

Abstract
Human immunodeficiency virus type 1 (HIV-1) transcripts have three fates: to serve as genomic RNAs, unspliced mRNAs, or spliced subgenomic mRNAs. Recent structural studies have shown that sequences near the 5′ end of HIV-1 RNA can adopt at least two alternate three-dimensional conformations, and that these structures dictate genome versus unspliced mRNA fates. HIV-1's use of alternate transcription start sites (TSS) can influence which RNA conformer is generated, and this choice, in turn, dictates the fate of the unspliced RNA. The structural context of HIV-1's major 5′ splice site differs in these two RNA conformers, suggesting that the conformers may differ in their ability to support HIV-1 splicing events. Here, we tested the hypothesis that TSS that shift the RNA monomer/dimer structural equilibrium away from the splice site sequestering dimer-competent fold would favor splicing. Consistent with this hypothesis, the results showed that the 5′ ends of spliced HIV-1 RNAs were enriched in 3GCap structures and depleted of 1GCap RNAs relative to the total intracellular RNA population. These findings expand the functional significance of HIV-1 RNA structural dynamics by demonstrating roles for RNA structure in defining all three classes of HIV-1 RNAs, and suggest that HIV-1 TSS choice initiates a cascade of molecular events that dictate the fates of nascent HIV-1 RNAs.
Keywords: HIV-1, 5′-leader, RNA structure, splicing, transcription start site
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) replication requires the generation of multiple species of RNA from a single transcription unit (Stoltzfus 2009; Emery et al. 2017; Sertznig et al. 2018). To achieve this, HIV-1 primary transcripts are processed to yield three distinct length classes of mature RNAs: unspliced (∼9 kb), partially spliced (∼4 kb), and multiply/completely spliced (∼1.8 kb) RNAs (Stoltzfus 2009; LeBlanc et al. 2013). Completely spliced RNAs are especially important during early phases of infection, as they include the mRNAs that encode the essential regulatory proteins Tat and Rev, which aid in transcription and subsequent export of unspliced and partially spliced transcripts from the nucleus (Karn and Stoltzfus 2012). Partially spliced transcripts encode Env and certain accessory factors. Unspliced RNAs play two distinct roles in HIV-1 replication: alternately serving as the genomic RNAs that will become encapsidated into progeny virions or as mRNAs for the structural and enzymatic Gag and Gag-Pol polyproteins (Tazi et al. 2010; LeBlanc et al. 2013).
To ensure the transmission of their intact genomes, retroviruses must favor the recruitment of unspliced RNAs over spliced isoforms during virion assembly. For gammaretroviruses, RNA packaging signals are located in intronic sequences that are removed from subgenomic RNAs. In contrast, some RNA elements that contribute to HIV-1 packaging specificity, albeit that are insufficient on their own, lie upstream of HIV-1's initial 5′ splice site and thus are not removed from spliced RNAs. Recent structural studies support earlier suggestions that, at least in part, HIV-1 solves the problem of how to discriminate against spliced RNAs during packaging and to direct its unspliced RNAs into more than one fate by forming multiple alternate RNA structures from sequences residing near the RNAs’ 5′ ends (Greatorex 2004; Abbink et al. 2005; Lu et al. 2011a).
The 5′ ends of unspliced HIV-1 RNAs can adopt at least two distinct folded conformations, and structural NMR has provided evidence that these adopt secondary structures that include features illustrated in Figure 1A,B (Abbink et al. 2005; Keane et al. 2015, 2016; Keane and Summers 2016). In the dimer-competent conformation that precedes packaging, HIV-1 RNAs display palindromic sequences in the hairpin loops of their so-called dimer initiation signal (DIS) hairpins (Fig. 1B). The DIS palindrome pairs intermolecularly with the corresponding sequences in a second RNA to initiate dimerization. Subsequent structural rearrangements result in the formation of the mature dimeric packaging signal that becomes encapsidated in HIV-1 particles (Abbink et al. 2005; Keane et al. 2015, 2016; Keane and Summers 2016).
FIGURE 1.
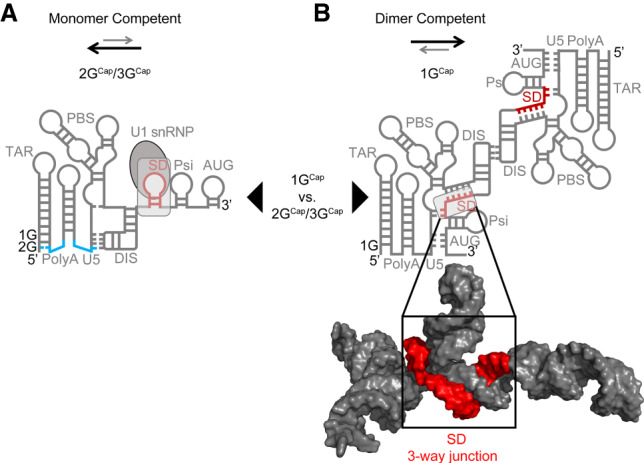
Predicted alternate secondary structure elements of HIV-1 RNAs’ 5′ ends. HIV-1 RNAs with 1GCap 5′ ends, which exist in the dimer-competent fold (B) and 2GCap or 3GCap 5′ RNAs, which adopt a constitutively monomeric form (A). The addition of the single guanosine that differentiates 2GCap from 1GCap RNAs allows for the restructuring of the bottom of the poly(A) hairpin stem as shown. Specifically, an additional 5′ G extends the TAR hairpin stem by recruiting a cytosine residue from the base of the poly(A) hairpin stem, thereby liberating a previously paired guanosine from the downstream arm of the poly(A) hairpin, which in turn extends Watson–Crick base-pairing between sequences in U5 and in the DIS hairpin loop. This restructuring affects the accessibility of the dimer initiation sequence (DIS) palindromic hairpin loop. An early step in the formation of the HIV-1 RNA dimer linkage is a loop–loop “kissing” interaction between the DIS palindrome on one HIV-1 RNA with an identical sequence on a second HIV-1 RNA, which will become its packaging partner. Arrows between each structure demonstrate two populations of the HIV-1 5′ leader that exist based upon heterogeneous transcription start sites (TSS). Arrows above each structure indicate the favored equilibrium between dimer versus monomer competent conformations. Lighter, gray arrows represent less favored and darker, heavier arrows represent more favored. Sequestration of the splice donor (SD) in a three-way junction structure is shown in red in the space-filling model of B.
