

Chromosomal and plasmid-borne AmpC cephalosporinases are a major resistance mechanism to β-lactams in Enterobacteriaceae and Pseudomonas aeruginosa. The new β-lactamase inhibitor avibactam effectively inhibits class C enzymes and can fully restore ceftazidime susceptibility. The conserved amino acid residue Asn346 of AmpC cephalosporinases directly interacts with the avibactam sulfonate. Disruption of this interaction caused by the N346Y amino acid substitution in Citrobacter freundii AmpC was previously shown to confer resistance to the ceftazidime-avibactam combination (CAZ-AVI).
KEYWORDS: avibactam, β-lactamase inhibitor, AmpC, cephalosporinase, ceftazidime, DHA, FOX, Enterobacter cloacae, Pseudomonas aeruginosa
ABSTRACT
Chromosomal and plasmid-borne AmpC cephalosporinases are a major resistance mechanism to β-lactams in Enterobacteriaceae and Pseudomonas aeruginosa. The new β-lactamase inhibitor avibactam effectively inhibits class C enzymes and can fully restore ceftazidime susceptibility. The conserved amino acid residue Asn346 of AmpC cephalosporinases directly interacts with the avibactam sulfonate. Disruption of this interaction caused by the N346Y amino acid substitution in Citrobacter freundii AmpC was previously shown to confer resistance to the ceftazidime-avibactam combination (CAZ-AVI). The aim of this study was to phenotypically and biochemically characterize the consequences of the N346Y substitution in various AmpC backgrounds. Introduction of N346Y into Enterobacter cloacae AmpC (AmpCcloacae), plasmid-mediated DHA-1, and P. aeruginosa PDC-5 led to 270-, 12,000-, and 79-fold decreases in the inhibitory efficacy (k2/Ki) of avibactam, respectively. The kinetic parameters of AmpCcloacae and DHA-1 for ceftazidime hydrolysis were moderately affected by the substitution. Accordingly, AmpCcloacae and DHA-1 harboring N346Y conferred CAZ-AVI resistance (MIC of ceftazidime of 16 μg/ml in the presence of 4 μg/ml of avibactam). In contrast, production of PDC-5 N346Y was associated with a lower MIC (4 μg/ml) since this β-lactamase retained a higher inactivation efficacy by avibactam in comparison to AmpCcloacae N346Y. For FOX-3, the I346Y substitution did not reduce the inactivation efficacy of avibactam and the substitution was highly deleterious for β-lactam hydrolysis, including ceftazidime, preventing CAZ-AVI resistance. Since AmpCcloacae and DHA-1 display substantial sequence diversity, our results suggest that loss of hydrogen interaction between Asn346 and avibactam could be a common mechanism of acquisition of CAZ-AVI resistance.
INTRODUCTION
Chromosomally encoded class C β-lactamases (cAmpCs) are ubiquitous in many clinically relevant Gram-negative bacteria such as Enterobacter cloacae, Citrobacter freundii, Morganella morganii, and Pseudomonas aeruginosa (1). These enzymes display broad substrate profiles, which include narrow-spectrum cephalosporins, cephamycins, and aminopenicillins (1). Derepression of cAmpC-encoding genes generally results in resistance to most β-lactams, except cefepime and carbapenems (1). Related members of the cAmpC family, designated extended-spectrum AmpC β-lactamases (ESACs), have evolved to include in their substrate profiles ceftazidime, other oxyimino-cephalosporins, and, in some instances, carbapenems (2–4). In the late 90s, the high diversity of class C β-lactamases further increased with the spread of AmpC-encoding genes by horizontal transfer of mobile genetic elements (plasmid-mediated AmpC or pAmpC) (5). These transfers have increased the burden of class C enzymes to include Gram-negative bacteria that are naturally devoid of any cAmpC, such as Klebsiella pneumoniae (5). Plasmid-born AmpCs are derivatives of the chromosomally encoded cephalosporinases from Enterobacter cloacae (ACT and MIR types), Citrobacter freundii (CMY-2, BIL, LAT, and CFE types), Morganella morganii (DHA type), Hafnia alvei (ACC type), and Aeromonas spp. (CMY-1, FOX, and MOX types). The expression of genes encoding pAmpCs is usually under the control of strong promoters, resulting in resistance levels similar to those found in cAmpC-derepressed isolates (6). The level of resistance to β-lactams conveyed by AmpC β-lactamases is determined by a complex combination of several factors, including the efficacy of drug hydrolysis, the level of AmpC production, and bacterial host factors such as porins, efflux pumps, and the drug targets, all of which may be mutationally altered under the selective pressure of β-lactams (7).
