

Summary
Motivated by the growing demand for reducing the chemical optimization burden of H2L, we developed auto in silico ligand directing evolution (AILDE, http://chemyang.ccnu.edu.cn/ccb/server/AILDE), an efficient and general approach for the rapid identification of drug leads in accessible chemical space. This computational strategy relies on minor chemical modifications on the scaffold of a hit compound, and it is primarily intended for identifying new lead compounds with minimal losses or, in some cases, even increases in ligand efficiency. We also described how AILDE greatly reduces the chemical optimization burden in the design of mesenchymal-epithelial transition factor (c-Met) kinase inhibitors. We only synthesized eight compounds and found highly efficient compound 5g, which showed an ∼1,000-fold improvement in in vitro activity compared with the hit compound. 5g also displayed excellent in vivo antitumor efficacy as a drug lead. We believe that AILDE may be applied to a large number of studies for rapid design and identification of drug leads.
Subject Areas: Pharmaceutical Science, Chemistry, Computational Chemistry
Graphical Abstract

Highlights
-
•
AILDE was developed for the rapid identification of drug leads
-
•
A potent drug lead targeted to c-Met was found by synthesizing only eight compounds
Pharmaceutical Science; Chemistry; Computational Chemistry
Introduction
Drug discovery is a time-consuming and resource-intensive process that requires large investments by pharmaceutical industry corporations as well as governments (Paul et al., 2010). Developing a new drug from the original idea to the launch of a finished product is usually a complicated process that takes 10–15 years and costs US$0.5 billion to US$2.6 billion (Hughes et al., 2011, Zhavoronkov et al., 2019). Considerations of all possible organic molecules has led to the concept of “chemical space,” which is considered to include at least 1060 molecules (Reymond et al., 2010). Only a small fraction of chemical space can be developed into final drugs (Medina-Franco et al., 2008). Exploring chemical space through synthesis and screening is the conventional method used when searching for new drugs, but it is also a large burden in drug discovery. Consequently, the development of strategies to efficiently explore chemical space for drug discovery is an important scientific issue.
Hit-to-lead (H2L) optimization while working in bioactive and accessible chemical space is crucial to the success of drug discovery and is thus currently a cutting edge research area in medicinal chemistry (Rebecca and Benoit, 2004, Zhao, 2007). H2L optimization involves evaluation and structural optimization of a hit compound to identify promising lead compounds (Keserű and Makara, 2006, Orita et al., 2011). The key to starting H2L optimization is to identify an appropriate scaffold with the desired basic bioactivity that can provide a sufficient number of high-quality analogues. Owing to the development of new technologies in drug design, such as virtual screening, combinatorial chemistry, and high-throughput screening, discovering an appropriate scaffold with the desired basic bioactivity is not difficult. However, 5,000–10,000 compounds still have to be synthesized in the laboratory and screened in the H2L stage to identify a clinical lead (Moridani and Harirforoosh, 2014). The large number of analogues that need to be synthesized and validated experimentally in this step make H2L, a complex multiple-property optimization process, rate-limiting in drug discovery (Hoffer et al., 2018).
To overcome this bottleneck, rational design approaches are widely used in the H2L stage of drug discovery, and many successful applications have been reported (Chéron et al., 2016, Erlanson et al., 2000, Erlanson et al., 2003, Hajduk et al., 1997, Hochguertel et al., 2002, Lin et al., 2019, Nienaber et al., 2000, Oltersdorf et al., 2005, Schulz et al., 2018, Vinkers et al., 2003). One major trend is fragment-based drug discovery (Erlanson et al., 2016, Hao et al., 2016). Graeme et al. developed a fragment-based method to optimize the pyrazole carboxylic ester scaffold with low affinity into a potent cyclic nucleotide phosphodiesterase IV (PDE4) inhibitor by synthesizing only 21 compounds (Card et al., 2005). Another example is the discovery of the selective B cell CLL/lymphoma 2 (BCL-2) drug ABT-199 by screening fragments on two distinct regions of BCL-2 (Souers et al., 2013). Artificial intelligence (AI) is another emerging trend. In a recent work, Zhavoronkov et al. developed a deep generative model for the rapid identification of potent discoidin domain receptor 1 (DDR1) kinase drug leads by synthesizing 40 compounds (Zhavoronkov et al., 2019). These rational design approaches are used to explore key interactions and to understand the relationships between structure and activity, and they are well suited for the discovery of new ligands with improved binding, selectivity, or other pharmacological properties. However, in the H2L stage, such approaches also encounter difficulties such as targeting unreachable chemical space, poor applicability of data-driven models, and inefficient deduction and decision-making on the next leads to be synthesized, which remain formidable challenges.
We developed an efficient and general approach called auto in silico ligand directing evolution (AILDE) for the rapid identification of drug leads in accessible chemical space. AILDE performs minor chemical modifications on the scaffold of a hit compound, and these modifications can result in minimal losses or, in some cases, even increases in ligand efficiency. Hence, this strategy can explore the chemical space around each hit in a series of compounds and drive the evolution of hit compounds into more “clinic-ready” lead structures. The deduction and decision-making on the leads, as well as the applicability of AILDE, have been rigorously validated on 19 drug targets with 157 ligands. The predicted binding affinities were linearly correlated (R2 = 0.82) and within 0.31 kcal mol−1 on average of the experimental values. We also describe how this approach was applied to discover a potent anticancer drug lead (5g) with surprisingly high efficiency.
Owing to their pivotal roles in signal transduction and the regulation of a range of cellular activities, mesenchymal-epithelial transition factor (c-Met) has been established as a promising drug target for the treatment of cancer (Gharwan and Groninger, 2016, Gross et al., 2015, Wu et al., 2016). A large number of c-Met kinase inhibitors have been reported over the past few decades (Basilico et al., 2013, Comoglio et al., 2008, Cui, 2014, Cui et al., 2011, Ma et al., 2005, Martens et al., 2006). However, few examples of the discovery of c-Met kinase inhibitor leads have achieved highly efficient optimization using computational design, synthesis, in vitro and in vivo assays, and cocrystallization validation. We applied AILDE to c-Met and successfully discovered a potent drug lead (5g) by synthesizing only eight compounds. Two steps of ligand-directed evolution were performed. Compound 5g ultimately showed an ∼1,000-fold activity improvement in the enzyme-based assay (IC50 = 9.7 nM) compared with 5a. 5g also exhibited potent in vitro inhibition in a cell-based assay (IC50 = 47.3 nM). Moreover, 5g induced dose-dependent tumor growth inhibition (TGI), with a minimum effective dose (MED/ED50, 50% TGI) of ∼8.3 mg/kg. At 25 mg/kg, 5g showed significant in vivo antitumor efficacy (TGI of 82%). The binding mode and interactions seen in the cocrystal structure of compound 5i (an analogue of 5g) with c-Met were highly consistent with our predicted result, which confirmed the reliability of our strategy. AILDE may improve the efficiency and effectiveness of the initial stages of drug discovery. We also developed a web service to allow medicinal chemists to easily use AILDE.