In contrast to its accessible position in the dimer-competent fold, the DIS palindrome is sequestered by intramolecular base-pairing in an alternate predicted monomeric conformer (Fig. 1A). Recent work has helped explain what dictates alternate 5′ leader folds by showing that HIV-1 transcription initiates within a cluster of three guanosine residues, and that TSS choice affects RNA structure. RNAs initiating with 1Gcap, 2GCap, and 3GCap are all observed in cells, but under native replication conditions, >90% of encapsidated genomic RNAs possess 1Gcap ends (Kharytonchyk et al. 2016; Masuda et al. 2016). Consistent with the hypothesis that the equilibrium between the monomer form of the HIV-1 5′ leader and the alternate, dimer-competent conformation is modulated by this TSS heterogeneity, unspliced transcripts containing a single guanosine plus 7meG (1GCap) at their 5′ ends preferentially adopt the dimer-competent fold in vitro, whereas RNAs possessing either 2GCap or 3GCap ends adopt an alternate fold in vitro and are found enriched on polysomes in cells (Tran et al. 2015; Kharytonchyk et al. 2016). Figure 1 illustrates how the equilibrium between RNA monomer and dimer-competent forms can be shifted by the single 5′ nucleotide variation between 1GCap and 2GCap RNAs; 3GCap RNAs adopt the same fold as 2GCap RNAs (Kharytonchyk et al. 2016).
Prior to the recent elucidation of the HIV-1 packaging signal's three-dimensional structure by NMR, folding algorithms and chemical probing experiments had predicted that HIV-1's 5′ splice site (5′ss; also called the major splice donor or D1), resides in the loop of an RNA hairpin that is sometimes called the SD (splice donor) hairpin (Abbink and Berkhout 2003; Kenyon et al. 2013). This predicted SD hairpin is represented in the monomeric 5′ leader drawing in Figure 1A.
Surprisingly, however, the three-dimensional structure of the packaging signal revealed that the D1 5′ss does not exist in an independently folded SD hairpin. Instead, the arms and loop sequences of this predicted hairpin join with more distal primary sequences and contribute to the body of a double three-way junction structure that is involved in genomic RNA recognition (Fig. 1B; LeBlanc et al. 2013; Keane et al. 2015; Sertznig et al. 2018). As a result, the D1 5′ss, which is used in the generation of every splicing product required during HIV-1 replication, is buried in a structural context, as shown in the space-filling model of Figure 1B. Its location within the packaging signal RNA structure suggests HIV-1's major 5′ss would be largely inaccessible for U1 snRNP binding and early spliceosome complex formation when RNAs adopt the dimeric packaging competent conformation.
Integrating findings that HIV-1 RNAs with 1GCap ends adopt the dimer-competent fold but that one- or two-base longer RNAs do not with the apparent inaccessibility of the D1 5′ss within the packaging structure fold predicts that HIV-1 spliced transcripts should be biased against RNAs with 1GCap 5′ ends. These observations further predict that spliced HIV-1 transcripts should exhibit an enrichment of 2GCap or 3GCap 5′ ends because unspliced RNAs with these 5′ ends adopt the alternate structure.
RESULTS AND DISCUSSION
To test the hypothesis that HIV-1 spliced RNAs are enriched for 2GCap/3GCap transcripts, the 5′ ends of specific classes of HIV-1 RNAs were compared at single-nucleotide resolution. To achieve this, specific spliced transcript populations were isolated from 293T cells transfected with an NL4-3 strain proviral clone that contained a catalytically inactivated integrase gene and a frameshift mutation in env. HIV-1 RNA splicing generates 40 or more different products and makes use of multiple combinations of 5′ and 3′ splice sites, with 5′ splice sites referred to as D1, D2, and so forth (for “splice donor”), while 3′ splice sites are designated as acceptor sites A1, A2, etc. Splice donor/acceptor pair D1/A5 (shown in red font in Fig. 2A) was selected for study because D1/A5 transcripts are the most abundant HIV-1 splicing products in cells (Emery et al. 2017), while D4/A7 (shown in purple) was also chosen because D4/A7 is the most abundant junction unique to multiply spliced HIV-1 RNAs.
FIGURE 2.
