

Abstract
Background
Meningiomas are the most common primary intracranial tumors in adults. They are initially detected with neuroimaging techniques, but definite histological diagnosis requires tumor surgery to collect tumor tissue. Gross total resection is an optimal and final treatment for the majority of patients, followed by radiotherapy in malignant or refractory cases. However, there are a lot of uncertainties about i.a. the need for intervention in incidental cases, estimation of growth kinetics, risk of malignant transformation, or response to radiotherapy. Therefore a new diagnostic approach is needed. It has already been shown that epigenetics plays a crucial role in cancer biology, development, and progression. DNA methylation, the presence of 5-methylcytosine in DNA, is one of the main elements of a broad epigenetic program in a eukaryotic cell, with superior regulatory significance. Therefore, we decided to look at meningioma through changes of 5-methylcytosine.
Methods
We performed an analysis of the total amount of 5-methylcytosine in DNA isolated from intracranial meningioma tissues and peripheral blood samples of the same patients. The separation and identification of radioactively labeled nucleotides were performed using thin-layer chromatography.
Results
We found that the 5-methylcytosine level in DNA from intracranial meningiomas is inversely proportional to the malignancy grade. The higher the tumor WHO grade is, the lower the total DNA methylation. The amount of 5-methylcytosine in tumor tissue and peripheral blood is almost identical.
Conclusions
We conclude that the total DNA methylation can be a useful marker for brain meningioma detection, differentiation, and monitoring. It correlates with tumor WHO grade, and the 5-methylcytosine level in peripheral blood reflects that in tumor tissue. Therefore it’s applicable for liquid biopsy.
Our study creates a scope for further research on epigenetic mechanisms in neurooncology and can lead to the development of new diagnostic methods in clinical practice.
Keywords: Meningioma, DNA methylation, 5-methylcytosine, Biomarker, DNA damage
Background
Meningiomas are the most frequent primary central nervous system tumors (37.6%), reaching an annual incidence of 8.56/100000 [1]. The occurrence rate for meningioma increases with age (adults age 65 years and older), is higher in females than in males (female: male ratio is 2.21), and in Blacks than in Whites [1]. They present with overall five-year survival of 68.2% for malignant and 88.0% for non-malignant cases, and a relative 10-year survival ranging from 61.7% for malignant and 83.7% for non-malignant meningiomas, respectively [1]. The well-validated risk factor for those tumors is ionizing radiation [1, 2]. The majority (80.5%) of meningiomas are low grade (I) tumors, but 17.7% are atypical (grade II), and 1.7% anaplastic (grade III) according to the WHO 2016 criteria [1, 3]. The WHO brain tumors’ classification lists 15 meningioma subtypes, most of them allotted to WHO grade I (Meningothelial, Fibrous/Fibroblastic, Transitional/Mixed, Psammomatous, Angiomatous, Microcystic, Secretory, Lymphoplasmacyte-rich, Metaplastic). Other variants, presenting with a higher likelihood of recurrence and aggressive behavior, belong either to grade II (Atypical, Clear-cell, Chordoid) or III (Anaplastic/Malignant, Rhabdoid, Papillary) [4]. Those subtypes are distinguished by their specific microscopic features. However, some genetic and clinical relevancies are also known, as the presence of NF2 mutations (up to 80% in fibroblastic and transitional meningiomas, only in 25% of meningothelial ones, and very rarely in the secretory), prevalence in specific locations (Clear-cell meningiomas in the spinal cord and posterior fossa, Chordoid - typically supratentorial), and age groups (as Papillary type in children) [4]. Grading is entirely based on histological features and does not include molecular markers. Mitotic count of 4 or more per 10 high power fields and brain invasion are diagnostic criteria of atypical meningioma, WHO grade II. The existing classification and grading system have prognostic value. However, they possess some shortcomings, such as ill-defined subtypes’ parameters and grading criteria that are susceptible to arbitrary judgment [3].
Watchful waiting and surgery are the first-line treatments of meningiomas. Adjuvant radiotherapy and radiosurgery are taken into account for atypical cases and indicated for anaplastic meningiomas [5]. The most of meningiomas may be cured by surgical resection. However, ca. 20% of tumors present aggressive clinical behavior with recurrence or progression, which results in significant morbidity and mortality of affected patients. Skull base meningiomas are usually the most challenging cases because of vascular and nervous structures involvement in and adjacent to the tumor, as well as resistance to radiation therapy. That limits the possibility of their total surgical removal and radiotherapeutic approach. Chemotherapy has not yet been proven to be effective in meningiomas, but there are several clinical trials ongoing [6].
The vital challenge for neuropathological evaluation of meningioma is not the identification of the entity, but its subtyping and grading. However, the standard WHO scheme does not allow for sufficient prediction of the recurrence of the tumor, overall prognosis, and systemic treatment options [7]. Therefore, for completion of the histological classification with therapeutic insights, the molecular characterization of these tumors is essential. However, the pathogenic mechanisms leading to meningioma development is still undefined. In 60–80% of sporadic meningiomas, the mutation in neurofibromatosis type 2 gene (NF2) leading to its inactivation occurs [8, 9]. The ongoing research involves the exploration of other genetic mutations, such as SMO, AKT1, TERT, BAP1, KLF4, TRAF7, as well as the methylation profile [7, 10]. TERT gene alterations, including promoter mutations, gene translocations, and DNA amplifications, appear to be a biomarker significantly predicting higher recurrence and mortality rate in meningiomas [11]. An accumulation of cytogenetic aberrations, usually 1p, 10, and 14q losses, was shown to be associated with higher malignancy and increased recurrence rate [4, 12, 13]. Grade II and III meningiomas display a more complex molecular and cytogenetic background than benign (grade I) ones. That includes the inactivation of tumor suppressor genes, oncogenes’ activation, and modifications in various genes taking part in multiple cellular processes [14, 15]. However, none of those alterations appeared to have prognostic relevance and biomarker potential. Moreover, most meningiomas do not present well defined genetic abnormalities, which may imply that other mechanisms, e.g., epigenetic aberrations, may influence tumor behavior [16]. However, the meningiomas’ epigenetic landscape remains incomplete.
DNA methylation, the presence of 5-methylcytosine (m5C) in DNA, is one of the main elements of a broad epigenetic program in a eukaryotic cell, with the regulatory significance. It results in silencing or reactivation of cancer-related genes [17]. Distinctiveness and specificity of m5C as an epigenetic marker lies in high stability of C-C bond in m5C, as well as lack of specific demethylating enzymes. Therefore the process of methyl group removal (demethylation) is carried out through enzymatic oxidation with Ten-Eleven Translocation (TET) enzymes or through spontaneous oxidation by reactive oxygen species (ROS) action [18]. It has been shown that cancer development and progression is the result of the disruption of the redox balance of the cell, which is induced by enhanced reactive oxygen species (ROS) generation, their accumulation, and antioxidant enzymes downregulation [19]. ROS cause damage to DNA and other cell components, promote epigenetic alterations, interact with oncogenes and tumor suppressor genes, and finally modulate immunological responses [20, 21]. ROS-induced m5C damage leads to its demethylation and deamination [22]. It results in the global (genomic) hypomethylation of cellular DNA. Therefore, total DNA methylation (m5C contents) is a sensitive marker for carcinogenesis as an effect of the oxidative stress, ROS formation, and damage reactions [23]. We have recently shown that DNA methylation status (hypomethylation) reflects the level of oxidative stress in the cell [24].