Results and Discussion
Small Group Library
The small group library includes 47 substituents that are from two fragment-based databases, PADFrag (http://chemyang.ccnu.edu.cn/ccb/database/PADFrag/) (Yang et al., 2018) and Molinspiration (https://www.molinspiration.com/). PADFrag is a searchable web-enabled resource that combines 1,652 FDA-approved drugs, 1,259 commercial pesticides, and 5,919 generated molecular fragments. It was designed for molecule design, and several functions are included in the server, such as viewing, sorting, and fragment extracting. Molinspiration offers a database of substituents and linkers obtained by substructure analysis of a collection of current drugs, development drugs, and other molecules. About 21,000 substituents from 17,000 entries are contained in the database. It has been successfully used in the area of virtual combinatorial chemistry, generation of bioactive molecules, bioisosteric design, and so on. The selection method of the 47 substituents is shown in Transparent Methods. The structures of the substituents are shown in Figure S1.
The Computational Protocol of Ligand-Directed Evolution
A well-designed and organized computational strategy is a powerful tool for improving computational accuracy and efficiency. AILDE, which combined one-step free energy perturbation (FEP) and molecular dynamics (MD) simulation strategies, was achieved by replacing hydrogen atoms with small groups on the hit compound to generate lead candidates with enhanced potency. MD simulation was first performed on the hit-receptor complex system. We collected 50 snapshots from the equilibrated MD trajectory to obtain a representative ensemble of the binding structures. Then, one-step FEP scanning was introduced. We replaced every hydrogen one-by-one by linking the constructed small groups in each snapshot. Each newly obtained ligand-receptor was refined by using two different minimization methods to obtain the final structures. We only performed molecular mechanics (MM) minimization with the steepest descent and conjugated gradient method in the first method. The second method was realized by adding an MD refinement step after MM minimization. Both methods were used to study the case mentioned previously. We used the MM-PBSA method to evaluate the binding free energies of the refined complex structures. The new lead compounds were finally ranked according to the binding free energy shifts between the lead-receptor and hit-receptor complexes. The details of the workflow of the computational strategy are shown in Figure 1. When evaluating a large number of changes to a parent ligand, AILDE was orders of magnitude faster than the conventional FEP-based method. The improvement in efficiency was greater when there were more changes on the ligand.
Figure 1.

The Calculation Workflow of AILDE
The computational process includes initialization, MD simulation, one-step FEP scanning, and free energy calculation. In the snapshot minimization, we use two different strategies for comparison.
Dataset Selection
A collection of protein-ligand structures and bioactivities is prerequisite to evaluate the performance of AILDE. The diverse data to construct a test set were collected according to three criteria: (1) the cocrystal structure of the protein and hit compound is available, (2) the bioactivities of the hit compound and its analogues are available, and (3) the ligands in the dataset show high flexibility. The 19 hit-receptor complexes were collected from published papers (Aronov et al., 2009, Carbain et al., 2014, Charrier et al., 2011, Congreve et al., 2012, Crawford et al., 2014, Demont et al., 2015, Denny et al., 2017, Efremov et al., 2012, Green et al., 2015, Lawhorn et al., 2016, Liu et al., 2017, Medina et al., 2011, Mirguet et al., 2013, Narumi et al., 2013, Tsukada et al., 2010, Wang et al., 2018, Woodhead et al., 2010). There were 157 inhibitors with their experimental bioactivity data in the test dataset. These data are shown in the format of IC50, Ki, Kd, or EC50. These proteins included HIV1-gp120, GSK3, PDK1, and Hsp90 (Table S1). They covered multiple target types, such as viral protein, transferase, kinase, signaling protein, hydrolase, and transcription targets. The hit compounds had different sizes and properties. They were evaluated based on their molecular weight (MW), number of heavy atoms, and number of rotatable bonds (Table S1). The MWs ranged from 184.10 to 460.25 Da. Between 14 and 34 heavy atoms were present. The number of rotatable bonds ranged from 0 to 8. Hence, we considered both rigid and flexible molecules in the dataset. The structures of the hit compounds are shown in Table S1. According to the above analysis, the compounds in our testing set cover a wide range of compound sizes, properties, and target types.
Performance Evaluation and Comparison
To evaluate the impact of an additional MD simulation on the prediction accuracy, we compared the performance of two minimization methods from the perspective of qualitative and quantitative accuracy. All 19 systems were submitted to AILDE for evaluation. We obtained all the binding free energy shift (ΔΔGcal) values between the hit-receptor complexes and the lead-receptor complexes (Table S2). The related experimental binding free energy shift (ΔΔGexp) values were calculated by using the collected activity data based on Equation 4. We found that an additional MD refinement may provide a more convergent structure and a more accurate result. We have offered a detailed application example of TNNI3K-benzenesulfonamide system (see Transparent Methods and Figure S2). Finally, we also compared several published H2L strategies with AILDE, and relative to the other methods, AILDE showed a better predictive performance but lower structural diversity.
