

Abstract
N6-methyladenosine (m6A) is an abundant post-transcriptional modification that can impact RNA fate via interactions with m6A-specific RNA binding proteins. Despite accumulating evidence that m6A plays an important role in modulating pluripotency, the influence of m6A reader proteins in pluripotency is less clear. Here, we report that YTHDF2, an m6A reader associated with mRNA degradation, is highly expressed in induced pluripotent stem cells (iPSCs) and down-regulated during neural differentiation. Through RNA sequencing, we identified a group of m6A-modified transcripts associated with neural development that are directly regulated by YTDHF2. Depletion of YTHDF2 in iPSCs leads to stabilization of these transcripts, loss of pluripotency, and induction of neural-specific gene expression. Collectively, our results suggest YTHDF2 functions to restrain expression of neural-specific mRNAs in iPSCs and facilitate their rapid and coordinated up-regulation during neural induction. These effects are both achieved by destabilization of the targeted transcripts.
Keywords: YTHDF2, induced pluripotent stem cells, m6A modification, mRNA decay, neural development
INTRODUCTION
The ability of stem cells to maintain a state of self-renewal, yet also rapidly differentiate in response to external signals requires complex and coordinated control of global gene expression. Although transcriptional changes are integral to this control, stem cells also utilize post-transcriptional regulation to achieve rapid remodeling of the transcriptome during differentiation (Ivanova et al. 2017; Zhao et al. 2017; Kami et al. 2018; Lloret-Llinares et al. 2018; Oh et al. 2018). The RNA modification N6-methyladenosine (m6A) is one post-transcriptional mechanism used by stem cells to regulate gene expression (Batista et al. 2014; Chen et al. 2015b; Geula et al. 2015; Li et al. 2015; Zhang et al. 2017; Heck and Wilusz 2019). m6A is by far the most prevalent internal modification RNA species can experience with ∼10,000 m6A sites documented in over a quarter of human transcripts (Dominissini et al. 2012; Meyer et al. 2012; Chen et al. 2015a). m6A modifications are deposited, or written, onto RNAs by the methyl-writer complex which can be divided into two sub-complexes; the m6A-METTL complex (MAC), consisting of METTL3, the catalytically active subunit of the complex, and METTL14 (Liu et al. 2014), and the m6A-METTL-associated complex (MACOM), comprising WTAP, ZC3H13, RBM15/15B, VIRMA and HAKAI (Ping et al. 2014; Ru˚žička et al. 2017; Guo et al. 2018; Knuckles et al. 2018; Yue et al. 2018). Conversely, m6A can also be erased by one of two demethylases, ALKBH5 or ALKBH9/FTO (Jia et al. 2011; Zheng et al. 2013), although the overall impact of demethylation remains open for debate (Ke et al. 2017; Mauer et al. 2017; Darnell et al. 2018; Zhao et al. 2018). Importantly, the ultimate impact of m6A methylation is dependent on which m6A-binding proteins (readers) interact with the modified RNA. The best-characterized m6A readers are the YTH (YT521-B homology) family of proteins which consists of YTHDF1-3 as well as YTHDC1 and 2 (Patil et al. 2018). Readers influence the fate of m6A-modified RNAs at the level of cleavage/polyadenylation (Kasowitz et al. 2018), splicing (Xiao et al. 2016), subcellular localization (Roundtree et al. 2017), translation (Wang et al. 2015) and decay (Wang et al. 2014a).
METTL3 knockout in mice is lethal by embryonic day 8.5 (E8.5) (Geula et al. 2015) demonstrating that methylation is essential for early development. In fact, m6A methylation is important at the very earliest steps, as it is needed for pluripotency in both human and mouse embryonic stem cells (ESCs) (Batista et al. 2014; Wang et al. 2014b; Aguilo et al. 2015; Chen et al. 2015b; Geula et al. 2015; Bertero et al. 2018; Wen et al. 2018). Moreover, a reduction in global m6A levels alters the self-renewal and differentiation capabilities of ESCs (Batista et al. 2014; Wang et al. 2014b; Geula et al. 2015; Wen et al. 2018). This is likely in part because mRNAs encoding core pluripotency factors such as SOX2, KLF4, NANOG and MYC (but not OCT4) are m6A-modified (Batista et al. 2014; Wang et al. 2014b; Aguilo et al. 2015; Bertero et al. 2018) leading to altered mRNA stability, translation, export and/or splicing when m6A modification is reduced. The studies described above largely focused on the influence of m6A and methyltransferase activities and did not address which m6A readers execute the coordinated, global changes in gene expression that are necessary to facilitate rapid remodeling of the transcriptome in response to external cues. Characterizing the impact of reader activity is critical to our understanding of how m6A influences pluripotency and development.
Relatively little is known regarding which m6A readers are active in the early embryo and specifically in embryonic stem cells. Of the YTH domain family of proteins, only YTHDF2 and YTHDC1 are embryonic lethal when knocked out, suggesting they play a prominent role in early development (Kasowitz et al. 2018; Li et al. 2018a). However, it is not clear whether these factors mediate the effects of m6A on pluripotency, as their role in stem cells has not been characterized. In differentiated cells, YTHDC1 modulates splicing and export from the nucleus (Xiao et al. 2016; Roundtree et al. 2017; Kasowitz et al. 2018), whereas YTHDF2 facilitates the degradation of m6A-modified transcripts by recruiting deadenylases and endonucleases (Wang et al. 2014a; Du et al. 2016; Park et al. 2019). Previous studies have suggested YTHDF2 plays an important role in developmental transitions such as the maternal-to-zygotic transition (Ivanova et al. 2017; Zhao et al. 2017), epithelial–mesenchymal transition (Chen et al. 2017) and differentiation of neural progenitor cells (NPCs) (Li et al. 2018a), where it facilitates the clearance of “old” RNAs to allow establishment of a new gene expression pattern. Based on the evidence outlined above, we hypothesized that YTHDF2 mediates the influence of m6A methylation on pluripotency and differentiation in pluripotent stem cells.
Here, we report that YTHDF2 helps to maintain a pluripotent state by targeting a group of key m6A-modified transcripts encoding neural-specific factors for degradation. When neural differentiation is induced, expression of YTHDF2 decreases. This leads to stabilization and increased abundance of neural transcripts which helps to drive differentiation. Depletion of YTHDF2 in self-renewing iPSCs leads to inappropriate expression of neural markers and loss of pluripotency. However, depletion of YTHDF2 during neural induction disrupts expression of both pluripotency and neural-specific factors. Taken together, our findings reveal pluripotent stem cells rely on YTHDF2 to restrain expression of neural-specific factors in order to maintain a pluripotent state until a signal to differentiate is received.
RESULTS
YTHDF2 is required for pluripotency in human iPSCs
YTHDF2 protein expression is approximately sevenfold higher in induced pluripotent stem cells (iPSCs) than in genetically matched human foreskin fibroblasts (HFFs) (Fig. 1A,B). This dramatic difference in expression between somatic and pluripotent cell types coupled with previous evidence indicating m6A plays an important role in pluripotency (Batista et al. 2014; Wang et al. 2014b; Aguilo et al. 2015; Chen et al. 2015b; Geula et al. 2015; Bertero et al. 2018; Wen et al. 2018) invited further investigation. Therefore, we depleted YTHDF2 in iPSCs using a previously validated siRNA (Wang et al. 2014a). We treated cells with siRNA over a period of 5 d and evaluated the effects on pluripotency. Efficient knockdown was verified at both protein and mRNA levels (Fig. 2A,B). Intriguingly, we observed cell and colony morphological changes. As seen in Figure 2C, YTHDF2 depletion resulted in a loss of the structural integrity of the colony, as the edges of the colony became poorly defined. Moreover, cells were no longer tightly packed within the colony, and individual cells and cell borders could be more easily visualized. These changes are all consistent with differentiation (Wakao et al. 2012; Nagasaka et al. 2017). Other cellular changes linked to differentiation, like enlargement of the nuclei (Rozwadowska et al. 2013) and reduced expression of pluripotency markers TRA1-60 (Grigor'eva et al. 2019; Vilà-González et al. 2019) and SSEA4 (Zhang et al. 2018a) were also observed (Fig. 2D,E; Supplemental Fig. S1). In summary, iPSCs depleted of YTHDF2 exhibit phenotypes consistent with loss of pluripotency.
FIGURE 1.

YTHDF2 protein is highly expressed in iPSCs. (A) Western blot to detect YTHDF2 in three independent iPSC and HFF extracts. GAPDH was used as a loading control. (B) Quantification of A. Asterisks indicate significant difference in the relative mean protein expression between iPSC and HFF samples ([**] P-value <0.005).
FIGURE 2.

Depletion of YTHDF2 in iPSCs results in morphological changes. (A) Representative western blot using iPSC extracts to demonstrate effective depletion of YTHDF2 following 2 d of siRNA treatment. GAPDH was used as a loading control. Percent protein remaining calculated from three independent biological replicates is indicated below the lanes. (B) RT-dPCR analysis of YTHDF2 mRNA abundance in siRNA negative control and YTHDF2 depleted samples normalized to GAPDH mRNA. Asterisks indicate significant difference in mean abundance between control and YTHDF2 KD samples ([***] P-value <0.0005). (C) Bright field images of siRNA negative control and YTHDF2-depleted iPSC colonies following 5 d of treatment. Scale bars indicate 100 µm. (D,E) iPSCs were subjected to 5 d of treatment with negative control or YTHDF2 siRNAs. The nucleus was stained with DAPI. (D) Nucleus size of siRNA negative control or YTHDF2 depleted cells (cells = 50) was quantified via ImageJ. (*) P-value <0.05. (E) The indicated protein was detected by immunofluorescence (IF) staining. Scale bars indicate 10 µm. n = 2.