The results of gene-candidate studies [25–29] and genome-wide analysis [30, 31] in meningiomas suggested that aberrant DNA promoter methylation may contribute to the initiation and progression of those tumors.
To evaluate the role of DNA methylation in the development and progression of meningiomas, we have analyzed the total DNA methylation level in tumor and peripheral blood samples from intracranial meningioma patients undergoing surgical resection. For the estimation of m5C contents, we used a sensitive and straightforward two-dimensional thin-layer chromatography (TLC) technique of radioactively labeled nucleotides separation ([γ32P] post labeling method) [32, 33]. Through a detailed analysis of the total DNA methylation in intracranial meningiomas, we propose a new epigenetic approach for meningioma characterization, that can potentially be practically applied in clinical diagnostics, and treatment monitoring.
Methods
Ethics approval and consent to participate
Blood and tissue molecular testing was approved by the Bioethical Committee of Karol Marcinkowski University of Medical Sciences, Poznan (896/9; 838/12). All participants provided informed and written consent to donate their peripheral blood and tumor tissue samples for research.
Collection of tumor and peripheral blood samples
The brain meningioma tissue samples were collected from 100 consecutive patients who underwent brain tumor surgery at the Department of Neurosurgery and Neurotraumatology of the Karol Marcinkowski University of Medical Sciences in Poznan between 2004 and 2012. In 29 of those patients, peripheral blood samples were also collected preoperatively. The peripheral blood samples were also taken from 30 generally healthy individuals (not related to the cases, volunteers, with no known pathologies, without regular drug intake, non-smokers) comprising the control group. Tumor and peripheral blood samples were immediately frozen, then stored at − 80 °C.
Demographic and clinical data were extracted from the patient’s medical records. Intracranial tumor samples were routinely neuropathologically evaluated to determine their histological types and grades.
DNA isolation from tumor tissue samples
The extraction of genomic DNA from tumor tissue samples was performed with a commercially available kit (A&A Biotechnology). The samples were first incubated with proteinase K, then with RNaseA. The supernatant, obtained after centrifugation, was applied to a mini-column. DNA elution was done with Tris-buffer pH 8.5. It was then stored at − 20 °C for further analysis. DNA UV absorbance was measured at 260 and 280 nm for checking DNA purity. The A260/A280 ratio was 2.0–2.1.
DNA isolation from peripheral blood samples
DNA from a peripheral blood sample (7.5 ml) was isolated by lysis with 30 ml of cold (4 °C) buffer of NH4Cl (155 mM), KHCO3 (10 mM), and Na2EDTA (0.1 mM), in pH 7.4 for 30 min., then centrifuged at 3000 rpm for 10 min. in 4 °C. The pellets were resuspended in 10 ml of the above-mentioned buffer and centrifuged again. The cell lysate was resuspended in 5 ml of buffer containing NaCl (75 mM), Na2EDTA (1 mM), in pH 8.0, and digested with protease K solution (25 μl, concentration 10 μg/μl), and 20% SDS (250 μl) for 16 h at 55 °C. After the incubation 5 M NaCl (1500 μl) was added, then tube shaken vigorously for 15 s, and centrifuged at 4000 rpm for 15 mins at room temperature. The DNA precipitation was done with two volumes of cold ethanol. The solution was centrifuged at 4000 rpm for 20 mins at room temperature. Ethanol was removed, and the precipitate dissolved in distilled water (100 μl). DNA UV absorbance was measured at 260 and 280 nm for checking DNA purity. The A260/A280 ratio was 2.0–2.1. The amount of DNA was calculated using the standard relation: 1 OD (absorbance at 260 nm) = 50 μg DNA. OD value was measured with the spectrophotometer.
DNA hydrolysis, Labelling and TLC chromatography
The amount of water solution containing 1 μg of DNA was placed in 1.5 ml Eppendorf tube and dehydrated in a vacuum concentrator. Dried DNA was dissolved in CaCl2 (10 mM) succinate buffer (pH 6.0), and then digested with the mixture of spleen phosphodiesterase II (0.001 units), and micrococcal nuclease (0.02 units) in the total volume of 3.5 μl for 5 h at 37 °C. DNA digest (0.17 μg) was labeled with [γ-32P] ATP (1 μCi; stock solution: 6000 Ci/mM; Hartmann Analytic GmbH), and T4 polynucleotide kinase (1.5 units) in 10 mM bicine-NaOH pH 9.7 (3 μl) buffer containing MgCl2 (10 mM), DTT (10 mM), and spermidine (1 mM). After 30 min. at 37 °C, apyrase (3 μl, 10 units/ml) in the same buffer was added, and the solution was incubated for another 30 min. The 3′ nucleotide phosphates were cleaved off with RNase P1 (0.2 μg) in ammonium acetate buffer (500 mM, pH 4.5). Identification of [γ32P]m5C was performed with a thin-layer chromatography (TLC) on cellulose plates (Merck, Germany) in two dimensions using solvent system: isobutyric acid: NH4OH:H2O (66:1:17 v/v) - first dimension, and 0.2 M sodium phosphate (pH 6.8)-ammonium sulfate-n-propyl alcohol (100 ml/60 g/1.5 ml) - second dimension. Radioactive spot detection was performed with the Phosphoimager Typhoon Screen (Pharmacia, Sweden), and ImageQuant Software (GE Healthcare, USA) was used for image analysis. The testing was repeated thrice for each probe, and the statistic software was used for results evaluation. The amount of radioactive material (spot intensity) corresponding to m5C, C (cytosine), and T (thymine) was used for calculations. The global DNA methylation was calculated as R = (m5C/(m5C + C + T)) × 100 (Fig. 1), because cytosine and thymine are formed in the m5C oxidation process [33].
Fig. 1.

Flowchart of total genomic m5C estimation. Isolated DNA is hydrolyzed to 3′-mononucleotides (Np, A - adenosine, G - guanosine, C - cytidine, T - thymidine). The hydrolysate is labeled with [γ-32P] ATP, dephosphorylated (detachment of 3′ phosphate), and separated in two dimensions with TLC. The chromatogram is evaluated with phosphoimager, and the spots’ intensities are measured. Those values are used for the calculation of the R coefficient according to the given equation [33]
Statistical analysis
Statistical analysis (descriptive statistics, ANOVA test, correlation) was performed with STATISTICA 13.3 (Statsoft Poland) software.
Results
Patients’ characteristics
The analyzed cohort consisted of 100 individuals diagnosed with brain meningioma, aged from 27 to 80 years. Patients within the age range of 51–60 years (37 individuals) comprised the largest subgroup, followed by the age group of 61–70 (27 individuals). The median age of patients at the time of tumor diagnosis was 57.4 ± 10.8 years. There were 30 (30.0%) males and 70 (70.0%) females. The histological types and grades (from I – least malignant, to III most malignant), as well as numeric data from total DNA methylation analysis, are shown in Table 1.
Table 1.
