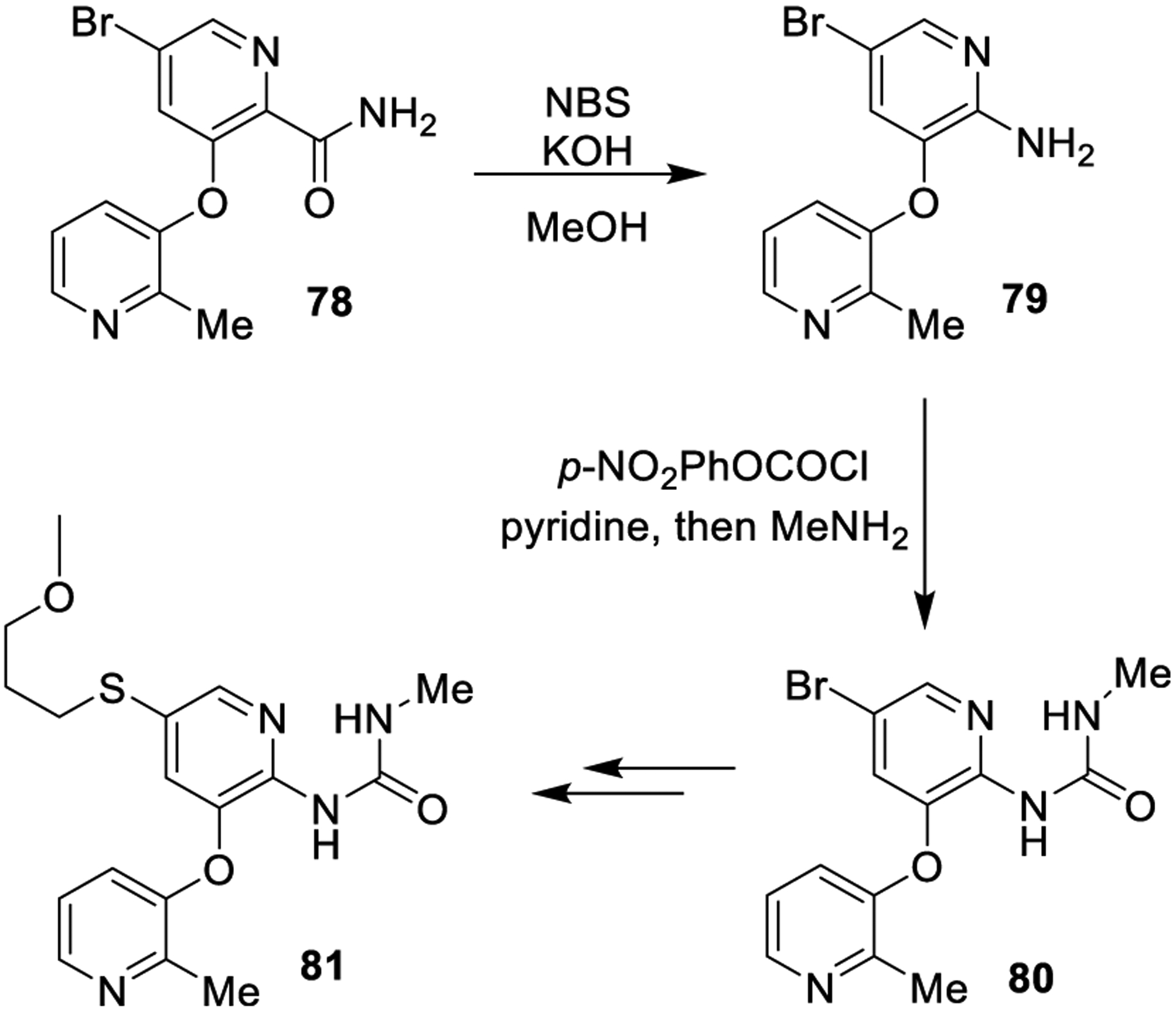
Formation of the Urea Derivative 81 Using a Key Hofmann Rearrangement
Curtius rearrangement of an acyl azide provides the corresponding isocyanate. This can undergo in situ reaction with a variety of nucleophiles, thus affording urea and carbamate derivatives or other N-acyl compounds. A one-pot Curtius rearrangement starting from carboxylic acid derivatives using diphenylphosphoryl azide (DPPA) provides the corresponding isocyanate derivatives for urea synthesis.147–150 The use of Curtius rearrangement for the synthesis of urea derivative 83 is shown in Scheme 13. Compound 83 is a potent inhibitor of human influenza virus replication. Carboxylic acid precursor 82 was reacted with DPPA and triethylamine, and subsequent reaction with morpholine provided urea derivative 83.151
Scheme 13.
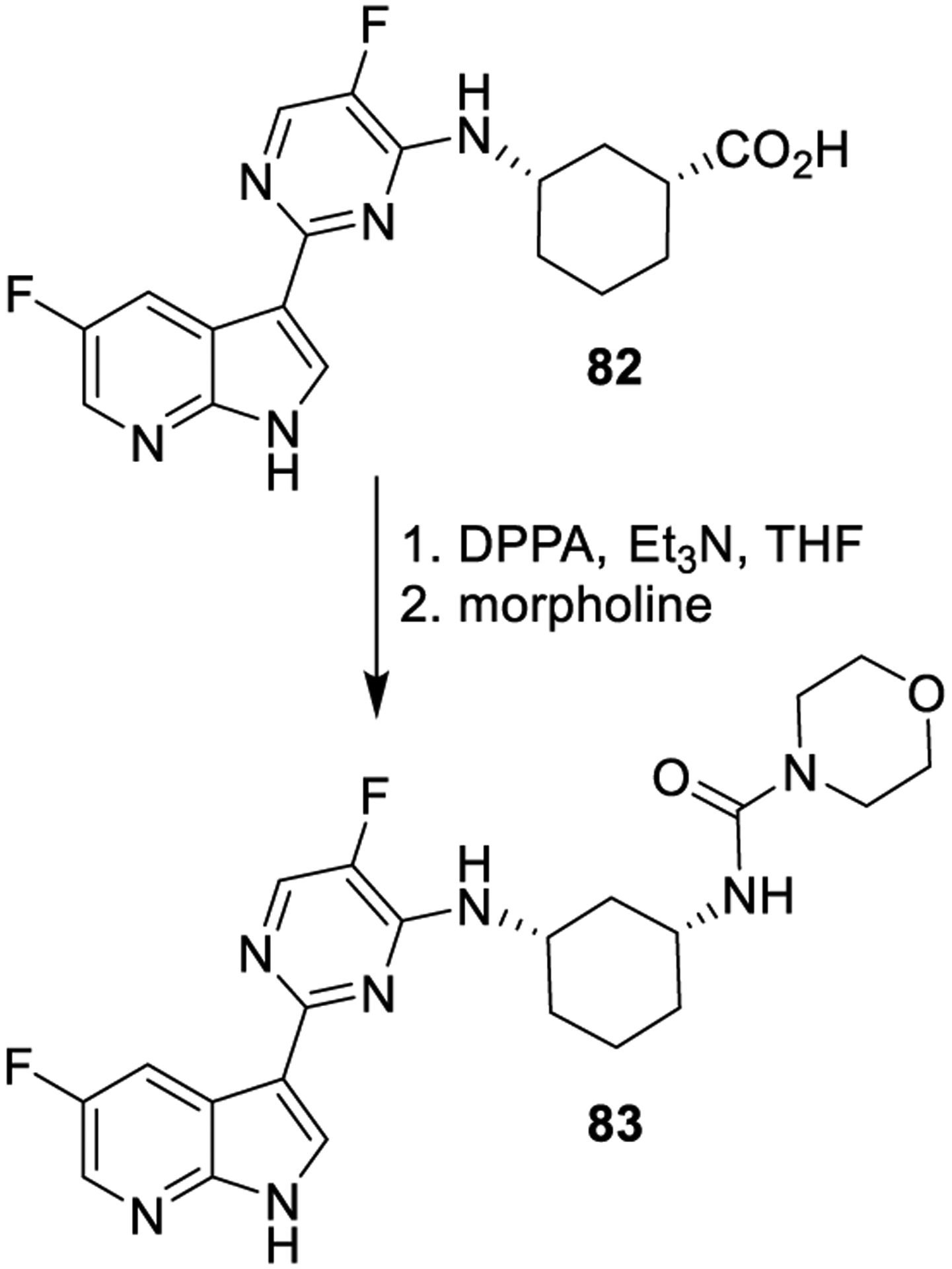
Formation of Urea Derivative 83 Using the Curtius Rearrangement
The Lossen rearrangement is the conversion of hydroxamic acids into isocyanates. The transformation is initiated by exposure of the hydroxamic acid to base or heat. The resulting anion is rapidly converted into the corresponding isocyanate. The isocyanate could then easily provide the desired urea derivatives. As shown in Scheme 14, valine-derived hydroxamic acid 84 was treated with alanine methyl ester 85 in the presence of EDCI, providing the urea derivative 86 in 81% yield.152
Scheme 14.
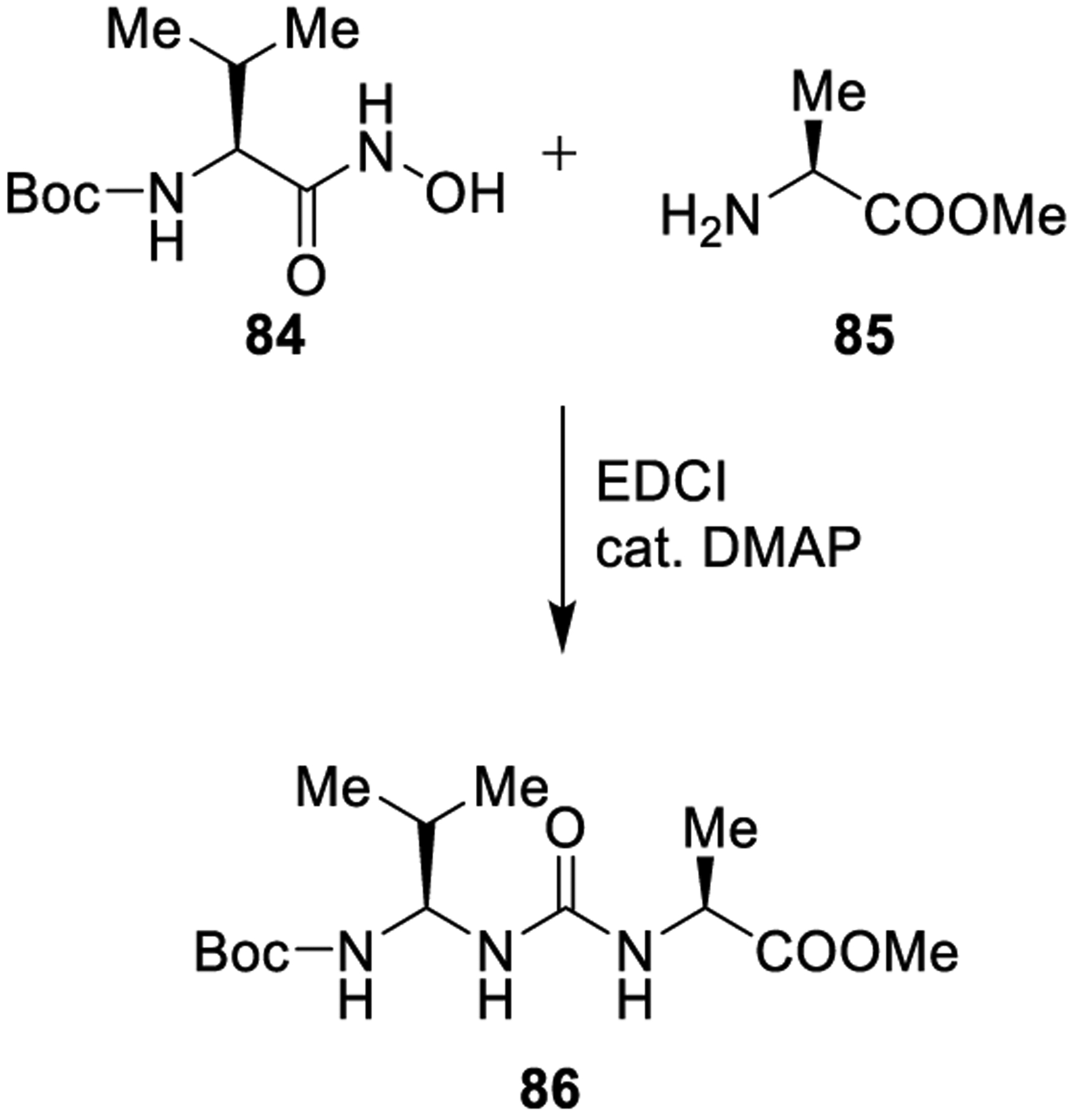
Formation of Urea Derivative 86 Using the Lossen Rearrangement
Dubé and collaborators explored the potential of an inventive CDI-mediated Lossen rearrangement. The proposed protocol is characterized by operational simplicity and mildness, leading to only imidazole and CO2 as the stoichiometric byproducts (Scheme 15). The authors proved the electronic nature of the transformation and provided a wide exploration of the reaction scope. Morpholine trapping of isocyanates quantitatively affords the corresponding urea derivatives. Therefore, this nucleophile was employed to examine the outcome of the protocol employing hydroxamates deriving from aryl, heteroaryl, and aliphatic hydroxamic acids. Diverse ureas were generated in good yields and acceptable purity grades according to this protocol, which also represents a green alternative to traditional Curtius and Hofmann rearrangements.153
Scheme 15.
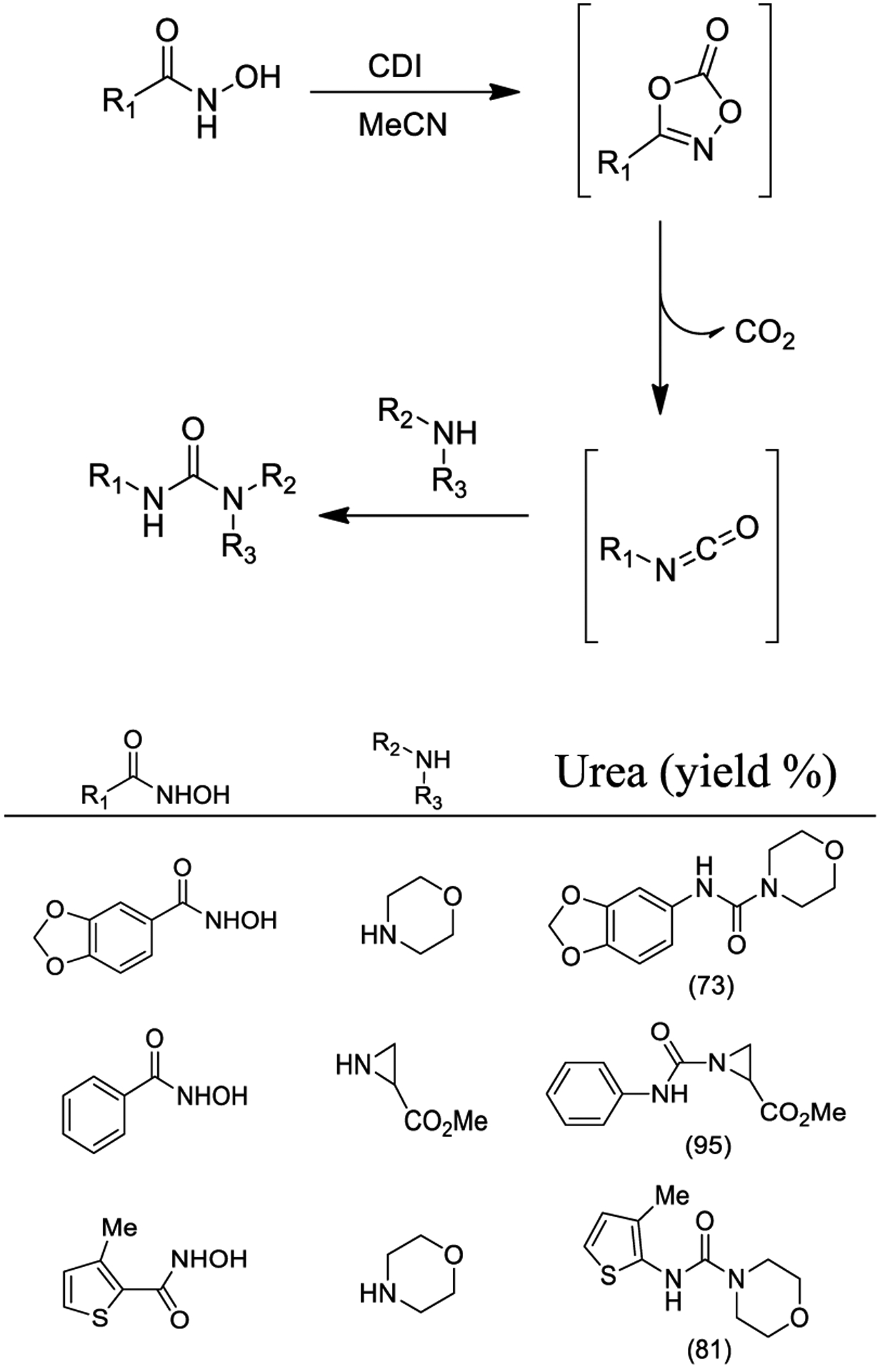
Synthesis of Urea Derivatives Using CDI-Mediated Lossen Rearrangement
Sureshbabu and co-workers reported the ultrasonication-mediated formation of hydroxamates from aromatic acids and N-protected amino acids using 1-propanephosphonic acid cyclic anhydride (87, T3P) as the carboxy activator as shown in Scheme 16. Compound 87 activates hydroxamates, to form isocyanates via a Lossen rearrangement, thus ultimately affording, upon reaction with suitable nucleophiles, the corresponding urea derivatives. The reported methodology was applied to the synthesis of several Nα-protected amino acid hydroxamates and for the preparation of peptidomimetics.154
Scheme 16.
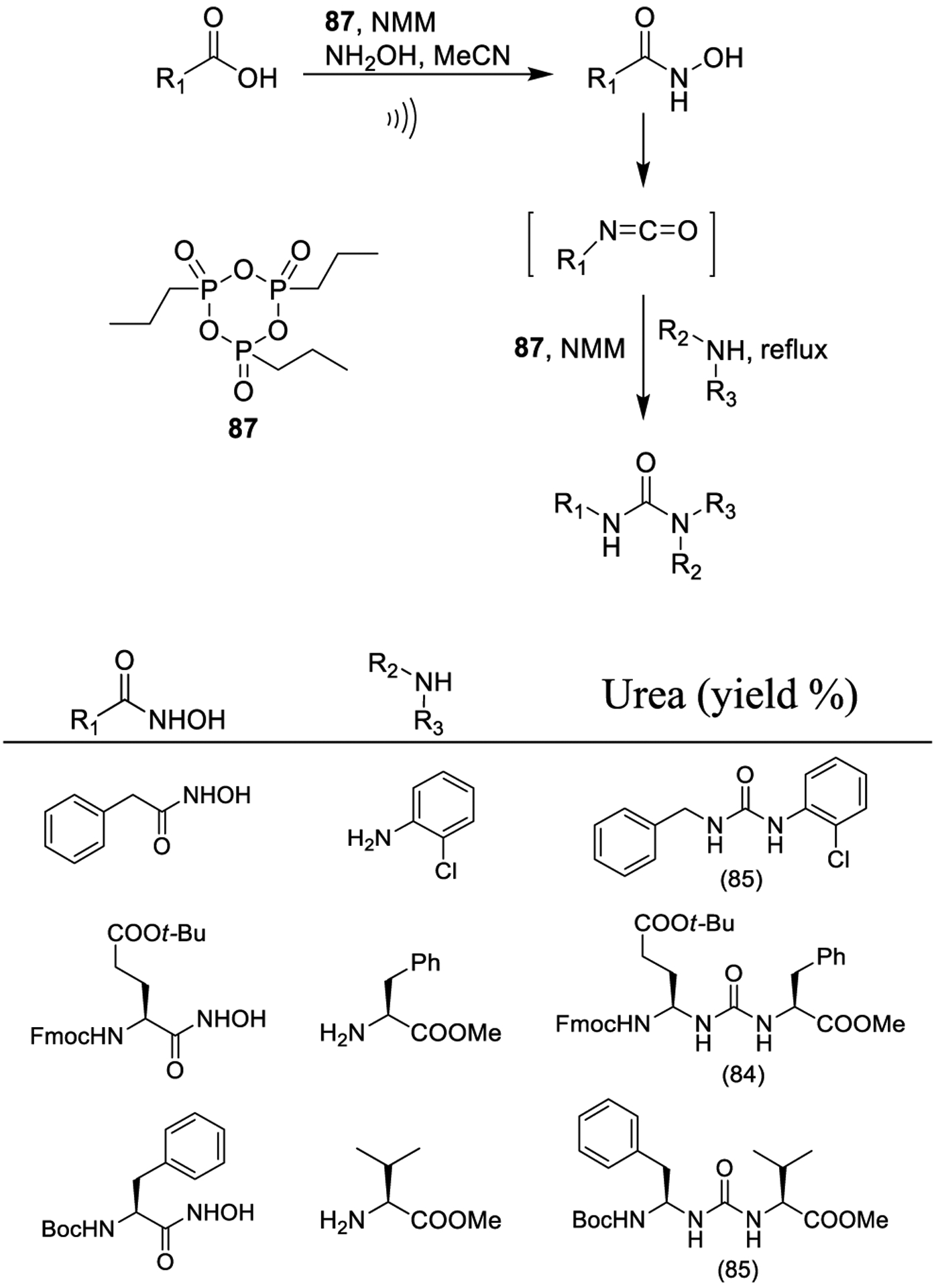
Synthesis of Urea Derivatives Using a Lossen Rearrangement Mediated by 1-Propanephosphonic Acid Cyclic Anhydride (87, T3P)
Mandal and co-workers proposed the use of 2-cyano-2-(4-nitrophenylsulfonyloxyimino)acetate (88, 4-NBsOXY) for both the generation of hydroxamic acids and the subsequent Lossen rearrangement. As shown in Scheme 17, activation of the carboxylic acids with reagent 88 (1 equiv) in the presence of DIPEA and catalytic DMAP followed by the addition of hydroxylamine led to the corresponding hydroxamic acid derivatives. For amino acids, there was no detectable racemization. Urea derivatives were obtained via Lossen rearrangement from the generated hydroxamic acids by reaction of 88 (1 equiv) with DIPEA (0 °C for 90 min) and subsequent addition of amine at room temperature. Interestingly, the side products 4-nitrobenzenesulfonic acid and Oxyma [ethyl 2-cyano-2-(hydroxyimino)acetate] can be used to regenerate 88.155
Scheme 17.
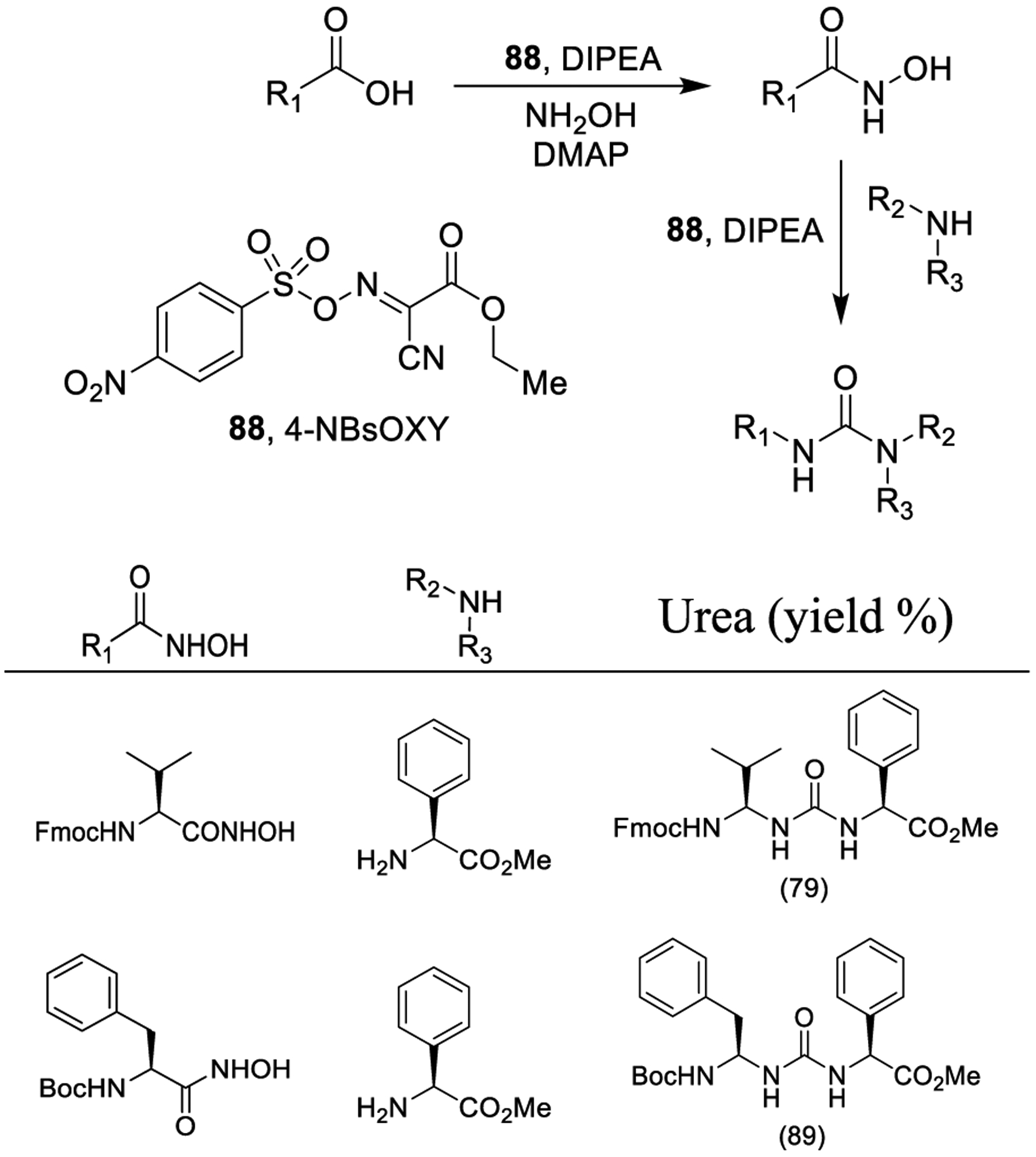
Synthesis of Urea Derivatives Using a Lossen Rearrangement Mediated by 2-Cyano-2-(4-nitrophenylsulfonyloxyimino)acetate (88, 4-NBsOXY)
Lebel and co-worker synthesized aromatic carbamates and urea derivatives from aromatic acids exploiting a Curtius rearrangement in the presence of di-tert-butyl dicarbonate or chloroformates and sodium azide as shown in Scheme 18. The reaction possibly involved the generation of an intermediate azidoformate. Final amine trapping of the acyl azides affords urea derivatives.156
Scheme 18.
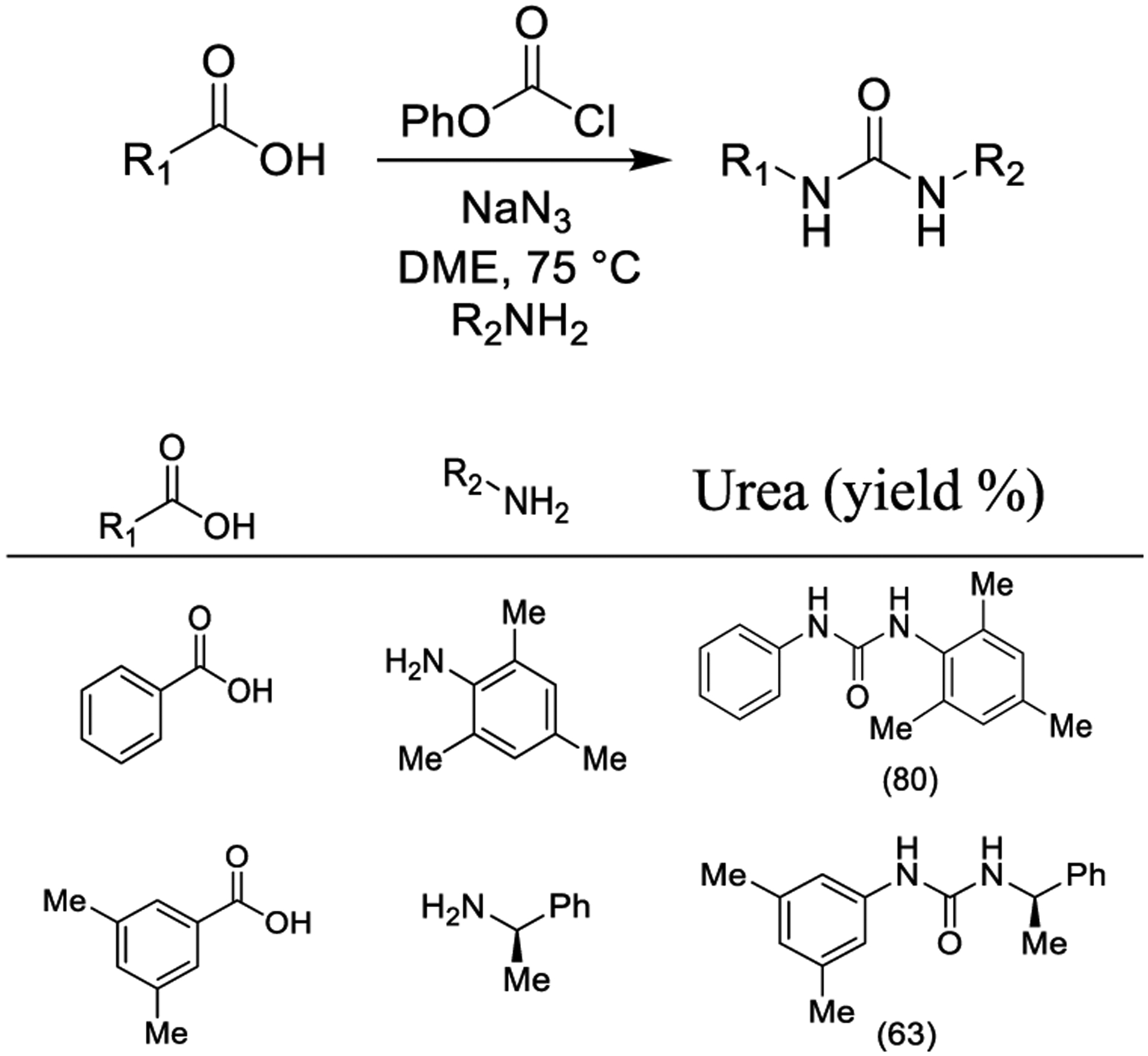
Synthesis of Urea Derivatives Using Curtius Rearrangement via Formation of the Azidoformate
Sureshbabu and collaborators developed a simple one-pot protocol for the preparation of neoglycopeptides and urea-containing peptidomimetics employing a Curtius rearrangement in the presence of Deoxo-Fluor (90, bis(2-methoxyethyl)-aminosulfur trifluoride) and TMSN3 under ultrasonication conditions as shown in Scheme 19. The method could involve four sequential steps: (i) generation of acyl fluoride, (ii) azide formation, (iii) rearrangement into isocyanate, and (iv) reaction with the selected amine to provide urea derivatives. The conditions represent a racemization-free procedure.157
Scheme 19.
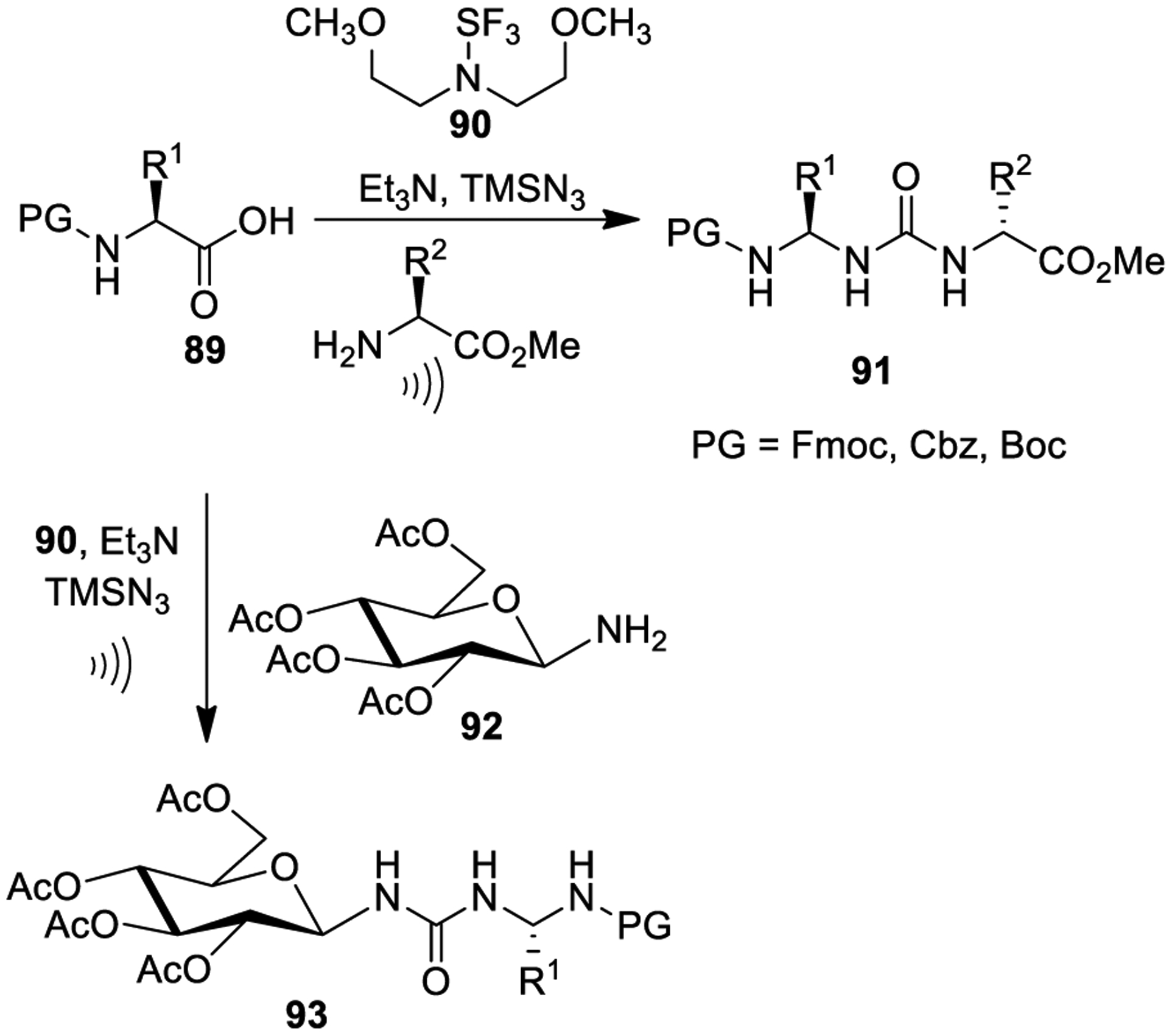
Synthesis of Urea Derivatives 91 and 93 Using Curtius Rearrangement under Ultrasonication Conditions
Thakur and co-workers reported a simple protocol for the preparation of unsymmetrical ureas starting from (hetero)-aromatic carboxylic acids and amines by using DPPA as outlined in Scheme 20. This one-pot, microwave-accelerated method was reported to provide urea derivatives in an extremely rapid fashion (1–5 min) and in excellent yields. The method was applied to the synthesis of cannabinoid 1 and α7 nicotinic receptor ligands (e.g., compound 94, PSNCBAM-1).158
Scheme 20.