YTHDF2 depletion affects transcripts required for neural development
To gain insights into the specific transcripts affected by YTHDF2 and the phenotype adopted following extended YTHDF2 knockdown, we performed RNA-sequencing on control and YTHDF2-depleted cells. We observed 1689 and 1066 transcripts were up- or down-regulated, respectively (Fig. 3A; Supplemental Table S1). As expected, YTHDF2 itself exhibited a large decrease in expression. To better understand the structure of the data set, we performed hierarchical clustering of the differentially expressed genes. Our analysis revealed up-regulated transcripts displayed more consistent levels of expression between biological replicates and more robust differences between treatment and control samples when compared to the down-regulated group of transcripts (Supplemental Fig. S2D). This is consistent with YTHDF2 functioning as a destabilizing RNA-binding protein, in that depletion is expected to result in increased abundance of target RNAs. To identify mRNAs that are likely to be direct targets of YTHDF2, we compared our sets of differentially expressed transcripts with transcripts that are m6A methylated in human embryonic stem cells (Batista et al. 2014). Importantly, almost 70% of the transcripts that were up-regulated following YTHDF2 knockdown were m6A methylated, far more than expected by chance (Fig. 3B).
FIGURE 3.

YTHDF2 depletion affects transcripts required for neural development. (A) Volcano plot generated from sequencing data showing the adjusted P-value (y-axis) plotted against the fold change (x-axis) for individual genes. Differentially expressed genes are shown in green (differential expression: read counts >5, Benjamini-Hochberg adjusted P-value <0.05, and log2-fold change >1.0, represented by gray dotted lines). Neural-specific transcripts evaluated in subsequent figures are labeled. (B–E) Venn diagrams showing the overlap between transcripts up-regulated (B,D) or down-regulated (C,E) following YTHDF2 depletion and transcripts previously shown to be m6A methylated in hESCs (B,D) (Batista et al. 2014) or bound by YTHDF2 (C,E) (Wang et al. 2014a). P-values were determined using a hypergeometric test. (F) Boxplot of the log2 fold change in expression (YTHDF2 KD/Control), taken from results generated by DESeq2, vs. number of m6A sites. Transcripts were binned based on the number of m6A sites identified in Batista et al. (2014). The central line of the box plot represents the median log2 fold change expression value. n denotes the number of genes in each bin. Binned groups were compared using an unpaired two-sided Student's t-test ([****] P-value <0.0001). (G–I) Functional annotation clustering of biological process (G), cellular component (H), and tissue expression (I) performed by DAVID on genes up-regulated following YTHDF2 depletion. Annotation clusters with the highest enrichment according to FDR P-value are listed. The number of genes in each cluster is shown next to the respective bar and fold enrichment over background is given in parentheses. (J) RT-dPCR analysis of mRNA abundances in control and YTHDF2 depleted samples normalized to GAPDH. Data are reported as fold change (YTHDF2 KD/Control). Asterisks indicate significant difference in mean abundance between control and YTHDF2 KD samples ([*] P-value <0.05, [**] P-value <0.005).
This, and the fact that only 31% of down-regulated RNAs showed evidence of m6A methylation (Fig. 3C) are consistent with previous observations that YTHDF2 recognizes m6A and recruits RNA decay enzymes (Wang et al. 2014a; Du et al. 2016; Park et al. 2019). Notably, transcripts with more than 1 m6A site show a significantly greater change in abundance following YTHDF2 knockdown than those with 0 or 1 sites (Fig. 3F). This is consistent with recent evidence that recruitment of multiple YTHDF2 proteins may be required for maximal effect (Ries et al. 2019). Overall, these data suggest that a significant proportion of m6A methylated transcripts in iPSCs are targeted by YTHDF2 and that the primary outcome of YTHDF2 binding is reduced RNA abundance, likely through destabilization.
We also compared our set of differentially expressed transcripts to those previously shown to be bound by YTHDF2 in HeLa cells (Wang et al. 2014a). Consistent with the results above, RNAs up-regulated following depletion of YTHDF2 in iPSCs were enriched among the set of transcripts bound by YTHDF2 (Fig. 3D). Moreover, there was no significant overlap between the list of down-regulated transcripts and YTHDF2 association (Fig. 3E). This is again expected given that YTHDF2 association results in destabilization of mRNAs.
To learn more about the pathways and processes that might be affected by depletion of YTHDF2, we performed Gene Ontology (GO) analyses via DAVID v8 (Huang et al. 2009a,b). Among the down-regulated genes, which are unlikely to be directly targeted by YTHDF2, we observed some enrichment of transcripts encoding proteins located in the nucleus and/or associated with mitosis but there was no overt connection to pluripotency or development (Supplmental Fig. S4). In contrast, analysis of GO-terms associated with up-regulated transcripts revealed an intriguing link with neural differentiation/development in several categories. Several of the top hits for biological processes (Fig. 3G) and cellular component (Fig. 3H) have strong associations with neural development. Furthermore, over half of the up-regulated transcripts (857 out of 1689) are commonly expressed in the brain (Fig. 3I). These results coincide with previous reports that m6A methylation (Yoon et al. 2017; Ma et al. 2018; Wang et al. 2018c; Chen et al. 2019; Zhuang et al. 2019), and YTHDF2 (Li et al. 2018a), play a role in modulating neural development in mammals. Based on these observations and the fact that up-regulated transcripts are more likely to be directly targeted by YTHDF2, we further characterized several of the most dramatically increased transcripts that encode factors with a role in neural development (Fig. 3A). We validated the observed changes in mRNA abundance via reverse transcription digital PCR (RT-dPCR). As can be seen in Figure 3J, we were able to independently reproduce our sequencing results, with statistical significance, for a majority of the identified transcripts (10 out of 13). In summary, our results are consistent with the idea that YTHDF2 plays a role in neural differentiation by modulating expression of mRNAs encoding factors associated with neural development.
YTHDF2 interacts with neural-associated transcripts
As an m6A reader, YTHDF2 interacts with target transcripts by binding to m6A via its YTH domain (Li et al. 2014; Zhu et al. 2014; Xu et al. 2015). Upon closer examination, all of the neural-associated transcripts we identified possess one or more m6A site(s) as defined via m6A-seq experiments performed on human and/or mouse samples (Zheng et al. 2018). We performed RNA immunoprecipitations (RIPs) in iPSCs to assess whether the neural-associated transcripts we identified were selectively bound to YTHDF2. Briefly, YTHDF2-specific and IgG control antibodies were used to retrieve YTHDF2 and associated mRNAs from iPSC extracts following formaldehyde crosslinking.
Western blot analysis of Input, IgG immunoprecipitation (IP) and YTHDF2 IP samples revealed YTHDF2 protein was detectable in Input and YTHDF2 IP samples, but not in the IgG IP sample (Fig. 4A), demonstrating effective isolation of YTHDF2. RNA was isolated from input, IgG IP and YTHDF2 IP samples and mRNA abundance assessed via RT-dPCR. GAPDH mRNA which has been previously established as a nonmethylated transcript that does not bind YTHDF2 (Batista et al. 2014; Wang et al. 2014a) showed no enrichment over IgG, while CREBBP mRNA, which is known to bind YTHDF2, was significantly enriched and served as a positive control (Fig. 4B; Wang et al. 2014a). Interestingly, all of the neural-associated transcripts were highly enriched (greater than sixfold over IgG) and were significantly enriched when compared to GAPDH (Fig. 4B). Overall, these results further validate the findings from our RNA-seq experiment and indicate that YTHDF2 directly binds to several neural-associated transcripts in pluripotent stem cells.
FIGURE 4.

YTHDF2 binds to neural-associated transcripts and modulates their half-lives in an m6A-dependent manner. (A) Representative western blot showing YTHDF2 protein abundance in Input, IgG IP, and YTHDF2 IP samples from RIP experiment. (B) RT-dPCR analysis of transcript abundance in IgG and YTHDF2 IP samples. Data are represented in a violin plot as fold enrichment over IgG (YTHDF2 IP/IgG IP). Blue dots indicate individual replicate values (n = 3 or 4). Asterisks indicate significant difference in mean fold enrichment between GAPDH and specified transcripts ([*] P-value <0.05). (C,D) Half-lives from cells treated for 2 d with siRNA negative control and YTHDF2 KD (C) or siRNA negative control and YTHDF2-METTL3-WTAP KD (D) were generated by RT-dPCR analysis of Total and Nascent samples using the formula from Rädle et al. (2013) and normalized to a 4sU-labeled synthetic RNA to account for experimental variation. Data are reported as fold change (KD/Control). Asterisks indicate significant difference in mean half-life between control and KD samples ([*] P-value <0.05, [**] P-value <0.005). Nonsignificant P-values are given above bars.
YTHDF2 modulates the half-lives of neural-associated transcripts in m6A dependent manner
As previously stated, YTHDF2 facilitates the degradation of target transcripts upon binding (Wang et al. 2014a; Du et al. 2016). Therefore, we predicted the set of neural-associated transcripts we identified should be stabilized following YTHDF2 depletion. We assessed changes in half-life using 4-thiouridine (4sU) to label and eventually isolate nascent RNAs (Russo et al. 2017). Half-lives were inferred based on the ratio of total to nascent RNA for each transcript (see Materials and Methods for details). As predicted, depletion of YTHDF2 resulted in stabilization for almost all of the neural-associated transcripts tested (Fig. 4C). This indicates that YTHDF2 targets neural-specific m6A-modified transcripts in order to facilitate their degradation.
If YTHDF2 is specifically targeting these transcripts through m6A, then loss of m6A should mimic the effect of YTHDF2 depletion. To investigate this, we simultaneously depleted the catalytically active subunit of the methyltransferase complex METTL3 and the major scaffolding subunit WTAP (Supplemental Fig. S5A,B). Previous studies have shown that depletion of either protein reduces global levels of m6A (Batista et al. 2014; Ping et al. 2014; Schwartz et al. 2014; Yoon et al. 2017; Wang et al. 2018a). As predicted, knockdown of METTL3 and WTAP increased the mRNA half-life for the vast majority (eight out of ten) of the transcripts (Supplemental Fig. S5C). Moreover, simultaneous depletion of all three factors (YTHDF2/METTL3/WTAP) did not notably further stabilize mRNAs (Fig. 4D; Supplemental Fig. S5D). Based on these observations, we conclude that when a cell is in a pluripotent state, YTHDF2 targets neural-associated transcripts for decay in an m6A-dependent manner.