The list of 100 patients with intracranial meningioma evaluated in this study. The histological types and grades were estimated in routine pathological report
| Case | Histological type | Grade | Age range | R tissue | SD R tissue |
R blood | SD R blood |
|---|---|---|---|---|---|---|---|
| 1. | Meningothelial meningioma | I | 21–30 | 1.63 | 0.02 | ||
| 2. | Meningothelial meningioma | I | 21–30 | 1.56 | 0.04 | 1.55 | 0.06 |
| 3. | Meningothelial meningioma | I | 41–50 | 1.63 | 0.09 | ||
| 4. | Meningothelial meningioma | I | 41–50 | 1.51 | 0.05 | ||
| 5. | Meningothelial meningioma | I | 41–50 | 1.65 | 0.03 | ||
| 6. | Meningothelial meningioma | I | 41–50 | 1.62 | 0.06 | 1.69 | 0.1 |
| 7. | Meningothelial meningioma | I | 41–50 | 1.57 | 0.09 | ||
| 8. | Meningothelial meningioma | I | 41–50 | 1.59 | 0.16 | ||
| 9. | Meningothelial meningioma | I | 41–50 | 1.64 | 0.08 | 1.46 | 0.06 |
| 10. | Meningothelial meningioma | I | 41–50 | 1.63 | 0.09 | ||
| 11. | Meningothelial meningioma | I | 41–50 | 1.59 | 0.08 | ||
| 12. | Meningothelial meningioma (brain parenchyma infiltration) | II | 41–50 | 1.61 | 0.04 | ||
| 13 | Meningothelial meningioma | I | 51–60 | 1.61 | 0.08 | ||
| 14. | Meningothelial meningioma | I | 51–60 | 1.59 | 0.1 | 1.75 | 0.12 |
| 15. | Meningothelial meningioma | I | 51–60 | 1.55 | 0.04 | ||
| 16. | Meningothelial meningioma | I | 51–60 | 1.55 | 0.09 | 1.60 | 0.12 |
| 17. | Meningothelial meningioma | I | 51–60 | 1.57 | 0.07 | 1.52 | 0.09 |
| 18. | Meningothelial meningioma | I | 51–60 | 1.51 | 0.1 | ||
| 19. | Meningothelial meningioma | I | 51–60 | 1.71 | 0.01 | ||
| 20. | Meningothelial meningioma | I | 51–60 | 1.59 | 0.11 | ||
| 21. | Meningothelial meningioma | I | 51–60 | 1.56 | 0.01 | ||
| 22. | Meningothelial meningioma | I | 51–60 | 1.48 | 0.05 | ||
| 23. | Meningothelial meningioma (brain parenchyma infiltration) | II | 51–60 | 1.13 | 0.04 | 1.48 | 0.08 |
| 24. | Meningothelial meningioma | I | 51–60 | 1.61 | 0.03 | 1.51 | 0.09 |
| 25. | Meningothelial meningioma | I | 51–60 | 1.56 | 0.03 | ||
| 26. | Meningothelial meningioma | I | 51–60 | 1.60 | 0.04 | ||
| 27. | Meningothelial meningioma | I | 51–60 | 1.58 | 0.09 | ||
| 28. | Meningothelial meningioma | I | 61–70 | 1.53 | 0.07 | ||
| 29. | Meningothelial meningioma | I | 61–70 | 1.58 | 0.07 | 1.63 | 0.02 |
| 30 | Meningothelial meningioma | I | 61–70 | 1.55 | 0.05 | ||
| 31. | Meningothelial meningioma | I | 61–70 | 1.53 | 0.06 | 1.63 | 0.04 |
| 32. | Meningothelial meningioma | I | 61–70 | 1.71 | 0.06 | ||
| 33. | Meningothelial meningioma | I | 61–70 | 1.73 | 0.03 | ||
| 34. | Meningothelial meningioma | I | 61–70 | 1.54 | 0.05 | ||
| 35 | Meningothelial meningioma | I | 61–70 | 1.56 | 0.05 | 1.68 | 0.02 |
| 36. | Meningothelial meningioma | I | 71–80 | 1.53 | 0.09 | ||
| 37. | Meningothelial meningioma | I | 71–80 | 1.57 | 0.07 | ||
| 38. | Meningothelial meningioma | I | 71–80 | 1.52 | 0.09 | ||
| 39. | Meningothelial meningioma, partially cellular | I | 31–40 | 1.54 | 0.03 | ||
| 40. | Meningothelial meningioma, partially cellular | I | 51–60 | 1.61 | 0.04 | ||
| 41. | Meningothelial meningioma | I | 51–60 | 1.72 | 0.07 | ||
| 42. | Meningothelial meningioma | I | 51–60 | 1.63 | 0.03 | ||
| 43. | Angiomatous and microcystic meningioma | I | 41–50 | 1.54 | 0.1 | ||
| 44. | Angiomatous meningioma | I | 41–50 | 1.52 | 0.09 | ||
| 45. | Angiomatous meningioma | I | 51–60 | 1.50 | 0.05 | 1.33 | 0.06 |
| 46. | Angiomatous meningioma | I | 51–60 | 1.46 | 0.09 | ||
| 47. | Angiomatous meningioma | I | 51–60 | 1.48 | 0.02 | 1.51 | 0.07 |
| 48. | Angiomatous meningioma | I | 61–70 | 1.49 | 0.02 | 1.45 | 0.04 |
| 49. | Angiomatous meningioma | I | 61–70 | 1.51 | 0.06 | ||
| 50. | Angiomatous meningioma | I | 61–70 | 1.46 | 0.09 | ||
| 51. | Angiomatous meningioma | I | 61–70 | 1.51 | 0.07 | 1.64 | 0.08 |
| 52. | Angiomatous meningioma | I | 61–70 | 1.54 | 0.06 | 1.59 | 0.1 |
| 53. | Angiomatous meningioma | I | 61–70 | 1.48 | 0.05 | ||
| 54. | Fibrous meningioma | I | 41–50 | 1.54 | 0.02 | ||
| 55. | Fibrous meningioma | I | 41–50 | 1.57 | 0.12 | ||
| 56. | Fibrous meningioma | I | 41–50 | 1.58 | 0.11 | ||
| 57. | Fibrous meningioma | I | 51–60 | 1.58 | 0.08 | ||
| 58. | Fibrous meningioma | I | 51–60 | 1.56 | 0.06 | ||
| 59. | Fibrous meningioma | I | 51–60 | 1.59 | 0.06 | ||
| 60. | Fibrous meningioma | I | 51–60 | 1.54 | 0.06 | 1.51 | 0.06 |
| 61. | Fibrous meningioma | I | 51–60 | 1.53 | 0.07 | ||
| 62. | Fibrous meningioma | I | 51–60 | 1.49 | 0.12 | ||
| 63. | Fibrous meningioma | I | 51–60 | 1.49 | 0.06 | ||
| 64. | Fibrous meningioma, partially psammomatous (brain parenchyma infiltration) | II | 61–70 | 1.41 | 0.06 | 1.57 | 0.04 |
| 65. | Fibrous meningioma | I | 61–70 | 1.71 | 0.09 | 1.67 | 0.04 |
| 66. | Fibrous meningioma | I | 61–70 | 1.52 | 0.11 | ||
| 67. | Fibrous meningioma | I | 61–70 | 1.53 | 0.12 | ||