The qualitative performance of AILDE was first evaluated in terms of specificity, sensitivity, and accuracy. In the dataset, we defined the positive samples (ΔΔGexp ≥ 0, indicates the derivate has lower or equal bioactivity compared with the hit compound) and negative samples (ΔΔGexp < 0, indicates the derivate has better bioactivity than the hit compound). The numerical values of sensitivity represent the probability of AILDE identifying samples that do in fact have improved bioactivity. The specificity represents the probability of AILDE recognizing samples without giving false-positive results. Accuracy is the proportion of true results, either true positive or true negative, in the total samples. Because we used two minimization methods for one-step FEP scanning, we can summarize the results as follows. (1) For the first minimization method, AILDE identified 75 of the 93 positive samples with a sensitivity (true positive rate) of 80.6% and 56 of 64 negative samples with a specificity (true negative rate) of 87.5%. A total of 131 samples were correctly classified, corresponding to an accuracy of 83.4%, and the predicted binding free energy changes were within 0.71 kcal mol−1 of the experimental value on average. (2) For the second minimization method, 85 of the 93 positive samples and 62 of the 64 negative samples were correctly identified, corresponding to a sensitivity of 91.4% and specificity of 96.9%, respectively. The accuracy was 93.6% based on identifying 147 samples correctly of the 157 samples in the dataset, and the predicted binding free energy changes were within 0.31 kcal mol−1 of the experimental values on average (Figure 2A). We observed that both methods achieved a classification accuracy over 80%. Moreover, the accuracy of the second method was over 90%. The second method had a 10% improvement in accuracy and a 0.4 kcal mol−1 decrease in deviation compared with the first method, which confirmed that the introduction of an MD refinement step can improve the prediction accuracy. The MD step may further optimize the snapshot toward a more reasonable and convergent state.
Figure 2.

Performance Evaluation of AILDE
(A) The details of the predictive results for the total samples under the two minimization strategies; a true positive, b false positive, c false negative, d true negative, e TPR = TP/(TP + FN), f FPR = FP/(FP + TN), g SPC = TN/(FP + TN), h FNR = FN/(TP + FN), i ACC = (TP + TN)/(TP + FP + FN + TN).
(B) The ROC curve showing the relationship between the sensitivity and 1-specificity under the two different minimization strategies (blue for strategy 1 and red for strategy 2) for classifying compounds with lower activity (positive samples) or better activity (negative samples) than the hit compound.
(C) Scatterplot diagram of the first strategy with correlation coefficient R2 = 0.64 (σ = 1.02 kcal mol−1) for 157 studied samples.
(D) Scatterplot diagram of the second strategy with correlation coefficient R2 = 0.82 (σ = 0.54 kcal mol−1) for 157 studied samples. The red lines in (C) and (D) correspond to ideal predictions, and the blue dashed lines mark the 1σ region. The values on the x axis and y axis represent the ΔΔGexp value and ΔΔGcal values, respectively.
We then assessed the classification ability of AILDE based on the receiver operating characteristic (ROC) curve and the area under the ROC curve (AUC). An ROC curve plots sensitivity versus (1-specificity) in the range of 0.0–1.0 according to different thresholds. An AUC value of 1.0 represents a perfect classifier and 0.5 represents a classifier that is no better than random. The ROC curves are shown in Figure 2B and are labeled with the AUC values. The first minimization method showed AUC = 0.875 and the second showed AUC = 0.959. The higher AUC value further confirms the better predictive performance achieved with the introduction of the MD refinement step.
A linear equation can be used to describe the relationship between two sets of variables and show how one variable changes in a linear manner as a function of changes in the other variable. For further study, the linear correlation between ΔΔGcal and ΔΔGexp was obtained with correlation coefficients R2 = 0.64 (SD [σ] = 1.02 kcal mol−1) and 0.82 (σ = 0.54 kcal mol−1) (Figures 2C and 2D) for the first and second strategies, respectively. The linear relationships (R2) of both methods are over 0.6, confirming the prediction reliability of our strategy. The second minimization method resulted in a stronger correlation. The dots in the scatterplot diagram of the second strategy (see Figures 2C and 2D) were more tightly clustered than those in the plot of the first strategy. Therefore, the introduction of an MD refinement step may improve the energy convergence result, which supports our previously mentioned conclusion.
We also compared the accuracies of published computational H2L strategies and AILDE (Carlsson et al., 2008, Chéron et al., 2016, Enyedy and Egan, 2008, Guimarães and Cardozo, 2008, Hsu et al., 2017, Montero-Torres et al., 2006, Moro et al., 2006, Pillai et al., 2005, Vyas et al., 2017, Zhou et al., 2001). The prediction results obtained with the different methods are summarized in Table S3. The accuracies are divided into quantitative and qualitative results. Quantitative accuracy is determined based on the linear correlation coefficient (R or R2) between the experimental and calculated results. Qualitative accuracy was indicated by the AUC value or the accuracy of the model to discriminate samples into positive or negative. Compared with the ligand-based H2L methods, AILDE possesses better accuracy both quantitatively and qualitatively. The R2 value (0.82) is higher than the highest R2 value (0.78) achieved with other ligand-based methods. Most importantly, our strategy is better suited for model expansion because it has a broader target selection scope and a larger number of samples than other ligand-based methods. Most structure-based H2L strategies are based on docking, MD, or MM-PB(GB)SA. The prediction accuracy of docking methods is not over 0.70, although the computational model can be applied on a larger number of systems. The accuracy of the MM-PB(GB)SA method was highest (0.81), but it was limited to the smallest number of samples. In order to compare the diversity of the structures generated by the strategies from a starting point to the potential lead compound, we calculated the Tanimoto coefficient between the initial skeleton and final most activated compound in every study (shown in Table S3) by using RDKit packages (Godden et al., 2000). The compound 5a (hit) and 5g (lead), discovered by us as c-Met inhibitors (seen in Figure 3), were used to calculate the Tanimoto coefficient for AILDE. It shows that AILDE has the highest Tanimoto coefficient (0.709) compared with other methods, which proves the less diversity of the structures generated by AILDE during the optimization process. By comparison, we can see that, although AILDE has higher prediction accuracy and is applicable to a considerably larger number of test samples relative to other structure-based strategies, AILDE has certain limitation compared with other methods in exploring more extensive chemical space.
Figure 3.

The Process of Structural Directed Evolution from Hit Compound 5a with Enzyme-Based IC50 Values
Discovery of c-Met Inhibitors
We applied AILDE to the H2L optimization of c-Met inhibitors to discover a potent anticancer lead. Compound 5a was identified in a previous virtual screening of an in-house database as a hit compound targeting c-Met with weak in vitro activity (IC50 = 9,279 nM). Two steps of ligand-directed evolution were performed, and the process is shown in Figure 3. In the first step, we replaced block A with other groups to improve the activity because no obvious interactions were detected between the thiophene and the surrounding residues and enough space was observed between the thieno[3,2-d]pyrimidine and the receptor (PDB: 3F82) in the docking experiment (Figure S3). The thieno[3,2-d]pyrimidin-4-yl motif (block A) in compound 5a was replaced with a total of 9,403 medicinal fragments in PADFrag (Yang et al., 2018). The prediction result provided new compounds with different blocks, and some of the top-ranked structures are shown in Figure S4. Compounds 5b (ΔΔGcal = −1.31 kcal mol−1) and 5c (ΔΔGcal = −1.45 kcal mol−1) were selected for the enzyme-based activity test. 5c, which possessed a quinazoline moiety, displayed the most promise for further modifications to improve druggability and a moderate c-Met inhibitory activity (IC50 = 602.5 nM). 5c showed 15-fold higher activity than 5a.