YTHDF2 expression decreases during neural differentiation
If stem cells are utilizing YTHDF2 to suppress neural-associated transcripts, as suggested by our results above (Figs. 3 and 4) then YTHDF2 expression or activity may decrease to favor differentiation down the neural pathway into multipotent NPCs. To investigate this idea, we induced differentiation of iPSCs into NPCs and measured the levels of YTHDF2 protein over time. OCT4, a major pluripotency factor, and PAX6, a marker for neural differentiation (Zhang et al. 2010), were used as controls to demonstrate that differentiation was occurring. As shown in Figure 5A, by day 6 expression of OCT4 protein was almost abolished (Fig. 5B) while PAX6 protein expression increased >50-fold (Fig. 5C). Notably, YTHDF2 protein expression exhibited a steady decline throughout the time course with a >60% reduction in protein expression by day 6 (Fig. 5A,D). In summary, YTHDF2 protein expression decreases as iPSCs differentiate into NPCs.
FIGURE 5.

Changes in YTHDF2 expression and m6A abundance after neural differentiation increase mRNA half-life and abundance of neural-associated transcripts. (A) Representative western blot of YTHDF2, OCT4, and PAX6 protein expression at 0, 2, 4, and 6 d after neural induction. GAPDH was used as a loading control. (B–D) Expression of OCT4 (B), PAX6 (C), and YTHDF2 (D) over the time course was normalized to GAPDH, and the 0 d time point was set to 100% relative protein expression for each replicate. Normalized protein expression from 2, 4, and 6 d after induction was reported as percent protein expression compared to 0 d time point. Asterisks indicate significant difference between the given time point and 0 d induction ([*] P-value <0.05, [**] P-value <0.005, [***] P-value <0.0005). (E) Half-lives from noninduced (control) and 6 d neural induced samples were generated by RT-dPCR analysis of Total and Nascent samples using the formula from Rädle et al. (2013) and normalized to a 4sU-labeled synthetic RNA to account for experimental variation. Data are reported as fold change (Neural Induced/Control). Asterisks indicate significant difference in mean half-life between control and neural induced samples ([*] P-value <0.05, [**] P-value <0.005). Nonsignificant P-values are given above bars. (F) RT-dPCR analysis of mRNA abundances, normalized to GAPDH, from samples that underwent no induction (control) and 6 d of neural induction. Data are reported as fold change (Neural Induced/Control). Asterisks indicate significant difference in mean abundance between control and neural induction samples ([*] P-value <0.05, [**] P-value <0.005). (G) The methylation ratio at four m6A positions quantified by MazF-dPCR using samples that underwent no induction (WT) and 6 d of neural induction. Asterisks indicate significant difference in mean methylation ratio between WT and neural induced samples ([*] P-value <0.05).
Stability of YTHDF2 target transcripts increases during neural differentiation
Based on the changes in mRNA half-life and abundance we observed following YTHDF2 depletion in iPSCs (Figs. 3 and 4) and the decrease in YTHDF2 expression over the course of neural induction (Fig. 5A,D), we predicted the group of neural-associated transcripts we identified as targets of YTHDF2 would exhibit increases in half-life and abundance during neural differentiation. To investigate, we induced neural differentiation for 6 d and then labeled with 4sU for 4 h before isolating and processing RNA as before. Neural differentiated samples were compared to noninduced (pluripotent) controls. We observed stabilization for nine out of ten neural-associated transcripts (Fig. 5E). Interestingly, stabilization did not uniformly result in elevated mRNA abundance reflecting that other types of regulation are likely also at play (Fig. 5F). Overall, these results show that differentiation of iPSCs into NPCs results in stabilization of several neural-associated transcripts, and a decrease in YTHDF2 expression during neural differentiation may facilitate this (Fig. 4).
The level of m6A deposition is not globally altered during neural differentiation
Previous studies have shown global m6A abundance and expression of the catalytically active subunit of the m6A writer complex, METTL3, decreases during differentiation (Aguilo et al. 2015; Geula et al. 2015; Wang et al. 2018a). To explore whether this might also influence expression of YTHDF2 substrates, we evaluated methylation during differentiation by modifying a recently published protocol which utilizes the MazF endoribonuclease to quantify m6A abundance at a specific site (MazF-RT-dPCR) (Garcia-Campos et al. 2019). Briefly, MazF cleaves RNA at ACA motifs and is blocked by N6-adenosine methylation. Thus, following treatment with MazF, RNA regions containing m6A remain intact while unmethylated regions are cleaved and no longer detected by RT-dPCR. The fraction of methylated RNA can be determined by comparing the abundance of a region containing an m6A site with the abundance of a different region lacking ACA (Supplemental Fig. S5E). Unfortunately, many of the neural transcripts associated with YTHDF2 did not contain m6A sites that were amenable to this analysis (i.e., did not have the ACA sequence or had multiple ACA sequences in close proximity) but we identified two m6A sites in the WDR62 transcript, and one each in NANOG and SON which were suitable. Interestingly, although we observed an approximately twofold drop in m6A deposition at one WDR62 m6A site after 5 d of neural induction the other three sites we examined, including a second site in WDR62, showed no significant difference in methylation ratio (Fig. 5G). This suggests that the primary reason for increased stabilization of mRNAs during differentiation is reduced YTHDF2 expression rather than reduced m6A deposition.
YTHDF2 depletion disrupts neural differentiation of iPSCs
We observed above that extended YTHDF2 depletion in iPSCs increased the size of the nucleus (Fig. 2D; Supplemental Fig. S1) and abrogated expression of two pluripotency markers, TRA-1-60 and SSEA4 (Fig. 2E). Both of these observations suggest depletion of YTHDF2 results in loss of pluripotency. Moreover, YTHDF2 binds to and regulates transcripts associated with neural development (Fig. 3). We therefore wondered if YTHDF2 depletion influences differentiation down the neural lineage. As an initial test, we again depleted YTHDF2 in iPSCs for 5 d and used immunofluorescence (IF) to examine the protein expression of three neural-specific markers, PAX6, CXCR4, NES (Nestin) and SOX1 (Feng et al. 2014; Malchenko et al. 2014). Intriguingly, the intensity of staining for the membrane-bound protein NES and the transcription factor SOX1 both exhibit increasing trends in expression in YTHDF2 depleted cells compared to controls (Fig. 6A,B). However, we were unable to detect expression of PAX6 or CXCR4 protein in control or YTHDF2 depleted samples although mRNA was present, and expression was detected in neural induced cells (Supplemental Fig. S6). These results suggest YTHDF2 depletion in iPSCs may prime cells for the neural gene expression program, but does not induce neural differentiation on its own.
FIGURE 6.

Depletion of YTHDF2 in iPSCs favors the neural-lineage and promotes a more robust differentiation response. (A) iPSCs were treated with negative control or YTHDF2 targeting siRNA for 5 d. The indicated protein was detected by immunofluorescence staining. The nucleus was stained with DAPI. White scale bar indicates 10 µm. n = 2. (B) Quantification of IF staining shown in A. (C) RT-dPCR analysis of mRNA abundances in control and YTHDF2 depleted samples prior to neural induction (0 d). Data are reported as fold change (YTHDF2 KD/Control). Asterisks indicate significant difference in mean abundance between control and YTHDF2 KD samples ([*] P-value <0.05). (D–G) mRNA expression of pluripotency (OCT4 and NANOG) and neural-lineage (PAX6 and NES) factors over the course of neural differentiation was quantified via RT-dPCR and normalized to GAPDH. Abundance at the 0 d time point was set to 100%. Asterisks indicate significant difference in the mean percent RNA remaining between control and YTHDF2 KD replicates at that time point ([*] P-value <0.05, [**] P-value <0.005).
If YTHDF2 depletion primes iPSCs for neural differentiation, we hypothesized this influence might be potentiated upon neural induction when compared to non-YTHDF2 depleted controls. To evaluate this, we depleted YTHDF2 prior to inducing differentiation and treated with siRNAs again the day neural induction began. We measured the abundances of several pluripotency factor mRNAs (OCT4 and NANOG), and neural-specific factors (PAX6 and NES). Prior to neural induction, OCT4 and NANOG were unaffected by YTHDF2 depletion, whereas PAX6 and NES exhibited modest decreases in mRNA abundance (Fig. 6C). In contrast, we observed a substantial increase in mRNA abundance for PAX6 at both 2 and 5 d after neural induction in the YTHDF2 depleted samples when compared to controls (Fig. 6D). Moreover, expression of both OCT4 and NANOG were significantly reduced after 5 d of neural induction in YTHDF2 depleted cells compared to controls, with NANOG also being significantly reduced after only 2 d of induction (Fig. 6E,F). There was no clear effect on NES expression during neural induction (Fig. 6G). Thus, it appears depletion of YTHDF2 in iPSCs primes them for neural differentiation. However, improper changes in YTHDF2 expression can disrupt the coordination necessary to achieve precise cell state transitions resulting in aberrant expression of pluripotency and neural-specific factors.
DISCUSSION
In the experiments described above, we have shown that YTHDF2 is important for pluripotency. Through RNA sequencing, we discovered that mRNAs encoding factors associated with neural development are up-regulated following YTHDF2 knockdown. YTHDF2 directly interacts with many of these neural-associated mRNAs, presumably targeting them for degradation in an m6A-dependent manner. Over the course of neural differentiation, YTHDF2 expression is decreased and neural-associated mRNAs are simultaneously stabilized. Reducing expression of YTHDF2 before neural induction disrupts the differentiation process resulting in aberrant expression of both neural and pluripotency factors. Overall, our data support the notion that YTHDF2 is a key m6A reader in pluripotent cells that specifically restrains neural differentiation.