| 68. | Fibrous meningioma | I | 61–70 | 1.57 | 0.1 | 1.68 | 0.08 |
| 69. | Fibrous meningioma | I | 61–70 | 1.56 | 0.08 | ||
| 70. | Fibrous meningioma | I | 61–70 | 1.45 | 0.07 | ||
| 71. | Fibrous meningioma | I | 61–70 | 1.52 | 0.02 | ||
| 72. | Fibrous meningioma | I | 71–80 | 1.62 | 0.02 | 1.67 | 0.12 |
| 73. | Fibrous meningioma | I | 71–80 | 1.55 | 0.03 | ||
| 74. | Fibrous meningioma | I | 71–80 | 1.54 | 0.07 | ||
| 75. | Fibrous meningioma | I | 51–60 | 1.61 | 0.09 | ||
| 76. | Psammomatous meningioma | I | 51–60 | 1.58 | 0.04 | ||
| 77. | Psammomatous meningioma | I | 51–60 | 1.52 | 0.07 | ||
| 78. | Psammomatous meningioma | I | 51–60 | 1.46 | 0.07 | ||
| 79. | Psammomatous meningioma | I | 71–80 | 1.59 | 0.05 | ||
| 80. | Atypical meningioma | II | 21–30 | 1.57 | 0.08 | ||
| 81. | Atypical meningioma | II | 51–60 | 1.46 | 0.11 | 1.45 | 0.04 |
| 82. | Atypical meningioma | II | 61–70 | 1.53 | 0.09 | 1.44 | 0.02 |
| 83. | Atypical meningioma | II | 61–70 | 1.25 | 0.09 | ||
| 84. | Atypical meningioma | II | 71–80 | 1.43 | 0.06 | 1.35 | 0.01 |
| 85. | Anaplastic meningioma | III | 41–50 | 1.42 | 0.06 | 1.26 | 0.03 |
| 86. | Anaplastic meningioma | III | 41–50 | 0.86 | 0.04 | ||
| 87. | Anaplastic meningioma | III | 71–80 | 0.99 | 0.04 | 0.95 | 0.04 |
| 88. | Anaplastic meningioma | III | 71–80 | 1.10 | 0.04 | ||
| 89. | Transitional meningioma | I | 31–40 | 1.56 | 0.08 | ||
| 90. | Transitional meningioma | I | 41–50 | 1.55 | 0.09 | ||
| 91. | Transitional meningioma | I | 41–50 | 1.66 | 0.08 | ||
| 92. | Transitional meningioma | I | 51–60 | 1.58 | 0.05 | ||
| 93. | Transitional meningioma | I | 51–60 | 1.65 | 0.11 | ||
| 94. | Transitional meningioma, recurrent | I | 51–60 | 1.66 | 0.14 | ||
| 95. | Meningothelial, transitional and angiomatous meningioma | I | 61–70 | 1.43 | 0.1 | ||
| 96. | Transitional meningioma | I | 51–60 | 1.46 | 0.09 | ||
| 97. | Meningothelial and transitional meningioma | II | 61–70 | 1.54 | 0.09 | 1.67 | 0.09 |
| 98. | Transitional meningioma, partially psammomatous | I | 61–70 | 1.53 | 0.06 | 1.63 | 0.12 |
| 99. | Transitional meningioma | I | 71–80 | 1.48 | 0.04 | 1.56 | 0.1 |
| 100. | Meningothelial, metaplastic and psammomatous meningioma | I | 71–80 | 1.46 | 0.06 |
The most abundant histological variant of meningioma in the analyzed cohort was meningothelial (42 cases), followed by fibrous, angiomatous, transitional, mixed, atypical, anaplastic, and psammomatous types (21, 10, 8, 6, 5, 4, and 4 cases respectively). There was no evident predilection of specific tumor variants to patients’ age or sex in the study. Tumor subtypes with the numerical amount and grade distribution are presented in Table 2. The control group consisted of 30 generally healthy persons, aged from 30 to 66 years (mean 49.8 ± 10.5 years), with 11 (36.7%) males and 19 (63.3%) females (Fig. 2).
Table 2.
Data summary of total DNA methylation in various intracranial meningioma subtypes with numerical amount and grade distribution in all patients evaluated in the study. Median age (in years) in the histological subclasses, and female to male (F:M) ratio is also given
| Histological type | Grade | No of cases | Median age [Y] | F:M ratio | R tissue | SD R tissue |
|---|---|---|---|---|---|---|
| Meningothelial meningioma | I | 40 | 54.9 | 26:14 | 1.59 | 0.006 |
| II | 2 | 53.0 | 1:1 | 1.37 | 0.34 | |
| Fibrous meningioma | I | 21 | 60.5 | 16:5 | 1.55 | 0.05 |
| Transitional meningioma | I | 8 | 51.5 | 7:1 | 1.58 | 0.08 |
| Psammomatous meningioma | I | 4 | 60.3 | 4:0 | 1.54 | 0.06 |
| Angiomatous meningioma | I | 10 | 60.9 | 7:3 | 1.50 | 0.03 |
| Mixed meningioma | I | 4 | 62.75 | 1:3 | 1.49 | 0.05 |
| II | 2 | 64.0 | 2:0 | 1.48 | 0.09 | |
| Atypical meningioma | II | 5 | 58.2 | 3:2 | 1.45 | 0.12 |
| Anaplastic meningioma | III | 4 | 60.0 | 3:1 | 1.09 | 0.24 |
Fig. 2.
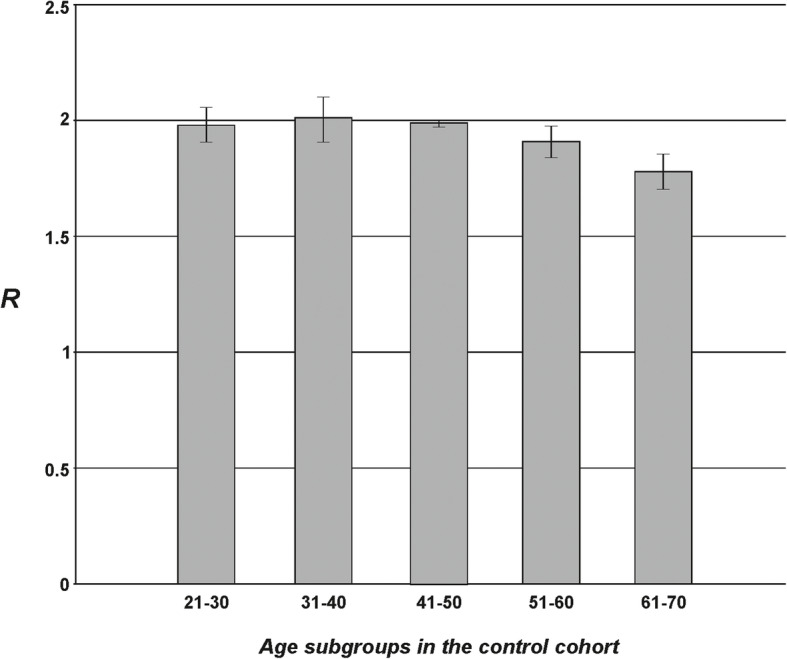
The total m5C contents (R) in DNA from peripheral blood samples in the cohort of generally healthy individuals, divided into age subgroups. One can observe that R ca. 2.0 is distinctive for the non-pathologic state
Global DNA methylation in tumor tissue samples
For all 100 patients, the total m5C amount in genomic DNA extracted from meningioma tumor tissue was analyzed (Tables 1 and 2). The total m5C amount expressed as R factor (see Materials and Methods), varies clearly between the patients (Table 1, Supplementary Figure 1), and the groups divided by the histological type (Table 2). The m5C level in intracranial meningioma DNA negatively correlates with tumor grade (Fig. 3). The less malignant tumors show higher m5C contents than more malignant, and the difference is statistically significant (F = 61.796 and p < 0.001). No correlation of total DNA methylation (R) was found with patients’ age (r = − 0.13) and sex (r = 0.21), as well as with specific meningioma subtypes.