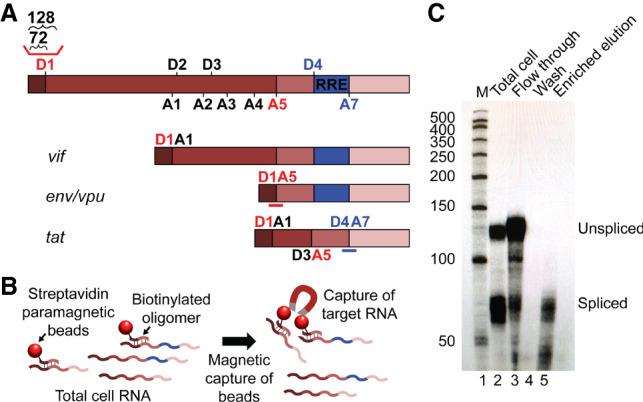
Purification of specific HIV-1 splicing products (A) HIV-1 RNA categories including unspliced and examples of partially spliced and completely spliced transcripts, with associated virus gene names (not to scale). Locations of donor (D) and acceptor (A) splice sites, as HIV-1 5′ and 3′ splice sites are called, are indicated. The red line above D1 indicates the location of the riboprobe used in C; lengths of fragments protected by unspliced and spliced RNAs are indicated above brackets. The red line under D1A5 under the env/vpu RNA represents the location of the D1/A5 capture probe, and the blue bar under the tat mRNA indicates the location of the D4/A7 capture probe. (B) Schematic overview of capture assay. Cell RNA is annealed with a biotinylated oligonucleotide selective (capture probe) specific for a single class of RNA followed by incubation and capture with streptavidin-coated paramagnetic beads. (C) RNase protection assay comparing total cell HIV-1 RNA before (lane 2) and after (lane 5) selective enrichment of specific spliced RNAs. The migration positions of unspliced and spliced RNA protected probe fragments are indicated. Total enrichment of spliced relative to unspliced RNA efficiencies varied between 83% and 98% across five capture assay repetitions.
Targeted spliced transcripts were selectively enriched from cells using biotinylated oligonucleotides complementary to the D1/A5 and D4/A7 splice site junctions, using the approach illustrated in Figure 2B. Selective enrichment after capture, as shown in Figure 2C, was evaluated via RNase protection assays (RPAs) using a probe that spanned the 5′ss D1. This 5′ splice site is believed to be used in the formation of all HIV-1 splicing products, and thus only unspliced RNAs will protect sequences that span D1. The results indicated that total RNA from cells protected both a short fragment (corresponding to spliced RNA) that includes sequences upstream of D1 and a longer fragment derived from unspliced RNA (Fig. 2C, lane 2). Importantly, HIV-1 RNAs captured using splice junction probes displayed no detectable unspliced RNA (Fig. 2C, lane 5).
After these specific splicing products were purified from total cell RNA, their 5′ ends were compared to those of total cell and virion-derived HIV-1 RNAs. This was initially done using a single base resolution RPA using previously described approaches (Fig. 3A; Kharytonchyk et al. 2016). Three capped synthetic RNAs, each with authentic HIV-1 5′ leader sequences and differing numbers of 5′ guanosines, as well as their uncapped derivatives, were used as standards to define the mobilities of 1GCap, 2GCap, and 3GCap transcription products (Fig. 3C displays a large portion of the gel that is presented in cropped form in Fig. 3D, lanes 1–6). Consistent with our previous results (Kharytonchyk et al. 2016), products of 1GCap and 2GCap standards each included two distinct protected bands, as visible in Figure 3D, lanes 2 and 4. This mixture of products reflects weak base-pairing between the RNAs’ 7-methylguanosine cap structure and the riboprobe. “Breathing” of this weak pair results in a lower protected band corresponding to the uncapped 1G structure, twinned with an upper band corresponding to the protection of two cytosines by 1GCap in the case of the 1GCap standard (Kharytonchyk et al. 2016). A similar double band pattern resulted when the 2GCap standard was probed. The 3GCap marker did not appear as a mixture of products because the riboprobe used here was complementary to the native U3/R junction (which contains 3Gs) and thus the same length fragment was protected by RNAs with both 3G and 3GCap 5′ ends. Riboprobe products protected by uncapped 1G, 2G, and 3G RNA standards are included for reference.
FIGURE 3.
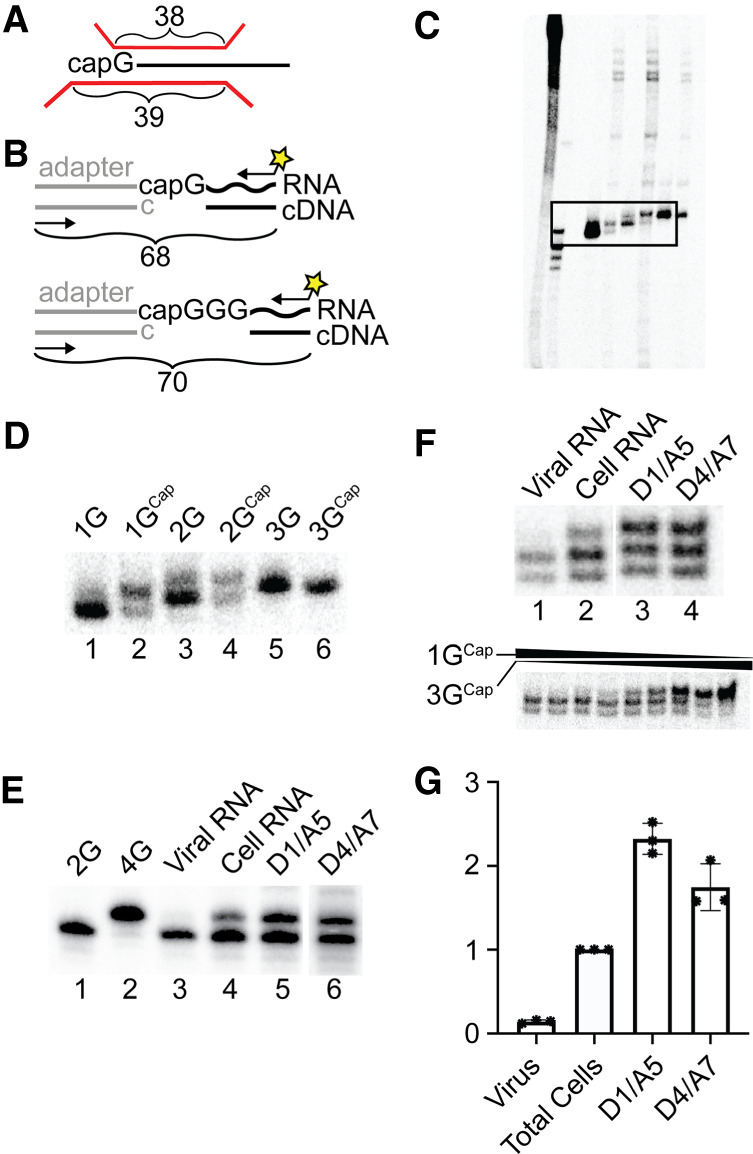