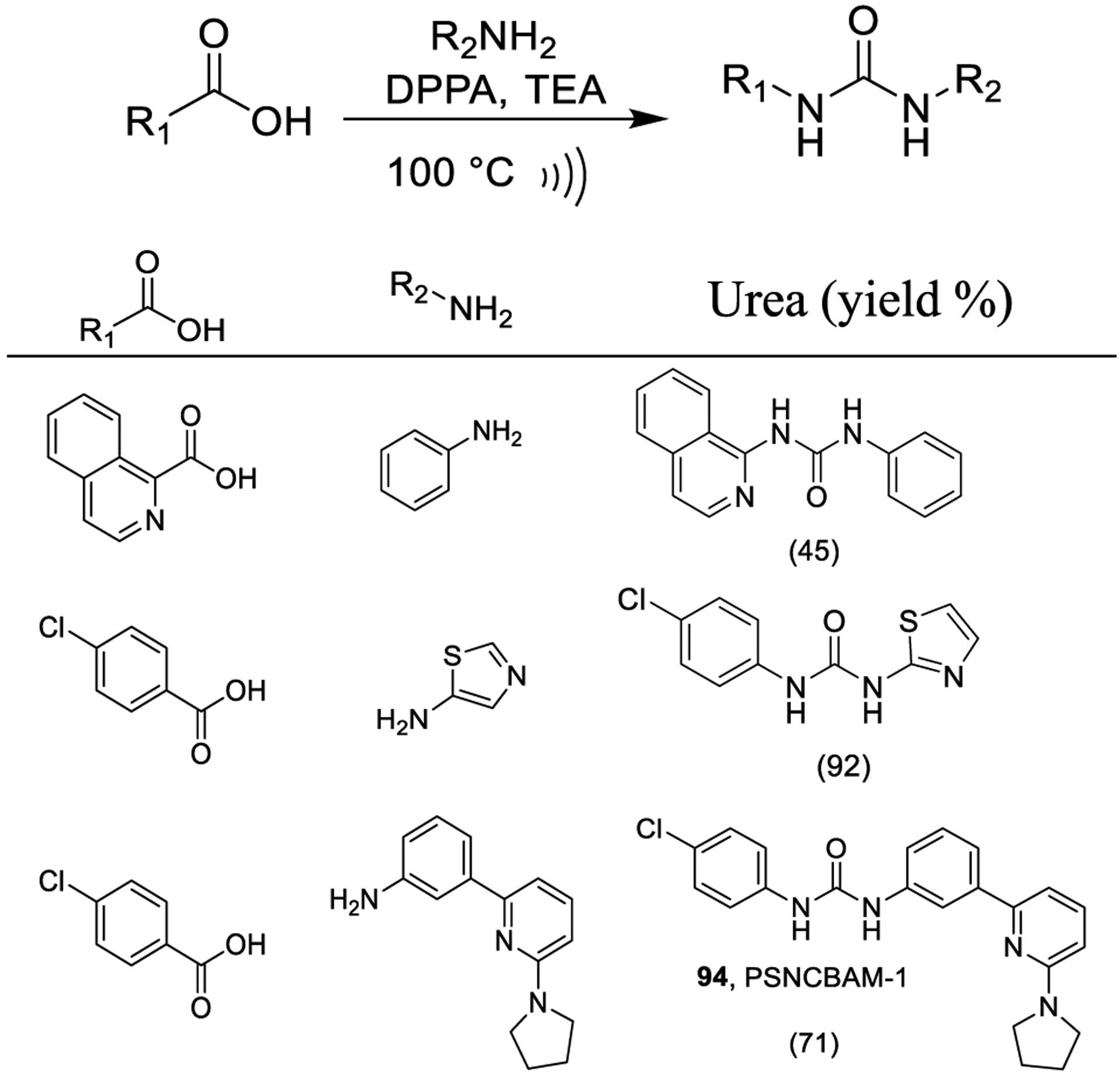
Synthesis of Urea Derivatives Using Curtius Rearrangement under Microwave Conditions
Hu and co-workers reported a Hofmann rearrangement using iodosylbenzene (PhIO) as the oxidizing agent to form the corresponding one-carbon shorter isocyanates from amide derivatives as shown in Scheme 21. Nucleophilic attack of amines finally generated the corresponding urea or carbamate derivatives. Several amides and amines were employed as substrates. Symmetric and asymmetric ureas were also prepared in good to excellent yield.159
Scheme 21.
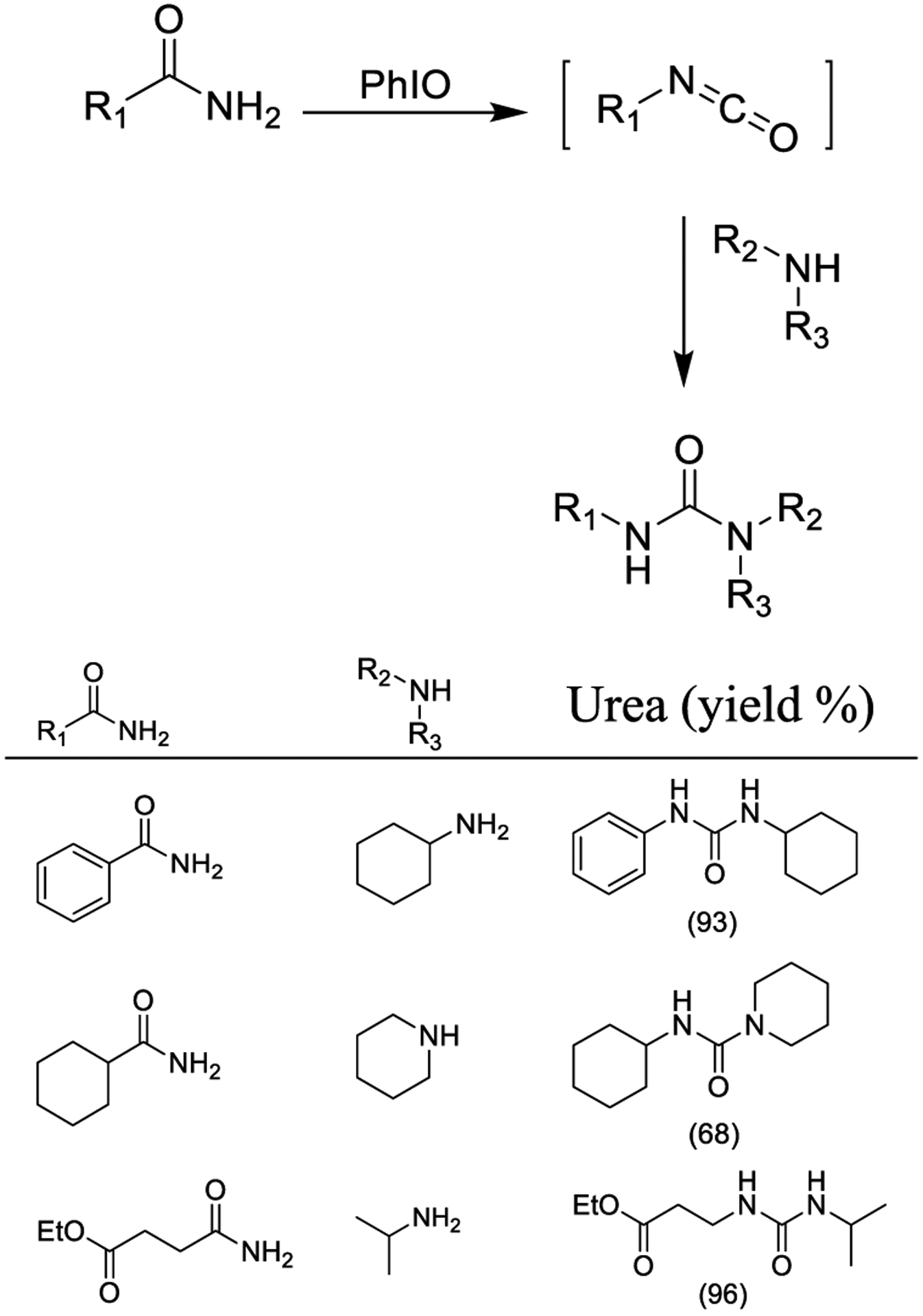
PhIO-Induced Hofmann Rearrangement
Chien and co-workers proposed the Tienmann rearrangement with benzenesulfonyl chlorides (TsCl or o-NsCl) to generate the corresponding N-substituted cyanamide derivatives as shown in Scheme 22. Acidic hydrolysis of the cyanamide derivatives would then afford N-monosubstituted ureas. N-monosubstituted ureas could also be prepared from nitriles in a one-pot fashion, encompassing the transformation of nitriles into amidoximes and subsequent Tienmann rearrangement followed by hydrolysis.160
Scheme 22.
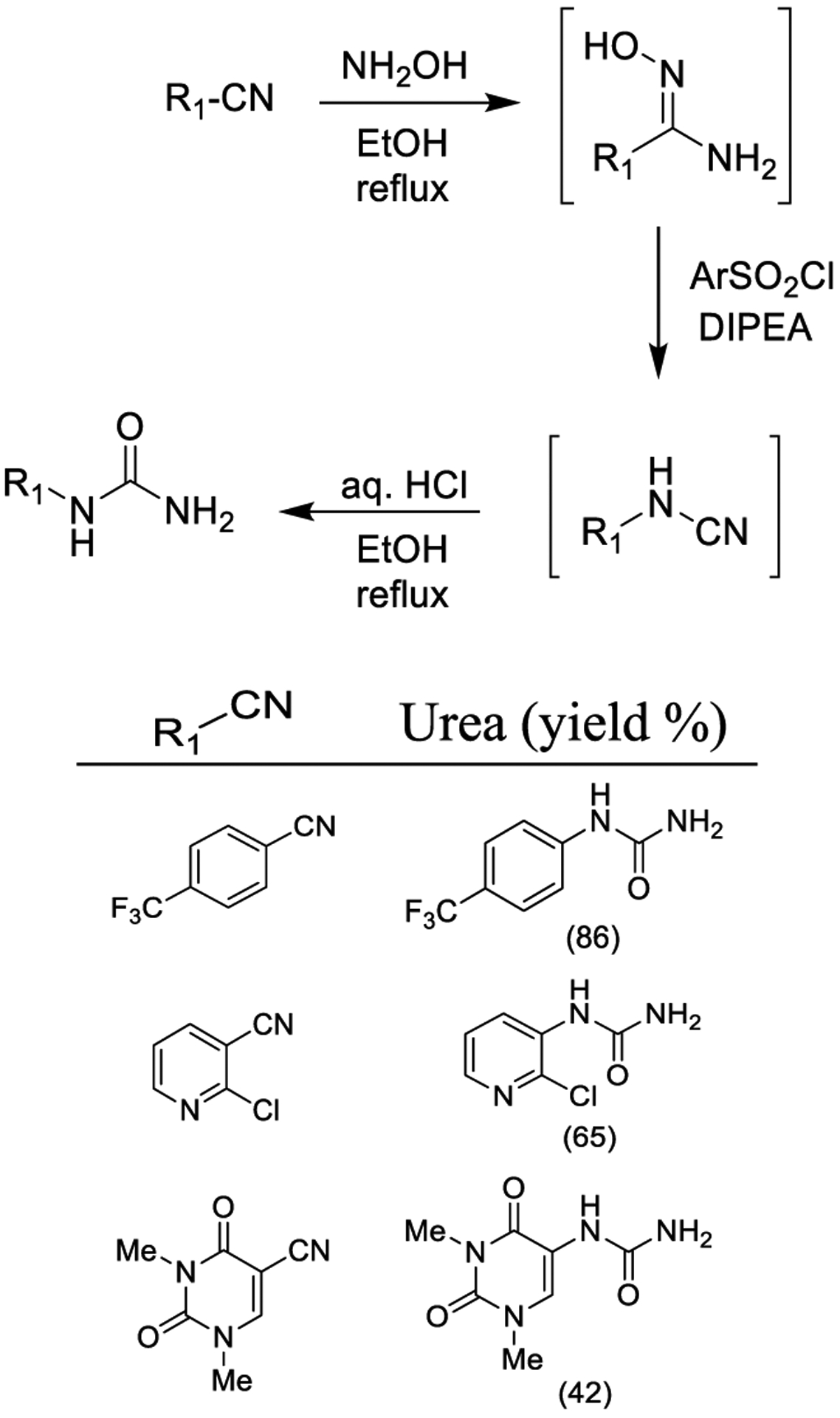
Synthesis of Urea Derivatives Using Tienmann Rearrangement
Recently, Sanz and co-workers reported the synthesis of novel benzylidene urea derivatives upon reaction of o-lithiated carbamates with nitriles according to a 1,5-O → N carbamoyl migration, namely a Snieckus-Fries-type rearrangement as outlined in Scheme 23. Several O-aryl N,N-diethylcarbamates were treated with (hetero)aromatic nitriles behaving as the electrophiles, affording the desired benzylidene ureas in high yields.161
Scheme 23.
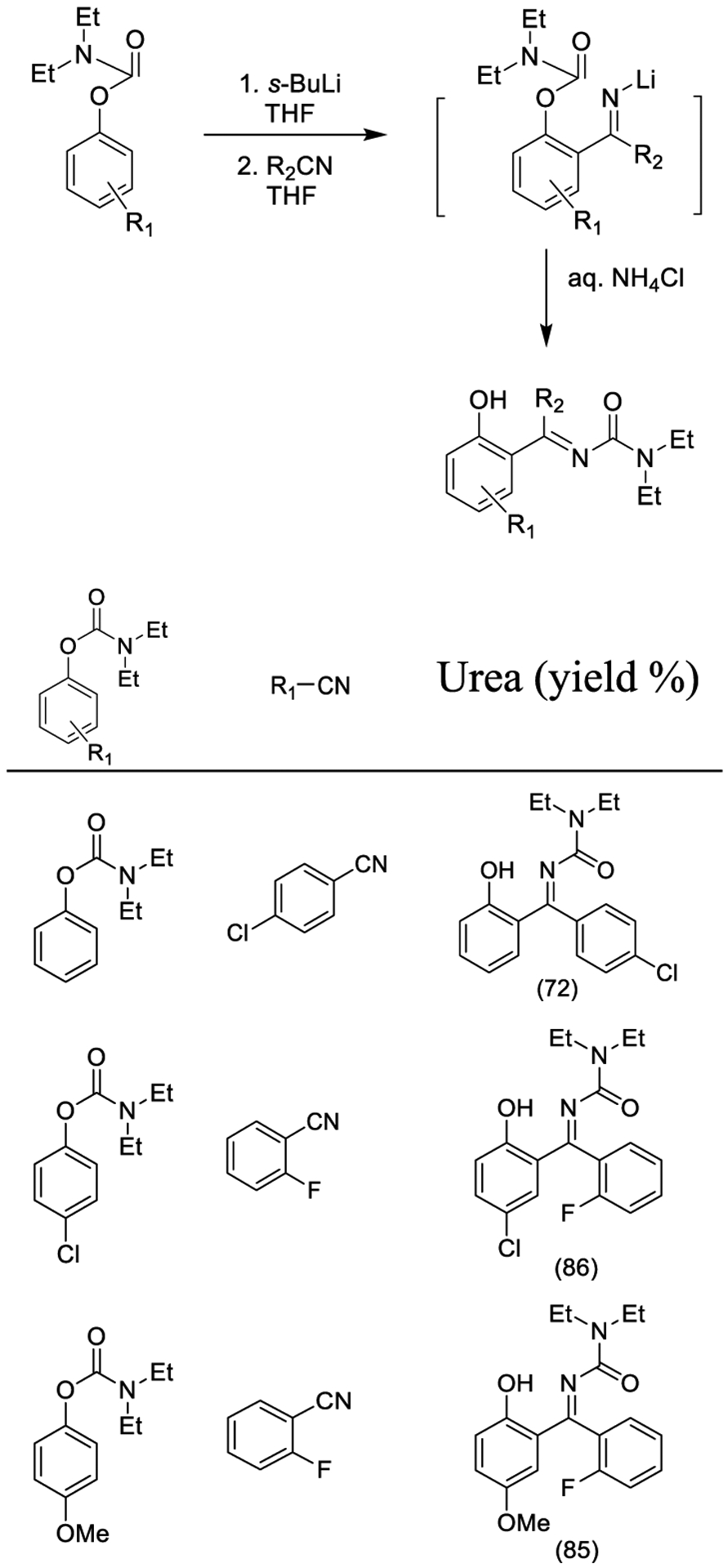
Synthesis of Urea Derivatives Using Snieckus-Fries-Type Rearrangement
6.4. Synthesis of Ureas via Metal-Catalyzed Methods.
Recently, many routes utilizing phosgene alternatives have been developed employing carbon dioxide or carbon monoxide as the carbonyl group source.162,163 Carbonylation of amines employing Pd-based catalysts has been broadly investigated since 1966, when it was first described.164 In this procedure, Pd(II) is reduced to Pd(0). Its reoxidation to the active form of the catalyst is a crucial step which is facilitated by the use of oxidizing agents such as a molecular oxygen, aromatic nitrocompounds, iodine, and quinones. The use of molecular oxygen as the oxidizing agent became attractive due to both sustainability and atom economy perspectives as water is the only coproduct formed. Accordingly, several methods encompassing the use of molecular oxygen are increasingly appealing. Unfortunately, most of the protocols take place in potentially harmful conditions, since they use carbon monoxide and molecular oxygen in volume proportions very close to the flammability range.165
Fukuoka and co-workers166 and Chaudhari and co-workers167 described the preparation of urea derivatives form alkylamines by exploiting oxidative carbonylation in the presence of Pd/C and molecular oxygen, promoted by iodide salts. Similar results were obtained by Gabriele and co-workers, who synthesized linear and cyclic ureas in moderate to good yields utilizing PdI2 and molecular oxygen.168 Mechanistically, the different reactivities of amine derivatives were ascribed to their capability to form the isocyanate intermediates upon conversion of the carbamoylpalladium complex generated in pre-equilibrium with starting materials. Pd(0) is then reoxidized to Pd(II) by addition of I2, which is in turn regenerated through oxygen-mediated oxidation of HI.163
In addition to Pd-based catalysts, Au, Co, Ni, Rh, Ru, and other transition metals have also proven useful in affording urea derivatives.163
As shown in Scheme 24, dicobalt octacarbonyl was used as a CO-free methodology to synthesize symmetrical and unsymmetrical urea derivatives under high-density microwave irradiation conditions. Reaction of cyclohexylamine 95 with dicobalt octacarbonyl led to the corresponding isocyanate derivative 96, which, upon reaction with an excess of secondary amine 97, provided trisubstituted unsymmetrical urea derivative 98 in moderate yields.5,169
Scheme 24.
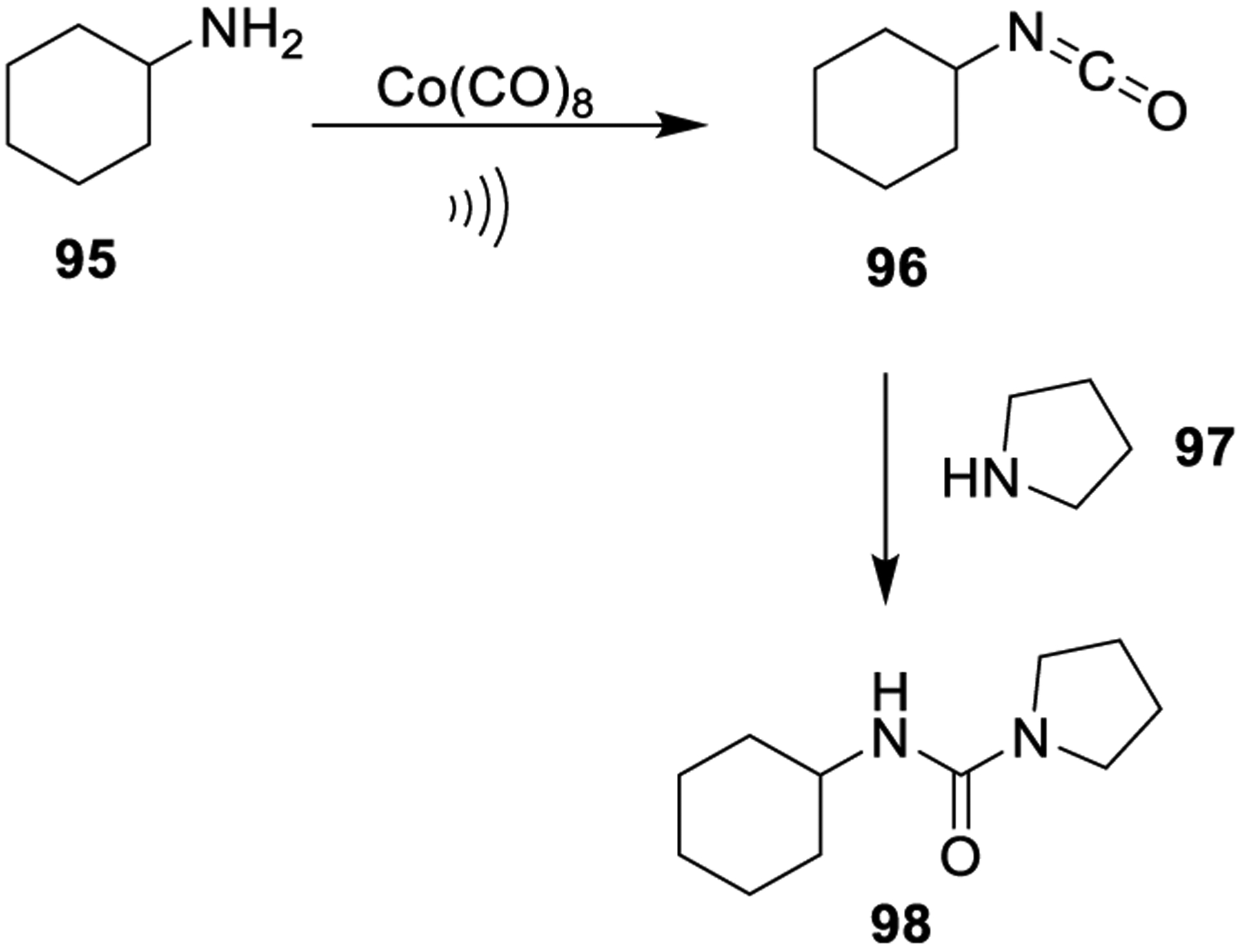
Dicobalt Octacarbonyl Promoted Generation of Urea Derivatives
Nitroarenes show a great potential for the preparation of ureas via reductive carbonylation. Homogeneous catalysts such as Pd and Ru are generally required for these reactions. In 1975, Heck described the synthesis of N,N-diarylureas through reaction of nitroarenes and CO with anilines in the presence of Pd(II) salts, organic phosphines, a tertiary amine, and tetrabutylammonium chloride at 90 °C. A combination of symmetrical and unsymmetrical ureas as well as amines derived from the reduction of the initial nitroarenes were isolated. Subsequently, it was found that portionwise addition of amines during the reaction could be a suitable strategy to increase the yield of unsymmetrical aryl-alkyl ureas. As shown in Scheme 25, nitroarene 99 was converted into urea derivative 100 using catalytic amounts of Pd(OAc)2 and dppp as ligand in the presence of acetic acid as the cocatalyst. The reaction yielded the desired urea in 98% conversion and 57% selectivity.170
Scheme 25.
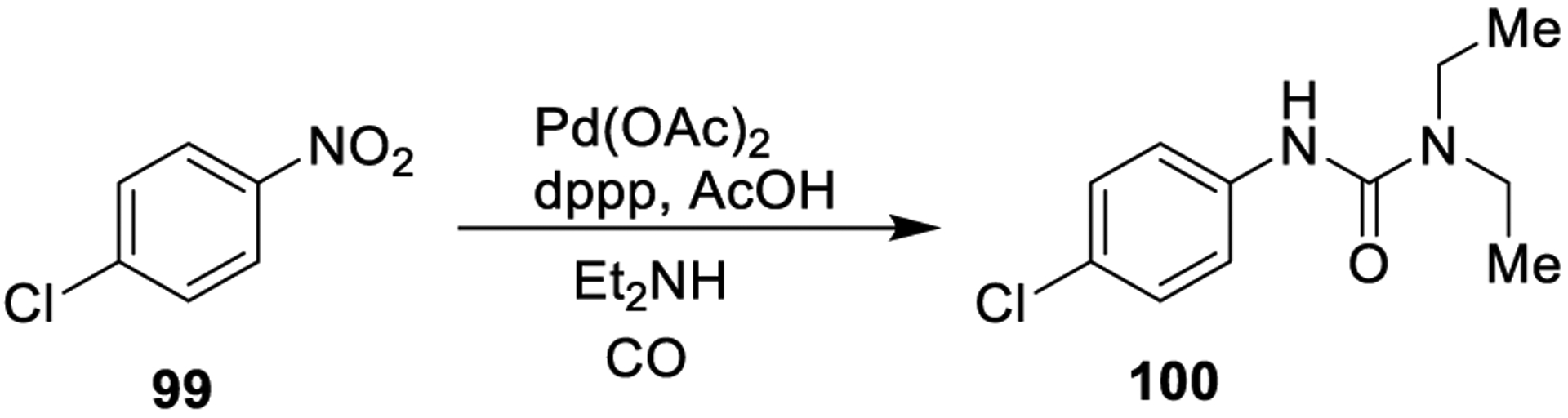
Generation of Urea Derivatives from Nitroarenes
Pd-catalyzed carbonylation is a renowned strategy to synthesize compounds containing carbonyl functionalities. Accordingly, transition metal mediated reaction of amines with CO and an oxidant provides a useful approach to generate urea derivatives.171 Buchwald and co-workers investigated a one-pot protocol for the preparation of aryl isocyanates employing a Pd-catalyzed cross coupling between aryl chlorides and triflates in the presence of sodium cyanate as depicted in Scheme 26.172 The resulting aryl isocyanates were reacted with an amine nucleophile to provide unsymmetrical N,N′-di- and N,N,N′-trisubstituted ureas. The methodology offers a broad substrate scope. The reaction protocol was applied in the synthesis of Omecamtiv Mecarbil (102), a phase-II clinical trial activator of cardiac myosin.
Scheme 26.
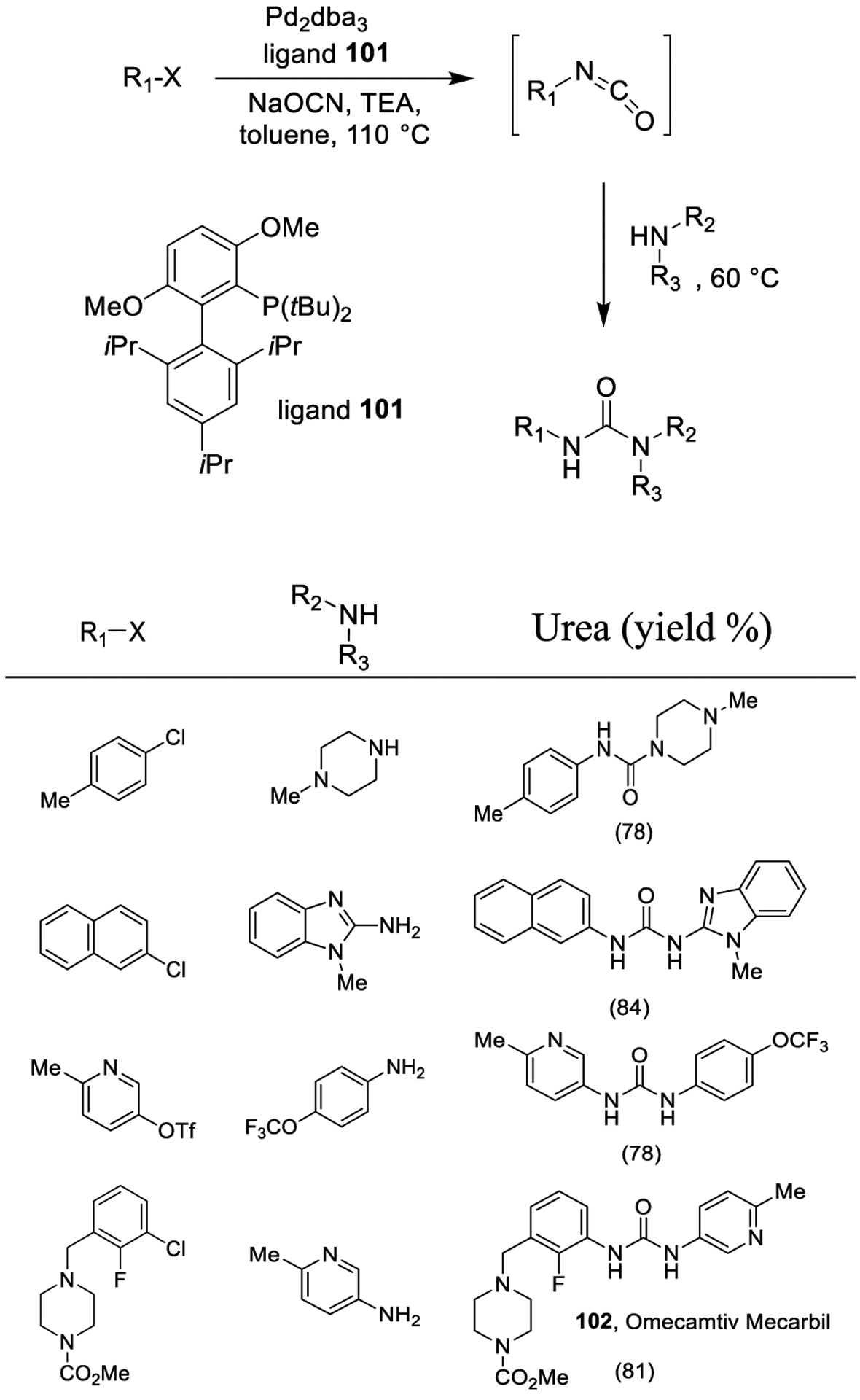
Synthesis of Urea Derivatives Using Cross Coupling of Aryl Chlorides and Triflates with Sodium Cyanate in the Presence of Ligand 101
Lemaire and co-workers developed a Pd-catalyzed procedure for the reductive alkylation of monosubstituted ureas using aldehydes and molecular hydrogen as a clean reducing agent as outlined in Scheme 27. The protocol used dry methanol, which behaves as a dehydrating agent and allows the generation of an imine from a hemiaminal precursor. Imine hydrogenation ultimately provides N,N-disubstituted urea derivatives. The reaction scope was investigated in optimized conditions (H2 at 5 bar, 5% Pd/C (2.5 mol %), MeOH as the solvent, 100 °C) employing decanal and diverse urea derivatives. The desired N,N-disubstituted ureas were smoothly formed in good to excellent yields. The methodology was also implemented in solvent-free conditions.92
Scheme 27.
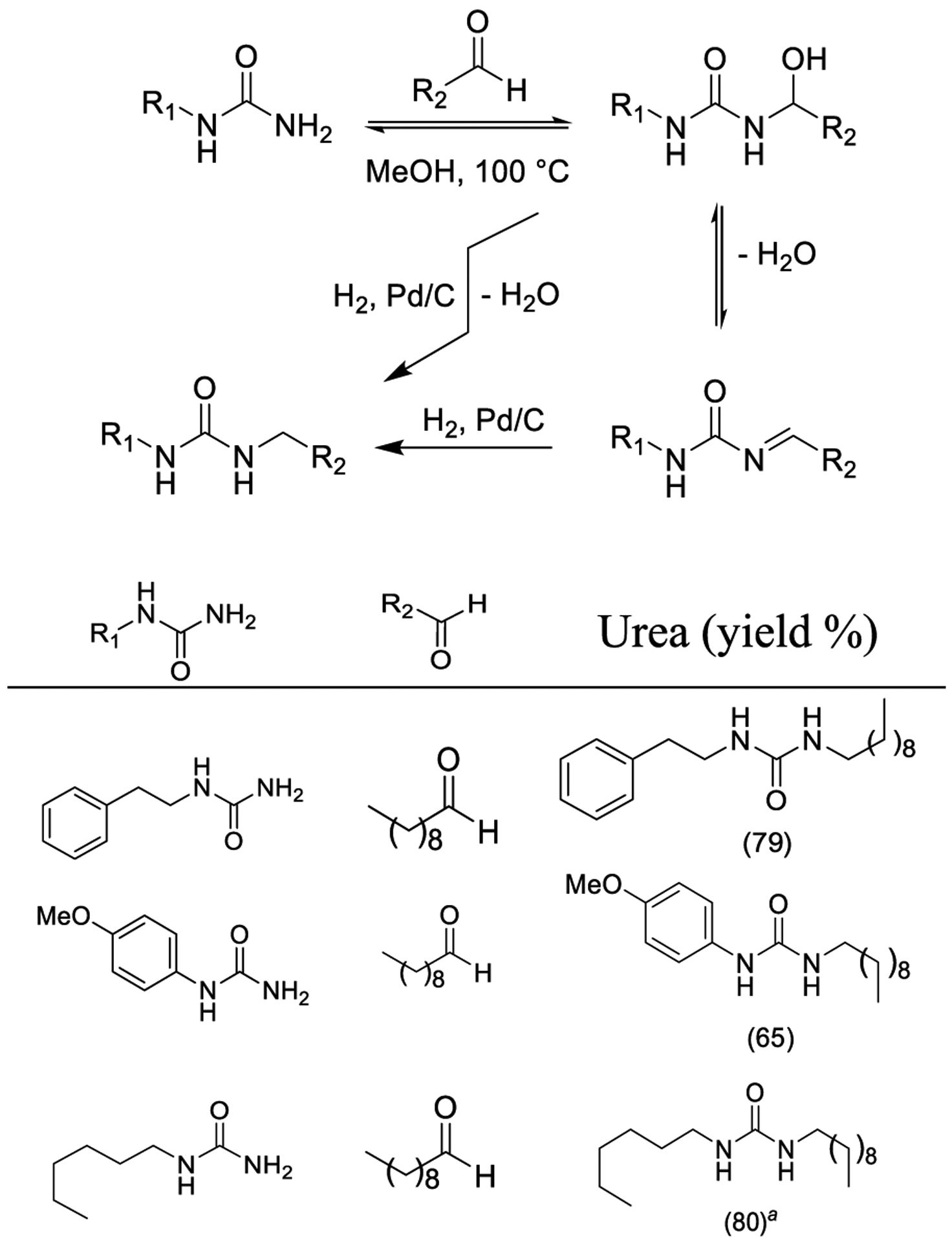
Pd-Catalyzed Reductive Alkylation of Urea Derivatives with Aldehydes
aSolvent free conditions.
The metal-catalyzed carbonylation of nitrenes with amines is gaining interest for the synthesis of unsymmetrical ureas. Recently, Zhang and co-workers described a Pd/C-catalyzed carbonylation of benzyl, alkyl, and aryl azides with amines to synthesize unsymmetrical urea derivatives as described in Scheme 28. The use of Pd/C offers advantages compared to general homogeneous catalysts in terms of safe handling, simple removal of catalyst by filtration, and recycling potential. Notably, the use of a phosphorus ligand such as XPhos increased the reaction yields, and toluene was selected as the best performing solvent. Due to the higher reactivity of aryl azides, reaction could be implemented at room temperature. This cross-coupling protocol proceeded even in an aqueous media containing 5% PhMe and 5% nBu4NCl.171
Scheme 28.