Previous studies have reported YTHDF2 influences maintenance and differentiation of multipotent stem cells from various lineages (Zhang et al. 2017; Li et al. 2018a,b; Wang et al. 2018b; Paris et al. 2019; Yao et al. 2019). Here, we show that YTHDF2 plays a role at the earliest stages of development; in pluripotent stem cells. Although mRNAs encoding both pluripotency factors and neural-specific factors can be m6A-modified (Batista et al. 2014), we find little evidence that YTHDF2 directly influences expression of pluripotency factors, as only neural-specific mRNAs are enriched in the set of transcripts up-regulated by YTHDF2 knockdown. This is consistent with YTHDF2 being required primarily to clear neural-specific transcripts as they are produced, thereby allowing the pluripotency gene expression program to dominate. Indeed, a similar observation was reported during the endothelial-to-hematopoietic transition in zebrafish where YTHDF2 facilitates the clearance of endothelial-specific mRNAs notch1a and rhoca to promote hematopoietic stem cell reprogramming (Zhang et al. 2017). Maintaining neural-specific mRNAs in an unstable state also facilitates their rapid and coordinated up-regulation following induction of the neural differentiation program, as unstable mRNAs reach a new steady state more rapidly when transcription changes (Bertero et al. 2018). When YTHDF2 is artificially depleted, iPSCs appear to inappropriately acquire some features of the neural cell gene expression program, but are unable to achieve effective differentiation down the neural pathway upon induction. These outcomes can both be explained by increased stability of neural-specific transcripts seen when either YTHDF2 or m6A itself are reduced. Importantly, once the neural gene expression program has been successfully engaged, long-term stabilization of neural transcripts may occur naturally through down-regulation of YTHDF2.
Interestingly, other studies also indicate that YTHDF2 and m6A are particularly important for neural gene expression programs. Specifically, Ythdf2 depletion in mice compromises neural development, and generates NPCs with reduced abilities to proliferate and differentiate (Li et al. 2018a). These effects correlate with stabilization of neural-specific mRNA targets. Furthermore, knockout of Mettl14 or depletion of Mettl3 in embryonic mouse brains alters expression of m6A-modified mRNAs encoding factors important for neurogenesis and neural differentiation (Yoon et al. 2017). Finally, deletion of Mettl14 in mouse NPCs reduced proliferation and induced premature differentiation (Wang et al. 2018c). It is unclear how methylated neural-specific transcripts might be selected by YTHDF2 given that a broad spectrum of transcripts is m6A-modified (Batista et al. 2014). A similar situation exists when m6A-modified maternal mRNAs are cleared from oocytes, while leaving zygotic m6A-modified transcripts intact (Ivanova et al. 2017; Zhao et al. 2017). It is possible that neural RNAs are somehow marked during transcription or other steps of RNA processing, perhaps by modification of the YTH protein or by recruitment of additional factors. Alternatively, it may be that the sequence context and/or distribution of m6A sites in neural-specific and other affected RNAs is more favorable for YTHDF2 binding.
Among the neural-specific targets of YTHDF2 we identified, WDR62, CHAC1 and NGFR stand out as encoding key factors for neural development. WDR62 regulates neurogenesis via the JNK1 signaling pathway (Xu et al. 2014) and its depletion disrupts mitotic progression and induces death of NPCs, which ultimately results in primary microcephaly (Bilgüvar et al. 2010; Nicholas et al. 2010; Chen et al. 2014). Moreover, WDR62 has been implicated in the specification of neural and glial progenitor cells during human pluripotent stem cell differentiation (Alshawaf et al. 2017). CHAC1 facilitates neurogenesis by antagonizing the Notch signaling pathway (Chi et al. 2012, 2014), which plays an important role in early neurodevelopment (Zhang et al. 2018b). Finally, the nerve growth factor receptor, NGFR has multiple roles throughout neural development and is a marker for in vitro differentiation of hESCs into NPCs (Lee et al. 2007; Pruszak et al. 2007). NGFR targets have been implicated in embryonic stem cell survival (Pyle et al. 2006) and neural lineage definition (Bibel et al. 2004). By targeting these key neural-specific transcripts, YTHDF2 can play a major role in coordinating gene expression changes during neural development.
In conclusion, the ability of m6A modification to influence self-renewal and neural differentiation of iPSCs is achieved through recruitment of YTHDF2 to neural-specific transcripts. Destabilization of neural-specific mRNAs is important for preventing inappropriate activation of neural gene expression and to permit rapid and coordinated differentiation upon neural induction.
MATERIALS AND METHODS
Cell culture and neural induction
A matched set of male human foreskin fibroblasts (HFF) and reprogrammed induced pluripotent stem cells (iPSC) were purchased from System Biosciences. iPSCs were grown on Matrigel (Corning). Plates were coated with MatriGel in accordance with manufacturer protocols, MatriGel was diluted in DMEM/F12 (+ L-glutamine, + 15 mM HEPES, - phenol red; Gibco, Dublin, Ireland). iPSCs were cultured in either exclusively mTesR1 media (STEMCELL Technologies) or a combination of mTesR1 and StemFlex (Thermo Fisher Scientific) media at 37°C in 5% CO2. Briefly, the combination culture consisted of passaging iPSCs in StemFlex for the first 1–2 d, to increase yield, then switch back to mTesR1 for the remainder of the culturing period. HFFs were cultured in DMEM (4.5 g/L glucose) supplemented with 2 mM L-glutamine, 0.1 mM nonessential amino acids and 10% NBCS (Peak Serum) at 37°C in 5% CO2.
Neural induced differentiation of iPSCs was performed using STEMdiff Neural Induction Medium (STEMCELL Technologies) in accordance with manufacturer's recommendations. Briefly, iPSCs were passaged onto new MatriGel coated plates in StemFlex and left to attach overnight. Approximately 24 h after passaging, media was removed and replaced with STEMdiff Neural Induction Medium. Cells were cultured in STEMdiff Neural Induction Medium for up to 7 d with media changed daily.
Transfection
Transient knockdown was achieved by transfecting iPSCs with siRNAs supplied by MilliporeSigma (see Supplemental Table S3 for detailed information about the siRNAs used). Transfections were carried out using Stemfect transfection reagent (Stemgent) in accordance with manufacturer's recommendations. Cells were transfected at a 50 µM concentration daily for 2–6 d, depending on the experiment, and harvested approximately 24 h after the final transfection.
RNA isolation and RT-dPCR
RNA isolation was performed using TRIzol Reagent (Thermo Fisher Scientific) in accordance with manufacturer's protocols. Total RNA was treated with DNase I (Thermo Fisher Scientific). Removal of DNase I was achieved by phenol/chloroform/isoamyl alcohol (25:24:1) extraction and ethanol precipitation.
Reverse transcription (RT) was performed in 20 μL reactions using Improm-II Reverse Transcriptase (Promega) with slight modifications to the manufacturer's protocol. Specifically, the reaction was primed with 0.5 μg of a 3:1 mixture of random hexamers:oligod(T) and RiboLock (Thermo Fisher Scientific) was included as an RNase inhibitor. The resulting cDNA was diluted such that the template copy number was between 10 and 100,000 and used in dPCR reactions with QX200 ddPCR EvaGreen Supermix (Bio-Rad) and transcript-specific primers. Detailed information about the primers used for each transcript can be found in Supplemental Table S3. Droplet generation was performed using a QX200 Droplet Generator (Bio-Rad) in accordance with manufacturer's protocols. Droplets were transferred to a 96-well plate and heat sealed with pierceable foil using the PX1 PCR Plate Sealer (Bio-Rad). A two-step amplification was performed in a C1000 Touch Thermal Cycler with 96-Deep Well Reaction Module (Bio-Rad) with annealing/extension at 60°C for 60 sec and denaturation at 95°C for 30 sec and a 2.5°C/sec ramp rate. Fluorescence of each droplet was interrogated using the QX200 Droplet Reader (Bio-Rad) and transcript copy numbers were determined using the QuantaSoft Software (Bio-Rad). Transcript copy numbers were normalized to GAPDH across compared samples and expression changes were reported as relative mRNA abundance or fold-change.
Protein preparation and western blotting
Colonies were detached using ReLeSR (STEM CELL Technologies), pelleted by centrifugation at 1000g and 4°C for 5 min and washed twice in 10 mL of cold PBS. After the second wash, cells were re-suspended in 100–500 μL of RIPA buffer (50 mM Tris-Cl pH 7.5, 1% NP40, 0.5% Sodium Deoxycholate, 0.05% SDS, 1 mM EDTA, 150 mM NaCl, 1 mM PMSF and 1 mM DTT), with Protease Inhibitor Cocktail (MilliporeSigma, # P8340), and incubated for 10 min at 4°C with gentle rotation. Cells were then lysed via sonication with 5 × 10 sec pulses at 6–7 W output with 1 min of rest on ice in between. After lysis, samples were centrifuged at 14,000 rpm for 10 min at 4°C. Whole cell lysates (supernatant) were quantified via Pierce Rapid Gold BCA kit (Thermo Fisher Scientific) in accordance with manufacturer's protocols.
20–40 μg of whole cell lysate was run on a 8% SDS polyacrylamide gel, transferred to an Immobilon PVDF membrane (EMD Millipore) and probed with primary antibodies at room temperature for 2 h (see Supplemental Table S3 for details). Membranes were washed, then incubated for 1 h at room temperature with HRP-conjugated anti-rabbit (1/5000; Bio-Rad) or anti-mouse (1/5000; Santa Cruz) antibodies. After washing, membranes were treated with SuperSignal West Dura Extended ECL reagent (Thermo Fisher Scientific) and luminescence was assessed on the ChemiDoc XRS+ system (Bio-Rad) or the Azure Sapphire Biomolecular Imager (Azure Biosystems). Band intensities were assessed using ImageLab software (Bio-Rad) and normalized to GAPDH.
4-Thiouridine (4sU) labeling and half-life analysis
4sU labeling and subsequent half-life analysis was performed as described previously (Russo et al. 2017). Briefly, cells treated with siRNAs for 2 d were labeled with 4sU at concentration of 400 μM for 4 h. RNA was collected and isolated using TRIzol reagent. Prior to fractionation, a 4sU-labeled synthetic transcript (Dharmacon) with the following sequence: 5′-AUUUAGGUGACACUAUAGGAUCCUCUAGAGUCGACCUUCUCCCUAUAGUGAGUCGUAUUAGCA[4-S-U]CAG-3′, was spiked into samples to assess fractionation efficiency. A total of 20–40 μg of total RNA and was labeled with MTSEA-biotin-XX (Biotium) and fractionated using μMACS streptavidin magnetic beads (Miltenyi Biotec). Transcript abundances were measured using RT-dPCR, and half-lives were calculated based on the ratio of labeled to unlabeled RNA (Rädle et al. 2013).