Single-nucleotide resolution analyses of RNA 5′ ends. (A) Schematic description of RNase protection assay, showing riboprobe in red and lengths of twinned fragments protected by 1G RNAs above, and by1GCap RNAs both above and below brackets. (B) Schematic of 7meG cap-dependent PCR approach for assessing 5′ end classes. A double-stranded adaptor with unpaired 5′ C is ligated to nascent cDNA in a cap-dependent manner. The yellow star represents the 32P radiolabel on one PCR primer used to amplify ligated adapter/cDNA products (C) RNase protection products of in vitro transcribed RNA size standards, including uncapped and capped 1G, 2G, and 3G 5′ ends, as described in panel A. An overexposed ∼25 cm portion of the 40 cm sequencing gel is visualized, with (left to right) undigested probe, molecular size standards, and experimental samples: the boxed data indicate regions enlarged in panel D. Panel C is included as a control to demonstrate what was cropped from subsequent panels. (D) Enlarged region from panel C. (E) 5′ end structural heterogeneity of HIV-1 spliced and unspliced RNAs in virions and cells. RNase protection products for viral RNA (lane 1), total cell RNA (lane 2), spliced D1/A5 RNA (lane 3), and spliced D4/A7 RNA (lane 4). This experiment was repeated five times, with similar patterns observed in each. The lower portion of panel E shows RNase protection products resulting from digestions of the following molar mixtures of in vitro synthesized 1GCap:3GCap standards (from left to right): 64:1, 32:1, 16:1, 8:1, 4:1, 2:1, 1:1, 1:2, and 1:4. (F) 5′ end structural heterogeneity of HIV-1 RNAs assessed using the cap-dependent PCR method outlined in panel B. PCR products for 2G or 4G controls (lanes 1 and 2), viral RNA (lane 3), total cell RNA (lane 4), captured spliced D1/A5 RNA (lane 5), and captured D4/A7 RNA (lane 6). Three technical replicates of this assay were performed. Note that because cDNA synthesis of 1GCap RNAs produces products with 2Gs during the cap-dependent PCR assay, the 2G and 4G controls generate fragments with mobilities diagnostic of 1GCap and 3GCap RNA products, respectively. (G) Bar graph and individual data points presenting the proportion of each indicated HIV-1 RNA species with 3GCap ends, as compared to total cell values; ratios determined for the total cell samples were set to 1. Data compiled from the experimental repetitions of panel F experiments, with standard deviations, are indicated.
We next compared the 5′ end contents of the specific HIV-1 splicing products to those of virion and total cell HIV-1 RNA samples (Fig. 3E). As noted above, cells contain a mixture of HIV-1 RNAs, with 1GCap, 2GCap, and 3GCap 5′ ends, with a large majority comprised of 1GCap and 3GCap ends (Kharytonchyk et al. 2016; Masuda et al. 2016). Earlier work performed under conditions used here showed that <5% of total HIV-1 RNAs in cells possess 2GCap 5′ ends, and thus the work here focused on 1GCap and 3GCap RNAs (Kharytonchyk et al. 2016). Because individual capped RNAs yield multiple bands in RPAs, as exemplified by the 1GCap standard's products visualized in lane 2 of Figure 3D, the comigration of probe fragments protected by capped RNAs and by uncapped RNAs one base longer unavoidably complicated quantifying the mixtures of RNase protection products generated by each sample. Nonetheless, a comparison of sample banding patterns in cells (Fig. 3E, lane 2) to those for specific splicing products (Fig. 3E, lanes 3 and 4), as well as visual comparison to RNase protection products generated from differing molar ratios of 1GCap and 3GCap standards (bottom portion of Fig. 3E), provided initial evidence for biases in TSS usage among spliced RNAs.
This analysis (Fig. 3E) revealed that, as reported previously, RNA isolated from virions was highly enriched in 1GCap RNA, while total cell RNA included HIV-1 RNAs with a mixture of 5′ 1GCap and one or two base longer 5′ ends (Fig. 3E, lanes 1 and 2). Consistent with predictions, results in lanes 3 and 4 demonstrated that both of the assayed purified splicing products—those with D1/A5 and with D4/A7 junctions—included a mixture of 5′ ends, with a higher proportion of 2GCap/3GCap 5′ ends than was observed for the total cell sample in lane 2. Visual comparison of splicing products’ 5′ ends (lanes 3 and 4) to those of experimental standards suggested a significant enrichment of the longer RNA product in these samples compared to total cell RNA (lane 2). The lower portion of panel E shows RNase protection assay banding patterns that result from varying proportions of synthetic RNA standards as a comparison. However, the overlapping twinned band patterns generated by capped RNAs differing by only one or two bases prevented reliable quantification of 5′ end ratios using this method.