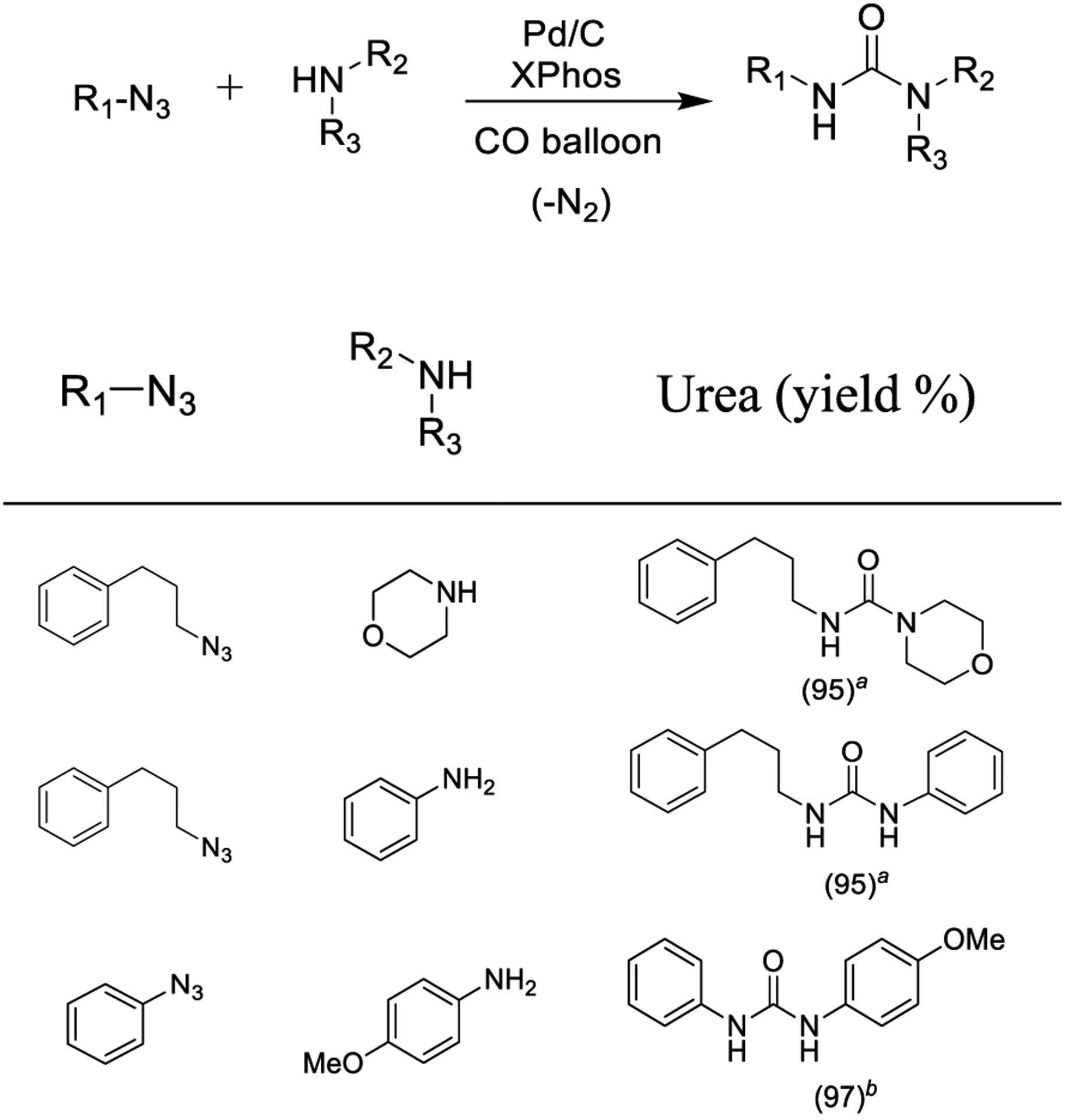
Pd/C-Catalyzed Carbonylation of Benzyl, Alkyl, and Aryl Azides with XPhos
aPhMe, 60 °C, 12 h. b5% nBu4NCl, H2O/PhMe 20:1, rt, 24 h.
Mancuso and co-workers reported a catalytic system involving the use of PdI2 in the presence of an excess of KI for the oxidative carbonylation of primary amines providing 1,2-disubstituted symmetric urea derivatives. The reaction with a mixture of a primary and a secondary amine leads to the formation of trisubstituted urea derivatives as outlined in Scheme 29. The protocol has been applied to synthesis of β-amino alcohols bearing a primary amine group for their conversion into the corresponding oxazolidinones. Conceivably, either the carba-moyl-palladium complex I or the isocyanate derivative II could provide the desired symmetric urea derivatives.172
Scheme 29.
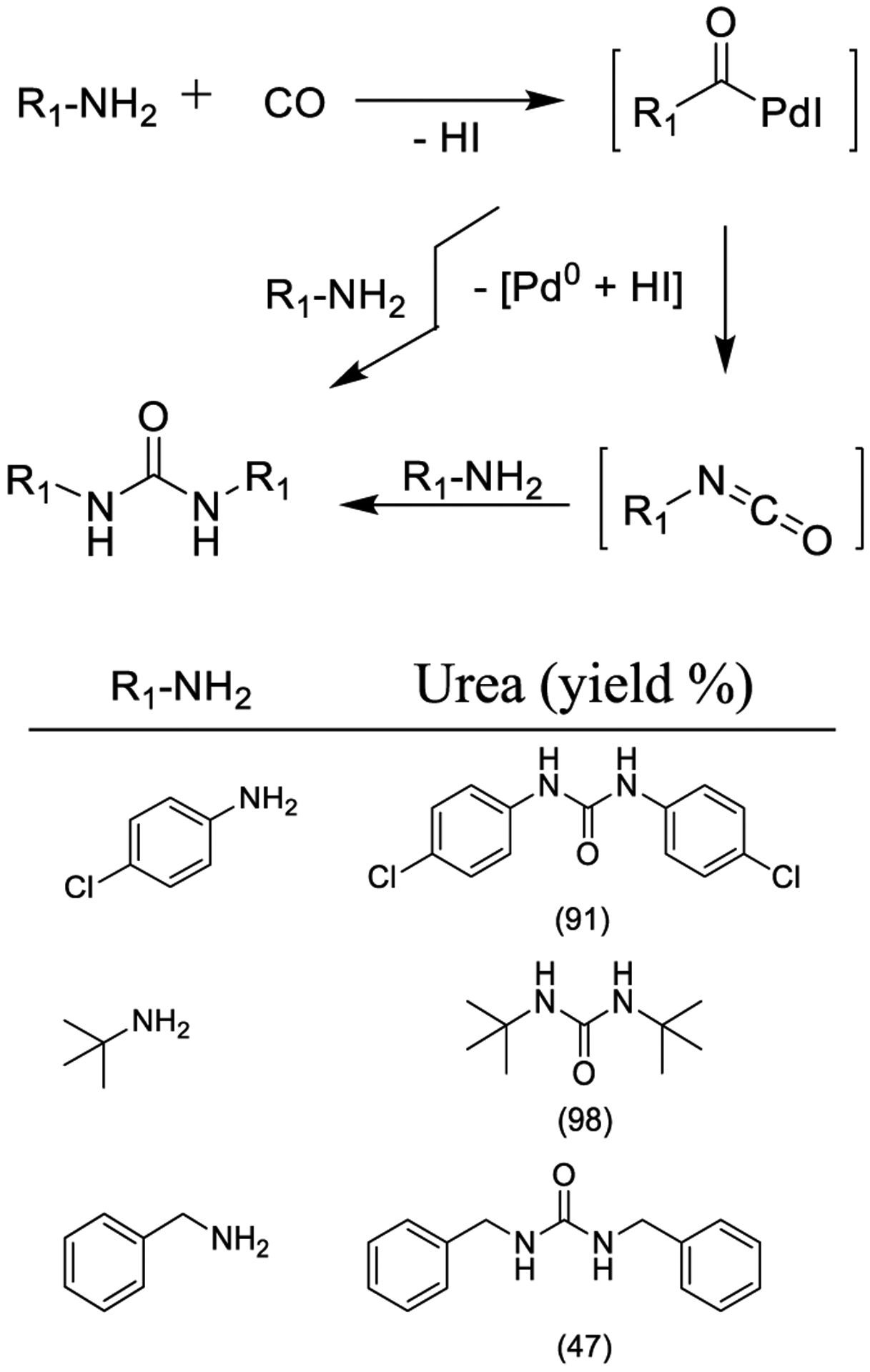
Synthesis of Urea Derivatives by Oxidative Carbonylation Using PdI2 and KI
The use of an appropriate ionic liquid such as 1-butyl-3-methylimidazolium tetrafluoroborate, ([bmim][BF4]) in the presence of the PdI2/KI catalytic system has been shown to provide tetrasubstituted oxamides from secondary amines through an oxidative double carbonylation.173 Also, carbonylation protocols employing a Pd(II)/Ag(I) catalytic system have been developed.174 Wu and collaborators developed several carbonylation protocols utilizing formic acid175–180 or Mo(CO)6 as a CO source.
The synthesis of unsymmetrical urea derivatives through a Pd/C-catalyzed carbonylation of nitroarenes using stoichio-metric Mo(CO)6 is shown in Scheme 30.181–183 An NaI additive was crucial for the conversion, but its exact function is not clear. Also, the use of Pd(OAc)2 in place of Pd/C provided slightly lower yields. Monodentate ligands performed better than bidentate ligands. From inexpensive and stable nitroarenes several unsymmetrical ureas, including the herbicide neburon (103), were synthesized in good yields. Beyond palladium, ruthenium is another metal of growing use for the synthesis of urea derivatives.
Scheme 30.
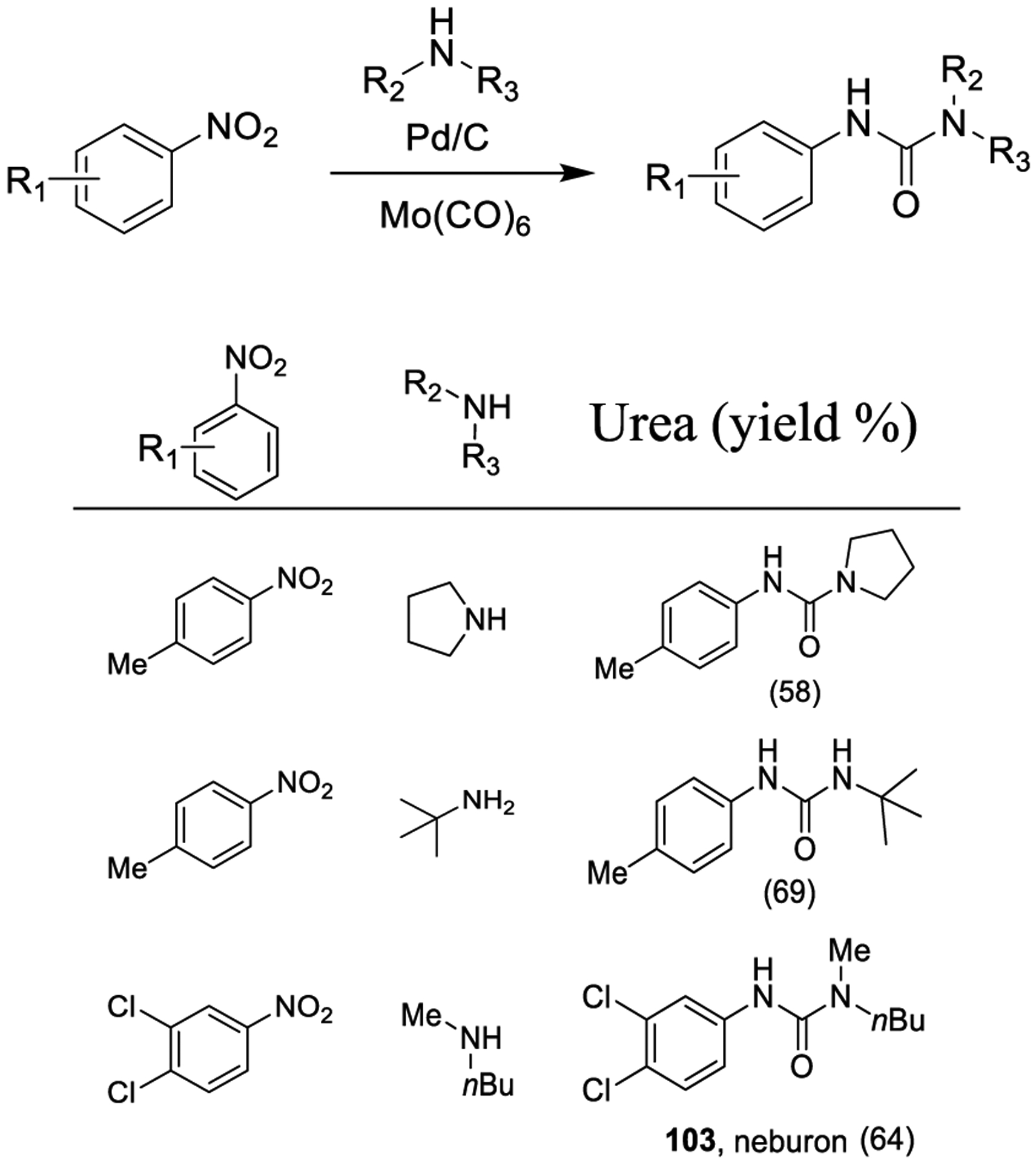
Synthesis of Urea Derivatives by Carbonylation with Mo(CO)6
Hong and co-workers very recently reported the synthesis of urea derivatives using ruthenium pincer complex as shown in Scheme 31.184 The method shows high atom economy and produces hydrogen as the only byproduct, and only low Ru catalyst loadings are required. The method was extended to the synthesis of unsymmetrical ureas via a sequential one-pot two-step protocol with initial generation of the formamide from an amine and methanol, and subsequent reaction of the formamide with a second amine derivative.184
Scheme 31.
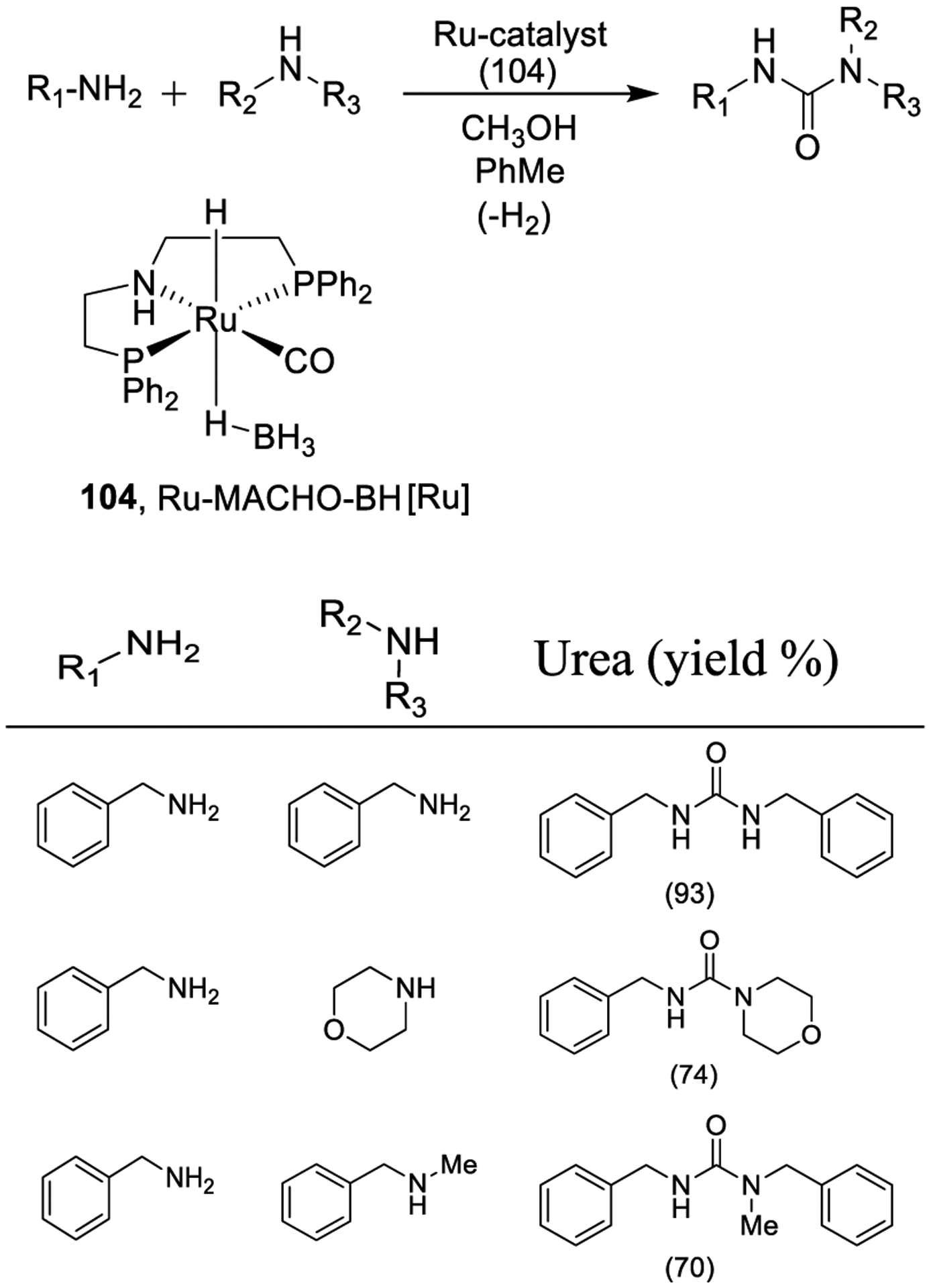
Synthesis of Urea Derivatives Using Ruthenium Pincer Complex (104, Ru-MACHO-BH) as the Precatalyst
Recently, Gunanathan and co-workers also reported “amine-amide” metal-ligand cooperation using a ruthenium pincer complex with DMF as the carbon monoxide surrogate as shown in Scheme 32. The use of 2 mol % of the catalyst 105 and 10 equiv of DMF provided the best reaction conditions for the generation of symmetrical arylmethyl-, arylalkyl-, and N,N-dialkylurea derivatives. Moreover, the protocol was extended to the preparation of unsymmetrical urea derivatives. After the generation of formamide derivatives, diverse amines were added and temperature was increased to efficiently provide the desired unsymmetrical urea derivatives.185
Scheme 32.
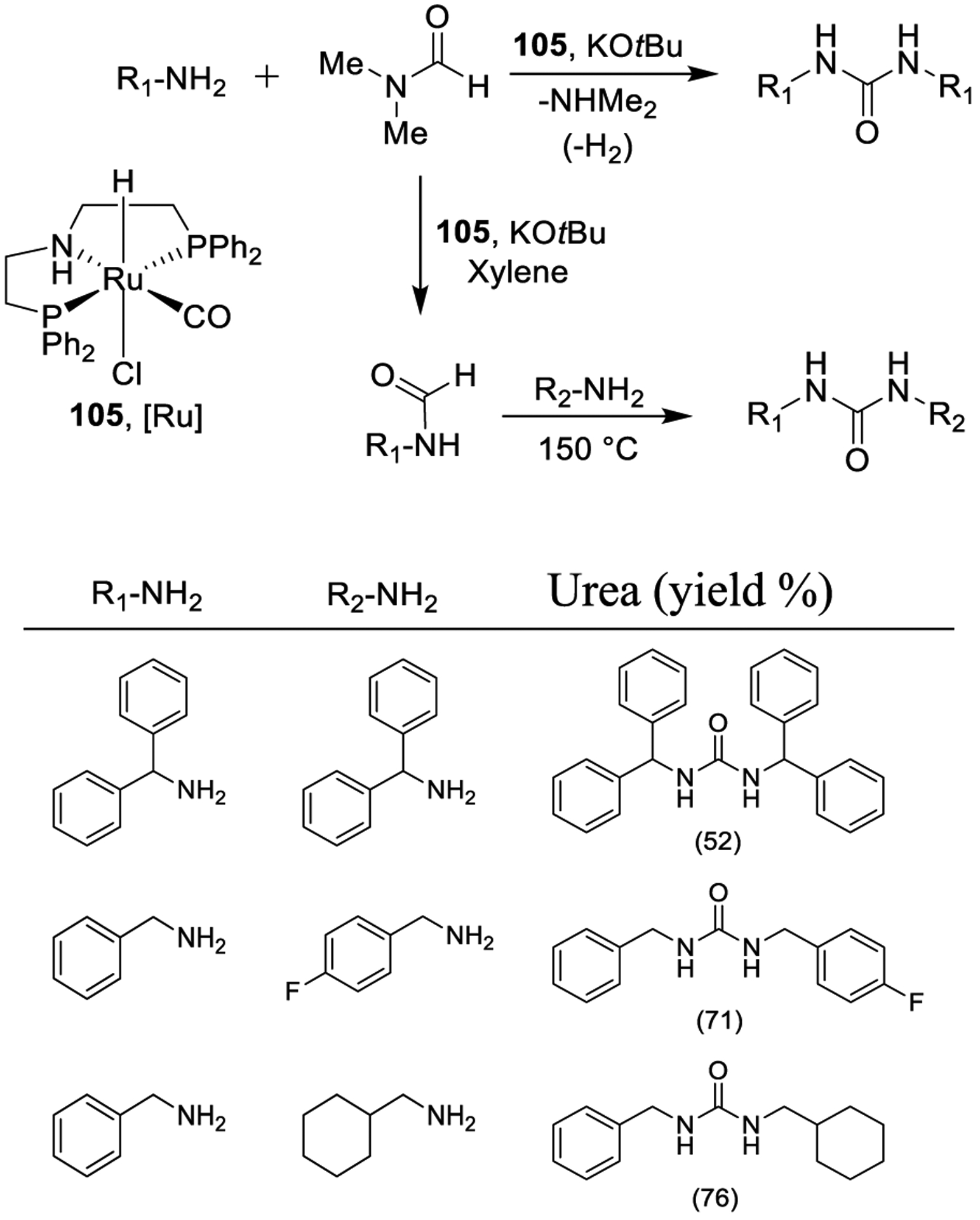
Synthesis of Urea Derivatives Using Ruthenium Pincer Complex 105 and DMF
Although Pd, Ru, and Rh have been the metals of choice for oxidative carbonylation, there are issues with the excessive cost of the catalysts and with the toxicity of the solvents. The use of other metals such as tungsten,186 cobalt,187 and copper-complex188 have been explored for the synthesis of cyclic urethanes. As shown in Scheme 33, Nacci and co-workers developed a copper-based system for the synthesis of 2-oxazolidinones, urea derivatives, and urethanes in an aqueous medium without the use of additives. Copper(II) chloride was the optimum catalyst, and the reactions proceeded efficiently in water under homogeneous conditions at 100 °C and in approximately 4 h.189
Scheme 33.
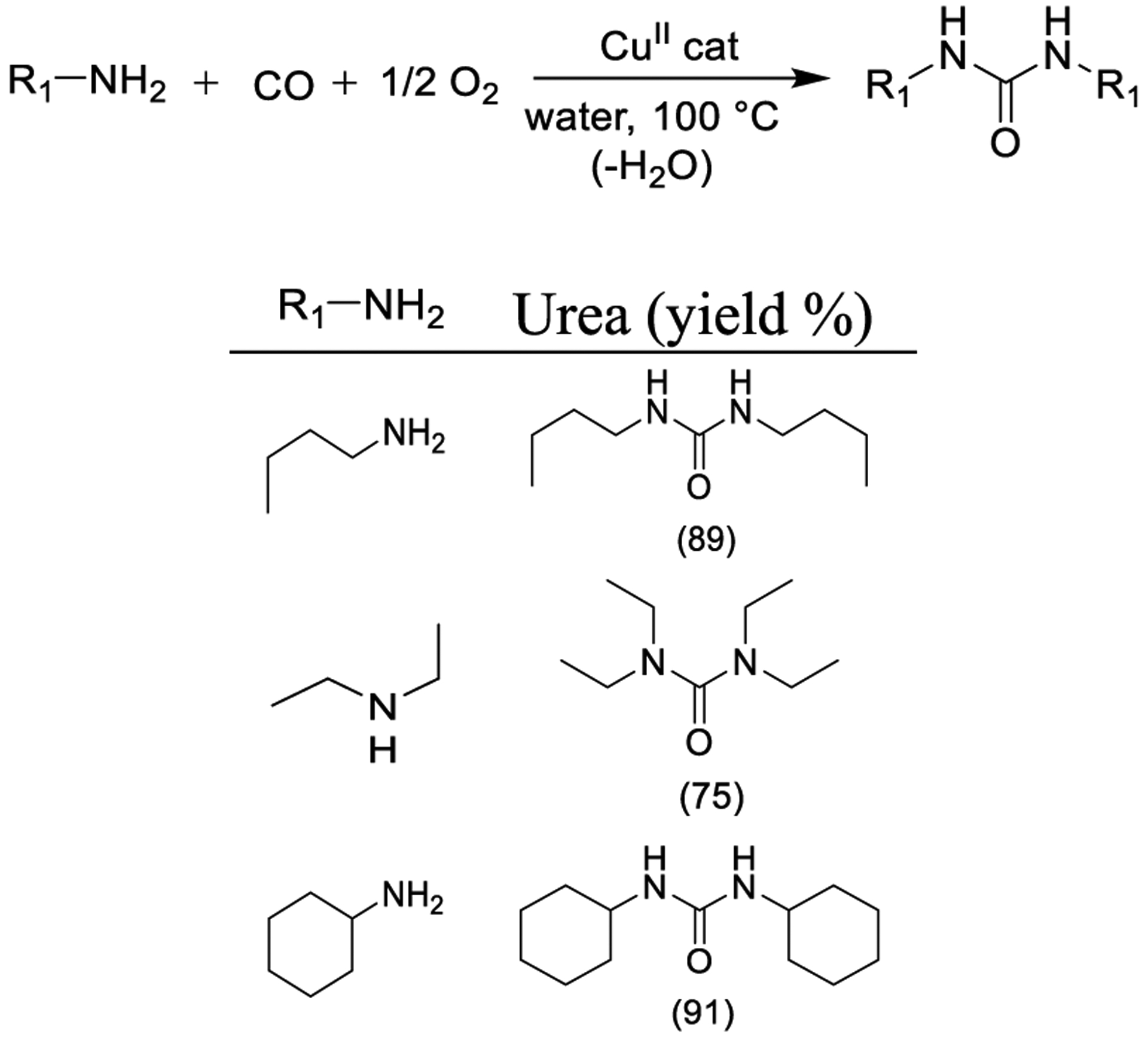
Synthesis of Urea Derivatives Using a Copper-Catalyzed Reaction
6.5. Synthesis of Urea Derivatives Using Urethanes and Isonitriles.
Syntheses of ureas starting from Boc-protected and Cbz-protected amines were carried out. As shown in Scheme 34, trifluoromethanesulfonyl anhydride was used to generate the isocyanate intermediate which provided the desired urea derivative upon reaction with a suitable amine.190,191
Scheme 34.
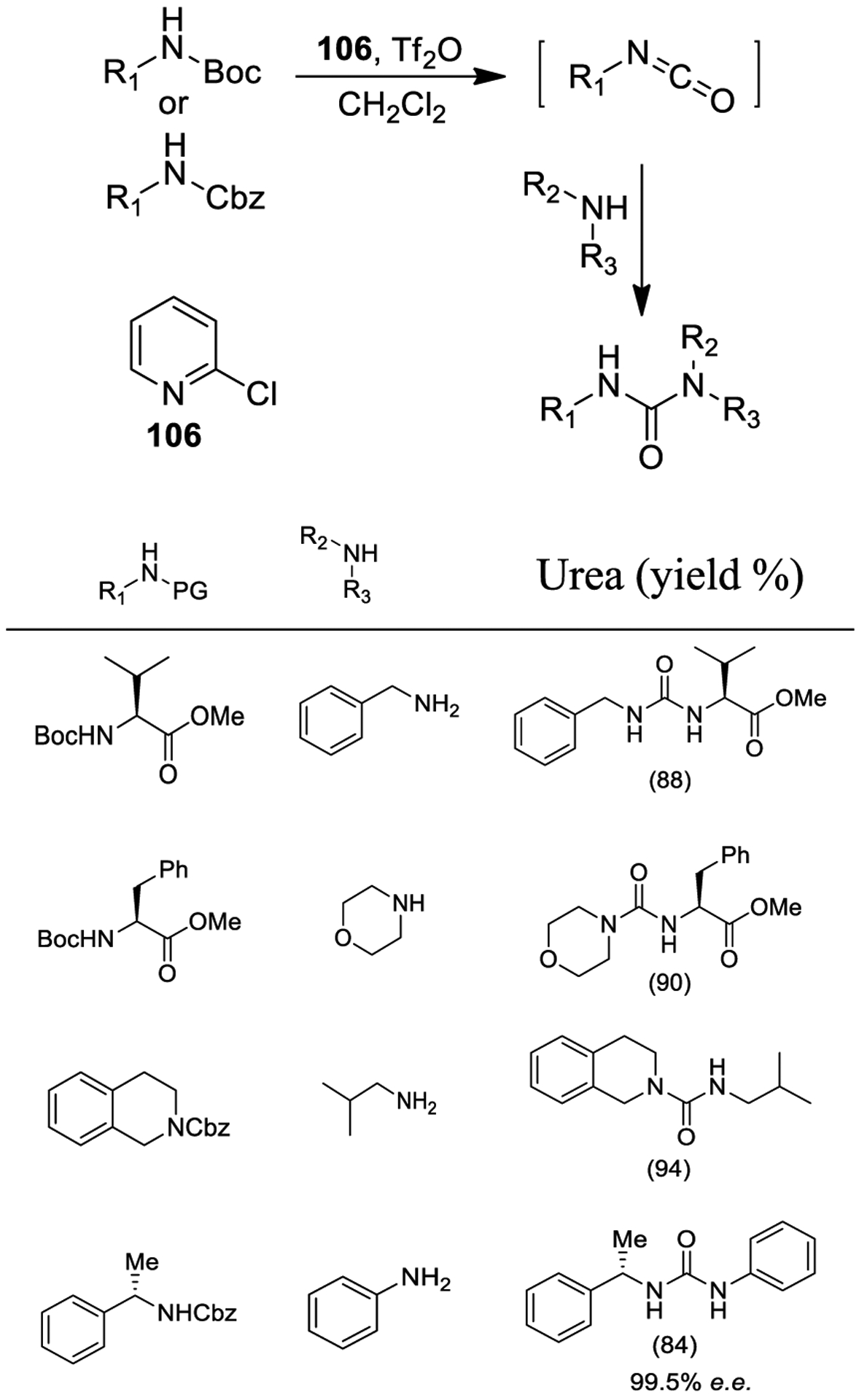
One-Pot Synthesis of Urea Derivatives from Boc-Amines and Cbz-Amines
The use of 2-substituted pyridines such as 2-chloropyridine (106) demonstrated high efficacy. In order to obtain satisfactory yields, the optimum amount of nucleophile is 3 equiv. The protocol was successfully applied to anilines, sterically hindered tert-butylamines, and secondary amines, affording trisubstituted ureas. Moreover, cyclic amines and noncyclic secondary amines led to high yields. The protocol was applied to chiral substrates with no racemization or epimerization.
Bana and co-workers designed a continuous-flow system involving two contiguous microreactors to synthesize non-symmetric ureas from Boc-protected amines under mild conditions as outlined in Scheme 35. In-line FT-IR was employed for monitoring the process. The setup was applied to the synthesis of a series of urea derivatives, and it was successfully used for the preparation of the active pharmaceutical ingredient cariprazine.192 Riesco-Domínguez and coworkers recently described the continuous flow synthesis of two libraries of urea derivatives containing a piperidin-4-one scaffold.193
Scheme 35.
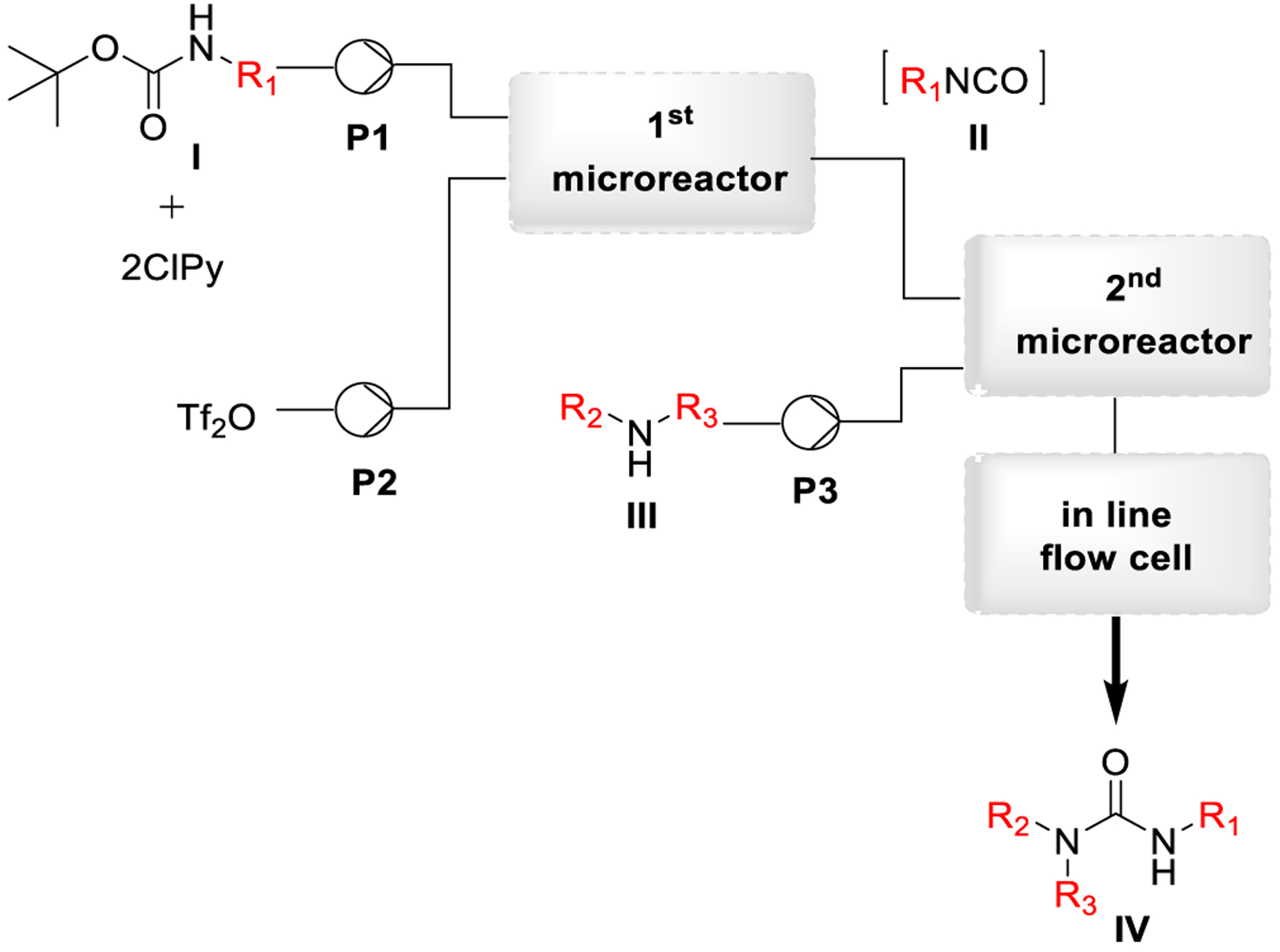
Synthesis of Urea Derivatives by a Continuous-Flow Process
Ganem and co-workers developed a simple and effective protocol for the oxidation of isonitrile derivatives to isocyanates employing DMSO as the oxidizing agent and trifluoroacetic anhydride as the catalyst as outlined in Scheme 36. Isocyanate formation is completed within a few minutes, with dimethyl sulfide being the sole byproduct. The resulting isocyanates were used to synthesize urea derivatives.194
Scheme 36.