RNA immunoprecipitation (RIP)
Cells were detached from 10-cm plates using ReLeSR (STEM CELL Technologies), pelleted by centrifugation at 1000g and 4°C for 5 min and washed twice in 10 mL of cold PBS. After the second wash, cells were re-suspended in 10 mL of cold PBS and RNA-protein crosslinking was performed by adding 0.3% formaldehyde. Cells were incubated in the crosslinking solution for 10 min at room temperature with gentle rocking. 1.4 mL of 2M glycine (pH 7.0) was added to quench the crosslinking reaction. Cells were then pelleted again at 1000g and 4°C for 5 min and washed twice in 10 mL of cold PBS. Cell pellets were stored at −80°C until further processing.
Cells were thawed and re-suspended in 1 mL of low stringency (LS) RIPA buffer (50 mM Tris-Cl pH 7.5, 1% NP40, 0.5% Sodium Deoxycholate, 0.05% SDS, 1 mM EDTA, 150 mM NaCl, 1 mM PMSF and 1 mM DTT) with Complete Mini EDTA-free Protease Inhibitor Cocktail tablets (MilliporeSigma) and RiboLock RNase inhibitor (Thermo Fisher Scientific). Cells were incubated in LS RIPA buffer for 10 min at 4°C with gentle rotation and then lysed by passing through a 25 gauge needle 10–12 times. Samples were spun down at 14,000 rpm for 10 min at 4°C. Cell extracts (supernatant) were transferred to new tubes and were precleared by incubation with 20 µL of magnetic protein G beads (NEB) for 60 min at 4°C with gentle rotation. Beads were removed from the extracts using a magnetic stand. After preclearing, a 100 µL aliquot was set aside to serve as the input. Then, 450 µL samples were incubated with 20 µL of either YTHDF2 or IgG antibodies with rotation at 4°C for 60 min. After incubation, 20 µL of magnetic protein G beads was added and samples were again incubated with rotation at 4°C for 60 min. Beads were then retrieved and resuspended in 500 µL of high stringency (HS) RIPA buffer (50 mM Tris-Cl pH 7.5, 1% NP40, 1% Sodium Deoxycholate, 0.1% SDS, 1 mM EDTA, 1 M NaCl, 1 M Urea, 1 mM PMSF and 1 mM DTT), with Complete Mini EDTA-free Protease Inhibitor Cocktail tablets and RiboLock RNase inhibitor included, and incubated with rotation at 4°C for 5 min. This was repeated for a total of five washes. To reverse the crosslinking, beads were re-suspended in 100 µL of TEDS buffer (50 mM Tris-Cl pH 7.0, 5 mM EDTA, 1% SDS, 10 mM DTT) and 50 µL of TEDS buffer was added to Input samples. IP and Input samples were then incubated at 70°C for 45 min. 300 and 450 µL of TRIzol was added to IP and Input samples, respectively. Samples were stored at −80°C until RNA could be extracted see RNA Isolation). Upon completion of RNA isolation, RNA samples were suspended in 4 µL of water and the full volume was taken into the reverse transcription reaction. RT-dPCR was used to measure RNA abundances in YTHDF2 IP, IgG IP and Input samples. Percent of Input (PoI) was determined and fold enrichments were calculated as YTHDF2 PoI/IgG PoI for a specific gene. Protein samples were obtained from independent RIPs and protein abundance was assessed via western blot to measure RIP efficiency.
Immunofluorescence staining
Circular coverslips were placed on 6- or 10-cm dishes and coated with MatriGel prior to seeding. Cells were seeded, cultured and treated in accordance with previously described protocols (see Cell Culture, Neural Induction and Transfection). After treatment, coverslips were collected and washed twice in 1 mL of PBS. Cells were fixed in a 3.7% formaldehyde PBS solution for 10 min at room temperature with slight agitation. After fixing, cells were washed twice in 1 mL of PBS and permeabilized in 70% EtOH overnight at 4°C. Cells were blocked by incubating in 1 mL of blocking buffer (20 mg/mL Fraction 5 BSA and 0.02% Triton X-100) overnight at 4°C. Following blocking, cells were probed with a 1:100 dilution of primary antibody in blocking buffer and placed in a humidity chamber at 37°C for 2 h (see Supplemental Table S3 for details). Cells were washed five times with PBS-T (0.02%) for 1 min with slight agitation. Cells were then probed with Alexa Fluor568-conjugated anti-rabbit (Abcam) or anti-mouse (Abcam) at a 1:500 dilution in blocking buffer and placed in a humidity chamber at 37°C for 2 h. After incubation, cells were again washed five times with PBS-T for 1 min with slight agitation. Slides were then mounted using one drop (∼100 μL) of ProLong Diamond Antifade Mountant with DAPI (Life Technologies, Carlsbad, CA) and left to cure in the dark at room temperature overnight. Slides were visualized using an Olympus IX71 inverted fluorescent microscope at 100× magnification using the 31000 DAPI/Hoechst filter (EX360, EM460) to visualize DAPI and the 41002 TRITC (Rhodamine)/Cy3 filter (EX535, EM610) to visualize Alexa568. Images were captured using Q Imaging Retiga 2000R camera. Background fluorescence was determined by measuring slides probed with secondary antibody only and the minimum threshold of images was adjusted accordingly. Nucleus size was analyzed via ImageJ (Schneider et al. 2012).
MazF digestion and analysis
MazF digestion and analysis was performed according to a published protocol (Garcia-Campos et al. 2019) with some modification. Briefly, 100 ng of total RNA was digested by 20 units of MazF enzyme (Takara Bio, Kusatsu, Japan) supplemented with 4 µL 5× buffer, 0.8 µL DMSO and 0.5 µL RNase inhibitor (Thermo Fisher Scientific) in a 20 µL reaction. Samples were denatured at 70°C for 5 min then incubated at 37°C for 30 min. Digested RNA was purified by phenol/chloroform/isoamyl alcohol (25:24:1) extraction and ethanol precipitation, then subjected to RT-dPCR as previously described (see RNA Isolation and RT-dPCR) with one exception. 100% random hexamer was used to prime the reaction instead of 3:1 mixture of random hexamers:oligod(T). Three primer sets were designed to create different products from the same transcript. The ACA product contained two unprotected ACA motifs, the control product contained no ACA motifs and the m6A product contained the methylated ACA motif of interest (Supplemental Fig. S5E). Methylation ratios were generated using the formula: m6A product abundance/Control product abundance. To compensate for enzymatic efficiency between samples and RT efficiency across different areas of the transcript, a normalization ratio was generated using the same formula, but with samples lacking MazF. The same analysis protocol was used with the ACA product to generate a background ratio. This background ratio was subtracted from the methylation ratio in order to get a more accurate quantification of methylation status.
Library preparation and RNA sequencing
Sequencing libraries were prepared from Control or YTHDF2 siRNA-treated iPSC total RNA samples (see RNA Isolation section, above). First strand cDNA was generated from 500 ng of total RNA using the SMARTer PCR cDNA Synthesis Kit (Takara Bio) in a 10 µL reaction per manual instructions. Reactions were incubated at 42°C for 90 min to achieve full-length cDNA synthesis. Second strand synthesis was performed using the Advantage 2 PCR kit (Takara Bio) in a 50 µL reaction with the following thermocycler conditions: 95°C for 15 sec, 65°C for 30 sec, 68°C for 6 min. Extension time was set at 6 min to accommodate the full-length cDNA synthesis performed during first strand synthesis. Amplification of cDNA was performed for four to six cycles. PCR products were isolated using 90 µL of Agencourt AMPure XP Beads (Beckman Coulter, Brea, CA) and eluted in 40 µL of TE buffer. DNA concentrations were quantified via the Qubit 3.0 fluorometer (Thermo Fisher Scientific) using the dsDNA HS kit (Thermo Fisher Scientific). DNA was tagmented using Tn5 transposase generously provided by Dr. Mark Stenglein. Preparation of the enzyme and reactions conditions were performed in accordance with a previously published protocol (Picelli et al. 2014). Five nanograms of dsDNA, 2 µL of Tn5 Tagmentation Enzyme at a 12.5 µM concentration and a 5× TAPS-DMF buffer (see Picelli et al. 2014 for recipe) were combined in a final volume of 20 µL. Reactions were incubated at 55°C for 10 min to achieve dsDNA fragments of approximately 200–300 base pairs in length. Reactions were terminated by adding 2.5 µL of 0.2% SDS and incubating at room temperature for 5 min. Tagmented DNA (5.8 µL) was used as a template for addition of full-length adapters in a 4-primer PCR reaction. This reaction was performed using the KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Wilmington, MA). For details on the tagmentation primers and index oligos used see Supplemental Table S3. PCR products were isolated using AMPure XP beads and eluted in 40 µL of TE buffer. A portion of each library was visualized on the Agilent TapeStation 2200 using D1000 ScreenTape (Agilent, Santa Clara, CA) and reagents to verify a median product size of 200–300 base pairs, and concentrations were measured fluorometrically via Qubit. Equal masses of DNA, 10 ng, from each sample were pooled, concentrated with AMPure XP beads and eluted in 40 µL of TE buffer. A portion of the pooled library was run on the Agilent TapeStation 2200 using D1000 ScreenTape and to ensure a median product size of 200–300 base pairs. Final library quantification was performed by the Colorado State University NGS Core using the Illumina library quantification kit (KAPA Biosystems) per manufacturer's protocols. Libraries were sequenced on an Illumina NextSeq 500 instrument using single-end 1 × 75 sequencing from a NextSeq 500/550 High Output Kit v2 (75 cycles) (Illumina) on two separate runs. The mean number of reads per sample was 25–50 million. Basecalling was done via RTA v2.0, and files were converted to fastq using bcl2fastq v2.20.0.422, consistent with standard Illumina protocols.