Fig. 3.

The total m5C contents (R) in DNA isolated from different intracranial meningioma tissues divided into the malignancy grades (WHO I represents the tumors of lowest malignancy, while WHO III relates to the most malignant tumors). Grade I tumors are characterized by mean R around 1.6, grade II – 1.4, and grade III – 1.1
Global DNA methylation in peripheral blood samples
For 29 patients, we also analyzed the total amount of m5C in genomic DNA from peripheral blood samples (Tables 1 and 3). The amount of m5C expressed as R coefficient varies clearly between the patients (Table 1, Supplementary Figure 2), and the groups divided by the histological type (Table 3). The level of m5C in DNA from peripheral blood samples negatively correlates with tumor grade (Fig. 4). Patients with more malignant tumors show lower total m5C contents in DNA from peripheral blood samples than less malignant, and the difference is statistically significant (F = 18.024 and p < 0.001). No correlation of total DNA methylation (R) was found with patients’ age (r = 0.34) and sex (r = 0.19), as well as with specific meningioma subtypes.
Table 3.
Data summary of total DNA methylation in tumor tissue and peripheral blood samples of 29 patients with various meningioma types with numerical amount and grade distribution. Median age (in years) in the histological subclasses, and female to male (F:M) ratio is also given
| Histological type | Grade | No of cases | Median age | F:M ratio | R tissue | SD R tissue | R blood | SD R blood |
|---|---|---|---|---|---|---|---|---|
| Meningothelial meningioma | I | 10 | 53.0 | 9:1 | 1.58 | 0.03 | 1.60 | 0.09 |
| II | 1 | 57.0 | 1:0 | 1.13 | 0.04 | 1.48 | 0.08 | |
| Fibrous meningioma | I | 4 | 63.75 | 2:2 | 1.61 | 0.07 | 1.63 | 0.08 |
| Transitional meningioma | I | 1 | 71.0 | 1:0 | 1.48 | 0.04 | 1.56 | 0.1 |
| Angiomatous meningioma | I | 5 | 61.0 | 3:2 | 1.50 | 0.02 | 1.50 | 0.12 |
| Mixed meningioma | I | 1 | 69.0 | 1:0 | 1.53 | 0.06 | 1.63 | 0.12 |
| II | 2 | 64.0 | 2:0 | 1.48 | 0.09 | 1.62 | 0.07 | |
| Atypical meningioma | II | 3 | 65.3 | 1:2 | 1.47 | 0.05 | 1.41 | 0.06 |
| Anaplastic meningioma | III | 2 | 58.0 | 2:0 | 1.21 | 0.03 | 1.11 | 0.22 |
Fig. 4.

Total amounts of m5C in DNA (R) from tumor (black bars) and peripheral blood (white bars) samples of the same subjects with intracranial meningiomas. Total DNA methylation level in liquid biopsy (blood) samples reflect that in tumor tissues allowing noninvasive diagnostics and monitoring
In the control group of generally healthy individuals, the mean R coefficient was 1.93 ± 0.10, so significantly higher than in any of the brain meningioma groups (Fig. 2). The ANOVA test for blood results in cancer patients and the control group showed F = 90.203 and p < 0.001.
Comparison of total m5C contents in genomic DNA from tumor and peripheral blood samples
Total contents of m5C in DNA from the intracranial meningioma and peripheral blood samples of the same patients were comparable. The one-way ANOVA test values were: F = 0.307 and p = 0.581, showing no statistically significant differences between the groups. The relations between the WHO malignancy groups analyzing mean values for global DNA methylation in tumor tissue and blood for the same patients are presented in Fig. 4. The calculated r correlation coefficient for the whole group of patients with the tissue-blood pair was 0.72 (Fig. 5).
Fig. 5.

Comparison of total genomic m5C contents in DNA from intracranial meningioma tissue and peripheral blood samples of the same subjects. Pearson r correlation factor is 0.72
Discussion
While meningiomas are in general benign tumors, difficulties in their management can arise because of surgical limitations, or from aggressive and invasive tumor characteristics, that need adjuvant treatment and radiotherapy [5]. Currently used WHO classification is very subjective and miss reliable markers identifying these recurrence-prone tumors [34]. Recent genome- and exome-wide sequencing approaches have described the meningiomas’ mutational landscape comprehensively defining potential drivers of malignant progression and suggesting potential therapeutic targets [35, 36]. Moreover, the higher rate of genomic disruption was observed among the grade II-III meningiomas than in grade I counterparts (higher rate of non-synonymous mutations and the proportion of the genome affected by somatic copy number alterations) [35].
The established meningioma risk factors (ionizing radiation, head trauma, hormone-replacement therapy, hypertension, and advanced age [37, 38] are all extrinsic and intrinsic stress-inducing factors that can result in oxidative damage of cellular components, also DNA. ROS-induced damage comprises an assembly of DNA lesions that includes base damage, single-strand breaks, and double-strand breaks [39]. Exposure of an organism to chronic stress can result in imbalances of tissue homeostasis and possible tumor formation [40].
Despite its totally low abundance in DNA (4–5% of the total cytosine) of mammalian cells, 5-methylcytosine can be oxidatively damaged and induce mutations. It results in GC → AT transitions at CpG dinucleotides on the genetic level. However, at the epigenetic level, its damage influences the transcription regulation, embryonic development, and other life processes, as well as carcinogenesis [41, 42]. Data are suggesting that global DNA hypomethylation, observed in cancer, increases the instability of the genome. Promoter hypermethylation results in the silencing of genes involved in DNA repair, regulation of cell cycle, initiation of apoptosis, and tumor signaling networks control. All those pathways reflect the hallmarks of cancer [43]. Most frequently, hypermethylated genes encode transcription factors taking part in development, and their methylation may cause a permanent silencing of a gene [44]. Our approach focuses on the estimation of the total amount of m5C, regardless of its place in the genome. Through that, we show total DNA damage that is happening on epigenetic (regulatory level) because the epigenetic changes occur earlier and more frequently than genetic ones [45]. Moreover, we’ve recently shown that a decrease in total DNA methylation is a marker of an increase of oxidative damage to the cell, a well-known carcinogenesis factor [24].
Using llumina Infinium HumanMethylation450 BeadChip in genomic-scale DNA methylation profiling Gao et al. identified significantly lower levels of methylation in DNA for malignant meningioma (WHO III) than for atypical (WHO II) or benign (WHO I) ones [29]. Further analysis of different gene regions showed that DNA hypomethylation occurs rather throughout the genome than is restricted to specific gene regions. Therefore, global DNA hypomethylation is observed in the malignant transformation of meningiomas. Changes in global DNA methylation cause gene expression deregulation. Promoter hypermethylation at CpG islands induced gene expression suppression in low and high grade meningiomas, which suggests that DNA methylation is a primary gene silencing mechanism in malignant meningiomas [29].