The second step was designed to perform the small-substituent-directed evolution because large modifications may reduce the ligand efficiency. The 10 most commonly used substituents (-CH3, -OH, -F, -Cl, -Br, -COOH, -CF3, -NH2, -NO2, and -OCH3) in the small group library were scanned in the R1 position of compound 5c. The five top-ranked and easily synthesizable compounds (5d ∼ h) were synthesized for further bioassay tests. The calculated binding free energy shifts compared with 5a are shown in Table 1. The synthetic route is shown in Figures S5 and S6. All the synthesized compounds were structurally characterized by 1H NMR and 13C NMR spectroscopy and HRMS (shown in Figures S7–S22). Resulting compounds 5d–h exhibited 8.6- to 62.1-fold increases in enzyme potency compared with 5c and 133.1- to 956.6-fold increases compared with 5a. In particular, compound 5g (R1 = 4-F) exhibited significant inhibition potency (IC50 = 9.7 and 47.3 nM) in both the enzyme-based and cell-based assays. By using AILDE, a c-Met inhibitor with great potential (5g) was finally found in a highly efficient manner, and it showed an ∼1,000-fold improvement in activity compared with the hit compound (5a).
Table 1.
Structures and c-Met Inhibitory Potencies of Compounds 5a–5h with Calculated ΔΔGcal Values Compared with 5a
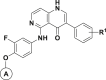 | |||||
|---|---|---|---|---|---|
| Compound | Block A | R1 | c-Met (IC50, nM) |
ΔΔGcal (kcal mol−1) | |
| Enzyme-Baseda | Cell-Basedb | ||||
| 5a |  |
4-OMe | 9,279 | -c | 0 |
| 5b |  |
4-OMe | 1,000 | -c | −1.31 |
| 5c |  |
4-OMe | 602.5 | -c | −1.45 |
| 5d | |
4-CF3 | 40.3 | 24,987 | −3.12 |
| 5e | |
2-F | 28.8 | 194.2 | −3.12 |
| 5f | |
3-F | 67.6 | 600.8 | −2.95 |
| 5g | |
4-F | 9.7 | 47.3 | −3.15 |
| 5h | |
-H | 69.7 | 358.4 | −3.03 |
In vitro kinase assays were performed with the indicated purified recombinant c-Met kinase domains (nM).
IC50 values (nM) for HGF-mediated autophosphoylation in MKN-45 cells.
Not tested.
Compound 5g was then subjected to pharmacokinetic (PK) evaluation and tumor growth inhibition studies in mice. As shown in Table 2, after oral administration (10 mg/kg) in rats, 5g displayed an excellent overall PK profile with a high Cmax (12.7 μg/mL), AUC0-∞ (481.2 μg·h/mL), and F (57%). Moreover, as shown in Figure 4, 5g induced dose-dependent TGI, with an ED50 of 8.3 mg/kg. At 25 mg/kg, 5g exhibited significant in vivo antitumor efficacy (TGI of 82%). More importantly, partial tumor regression (PR 2/6, TGI of 97%) was observed at a higher dose of 5g (75 mg/kg), and the PKs of 5g in beagle dogs and cynomolgus monkeys were outstanding with favorable oral bioavailabilities (F = 73 and 48.0%, respectively, shown in Table S4) after oral administration. In addition, the 28-day repeat-dose toxicity studies on rats showed that 5g has a good safety performance (safety index >40-fold shown in the Transparent Methods) (shown in Figure S23). Taken together, although numerous c-Met inhibitors have been reported, compound 5g demonstrated a favorable in vitro potency, in vivo efficacy, PK profile, and safety profile. Therefore, 5g has been advanced to preclinical studies.
Table 2.
In Vivo PK Profiles of Compound 5g in Rats
| Compound | Route | Dose (mg/kg) | Cmax (μg/mL) | Tmax (h) | T1/2 (h) | AUC0-∞ (μg∗ h/mL) | CL (L/h/kg) | Vz (L/kg) | F (%) |
|---|---|---|---|---|---|---|---|---|---|
| 5g | p.o. | 10 | 12.7 | 2.0 | 26.7 | 481.2 | 0.02 | 0.8 | 57 |
| i.v. | 5 | 30.7 | 0.03 | 16.8 | 420.6 | 0.01 | 0.3 |
Vehicle: 70% PEG400-30% water. Cmax, maximum concentration; Tmax, time of maximum concentration; T1/2, half-life; AUC0-∞, area under the plasma concentration time curve; CL, clearance; VZ, volume of distribution; F, oral bioavailability.
Reported data are the average of six animals.
Figure 4.

In Vivo Bioassay of Compound 5g
(A) In vivo TGI activity of compound 5g in rats. a Administered orally at corresponding dose once daily for 21 consecutive days (70% PEG-400/H2O); b tumor growth inhibition value, ∗:p < 0.05, ∗∗:p < 0.01; c partial regression.
(B) Tumor growth inhibition of 5g in the U87-MG xenograft model in mice.
(C) Tumor pictures at the end of the in vivo antitumor activity assay.
Cocrystal Structure of c-Met and the Inhibitor
To validate the accuracy of the simulated structure, the cocrystal structure of c-Met and the inhibitor was obtained. We have tried to cultivate the cocrystal structure of 5g and c-Met but failed because of the slightly poor solubility of 5g. Therefore, we synthesized compound 5i (IC50 = 27.1 nM, enzyme inhibition activity), which is closely related to the structure of 5g but has better solubility, to obtain the cocrystal structure with c-Met. The only difference between 5i and 5g is that 5i has a morpholinomethyl group linked to the methoxy moiety of 5g. We finally determined the X-ray crystal structure of compound 5i bound to c-Met at a resolution of 1.80 Å. As shown in Figure 5A, the quinazoline group lies in the adenine pocket and forms an H-bond with the backbone N of Met1160 in the hinge. The quinazoline-linked fluorophenoxy group forms a π-π interaction with residue Phe1223 in the DFG (Asp1222-Phe1223-Gly1224) motif. The DFG-out conformation opens a hydrophobic allosteric pocket that is occupied by the terminal 4-fluorophenyl group. In addition to forming an H-bond with residue Lys1110 in the N-lobe, the naphthyridin-4-one group also binds with an allosteric channel and directly interacts with DFG residue Asp1222 through the formation of an H-bond. The morpholine group extends into the solvent. We compared our predicted binding mode of 5g with the X-ray crystal model of 5i and found that the binding model of 5g was very close to that of 5i (Figure 5B). The same parts of their skeleton have a root-mean-square deviation (RMSD) of 0.25 Å. 5g has the same H-bonding and hydrophobic interactions as 5i in the crystal complex. The high consistency of the binding modes and interactions in the predicted and experimental crystal structures confirmed the reliability of our computational strategy.