Thus, an alternate, more readily quantifiable cap-dependent PCR method for determining 5′ end ratios was developed. Unlike standard 5′ RACE, which can artifactually yield several amplification products from a single species of RNA if the studied RNA includes broken molecules, if RNA structure or other factors interfere with cDNA synthesis reaching the RNAs’ 5′ ends, or if the polymerase used adds nontemplated terminal nucleotides, this new method has enhanced specificity for completed cDNAs synthesized on 5′ capped RNAs. The technique is outlined in Figure 3B and the results displayed in panels F and G. Results using this new PCR-based assay confirmed the trends evident in the unamplified 5′ end protection assay shown in Figure 3E, and indicated that D1/A5 spliced RNAs displayed a more than twofold enrichment of 3GCap 5′ ends relative to total cellular HIV-1 RNA. Note that cellular RNA included a mixture of unspliced and spliced RNAs: an undetermined fraction of which contained the D1/A5 splice junction.
This apparent de-enrichment of 1GCap RNAs within the populations of specific splicing products, as indicated by the reduced fraction of RNAs with 1GCap ends as compared to total cell RNA, was consistent with the hypothesis that 1GCap RNAs favor a conformation that is relatively refractory to splicing. These findings add to existing examples in nature, such as riboswitches, where RNA conformational heterogeneity drives alternate functions (Al-Hashimi and Walter 2008; Mustoe et al. 2014; Sherwood and Henkin 2016). The equilibrium between monomeric and dimeric conformations of the HIV-1 RNA 5′ leader has been shown to impact the packaging versus translation fate decision of unspliced transcripts (Dirac et al. 2001; Huthoff and Berkhout 2001a,b; Ooms et al. 2004a,b; Abbink et al. 2005; Lu et al. 2011b). Previous reports have provided data that suggests modulation of secondary structure in the vicinity of the major splice donor can perturb HIV-1 splicing (Abbink and Berkhout 2008; Mueller et al. 2014). Now, with insight from the NMR structure of the packaging signal, findings that a key modulator of the fine-tuned equilibrium of RNA structures is TSS heterogeneity, paired with the new data here, provides a more complete structurally driven model wherein TSS differences also modulate 5′ splice site accessibility (Lu et al. 2011a,b; Tran et al. 2015; Keane and Summers 2016; Keane et al. 2016).
In this model, recognition and binding of U1 snRNP to the major 5′ss (D1) is inhibited when RNAs adopt the dimeric conformation, as is favored by 1GCap RNAs. In contrast, the SD is within more accessible sequences in RNAs that adopt the monomeric fold, which is strongly favored for RNAs initiating with 2GCap or 3GCap, thus allowing recognition by U1 snRNP and efficient splicing. In agreement with the structural models depicted in Figure 1, the spliced HIV RNAs studied here, including D1/A5 and D4/A7 transcripts, showed 5′ end structures biased toward those with 3GCap ends when compared to total cell RNA, whereas unspliced viral genomic RNAs were enriched for 1GCap 5′ ends.
The finding here that the 5′ ends of spliced RNAs displayed an enrichment for a subset of total TSS products, but did not exclude the 1GCap products associated with packaging, is consistent with earlier findings about the RNA populations that associate with polysomes (Kharytonchyk et al. 2016). Understanding of the factors that contribute to these TSS product biases, which may include differential RNA stability and cotranscriptional versus post-transcriptional splicing, will require additional study (Girard et al. 2012; Martin et al. 2013; Merkhofer et al. 2014). Nonetheless, our results demonstrate a marked enrichment of 3GCap 5′ end structures among spliced RNAs, thus supporting the hypothesis that a TSS-enabled structural equilibrium shift in the nascent 5′ leader enhances its splicing competence, and that TSS choice delimits the ability of an HIV-1 RNA to adopt each of its three fates.
The results here describe the properties of RNAs generated by a single common laboratory subtype B isolate of HIV-1, NL4-3, and only examine RNAs produced in cultured cells in vitro. In work currently ongoing, we have demonstrated that both the first and third of three guanosines at the U3-R junction in the subtype A strain, HIV-1MAL, also are used as TSS and that, as is the case for NL4-3, the vast majority of encapsidated RNAs possess 1GCap 5′ ends (Brown et al. 2020). Most HIV-1 strains contain the GGG sequence signature at their TSS, and most Pol II transcripts initiate with purine residues. However, addressing how universal alternate TSS use is, and how often biased 5′ end populations correlate with HIV-1 RNA fates, remain to be determined. Now, with our newly developed cap-dependent 5′ end mapping assay, we are well positioned to address the extents to which these findings pertain to viruses propagated under more physiologic conditions, to other retrovirus isolates, and possibly to variation in transcription elongation outcomes during host gene expression as well.
MATERIALS AND METHODS
Plasmids
pNL4-3, which contains the full genome of HIV-1 strain NL4-3 (Adachi et al. 1986; Delwart et al. 1992; Freed et al. 1992) was modified to generate HIV-Int, which encodes all HIV proteins except Env and IN. The mutation in Env was designed to attenuate this construct for use in a BSL-2 laboratory and consists of a frameshift generated by fill-in of a Kpn I site in env. HIV-Int contains an additional mutation generated by overlap extension PCR within integrase coding sequences, that consists of the insertion of a 376 bp fragment of the murine leukemia virus genome, which contains an oligo binding domain sequence used in previous work and empirically determined to be efficiently selected in capture assays (Flynn and Telesnitsky 2006).