β-lactam-based first-generation β-lactamase inhibitors, clavulanic acid, tazobactam, and sulbactam, are generally not active against AmpC enzymes (1). Ceftazidime combined with avibactam, a second-generation inhibitor based on the diazabicyclooctane scaffold (8), has been proposed as a therapeutic option against AmpC-overproducing multidrug-resistant strains of Gram-negative bacteria (9). The AmpC residues that determine the efficacy of enzyme inhibition by avibactam and the risk of resistance acquisition by modification of these residues have not been extensively investigated. In vitro selection of ceftazidime-avibactam-resistant mutants of P. aeruginosa strains producing derepressed AmpC led to deletions of 5 to 19 amino acid residues of the Ω loop. Purification of a representative enzyme with a 5-residue deletion showed that resistance was due both to an increase in the efficacy of ceftazidime hydrolysis and to a reduction of the efficacy of inhibition by avibactam (10). One of the mutants harbored a G183D substitution located outside the Ω loop, but the corresponding enzyme was not biochemically characterized (10). In another study, also based on in vitro selection of mutants, resistance or decreased susceptibility to the ceftazidime-avibactam combination in derepressed AmpC-producing Enterobacteriaceae was found to result from the Arg168Pro/His, Gly176Arg/Asp, or Asn366Tyr substitution or from small deletions around positions 309 to 314 (11). These enzymes were not kinetically characterized. Crystallographic structures revealed that eight residues are in direct contact with avibactam, including Ser64 and Lys67 (Ambler amino-acid numbering scheme) from the SXXK motif, Tyr150 and Asn152 from the YXN motif, Lys315 and Thr316 from the KTG motif, Gln120, and Asn346 (10, 12, 13). Among these residues, Ser64 from the SXXK motif forms a covalent adduct with avibactam (carbamoyl-enzyme) leading to reversible enzyme inactivation (12, 13). Residues from the conserved SXXK, YXN, and KTG motifs are invariant in AmpC β-lactamases, whereas positions 120 (Gln or Phe) and 346 (Asn or Ile) display limited variations (See Fig. S1 in the supplemental material for a sequence alignment). In the latter position, an N346Y substitution in cAmpC was previously reported to confer resistance to the ceftazidime-avibactam combination in C. freundii (11, 13). This prompted us to investigate the impact of replacing asparagine or isoleucine at Ambler position 346 by tyrosine (N346Y and I346Y substitutions, respectively) in four highly divergent AmpC β-lactamases (See Fig. S2 for an evolutionary tree). This panel of four sequences included two chromosome-mediated cephalosporinases (AmpCcloacae from Enterobacter cloacae P99 and PDC-5 from P. aeruginosa ATCC 27853) and two plasmid-mediated cephalosporinases (DHA-1 and FOX-3, which derive from chromosomally encoded enzymes from Morganella morganii and Aeromonas spp, respectively). The phenotypic consequences of the substitutions were evaluated by constructing isogenic strains of Escherichia coli producing AmpCcloacae, PDC-5, DHA-1, FOX-3, and derivatives of these four β-lactamases containing a Tyr residue at position 346. The impact of the substitutions was also evaluated by purifying soluble forms of the β-lactamases in order to compare efficacies of β-lactam hydrolysis and of inactivation by avibactam. In parallel, we investigated whether exposure of the recombinant E. coli strains producing wild-type AmpCcloacae, PDC-5, DHA-1, or FOX-3 could lead to the selection of mutations resulting in the N346Y or I346Y substitution in these enzymes. We show that the N346Y substitution, but not I346Y, is a likely route of acquisition of resistance to the ceftazidime-avibactam combination in AmpC β-lactamases as it drastically reduced the efficacy of enzyme carbamoylation by avibactam while preserving the efficacy of ceftazidime hydrolysis. This conclusion applies to various members of the AmpC family despite substantial sequence divergence.