RNA-seq alignment
Quality of sequence reads was assessed via FastQC v0.11.5. Reads were trimmed of adapter sequences and filtered for low-complexity or low-quality reads via Trimmomatic v0.32 (Bolger et al. 2014). rRNA reads were filtered out using Bowtie2 v2.3.4.3 (Langmead and Salzberg 2012) and a reference fasta file of mature 5S, 18S and 28S rRNA sequence obtained from NCBI RefSeq (mature_rRNA.fasta.gz). Filtered reads were mapped to the hg19 genome using TopHat2 v2.0.12 (Kim et al. 2013) under default parameters. Read counts from aligned reads were calculated using HTSeq v0.11.1 (Anders et al. 2015) with the parameters ‐‐mode = union ‐‐stranded=no ‐‐minaqual=20, Samtools v1.9 (Li et al. 2009) and the hg19 genome feature file obtained from GENCODE (hg19, Ensembl 74, hg19_gene_annotations.gtf.gz). Raw and processed RNA-seq data generated in this study, as well as the genome feature file and rRNA fasta file used, were uploaded to the NCBI Gene Expression Omnibus (GEO) database under the accession number GSE133898 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133898).
Differential expression analysis
Principle component analysis (PCA) of the raw RNA-seq count data indicated the presence of unwanted variation in the sequencing data (Supplemental Fig. S2A). To remove the unwanted variation, raw RNA-seq count data was filtered for detectable genes (>5 counts across all samples) and normalized via the RUVSeq package using the RUVr option in accordance with the published protocol (Risso et al. 2014). PCA was performed on the normalized data to evaluate the removal of unwanted variation (Supplemental Fig. S2B). Normalized count data was analyzed using DESeq2 v1.22.1 (Love et al. 2014) under default parameters. To assess the correlations between samples, sample-to-sample Euclidian distances were calculated on normalized, variance-stabilized, log-transformed count data (Supplemental Fig. S2C). Thresholds were set at Benjamin-Hochberg corrected P-value <0.05 and log2-fold change >1.0 for up-regulated genes and log2-fold change <1.0 for down-regulated genes. Transcripts that met both thresholds were classified as differentially expressed (Supplemental Table S1). Gene symbol, Entrez Gene ID and gene biotype were retrieved using the biomaRt package v2.38.0 (Durinck et al. 2009) and the hsapiens_gene_ensembl data set. Hierarchical clustering of differentially expressed genes was performed and plotted on Z-scored intensity values using the heatmap.2 function of the gplots package v3.0.1 (Warnes et al. 2016). Distances were calculated based on the Pearson correlation coefficient and clustering was performed using the unweighted pair group method with arithmetic mean.
Functional annotation analysis
Gene enrichment analysis was performed using the online functional annotation tool DAVID v6.8 (Huang et al. 2009a,b). Separate lists of Ensembl Gene IDs corresponding to differentially expressed transcripts, either up- or down-regulated, were uploaded and mapped to DAVID gene IDs. Functional annotation cluster was performed to determine overrepresented Gene Ontology biological processes, cellular components and molecular functions. Analysis of overrepresented tissue expression profiles was also performed for the up-regulated list. The top four to five nonredundant terms from each analysis, defined by the lowest FDR corrected P-values were reported along with the number of genes associated with each term and the fold enrichment over background (Fig. 2).
Overlap analysis
Data for transcripts previously reported to be m6A-modified via m6A RIP-seq in hESCs (Batista et al. 2014) or bound by YTHDF2 via PAR-CLIP seq in HeLa cells (Wang et al. 2014a) were obtained through publically available data sets. Lists of gene names for transcripts up-regulated, down-regulated, m6A-modified and YTHDF2-bound along with all genes detected in each experiment were imported into R v3.5.1 (R Core Team 2018). Overlap analysis was performed using the packages plyr v1.8.4 (Wickham 2011), VennDiagram v1.6.20 (Chen 2018) and futile.logger v1.4.3 (Rowe 2016). Background filtering was performed to ensure only genes that were detected in both experiments were included in the overlap analysis (Supplemental Fig. S3A). Lists of the genes that were identified in each section of the Venn diagrams from Figure 2 and Supplemental Figure S3 can be found in Supplmental Table S2. P-values were calculated via hypergeometric test using the phyper function of the stats package.
Quantification and statistical analysis
Three independent biological replicates were performed for each sample/treatment in an experiment and data are reported with center values as mean and error bars as s.e.m. unless noted otherwise. Statistical analyses were performed using GraphPad Prism 8 software (GraphPad Software, Inc.). For RIP analyses, a one-tailed t-test was used. For all other comparisons, a two-tailed t-test was used and a false discovery rate (FDR) or q-value was calculated using the two-stage step-up method of Benjamini, Krieger and Yekutieli (Benjamini et al. 2006) to correct for multiple comparisons, when applicable. P-values from t-tests and q-values from FDR tests of less than 0.05 were considered significant.
DATA DEPOSITION
The accession number for the RNA-seq data reported in this study is GEO: GSE133898. All scripts/codes used for computational analysis are available upon request.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Dr. Mark Stenglein, Dr. Justin Lee, and the CSU Next Generation Sequencing Core for their help in library preparation, sequencing runs, and data analysis and the CSU Molecular Quantification Core for access to digital PCR. This work was supported by the National Institutes of Health, National Institute for General Medical Sciences award #GM114247 to C.J.W. and J.W. A.M.H. was supported in part through NSF-NRT (National Science Foundation-NSF Research Traineeship) award #1450032 (PrincipaI Investigator: Tom Chen) and an NSF Graduate Research Fellowship.
Author contributions: Conceptualization: C.J.W. and J.W.; methodology: A.M.H. and J.R.; investigation: A.M.H. and J.R.; formal analysis: A.M.H.; visualization: A.M.H. and C.J.W.; writing—original draft: A.M.H. and C.J.W.; writing—reviewing and editing: A.M.H., J.R., J.W., E.O.N., and C.J.W.; supervision: C.J.W., J.W., and E.O.N.; project administration: C.J.W.; funding acquisition: A.M.H., J.W., and C.J.W.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.073502.119.
REFERENCES
- Aguilo F, Zhang F, Sancho A, Fidalgo M, Di Cecilia S, Vashisht A, Lee D-F, Chen C-H, Rengasamy M, Andino B, et al. 2015. Coordination of m6A mRNA methylation and gene transcription by ZFP217 regulates pluripotency and reprogramming. Cell Stem Cell 17: 689–704. 10.1016/j.stem.2015.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshawaf AJ, Antonic A, Skafidas E, Ng DCH, Dottori M. 2017. WDR62 regulates early neural and glial progenitor specification of human pluripotent stem cells. Stem Cells Int 2017: 1–9. 10.1155/2017/7848932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. 2014. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15: 707–719. 10.1016/j.stem.2014.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Krieger AM, Yekutieli D. 2006. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93: 491–507. 10.1093/biomet/93.3.491 [DOI] [Google Scholar]
- Bertero A, Brown S, Madrigal P, Osnato A, Ortmann D, Yiangou L, Kadiwala J, Hubner NC, de los Mozos IR, Sadée C, et al. 2018. The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature 555: 256–259. 10.1038/nature25784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel M, Richter J, Schrenk K, Tucker KL, Staiger V, Korte M, Goetz M, Barde YA. 2004. Differentiation of mouse embryonic stem cells into a defined neuronal lineage. Nat Neurosci 7: 1003–1009. 10.1038/nn1301 [DOI] [PubMed] [Google Scholar]
- Bilgüvar K, Öztürk AK, Louvi A, Kwan KY, Choi M, Tatli B, Yalnizoǧlu D, Tüysüz B, Çaǧlayan AO, Gökben S, et al. 2010. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467: 207–210. 10.1038/nature09327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. 2018. VennDiagram: generate high-resolution Venn and Euler PlotsPackage “VennDiagram.” https://cran.r-project.org/package=VennDiagram
- Chen JF, Zhang Y, Wilde J, Hansen KC, Lai F, Niswander L. 2014. Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size. Nat Commun 5: 3885 10.1038/ncomms4885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Lu Z, Wang X, Fu Y, Luo G-Z, Liu N, Han D, Dominissini D, Dai Q, Pan T, et al. 2015a. High-resolution N6-methyladenosine (m6A) map using photo-crosslinking-assisted m6A sequencing. Angew Chem Int Ed 54: 1587–1590. 10.1002/anie.201410647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Hao Y-J, Zhang Y, Li M-M, Wang M, Han W, Wu Y, Lv Y, Hao J, Wang L, et al. 2015b. m6A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 16: 289–301. 10.1016/j.stem.2015.01.016 [DOI] [PubMed] [Google Scholar]