Viruses and cells
For transient transfection with the HIV-Int vector, HEK 293T cells were grown in 10 cm2 culture dishes to a confluence of 80% in 10 mL of DMEM containing 10% FBS, 50 µg/mL gentamicin, and 0.33 µg/mL amphotericin B. These cells were transfected with pHIV-Int at a scale of 5 µg of plasmid and 20 µg PEI (4 µg PEI per microgram of DNA used) per plate. Plasmid DNA was dissolved in Tris–EDTA, pH 8 (TE) buffer and PEI (Polysciences, Inc.) was prepared in water and the pH adjusted to 7. Cells and viral media were harvested 48 h after transfection and reverse transcriptase (RT) activity in the media was quantified via a real-time PCR assay (RT assay) (Vermeire et al. 2012) including media with a known concentration of CA-p24 as a standard.
RNA extraction
Viral RNA was obtained by filtering transfected cells’ media through 0.22 µm filters followed by ultracentrifugation through a 20% sucrose cushion. To lyse virus-containing pellets, TRIzol reagent (Ambion) was used followed by chloroform/isoamyl alcohol extraction and isopropanol precipitation. Following precipitation, RNA pellets were dissolved in Tris-EDTA (TE) buffer, treated with RQ1 DNase (Promega), then phenol/chloroform/isoamyl alcohol extracted and ethanol precipitated. Final RNA pellets were dissolved in TE and nucleic acid concentration was determined by nanodrop measurement. To obtain total cell RNA from transfected 293T cells, the culture medium was removed and 2.4 mL of TRIzol reagent (Ambion) was added to each 10 cm2 culture dish. The cell RNA was then extracted from the TRIzol-cell mixture using chloroform/isoamyl alcohol as described above. Subsequent steps to DNase treat and extract/precipitate RNA were performed as above.
Capture assays
Sequence-specific biotinylated DNA oligo capture probes (Invitrogen) were designed to enrich for completely spliced (D4/A7) or partially spliced (D1/A5) HIV-1 RNAs. The biotinylated D1/A5 capture probe sequence was 5′ CTCCGCTTCTTCCAGTCGCCGCC, and the D4/A7 probe was 5′ TCGGGATTGGGAGGTGGGTTGCTTTGA. For spliced capture assays, RNA was first poly(A) purified using a Dynabead mRNA micro purification kit (Invitrogen). Then, 2–4 µg of poly(A) purified or total cell RNA was mixed with capture probe (final concentration 2 µM), 20× sodium chloride/sodium citrate buffer (SSC; 3 M NaCl, 0.3 M Na3C6H5O7) plus water to a final SSC concentration of 2×, in a total volume of 100 µL. For hybridization, the samples were incubated at 65°C for 15 min followed by incubation on ice for 3 min before the addition of 500 µL per sample Streptavidin MagneSphere Paramagnetic beads (Promega). The beads were washed with 0.5× SSC three times and suspended in 100 µL of 0.5× SSC prior to addition to RNA samples. The beads and RNA samples were incubated for 10 min at room temperature followed by collection of the beads using a magnetic stand and removal of the “flow through” sample. Subsequently, the beads were washed four times with 0.5× SSC and the final wash was collected for analysis. Finally, the bound RNA was eluted by the addition of 250 µL water and heating at 95°C for 5 min. The flow through, final wash, and elution samples were ethanol precipitated and pellets were dissolved in 5 µL of TE buffer followed by concentration determination via nanodrop measurement. RPAs were used to evaluate the success of the capture assay.
RNase protection assay
RPAs were performed according to previously published protocols (Flynn and Telesnitsky 2006; Kharytonchyk et al. 2016). Briefly, the riboprobe template for evaluation of spliced and unspliced HIV RNAs (pSKh65) is a pBluescript KS+ derivative that contains a 128 nt PCR fragment from pNL4-3 that flanks the D1 5′ splice site (RNA map positions 217–344) and thus protects 128 nt of HIV-1 unspliced RNA and 72 nt upstream of D1 for all spliced RNAs. The riboprobe was 32P body-labeled by the synthesis in the presence of α-CTP and transcribed using T3 RNA polymerase and a linearized plasmid template. For the digestion of unhybridized RNAs, both RNase A and RNase T1 were used. Samples were run on an 8% denaturing gel made from Sequel A and Sequel B solutions (AmericanBio) at 1000 V for 1.5 h. For samples analyzed on 40 cm sequencing gels, a PCR generated template was transcribed using SP6 RNA polymerase to generate a body-labeled riboprobe. For digestion, only RNase A was used. The resolution of products was achieved using a 15% denaturing PAGE (29:1 acrylamide/bis-acrylamide) at 1700 V for 5–6 h. Uncapped and 5′ capped synthetic RNAs used as standards were provided by the Summers laboratory and prepared as previously described (De la Peña et al. 2007; Kharytonchyk et al. 2016).