RESULTS AND DISCUSSION
Resistance to ceftazidime and to the ceftazidime-avibactam combination mediated by the AmpC cephalosporinases.
MICs were determined for isogenic strains of E. coli TOP10 producing AmpCcloacae, PDC-5, DHA-1, and FOX-3 (Table 1). Production of these four β-lactamases conferred ceftazidime resistance to the E. coli host with 64- to 1,024-fold increases in the MIC of this antibiotic (from 0.25 μg/ml to 16 to 256 μg/ml). Avibactam fully restored the activity of ceftazidime against TOP10 derivatives producing AmpCcloacae and DHA-1 (MIC of ceftazidime = 0.25 μg/ml in the presence of 4 μg/ml of avibactam). Inhibition of PDC-5 and FOX-3 was almost complete (ceftazidime MIC in the presence of avibactam of 0.5 μg/ml and 2 μg/ml for PDC-5 and FOX-3, respectively). As expected, these results indicate that avibactam restored completely (AmpCcloacae and DHA-1) or partially (PDC-5 and FOX-3) the activity of ceftazidime against the four isogenic strains of E. coli producing AmpC β-lactamases.
TABLE 1.
MIC of β-lactams against E. coli TOP10 isogenic strains producing various AmpC β-lactamases
| AmpC β-lactamase | MIC (μg/ml)a |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CAZ | CAZ-AVI | AMX | AMC | PIP | ETP | MEM | CF | MA | FOX | CTX | ATM | FEP | |
| Noneb | 0.25 | 0.25 | 2 | 2 | 2 | ≤0.06 | ≤0.06 | 8 | 1 | 4 | 0.06 | 0.25 | 0.12 |
| AmpCcloacae | 64 | 0.25 | >256 | >256 | 128 | 0.25 | ≤0.06 | >256 | 256 | 128 | 64 | 32 | 1 |
| AmpCcloacae N346Y | 128 | 16 | >256 | >256 | 16 | ≤0.06 | ≤0.06 | 128 | 128 | 16 | 32 | 32 | 0.5 |
| PDC-5 | 16 | 0.5 | >256 | >256 | 256 | ≤0.06 | ≤0.06 | >256 | 256 | 128 | 32 | 16 | 0.5 |
| PDC-5 N346Y | 8 | 4 | 64 | 32 | 4 | 0.12 | ≤0.06 | 128 | 16 | 128 | 4 | 1 | 0.25 |
| DHA-1 | 256 | 0.25 | >256 | >256 | 256 | ≤0.06 | ≤0.06 | 256 | 128 | 16 | 16 | 16 | 0.12 |
| DHA-1 N346Y | 64 | 16 | 128 | 128 | 8 | ≤0.06 | ≤0.06 | 16 | 4 | 8 | 2 | 16 | 0.12 |
| FOX-3 | 256 | 2 | 128 | 128 | 8 | ≤0.06 | ≤0.06 | >256 | 256 | 256 | 16 | 8 | 0.5 |
| FOX-3 I346Y | 16 | 1 | 32 | 32 | 1 | ≤0.06 | ≤0.06 | 32 | 1 | 8 | 0.25 | 0.5 | 0.12 |
CAZ, ceftazidime; CAZ-AVI, ceftazidime combined with avibactam (4 μg/ml); AMX, amoxicillin; AMC, amoxicillin combined with clavulanate (2 μg/ml); PIP, piperacillin; ETP, ertapenem; MEM, meropenem; CF, cephalothin; MA, cefamandole; FOX, cefoxitin; CTX, cefotaxime; ATM, aztreonam; FEP, cefepime.