On the contrary, Harmanci et al. showed a statistically significant positive correlation between the degree of chromosomal alterations and the amount of genome-wide DNA hypermethylation [37]. NF2 mutant atypical meningiomas displayed a hypermethylated phenotype. There was enrichment in methylation of Polycomb Repressive Complex 2 and Homeobox sites across benign meningiomas compared with control meninges, albeit less than that observed in atypical samples [37].
One should keep in mind that Infinium HumanMethylation450 is a widely-used tool to perform large-scale DNA methylation profiling. However, that method is focused on protein-coding, cancer-associated, and mitochondrial-related genes. The coverage of total CpG sites (the site of high 5-methylcytosine abundance) is low (only around 2%), which means that some features, such as enhancers, are only barely or not at all covered [46].
A multicenter evaluation of genome-wide DNA methylation patterns in 497 samples from meningiomas revealed six different methylation classes presenting with typical mutational, cytogenetic, and gene expression patterns, as well as with clinical relevance [47]. They identified patients with increased risk of tumor progression in the WHO I group, as well as patients with decreased probability of recurrence within the WHO II group. That meningioma classification, based on genome-wide DNA methylation, captured groups that are more homogenous clinically, therefore is more potent than the WHO classification in tumor recurrence prediction and prognosis estimation [47].
Earlier, Kishida et al. distinguished three meningioma clusters based on the methylation status of 6157 genes in 30 meningioma samples [31]. Clusters 2 and 3 included more males compared with cluster 1. The recurrent cases tended to accumulate in cluster 3 compared with clusters 1 and 2. Clusters 2 and 3 were generally hypermethylated, while cluster 1 showed the low methylator phenotype. The proportion of the WHO grades did not show a significant correlation with cluster formation [31].
The recent work of Nassiri et al. presents the methylome-based algorithm that predicts tumor recurrence more reliably than histologic grading, and independently of established clinical and molecular factors [48].
The results of the study presented in this paper show decreasing total DNA methylation with increasing tumor malignancy (Figs. 3 and 4). The method used (TLC separation of radioactively labeled nucleotides from the whole sample’s DNA) allows the estimation of total m5C amount in DNA in relation to the total contents of pyrimidines (Fig. 1). The final results come from straightforward calculations, and no data processing is needed. Our approach gives a numerical result, which is reversely correlated with tumor grade. The more malignant the tumor is, the smaller R-value (expressing total DNA methylation) is observed (Figs. 3 and 4). Grade I tumors are characterized by mean R around 1.6, grade II – 1.4, and grade III – 1.1. That tendency is comparable to our previous results for brain gliomas [33] and metastases [49]. However, the mean total DNA methylation levels (R) in those cases were generally lower compared to meningiomas reflecting the known higher malignancy of gliomas and metastasis.
The ultimate goal of every tumor molecular characterization is the establishment of a feature that could serve as a diagnostic or prognostic marker, as well as a therapeutic target [10]. There is limited access to the central nervous system (CNS) tumor tissue just for diagnostic purposes. Unlike many other tumors, the biopsy of intracranial lesions confers a non-negligible degree of risk related to the procedure. A liquid biopsy would facilitate our ability to follow patients longitudinally following initial diagnosis and treatment. Taking into account the aspect of invasiveness, lumbar puncture for cerebrospinal fluid sampling is also a not optimal solution. Therefore, peripheral blood samples are the most obvious choice. The most important question is why and how the global DNA methylation could be the same in tumor and blood samples. The possible answer lies in the mechanism through which DNA methylation is affected by ROS. Because it is a random and global process, not limited to a certain area or cell type, the disease signs can be found in the whole body, not only in the foci where tumors are localized [24, 50]. So far, only a few studies have evaluated the serum samples in CNS neoplasms in the search for DNA/RNA biomarker, mainly for glioma and brain metastasis [51–53], and only single cases for meningiomas in recent years [54–56]. In our previous study, we have analyzed the profile of aberrant methylation of MGMT, RASSF1A, p15INK4B, and p14ARF genes in serum free-circulating DNA and corresponding tumor tissue in a group of CNS cancer patients. The comparison of the results obtained for paired serum and tumor samples (also for meningioma) allowed the conclusion that the global concordance of results between these two sample sources is fairly high [57]. Moreover, we performed earlier total DNA methylation analysis with the TLC method for different brain tumors, breast and colon cancers, as well as arterial hypertension. The R values for tumor and blood DNA were comparable, showing that peripheral blood is an adequate source of DNA for complete methylation analysis, and can reflect the state of tumor tissue [58]. In the present study, the total m5C contents in DNA from tumor tissue and peripheral blood samples of the same intracranial meningioma patients were almost identical (Figs. 4 and 5). Therefore, it seems that methylation level of DNA from peripheral blood can be used as a diagnostic tool in neurooncology.
Conclusions
We showed a reciprocal relation of total m5C contents in tumor DNA to its malignancy grade. Therefore total DNA methylation of intracranial meningiomas can serve as a tool for their characteristics and malignancy estimation. Moreover, the correlation between total DNA methylation in tumor tissue and peripheral blood samples from the same patient was observed. That puts a light on the tumor pathogenesis (oxidative stress) but also enables the use of peripheral blood as a sample for detecting the tumor or monitoring of the disease. The identification of biomolecular parameters that could improve meningioma classification may optimize the indications for possible adjuvant therapies and closer follow-up.
Supplementary information
Additional file 1: Supplementary Figure 1. The total m5C contents (R) in DNA from meningioma tissues of 100 patients evaluated in this study. The bar graph visualizes heterogeneity among the patients. The patients’ order reflects that in Table 1 (where SD values for each data point are included). Meningioma variant names are accompanied by relevant malignancy grade. Grey bars correspond to WHO I, yellow – WHO II, and red – WHO III.
Additional file 2: Supplementary Figure 2. The total m5C contents (R) in DNA from peripheral blood samples of 29 meningioma patients evaluated in this study. The bar graph visualizes heterogeneity among the patients. The patients’ order reflects that in Table 1 (where SD values for each data point are included). Meningioma variant names are accompanied by relevant malignancy grade. Grey bars correspond to WHO I, yellow – WHO II, and red – WHO III.
Acknowledgements
The author would like to thank Dr. P. M. Perrigue for reading of the manuscript and language corrections.
Abbreviations
- C
Cytosine
- CNS
Central nervous system
- m5C
5-methylcytosine
- ROS
Reactive oxygen species
- T
Thymine
- TLC
Thin layer chromatography
- WHO
World Health Organisation
Author’s contributions
AMB collected data and samples, performed the experiments, and wrote the manuscript. The author(s) read and approved the final manuscript.
Funding
This study has been supported by the Chair and Department of Neurosurgery and Neurotraumatology, Karol Marcinkowski University of Medical Sciences.
The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
Availability of data and materials
The data generated or analyzed during this study are included in the published article. Further details are available from the corresponding author upon request.
Ethics approval and consent to participate
Blood and tissue molecular analysis was approved by the Bioethical Committee of Karol Marcinkowski University of Medical Sciences, Poznan (896/9; 838/12). All participants provided informed and written consent to donate their peripheral blood and tumor tissue samples for research.