Figure 5.

X-ray Structure of 5i and Modeled Structure of 5g Bound to c-Met
(A) The X-ray structure of 5i bound to c-Met, 5i is shown with yellow ball-and-stick model, residues from c-Met are shown as magenta sticks, H-bonds are shown as red, dotted lines.
(B) The superimposition of the modeled and X-ray crystal structures of c-Met in complex with ligands (5g and 5i). The modeled structure of 5g is shown in green lines, and the receptor (PDB: 3F82) is shown in blue sticks and cartoon. The X-ray structure of 5i is given by the yellow lines, with the receptor as magenta sticks and cartoon.
AILDE Server Implementation
A freely accessible web server also named AILDE was constructed to allow medicinal chemists to easily use AILDE. The AILDE web server includes the front end and background applications. The architecture of the AILDE web server is shown in Figure S24. The front end consists of a client layer and a resulting browse layer. The client layer provides a user-friendly entry for the user to submit computational tasks. Users may also obtain comprehensive instruction about how to use AILDE from the client layer. The resulting browsing layer offers output formats for users to better understand the results. The background was coded in Python to finish the computational process. The server is managed by Maui-3.3.1 and Torque-6.0.1 on a supercomputing cluster in our laboratory (including 48 computing nodes with 40 CPUs each, 20 computing nodes with 4 GPUs each, 8 computing nodes with 40 CPUs and 256 GB large memory each, 3 storage servers, and 1 management server). The calculation time for a job depends largely on the size of the complex and the hardware status. Take the example job of the server as an example, the protein has 380 residues in total, the MD simulation step (about 6 ns) takes about 1.5 h (4 GPU parallel), the hydrogen replacement and minimization step (for first minimization strategy) takes 3 min per newly obtained lead compound (25 CPUs parallel for 50 snapshots of every compound). The free energy calculation step takes 4 min per compound (4 CPUs parallel). The compound in the example has 17 hydrogens and there are 10 substituents on the server. It will generate 170 new compounds in total. The total time cost of the example job is 1.5 h + 3 min ∗ 170 + 4 min ∗ 170 = 21.3 h and it takes less than 1 day to finish the job. The wizard of the AILDE server is shown in Figure 6, and a detailed description of how to use the server can be found in the Transparent Methods. To enable users to use AILDE in a more private way, we have also provided an offline version of our code on GitHub (https://github.com/fwangccnu/AILDE) for scientists to download and use.
Figure 6.

The Detailed Wizard of AILDE Server to Offer a Intuitive View for Users to Explore It
AILDE server wizard: (1) uploading of a hit-protein complex; (2) selection of a hit compound; (3) hydrogen serial numbers; (4) table with details about the new leads, ΔΔG and activity fold-changes; (5) heatmap showing the relationship matrix between the substituent positions and substituents (darker red colors indicate compounds with higher activities); (6) histogram illustrating the overall result based on substituent positions, which helps to elucidate which positions possess the most potential for transformation; (7) 3D viewer showing the binding mode of every compound with the target protein.
Limitations of the Study
AILDE reasonably relies on the resemblance assumption between the binding mode of hit-receptor and lead-receptor and adequacy of the conformational sampling, so it is the reliable prediction results when binding mode of the lead compound relative to the hit compound does not change much. We can also see that AILDE has certain limitation compared with other methods in exploring more extensive chemical space, although it has higher prediction accuracy and is applicable to a considerably larger number of test samples.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gefei Hao (gefei_hao@foxmail.com).
Materials Availability
All unique/stable reagents generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
The online web service of AILDE can be accessed from http://chemyang.ccnu.edu.cn/ccb/server/AILDE. The offline version of AILDE program can be downloaded from https://github.com/fwangccnu/AILDE.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This research was supported in part by the National Key R&D Program (2017YFD0200501) and the National Natural Science Foundation of China (No. 21837001, 21772059, and 91853127).
Author Contributions
F.W. conducted molecular simulation, data analysis, and results discussion. L.Z. conducted molecular synthesis. F.W. conducted the AILDE server design. W.H., G.H., and G.Y. supervised the study, designed the experiments, and carried out the results discussion.
Declaration of Interests
The authors declare no competing interests.
Published: June 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101179.
Contributor Information
Wei Huang, Email: weihuangwuhan@126.com.
Gefei Hao, Email: gefei_hao@foxmail.com.
Guangfu Yang, Email: gfyang@mail.ccnu.edu.cn.
Supplemental Information
a Collected experimental represented by IC50, EC50, Ki or Kd and the related publications. b The calculation data with two minimization strategies, ΔΔE is the shift of enthalpy change that is the combination of gas-phase binding energy (ΔEMM) and solvation free energy (ΔGsol) (shown in Equation 1 of this Supplemental Information), -TΔΔS is the shift of entropy change, ΔΔG is the shift of binding free energy, which is obtained by adding ΔΔE and –TΔΔS.