None, control pTRC-99k vector without any ampC-encoding gene.
Impact of N346Y and I346Y on AmpC-mediated resistance to the ceftazidime-avibactam combination.
In the AmpCcloacae and DHA-1 backgrounds, the N346Y substitution was associated with a 64-fold increase in the MIC of ceftazidime in the presence of avibactam (from 0.25 μg/ml to 16 μg/ml) (Table 1). Introduction of N346Y in PDC-5 produced a lower increase (8-fold) in the MIC of ceftazidime in the presence of avibactam (from 0.5 μg/ml to 4 μg/ml). For these three β-lactamases (AmpCcloacae, DHA-1, and PDC-5), the substitution had a marginal impact on the MIC of ceftazidime alone. These results suggest that reduced activity of the ceftazidime-avibactam combination was due to impaired inhibition of these enzymes by avibactam but not to improved hydrolysis of ceftazidime.
The activity of the ceftazidime-avibactam combination was compromised by the introduction of a tyrosine residue at position 346 of AmpC cephalosporinases from various origins, AmpCcloacae, DHA-1, and PDC-5, which contain an asparagine at this position (above). In contrast, for FOX-3, which contains an isoleucine at this position, tyrosine introduction did not lead to any increase in the MIC of ceftazidime in the presence of avibactam. The I346Y substitution in FOX-3 impaired expression of ceftazidime resistance (a 16-fold decrease in the MIC of ceftazidime, from 256 to 16 μg/ml). Impaired hydrolysis of ceftazidime by FOX-3 I346Y may therefore account for the lack of resistance to the ceftazidime-avibactam combination.
Impact of N346Y and I346Y on AmpC-mediated resistance to other β-lactams.
The substitutions led to significant decreases of the MICs of certain β-lactams for the four AmpC cephalosporinases (Table 1). For AmpCcloacae, the antibiotics most impacted were piperacillin, cephalotin, and cefoxitin with 8-, ≥4-, and 8-fold MIC decreases, respectively. For DHA-1, the MIC of amoxicillin, piperacillin, cephalotin, and cefotaxime were reduced ≥4-, 32-, 16-, and 8-fold, respectively. For PDC-5, resistance was impaired for amoxicillin (≥8-fold), piperacillin (64-fold), cephalotin (≥4-fold), cefamandole (16-fold), aztreonam (16-fold), and cefotaxime (8-fold). For FOX-3, the I346Y substitution abolished resistance to piperacillin, cefamandole, cefoxitin, cefotaxime, and aztreonam. In conclusion, the N346Y substitutions in AmpCcloacae, DHA-1, and PDC-5 impaired but did not abolish resistance to most β-lactams, while I346Y in FOX-3 was highly deleterious. As expected (7), clavulanate did not affect the MIC of amoxicillin against E. coli strains producing the four AmpC cephalosporinases or their derivatives with substitutions at position 346.
Substrate profile of purified wild-type AmpC cephalosporinases.
Soluble forms of AmpCcloacae, PDC-5, DHA-1, and FOX-3 that lacked the N-terminal signal peptides were produced in E. coli and purified by metal affinity and size-exclusion chromatography. The catalytic efficacy (kcat/Km) of the four AmpC cephalosporinases was high for amoxicillin (>104 M−1 s−1), first-generation cephalosporins (>107 M−1 s−1), and second-generation cephalosporins (>105 M−1 s−1) (Table 2). For third-generation cephalosporins, hydrolysis efficacy was lower for ceftazidime (from 1.3 × 103 to 1.6 × 105 M−1 s−1) than for cefotaxime (from 3.6 × 105 to 1.7 × 107 M−1 s−1). Among cephalosporins, the lowest hydrolysis efficacy was observed for the fourth-generation drug cefepime (3.0 × 102 to 4.8 × 103 M−1 s−1). Aztreonam was not a substrate of the purified enzymes.
TABLE 2.