Consent for publication
Not applicable. The patients‘clinical details presented in the paper do not compromise anonymity.
Competing interests
The author declares that she has no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12885-020-06982-3.
References
- 1.Ostrom QT, Patil N, Gittleman H, Truitt G, Waite K, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012-2016. Neuro-Oncology. 2019;21(Suppl 5):v1–v100. doi: 10.1093/neuonc/noz150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holleczek B, Zampella D, Urbschat S, Sahm F, von Deimling A, Oertel J, et al. Incidence, mortality and outcome of meningiomas: a population-based study from Germany. Cancer Epidemiol. 2019;62:101562. doi: 10.1016/j.canep.2019.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 4.Riemenschneider MJ, Perry A, Reifenberger G. Histological classification and molecular genetics of meningiomas. Lancet Neurol. 2006;5:1045–1054. doi: 10.1016/S1474-4422(06)70625-1. [DOI] [PubMed] [Google Scholar]
- 5.Walcott BP, Nahed BV, Brastianos PK, Loeffler JS. Radiation treatment for WHO grade II and III Meningiomas. Front Oncol. 2013;3:227. doi: 10.3389/fonc.2013.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta S, Bi WL, Dunn IF. Medical management of meningioma in the era of precision medicine. Neurosurg Focus. 2018;44:E3. doi: 10.3171/2018.1.FOCUS17754. [DOI] [PubMed] [Google Scholar]
- 7.Horbinski C, Ligon KL, Brastianos P, Huse JT, Venere M, Chang S, et al. The medical necessity of advanced molecular testing in the diagnosis and treatment of brain tumor patients. Neuro-Oncology. 2019;21:1498–1508. doi: 10.1093/neuonc/noz119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Apra C, Peyre M, Kalamarides M. Current treatment options for meningioma. Expert Rev Neurother. 2018;18:241–249. doi: 10.1080/14737175.2018.1429920. [DOI] [PubMed] [Google Scholar]
- 9.Zang KD, Singer H. Chromosomal constitution of meningiomas. Nature. 1967;216:84–85. doi: 10.1038/216084a0. [DOI] [PubMed] [Google Scholar]
- 10.Proctor DT, Ramachandran S, Lama S, Sutherland GR. Towards molecular classification of meningioma: evolving treatment and diagnostic paradigms. World Neurosurg. 2018;119:366–373. doi: 10.1016/j.wneu.2018.08.019. [DOI] [PubMed] [Google Scholar]
- 11.Mirian C, Duun-Henriksen AK, Juratli T, Sahm F, Spiegl-Kreinecker S, Peyre M, et al. Poor prognosis associated with TERT gene alterations in meningioma is independent of the WHO classification: an individual patient data meta-analysis. J Neurol Neurosurg Psychiatry. 2020;91:378–387. doi: 10.1136/jnnp-2019-322257. [DOI] [PubMed] [Google Scholar]
- 12.Alexiou GA, Markoula S, Gogou P, Kyritsis AP. Genetic and molecular alterations in meningiomas. Clin Neurol Neurosurg. 2011;113:261–267. doi: 10.1016/j.clineuro.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Ketter R, Henn W, Niedermayer I, Steilen-Gimbel H, König J, Zang KD, et al. Predictive value of progression-associated chromosomal aberrations for the prognosis of meningiomas: a retrospective study of 198 cases. J Neurosurg. 2001;95:601–607. doi: 10.3171/jns.2001.95.4.0601. [DOI] [PubMed] [Google Scholar]
- 14.Espinosa AB, Tabernero MD, Maíllo A, Sayagués JM, Ciudad J, Merino M, et al. The cytogenetic relationship between primary and recurrent meningiomas points to the need for new treatment strategies in cases at high risk of relapse. Clin Cancer Res. 2006;12:772–780. doi: 10.1158/1078-0432.CCR-05-1480. [DOI] [PubMed] [Google Scholar]
- 15.Ketter R, Urbschat S, Henn W, Feiden W, Beerenwinkel N, Lengauer T, et al. Application of oncogenetic trees mixtures as a biostatistical model of the clonal cytogenetic evolution of meningiomas. Int J Cancer. 2007;121:1473–1480. doi: 10.1002/ijc.22855. [DOI] [PubMed] [Google Scholar]
- 16.Suvà ML, Louis DN. Next-generation molecular genetics of brain tumours. Curr Opin Neurol. 2013;26:681–687. doi: 10.1097/WCO.0000000000000027. [DOI] [PubMed] [Google Scholar]
- 17.Breiling A, Lyko F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenetics Chromatin. 2015;8:24. doi: 10.1186/s13072-015-0016-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen L, Song CX, He C, Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem. 2014;83:585–614. doi: 10.1146/annurev-biochem-060713-035513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toraño EG, García MG, Fernández-Morera JL, Niño-García P, Fernández AF. The impact of external factors on the Epigenome: in utero and over lifetime. Biomed Res Int. 2016;2016:2568635. doi: 10.1155/2016/2568635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Panieri E, Santoro MM. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016;7:e2253. doi: 10.1038/cddis.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Q, Ni X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets. 2015;16:13–19. doi: 10.2174/1389450116666150113121054. [DOI] [PubMed] [Google Scholar]
- 22.Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: moving forward. PLoS Genet. 2018;14:e1007362. doi: 10.1371/journal.pgen.1007362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong Z, Zhu JK. Active DNA demethylation by oxidation and repair. Cell Res. 2011;21:1649–1651. doi: 10.1038/cr.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barciszewska AM, Giel-Pietraszuk M, Perrigue PM, Naskręt-Barciszewska M. Total DNA methylation changes reflect random oxidative DNA damage in gliomas. Cells. 2019;8:E1065. doi: 10.3390/cells8091065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 26.Barski D, Wolter M, Reifenberger G, Riemenschneider MJ. Hypermethylation and transcriptional downregulation of the TIMP3 gene is associated with allelic loss on 22q12.3 and malignancy in meningiomas. Brain Pathol. 2010;20:623–631. doi: 10.1111/j.1750-3639.2009.00340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kandenwein JA, Park-Simon TW, Schramm J, Simon M. uPA/PAI-1 expression and uPA promoter methylation in meningiomas. J Neuro-Oncol. 2011;103:533–539. doi: 10.1007/s11060-010-0411-6. [DOI] [PubMed] [Google Scholar]
- 28.Di Vinci A, Brigati C, Casciano I, Banelli B, Borzì L, Forlani A, et al. HOXA7, 9, and 10 are methylation targets associated with aggressive behavior in meningiomas. Transl Res. 2012;160:355–362. doi: 10.1016/j.trsl.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 29.Gao F, Shi L, Russin J, Zeng L, Chang X, He S, et al. DNA methylation in the malignant transformation of meningiomas. PLoS One. 2013;8:e54114. doi: 10.1371/journal.pone.0054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majchrzak-Celińska A, Paluszczak J, Szalata M, Barciszewska AM, Nowak S, Baer-Dubowska W. DNA methylation analysis of benign and atypical meningiomas: correlation between RUNX3 methylation and WHO grade. J Cancer Res Clin Oncol. 2015;141:1593–1601. doi: 10.1007/s00432-015-1930-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kishida Y, Natsume A, Kondo Y, Takeuchi I, An B, Okamoto Y, et al. Epigenetic subclassification of meningiomas based on genome-wide DNA methylation analyses. Carcinogenesis. 2012;33:436–441. doi: 10.1093/carcin/bgr260. [DOI] [PubMed] [Google Scholar]