References
- Aronov A.M., Tang Q., Martinez-Botella G., Bemis G.W., Cao J.R., Chen G.J., Ewing N.P., Ford P.J., Germann U.A., Green J. Structure-guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal-regulated kinase (ERK) using conformational control. J. Med. Chem. 2009;52:6362–6368. doi: 10.1021/jm900630q. [DOI] [PubMed] [Google Scholar]
- Basilico C., Pennacchietti S., Vigna E., Chiriaco C., Arena S., Bardelli A., Valdembri D., Serini G., Michieli P. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin. Cancer Res. 2013;19:2381–2392. doi: 10.1158/1078-0432.CCR-12-3459. [DOI] [PubMed] [Google Scholar]
- Carbain B., Paterson D.J., Anscombe E., Campbell A.J., Cano C., Echalier A., Endicott J.A., Golding B.T., Haggerty K., Hardcastle I.R. 8-Substituted O6-cyclohexylmethylguanine CDK2 inhibitors: using structure-based inhibitor design to optimize an alternative binding mode. J. Med. Chem. 2014;57:56–70. doi: 10.1021/jm401555v. [DOI] [PubMed] [Google Scholar]
- Card G.L., Blasdel L., England B.P., Zhang C., Suzuki Y., Gillette S., Fong D., Ibrahim P.N., Artis D.R., Bollag G. A family of phosphodiesterase inhibitors discovered by cocrystallography and scaffold-based drug design. Nat. Biotechnol. 2005;23:201–207. doi: 10.1038/nbt1059. [DOI] [PubMed] [Google Scholar]
- Carlsson J., Boukharta L., Åqvist J. Combining docking, molecular dynamics and the linear interaction energy method to predict binding modes and affinities for non-nucleoside inhibitors to HIV-1 reverse transcriptase. J. Med. Chem. 2008;51:2648–2656. doi: 10.1021/jm7012198. [DOI] [PubMed] [Google Scholar]
- Charrier J.D., Miller A., Kay D.P., Brenchley G., Twin H.C., Collier P.N., Ramaya S., Keily S.B., Durrant S.J., Knegtel R.M.A. Discovery and structure−activity relationship of 3-Aminopyrid-2-ones as potent and selective interleukin-2 inducible T-cell kinase (Itk) inhibitors. J. Med. Chem. 2011;54:2341–2350. doi: 10.1021/jm101499u. [DOI] [PubMed] [Google Scholar]
- Chéron N., Jasty N., Shakhnovich E.I. OpenGrowth: an automated and rational algorithm for finding new protein ligands. J. Med. Chem. 2016;59:4171–4188. doi: 10.1021/acs.jmedchem.5b00886. [DOI] [PubMed] [Google Scholar]
- Comoglio P.M., Giordano S., Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008;7:504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- Congreve M., Andrews S.P., Doré A.S., Hollenstein K., Hurrell E., Langmead C.J., Mason J.S., Ng I.W., Tehan B., Zhukov A. Discovery of 1,2,4-Triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem. 2012;55:1898–1903. doi: 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford T.D., Ndubaku C.O., Chen H., Boggs J.W., Bravo B.J., DeLaTorre K., Giannetti A.M., Gould S.E., Harris S.F., Magnuson S.R. Discovery of selective 4-Amino-pyridopyrimidine inhibitors of MAP4K4 using fragment-based lead identification and optimization. J. Med. Chem. 2014;57:3484–3493. doi: 10.1021/jm500155b. [DOI] [PubMed] [Google Scholar]
- Cui J.J. Targeting receptor tyrosine kinase MET in cancer: small molecule inhibitors and clinical progress. J. Med. Chem. 2014;57:4427–4453. doi: 10.1021/jm401427c. [DOI] [PubMed] [Google Scholar]
- Cui J.J., Tran-Dube M., Shen H., Nambu M., Kung P.-P., Pairish M., Jia L., Meng J., Funk L., Botrous I. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK) J. Med. Chem. 2011;54:6342–6363. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- Demont E.H., Chung C., Furze R.C., Grandi P., Michon A.-M., Wellaway C., Barrett N., Bridges A.M., Craggs P.D., Diallo H. Fragment-based discovery of low-micromolar ATAD2 bromodomain inhibitors. J. Med. Chem. 2015;58:5649–5673. doi: 10.1021/acs.jmedchem.5b00772. [DOI] [PubMed] [Google Scholar]
- Denny R.A., Flick A.C., Coe J., Langille J., Basak A., Liu S., Stock I., Sahasrabudhe P., Bonin P., Hay D.A. Structure-based design of highly selective inhibitors of the CREB binding protein bromodomain. J. Med. Chem. 2017;60:5349–5363. doi: 10.1021/acs.jmedchem.6b01839. [DOI] [PubMed] [Google Scholar]
- Efremov I.V., Vajdos F.F., Borzilleri K.A., Capetta S., Chen H., Dorff P.H., Dutra J.K., Goldstein S.W., Mansour M., McColl A. Discovery and optimization of a novel spiropyrrolidine inhibitor of β-secretase (BACE1) through fragment-based drug design. J. Med. Chem. 2012;55:9069–9088. doi: 10.1021/jm201715d. [DOI] [PubMed] [Google Scholar]
- Enyedy I.J., Egan W.J. Can we use docking and scoring for hit-to-lead optimization? J. Comput. Aided Mol. Des. 2008;22:161–168. doi: 10.1007/s10822-007-9165-4. [DOI] [PubMed] [Google Scholar]
- Erlanson D.A., Braisted A.C., Raphael D.R., Randal M., Stroud R.M., Gordon E.M., Wells J.A. Site-directed ligand discovery. Proc. Natl. Acad. Sci. U S A. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlanson D.A., Fesik S.W., Hubbard R.E., Jahnke W., Jhoti H. Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016;15:605–619. doi: 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- Erlanson D.A., Lam J.W., Wiesmann C., Luong T.N., Simmons R.L., DeLano W.L., Choong I.C., Burdett M.T., Flanagan W.M., Lee D. In situ assembly of enzyme inhibitors using extended tethering. Nat. Biotechnol. 2003;21:308–314. doi: 10.1038/nbt786. [DOI] [PubMed] [Google Scholar]
- Gharwan H., Groninger H. Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nat. Rev. Clin. Oncol. 2016;13:209–227. doi: 10.1038/nrclinonc.2015.213. [DOI] [PubMed] [Google Scholar]
- Godden J.W., Xue L., Bajorath J. Combinatorial preferences affect molecular similarity/diversity calculations using binary fingerprints and Tanimoto coefficients. J. Chem. Inf. Comp. Sci. 2000;40:163–166. doi: 10.1021/ci990316u. [DOI] [PubMed] [Google Scholar]