- Chen J, Sun Y, Xu X, Wang D, He J, Zhou H, Lu Y, Zeng J, Du F, Gong A, et al. 2017. YTH domain family 2 orchestrates epithelial-mesenchymal transition/proliferation dichotomy in pancreatic cancer cells. Cell Cycle 16: 2259–2271. 10.1080/15384101.2017.1380125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang YC, Huang C, Shen H, Sun B, Cheng X, Zhang YJ, Yang YG, Shu Q, Yang Y, et al. 2019. m6A regulates neurogenesis and neuronal development by modulating histone methyltransferase Ezh2. Genomics Proteomics Bioinformatics 17: 154–168. 10.1016/j.gpb.2018.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Z, Zhang J, Tokunaga A, Harraz MM, Byrne ST, Dolinko A, Xu J, Blackshaw S, Gaiano N, Dawson TM, et al. 2012. Botch promotes neurogenesis by antagonizing Notch. Dev Cell 22: 707–720. 10.1016/j.devcel.2012.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Z, Byrne ST, Dolinko A, Harraz MM, Kim MS, Umanah G, Zhong J, Chen R, Zhang J, Xu J, et al. 2014. Botch is a γ-glutamyl cyclotransferase that deglycinates and antagonizes Notch. Cell Rep 7: 681–688. 10.1016/j.celrep.2014.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell RB, Ke S, Darnell JE. 2018. Pre-mRNA processing includes N6 methylation of adenosine residues that are retained in mRNA exons and the fallacy of “RNA epigenetics.” RNA 24: 262–267. 10.1261/rna.065219.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485: 201–206. 10.1038/nature11112 [DOI] [PubMed] [Google Scholar]
- Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, Ma J, Wu L. 2016. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4–NOT deadenylase complex. Nat Commun 7: 12626 10.1038/ncomms12626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, Huber W. 2009. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4: 1184–1191. 10.1038/nprot.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng N, Han Q, Li J, Wang S, Li H, Yao X, Zhao RC. 2014. Generation of highly purified neural stem cells from human adipose-derived mesenchymal stem cells by Sox1 activation. Stem Cells Dev 23: 515–529. 10.1089/scd.2013.0263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Campos MA, Edelheit S, Toth U, Safra M, Shachar R, Viukov S, Winkler R, Nir R, Lasman L, Brandis A, et al. 2019. Deciphering the “m6A Code” via antibody-independent quantitative profiling. Cell 178: 731–747.e16. 10.1016/j.cell.2019.06.013 [DOI] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. 2015. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347: 1002–1006. 10.1126/science.1261417 [DOI] [PubMed] [Google Scholar]
- Grigor'eva EV, Malankhanova TB, Surumbayeva A, Minina JM, Morozov VV, Abramycheva NY, Illarioshkin SN, Malakhova AA, Zakian SM. 2019. Generation of induced pluripotent stem cell line, ICGi007-A, by reprogramming peripheral blood mononuclear cells from a patient with Huntington's disease. Stem Cell Res 34: 101382 10.1016/j.scr.2018.101382 [DOI] [PubMed] [Google Scholar]
- Guo J, Tang H-W, Li J, Perrimon N, Yan D. 2018. Xio is a component of the Drosophila sex determination pathway and RNA N6-methyladenosine methyltransferase complex. Proc Natl Acad Sci 115: 3674–3679. 10.1073/pnas.1720945115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck AM, Wilusz CJ. 2019. Small changes, big implications: the impact of m6A RNA methylation on gene expression in pluripotency and development. Biochim Biophys Acta Gene Regul Mech 1862: 194402 10.1016/j.bbagrm.2019.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Ivanova I, Much C, Di Giacomo M, Azzi C, Morgan M, Moreira PN, Monahan J, Carrieri C, Enright AJ, O'Carroll D. 2017. The RNA m6A reader YTHDF2 is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol Cell 67: 1059–1067.e4. 10.1016/j.molcel.2017.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang Y-G, et al. 2011. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7: 885–887. 10.1038/nchembio.687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kami D, Kitani T, Nakamura A, Wakui N, Mizutani R, Ohue M, Kametani F, Akimitsu N, Gojo S. 2018. The DEAD-box RNA-binding protein DDX6 regulates parental RNA decay for cellular reprogramming to pluripotency. PLoS One 13: e0203708 10.1371/journal.pone.0203708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasowitz SD, Ma J, Anderson SJ, Leu NA, Xu Y, Gregory BD, Schultz RM, Wang PJ. 2018. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet 14: e1007412 10.1371/journal.pgen.1007412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vågbø CB, Geula S, Hanna JH, Black DL, Darnell JE, Darnell RB. 2017. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31: 990–1006. 10.1101/gad.301036.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, Masiello I, Hares T, Villaseñor R, Hess D, et al. 2018. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m6A machinery component Wtap/Fl(2)d. Genes Dev 32: 415–429. 10.1101/gad.309146.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Kim H, Elkabetz Y, Al Shamy G, Panagiotakos G, Barberi T, Tabar V, Studer L. 2007. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat Biotechnol 25: 1468–1475. 10.1038/nbt1365 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Zhao D, Wu J, Shi Y. 2014. Structure of the YTH domain of human YTHDF2 in complex with an m6A mononucleotide reveals an aromatic cage for m6A recognition. Cell Res 24: 1490–1492. 10.1038/cr.2014.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang Y, Zhang Z, Zamudio AV, Zhao JC. 2015. Genome-wide detection of high abundance N6-methyladenosine sites by microarray. RNA 21: 1511–1518. 10.1261/rna.051474.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zhao X, Wang W, Shi H, Pan Q, Lu Z, Perez SP, Suganthan R, He C, Bjørås M, et al. 2018a. Ythdf2-mediated m6A mRNA clearance modulates neural development in mice. Genome Biol 19: 69 10.1186/s13059-018-1436-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Qian P, Shao W, Shi H, He XC, Gogol M, Yu Z, Wang Y, Qi M, Zhu Y, et al. 2018b. Suppression of m6A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res 28: 904–917. 10.1038/s41422-018-0072-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. 2014. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10: 93–95. 10.1038/nchembio.1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloret-Llinares M, Karadoulama E, Chen Y, Wojenski LA, Villafano GJ, Bornholdt J, Andersson R, Core L, Sandelin A, Jensen TH. 2018. The RNA exosome contributes to gene expression regulation during stem cell differentiation. Nucleic Acids Res 46: 11502–11513. 10.1093/nar/gky817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Chang M, Lv H, Zhang Z-W, Zhang W, He X, Wu G, Zhao S, Zhang Y, Wang D, et al. 2018. RNA m6A methylation participates in regulation of postnatal development of the mouse cerebellum. Genome Biol 19: 68 10.1186/s13059-018-1435-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malchenko S, Xie J, de Fatima Bonaldo M, Vanin EF, Bhattacharyya BJ, Belmadani A, Xi G, Galat V, Goossens W, Seftor REB, et al. 2014. Onset of rosette formation during spontaneous neural differentiation of hESC and hiPSC colonies. Gene 534: 400–407. 10.1016/j.gene.2013.07.101 [DOI] [PubMed] [Google Scholar]
- Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur J-J, Chen Q, et al. 2017. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 541: 371–375. 10.1038/nature21022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646. 10.1016/j.cell.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasaka R, Matsumoto M, Okada M, Sasaki H, Kanie K, Kii H, Uozumi T, Kiyota Y, Honda H, Kato R. 2017. Visualization of morphological categories of colonies for monitoring of effect on induced pluripotent stem cell culture status. Regen Ther 6: 41–51. 10.1016/j.reth.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas AK, Khurshid M, Désir J, Carvalho OP, Cox JJ, Thornton G, Kausar R, Ansar M, Ahmad W, Verloes A, et al. 2010. WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat Genet 42: 1010–1014. 10.1038/ng.682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Park J, Kim J-I, Chang M-Y, Lee S-H, Cho Y-H, Hwang J. 2018. Lin28B and miR-142-3p regulate neuronal differentiation by modulating Staufen1 expression. Cell Death Differ 25: 432–443. 10.1038/cdd.2017.182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, Mapperley C, Lawson H, Wotherspoon DA, Sepulveda C, et al. 2019. Targeting the RNA m6A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell 25: 137–148.e6. 10.1016/j.stem.2019.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park OH, Ha H, Lee Y, Boo SH, Kwon DH, Song HK, Kim YK. 2019. Endoribonucleolytic cleavage of m6A-containing RNAs by RNase P/MRP complex. Mol Cell 74: 494–507.e8. 10.1016/j.molcel.2019.02.034 [DOI] [PubMed] [Google Scholar]