Cap-dependent PCR assay of RNA 5′ ends
To analyze 5′ end structures of capped viral RNAs, RNA samples from the indicated sources were purified as described in the RNA extraction section above and used as templates for cDNA synthesis using the TeloPrime Full-Length cDNA Amplification Kit V2 (Lexogen) according to the manufacturer's protocol. cDNA synthesis was primed using an HIV-1 specific primer 5′ CGCTTCAGCAAGCCGAGTCC (corresponding to positions 239–258 on viral RNA) using 250 ng RNA as template in each reaction. After cap-depended adaptor ligation, second-strand DNA synthesis was performed according to the manufacturer's instructions. Based on empirical trials to optimize detection, viral cDNA was amplified for 15 PCR cycles with Phusion polymerase (NEB) and primers 5′ TGGATTGATATGTAATACGACTCACTATAGG (complementary to ligated adaptor) and 5′ AGAGCTCCCAGGCTCAGATC (complementary to HIV-1 R region). In initial work to confirm specific amplification of RNA 5′ ends using this approach, PCR products obtained from total cell RNA were cloned into pGEM T-easy (Promega). Twelve individual plasmid clones were sequenced and, as anticipated, all were found to contain either 2G or 4G nucleotides at the positions corresponding to the 5′ end of HIV-1 RNA. Subsequently, two plasmid clones, one containing 2G and the other 4G, were used as templates for PCR amplification to generate size standards. The same amplification conditions used to generate cloneable fragments were used for the PCR reactions visualized by autoradiography, with the exception that the HIV-1 specific primer was first radiolabeled with [γ-32P]-ATP and T4 PNK (NEB), using the NEB protocol. PCR products were resolved by denaturing 15% PAGE (29:1 acrylamide/bis-acrylamide) at 1700 V for ∼5 h and quantified by phosphorimaging using ImageQuant software. Autoradiogram shown is one of three technical replicates; all data points obtained are indicated in Figure 3G, and bar graph represents values and standard deviations from these three experiments.
ACKNOWLEDGMENTS
We thank Joshua Brown and the Summers laboratory (University of Maryland Baltimore County—UMBC) for providing unpublished data and in vitro capped RNA size standards, and Cleo Burnett for help with the artwork. This work was supported by National Institutes of Health grants U54 AI150470 and R01 AI50498 (A.T.) and T32 AI007528 (J.M.E.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.073650.119.
REFERENCES
- Abbink TE, Berkhout B. 2003. A novel long distance base-pairing interaction in human immunodeficiency virus type 1 RNA occludes the Gag start codon. J Biol Chem 278: 11601–11611. 10.1074/jbc.M210291200 [DOI] [PubMed] [Google Scholar]
- Abbink TEM, Berkhout B. 2008. RNA structure modulates splicing efficiency at the human immunodeficiency virus type I major splice donor. J Virol 82: 3090–3098. 10.1128/JVI.01479-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbink TEM, Ooms M, Haasnoot PCJ, Berkhout B. 2005. The HIV-1 leader RNA conformational switch regulates RNA dimerization but does not regulate mRNA translation. Biochemistry 44: 9058–9066. 10.1021/bi0502588 [DOI] [PubMed] [Google Scholar]
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59: 284–291. 10.1128/JVI.59.2.284-291.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hashimi HM, Walter NG. 2008. RNA dynamics: it is about time. Curr Opin Struct Biol 18: 321–329. 10.1016/j.sbi.2008.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JD, Kharytonchyk S, Chaudry I, Iyer AS, Carter H, Becker G, Desai Y, Glang L, Choi SH, Singh K, et al. 2020. Structural basis for transcriptional start site control of HIV-1 RNA fate. Science (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Peña M, Kyrieleis OJP, Cusack S. 2007. Structural insights into the mechanism and evolution of the vaccinia virus mRNA cap N7 methyl-transferase. EMBO J 26: 4913–4925. 10.1038/sj.emboj.7601912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delwart EL, Buchschacher GL, Freed EO, Panganiban AT. 1992. Analysis of Hiv-1 envelope mutants and pseudotyping of replication-defective Hiv-1 vectors by genetic complementation. AIDS Res Hum Retrov 8: 1669–1677. 10.1089/aid.1992.8.1669 [DOI] [PubMed] [Google Scholar]
- Dirac AM, Huthoff H, Kjems J, Berkhout B. 2001. The dimer initiation site hairpin mediates dimerization of the human immunodeficiency virus, type 2 RNA genome. J Biol Chem 276: 32345–32352. 10.1074/jbc.M103462200 [DOI] [PubMed] [Google Scholar]
- Emery A, Zhou ST, Pollom E, Swanstrom R. 2017. Characterizing HIV-1 splicing by using next- generation sequencing. J Virol 91: e02515-16 10.1128/JVI.02515-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JA, Telesnitsky A. 2006. Two distinct Moloney murine leukemia virus RNAs produced from a single locus dimerize at random. Virology 344: 391–400. 10.1016/j.virol.2005.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Delwart EL, Buchschacher GL, Panganiban AT. 1992. A mutation in the human-immunodeficiency-virus type-1 transmembrane glycoprotein-Gp41 dominantly interferes with fusion and infectivity. Proc Natl Acad Sci 89: 70–74. 10.1073/pnas.89.1.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard C, Will CL, Peng J, Makarov EM, Kastner B, Lemm I, Urlaub H, Hartmuth K, Lührmann R. 2012. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat Commun 3: 994 10.1038/ncomms1998 [DOI] [PubMed] [Google Scholar]