- Green J., Cao J., Bandarage U.K., Gao H., Court J., Marhefka C., Jacobs M., Taslimi P., Newsome D., Nakayama T. Design, synthesis, and structure–activity relationships of pyridine-based Rho kinase (ROCK) inhibitors. J. Med. Chem. 2015;58:5028–5037. doi: 10.1021/acs.jmedchem.5b00424. [DOI] [PubMed] [Google Scholar]
- Gross S., Rahal R., Stransky N., Lengauer C., Hoeflich K.P. Targeting cancer with kinase inhibitors. J. Clin. Invest. 2015;125:1780–1789. doi: 10.1172/JCI76094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimarães C.R.W., Cardozo M. MM-GB/SA rescoring of docking poses in structure-based lead optimization. J. Chem. Inf. Model. 2008;48:958–970. doi: 10.1021/ci800004w. [DOI] [PubMed] [Google Scholar]
- Hajduk P.J., Sheppard G., Nettesheim D.G., Olejniczak E.T., Shuker S.B., Meadows R.P., Steinman D.H., Carrera G.M., Marcotte P.A., Severin J. Discovery of potent nonpeptide inhibitors of stromelysin using SAR by NMR. J. Am. Chem. Soc. 1997;119:5818–5827. [Google Scholar]
- Hao G.F., Jiang W., Ye Y.N., Wu F.X., Zhu X.L., Guo F.B., Yang G.F. ACFIS: a web server for fragment-based drug discovery. Nucleic Acids Res. 2016;44:W550–W556. doi: 10.1093/nar/gkw393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochguertel M., Kroth H., Piecha D., Hofmann M.W., Nicolau C., Krause S., Schaaf O., Sonnenmoser G., Eliseev A.V. Target-induced formation of neuraminidase inhibitors from in vitro virtual combinatorial libraries. Proc. Natl. Acad. Sci. U S A. 2002;99:3382–3387. doi: 10.1073/pnas.052703799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffer L., Muller C., Roche P., Morelli X. Chemistry-driven hit-to-lead optimization guided by structure-based approaches. Mol. Inform. 2018;37:e1800059. doi: 10.1002/minf.201800059. [DOI] [PubMed] [Google Scholar]
- Hsu H.H., Hsu Y.C., Chang L.J., Yang J.M. An integrated approach with new strategies for QSAR models and lead optimization. BMC Genomics. 2017;18:104–112. doi: 10.1186/s12864-017-3503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J.P., Rees S., Kalindjian S.B., Philpott K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011;162:1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keserű G.M., Makara G.M. Hit discovery and hit-to-lead approaches. Drug Discov. Today. 2006;11:741–748. doi: 10.1016/j.drudis.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Lawhorn B.G., Philp J., Graves A.P., Holt D.A., Gatto G.J., Kallander L.S. Substituent effects on drug–receptor H-bond interactions: correlations useful for the design of kinase inhibitors. J. Med. Chem. 2016;59:10629–10641. doi: 10.1021/acs.jmedchem.6b01342. [DOI] [PubMed] [Google Scholar]
- Lin H.Y., Chen X., Chen J.N., Wang D.W., Wu F.X., Lin S.Y., Zhan C.G., Wu J.W., Yang W.C., Yang G.F. Crystal structure of 4-hydroxyphenylpyruvate dioxygenase in complex with substrate reveals a new starting point for herbicide discovery. Research (Wash. D C) 2019;2019:2602414. doi: 10.34133/2019/2602414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Huang F., Yuan X., Wang K., Zou Y., Shen J., Xu Y. Structure-guided discovery of novel, potent, and orally bioavailable inhibitors of lipoprotein-associated phospholipase A2. J. Med. Chem. 2017;60:10231–10244. doi: 10.1021/acs.jmedchem.7b01530. [DOI] [PubMed] [Google Scholar]
- Ma P.C., Schaefer E., Christensen J.G., Salgia R. A selective small molecule c-MET inhibitor, PHA665752, cooperates with rapamycin. Clin. Cancer Res. 2005;11:2312–2319. doi: 10.1158/1078-0432.CCR-04-1708. [DOI] [PubMed] [Google Scholar]
- Martens T., Schmidt N.-O., Eckerich C., Fillbrandt R., Merchant M., Schwall R., Westphal M., Lamszus K. A novel one-armed anti-c-met antibody inhibits glioblastoma growth in vivo. Clin. Cancer Res. 2006;12:6144–6152. doi: 10.1158/1078-0432.CCR-05-1418. [DOI] [PubMed] [Google Scholar]
- Medina-Franco J.L., Martinez-Mayorga K., Giulianotti M.A., Houghten R.A., Pinilla C. Visualization of the chemical space in drug discovery. Curr. Comput. Aided Drugs. 2008;4:322–333. [Google Scholar]
- Medina J.R., Becker C.J., Blackledge C.W., Duquenne C., Feng Y.H., Grant S.W., Heerding D., Li W.H., Miller W.H., Romeril S.P. Structure-based design of potent and selective 3-phosphoinositide-dependent kinase-1 (PDK1) inhibitors. J. Med. Chem. 2011;54:1871–1895. doi: 10.1021/jm101527u. [DOI] [PubMed] [Google Scholar]
- Mirguet O., Gosmini R., Toum J., Clement C.A., Barnathan M., Brusq J.-M., Mordaunt J.E., Grimes R.M., Crowe M., Pineau O. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem. 2013;56:7501–7515. doi: 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- Montero-Torres A., García-Sánchez R.N., Marrero-Ponce Y., Machado-Tugores Y., Nogal-Ruiz J.J., Martínez-Fernández A.R., Arán V.J., Ochoa C., Meneses-Marcel A., Torrens F. Non-stochastic quadratic fingerprints and LDA-based QSAR models in hit and lead generation through virtual screening: theoretical and experimental assessment of a promising method for the discovery of new antimalarial compounds. Eur. J. Med. Chem. 2006;41:483–493. doi: 10.1016/j.ejmech.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Moridani M., Harirforoosh S. Drug development and discovery: challenges and opportunities. Drug Discov. Today. 2014;19:1679–1681. doi: 10.1016/j.drudis.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Moro S., Bacilieri M., Cacciari B., Bolcato C., Cusan C., Pastorin G., Klotz K.-N., Spalluto G. The application of a 3D-QSAR (autoMEP/PLS) approach as an efficient pharmacodynamic-driven filtering method for small-sized virtual library: application to a lead optimization of a human A3 adenosine receptor antagonist. Bioorg. Med. Chem. 2006;14:4923–4932. doi: 10.1016/j.bmc.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Narumi T., Arai H., Yoshimura K., Harada S., Hirota Y., Ohashi N., Hashimoto C., Nomura W., Matsushita S., Tamamura H. CD4 mimics as HIV entry inhibitors: lead optimization studies of the aromatic substituents. Bioorg. Med. Chem. 2013;21:2518–2526. doi: 10.1016/j.bmc.2013.02.041. [DOI] [PubMed] [Google Scholar]