- Patil DP, Pickering BF, Jaffrey SR. 2018. Reading m6A in the transcriptome: m6A-binding proteins. Trends Cell Biol 28: 113–127. 10.1016/j.tcb.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Björklund ÅK, Reinius B, Sagasser S, Winberg G, Sandberg R. 2014. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 24: 2033–2040. 10.1101/gr.177881.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping X-L, Sun B-F, Wang L, Xiao W, Yang X, Wang W-J, Adhikari S, Shi Y, Lv Y, Chen Y-S, et al. 2014. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24: 177–189. 10.1038/cr.2014.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruszak J, Sonntag K-C, Aung MH, Sanchez-Pernaute R, Isacson O. 2007. Markers and methods for cell sorting of human embryonic stem cell-derived neural cell populations. Stem Cells 25: 2257–2268. 10.1634/stemcells.2006-0744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle AD, Lock LF, Donovan PJ. 2006. Neurotrophins mediate human embryonic stem cell survival. Nat Biotechnol 24: 344–350. 10.1038/nbt1189 [DOI] [PubMed] [Google Scholar]
- R Core Team. 2018. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Rädle B, Rutkowski AJ, Ruzsics Z, Friedel CC, Koszinowski UH, Dölken L. 2013. Metabolic labeling of newly transcribed RNA for high resolution gene expression profiling of RNA synthesis, processing and decay in cell culture. J Vis Exp e50195 10.3791/50195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries RJ, Zaccara S, Klein P, Olarerin-George A, Namkoong S, Pickering BF, Patil DP, Kwak H, Lee JH, Jaffrey SR. 2019. m6A enhances the phase separation potential of mRNA. Nature 571: 424–428. 10.1038/s41586-019-1374-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, Dudoit S. 2014. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 32: 896–902. 10.1038/nbt.2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Luo G-Z, Zhang Z, Wang X, Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P, et al. 2017. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife 6 10.7554/eLife.31311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe BLY. 2016. futile.logger: A Logging Utility for R. https://CRAN.R-project.org/package=futile.logger
- Rozwadowska N, Kolanowski T, Wiland E, Siatkowski M, Pawlak P, Malcher A, Mietkiewski T, Olszewska M, Kurpisz M. 2013. Characterisation of nuclear architectural alterations during in vitro differentiation of human stem cells of myogenic origin. PLoS One 8: e73231 10.1371/journal.pone.0073231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo J, Heck AM, Wilusz J, Wilusz CJ. 2017. Metabolic labeling and recovery of nascent RNA to accurately quantify mRNA stability. Methods 120: 39–48. 10.1016/j.ymeth.2017.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru˚žička K, Zhang M, Campilho A, Bodi Z, Kashif M, Saleh M, Eeckhout D, El-Showk S, Li H, Zhong S, et al. 2017. Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol 215: 157–172. 10.1111/nph.14586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. 2014. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8: 284–296. 10.1016/j.celrep.2014.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilà-González M, Kelaini S, Magee C, Caines R, Campbell D, Eleftheriadou M, Cochrane A, Drehmer D, Tsifaki M, O'Neill K, et al. 2019. Enhanced function of induced pluripotent stem cell-derived endothelial cells through ESM1 signaling. Stem Cells 37: 226–239. 10.1002/stem.2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakao S, Kitada M, Kuroda Y, Ogura F, Murakami T, Niwa A, Dezawa M. 2012. Morphologic and gene expression criteria for identifying human induced pluripotent stem cells. PLoS One 7: e48677 10.1371/journal.pone.0048677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. 2014a. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505: 117–120. 10.1038/nature12730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. 2014b. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16: 191–198. 10.1038/ncb2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. 2015. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161: 1388–1399. 10.1016/j.cell.2015.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C-X, Cui G-S, Liu X, Xu K, Wang M, Zhang X-X, Jiang L-Y, Li A, Yang Y, Lai W-Y, et al. 2018a. METTL3-mediated m6A modification is required for cerebellar development. PLoS Biol 16: e2004880 10.1371/journal.pbio.2004880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zuo H, Liu J, Wen F, Gao Y, Zhu X, Liu B, Xiao F, Wang W, Huang G, et al. 2018b. Loss of YTHDF2-mediated m6A-dependent mRNA clearance facilitates hematopoietic stem cell regeneration. Cell Res 28: 1035–1038. 10.1038/s41422-018-0082-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler-Reya RJ, Zhang Z, Ogawa Y, Kellis M, Duester G, et al. 2018c. N6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci 21: 195–206. 10.1038/s41593-017-0057-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes GR, Bolker B, Bonebakker L, Gentleman R, Liaw WHA, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, et al. 2016. Package “gplots”: Various R programming tools for plotting data. R Package version 2170 10.1111/j.0022-3646.1997.00569.x. [DOI] [Google Scholar]
- Wen J, Lv R, Ma H, Shen H, He C, Wang J, Jiao F, Liu H, Yang P, Tan L, et al. 2018. Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Mol Cell 69: 1028–1038.e6. 10.1016/j.molcel.2018.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2011. The split-apply-combine strategy for data analysis. J Stat Softw 40: 1–29. 10.18637/jss.v040.i01 [DOI] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen Y-S, Hao Y-J, Sun B-F, Sun H-Y, Li A, Ping X-L, Lai W-Y, et al. 2016. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell 61: 507–519. 10.1016/j.molcel.2016.01.012 [DOI] [PubMed] [Google Scholar]
- Xu D, Zhang F, Wang Y, Sun Y, Xu Z. 2014. Microcephaly-associated protein WDR62 regulates neurogenesis through JNK1 in the developing neocortex. Cell Rep 6: 104–116. 10.1016/j.celrep.2013.12.016 [DOI] [PubMed] [Google Scholar]
- Xu C, Liu K, Ahmed H, Loppnau P, Schapira M, Min J. 2015. Structural basis for the discriminative recognition of N6-methyladenosine RNA by the human YT521-B homology domain family of proteins. J Biol Chem 290: 24902–24913. 10.1074/jbc.M115.680389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Bi Z, Wu R, Zhao Y, Liu Y, Liu Q, Wang Y, Wang X. 2019. METTL3 inhibits BMSC adipogenic differentiation by targeting the JAK1/STAT5/C/EBPβ pathway via an m6A-YTHDF2–dependent manner. FASEB J 33: 7529–7544. 10.1096/fj.201802644R [DOI] [PubMed] [Google Scholar]
- Yoon K-J, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D, Su Y, Kim N-S, Zhu Y, Zheng L, et al. 2017. Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell 171: 877–889.e17. 10.1016/j.cell.2017.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H, et al. 2018. VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov 4: 10 10.1038/s41421-018-0019-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Huang CT, Chen J, Pankratz MT, Xi J, Li J, Yang Y, LaVaute TM, Li XJ, Ayala M, et al. 2010. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell 7: 90–100. 10.1016/j.stem.2010.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, Lv J, Heng J, Ding Y, Xue Y, et al. 2017. m6A modulates haematopoietic stem and progenitor cell specification. Nature 549: 273–276. 10.1038/nature23883 [DOI] [PubMed] [Google Scholar]
- Zhang J, Nie D, Rocha JL, Hogan M V, Wang JH-C. 2018a. Characterization of the structure, cells, and cellular mechanobiological response of human plantar fascia. J Tissue Eng 9: 204173141880110 10.1177/2041731418801103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Engler A, Taylor V. 2018b. Notch: an interactive player in neurogenesis and disease. Cell Tissue Res 371: 73–89. 10.1007/s00441-017-2641-9 [DOI] [PubMed] [Google Scholar]
- Zhao BS, Wang X, Beadell A V, Lu Z, Shi H, Kuuspalu A, Ho RK, He C. 2017. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542: 475–478. 10.1038/nature21355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Nachtergaele S, Roundtree IA, He C. 2018. Our views of dynamic N6-methyladenosine RNA methylation. RNA 24: 268–272. 10.1261/rna.064295.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, Vågbø CB, Shi Y, Wang W-L, Song S-H, et al. 2013. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18–29. 10.1016/j.molcel.2012.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Nie P, Peng D, He Z, Liu M, Xie Y, Miao Y, Zuo Z, Ren J. 2018. m6AVar: a database of functional variants involved in m6A modification. Nucleic Acids Res 46: D139–D145. 10.1093/nar/gkx895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T, Roundtree IA, Wang P, Wang X, Wang L, Sun C, Tian Y, Li J, He C, Xu Y. 2014. Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res 24: 1493–1496. 10.1038/cr.2014.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang M, Li X, Zhu J, Zhang J, Niu F, Liang F, Chen M, Li D, Han P, Ji S-JJ. 2019. The m6A reader YTHDF1 regulates axon guidance through translational control of Robo3.1 expression. Nucleic Acids Res 47: 4765–4777. 10.1093/nar/gkz157 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
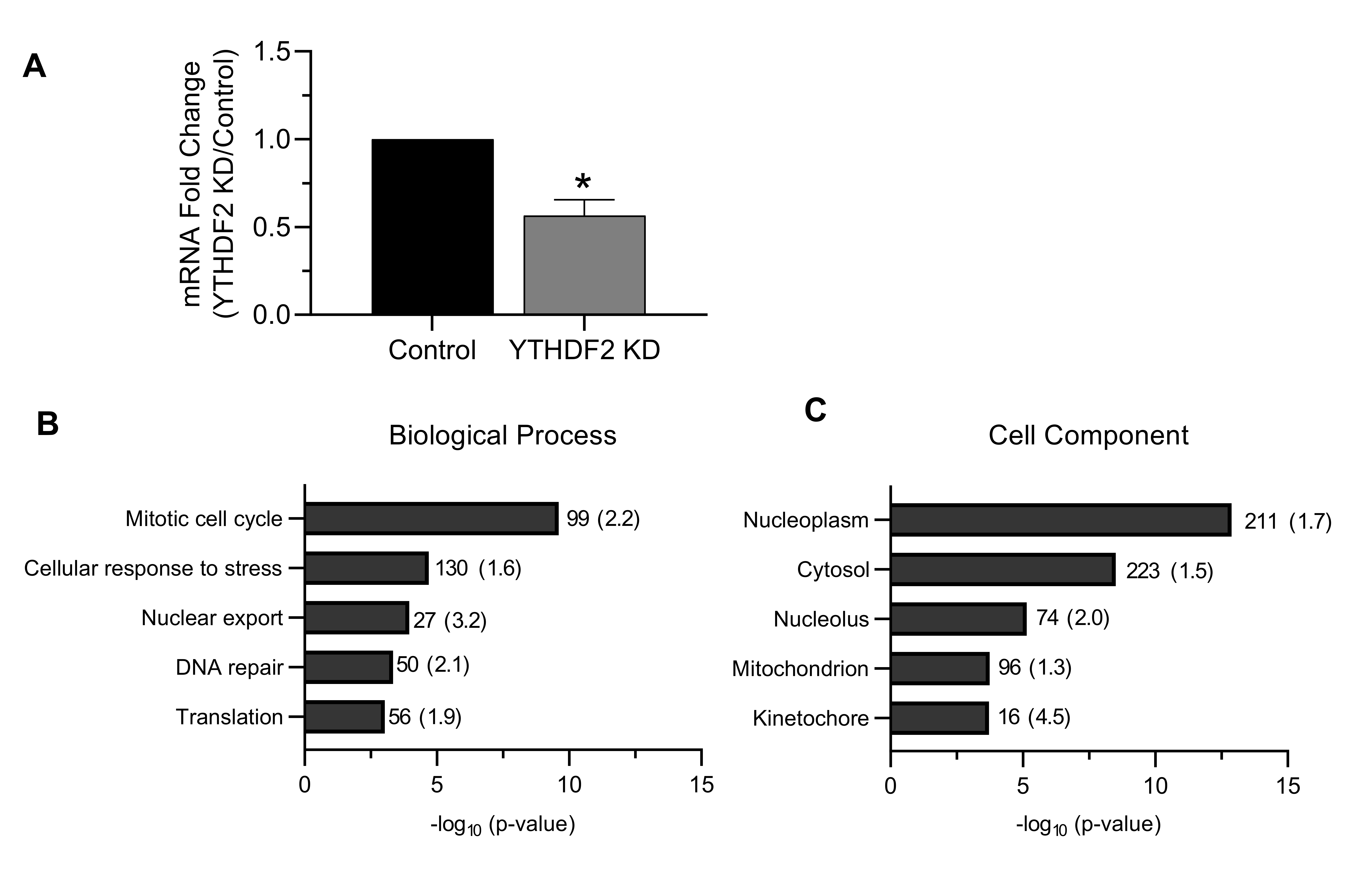{kind=link}
{kind=link}
{kind=link}