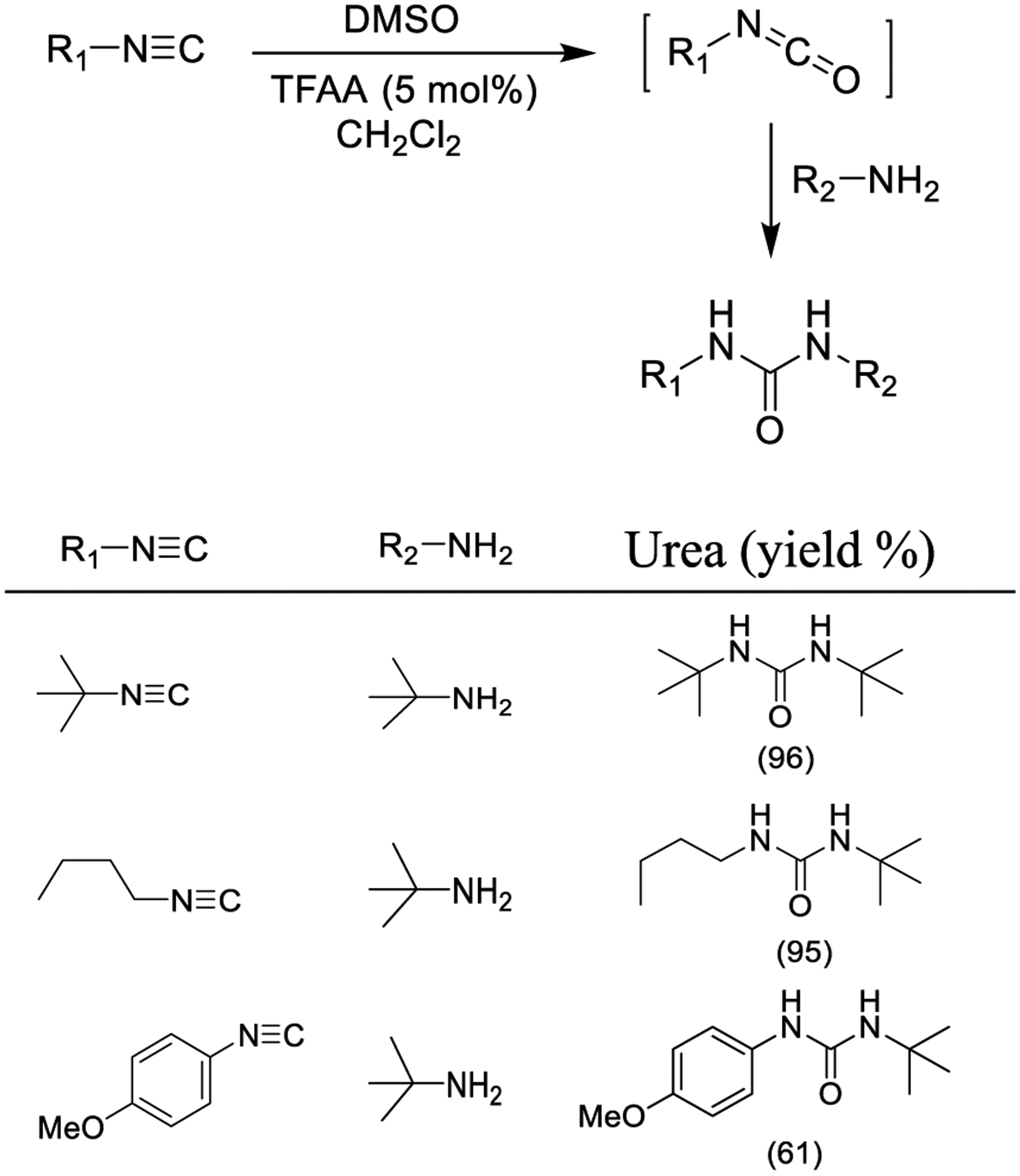
Synthesis of Ureas from Isonitriles via the Intermediate Isocyanates
Recently, Wang and co-workers reported the synthesis of urea derivatives employing oximes as recyclable starting materials by an intermolecular oxidative C-O bond formation between oxime derivatives and isocyanides as shown in Scheme 37.195
Scheme 37.
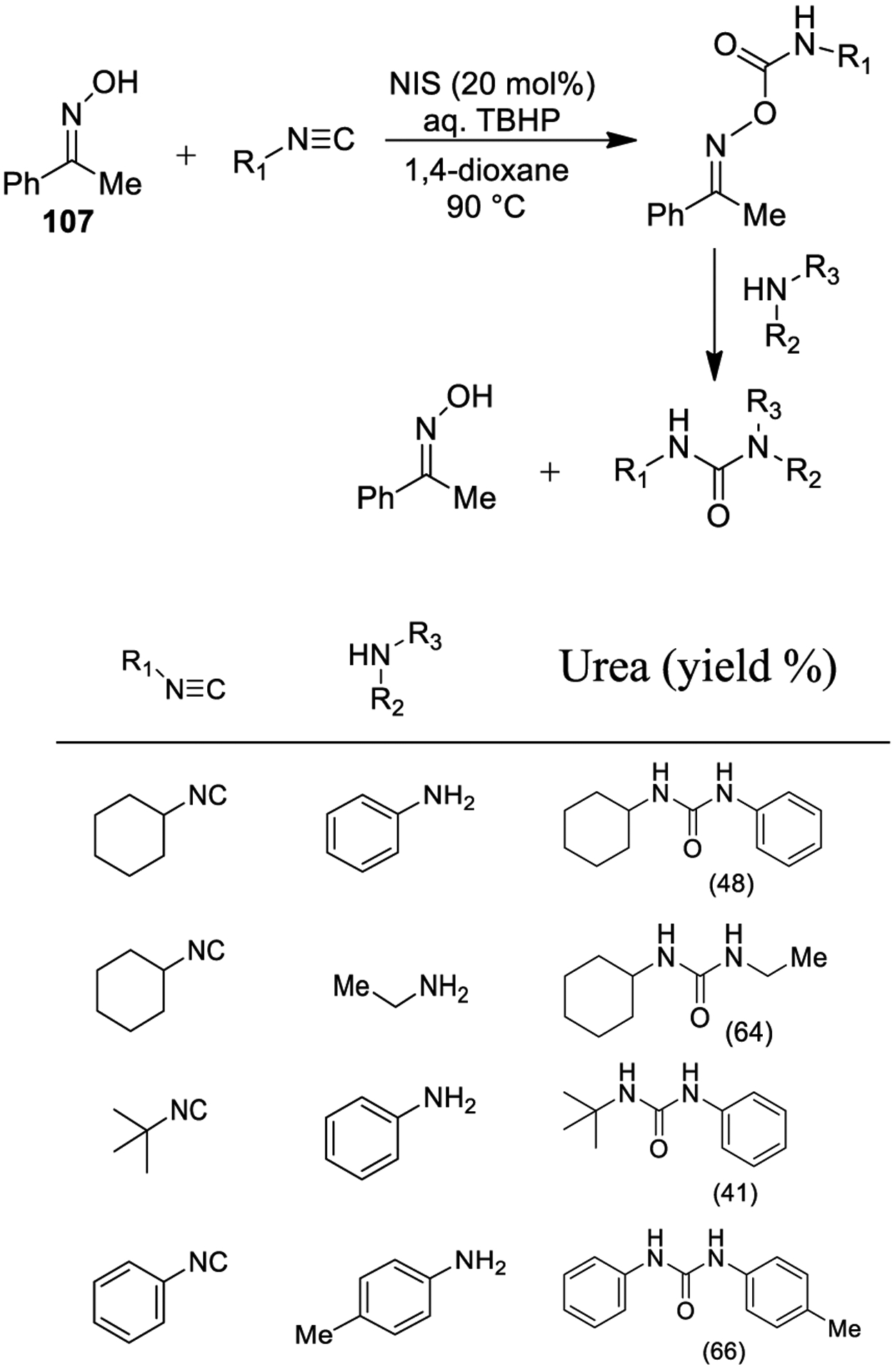
Synthesis of Urea Derivatives Using Oximes
1-Phenylethan-1-one oxime (107) and cyclohexyl isocyanide were used to set up optimum reaction conditions to form the corresponding carbamoyl oxime as shown. The use of 20% NIS in the presence of aqueous TBHP, with 1,4-dioxane as the solvent at 90 °C, represented the best reaction conditions.
Peterson and co-workers reported the synthesis of urea libraries starting from carbamic acids prepared from the DBU-catalyzed reaction of amines with gaseous carbon dioxide as outlined in Scheme 38. The desired urea derivatives were synthesized by reacting carbamic acids from primary amines with Mitsunobu reagents to generate isocyanates in situ, which then reacted with primary and secondary amines. The resulting isocyanates efficiently formed trisubstituted urea derivatives upon treatment with secondary amines.196 The methodology was used successfully with benzylamine, and then extended to anilines.
Scheme 38.
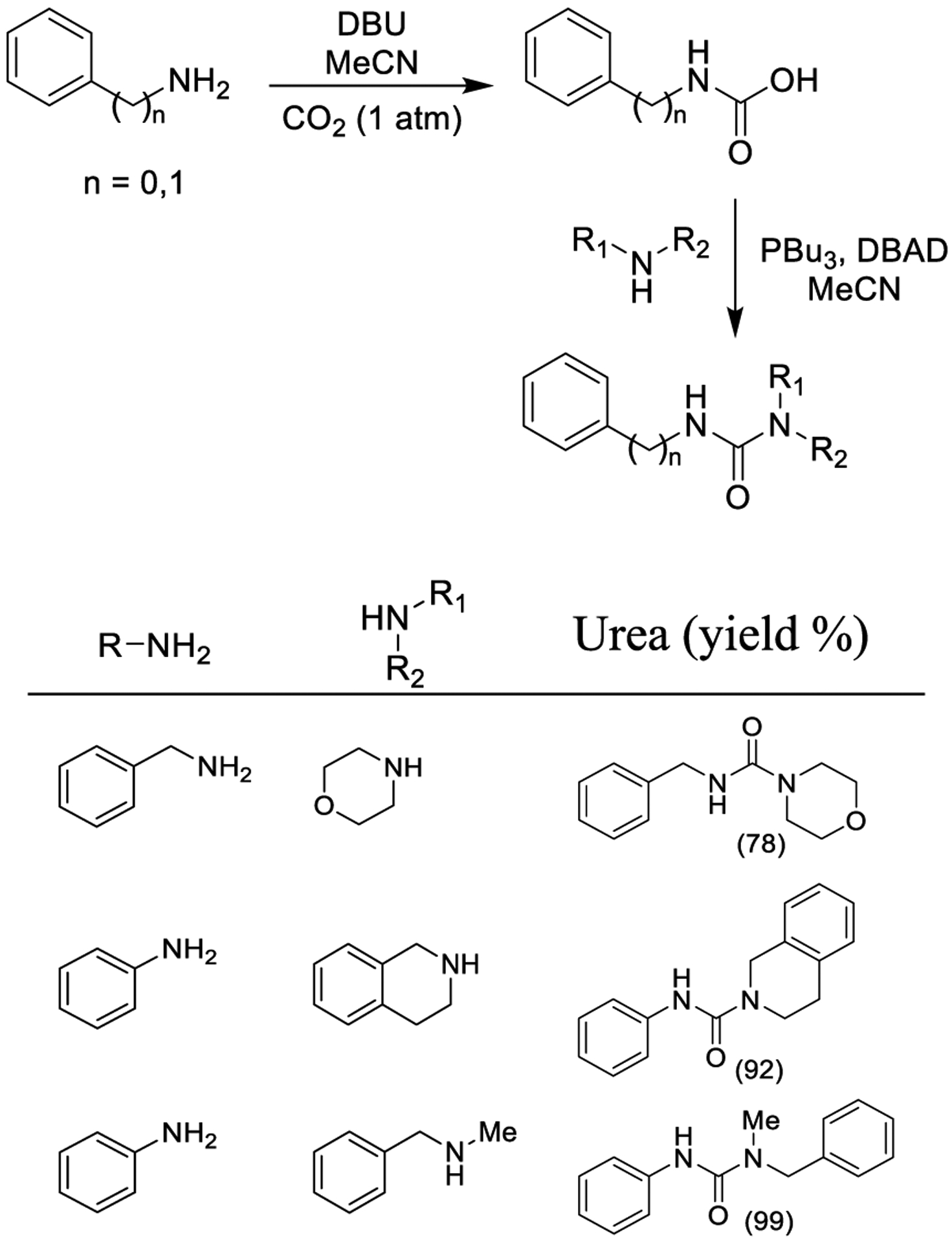
Synthesis of Urea Derivatives via Carbamic Acids
7. RECENT DESIGN OF UREA DERIVATIVES IN DRUG DISCOVERY
As described earlier, urea functionality is a structural feature in a variety of FDA approved drugs. In this section, we cover design of urea-based compounds in medicinal chemistry and drug discovery in the past 10 years in a range of medicinal chemistry fields.
7.1. Urea-Containing Anticancer Agents.
Urea derivatives have been designed and synthesized to show broad biological properties, particularly potent antitumor activity. In particular, diarylurea derivatives have been designed as anticancer agents, targeting protein kinase or microtubules.
7.1.1. Kinase Inhibitors.
In recent years, a series of kinase inhibitors containing aryl urea substructures have been introduced in the market for the treatment of cancers or tested clinically. Among them, linifanib was approved for the therapy of colorectal cancer,197 Sorafenib is used for advanced renal cell carcinoma,198 and tandutinib is employed in the treatment of kidney cancer199 and acute leukemia.200 Meanwhile, Gedatolisib (PKI-587) is in phase II trials for treatment of acute myeloid leukemia.201 In recent years, a considerable effort has been directed toward the design of antitumor agents containing urea derivatives, especially in the fields of kinase inhibitors.
Epidermal growth factor receptor (EGFR) overexpression has been observed in many cancer types.202 Homo- or heterodimerization are able to activate EGFR; subsequent autophosphorylation of tyrosine at the intracellular kinase domain stimulates Ras/Raf/MAPK and the PI3K/AKt/mTOR pathways.203,204 Several small molecule EGFR inhibitors have been developed for antitumor therapy. EGFR inhibitors belonging to the second generation feature a Michael acceptor group and show higher selectivity compared to first generation inhibitors. They are more prone to overcoming drug resistance.
Mowafy and collaborators reported a series of inhibitors incorporating a urea moiety at the C-6 position of the quinazoline core that behaves as a hydrogen bond acceptor. A number of derivatives were potent EGFR inhibitors. 6-Ureidoanilinoquinazoline derivative 109 (Figure 20) displayed an IC50 value of 0.061 μM, showing remarkable growth inhibitory activity against tumor cells characterized by EGFR over-expression, namely nonsmall cell lung cancer (EKVX NCIH322 M cell line), renal cancer (A498, TK-10 cell lines), and breast cancer (MDA-MB-468 cell line). Docking studies unveiled a binding mode similar to gefitinib and supported an extra interaction with Cys-773 residue at the kinase gate-keeper.205
Figure 20.
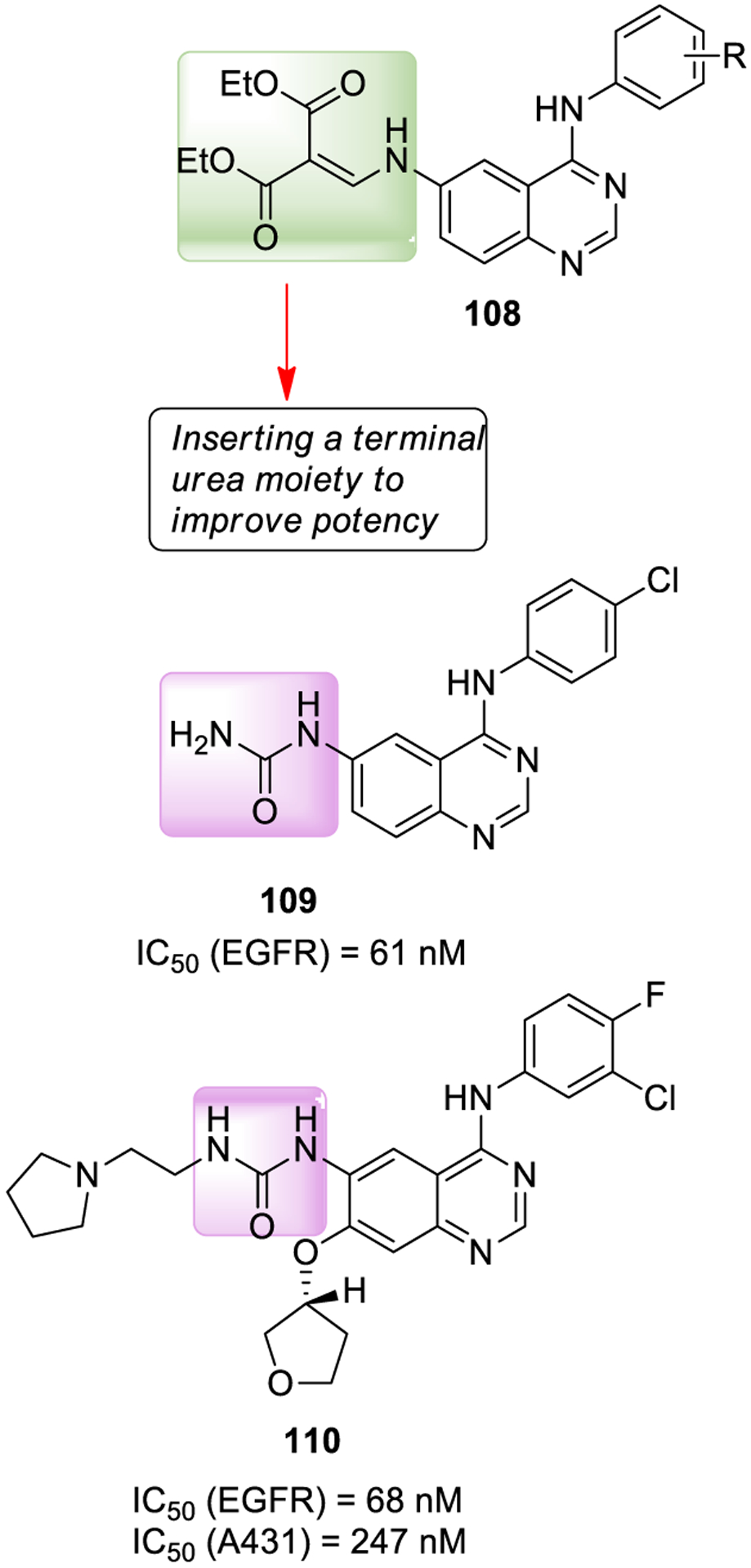
EGFR inhibitor 108 and urea-containing EGFR inhibitors 109 and 110.
SAR studies show that replacement of the amino-2-cyanoacrylate with amino-methylene malonate resulted in increased potency for compound 108. Further substitution with urea resulted in the very potent compound 109. The urea moiety of compound 109 established a key hydrogen bonding interaction with Cys793 which is absent in gefitinib. This hydrogen bonding interaction may be responsible for the enhanced binding affinity and also for particular ligand positioning.
Further modification of the anilinoquinazoline scaffold containing a C-6 urea and a C-7 tetrahydrofuranyl ether led to compound 110, which is a reversible inhibitor of EGFR. Compound 110 effectively inhibited EGFR and its autophosphorylation in A431 human epithelial carcinoma cells. Further studies later established that compound 109 behaved as an irreversible EGFR inhibitor.206
A series of potent multikinase inhibitors was designed utilizing established pharmacophores for kinase inhibition such as 4-anilinoquinazoline and unsymmetrical diarylurea derivatives. Among them, compound 111 (Figure 21) exhibited significant potency toward BRAF, BRAF V600E, EGFR, and VEGFR-2. Docking studies showed that the compound might nicely fit the DFG-out conformation of BRAF.207
Figure 21.
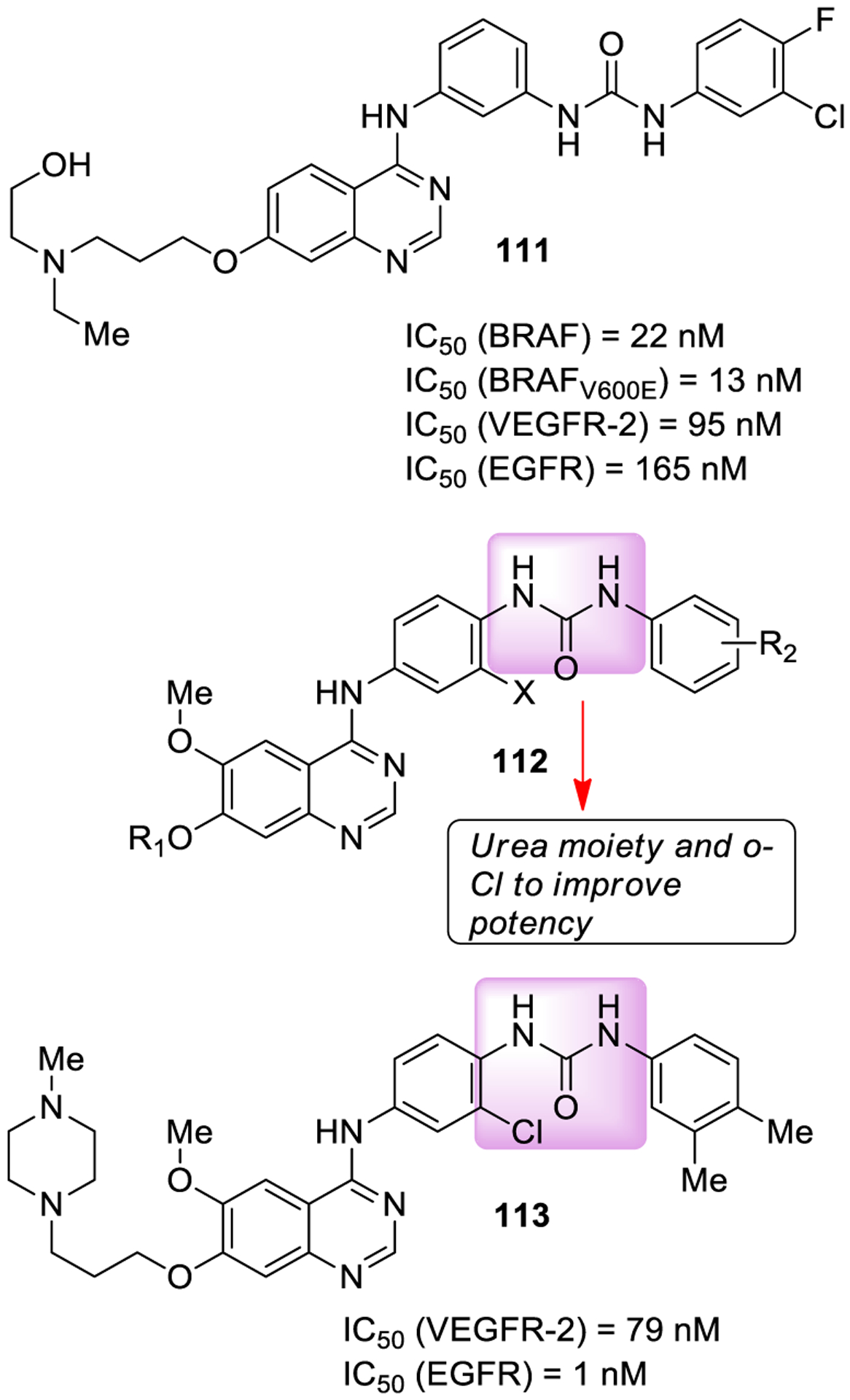
Urea-containing multikinase inhibitors 111–113.
Further modification on the aryl group resulted in diaryl ureas (112) as dual EGFR and VEGFR-2 inhibitors. The SAR studies established that a terminal diaryl urea moiety with a chlorine atom at the ortho-position of the urea group was optimal for high inhibitory potency. Compound 113 exhibited potent inhibition of EGFR (IC50 = 1 nM) and VEGFR-2 (IC50 = 79 nM), and good antiproliferative activities on HT-29 (colorectal adeno-carcinoma), H460 (lung cancer), and MCF-7 (breast cancer) cell lines. Docking studies suggested binding of 113 with the DFG-out conformation of VEGFR-2.208
Angiogenesis is a complex process triggered by the production of a range of pro-angiogenic and antiangiogenic factors.209 The altered equilibrium between these factors is implicated in the etiology of different diseases, such as cancer.210 The discovery of compounds that are able to block the autophosphorylation of the pro-angiogenic VEGFR-2 has represented a major area of therapeutic intervention for the treatment of several cancers.211
Eldehna and co-workers developed 1-(4-((2-oxoindolin-3-ylidene)amino)phenyl)-3-arylureas as novel agents that are potentially effective against HepG2 hepatocellular carcinoma through VEGFR-2 inhibition. Urea derivative 114 (Figure 22) emerged as the most active compound (IC50 = 0.31 μM), exhibiting good activity against HepG2 cells as well. Docking studies suggested the involvement of the urea linker in two key hydrogen bonding interactions.212
Figure 22.
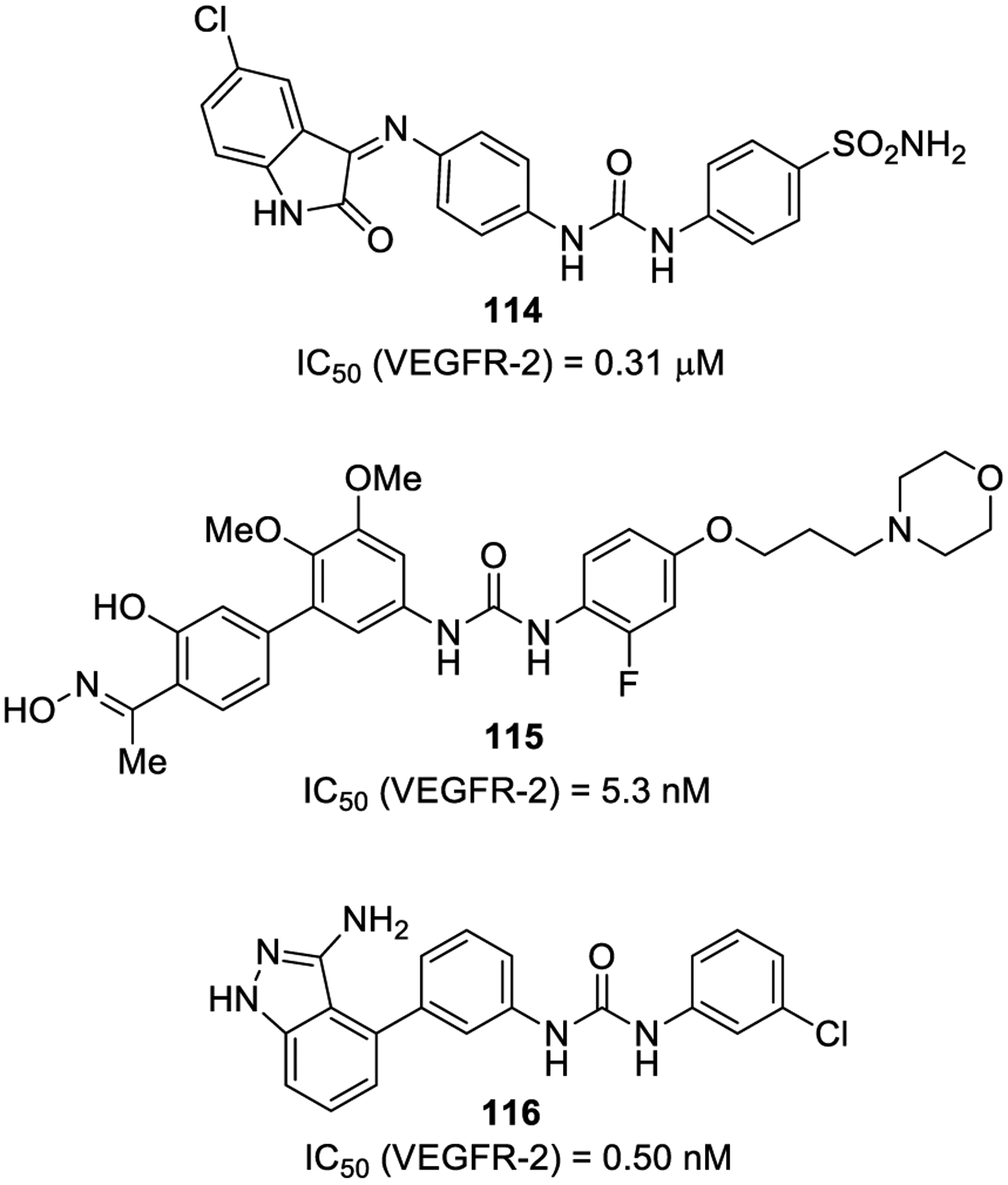
Urea-containing VEGFR-2 inhibitors 114–116.
Gao and co-workers reported a set of VEGFR-2 inhibitors containing a biphenyl urea and an oxime moiety. Urea derivative 115 showed potent enzyme inhibitory activity (IC50 = 5.3 nM) and excellent antiproliferative potency against several cancer cell lines (among them A549, MCF-7, and HT29 cells).212 Structurally, the salicylaldoxime portion allowed the generation of a pseudo six-membered ring formed through intramolecular hydrogen bond interaction between the oxime functionality and the adjacent hydroxyl group in order to mimic the planar quinazoline of known ATP-competitive inhibitors. The intra-molecular hydrogen bond stabilizes the planar conformation and confers structural rigidity. The conformational similarity of salicylaldoxime to quinazoline led to the hypothesis that both of them could act with the hinge region of VEGFR-2. Docking studies highlighted three crucial hydrogen bonding interactions of urea and salicylaldoxime moieties with VEGFR-2.
Further modification and introduction of aromatic-hetero-cyclic templates as hinge-binding fragments via a core-refining approach led to potent inhibitor 116, which showed excellent VEGFR-2 inhibitory activity (IC50 = 0.50 nM) and significant antiproliferative activity (A549 and SMMC-7721 cell lines).205
Ravez and co-workers reported multityrosine kinase inhibitors based on a 7-aminoalkoxy-4-aryloxy-quinazoline urea derivative. Compound 117 (Figure 23) displayed one-digit nanomolar inhibition toward VEGFR-1 (IC50 = 5 nM), VEGFR-2 (IC50 = 4 nM), VEGFR-3 (IC50 = 8 nM), PDGFR-ß (IC50 = 9 nM), and c-Kit (IC50 = 9 nM). This compound also showed excellent activity on PC3, MCF7, and HT29 tumor cell lines and HUVEC (human umbilical vein endothelial cells). The potential of compound 117 to reduce the induction of a weblike network of capillary tubes and invasion was also evaluated in HUVEC cells.213
Figure 23.
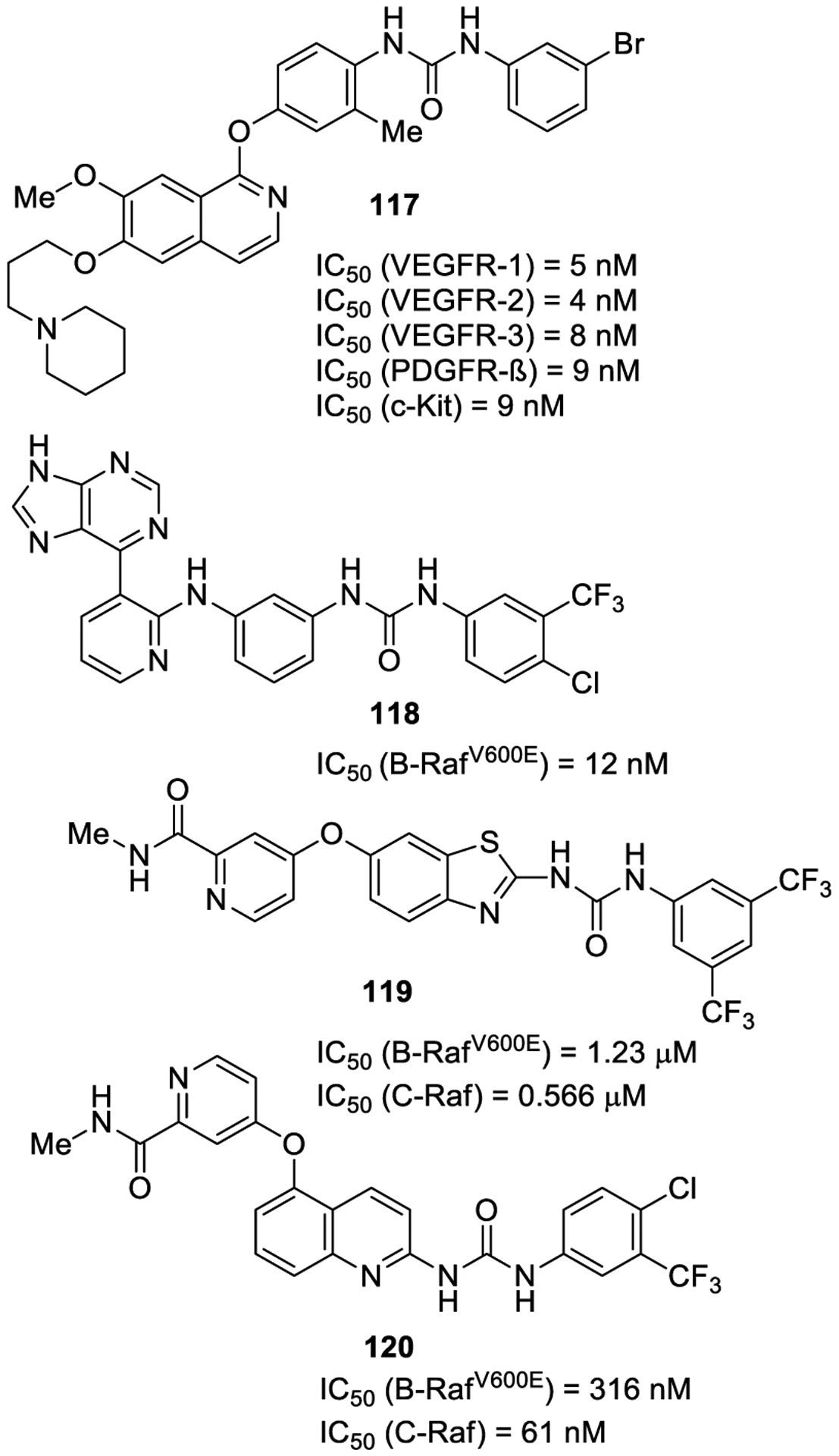
Urea-containing kinase inhibitors as antitumor agents 117–120.
Raf kinases, namely A-Raf, B-Raf, and C-Raf (Raf-1), play a crucial role in the MAPK cascade (Ras-Raf-MEK-ERK), thus regulating cell survival and proliferation.214 In this context, BRaf represents an attractive target for cancer.215 Substitution of a glutamic acid with a valine at residue 600 (V600E) represents the most common aberration in B-Raf (>90%).216 This point mutation is mainly related to melanoma insurgence with a 60% frequency.217
Yang and co-workers developed a series of bis-aryl ureas based upon a 2-amino-3-purinylpyridine moiety as novel DFG-out BRafV600E inhibitors. Compound 118 displayed good inhibitory activity against B-RafV600E (IC50 = 12 nM) and potent antiproliferative activities toward the melanoma A375 cell line (B-RafV600E, IC50 = 27.27 μM). The compound was further optimized and led to a subsequent series of bis-aryl amides active in an A375 xenograft mouse model.218
El-Damasy and collaborators developed benzothiazole amides and ureas linked to a pyridylamide moiety by an aryl-ether tether at the 6-position of the benzothiazole ring. The best compound of the series was the 3,5-bis-trifluoromethylphenyl urea 119, which effectively inhibited B-RafV600E and C-Raf with an IC50 of 1.23 μM and 0.566 μM, respectively. The compound also showed antiproliferative activity on 57 human tumor cell lines. Docking studies indicated comparable binding mode with the two homologous kinases and the role of the urea moiety in forming crucial hydrogen bonding interactions, as well. Profiling on CYP450 and hERG channel ruled out the possibility of drug-drug interactions and cardiac toxicity.219
The same authors further designed 2-amido and ureido quinoline derivatives bearing a 2-N-methylamido-pyridin-4-yloxy group at the 5-position of quinoline. The 4-chloro-3-trifluoromethylphenyl urea derivative 120 was one of the best compounds of the series against BRAFV600E (IC50 values of 316 nM) and C-Raf kinases (IC50 values of 61 nM). Compound 120 showed antiproliferative activity on several cancer cell lines such as RCC and NSCL.