- Nienaber V.L., Richardson P.L., Klighofer V., Bouska J.J., Giranda V.L., Greer J. Discovering novel ligands for macromolecules using X-ray crystallographic screening. Nat. Biotechnol. 2000;18:1105–1108. doi: 10.1038/80319. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T., Elmore S.W., Shoemaker A.R., Armstrong R.C., Augeri D.J., Belli B.A., Bruncko M., Deckwerth T.L., Dinges J., Hajduk P.J. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Orita M., Ohno K., Warizaya M., Amano Y., Niimi T. Chapter fifteen - lead generation and examples: opinion regarding how to follow up hits. Method. Enzymol. 2011;493:383–419. doi: 10.1016/B978-0-12-381274-2.00015-7. [DOI] [PubMed] [Google Scholar]
- Paul S.M., Mytelka D.S., Dunwiddie C.T., Persinger C.C., Munos B.H., Lindborg S.R., Schacht A.L. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat. Rev. Drug Discov. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- Pillai A.D., Rani S., Rathod P.D., Xavier F.P., Vasu K.K., Padh H., Sudarsanam V. QSAR studies on some thiophene analogs as anti-inflammatory agents: enhancement of activity by electronic parameters and its utilization for chemical lead optimization. Bioorg. Med. Chem. 2005;13:1275–1283. doi: 10.1016/j.bmc.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Rebecca D.P., Benoit D. Facts, figures and trends in lead generation. Curr. Top. Med. Chem. 2004;4:569–580. doi: 10.2174/1568026043451168. [DOI] [PubMed] [Google Scholar]
- Reymond J.L., van Deursen R., Blum L.C., Ruddigkeit L. Chemical space as a source for new drugs. Medchemcomm. 2010;1:30–38. [Google Scholar]
- Schulz R., Atef A., Becker D., Gottschalk F., Tauber C., Wagner S., Arkona C., Abdel-Hafez A.A., Farag H.H., Rademann J. Phenylthiomethyl ketone-based fragments show selective and irreversible inhibition of enteroviral 3C proteases. J. Med. Chem. 2018;61:1218–1230. doi: 10.1021/acs.jmedchem.7b01440. [DOI] [PubMed] [Google Scholar]
- Souers A.J., Leverson J.D., Boghaert E.R., Ackler S.L., Catron N.D., Chen J., Dayton B.D., Ding H., Enschede S.H., Fairbrother W.J. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Tsukada T., Takahashi M., Takemoto T., Kanno O., Yamane T., Kawamura S., Nishi T. Structure-based drug design of tricyclic 8H-indeno 1,2-d 1,3 thiazoles as potent FBPase inhibitors. Bioorg. Med. Chem. Lett. 2010;20:1004–1007. doi: 10.1016/j.bmcl.2009.12.056. [DOI] [PubMed] [Google Scholar]
- Vinkers H.M., de Jonge M.R., Daeyaert F.F.D., Heeres J., Koymans L.M.H., van Lenthe J.H., Lewi P.J., Timmerman H., Van Aken K., Janssen P.A.J. SYNOPSIS: synthesize and optimize system in silico. J. Med. Chem. 2003;46:2765–2773. doi: 10.1021/jm030809x. [DOI] [PubMed] [Google Scholar]
- Vyas V.K., Shah S., Ghate M. Generation of new leads as HIV-1 integrase inhibitors: 3D QSAR, docking and molecular dynamics simulation. Med. Chem. Res. 2017;26:532–550. [Google Scholar]
- Wang F., Jeon K.O., Salovich J.M., Macdonald J.D., Alvarado J., Gogliotti R.D., Phan J., Olejniczak E.T., Sun Q., Wang S. Discovery of potent 2-Aryl-6,7-dihydro-5H-pyrrolo[1,2-a]imidazoles as WDR5-WIN-site inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2018;61:5623–5642. doi: 10.1021/acs.jmedchem.8b00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhead A.J., Angove H., Carr M.G., Chessari G., Congreve M., Coyle J.E., Cosme J., Graham B., Day P.J., Downham R. Discovery of (2,4-dihydroxy-5-isopropylphenyl)- 5-(4-methylpiperazin-1-ylmethyl)-1,3- dihydroisoindol-2-yl methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design. J. Med. Chem. 2010;53:5956–5969. doi: 10.1021/jm100060b. [DOI] [PubMed] [Google Scholar]
- Wu P., Nielsen T.E., Clausen M.H. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov. Today. 2016;21:5–10. doi: 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Yang J.F., Wang F., Jiang W., Zhou G.Y., Li C.Z., Zhu X.L., Hao G.F., Yang G.F. PADFrag: a database built for the exploration of bioactive fragment space for drug discovery. J. Chem. Inf. Model. 2018;58:1725–1730. doi: 10.1021/acs.jcim.8b00285. [DOI] [PubMed] [Google Scholar]
- Zhao H. Scaffold selection and scaffold hopping in lead generation: a medicinal chemistry perspective. Drug Discov. Today. 2007;12:149–155. doi: 10.1016/j.drudis.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Zhavoronkov A., Ivanenkov Y.A., Aliper A., Veselov M.S., Aladinskiy V.A., Aladinskaya A.V., Terentiev V.A., Polykovskiy D.A., Kuznetsov M.D., Asadulaev A. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019;37:1038–1040. doi: 10.1038/s41587-019-0224-x. [DOI] [PubMed] [Google Scholar]
- Zhou R., Friesner R.A., Ghosh A., Rizzo R.C., Jorgensen W.L., Levy R.M. New linear interaction method for binding affinity calculations using a continuum solvent model. J. Phys. Chem. B. 2001;105:10388–10397. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
a Collected experimental represented by IC50, EC50, Ki or Kd and the related publications. b The calculation data with two minimization strategies, ΔΔE is the shift of enthalpy change that is the combination of gas-phase binding energy (ΔEMM) and solvation free energy (ΔGsol) (shown in Equation 1 of this Supplemental Information), -TΔΔS is the shift of entropy change, ΔΔG is the shift of binding free energy, which is obtained by adding ΔΔE and –TΔΔS.
Data Availability Statement
The online web service of AILDE can be accessed from http://chemyang.ccnu.edu.cn/ccb/server/AILDE. The offline version of AILDE program can be downloaded from https://github.com/fwangccnu/AILDE.