- 32.Jin Z, Liu Y. DNA methylation in human diseases. Genes Dis. 2018;5:1–8. doi: 10.1016/j.gendis.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zukiel R, Nowak S, Barciszewska AM, Gawronska I, Keith G, Barciszewska MZ. A simple epigenetic method for the diagnosis and classification of brain tumors. Mol Cancer Res. 2004;2:196–202. [PubMed] [Google Scholar]
- 34.Brokinkel B, Hess K, Mawrin C. Brain invasion in meningiomas-clinical considerations and impact of neuropathological evaluation: a systematic review. Neuro-Oncology. 2017;19:1298–1307. doi: 10.1093/neuonc/nox071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet. 2013;45:285–289. doi: 10.1038/ng.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339:1077–1080. doi: 10.1126/science.1233009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harmancı AS, Youngblood MW, Clark VE, Coşkun S, Henegariu O, Duran D, et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat Commun. 2017;8:14433. doi: 10.1038/ncomms14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muskens IS, Wu AH, Porcel J, Cheng I, Le Marchand L, Wiemels JL, et al. Body mass index, comorbidities, and hormonal factors in relation to meningioma in an ethnically diverse population: the multiethnic cohort. Neuro-Oncology. 2019. 10.1093/neuonc/noz005. [DOI] [PMC free article] [PubMed]
- 39.Wiemels J, Wrensch M, Claus EB. Epidemiology and etiology of meningioma. J Neuro-Oncol. 2010;99:307–314. doi: 10.1007/s11060-010-0386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hegde ML, Mantha AK, Hazra TK, Bhakat KK, Mitra S, Szczesny B. Oxidative genome damage and its repair: implications in aging and neurodegenerative diseases. Mech Ageing Dev. 2012;133:157–168. doi: 10.1016/j.mad.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res. 2011;711:193–201. doi: 10.1016/j.mrfmmm.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 42.Madugundu GS, Cadet J, Wagner JR. Hydroxyl-radical-induced oxidation of 5-methylcytosine in isolated and cellular DNA. Nucleic Acids Res. 2014;42:7450–7460. doi: 10.1093/nar/gku334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ito S, Kuraoka I. Epigenetic modifications in DNA could mimic oxidative DNA damage: a double-edged sword. DNA Repair (Amst) 2015;32:52–57. doi: 10.1016/j.dnarep.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 44.Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res. 2017;7:1016–1036. [PMC free article] [PubMed] [Google Scholar]
- 45.Kanherkar RR, Bhatia-Dey N, Csoka AB. Epigenetics across the human lifespan. Front Cell Dev Biol. 2014;2:49. doi: 10.3389/fcell.2014.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morris TJ, Beck S. Analysis pipelines and packages for infinium humanmethylation450 beadchip (450k) data. Methods. 2015;72:3–8. doi: 10.1016/j.ymeth.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sahm F, Schrimpf D, Stichel D, Jones DTW, Hielscher T, Schefzyk S, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 2017;18:682–694. doi: 10.1016/S1470-2045(17)30155-9. [DOI] [PubMed] [Google Scholar]
- 48.Nassiri F, Mamatjan Y, Suppiah S, Badhiwala JH, Mansouri S, Karimi S, et al. DNA methylation profiling to predict recurrence risk in meningioma: development and validation of a nomogram to optimize clinical management. Neuro-Oncology. 2019;21:901–910. doi: 10.1093/neuonc/noz061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barciszewska AM. Global DNA demethylation as an epigenetic marker of human brain metastases. Biosci Rep. 2018;38:BSR20180731. doi: 10.1042/BSR20180731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rani V, Deep G, Singh RK, Palle K, Yadav UC. Oxidative stress and metabolic disorders: pathogenesis and therapeutic strategies. Life Sci. 2016;148:183–193. doi: 10.1016/j.lfs.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 51.Al-Moghrabi N, Nofel A, Al-Yousef N, Madkhali S, Bin Amer SM, Alaiya A. The molecular significance of methylated BRCA1 promoter in white blood cells of cancer-free females. BMC Cancer. 2014;14:830. doi: 10.1186/1471-2407-14-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lavon I, Refael M, Zelikovitch B, Shalom E, Siegal T. Serum DNA can define tumor-specific genetic and epigenetic markers in gliomas of various grades. Neuro-Oncology. 2010;12:173–180. doi: 10.1093/neuonc/nop041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wakabayashi T, Natsume A, Hatano H, Fujii M, Shimato S, Ito M, et al. p16 promoter methylation in the serum as a basis for the molecular diagnosis of gliomas. Neurosurgery. 2009;64:455–461. doi: 10.1227/01.NEU.0000340683.19920.E3. [DOI] [PubMed] [Google Scholar]
- 54.Weaver KD, Grossman SA, Herman JG. Methylated tumor-specific DNA as a plasma biomarker in patients with glioma. Cancer Investig. 2006;24:35–40. doi: 10.1080/07357900500449546. [DOI] [PubMed] [Google Scholar]
- 55.Zhi F, Shao N, Li B, Xue L, Deng D, Xu Y, et al. A serum 6-miRNA panel as a novel non-invasive biomarker for meningioma. Sci Rep. 2016;6:32067. doi: 10.1038/srep32067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mashayekhi F, Saberi A, Mashayekhi S. Serum TIMP1 and TIMP2 concentration in patients with different grades of meningioma. Clin Neurol Neurosurg. 2018;170:84–87. doi: 10.1016/j.clineuro.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 57.Majchrzak-Celińska A, Paluszczak J, Kleszcz R, Magiera M, Barciszewska AM, Nowak S, et al. Detection of MGMT, RASSF1A, p15INK4B, and p14ARF promoter methylation in circulating tumor-derived DNA of central nervous system cancer patients. J Appl Genet. 2013;54:335–344. doi: 10.1007/s13353-013-0149-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barciszewska AM, Nowak S, Naskręt-Barciszewska MZ. The degree of global DNA hypomethylation in peripheral blood correlates with that in matched tumor tissues in several neoplasia. PLoS One. 2014;9:e92599. doi: 10.1371/journal.pone.0092599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Figure 1. The total m5C contents (R) in DNA from meningioma tissues of 100 patients evaluated in this study. The bar graph visualizes heterogeneity among the patients. The patients’ order reflects that in Table 1 (where SD values for each data point are included). Meningioma variant names are accompanied by relevant malignancy grade. Grey bars correspond to WHO I, yellow – WHO II, and red – WHO III.
Additional file 2: Supplementary Figure 2. The total m5C contents (R) in DNA from peripheral blood samples of 29 meningioma patients evaluated in this study. The bar graph visualizes heterogeneity among the patients. The patients’ order reflects that in Table 1 (where SD values for each data point are included). Meningioma variant names are accompanied by relevant malignancy grade. Grey bars correspond to WHO I, yellow – WHO II, and red – WHO III.
Data Availability Statement
The data generated or analyzed during this study are included in the published article. Further details are available from the corresponding author upon request.