Recently, researchers from Eli Lilly developed the urea-based clinical candidate LY3009120 (121, Figure 24).220 Compound 121 behaves as a type IIa kinase inhibitor toward A-Raf, B-Raf, and C-Raf kinases with an IC50 of 44, 31–47, and 42 nM, respectively. Unlike more selective BRAF inhibitors, compound 121 displayed minimal paradoxical pathway activation in Raf wild-type cells. The compound was found to be active in vivo against B-Raf and K-Ras mutated colorectal cancer xenograft models.221 The compound is currently under evaluation in phase I clinical trials.
Figure 24.
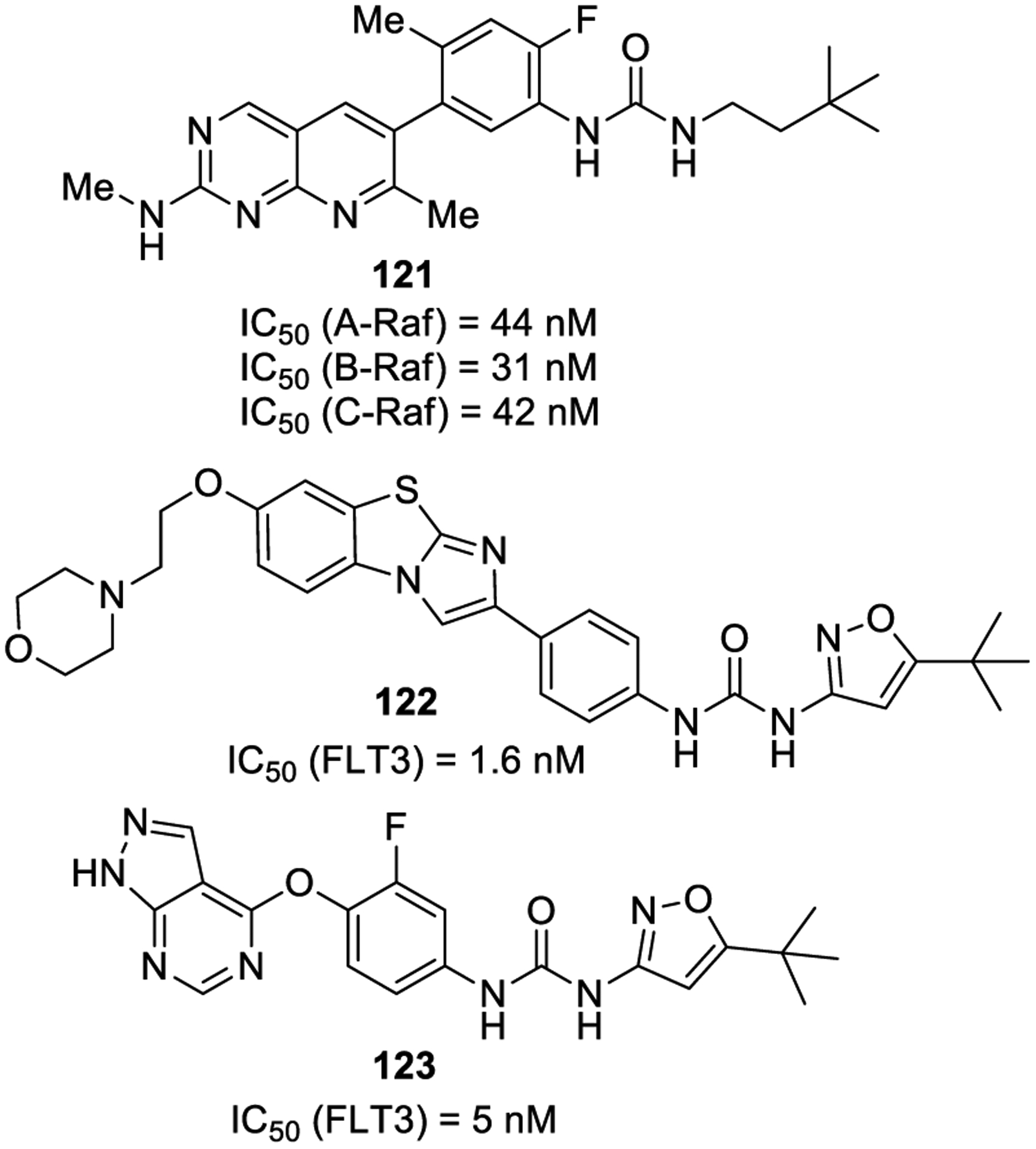
Urea derivatives as kinase inhibitors 121–123.
FMS-like tyrosine kinase 3 (FLT3) belongs to the type III split-kinase domain family of receptor tyrosine kinases (RTKs).222 Upon binding of FLT3 ligand to the receptor, dimerization and autophosphorylation occur, thus prompting FLT3 kinase activation and promoting proliferation of leukemic cells. Therefore, small molecule inhibitors of FLT3 have been explored as a viable therapeutic option for AML.223
Urea derivative 122 (Quizartinib, AC220) has been developed as an exceedingly potent and selective FLT3 inhibitor (IC50 = 1.6 nM) with favorable drug properties and good tolerability in tumor xenografts. Compound 122 has also demonstrated a comparable efficacy and tolerability profile in humans and is presently in phase II clinical trials.224,225
Increased levels of FLT3+ dendritic cells (DCs) are seen in human psoriatic lesions.226 Therefore, inhibition of FLT3 could be potentially exploited as a therapeutic option for psoriasis through interference with DCs. Recently, Li and colleagues developed compound 123 as a potent FLT3 inhibitor with an IC50 value of 5 nM. In vitro studies indicated the ability of 123 to lower the production of DCs and reduce cytokine secretion of DCs. The compound demonstrated promising antipsoriatic effects in vivo in the murine K14-VEGF model, displaying no recurrence 15 days after the last administration.227
7.1.2. Microtubule-Targeting Agents.
Microtubules (MTs) are highly dynamic structures containing heterodimers of α- and β-tubulin. The MT polymerization and depolymerization balance plays a key role in mitosis and vesicular transport.228 Disruption and interference with MT dynamics would lead to mitotic catastrophe causing cell death.229 Several clinical anticancer agents target MTs, including some natural derivatives. Urea functionality has been incorporated in a variety of microtubule targeting agents (MTAs).
Song and co-workers synthesized a series of 3-haloacylamino benzoylurea derivatives as MTAs. 6-Fluoro derivative 124 (Figure 25) showed excellent potency toward nine human cancer cell lines, including Bel-7402 (hepatoma), MCF-7, DU-145, and PC-3 (prostate cancer) and LOVO (colon cancer), DND-1A (melanoma), and MIA Paca (pancreatic cancer) with IC50 values ranging from 0.01 to 0.30 μM. This compound showed a strong anticancer effect against human hepatocarcinoma in a mouse model with over 86% tumor growth inhibition. The direct effects of 124 were assessed on purified tubulin, demonstrating that the compound selectively inhibited the process of MT assembly without affecting the disassembly process.230
Figure 25.
Urea-containing microtubule targeting agents 124–127.
Diaryl urea derivative 125 was reported as a dual acting compound displaying both receptor tyrosine kinase (Flt3, FGFR1, cKit, and PDGFRβ) inhibitory activity and tubulin polymerization enhancer properties. The compound demonstrated antiproliferative effects in several cancer cell lines and inhibited tumor growth in LoVo and HT29 mice xenografts. Compound 125 showed significant antiangiogenic effect, and this may be due to combination of microtubule disruption and VEGFR inhibition.231
Boger and co-workers reported several formerly inaccessible C20′ urea derivatives of vinblastine with the aim of improving their affinity for tubulin and cellular activity. A very interesting finding was the steric tolerance for the size of a C20′ substituent. The importance of the presence of a hydrogen bond donor on the C20′ position was also demonstrated. Moreover, the insertion of a sterically demanding urea functionality is responsible for enhancing potency as much as 10-fold, and can be of great utility to modulate the physicochemical properties of these compounds. Representative compound 126, bearing the large biphenyl urea moiety, binds to tubulin with higher affinity than vinblastine. The compound also displayed good cell growth inhibitory activity against the HCT116 (human colon cancer) and HCT116/VM46 (resistant human colon cancer) cancer cell lines.232 Gagné-Boulet and co-workers incorporated cyclic and acyclic urea substructures while developing combretastatin analogues. Representative compound 127, containing an imidazoline-2-one moiety and a trimethoxyphenyl group, displayed antiproliferative activity in the low nanomolar range on a variety of tumor cell lines (e.g., HT29, M21, MCF7, and HT1080). The compound efficiently arrested the cell cycle in the G2/M phase by binding to the colchicine-binding site and leading to the depolymerization of MTs.233
7.1.3. Urea-Derived Anticancer Agents with Unknown Mechanisms of Actions.
Post-translational modifications of histones can influence chromatin architecture, thus holding a key importance in the regulation of gene expression. Histone acetyltransferases (HATs) and histone deacetylases (HDAC) act on histone lysine tails by attaching or removing acetyl groups, respectively.234 These processes play a key role in controlling cell division, cell-cell communication, survival, and invasion. Therefore, their inhibition is an attractive anticancer strategy.235
Kozikowski and co-workers developed a series of selective urea-based HDAC6 inhibitors. Compound 128 (Figure 26) shows potent inhibitory potency against HDAC6 with an IC50 value of 5.02 nM and a selectivity of ~600-fold with respect to the HDAC1 isoform. This compound inhibited the growth of B16 melanoma cells.236
Figure 26.
Urea containing anticancer agents 128–131.
Schnekenburger and co-workers discovered urea derivative 129 as an HDAC class III sirtuins 1 and 2 (SIRT1/2) inhibitor with IC50 values of 6.2 and 4.2 μM on SIRT1 and 2, respectively. Compound 129 showed comparable potency and was evaluated on the NCI-60 cell line and exhibited promising antiproliferative activity on several cancer cell lines. Moreover, neither mutation of p53 nor overexpression of MDR efflux pumps significantly reduced the anticancer activity of 129. The therapeutic efficacy of 129 was confirmed in 3D glioblastoma spheroids and in zebrafish xenografts.237
Prostate-specific membrane antigen (PSMA) is a well-known biomarker for prostate cancer.238 PSMA expression in prostate cancer is generally 10–100-fold higher than in healthy prostate tissue.239 Harada and co-workers developed unsymmetrical urea-containing compounds targeting PSMA through a nucleophilic conjugate addition between cysteine- and maleimide-containing reagents. Compound 130 ([123I]-IGLCE) displayed high affinity for PSMA, thus supporting the importance of the phenyl ring and succinimidyl moiety.240
Hepsin is a cell surface serine protease overexpressed in the vast majority of prostate tumors.241 PSMA and hepsin may constitute effective targets for imaging in prostate cancer since both of them feature an extracellular enzymatic active site. Azam and co-workers developed the heterobivalent urea containing compound 131, targeting both hepsin and PSMA with IC50 values of 2.8 μM and 28 nM, respectively. In vitro imaging studies demonstrated selective binding and retention to both PSMA and hepsin in cells highly expressing PC3/ML-PSMAHPN.242
7.2. Urea Derivatives for Neurological and Neuro-degenerative Diseases.
Urea derivatives are structural features in many compounds for the treatment of neurological and neurodegenerative diseases such as Parkinson disease (PD), Alzheimer’s disease (AD), and many other CNS disorders. Azam and co-workers designed 3-phenyl/ethyl-2-thioxo-2,3-dihydrothiazolo[4,5-d]pyrimidin-7-yl urea derivatives that reduced haloperidol-induced catalepsy and oxidative stress in a PD animal model. Compound 132 (Figure 27), administered 30 min prior to haloperidol, was able to reduce oxidative stress, restore enzyme activities, and reinstate the levels of glutathione and malondialdehyde.243 Ghiron and co-workers developed urea containing selective agonists of α7 nicotinic acetylcholine receptor for possible treatment of schizophrenia and AD. Compound 133 displayed good in vitro profile. It was investigated in diverse in vivo behavioral models, showing efficacy after oral administration on perceptual processing and cognition and ability to reduce pharmacologically induced deficits.244,245 Further optimization of drug-like properties led to compound 134, which displayed the desired activity/selectivity and showed a good preliminary ADME profile.246
Figure 27.
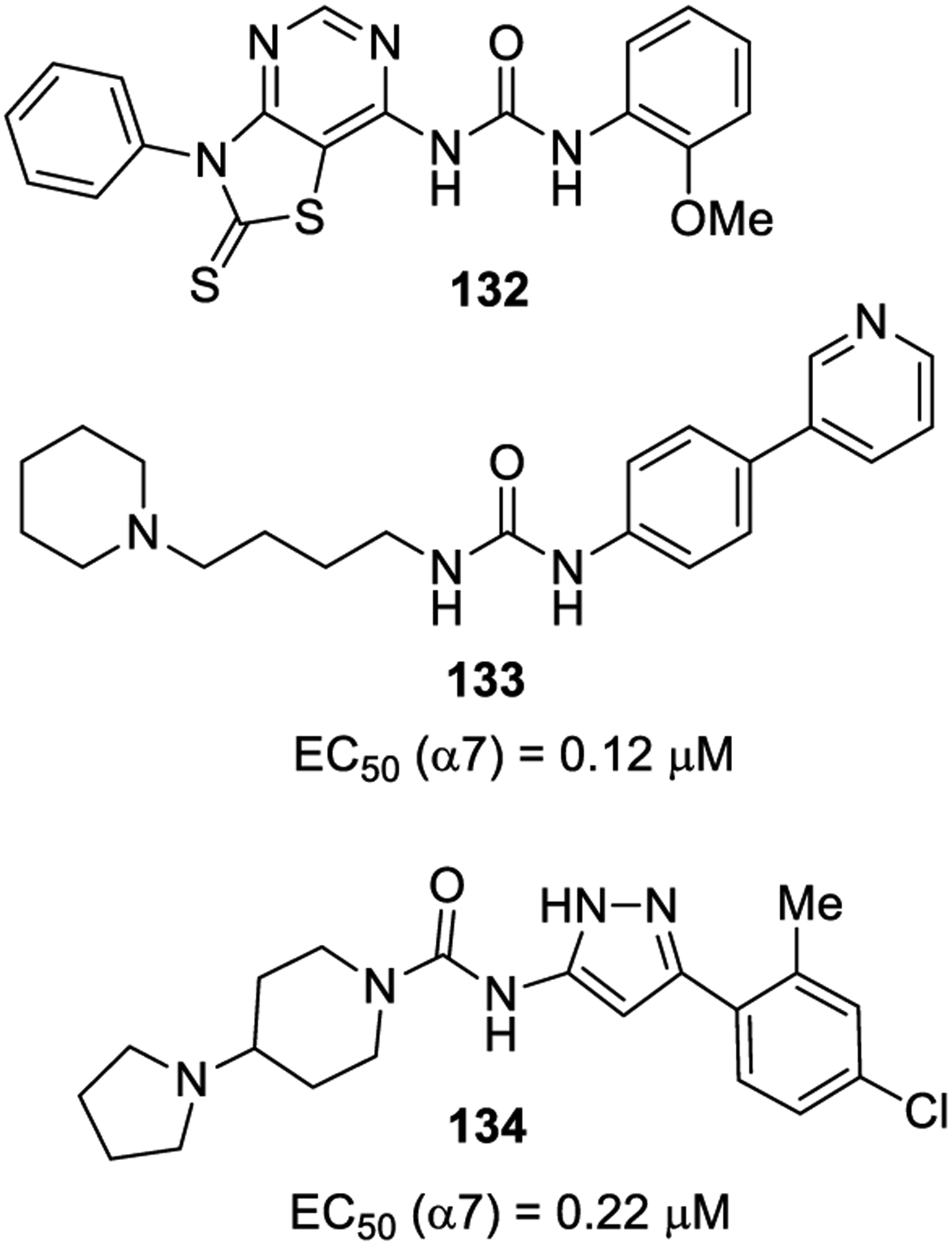
Urea derivatives 132–134 for neurodegenerative diseases.
Elkhamhawy and co-workers developed a series of quinazo-line urea derivatives that block β-amyloid peptide (Aβ)-mediated mitochondrial permeability transition pore (mPTP) opening as evaluated by JC-1 assay. Compound 135 (Figure 28) is the most active nonpeptidyl mPTP blocker possessing improved inhibitory profile with respect to the reference cyclosporin A (CsA), showing potential as a novel AD treatment. Compound 135 showed very low toxicity and excellent cell viability in an MTT assay.247
Figure 28.
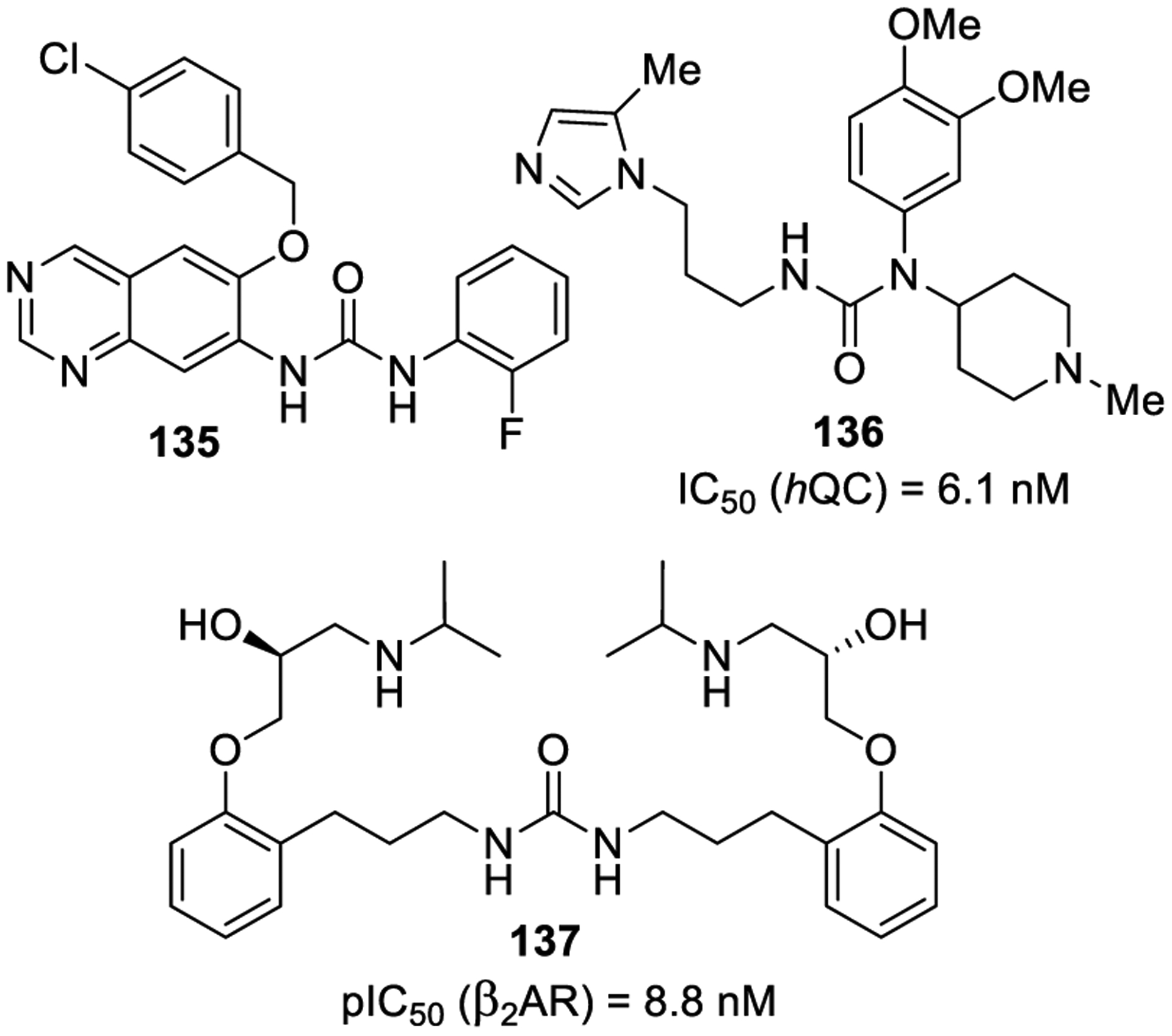
Urea derivatives 135–137 for neurodegenerative diseases.
Recently, Hoang and co-workers developed human glutaminyl cyclase (QC) inhibitors as potent anti-AD agents. Based on the putative active conformation of a prototype inhibitor, novel N-substituted urea derivatives were developed as conformationally restricted analogues, potentially favoring a bent bioactive conformation (Z-E conformer). Among them, compound 136 showed the best inhibitory profile on human QC (IC50 = 6.1 nM) and favorable drug disposition properties.
When assayed in the 5XFAD mouse model, compound 136 effectively reduced the concentrations of pyroform Aβ in the brain after 4 weeks of i.p. administration.248
Disfunction of beta 2-adrenergic receptor (β2AR) has been recently related to AD and other neurodegenerative disorders, although the mechanisms involved are still poorly understood. Gaiser and co-workers recently developed bitopic urea-based ligands in order to target the orthosteric binding site (OBS) and a metastable binding site (MBS) on β2AR. The compounds were designed on the basis of docking studies involving the antagonist (S)-alprenolol into the OBS and a MBS. In general, the conceived ligands showed potency and affinity comparable to a (S)-alprenolol. Among them, bitopic ligand 137 demonstrated pIC50 of 8.8 on β2AR, as determined by a cell-based cAMP assay.249
Kurt and co-workers developed new chemotypes as multi-functional agents in AD. In particular, urea substituted benzofuranylthiazole derivatives were shown to inhibit AChE and BuChE and behave as antioxidant agents. Among them, compound 138 (Figure 29) showed the best inhibitory potency against BuChE with an IC50 value of 2.03 μM, a value 8.5-fold more potent than that of galanthamine. Also, a number of compounds showed good ABTS cation radical scavenging ability.250
Figure 29.
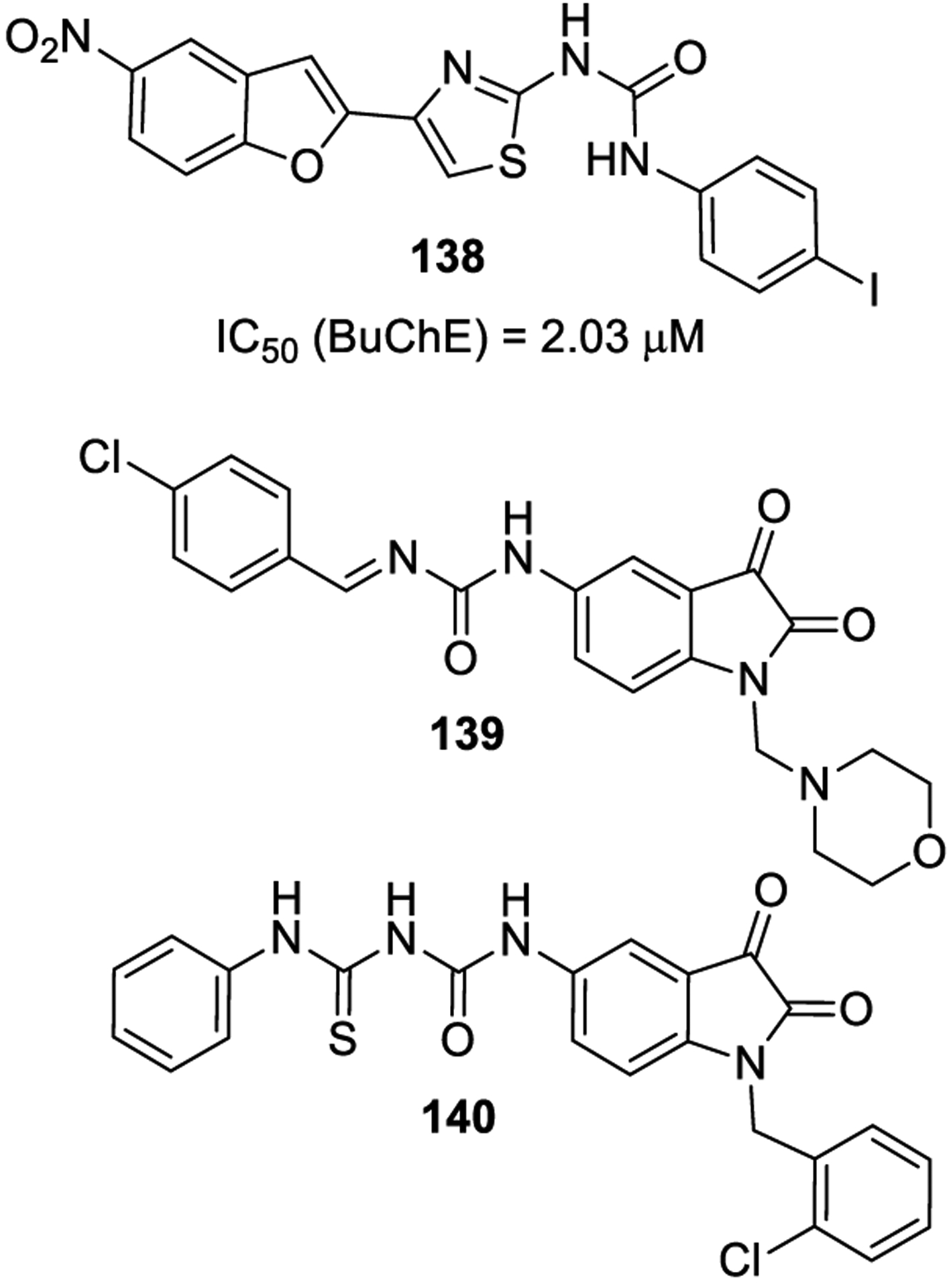
Urea derivatives 138–140 for neurobiological and neurodegenerative diseases.
Prakash and co-workers developed a series of urea derivatives as potential anticonvulsant agents. Compound 139 showed significant activity in the mouse model of epilepsy (MES) revealing protection in the electrically induced seizures at 30 mg/kg and 100 mg/kg 0.5 and 4 h after IP administration, respectively. This compound also displayed protective potential in the subcutaneous pentylenetetrazole model (scPTZ) (at 300 mg/kg at both time intervals), thus emerging as a promising wide-spectrum antiepileptic lead devoid of neurotoxic effects.251 Siddiqui and co-workers reported a series of urea derivatives with anticonvulsant activity. Compound 140 showed relevant anticonvulsant potency at a dose of 100 and 300 mg/kg in both screenings without displaying neurotoxic effects.252
Recently, Amato and co-workers reported the diarylurea-containing compound 141 (VU0463841, Figure 30) as a potent and selective mGluR5 (IC50 = 13 nM, other mGlus >30 μM) negative allosteric modulator with favorable CNS penetration in rats. The efficacy of 141 was demonstrated in suitable rat models of cocaine addiction. In particular, IP dosing of 141 in rats led to therapeutically relevant brain exposures, with 141 being able to dose-dependently reduce cocaine self-administration and cocaine place preference.253 Xu and co-workers identified urea-based compound 142 as a mean CNS-penetrating CXCR2 antagonist. The compound demonstrated promising activity upon oral administration in a cuprizone model of demyelination, thus validating the role of CXCR2 as promising target for the treatment of multiple sclerosis.254
Figure 30.
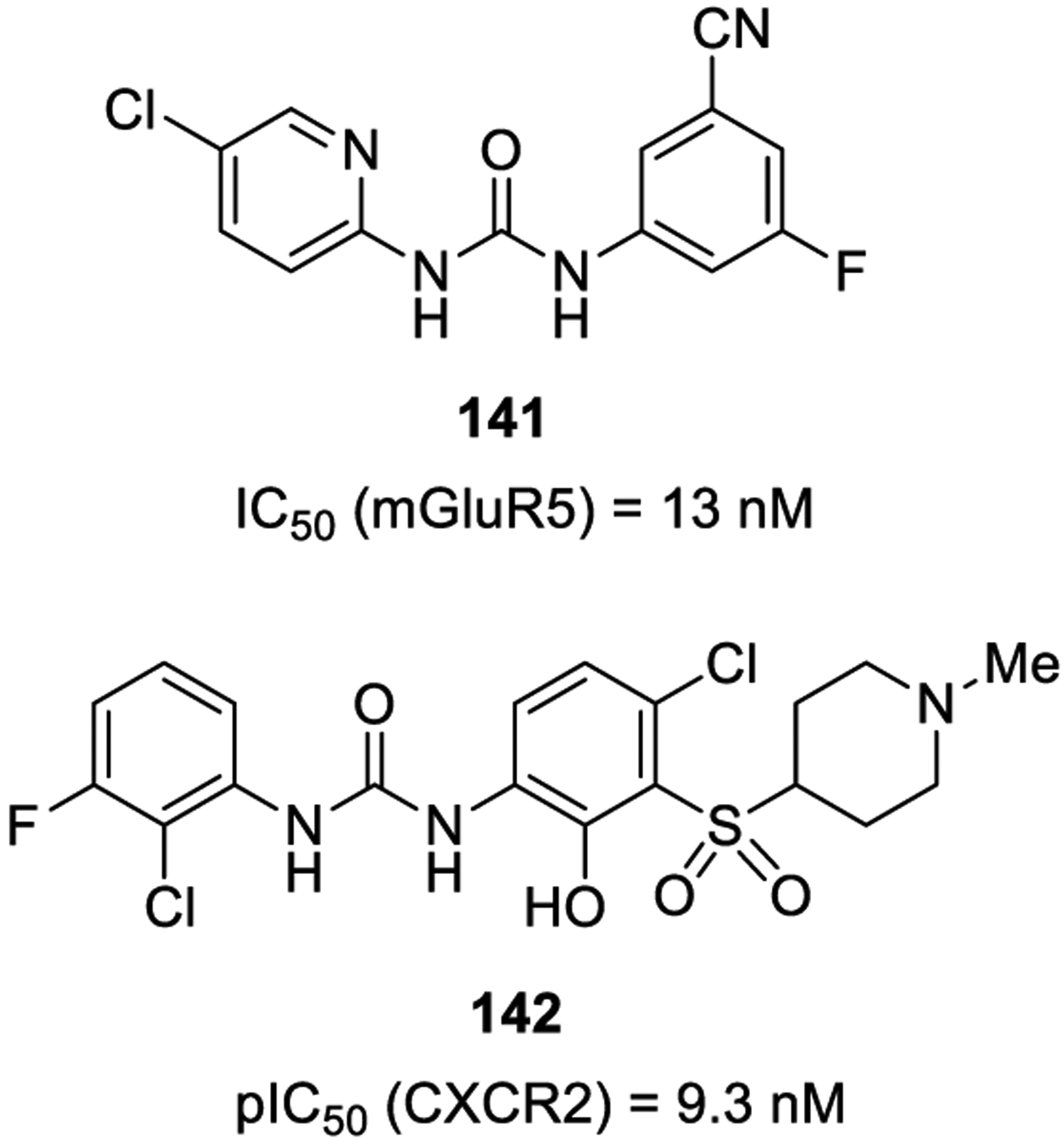
Urea derivatives 141 and 142 for neurodegenerative diseases.
7.3. Urea-Containing Antiviral, Antibacterial, and Antiparasitic Compounds.
The urea functionality is a structural feature in a variety of antiviral, antibacterial, and antiparasitic agents. We provide recent examples of urea containing compounds in the design and development of these agents.
7.3.1. Antiviral Agents.
Duan and co-workers reported the design of CCR5 antagonists based on a cyclic urea structure. These derivatives were designed relying upon the rationale that five- and six-membered cyclic ureas would function as stable pharmacophores. The transition from acyclic to cyclic ureas not only substantially improved the chemical stability but also led to the development of compound 143 (Figure 31) that showed very good in vivo pharmacokinetic properties.255 A number of compounds showed high antiviral potency against HIV-1 in both human osteosarcoma (HOS) cell and peripheral blood lymphocyte (PBL) cell assays.
Figure 31.
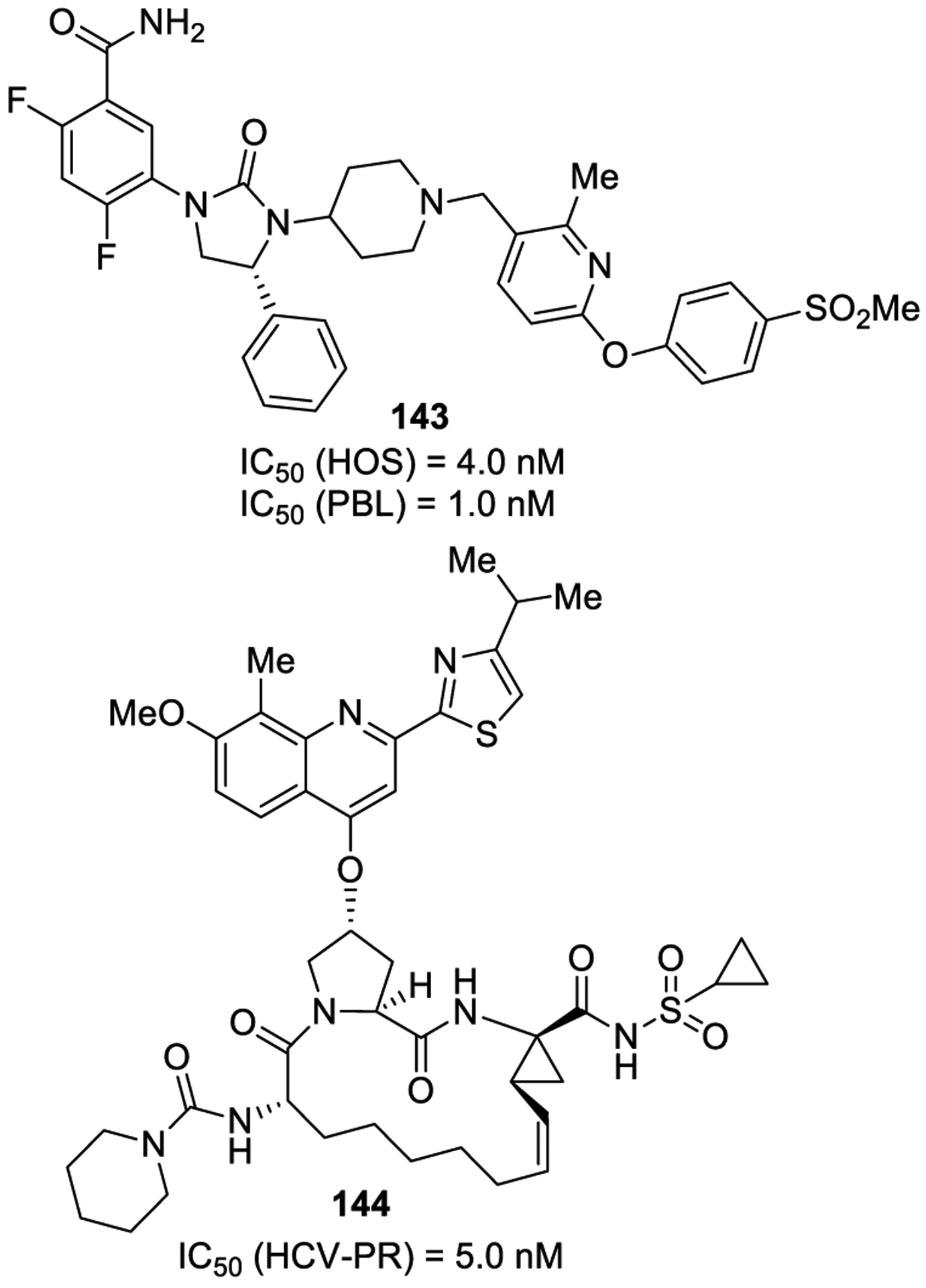
Urea-containing antiviral agents 143 and 144.