- Greatorex J. 2004. The retroviral RNA dimer linkage: different structures may reflect different roles. Retrovirology 1: 22 10.1186/1742-4690-1-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huthoff H, Berkhout B. 2001a. Mutations in the TAR hairpin affect the equilibrium between alternative conformations of the HIV-1 leader RNA. Nucleic Acids Res 29: 2594–2600. 10.1093/nar/29.12.2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huthoff H, Berkhout B. 2001b. Two alternating structures of the HIV-1 leader RNA. RNA 7: 143–157. 10.1017/S1355838201001881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn J, Stoltzfus CM. 2012. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb Perspect Med 2: a006916 10.1101/cshperspect.a006916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane SC, Summers MF. 2016. NMR studies of the structure and function of the HIV-1 5′-leader. Viruses-Basel 8: 338 10.3390/v8120338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane SC, Heng X, Lu K, Kharytonchyk S, Ramakrishnan V, Carter G, Barton S, Hosic A, Florwick A, Santos J, et al. 2015. Structure of the HIV-1 RNA packaging signal. Science 348: 917–921. 10.1126/science.aaa9266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane SC, Van V, Frank HM, Sciandra CA, McCowin S, Santos J, Heng X, Summers MF. 2016. NMR detection of intermolecular interaction sites in the dimeric 5′-leader of the HIV-1 genome. Proc Natl Acad Sci 113: 13033–13038. 10.1073/pnas.1614785113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon JC, Prestwood LJ, Le Grice SFJ, Lever AML. 2013. In-gel probing of individual RNA conformers within a mixed population reveals a dimerization structural switch in the HIV-1 leader. Nucleic Acids Res 41: e174 10.1093/nar/gkt690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharytonchyk S, Monti S, Smaldino PJ, Van V, Bolden NC, Brown JD, Russo E, Swanson C, Shuey A, Telesnitsky A, et al. 2016. Transcriptional start site heterogeneity modulates the structure and function of the HIV-1 genome. Proc Natl Acad Sci 113: 13378–13383. 10.1073/pnas.1616627113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc J, Weil J, Beemon K. 2013. Posttranscriptional regulation of retroviral gene expression: primary RNA transcripts play three roles as pre-mRNA. mRNA, and genomic RNA. Wires RNA 4: 567–580. 10.1002/wrna.1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Heng X, Garyu L, Monti S, Garcia EL, Kharytonchyk S, Dorjsuren B, Kulandaivel G, Jones S, Hiremath A, et al. 2011a. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science 334: 242–245. 10.1126/science.1210460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Heng X, Summers MF. 2011b. Structural determinants and mechanism of HIV-1 genome packaging. J Mol Biol 410: 609–633. 10.1016/j.jmb.2011.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RM, Rino J, Carvalho C, Kirchhausen T, Carmo-Fonseca M. 2013. Live-cell visualization of pre-mRNA splicing with single-molecule sensitivity. Cell Rep 4: 1144–1155. 10.1016/j.celrep.2013.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Sato Y, Huang Y-L, Koi S, Takahata T, Hasegawa A, Kawai G, Kannagi M. 2016. Fate of HIV-1 cDNA intermediates during reverse transcription is dictated by transcription initiation site of virus genomic RNA. Sci Rep 5: 17680 10.1038/srep17680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkhofer EC, Hu P, Johnson TL. 2014. Introduction to cotranscriptional RNA splicing. Methods Mol Biol 1126: 83–96. 10.1007/978-1-62703-980-2_6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller N, van Bel N, Berkhout B, Das AT. 2014. HIV-1 splicing at the major splice donor site is restricted by RNA structure. Virology 468–470: 609–620. 10.1016/j.virol.2014.09.018 [DOI] [PubMed] [Google Scholar]
- Mustoe AM, Brooks CL, Al-Hashimi HM. 2014. Hierarchy of RNA functional dynamics. Annu Rev Biochem 83: 441–466. 10.1146/annurev-biochem-060713-035524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms M, Huthoff H, Russell R, Liang C, Berkhout B. 2004a. A riboswitch regulates RNA dimerization and packaging in human immunodeficiency virus type 1 virions. J Virol 78: 10814–10819. 10.1128/JVI.78.19.10814-10819.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms M, Verhoef K, Southern E, Huthoff H, Berkhout B. 2004b. Probing alternative foldings of the HIV-1 leader RNA by antisense oligonucleotide scanning arrays. Nucleic Acids Res 32: 819–827. 10.1093/nar/gkh206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sertznig H, Hillebrand F, Erkelenz S, Schaal H, Widera M. 2018. Behind the scenes of HIV-1 replication: alternative splicing as the dependency factor on the quiet. Virology 516: 176–188. 10.1016/j.virol.2018.01.011 [DOI] [PubMed] [Google Scholar]
- Sherwood AV, Henkin TM. 2016. Riboswitch-mediated gene regulation: novel RNA architectures dictate gene expression responses. Annu Rev Microbiol 70: 361–374. 10.1146/annurev-micro-091014-104306 [DOI] [PubMed] [Google Scholar]
- Stoltzfus CM. 2009. Regulation of HIV-1 alternative RNA splicing and its role in virus replication. Adv Virus Res 74: 1–40. 10.1016/S0065-3527(09)74001-1 [DOI] [PubMed] [Google Scholar]
- Tazi J, Bakkour N, Marchand V, Ayadi L, Aboufirassi A, Branlant C. 2010. Alternative splicing: regulation of HIV-1 multiplication as a target for therapeutic action. FEBS J 277: 867–876. 10.1111/j.1742-4658.2009.07522.x [DOI] [PubMed] [Google Scholar]
- Tran T, Liu Y, Marchant J, Monti S, Seu M, Zaki J, Yang AL, Bohn J, Ramakrishnan V, Singh R, et al. 2015. Conserved determinants of lentiviral genome dimerization. Retrovirology 12: 83 10.1186/s12977-015-0209-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeire J, Naessens E, Vanderstraeten H, Landi A, Iannucci V, Van Nuffel A, Taghon T, Pizzato M, Verhasselt B. 2012. Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS One 7: e50859 10.1371/journal.pone.0050859 [DOI] [PMC free article] [PubMed] [Google Scholar]