Kazmierski and co-workers developed urea-based potent HCV protease inhibitors. A number of derivatives, including 144, established improved potency in HCV replicon assays with respect to second generation inhibitor, such as danoprevir. Oral administration of compound 144 showed favorable drug concentrations in rat liver, suggesting promising anti-HCV and pharmacokinetic profiles.256
7.3.2. Antibacterial Agents.
DNA gyrase and topoisomerase IV (topoIV) attracted great attention as targets for effective antibiotics.257 They are heterotetrameric type II topoisomerase enzymes that can modify chromosome structure through breaking and joining DNA double strands. These enzymes are also important for bacterial DNA replication.258,259 Charifson and co-workers developed benzimidazole ureas as dual targeting agents exerting simultaneous inhibition of the ATPase function of topoisomerase IV and gyrase. Urea-derivative 145 (Figure 32) showed an E. coli Gyrase Ki value of 6 nM and S. aureus TopoIV Ki value of 6 nM. Several related compounds displayed strong antibacterial activity toward clinically relevant, multidrug resistant strains. Inhibitor 145 possesses low in vitro spontaneous resistance frequencies and significant bactericidal activity. The compound was also shown to be effective upon oral and intravenous administration in mouse models of pneumonia and skin infections.260
Figure 32.
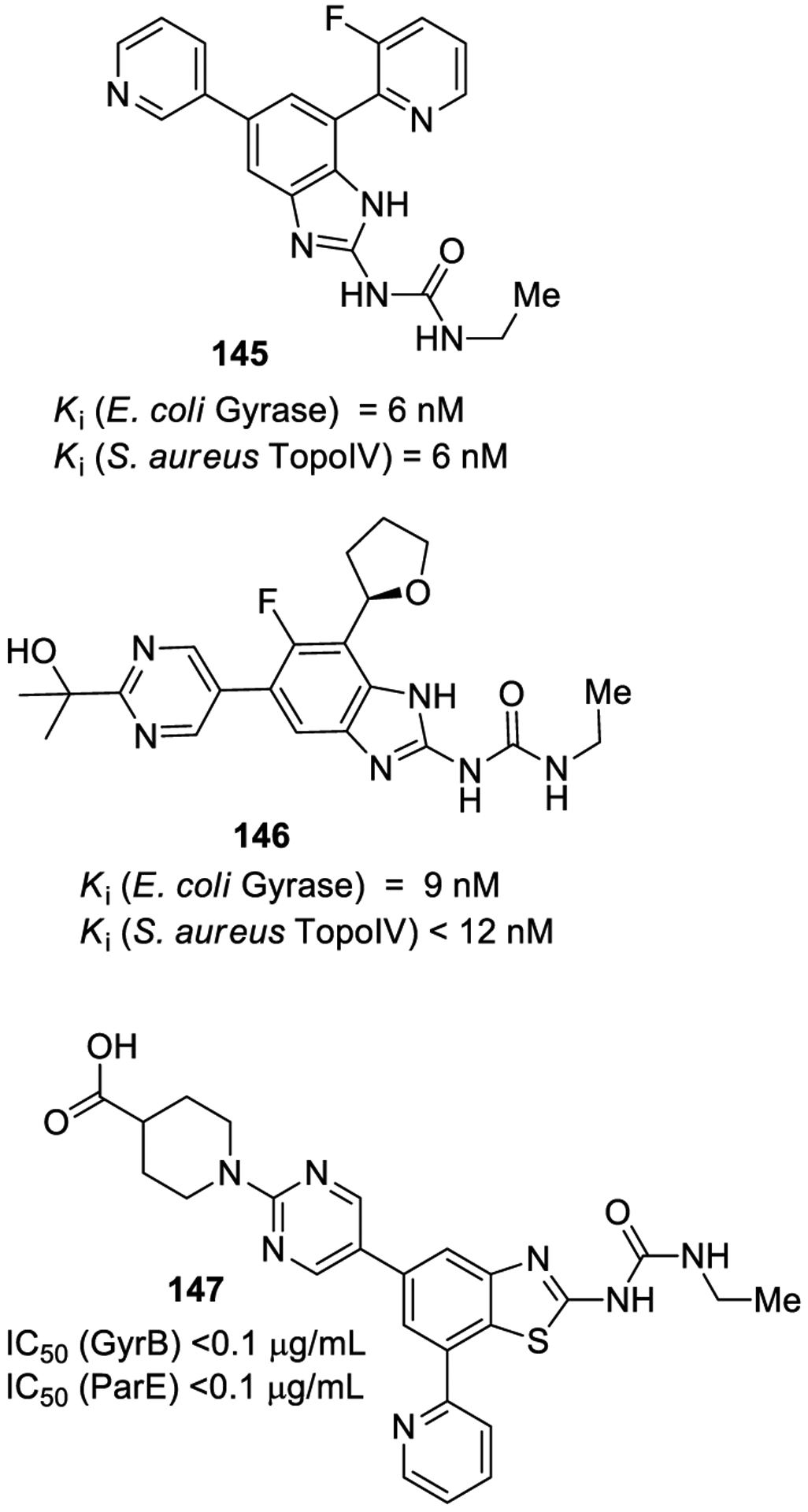
Urea-derivatives 145–147 as antibacterial agents.
Compound 145 behaved as a potent CYP3A4 inhibitor and displayed poor drug disposition properties. Subsequently, a number of second-generation aminobenzimidazole ureas were designed to address these issues. Compound 146 provided a dose- and time-dependent antibacterial effect. Also, investigation of metabolic properties of 146 showed that unlike compound 145, metabolic degradation occurs away from the urea functionality.261
Stokes and co-workers replaced the aminobenzimidazole substructure and developed benzothiazole ethyl urea derivatives such as compound 147 which inhibited both DNA gyrase (GyrB) and topoisomerase IV (ParE) with IC50 values of <0.1 μg/mL. The compound did not display affinity for human topoisomerase II. Moreover, no cytotoxicity toward HepG2 cells was observed. Compound 147 showed antibacterial activity against several Gram-positive bacterial strains with minimal inhibitory concentrations (MICs) ranging from 0.008 to 0.12 μg/mL. However, it showed contrasting outcomes against Gram-negative organisms.262
Yule and co-workers reported the synthesis and SAR of pyridine-3-carboxamide-6-yl ureas targeting DNA gyrase. Compound 148 (Figure 33) demonstrated broad-spectrum activity on Gram positive microorganisms and antibacterial activity against a Gram-negative efflux pump mutant and single-step GyrB/ParE mutants.263 Linezolid was the first FDA-approved oxazolidinone antibiotic approved in 2000, and tedizolid followed after (2014). Linezolid is used to treat serious Gram-positive infections such as those caused by vancomycin-resistant Enterococcus faecalis (VRE) and methicillin-resistant Staphylococcus aureus (MRSA).264 However, linezolid-resistant strains have been described.265–267 In an effort to develop more effective oxazolidinone antibiotics, Suzuky and co-workers reported urea-substituted [1,2,5]-triazepane or [1,2,5]oxadiazepine derivatives of linezolid. A number of derivatives showed potent antibacterial activity in vitro, and a subset underwent in vivo testing on a mouse model infected with S. aureus SR3637 strain. Compound 149 showed the best in vivo therapeutic effect. Furthermore, compound 149 displayed negligible monoamine oxidase (MAO)-inhibitory activity. The seven-membered heterocycles linked to a urea moiety appear to be a valuable structural template.268
Figure 33.
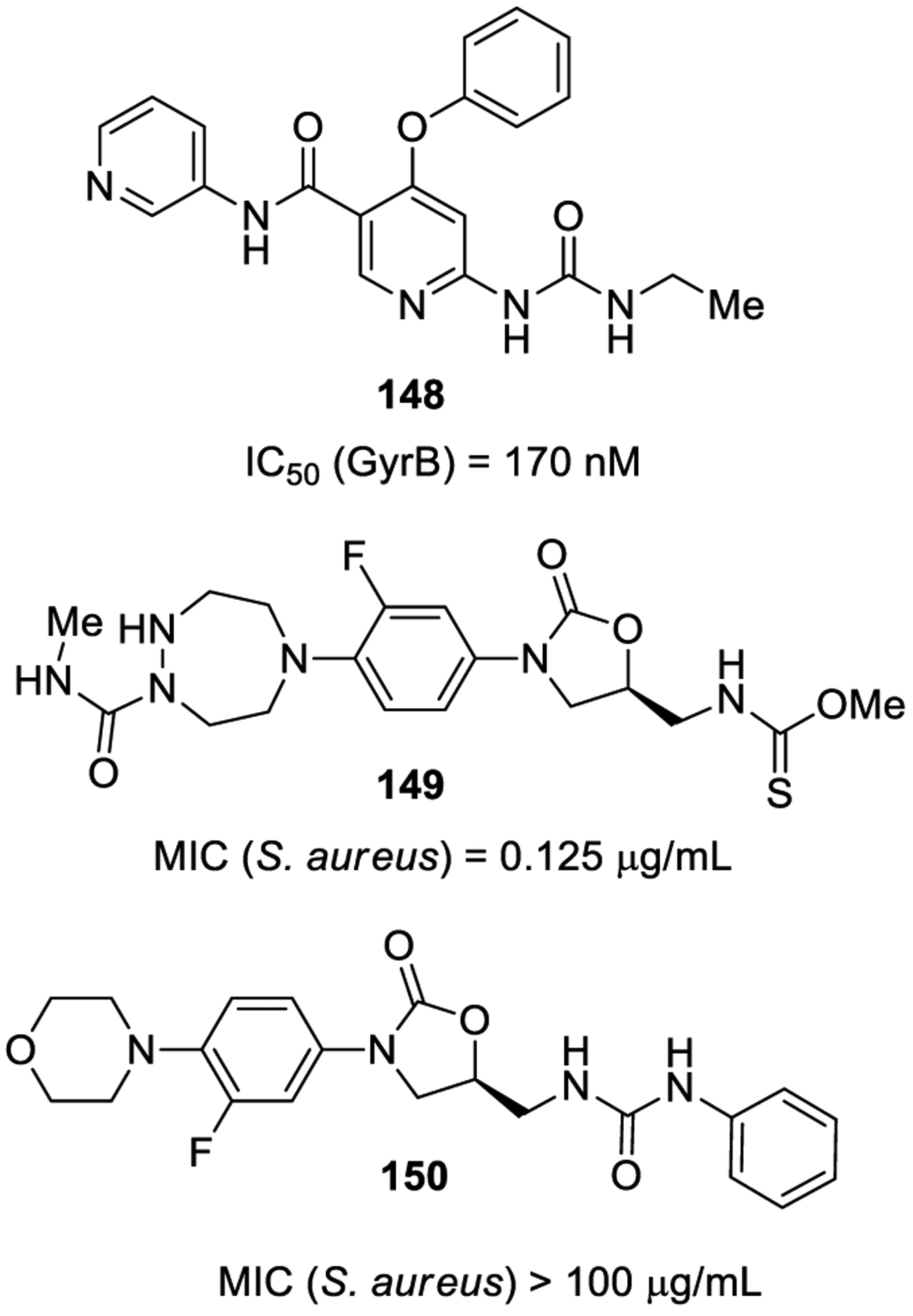
Urea-containing antibacterial agents 148–150.
De Rosa and co-workers also reported linezolid analogues.269 They performed molecular modeling studies in order to design derivatives with improved binding, particularly to ribosomes carrying mutations conferring partial or complete resistance to linezolid. In general, thiourea derivatives displayed better activities than their urea counterparts. Representative compound 150 showed a MIC higher than 100 μg/mL.
7.3.3. Antiparasitic Agents.
Malaria is a parasitic disease caused by parasites of the genus Plasmodium, and it represents a major global health and economic problem. Although several drugs are used therapeutically to treat malaria, all have now succumbed to parasite drug resistance, therefore displaying significantly decreased efficacy.270–272
Several alkylated (bis)urea polyamine derivatives were reported by Verlinden and co-workers.273 Compounds were tested for antimalarial potency against chloroquine (CQ)-sensitive and CQ-resistant strains of Plasmodium falciparum. The majority of compounds are structural analogs of compound 151 (Figure 34). These compounds displayed inhibitory activity against P. falciparum in the 100–650 nM range. Compound 151 was active against CQ-resistant P. falciparum. The compounds in this class also showed excellent selectivity toward the parasite (>7000-fold lower IC50 against P. falciparum) compared to the HepG2 mammalian cell line.
Figure 34.
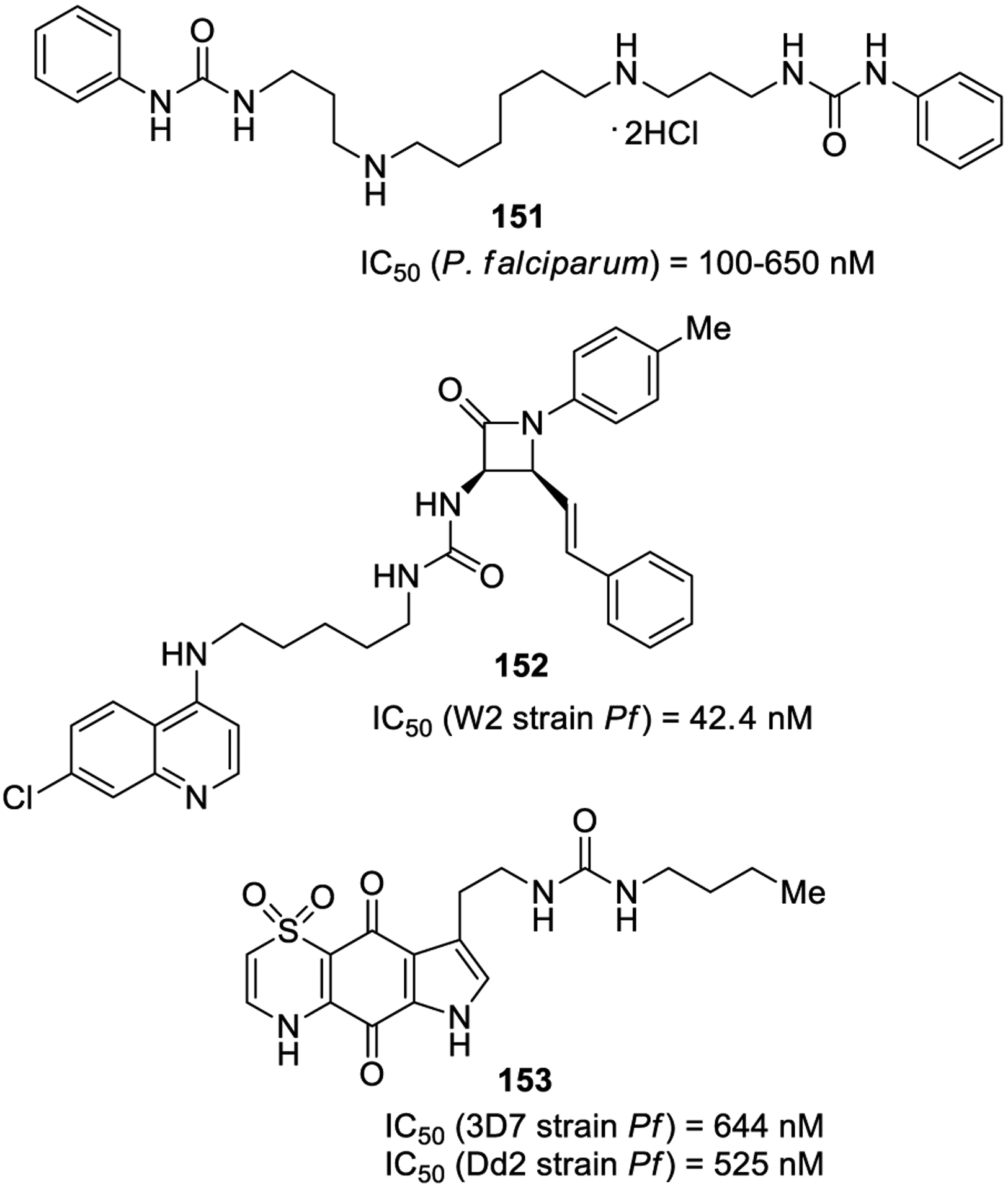
Urea-containing antimalarial agents 151–153.
Recently, Singh and co-workers reported β-lactam-4-amino-quinoline conjugates containing urea/oxalamide linkers as antimalarial agents.274 The presence of a substituent on the N-1 nitrogen of the β-lactam ring, along with the linker structure, and the nature and length of the alkyl chain influenced the activity profiles as well as the physicochemical properties. Urea derivative 152 is the most potent compound and displayed an IC50 of 42.4 nM against the CQ-resistant W2 strain of P. falciparum.
A series of urea derivatives based upon the thiaplakortone A tricyclic scaffold was reported by Schwartz and co-workers.275 Compounds were assayed for their in vitro antimalarial potential and mammalian cell toxicity. ADME profiling was also evaluated for several analogues. Compound 153 showed an IC50 value of 644 nM against 3D7 strain. This compound was well tolerated in a mouse model when administered subcutaneously at 32 mg/kg twice daily for 4 days. At this dose, compound 153 also suppressed blood stage P. berghei by 26%. However, oral administration at the same dose did not suppress parasitemia, most likely due to poor oral bioavailability.
Human African trypanosomiasis (HAT) is a parasitic disease caused by Trypanosoma brucei gambiense or Trypanosoma brucei rhodesiense. The World Health Organization (WHO) currently counts 20000 cases with 65 million people at infection risk.276 There is an urgent need for innovative anti-HAT drugs.
Patrick and co-workers reported 2-(2-benzamido)ethyl-4-phenylthiazole derivative 154 (Figure 35), as a promising hit against Trypanosoma brucei.277 However, 154 displayed poor metabolic stability. Further optimization led to urea-containing compound 47, which exhibited good in vitro potency with an EC50 value of 34.8 nM. This compound did not show any significant toxicity toward CRL-8155 and HepG2 human cell lines. It showed excellent half-lives in human and mouse liver microsomes (at least 60 min). Compound 47 also demonstrated promising oral bioavailability and optimum brain penetration in mice. Following oral administration, compound 47 led to 5/5 cures in both acute and chronic HAT murine models.124
Figure 35.
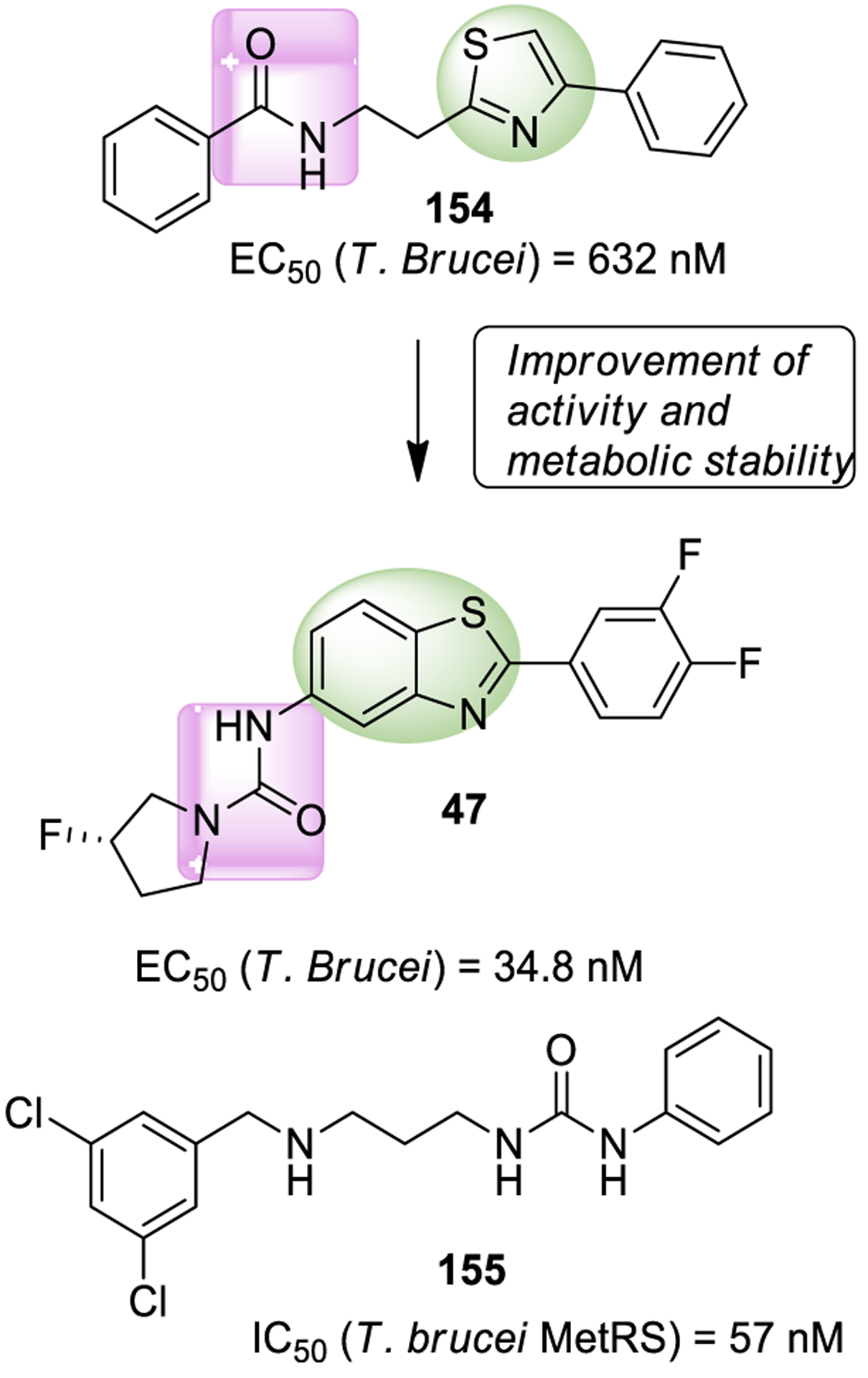
Urea-containing antitripanosomal agents 47, 154, and 155.
Shibata and co-workers reported methionyl-tRNA synthetase (MetRS) inhibitors based on a urea structure and assessed their potential against HAT.278 Urea derivative 155 showed high preference for the parasitic enzyme with respect to the closest human homologue (IC50 T. brucei MetRS = 57 nM, IC50 human mitochondrial MetRS > 10 μM). Compound 155 also inhibited parasite growth (EC50 = 450 nM) while displaying low toxicity toward mammalian cells (CRL-8155 and HepG2). Furthermore, compound 155 showed CNS penetration in mice and also showed optimum membrane permeation ability when tested in the MDR1-MDCKII model.
8. CONCLUSION
The historic first synthesis of urea by Wöhler occurred nearly 200 years ago. Today, urea and its derivatives are substructures in numerous approved drugs. The many applications of urea derivatives in modern medicinal chemistry and drug development demonstrate the broad scope and utility of urea containing compounds. The urea functionality is quite unique, and it is involved in critical drug-target interactions and modulation of drug properties. In this perspective, we highlighted the role of the urea functionality in medicinal chemistry, provided an overview of physiochemical properties, and emphasized its involvement in the formation of donor-acceptor hydrogen bonding interactions with the target enzymes or receptors. We outlined the predominant role of the urea substructure in approved drugs and compounds with clinical potential. Furthermore, we showcased recent designs and applications of urea derivatives in the development of numerous anticancer and anti-infective agents as well as agents for the potential treatment of neurodegenerative diseases. We hope that the present perspective will further stimulate the use of urea in modern medicinal chemistry and drug design.
ACKNOWLEDGMENTS
Financial support of this research by the National Institutes of Health (AI150466, AKG) and Purdue University is gratefully acknowledged. We also acknowledge MIUR Grant Dipartimento di Eccellenza (Department of Excellence, MB) 2018-2022 to the Department of Pharmacy, University of Naples Federico II. We would like to thank Suzanne Snoeberger for her help with the manuscript preparation. The authors also thank William Robinson, Josh Born, and Hannah Simpson (Purdue University) for helpful discussions.
ABBREVIATIONS USED
- Aβ
amyloid beta
- AD
Alzheimer disease
- AKR
aldo-keto reductase
- AML
acute myeloid leukemia
- BBB
blood-brain barrier
- [bmim][BF4]
1-butyl-3-methylimidazolium tetrafluoroborate
- BTC
bis(trichloromethyl)carbonate
- CCR5
chemokine receptor 5
- CDI
N,N′-carbonyldiimidazole
- CGRP
calcitonin gene-related peptide
- CNS
central nervous system
- CQ
chloroquine
- CsA
cyclosporin A
- CYP450
cytochrome P450
- DBAD
dibenzyl azodicarboxylate
- DBU
1,8-diazabicyclo(5.4.0)undec-7-ene
- DC
dendritic cell
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DIPPU
N,N’-di(2,6-diisopropylphenyl)urea
- DMAP
4-(dimethylamino)pyridine
- DMF
dimethylformamide
- DMDTC
S,S-dimethyldithiocarbonate
- DMSO
dimethyl sulf-oxide
- DNA
DNA
- DPPA
diphenylphosphoryl azide
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- sHE
soluble epoxide hydrolase
- EGFR
epidermal growth factor receptor
- FAAH
fatty acid amide hydrolase
- FDA
Food & Drug Administration (US)
- FLT3
FMS-like tyrosine kinase 3
- FTIR
Fourier transform infrared
- HATs
histone acetyltransferase
- HAT
Human African trypanosomiasis
- HCV
hepatitis C virus
- HDAC
histone deacetylases
- HIV
human immunodeficiency virus
- HOMO
highest occupied molecular orbital
- HOS
human osteosarcoma
- HSDD
hypoactive sexual desire disorder
- IR
infrared
- KHMDS
potassium bis(trimethylsilyl)amide
- LDA
lithium diisopropylamide
- 5-LOX
5-lipoxygenase
- LUMO
lowest unoccupied molecular orbital
- MAGL
moanoacylglycerol lipase
- MAO
monoamine oxidase
- MDR
multidrug resistant
- MES
maximal electroshock seizure
- MetRS
methionyl-tRNA synthetase
- MIC
minimal inhibitory concentration
- MRSA
methicillin-resistant Staphylococcus aureus
- MT
micro-tubule
- MTA
microtubule targeting agent
- NBS
N-bromosuccinimide
- 4-NBsOXY
2-cyano-2-(4 -nitrophenylsulfonyloxyimino)acetate
- NMR
nuclear magnetic resonance
- NS3
nonstructural protein 3
- PBL
peripheral blood lymphocytes
- PD
Parkinson’s disease
- PDGFR
platelet derived growth factor receptor
- P-gp
P-glycoprotein
- PSMA
prostate-specific membrane antigen
- PTP
permeability transition pore
- RNA
ribonucleic acid
- RTK
receptor tyrosine kinase
- SAR
structure-activity relationships
- scPTZ
subcutaneous pentylenetetrazole
- SET
single-electron transfer
- SIRT
sirtuin
- SPECT
single photon emission computed tomography
- TBHP
tert-butyl hydroperoxide
- TEA
triethylamine
- TFA
trifluoroacetic acid
- TFAA
trifluoroacetic anhydride
- THF
tetrahydrofuran
- T3P
1-propanephosphonic acid cyclic anhydride
- UGT1A9
UDP-glucuronosyltransferase 1A9
- VEGFR2
vascular endothelial growth factor receptor-2
- VRE
vancomycin-resistant Enterococcus faecalis
- WHO
World Health Organization
Biographies
Arun K. Ghosh obtained his BS and MS with honors in Chemistry from the University of Calcutta and Indian Institute of Technology, Kanpur, India, respectively. He received his Ph.D. in chemistry from the University of Pittsburgh (1985). He then carried out postdoctoral research in Professor E. J. Corey’s laboratory at Harvard University (1985–1988). He was a research fellow at Merck prior to joining the chemistry faculty at the University of Illinois, Chicago as an Assistant Professor in 1994. In 2005, he joined the Department of Chemistry and Medicinal Chemistry at Purdue University, West Lafayette. In 2009, he became the Ian P. Rothwell Distinguished Professor of Chemistry and Medicinal Chemistry. His research interests are in broad areas of synthetic organic, bioorganic, and medicinal chemistry.
Margherita Brindisi graduated cum laude from the University of Siena, where she also received her Ph.D. in Pharmaceutical Sciences in 2008. In 2010–2011, she worked as a postdoctoral fellow in the group of Professor Arun Ghosh at Purdue University on the development of aspartyl protease inhibitors. From 2012 to 2015 she worked as a fixed-term Researcher at the Department of Biotechnology, Chemistry and Pharmacy at the University of Siena on the development of novel treatments for cancer, parasitic diseases, and brain disorders. In 2016–2017, she again moved to Purdue University as a Visiting Researcher. Dr. Brindisi currently holds a position as Assistant Professor at the Department of Excellence of Pharmacy at the University of Naples Federico II.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Wöhler F Ueber künstliche bildung des harnstoffs. Ann. Phys 1828, 88, 253–256. [Google Scholar]
- (2).Ramberg PJ The death of vitalism and the birth of organic chemistry: Wohler’s urea synthesis and the disciplinary identity of organic chemistry. Ambix 2000, 47, 170–195. [DOI] [PubMed] [Google Scholar]
- (3).Burger A History and Economics of Medicinal Chemistry In Medicinal Chemistry, 3rd ed.; Burger A, Ed.; John Wiley & Sons: Hoboken, NJ, 1970; Vol 1, pp 4–19. [Google Scholar]
- (4).Lombardino JG; Lowe JA 3rd The role of the medicinal chemist in drug discovery-then and now. Nat. Rev. Drug Discovery 2004, 3, 853–862. [DOI] [PubMed] [Google Scholar]
- (5).Gallou I Unsymmetrical ureas. Synthetic methodologies and application in drug design. Org. Prep. Proced. Int 2007, 39, 355–383. [Google Scholar]
- (6).Regan J; Breitfelder S; Cirillo P; Gilmore T; Graham AG; Hickey E; Klaus B; Madwed J; Moriak M; Moss N; Pargellis C; Pav S; Proto A; Swinamer A; Tong L; Torcellini C Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate. J. Med. Chem 2002, 45, 2994–3008. [DOI] [PubMed] [Google Scholar]
- (7).Ghosh AK; Gemma S Structure-Based Design of Drugs and Other Bioactive Molecules: Tools and Strategies; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- (8).Hubbard RE Structure-Based Drug Discovery: An overview; RSC Publishing: London, England, 2006. [Google Scholar]
- (9).Steverding D The development of drugs for treatment of sleeping sickness: a historical review. Parasites Vectors 2010, 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Williamson J In Review of Chemotherapeutic and Chemoprophylactic Agents. The African trypanosomiasis; Mulligan HW, Ed.; Allen & Unwin: London, 1970. [Google Scholar]
- (11).Katsilambros N; Diakoumopoulou E; Ioannidis I; Liatis S; Makrilakis K; Tentolouris N; Tsapogas P Diabetes in Clinical Practice: Questions and Answers from Case Studies; John Wiley & Sons: Hoboken, NJ, 2007; p 342. [Google Scholar]
- (12).Fabian MA; Biggs WH 3rd; Treiber DK; Atteridge CE; Azimioara MD; Benedetti MG; Carter TA; Ciceri P; Edeen PT; Floyd M; Ford JM; Galvin M; Gerlach JL; Grotzfeld RM; Herrgard S; Insko DE; Insko MA; Lai AG; Lelias JM; Mehta SA; Milanov ZV; Velasco AM; Wodicka LM; Patel HK; Zarrinkar PP; Lockhart DJ A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- (13).Mori S; Cortes J; Kantarjian H; Zhang W; Andreef M; Ravandi F Potential role of sorafenib in the treatment of acute myeloid leukemia. Leuk. Lymphoma 2008, 49, 2246–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Malcolm BA; Liu R; Lahser F; Agrawal S; Belanger B; Butkiewicz N; Chase R; Gheyas F; Hart A; Hesk D; Ingravallo P; Jiang C; Kong R; Lu J; Pichardo J; Prongay A; Skelton A; Tong X; Venkatraman S; Xia E; Girijavallabhan V; Njoroge FG SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob. Agents Chemother 2006, 50, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jensen DM A new era of hepatitis C therapy begins. N. Engl. J. Med 2011, 364, 1272–1274. [DOI] [PubMed] [Google Scholar]
- (16).Nowick JS; Mahrus S; Smith EM; Ziller JW Triurea derivatives of diethylenetriamine as potential templates for the formation of artificial beta-sheets. J. Am. Chem. Soc 1996, 118, 1066–1072. [Google Scholar]
- (17).Holmes DL; Smith EM; Nowick JS Solid-phase synthesis of artificial beta-sheets. J. Am. Chem. Soc 1997, 119, 7665–7669. [Google Scholar]
- (18).Claudon P; Violette A; Lamour K; Decossas M; Fournel S; Heurtault B; Godet J; Mely Y; Jamart-Gregoire B; Averlant-Petit MC; Briand JP; Duportail G; Monteil H; Guichard G Consequences of isostructural main-chain modifications for the design of antimicrobial foldamers: helical mimics of host-defense peptides based on a heterogeneous amide/urea backbone. Angew. Chem., Int. Ed 2010, 49, 333–336. [DOI] [PubMed] [Google Scholar]
- (19).Fischer L; Claudon P; Pendem N; Miclet E; Didierjean C; Ennifar E; Guichard G The canonical helix of urea oligomers at atomic resolution: insights into folding-induced axial organization. Angew. Chem., Int. Ed 2010, 49, 1067–1070. [DOI] [PubMed] [Google Scholar]
- (20).Chen YY; Chang LT; Chen HW; Yang CY; Hsin LW Fast and facile synthesis of 4-nitrophenyl 2-azidoethylcarbamate derivatives from N-Fmoc-protected alpha-amino acids as activated building blocks for urea moiety-containing compound library. ACS Comb. Sci 2017, 19, 131–136. [DOI] [PubMed] [Google Scholar]
- (21).Burgess K; Linthicum DS; Shin HW Solid-phase syntheses of unnatural biopolymers containing repeating urea units. Angew. Chem., Int. Ed. Engl 1995, 34, 907–909. [Google Scholar]
- (22).Zhang Z; Schreiner PR (Thio)urea organocatalysis-what can be learnt from anion recognition? Chem. Soc. Rev 2009, 38, 1187–1198. [DOI] [PubMed] [Google Scholar]
- (23).Serdyuk OV; Heckel CM; Tsogoeva SB Bifunctional primary amine-thioureas in asymmetric organocatalysis. Org. Biomol. Chem 2013, 11, 7051–7071. [DOI] [PubMed] [Google Scholar]
- (24).Wu CY; Cheng HY; Liu RX; Wang QA; Hao YF; Yu YC; Zhao FY Synthesis of urea derivatives from amines and CO2 in the absence of catalyst and solvent. Green Chem. 2010, 12, 1811–1816. [Google Scholar]
- (25).Jagtap AD; Kondekar NB; Sadani AA; Chern JW Ureas: applications in drug design. Curr. Med. Chem 2017, 24, 622–651. [DOI] [PubMed] [Google Scholar]
- (26).Sikka P; Sahu JK; Mishra AK; Hashim SR Role of aryl urea containing compounds in medicinal chemistry. Med. Chem 2015, 5, 479–483. [Google Scholar]
- (27).Ganis P; Avitabile G; Benedetti E; Pedone C; Goodman M Crystal and molecular structure of N,N’-diethyl- N,N’-diphenylurea. Proc. Natl. Acad. Sci. U. S. A 1970, 67, 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Matsumura M; Tanatani A; Azumaya I; Masu H; Hashizume D; Kagechika H; Muranaka A; Uchiyama M Unusual conformational preference of an aromatic secondary urea: solvent-dependent open-closed conformational switching of N,N’-bis-(porphyrinyl)urea. Chem. Commun. (Cambridge, U. K.) 2013, 49, 2290–2292. [DOI] [PubMed] [Google Scholar]
- (29).Bohmer V; Meshcheryakov D; Thondorf I; Bolte M 3-oxa-6,8-diaza-1,2:4,5-dibenzo-cycloocta-1,4-dien-7-one: a three-dimensional network assembled by hydrogen-bonding, pi-pi and edge-to-face interactions. Acta Crystallogr., Sect. C: Cryst. Struct. Commun 2004, 60, O136–O139. [DOI] [PubMed] [Google Scholar]
- (30).Meshcheryakov D; Arnaud-Neu F; Bohmer V; Bolte M; Hubscher-Bruder V; Jobin E; Thondorf I; Werner S Cyclic triureas-synthesis, crystal structures and properties. Org. Biomol. Chem 2008, 6, 1004–1014. [DOI] [PubMed] [Google Scholar]
- (31).Lepore G; Migdal S; Blagdon DE; Goodman M Conformations of substituted arylureas in solution. J. Org. Chem 1973, 38, 2590–2594. [Google Scholar]
- (32).Lepore U; Castronuovo Lepore G; Ganis P; Germain G; Goodman M Conformation of substituted arylureas. Crystal structures of N,N’-dimethyl-N,N’-di(p-nitrophenyl)urea and N,N’-dimethyl-N,N’-di(2,4-dinitrophenyl)urea. J. Org. Chem 1976, 41, 2134–2137. [Google Scholar]
- (33).Galan JF; Germany E; Pawlowski A; Strickland L; Galinato MG Theoretical and spectroscopic analysis of N,N’-diphenylurea and N,N’-dimethyl-N,N’-diphenylurea conformations. J. Phys. Chem. A 2014, 118, 5304–5315. [DOI] [PubMed] [Google Scholar]
- (34).Clayden J; Hennecke U; Vincent MA; Hillier IH; Helliwell M The origin of the conformational preference of N,N’-diaryl-N,N’-dimethyl ureas. Phys. Chem. Chem. Phys 2010, 12, 15056–15064. [DOI] [PubMed] [Google Scholar]
- (35).Yamaguchi K; Shudo K Crystallographic studies on urea cytokinins. J. Agric. Food Chem 1991, 39, 793–796. [Google Scholar]
- (36).Yamaguchi K; Matsumura G; Kagechika H; Azumaya I; Ito Y; Itai A; Shudo K Aromatic architecture - use of the N-methylamide structure as a molecular splint. J. Am. Chem. Soc 1991, 113, 5474–5475. [Google Scholar]
- (37).Hill DJ; Mio MJ; Prince RB; Hughes TS; Moore JS A field guide to foldamers. Chem. Rev 2001, 101, 3893–4012. [DOI] [PubMed] [Google Scholar]
- (38).Clayden J; Pickworth M; Jones LH Relaying stereochemistry through aromatic ureas: 1,9 and 1,15 remote stereocontrol. Chem. Commun. (Cambridge, U. K.) 2009, 547–549. [DOI] [PubMed] [Google Scholar]
- (39).Lewis FD; Kurth TL; Hattan CM; Reiter RC; Stevenson CD Polyaryl anion radicals via alkali metal reduction of arylurea oligomers. Org. Lett 2004, 6, 1605–1608. [DOI] [PubMed] [Google Scholar]
- (40).Kudo M; Hanashima T; Muranaka A; Sato H; Uchiyama M; Azumaya I; Hirano T; Kagechika H; Tanatani A Identification of absolute helical structures of aromatic multilayered oligo(mphenylurea)s in solution. J. Org. Chem 2009, 74, 8154–8163. [DOI] [PubMed] [Google Scholar]
- (41).Thomas G Fundamentals of Medicinal Chemistry; John Wiley and Sons, Ltd.: West Sussex, U.K., 2003; pp 56–70. [Google Scholar]
- (42).Pajouhesh H; Lenz GR Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Mouritsen OG; Jorgensen K A new look at lipid-membrane structure in relation to drug research. Pharm. Res 1998, 15, 1507–1519. [DOI] [PubMed] [Google Scholar]
- (44).Urea: Synthesis, Properties and Uses; Nova Science Publishers, Inc.: New York, 2012. [Google Scholar]
- (45).Herbig ME; Evers DH Correlation of hydrotropic solubilization by urea with logD of drug molecules and utilization of this effect for topical formulations. Eur. J. Pharm. Biopharm 2013, 85, 158–160. [DOI] [PubMed] [Google Scholar]
- (46).Cui Y Hydrotropic solubilization by urea derivatives: a molecular dynamics simulation study. J. Pharm. (N. Y., NY, U. S.) 2013, 2013, 791370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lortie F; Boileau S; Bouteiller LN,N’-disubstituted ureas: influence of substituents on the formation of supramolecular polymers. Chem. - Eur. J 2003, 9, 3008–3014. [DOI] [PubMed] [Google Scholar]
- (48).Alex A; Millan DS; Perez M; Wakenhut F; Whitlock GA Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. MedChemComm 2011, 2, 669–674. [Google Scholar]
- (49).Kuhn B; Mohr P; Stahl M Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem 2010, 53, 2601–2611. [DOI] [PubMed] [Google Scholar]
- (50).Furet P; Caravatti G; Guagnano V; Lang M; Meyer T; Schoepfer J Entry into a new class of protein kinase inhibitors by pseudo ring design. Bioorg. Med. Chem. Lett 2008, 18, 897–900. [DOI] [PubMed] [Google Scholar]
- (51).Ishikawa M; Hashimoto Y Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem 2011, 54, 1539–1554. [DOI] [PubMed] [Google Scholar]
- (52).Wilkerson WW; Dax S; Cheatham WW Nonsymmetrically substituted cyclic urea HIV protease inhibitors. J. Med. Chem 1997, 40, 4079–4088. [DOI] [PubMed] [Google Scholar]
- (53).Scully SS; Tang AJ; Lundh M; Mosher CM; Perkins KM; Wagner BK Small-molecule inhibitors of cytokine-mediated STAT1 signal transduction in beta-cells with improved aqueous solubility. J. Med. Chem 2013, 56, 4125–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Yin Y; Zheng K; Eid N; Howard S; Jeong JH; Yi F; Guo J; Park CM; Bibian M; Wu W; Hernandez P; Park H; Wu Y; Luo JL; LoGrasso PV; Feng Y Bis-aryl urea derivatives as potent and selective LIM kinase (Limk) inhibitors. J. Med. Chem 2015, 58, 1846–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Giannicchi I; Jouvelet B; Isare B; Linares M; Dalla Cort A; Bouteiller L Orthohalogen substituents dramatically enhance hydrogen bonding of aromatic ureas in solution. Chem. Commun. (Cambridge, U. K.) 2014, 50, 611–613. [DOI] [PubMed] [Google Scholar]
- (56).Creighton TE Stability of folded conformations. Curr. Opin. Struct. Biol 1991, 1, 5–16. [Google Scholar]
- (57).Gill SJ; Hutson J; Clopton JR; Downing M Solubility of diketopiperazine in aqueous solutions of urea. J. Phys. Chem 1961, 65, 1432–1435. [Google Scholar]
- (58).Sliwoski G; Kothiwale S; Meiler J; Lowe EW Computational methods in drug discovery. Pharmacol. Rev 2014, 66, 334–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Fradera X; Vu D; Nimz O; Skene R; Hosfield D; Wynands R; Cooke AJ; Haunsø A; King A; Bennett DJ; McGuire R; Uitdehaag JC X-ray structures of the LXR-alpha LBD in its homodimeric form and implications for heterodimer signaling. J. Mol. Biol 2010, 399, 120–132. [DOI] [PubMed] [Google Scholar]
- (60).Madej T; Lanczycki CJ; Zhang D; Thiessen PA; Geer RC; Marchler-Bauer A; Bryant SH MMDB and VAST+: tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014, 42, D297–D303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Padhi S; Priyakumar UD Urea-aromatic stacking and concerted urea transport: conserved mechanisms in urea transporters revealed by molecular dynamics. J. Chem. Theory Comput 2016, 12, 5190–5200. [DOI] [PubMed] [Google Scholar]
- (62).Goyal S; Chattopadhyay A; Kasavajhala K; Priyakumar UD Role of urea-aromatic stacking interactions in stabilizing the aromatic residues of the protein in urea-induced denatured state. J. Am. Chem. Soc 2017, 139, 14931–14946. [DOI] [PubMed] [Google Scholar]
- (63).Esteva-Font C; Anderson MO; Verkman AS Urea transporter proteins as targets for small-molecule diuretics. Nat. Rev. Nephrol 2015, 11, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Jaganade T; Chattopadhyay A; Pazhayam NM; Priyakumar UD Energetic, structural and dynamic properties of nucleobase-urea interactions that aid in urea assisted RNA unfolding. Sci. Rep 2019, 9, 8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Priyakumar UD; Hyeon C; Thirumalai D; MacKerell AD Urea destabilizes RNA by forming stacking interactions and multiple hydrogen bonds with nucleic acid bases. J. Am. Chem. Soc 2009, 131, 17759–17761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Maher TJ; Johson DA Receptors and Drug Action In Foye’s Principles of Medicinal Chemistry; Lippincott Williams and Wilkins: Philadelphia, PA, 2008. [Google Scholar]
- (67).Getman DP; DeCrescenzo GA; Heintz RM; Reed KL; Talley JJ; Bryant ML; Clare M; Houseman KA; Marr JJ; Mueller RA; Vazquez ML; Shieh H-S; Stallings WC; Stegeman RA Discovery of a novel class of potent HIV-1 protease inhibitors containing the (R)-(hydroxyethyl)urea isostere. J. Med. Chem 1993, 36, 288–291. [DOI] [PubMed] [Google Scholar]
- (68).Lam PY; Jadhav PK; Eyermann CJ; Hodge CN; Ru Y; Bacheler LT; Meek JL; Otto MJ; Rayner MM; Wong YN; Chang C-H; Weber PC; Jackson DA; Sharpe TR; Erickson-Viitanen S Rational design of potent, bioavailable, nonpeptide cyclic ureas as HIV protease inhibitors. Science 1994, 263, 380–384. [DOI] [PubMed] [Google Scholar]
- (69).Hodge CN; Aldrich PE; Bacheler LT; Chang CH; Eyermann CJ; Garber S; Grubb M; Jackson DA; Jadhav PK; Korant B; Lam PY; Maurin MB; Meek JL; Otto MJ; Rayner MM; Reid C; Sharpe TR; Shum L; Winslow DL; Erickson-Viitanen S Improved cyclic urea inhibitors of the HIV-1 protease: synthesis, potency, resistance profile, human pharmacokinetics and X-ray crystal structure of DMP 450. Chem. Biol 1996, 3, 301–314. [DOI] [PubMed] [Google Scholar]
- (70).Okamoto K; Ikemori-Kawada M; Jestel A; von Konig K; Funahashi Y; Matsushima T; Tsuruoka A; Inoue A; Matsui J Distinct binding mode of multikinase inhibitor lenvatinib revealed by biochemical characterization. ACS Med. Chem. Lett 2015, 6, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Moghaddam MF; Grant DF; Cheek JM; Greene JF; Williamson KC; Hammock BD Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat. Med 1997, 3, 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Hwang SH; Tsai HJ; Liu JY; Morisseau C; Hammock BD Orally bioavailable potent soluble epoxide hydrolase inhibitors. J. Med. Chem 2007, 50, 3825–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Kim IH; Tsai HJ; Nishi K; Kasagami T; Morisseau C; Hammock BD 1,3-disubstituted ureas functionalized with ether groups are potent inhibitors of the soluble epoxide hydrolase with improved pharmacokinetic properties. J. Med. Chem 2007, 50, 5217–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Burmistrov V; Morisseau C; Lee KS; Shihadih DS; Harris TR; Butov GM; Hammock BD Symmetric adamantyl-diureas as soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett 2014, 24, 2193–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Huang SX; Cao B; Morisseau C; Tin Y; Hammock BD; Long YQ Structure-based optimization of the piperazino-containing 1,3-disubstituted ureas affording sub-nanomolar inhibitors of soluble epoxide hydrolase. MedChemComm 2012, 3, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Gomez GA; Morisseau C; Hammock BD; Christianson DW Human soluble epoxide hydrolase: structural basis of inhibition by 4-(3-cyclohexylureido)-carboxylic acids. Protein Sci. 2006, 15, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Morisseau C; Goodrow MH; Dowdy D; Zheng J; Greene JF; Sanborn JR; Hammock BD Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 8849–8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Lonsdale R; Hoyle S; Grey DT; Ridder L; Mulholland AJ Determinants of reactivity and selectivity in soluble epoxide hydrolase from quantum mechanics/molecular mechanics modeling. Biochemistry 2012, 51, 1774–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Johnson DS; Stiff C; Lazerwith SE; Kesten SR; Fay LK; Morris M; Beidler D; Liimatta MB; Smith SE; Dudley DT; Sadagopan N; Bhattachar SN; Kesten SJ; Nomanbhoy TK; Cravatt BF; Ahn K Discovery of PF-04457845: a highly potent, orally bioavailable, and selective urea FAAH inhibitor. ACS Med. Chem. Lett 2011, 2, 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Cravatt BF; Giang DK; Mayfield SP; Boger DL; Lerner RA; Gilula NB Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [DOI] [PubMed] [Google Scholar]
- (81).Kathuria S; Gaetani S; Fegley D; Valino F; Duranti A; Tontini A; Mor M; Tarzia G; La Rana G; Calignano A; Giustino A; Tattoli M; Palmery M; Cuomo V; Piomelli D Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med 2003, 9, 76–81. [DOI] [PubMed] [Google Scholar]
- (82).Lichtman AH; Leung D; Shelton CC; Saghatelian A; Hardouin C; Boger DL; Cravatt BF Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J. Pharmacol. Exp. Ther 2004, 311, 441–448. [DOI] [PubMed] [Google Scholar]
- (83).Ahn K; Johnson DS; Mileni M; Beidler D; Long JZ; McKinney MK; Weerapana E; Sadagopan N; Liimatta M; Smith SE; Lazerwith S; Stiff C; Kamtekar S; Bhattacharya K; Zhang Y; Swaney S; Van Becelaere K; Stevens RC; Cravatt BF Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol 2009, 16, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Mileni M; Johnson DS; Wang Z; Everdeen DS; Liimatta M; Pabst B; Bhattacharya K; Nugent RA; Kamtekar S; Cravatt BF; Ahn K; Stevens RC Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 12820–12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).D’Souza DC; Cortes-Briones J; Creatura G; Bluez G; Thurnauer H; Deaso E; Bielen K; Surti T; Radhakrishnan R; Gupta A; Gupta S; Cahill J; Sherif MA; Makriyannis A; Morgan PT; Ranganathan M; Skosnik PD Efficacy and safety of a fatty acid amide hydrolase inhibitor (PF-04457845) in the treatment of cannabis withdrawal and dependence in men: a double-blind, placebo-controlled, parallel group, phase 2a single-site randomised controlled trial. Lancet Psychiatry 2019, 6, 35–45. [DOI] [PubMed] [Google Scholar]
- (86).Hsu CH; Shen YC; Shao YY; Hsu C; Cheng AL Sorafenib in advanced hepatocellular carcinoma: current status and future perspectives. J. Hepatocell Carcinoma 2014, 1, 85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Lathia C; Lettieri J; Cihon F; Gallentine M; Radtke M; Sundaresan P Lack of effect of ketoconazole-mediated CYP3A inhibition on sorafenib clinical pharmacokinetics. Cancer Chemother. Pharmacol 2006, 57, 685–692. [DOI] [PubMed] [Google Scholar]
- (88).Peer CJ; Sissung TM; Kim A; Jain L; Woo S; Gardner ER; Kirkland CT; Troutman SM; English BC; Richardson ED; Federspiel J; Venzon D; Dahut W; Kohn E; Kummar S; Yarchoan R; Giaccone G; Widemann B; Figg WD Sorafenib is an inhibitor of UGT1A1 but is metabolized by UGT1A9: implications of genetic variants on pharmacokinetics and hyperbilirubinemia. Clin. Cancer Res 2012, 18, 2099–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Ghosal A; Yuan Y; Tong W; Su AD; Gu C; Chowdhury SK; Kishnani NS; Alton KB Characterization of human liver enzymes involved in the biotransformation of boceprevir, a hepatitis C virus protease inhibitor. Drug Metab. Dispos 2011, 39, 510–521. [DOI] [PubMed] [Google Scholar]
- (90).New drugs-reports of new drugs recently approved by the FDA. Ritonavir. Bioorg. Med. Chem 1997, 5, 461–462. [PubMed] [Google Scholar]
- (91).Sevrioukova IF; Poulos TL Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 18422–18427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Hofmann C; Penner U; Dorow R; Pertz HH; Jahnichen S; Horowski R; Latte KP; Palla D; Schurad B Lisuride, a dopamine receptor agonist with 5-HT2B receptor antagonist properties: absence of cardiac valvulopathy adverse drug reaction reports supports the concept of a crucial role for 5-HT2B receptor agonism in cardiac valvular fibrosis. Clin. Neuropharmacol 2006, 29, 80–86. [DOI] [PubMed] [Google Scholar]
- (93).Humpel M; Krause W; Hoyer GA; Wendt H; Pommerenke G The pharmacokinetics and biotransformation of 14C-lisuride hydrogen maleate in rhesus monkey and in man. Eur. J. Drug Metab. Pharmacokinet 1984, 9, 347–357. [DOI] [PubMed] [Google Scholar]
- (94).Agai-Csongor E; Domany G; Nogradi K; Galambos J; Vago I; Keseru GM; Greiner I; Laszlovszky I; Gere A; Schmidt E; Kiss B; Vastag M; Tihanyi K; Saghy K; Laszy J; Gyertyan I; Zajer-Balazs M; Gemesi L; Kapas M; Szombathelyi Z Discovery of cariprazine (RGH-188): a novel antipsychotic acting on dopamine D3/ D2 receptors. Bioorg. Med. Chem. Lett 2012, 22, 3437–3440. [DOI] [PubMed] [Google Scholar]
- (95).Kiss B; Horvath A; Nemethy Z; Schmidt E; Laszlovszky I; Bugovics G; Fazekas K; Hornok K; Orosz S; Gyertyan I; Agai-Csongor E; Domany G; Tihanyi K; Adham N; Szombathelyi Z Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/ D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J. Pharmacol. Exp. Ther 2010, 333, 328–340. [DOI] [PubMed] [Google Scholar]
- (96).Caccia S; Invernizzi RW; Nobili A; Pasina L A new generation of antipsychotics: pharmacology and clinical utility of cariprazine in schizophrenia. Ther. Clin. Risk Manage 2013, 9, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Citrome L Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin. Drug Metab. Toxicol 2013, 9, 193–206. [DOI] [PubMed] [Google Scholar]
- (98).Ho TW; Ferrari MD; Dodick DW; Galet V; Kost J; Fan X; Leibensperger H; Froman S; Assaid C; Lines C; Koppen H; Winner PK Efficacy and tolerability of MK-0974 (telcagepant), a new oral antagonist of calcitonin gene-related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo-controlled, parallel-treatment trial. Lancet 2008, 372, 2115–2123. [DOI] [PubMed] [Google Scholar]
- (99).Ho TW; Connor KM; Zhang Y; Pearlman E; Koppenhaver J; Fan X; Lines C; Edvinsson L; Goadsby PJ; Michelson D Randomized controlled trial of the CGRP receptor antagonist telcagepant for migraine prevention. Neurology 2014, 83, 958–966. [DOI] [PubMed] [Google Scholar]
- (100).Roller S; Cui D; Laspina C; Miller-Stein C; Rowe J; Wong B; Prueksaritanont T Preclinical pharmacokinetics of MK-0974, an orally active calcitonin-gene related peptide (CGRP)-receptor antagonist, mechanism of dose dependency and species differences. Xenobiotica 2009, 39, 33–45. [DOI] [PubMed] [Google Scholar]
- (101).Peters-Golden M; Henderson WR Jr. Leukotrienes. N. Engl. J. Med 2007, 357, 1841–1854. [DOI] [PubMed] [Google Scholar]
- (102).Rossi A; Pergola C; Koeberle A; Hoffmann M; Dehm F; Bramanti P; Cuzzocrea S; Werz O; Sautebin L The 5-lipoxygenase inhibitor, zileuton, suppresses prostaglandin biosynthesis by inhibition of arachidonic acid release in macrophages. Br. J. Pharmacol 2010, 161, 555–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Machinist JM; Kukulka MJ; Bopp BA In vitro plasma protein binding of zileuton and its N-dehydroxylated metabolite. Clin. Pharmacokinet 1995, 29 (Suppl 2), 34–41. [DOI] [PubMed] [Google Scholar]
- (104).Perrone MH; Barrett JA Preclinical pharmacology of celiprolol: a cardioselective beta-adrenergic antagonist and mild vasodilator. Am. Heart J 1991, 121, 677–683. [DOI] [PubMed] [Google Scholar]
- (105).Nawarskas JJ; Cheng-Lai A; Frishman WH Celiprolol: a unique selective adrenoceptor modulator. Cardiol Rev. 2017, 25, 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Caruso FS; Doshan HD; Hernandez PH; Costello R; Applin W; Neiss ES Celiprolol: pharmacokinetics and duration of pharmacodynamic activity. Br J. Clin Pract Suppl 1985, 40, 12–16. [PubMed] [Google Scholar]
- (107).Sakamoto J; Oba K; Matsui T; Kobayashi M Efficacy of oral anticancer agents for colorectal cancer. Dis. Colon Rectum 2006, 49, S82–91. [DOI] [PubMed] [Google Scholar]
- (108).Shelton J; Lu X; Hollenbaugh JA; Cho JH; Amblard F; Schinazi RF Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs. Chem. Rev 2016, 116, 14379–14455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Yamamoto M; Arii S; Sugahara K; Tobe T Adjuvant oral chemotherapy to prevent recurrence after curative resection for hepatocellular carcinoma. Br. J. Surg 1996, 83, 336–340. [DOI] [PubMed] [Google Scholar]
- (110).Matsui J; Funahashi Y; Uenaka T; Watanabe T; Tsuruoka A; Asada M Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin. Cancer Res 2008, 14, 5459–5465. [DOI] [PubMed] [Google Scholar]
- (111).Matsui J; Yamamoto Y; Funahashi Y; Tsuruoka A; Watanabe T; Wakabayashi T; Uenaka T; Asada M E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int. J. Cancer 2008, 122, 664–671. [DOI] [PubMed] [Google Scholar]
- (112).Okamoto K; Kodama K; Takase K; Sugi NH; Yamamoto Y; Iwata M; Tsuruoka A Antitumor activities of the targeted multi-tyrosine kinase inhibitor lenvatinib (E7080) against RET gene fusion-driven tumor models. Cancer Lett. 2013, 340, 97–103. [DOI] [PubMed] [Google Scholar]
- (113).Cabanillas ME; Habra MA Lenvatinib: Role in thyroid cancer and other solid tumors. Cancer Treat. Rev 2016, 42, 47–55. [DOI] [PubMed] [Google Scholar]
- (114).Dubbelman AC; Rosing H; Thijssen B; Gebretensae A; Lucas L; Chen H; Shumaker R; Schellens JH; Beijnen JH Development and validation of LC-MS/MS assays for the quantification of E7080 and metabolites in various human biological matrices. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci 2012, 887–888, 25–34. [DOI] [PubMed] [Google Scholar]
- (115).Hussein Z; Mizuo H; Hayato S; Namiki M; Shumaker R Clinical pharmacokinetic and pharmacodynamic profile of lenvatinib, an orally active, small-molecule, multitargeted tyrosine kinase inhibitor. Eur. J. Drug Metab. Pharmacokinet 2017, 42, 903–914. [DOI] [PubMed] [Google Scholar]
- (116).Vu B; Wovkulich P; Pizzolato G; Lovey A; Ding Q; Jiang N; Liu JJ; Zhao C; Glenn K; Wen Y; Tovar C; Packman K; Vassilev L; Graves B Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett 2013, 4, 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Andreeff M; Kelly KR; Yee K; Assouline S; Strair R; Popplewell L; Bowen D; Martinelli G; Drummond MW; Vyas P; Kirschbaum M; Iyer SP; Ruvolo V; Gonzalez GM; Huang X; Chen G; Graves B; Blotner S; Bridge P; Jukofsky L; Middleton S; Reckner M; Rueger R; Zhi J; Nichols G; Kojima K Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin. Cancer Res 2016, 22, 868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Patnaik A; Tolcher A; Beeram M; Nemunaitis J; Weiss GJ; Bhalla K; Agrawal M; Nichols G; Middleton S; Beryozkina A; Sarapa N; Peck R; Zhi J Clinical pharmacology characterization of RG7112, an MDM2 antagonist, in patients with advanced solid tumors. Cancer Chemother. Pharmacol 2015, 76, 587–595. [DOI] [PubMed] [Google Scholar]
- (119).Nowick JS; Powell NA; Nguyen TM; Noronha G An improved method for the synthesis of enantiomerically pure amino-acid ester isocyanates. J. Org. Chem 1992, 57, 7364–7366. [Google Scholar]
- (120).Groszek G A convenient method of synthesis of unsym-metrical urea derivatives. Org. Process Res. Dev 2002, 6, 759–761. [Google Scholar]
- (121).Brindisi M; Maramai S; Gemma S; Brogi S; Grillo A; Di Cesare Mannelli L; Gabellieri E; Lamponi S; Saponara S; Gorelli B; Tedesco D; Bonfiglio T; Landry C; Jung KM; Armirotti A; Luongo L; Ligresti A; Piscitelli F; Bertucci C; Dehouck MP; Campiani G; Maione S; Ghelardini C; Pittaluga A; Piomelli D; Di Marzo V; Butini S Development and pharmacological characterization of selective blockers of 2-arachidonoyl glycerol degradation with efficacy in rodent models of multiple sclerosis and pain. J. Med. Chem 2016, 59, 2612–2632. [DOI] [PubMed] [Google Scholar]
- (122).Cotarca L; Geller T; Repasi J Bis(trichloromethyl)carbonate (BTC, triphosgene): a safer alternative to phosgene? Org. Process Res. Dev 2017, 21, 1439–1446. [Google Scholar]
- (123).Majer P; Randad RS A safe and efficient method for preparation of N,N’-unsymmetrically disubstituted ureas utilizing triphosgene. J. Org. Chem 1994, 59, 1937–1938. [Google Scholar]
- (124).Patrick DA; Gillespie JR; McQueen J; Hulverson MA; Ranade RM; Creason SA; Herbst ZM; Gelb MH; Buckner FS; Tidwell RR Urea derivatives of 2-aryl-benzothiazol-5-amines: a new class of potential drugs for human African trypanosomiasis. J. Med. Chem 2017, 60, 957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Patel M; Kaltenbach RF 3rd; Nugiel DA; McHugh RJ Jr.; Jadhav PK; Bacheler LT; Cordova BC; Klabe RM; Erickson-Viitanen S; Garber S; Reid C; Seitz SP The synthesis of symmetrical and unsymmetrical P1/P1’ cyclic ureas as HIV protease inhibitors. Bioorg. Med. Chem. Lett 1998, 8, 1077–1082. [DOI] [PubMed] [Google Scholar]
- (126).Matzen L; van Amsterdam C; Rautenberg W; Greiner HE; Harting J; Seyfried CA; Bottcher H 5-HT reuptake inhibitors with 5-HT(1B/1D) antagonistic activity: a new approach toward efficient antidepressants. J. Med. Chem 2000, 43, 1149–1157. [DOI] [PubMed] [Google Scholar]
- (127).Hammill JT; Bhasin D; Scott DC; Min J; Chen Y; Lu Y; Yang L; Kim HS; Connelly MC; Hammill C; Holbrook G; Jeffries C; Singh B; Schulman BA; Guy RK Discovery of an orally bioavailable inhibitor of defective in cullin neddylation 1 (DCN1)-mediated cullin neddylation. J. Med. Chem 2018, 61, 2694–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Katritzky AR; Pleynet DPM; Yang BZ A general synthesis of unsymmetrical tetrasubstituted ureas. J. Org. Chem 1997, 62, 4155–4158. [Google Scholar]
- (129).Gallou I; Eriksson M; Zeng X; Senanayake C; Farina V Practical synthesis of unsymmetrical ureas from isopropenyl carbamates. J. Org. Chem 2005, 70, 6960–6963. [DOI] [PubMed] [Google Scholar]
- (130).Kitteringham J; Shipton MR; Voyle M A simple method for the synthesis of unsymmetrical ureas. Synth. Commun 2000, 30, 1937–1943. [Google Scholar]
- (131).Ghosh AK; Brindisi M Organic carbamates in drug design and medicinal chemistry. J. Med. Chem 2015, 58, 2895–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Anbazhagan M; Deshmukh ARAS; Rajappa S Conversion of carbonimidodithioates into unsymmetrical di- and tri-substituted ureas including urea dipeptides. Tetrahedron Lett. 1998, 39, 3609–3612. [Google Scholar]
- (133).Batey RA; Santhakumar V; Yoshina-Ishii C; Taylor SD An efficient new protocol for the formation of unsymmetrical tri- and tetrasubstituted ureas. Tetrahedron Lett. 1998, 39, 6267–6270. [Google Scholar]
- (134).Grzyb JA; Shen M; Yoshina-Ishii C; Chi W; Brown RS; Batey RA Carbamoylimidazolium and thiocarbamoylimidazolium salts: novel reagents for the synthesis of ureas, thioureas, carbamates, thiocarbamates and amides. Tetrahedron 2005, 61, 7153–7175. [Google Scholar]
- (135).Artuso E; Degani L; Fochi R; Magistris C Preparation of mono-, di-, and trisubstituted ureas by carbonylation of aliphatic amines with S,S-dimethyl dithiocarbonate. Synthesis 2007, 2007, 3497–3506. [Google Scholar]
- (136).Lee HG; Kim MJ; Park SE; Kim JJ; Kim BR; Lee SG; Yoon YJ Phenyl 4,5-dichloro-6-oxopyridazine-1(6H)-carboxylate as carbonyl source: facile and selective synthesis of carbamates and ureas under mild conditions. Synlett 2009, 2009, 2809–2814. [Google Scholar]
- (137).Padiya KJ; Gavade S; Kardile B; Tiwari M; Bajare S; Mane M; Gaware V; Varghese S; Harel D; Kurhade S Unprecedented ″in water″ imidazole carbonylation: paradigm shift for preparation of urea and carbamate. Org. Lett 2012, 14, 2814–2817. [DOI] [PubMed] [Google Scholar]
- (138).Bogolubsky AV; Moroz YS; Mykhailiuk PK; Granat DS; Pipko SE; Konovets AI; Doroschuk R; Tolmachev A Bis(2,2,2-trifluoroethyl) carbonate as a condensing agent in one-pot parallel synthesis of unsymmetrical aliphatic ureas. ACS Comb. Sci 2014, 16, 303–308. [DOI] [PubMed] [Google Scholar]
- (139).Matsumura Y; Maki T; Satoh Y Electrochemically induced Hofmann rearrangement. Tetrahedron Lett. 1997, 38, 8879–8882. [Google Scholar]
- (140).Moriarty RM; Chany CJ; Vaid RK; Prakash O; Tuladhar SM Preparation of methyl carbamates from primary alkylcarboxamides and arylcarboxamides using hypervalent iodine. J. Org. Chem 1993, 58, 2478–2482. [Google Scholar]
- (141).Radlick P; Brown LR A versatile modification of the Hofmann rearrangement. Synthesis 1974, 4, 290–292. [Google Scholar]
- (142).Huang XC; Keillor JW Preparation of methyl carbamates via a modified Hofmann rearrangement. Tetrahedron Lett. 1997, 38, 313–316. [Google Scholar]
- (143).Senanayake CH; Fredenburgh LE; Reamer RA; Larsen RD; Verhoeven TR; Reider PJ Nature of N-bromosuccinimide in basic-media - the true oxidizing species in the Hofmann rearrangement. J. Am. Chem. Soc 1994, 116, 7947–7948. [Google Scholar]
- (144).Baumgarten HE; Smith HL; Staklis A Reactions of amines. XVIII. Oxidative rearrangement of amides with lead tetraacetate. J. Org. Chem 1975, 40, 3554–3561. [Google Scholar]
- (145).Kajigaeshi S; Asano K; Fujisaki S; Kakinami T; Okamoto T Oxidation using quaternary ammonium polyhalides 0.1. An efficient method for the Hofmann degradation of amides by use of benzyltrimethylammonium tribromide. Chem. Lett 1989, 18, 463–464. [Google Scholar]
- (146).Hinklin RJ; Aicher TD; Anderson DA; Baer BR; Boyd SA; Condroski KR; DeWolf WE Jr.; Kraser CF; McVean M; Rhodes SP; Sturgis HL; Voegtli WC; Williams L; Houze JB Discovery of 2-pyridylureas as glucokinase activators. J. Med. Chem 2014, 57, 8180–8186. [DOI] [PubMed] [Google Scholar]
- (147).Ma D; Sun H General route to 2,4,5-trisubstituted piperidines from enantiopure beta-amino esters. Total synthesis of pseudodistomin B triacetate and pseudodistomin F. J. Org. Chem 2000, 65, 6009–6016. [DOI] [PubMed] [Google Scholar]
- (148).Migawa MT; Swayze EE A solid-phase synthesis of N,N’-disubstituted ureas and Perhydroimidazo. Org. Lett 2000, 2, 3309–3311. [DOI] [PubMed] [Google Scholar]
- (149).Ghosh AK; Brindisi M; Sarkar A The Curtius rearrangement: applications in modern drug discovery and medicinal chemistry. ChemMedChem 2018, 13, 2351–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Ghosh AK; Sarkar A; Brindisi M The Curtius rearrangement: mechanistic insight and recent applications in natural product syntheses. Org. Biomol. Chem 2018, 16, 2006–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Liang JL; Cochran JE; Dorsch WA; Davies I; Clark MP Development of a scalable synthesis of an azaindolyl-pyrimidine inhibitor of influenza virus replication. Org. Process Res. Dev 2016, 20, 965–969. [Google Scholar]
- (152).Narendra N; Chennakrishnareddy G; Sureshbabu VV Application of carbodiimide mediated Lossen rearrangement for the synthesis of alpha-ureidopeptides and peptidyl ureas employing N-urethane alpha-amino/peptidyl hydroxamic acids. Org. Biomol. Chem 2009, 7, 3520–3526. [DOI] [PubMed] [Google Scholar]
- (153).Dube P; Nathel NF; Vetelino M; Couturier M; Aboussafy CL; Pichette S; Jorgensen ML; Hardink M Carbonyldiimidazole-mediated Lossen rearrangement. Org. Lett 2009, 11, 5622–5625. [DOI] [PubMed] [Google Scholar]
- (154).Vasantha B; Hemantha HP; Sureshbabu VV 1-Propanephosphonic acid cyclic anhydride (T3P) as an efficient promoter for the Lossen rearrangement: application to the synthesis of urea and carbamate derivatives. Synthesis 2010, 2010, 2990–2996. [Google Scholar]
- (155).Thalluri K; Manne SR; Dev D; Mandal B Ethyl 2-cyano-2-(4-nitrophenylsulfonyloxyimino)acetate-mediated Lossen rearrangement: single-pot racemization-free synthesis of hydroxamic acids and ureas from carboxylic acids. J. Org. Chem 2014, 79, 3765–3775. [DOI] [PubMed] [Google Scholar]
- (156).Lebel H; Leogane O Curtius rearrangement of aromatic carboxylic acids to access protected anilines and aromatic ureas. Org. Lett 2006, 8, 5717–5720. [DOI] [PubMed] [Google Scholar]
- (157).Hemantha HP; Chennakrishnareddy G; Vishwanatha TM; Sureshbabu VV One-pot synthesis of ureido peptides and urea tethered glycosylated amino acids employing Deoxo-Fluor and TMSN3. Synlett 2009, 2009, 407–410. [Google Scholar]
- (158).Kulkarni AR; Garai S; Thakur GA Scalable, one-pot, microwave-accelerated tandem synthesis of unsymmetrical urea derivatives. J. Org. Chem 2017, 82, 992–999. [DOI] [PubMed] [Google Scholar]
- (159).Liu P; Wang ZM; Hu XM Highly efficient synthesis of ureas and carbamates from amides by iodosylbenzene-induced Hofmann rearrangement. Eur. J. Org. Chem 2012, 2012, 1994–2000. [Google Scholar]
- (160).Wang CH; Hsieh TH; Lin CC; Yeh WH; Lin CA; Chien TC One-pot synthesis of N-monosubstituted ureas from nitriles via Tiemann rearrangement. Synlett 2015, 26, 1823–1826. [Google Scholar]
- (161).Feberero C; Suarez-Pantiga S; Cabello Z; Sanz R 1,5-O -> N Carbamoyl Snieckus-Fries-type rearrangement. Org. Lett 2018, 20, 2437–2440. [DOI] [PubMed] [Google Scholar]
- (162).Bigi F; Conforti ML; Maggi R; Piccinno A; Sartori G Clean synthesis in water: uncatalysed preparation of ylidenemalononitriles. Green Chem. 2000, 2, 101–103. [Google Scholar]
- (163).Diaz DJ; Darko AK; McElwee-White L Transition metal-catalyzed oxidative carbonylation of amines to ureas. Eur. J. Org. Chem 2007, 2007, 4453–4465. [Google Scholar]
- (164).Tsuji J; Iwamoto N Organic synthesis by means of noble-metal compounds. Palladium-catalyzed carbonylation of amines. Chem. Commun 1966, 380. [Google Scholar]
- (165).Gabriele B; Mancuso R; Salerno G Oxidative carbonylation as a powerful tool for the direct synthesis of carbonylated heterocycles. Eur. J. Org. Chem 2012, 2012, 6825–6839. [Google Scholar]
- (166).Fukuoka S; Chono M; Kohno M A novel catalytic synthesis of carbamates by oxydative alkoxycarbonylation of amines in the presence of palladium and iodide. J. Chem. Soc., Chem. Commun 1984, 399–400. [Google Scholar]
- (167).Kelkar AA; Kolhe DS; Kanagasabapathy S; Chaudhari RV Selectivity behavior in catalytic oxidative carbonylation of alkyl-amines. Ind. Eng. Chem. Res 1992, 31, 172–176. [Google Scholar]
- (168).Gabriele B; Salerno G; Brindisi D; Costa M; Chiusoli GP Synthesis of 2-oxazolidinones by direct palladium-catalyzed oxidative carbonylation of 2-amino-1-alkanols. Org. Lett 2000, 2, 625–627. [DOI] [PubMed] [Google Scholar]
- (169).Enquist PA; Nilsson P; Edin J; Larhed M Super fast cobalt carbonyl-mediated synthesis of ureas. Tetrahedron Lett. 2005, 46, 3335–3339. [Google Scholar]
- (170).Tafesh AM; Weiguny J A review of the selective catalytic reduction of aromatic nitro compounds into aromatic amines, isocyanates, carbamates, and ureas using CO. Chem. Rev 1996, 96, 2035–2052. [DOI] [PubMed] [Google Scholar]
- (171).Zhao J; Li Z; Yan S; Xu S; Wang MA; Fu B; Zhang Z Pd/C Catalyzed carbonylation of azides in the presence of amines. Org. Lett 2016, 18, 1736–1739. [DOI] [PubMed] [Google Scholar]
- (172).Vinogradova EV; Fors BP; Buchwald SL Palladium-catalyzed cross-coupling of aryl chlorides and triflates with sodium cyanate: a practical synthesis of unsymmetrical ureas. J. Am. Chem. Soc 2012, 134, 11132–11135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Mancuso R; Raut DS; Della Ca N; Fini F; Carfagna C; Gabriele B Catalytic oxidative carbonylation of amino moieties to ureas, oxamides, 2-oxazolidinones, and benzoxazolones. ChemSusChem 2015, 8, 2204–2211. [DOI] [PubMed] [Google Scholar]
- (174).Youn SW; Kim YH Pd(II)/Ag(I)-Promoted one-pot synthesis of cyclic ureas from (hetero)aromatic amines and isocyanates. Org. Lett 2016, 18, 6140–6143. [DOI] [PubMed] [Google Scholar]
- (175).Qi X; Li CL; Wu XF A convenient palladium-catalyzed reductive carbonylation of aryl iodides with dual role of formic acid. Chem. - Eur. J 2016, 22, 5835–5838. [DOI] [PubMed] [Google Scholar]
- (176).Qi X; Jiang LB; Li CL; Li R; Wu XF Palladium-catalyzed one-pot carbonylative Sonogashira reaction employing formic acid as the CO source. Chem. - Asian J 2015, 10, 1870–1873. [DOI] [PubMed] [Google Scholar]
- (177).Qi X; Jiang LB; Li HP; Wu XF A convenient palladium-catalyzed carbonylative Suzuki coupling of aryl halides with formic acid as the carbon monoxide source. Chem. - Eur. J 2015, 21, 17650–17656. [DOI] [PubMed] [Google Scholar]
- (178).Qi XX; Li CL; Jiang LB; Zhang WQ; Wu XF Palladium-catalyzed alkoxycarbonylation of aryl halides with phenols employing formic acid as the CO source. Catal. Sci. Technol 2016, 6, 3099–3107. [Google Scholar]
- (179).Peng JB; Wu FP; Li CL; Qi X; Wu XF A convenient and efficient palladium-catalyzed carbonylative Sonogashira transformation with formic acid as the CO source. Eur. J. Org. Chem 2017, 2017, 1434–1437. [Google Scholar]
- (180).Li CL; Zhang WQ; Qi XX; Peng JB; Wu XF Palladium-catalyzed carbonylative synthesis of alkynones from aryl iodides and phenylpropiolic acid employing formic acid as the CO source. J. Organomet. Chem 2017, 838, 9–11. [Google Scholar]
- (181).Wang Z; Yin Z; Wu XF Pd/C-Catalyzed amino-carbonylation of aryl iodides with anthranils in water using Mo(CO)6 as the CO source. Chem. - Eur. J 2017, 23, 15026–15029. [DOI] [PubMed] [Google Scholar]
- (182).He L; Sharif M; Neumann H; Beller M; Wu XF A convenient palladium-catalyzed carbonylative synthesis of 4(3H)-quinazolinones from 2-bromoformanilides and organo nitros with Mo(CO)6 as a multiple promoter. Green Chem. 2014, 16, 3763–3767. [Google Scholar]
- (183).Wu XF; Oschatz S; Sharif M; Langer P Palladium-catalyzed carbonylative synthesis of N-benzoylindoles with Mo(CO)6 as the carbon monoxide source. Synthesis 2015, 47, 2641–2646. [Google Scholar]
- (184).Kim SH; Hong SH Ruthenium-catalyzed urea synthesis using methanol as the C1 source. Org. Lett 2016, 18, 212–215. [DOI] [PubMed] [Google Scholar]
- (185).Krishnakumar V; Chatterjee B; Gunanathan C Rutheniumcatalyzed urea synthesis by N-H activation of amines. Inorg. Chem 2017, 56, 7278–7284. [DOI] [PubMed] [Google Scholar]
- (186).Zhang L; Darko AK; Johns JI; McElwee-White L Catalytic oxidative carbonylation of arylamines to ureas with W(CO) (6)/I-2 as catalyst. Eur. J. Org. Chem 2011, 2011, 6261–6268. [Google Scholar]
- (187).Liu JM; Peng XG; Liu JH; Zheng SZ; Sun W; Xia CG Synthesis of 2-oxazolidinones by salen-Co-complexes catalyzed oxidative carbonylation of beta-amino alcohols. Tetrahedron Lett 2007, 48, 929–932. [Google Scholar]
- (188).Zheng SZ; Li FW; Liu JM; Xia CG A novel and efficient (NHC)Cu-I (NHC = N-heterocyclic carbene) catalyst for the oxidative carbonylation of amino compounds. Tetrahedron Lett. 2007, 48, 5883–5886. [Google Scholar]
- (189).Casiello M; Iannone F; Cotugno P; Monopoli A; Cioffi N; Ciminale F; Trzeciak AM; Nacci A Copper(II)-catalysed oxidative carbonylation of aminols and amines in water: A direct access to oxazolidinones, ureas and carbamates. J. Mol. Catal. A: Chem 2015, 407, 8–14. [Google Scholar]
- (190).Spyropoulos C; Kokotos CG One-pot synthesis of ureas from Boc-protected amines. J. Org. Chem 2014, 79, 4477–4483. [DOI] [PubMed] [Google Scholar]
- (191).Kim HK; Lee A Facile one-pot synthesis of unsymmetrical ureas, carbamates, and thiocarbamates from Cbz-protected amines. Org. Biomol. Chem 2016, 14, 7345–7353. [DOI] [PubMed] [Google Scholar]
- (192).Bana P; Lako A; Kiss NZ; Beni Z; Szigetvari A; Koti J; Turos GI; Eles J; Greiner I Synthesis of urea derivatives in two sequential continuous-flow reactors. Org. Process Res. Dev 2017, 21, 611–622. [Google Scholar]
- (193).Riesco-Dominguez A; Blanco-Ania D; Rutjes FPJT Continuous flow synthesis of urea-containing compound libraries based on the piperidin-4-one scaffold. Eur. J. Org. Chem 2018, 2018, 1312–1320. [Google Scholar]
- (194).Le HV; Ganem B Trifluoroacetic anhydride-catalyzed oxidation of isonitriles by DMSO: a rapid, convenient synthesis of isocyanates. Org. Lett 2011, 13, 2584–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Senadi GC; Mutra MR; Lu TY; Wang JJ Oximes as reusable templates for the synthesis of ureas and carbamates by an in situ generation of carbamoyl oximes. Green Chem 2017, 19, 4272–4277. [Google Scholar]
- (196).Peterson SL; Stucka SM; Dinsmore CJ Parallel synthesis of ureas and carbamates from amines and CO2 under mild conditions. Org. Lett 2010, 12, 1340–1343. [DOI] [PubMed] [Google Scholar]
- (197).Aversa C; Leone F; Zucchini G; Serini G; Geuna E; Milani A; Valdembri D; Martinello R; Montemurro F Linifanib: current status and future potential in cancer therapy. Expert Rev. Anticancer Ther 2015, 15, 677–687. [DOI] [PubMed] [Google Scholar]
- (198).Ratain MJ; Eisen T; Stadler WM; Flaherty KT; Kaye SB; Rosner GL; Gore M; Desai AA; Patnaik A; Xiong HQ; Rowinsky E; Abbruzzese JL; Xia C; Simantov R; Schwartz B; O’Dwyer PJ Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J. Clin. Oncol 2006, 24, 2505–2512. [DOI] [PubMed] [Google Scholar]
- (199).Shepard DR; Cooney MM; Elson P; Bukowski RM; Dreicer R; Rini BI; Garcia JA A phase II study of tandutinib (MLN518), a selective inhibitor of type III tyrosine receptor kinases, in patients with metastatic renal cell carcinoma. Invest. New Drugs 2012, 30, 364–367. [DOI] [PubMed] [Google Scholar]
- (200).DeAngelo DJ; Stone RM; Heaney ML; Nimer SD; Paquette RL; Klisovic RB; Caligiuri MA; Cooper MR; Lecerf JM; Karol MD; Sheng S; Holford N; Curtin PT; Druker BJ; Heinrich MC Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics. Blood 2006, 108, 3674–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).https://www.clinicaltrials.gov/ct2/show/NCT02438761 (accessed April 19, 2019).
- (202).Kim H; Muller WJ The role of the epidermal growth factor receptor family in mammary tumorigenesis and metastasis. Exp. Cell Res 1999, 253, 78–87. [DOI] [PubMed] [Google Scholar]
- (203).Yarden Y; Sliwkowski MX Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol 2001, 2, 127–137. [DOI] [PubMed] [Google Scholar]
- (204).Saxena R; Dwivedi A ErbB family receptor inhibitors as therapeutic agents in breast cancer: current status and future clinical perspective. Med. Res. Rev 2012, 32, 166–215. [DOI] [PubMed] [Google Scholar]
- (205).Mowafy S; Farag NA; Abouzid KA Design, synthesis and in vitro anti-proliferative activity of 4,6-quinazolinediamines as potent EGFR-TK inhibitors. Eur. J. Med. Chem 2013, 61, 132–145. [DOI] [PubMed] [Google Scholar]
- (206).Zhang X; Peng T; Ji X; Li J; Tong L; Li Z; Yang W; Xu Y; Li M; Ding J; Jiang H; Xie H; Liu H Design, synthesis and biological evaluation of novel 4-anilinoquinazolines with C-6 urea-linked side chains as inhibitors of the epidermal growth factor receptor. Bioorg. Med. Chem 2013, 21, 7988–7998. [DOI] [PubMed] [Google Scholar]
- (207).Zhang QW; Diao YY; Wang F; Fu Y; Tang F; You QD; Zhou HY Design and discovery of 4-anilinoquinazoline ureas as multikinase inhibitors targeting BRAF, VEGFR-2 and EGFR. MedChemComm 2013, 4, 979–986. [Google Scholar]
- (208).Zhang HQ; Gong FH; Ye JQ; Zhang C; Yue XH; Li CG; Xu YG; Sun LP Design and discovery of 4-anilinoquinazo-line-urea derivatives as dual TK inhibitors of EGFR and VEGFR-2. Eur. J. Med. Chem 2017, 125, 245–254. [DOI] [PubMed] [Google Scholar]
- (209).Hanahan D; Weinberg RA The hallmarks of cancer. Cell 2000, 100, 57–70. [DOI] [PubMed] [Google Scholar]
- (210).Pralhad T; Madhusudan S; Rajendrakumar K Concept, mechanisms and therapeutics of angiogenesis in cancer and other diseases. J. Pharm. Pharmacol 2003, 55, 1045–1053. [DOI] [PubMed] [Google Scholar]
- (211).Huang L; Huang Z; Bai Z; Xie R; Sun L; Lin K Development and strategies of VEGFR-2/KDR inhibitors. Future Med. Chem 2012, 4, 1839–1852. [DOI] [PubMed] [Google Scholar]
- (212).Eldehna WM; Fares M; Ibrahim HS; Aly MH; Zada S; Ali MM; Abou-Seri SM; Abdel-Aziz HA; Abou El Ella DA Indoline ureas as potential anti-hepatocellular carcinoma agents targeting VEGFR-2: Synthesis, in vitro biological evaluation and molecular docking. Eur. J. Med. Chem 2015, 100, 89–97. [DOI] [PubMed] [Google Scholar]
- (213).Ravez S; Barczyk A; Six P; Cagnon A; Garofalo A; Goossens L; Depreux P Inhibition of tumor cell growth and angiogenesis by 7-aminoalkoxy-4-aryloxy-quinazoline ureas, a novel series of multi-tyrosine kinase inhibitors. Eur. J. Med. Chem 2014, 79, 369–381. [DOI] [PubMed] [Google Scholar]
- (214).Peyssonnaux C; Eychene A The Raf/MEK/ERK pathway: new concepts of activation. Biol. Cell 2001, 93, 53–62. [DOI] [PubMed] [Google Scholar]
- (215).Sawyers C Targeted cancer therapy. Nature 2004, 432, 294–297. [DOI] [PubMed] [Google Scholar]
- (216).Wan PT; Garnett MJ; Roe SM; Lee S; Niculescu-Duvaz D; Good VM; Jones CM; Marshall CJ; Springer CJ; Barford D; Marais R Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [DOI] [PubMed] [Google Scholar]
- (217).Cohen Y; Xing M; Mambo E; Guo Z; Wu G; Trink B; Beller U; Westra WH; Ladenson PW; Sidransky D BRAF mutation in papillary thyroid carcinoma. J. Natl. Cancer Inst 2003, 95, 625–627. [DOI] [PubMed] [Google Scholar]
- (218).Yang W; Chen Y; Zhou X; Gu Y; Qian W; Zhang F; Han W; Lu T; Tang W Design, synthesis and biological evaluation of bisaryl ureas and amides based on 2-amino-3-purinylpyridine scaffold as DFG-out B-Raf kinase inhibitors. Eur. J. Med. Chem 2015, 89, 581–596. [DOI] [PubMed] [Google Scholar]
- (219).El-Damasy AK; Lee JH; Seo SH; Cho NC; Pae AN; Keum G Design and synthesis of new potent anticancer benzothiazole amides and ureas featuring pyridylamide moiety and possessing dual BRaf(V600E) and C-Raf kinase inhibitory activities. Eur. J. Med. Chem 2016, 115, 201–216. [DOI] [PubMed] [Google Scholar]
- (220).Henry JR; Kaufman MD; Peng SB; Ahn YM; Caldwell TM; Vogeti L; Telikepalli H; Lu WP; Hood MM; Rutkoski TJ; Smith BD; Vogeti S; Miller D; Wise SC; Chun L; Zhang X; Zhang Y; Kays L; Hipskind PA; Wrobleski AD; Lobb KL; Clay JM; Cohen JD; Walgren JL; McCann D; Patel P; Clawson DK; Guo S; Manglicmot D; Groshong C; Logan C; Starling JJ; Flynn DL Discovery of 1-(3,3-dimethylbutyl)-3-(2-fluoro-4-methyl-5-(7-methyl-2-(methylamino)pyrido[2,3- d]pyrimidin-6-yl)phenyl)-urea (LY3009120) as a pan-RAF inhibitor with minimal paradoxical activation and activity against BRAF or RAS mutant tumor cells. J. Med. Chem 2015, 58, 4165–4179. [DOI] [PubMed] [Google Scholar]
- (221).Vakana E; Pratt S; Blosser W; Dowless M; Simpson N; Yuan XJ; Jaken S; Manro J; Stephens J; Zhang Y; Huber L; Peng SB; Stancato LF LY3009120, a panRAF inhibitor, has significant anti-tumor activity in BRAF and KRAS mutant preclinical models of colorectal cancer. Oncotarget 2017, 8, 9251–9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Drexler HG Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia 1996, 10, 588–599. [PubMed] [Google Scholar]
- (223).Heinrich MC Targeting FLT3 kinase in acute myelogenous leukemia: progress, perils, and prospects. Mini-Rev. Med. Chem 2004, 4, 255–271. [DOI] [PubMed] [Google Scholar]
- (224).Chao Q; Sprankle KG; Grotzfeld RM; Lai AG; Carter TA; Velasco AM; Gunawardane RN; Cramer MD; Gardner MF; James J; Zarrinkar PP; Patel HK; Bhagwat SS Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)-imidazo[2,1-b][1, 3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor. J. Med. Chem 2009, 52, 7808–7816. [DOI] [PubMed] [Google Scholar]
- (225).https://clinicaltrials.gov/ct2/results?term=quizartinib&Search=Search (accessed April 19, 2019).
- (226).Yan HX; Li WW; Zhang Y; Wei XW; Fu LX; Shen GB; Yin T; Li XY; Shi HS; Wan Y; Zhang QY; Li J; Yang SY; Wei YQ Accumulation of FLT3(+) CD11c (+) dendritic cells in psoriatic lesions and the anti-psoriatic effect of a selective FLT3 inhibitor. Immunol. Res 2014, 60, 112–126. [DOI] [PubMed] [Google Scholar]
- (227).Li GB; Ma S; Yang LL; Ji S; Fang Z; Zhang G; Wang LJ; Zhong JM; Xiong Y; Wang JH; Huang SZ; Li LL; Xiang R; Niu D; Chen YC; Yang SY Drug discovery against psoriasis: identification of a new potent FMS-like tyrosine kinase 3 (FLT3) inhibitor, 1-(4-((1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)-3-fluorophenyl)-3-(5-(tert-butyl)iso xazol-3-yl)urea, that showed potent activity in a psoriatic animal model. J. Med. Chem 2016, 59, 8293–8305. [DOI] [PubMed] [Google Scholar]
- (228).Akhmanova A; Steinmetz MO Control of microtubule organization and dynamics: two ends in the limelight. Nat. Rev. Mol. Cell Biol 2015, 16, 711–726. [DOI] [PubMed] [Google Scholar]
- (229).Kueh HY; Mitchison TJ Structural plasticity in actin and tubulin polymer dynamics. Science 2009, 325, 960–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Song DQ; Wang Y; Wu LZ; Yang P; Wang YM; Gao LM; Li Y; Qu JR; Wang YH; Li YH; Du NN; Han YX; Zhang ZP; Jiang JD Benzoylurea derivatives as a novel class of antimitotic agents: synthesis, anticancer activity, and structure-activity relationships. J. Med. Chem 2008, 51, 3094–3103. [DOI] [PubMed] [Google Scholar]
- (231).Zhang J; Zhou J; Ren X; Diao Y; Li H; Jiang H; Ding K; Pei D A new diaryl urea compound, D181, induces cell cycle arrest in the G1 and M phases by targeting receptor tyrosine kinases and the microtubule skeleton. Invest. New Drugs 2012, 30, 490–507. [DOI] [PubMed] [Google Scholar]
- (232).Leggans EK; Duncan KK; Barker TJ; Schleicher KD; Boger DL A remarkable series of vinblastine analogues displaying enhanced activity and an unprecedented tubulin binding steric tolerance: C20’ urea derivatives. J. Med. Chem 2013, 56, 628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Gagne-Boulet M; Fortin S; Lacroix J; Lefebvre CA; Cote MF; R CG Styryl-N-phenyl-N’-(2-chloroethyl)ureas and styrylphenylimidazolidin-2-ones as new potent microtubule-disrupting agents using combretastatin A-4 as model. Eur. J. Med. Chem 2015, 100, 34–43. [DOI] [PubMed] [Google Scholar]
- (234).Peserico A; Simone C Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol 2011, 2011, 371832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Yang XJ; Seto E HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [DOI] [PubMed] [Google Scholar]
- (236).Bergman JA; Woan K; Perez-Villarroel P; Villagra A; Sotomayor EM; Kozikowski AP Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J. Med. Chem 2012, 55, 9891–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Schnekenburger M; Goffin E; Lee JY; Jang JY; Mazumder A; Ji S; Rogister B; Bouider N; Lefranc F; Miklos W; Mathieu V; de Tullio P; Kim KW; Dicato M; Berger W; Han BW; Kiss R; Pirotte B; Diederich M Discovery and characterization of R/S-N-3-cyanophenyl-N’-(6-tert-butoxycarbonylamino-3,4-dihydro-2,2-dimethyl-2H-1- benzopyran-4-yl)urea, a new histone deacetylase class III inhibitor exerting antiproliferative activity against cancer cell lines. J. Med. Chem 2017, 60, 4714–4733. [DOI] [PubMed] [Google Scholar]
- (238).Barinka C; Rojas C; Slusher B; Pomper M Glutamate carboxypeptidase II in diagnosis and treatment of neurologic disorders and prostate cancer. Curr. Med. Chem 2012, 19, 856–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Silver DA; Pellicer I; Fair WR; Heston WD; Cordon-Cardo C Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res 1997, 3, 81–85. [PubMed] [Google Scholar]
- (240).Harada N; Kimura H; Ono M; Saji H Preparation of asymmetric urea derivatives that target prostate-specific membrane antigen for SPECT imaging. J. Med. Chem 2013, 56, 7890–7901. [DOI] [PubMed] [Google Scholar]
- (241).Tang X; Mahajan SS; Nguyen LT; Beliveau F; Leduc R; Simon JA; Vasioukhin V Targeted inhibition of cell-surface serine protease Hepsin blocks prostate cancer bone metastasis. Oncotarget 2014, 5, 1352–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Subedi M; Minn I; Chen J; Kim Y; Ok K; Jung YW; Pomper MG; Byun Y Design, synthesis and biological evaluation of PSMA/hepsin-targeted heterobivalent ligands. Eur. J. Med. Chem 2016, 118, 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Azam F; Alkskas IA; Ahmed MA Synthesis of some urea and thiourea derivatives of 3-phenyl/ethyl-2-thioxo-2,3-dihydrothiazolo[4,5-d]pyrimidine and their antagonistic effects on haloperidol-induced catalepsy and oxidative stress in mice. Eur. J. Med. Chem 2009, 44, 3889–3897. [DOI] [PubMed] [Google Scholar]
- (244).Haydar SN; Ghiron C; Bettinetti L; Bothmann H; Comery TA; Dunlop J; La Rosa S; Micco I; Pollastrini M; Quinn J; Roncarati R; Scali C; Valacchi M; Varrone M; Zanaletti R SAR and biological evaluation of SEN12333/WAY-317538: Novel alpha 7 nicotinic acetylcholine receptor agonist. Bioorg. Med. Chem 2009, 17, 5247–5258. [DOI] [PubMed] [Google Scholar]
- (245).Ghiron C; Haydar SN; Aschmies S; Bothmann H; Castaldo C; Cocconcelli G; Comery TA; Di L; Dunlop J; Lock T; Kramer A; Kowal D; Jow F; Grauer S; Harrison B; La Rosa S; Maccari L; Marquis KL; Micco I; Nencini A; Quinn J; Robichaud AJ; Roncarati R; Scali C; Terstappen GC; Turlizzi E; Valacchi M; Varrone M; Zanaletti R; Zanelli U Novel alpha-7 nicotinic acetylcholine receptor agonists containing a urea moiety: identification and characterization of the potent, selective, and orally efficacious agonist 1-[6-(4-fluorophenyl)pyridin-3-yl]-3-(4-piperidin-1-ylbutyl) urea (SEN34625/WYE-103914). J. Med. Chem 2010, 53, 4379–4389. [DOI] [PubMed] [Google Scholar]
- (246).Nencini A; Castaldo C; Comery TA; Dunlop J; Genesio E; Ghiron C; Haydar S; Maccari L; Micco I; Turlizzi E; Zanaletti R; Zhang J Design and synthesis of a hybrid series of potent and selective agonists of alpha7 nicotinic acetylcholine receptor. Eur. J. Med. Chem 2014, 78, 401–418. [DOI] [PubMed] [Google Scholar]
- (247).Elkamhawy A; Lee J; Park BG; Park I; Pae AN; Roh EJ Novel quinazoline-urea analogues as modulators for Abeta-induced mitochondrial dysfunction: design, synthesis, and molecular docking study. Eur. J. Med. Chem 2014, 84, 466–475. [DOI] [PubMed] [Google Scholar]
- (248).Hoang VH; Ngo VTH; Cui M; Manh NV; Tran PT; Ann J; Ha HJ; Kim H; Choi K; Kim YH; Chang H; Macalino SJY; Lee J; Choi S Discovery of conformationally restricted human glutaminyl cyclase inhibitors as potent anti-Alzheimer’s agents by structure-based design. J. Med. Chem 2019, 62, 8011–8027. [DOI] [PubMed] [Google Scholar]
- (249).Gaiser BI; Danielsen M; Marcher-Rorsted E; Ropke Jorgensen K; Wrobel TM; Frykman M; Johansson H; Brauner-Osborne H; Gloriam DE; Mathiesen JM; Sejer Pedersen D Probing the existence of a metastable binding site at the beta2-adrenergic receptor with homobivalent bitopic ligands. J. Med. Chem 2019, 62, 7806–7839. [DOI] [PubMed] [Google Scholar]
- (250).Kurt BZ; Gazioglu I; Basile L; Sonmez F; Ginex T; Kucukislamoglu M; Guccione S Potential of aryl-urea-benzofuranylthiazoles hybrids as multitasking agents in Alzheimer’s disease. Eur. J. Med. Chem 2015, 102, 80–92. [DOI] [PubMed] [Google Scholar]
- (251).Prakash CR; Raja S Design, synthesis and antiepileptic properties of novel 1-(substituted benzylidene)-3-(1-(morpholino/ piperidino methyl)-2,3-dioxoindolin-5-yl)urea derivatives. Eur. J. Med. Chem 2011, 46, 6057–6065. [DOI] [PubMed] [Google Scholar]
- (252).Siddiqui N; Alam MS; Stables JP Synthesis and anticonvulsant properties of 1-(amino-N-arylmethanethio)-3-(1-substituted benzyl-2, 3-dioxoindolin-5-yl) urea derivatives. Eur. J. Med. Chem 2011, 46, 2236–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Amato RJ; Felts AS; Rodriguez AL; Venable DF; Morrison RD; Byers FW; Daniels JS; Niswender CM; Conn PJ; Lindsley CW; Jones CK; Emmitte KA Substituted 1-Phenyl-3-(pyridin-2-yl)urea negative allosteric modulators of mGlu5: discovery of a new tool compound VU0463841 with activity in rat models of cocaine addiction. ACS Chem. Neurosci 2013, 4, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Xu H; Lu H; Xu Z; Luan L; Li C; Xu Y; Dong K; Zhang J; Li X; Li Y; Liu G; Gong S; Zhao YG; Liu A; Zhang Y; Zhang W; Cai X; Xiang JN; Elliott JD; Lin X Discovery of CNS penetrant CXCR2 antagonists for the potential treatment of CNS demyelinating disorders. ACS Med. Chem. Lett 2016, 7, 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Duan M; Kazmierski WM; Tallant M; Jun JH; Edelstein M; Ferris R; Todd D; Wheelan P; Xiong Z Discovery of a novel series of cyclic urea as potent CCR5 antagonists. Bioorg. Med. Chem. Lett 2011, 21, 6381–6385. [DOI] [PubMed] [Google Scholar]
- (256).Kazmierski WM; Hamatake R; Duan M; Wright LL; Smith GK; Jarvest RL; Ji JJ; Cooper JP; Tallant MD; Crosby RM; Creech K; Wang A; Li X; Zhang S; Zhang YK; Liu Y; Ding CZ; Zhou Y; Plattner JJ; Baker SJ; Bu W; Liu L Discovery of novel urea-based hepatitis C protease inhibitors with high potency against protease-inhibitor-resistant mutants. J. Med. Chem 2012, 55, 3021–3026. [DOI] [PubMed] [Google Scholar]
- (257).Drlica K; Zhao X DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol. Biol. Rev 1997, 61, 377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Champoux JJ DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem 2001, 70, 369–413. [DOI] [PubMed] [Google Scholar]
- (259).Maxwell A; Lawson DM The ATP-binding site of type II topoisomerases as a target for antibacterial drugs. Curr. Top. Med. Chem 2003, 3, 283–303. [DOI] [PubMed] [Google Scholar]
- (260).Charifson PS; Grillot AL; Grossman TH; Parsons JD; Badia M; Bellon S; Deininger DD; Drumm JE; Gross CH; LeTiran A; Liao Y; Mani N; Nicolau DP; Perola E; Ronkin S; Shannon D; Swenson LL; Tang Q; Tessier PR; Tian SK; Trudeau M; Wang T; Wei Y; Zhang H; Stamos D Novel dual-targeting benzimidazole urea inhibitors of DNA gyrase and topoisomerase IV possessing potent antibacterial activity: intelligent design and evolution through the judicious use of structure-guided design and structure-activity relationships. J. Med. Chem 2008, 51, 5243–5263. [DOI] [PubMed] [Google Scholar]
- (261).Grillot AL; Le Tiran A; Shannon D; Krueger E; Liao Y; O’Dowd H; Tang Q; Ronkin S; Wang T; Waal N; Li P; Lauffer D; Sizensky E; Tanoury J; Perola E; Grossman TH; Doyle T; Hanzelka B; Jones S; Dixit V; Ewing N; Liao S; Boucher B; Jacobs M; Bennani Y; Charifson PS Second-generation antibacterial benzimidazole ureas: discovery of a preclinical candidate with reduced metabolic liability. J. Med. Chem 2014, 57, 8792–8816. [DOI] [PubMed] [Google Scholar]
- (262).Stokes NR; Thomaides-Brears HB; Barker S; Bennett JM; Berry J; Collins I; Czaplewski LG; Gamble V; Lancett P; Logan A; Lunniss CJ; Peasley H; Pommier S; Price D; Smee C; Haydon DJ Biological evaluation of benzothiazole ethyl urea inhibitors of bacterial type II topoisomerases. Antimicrob. Agents Chemother 2013, 57, 5977–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Yule IA; Czaplewski LG; Pommier S; Davies DT; Narramore SK; Fishwick CW Pyridine-3-carboxamide-6-yl-ureas as novel inhibitors of bacterial DNA gyrase: structure based design, synthesis, SAR and antimicrobial activity. Eur. J. Med. Chem 2014, 86, 31–38. [DOI] [PubMed] [Google Scholar]
- (264).Moellering RC Linezolid: the first oxazolidinone antimicrobial. Ann. Intern. Med 2003, 138, 135–142. [DOI] [PubMed] [Google Scholar]
- (265).Tsiodras S; Gold HS; Sakoulas G; Eliopoulos GM; Wennersten C; Venkataraman L; Moellering RC; Ferraro MJ Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet 2001, 358, 207–208. [DOI] [PubMed] [Google Scholar]
- (266).Gonzales RD; Schreckenberger PC; Graham MB; Kelkar S; DenBesten K; Quinn JP Infections due to vancomycin-resistant Enterococcus faecium resistant to linezolid. Lancet 2001, 357, 1179. [DOI] [PubMed] [Google Scholar]
- (267).Toh SM; Xiong L; Arias CA; Villegas MV; Lolans K; Quinn J; Mankin AS Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol. Microbiol 2007, 64, 1506–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Suzuki H; Utsunomiya I; Shudo K; Fukuhara N; Iwaki T; Yasukata T Synthesis and in vitro/in vivo antibacterial activity of oxazolidinones having thiocarbamate at C-5 on the A-ring and an amide- or urea-substituted [1,2,5]triazepane or [1,2,5]oxadiazepane as the C-ring. Eur. J. Med. Chem 2013, 69, 262–277. [DOI] [PubMed] [Google Scholar]
- (269).De Rosa M; Zanfardino A; Notomista E; Wichelhaus TA; Saturnino C; Varcamonti M; Soriente A Novel promising linezolid analogues: rational design, synthesis and biological evaluation. Eur. J. Med. Chem 2013, 69, 779–785. [DOI] [PubMed] [Google Scholar]
- (270).Dondorp AM; Nosten F; Yi P; Das D; Phyo AP; Tarning J; Lwin KM; Ariey F; Hanpithakpong W; Lee SJ; Ringwald P; Silamut K; Imwong M; Chotivanich K; Lim P; Herdman T; An SS; Yeung S; Singhasivanon P; Day NP; Lindegardh N; Socheat D; White NJ Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med 2009, 361, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Wongsrichanalai C Artemisinin resistance or artemisinin-based combination therapy resistance? Lancet Infect. Dis 2013, 13, 114–115. [DOI] [PubMed] [Google Scholar]
- (272).Sibley CH Tracking artemisinin resistance in Plasmodium falciparum. Lancet Infect. Dis 2013, 13, 999–1000. [DOI] [PubMed] [Google Scholar]
- (273).Verlinden BK; Niemand J; Snyman J; Sharma SK; Beattie RJ; Woster PM; Birkholtz LM Discovery of novel alkylated (bis)urea and (bis)thiourea polyamine analogues with potent antimalarial activities. J. Med. Chem 2011, 54, 6624–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Singh P; Raj R; Gut J; Rosenthal PJ; Kumar V Urea/ oxalamide tethered beta-lactam-7-chloroquinoline conjugates: synthesis and in vitro antimalarial evaluation. Eur. J. Med. Chem 2014, 71, 128–134. [DOI] [PubMed] [Google Scholar]
- (275).Schwartz BD; Skinner-Adams TS; Andrews KT; Coster MJ; Edstein MD; MacKenzie D; Charman SA; Koltun M; Blundell S; Campbell A; Pouwer RH; Quinn RJ; Beattie KD; Healy PC; Davis RA Synthesis and antimalarial evaluation of amide and urea derivatives based on the thiaplakortone A natural product scaffold. Org. Biomol. Chem 2015, 13, 1558–1570. [DOI] [PubMed] [Google Scholar]
- (276).http://www.who.int/mediacentre/factsheets/fs259/en/ (accessed April 19, 2019).
- (277).Patrick DA; Wenzler T; Yang S; Weiser PT; Wang MZ; Brun R; Tidwell RR Synthesis of novel amide and urea derivatives of thiazol-2-ethylamines and their activity against Trypanosoma brucei rhodesiense. Bioorg. Med. Chem 2016, 24, 2451–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Shibata S; Gillespie JR; Ranade RM; Koh CY; Kim JE; Laydbak JU; Zucker FH; Hol WG; Verlinde CL; Buckner FS; Fan E Urea-based inhibitors of Trypanosoma brucei methionyl-tRNA synthetase: selectivity and in vivo characterization. J. Med. Chem 2012, 55, 6342–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]