Impact of the N346Y and I346Y substitutions on the hydrolysis of β-lactams by AmpCcloacae, PDC-5, DHA-1, and FOX-3
| Antibiotic and parameter | Values of kinetic parameters (mean ± SD) for indicated β-lactamases |
|||||||
|---|---|---|---|---|---|---|---|---|
| AmpCcloacae | AmpCcloacae N346Y | PDC-5 | PDC-5 N346Y | DHA-1 | DHA-1 N346Y | FOX-3 | FOX-3 I346Y | |
| Nitrocefin | ||||||||
| Km (μM) | 50 ± 10 | 18 ± 3 | 15 ± 4 | 5 ± 3 | 11 ± 1 | 1.7 ± 0.4 | 43 ± 8 | 23 ± 6 |
| kcat (s−1) | 790 ± 50 | 290 ± 10 | 240 ± 20 | 93 ± 8 | 240 ± 10 | 7.1 ± 0.6 | (1.3 ± 0.1) × 103 | 580 ± 40 |
| kcat/Km (M−1 s−1) | (1.6 ± 0.4) × 107 | (1.6 ± 0.3) × 107 | (1.6 ± 0.6) × 107 | (1.9 ± 1.3) × 107 | (2.3 ± 0.3) × 107 | (4.1 ± 1.6) × 106 | (3.1 ± 0.8) × 107 | (2.5 ± 0.6) × 107 |
| Amoxicillin | ||||||||
| Km (μM) | 2.4 ± 0.9a | 6.9 ± 1.3a | 4.4 ± 1.4a | 67 ± 19 | 0.32 ± 0.10a | 7.4 ± 5.4a | 9.7 ± 2.7a | 0.47 ± 0.03a |
| kcat (s−1) | 0.40 ± 0.04 | 0.10 ± 0.01 | 2.2 ± 0.3 | 2.1 ± 0.1 | 2.0 ± 0.2 | 0.26 ± 0.01 | 0.22 ± 0.02 | 0.19 ± 0.01 |
| kcat/Km (M−1 s−1) | (1.6 ± 0.8) × 105 | (1.4 ± 0.4) × 104 | (4.9 ± 2.3) × 105 | (3.1 ± 1.0) × 104 | (6.2 ± 2.6) × 106 | (3.5 ± 2.7) × 104 | (2.3 ± 0.8) × 104 | (4.0 ± 0.5) × 105 |
| Cephalothin | ||||||||
| Km (μM) | 23 ± 7 | 5.5 ± 1.3a | 12 ± 4 | 47 ± 12 | 8.7 ± 3.6a | 2.0 ± 1.0a | 48 ± 16 | 40 ± 11 |
| kcat (s−1) | 370 ± 60 | 83 ± 10 | 180 ± 10 | 230 ± 20 | 170 ± 20 | 4.3 ± 0.2 | (1.9 ± 0.2) × 103 | (2.5 ± 0.2) × 103 |
| kcat/Km (M−1 s−1) | (1.6 ± 0.8) × 107 | (1.5 ± 0.5) × 107 | (1.5 ± 0.6) × 107 | (4.9 ± 1.6) × 106 | (2.0 ± 1.0) × 107 | (2.1 ± 1.2) × 106 | (4.0 ± 1.8) × 107 | (6.3 ± 2.3) × 107 |
| Cefamandole | ||||||||
| Km (μM) | 3.1 ± 1.1a | 1.6 ± 0.3a | 0.6 ± 0.2a | 57 ± 13 | 1.2 ± 0.4a | 2.3 ± 1.4a | 41 ± 7 | 17 ± 2 |
| kcat (s−1) | 33 ± 1 | 4.1 ± 0.2 | 4.9 ± 0.5 | 32 ± 2 | 12 ± 1 | 0.50 ± 0.03 | 500 ± 20 | 110 ± 10 |
| kcat/Km (M−1 s−1) | (1.1 ± 0.4) × 107 | (2.6 ± 0.6) × 106 | (8.1 ± 3.6) × 106 | (5.6 ± 1.6) × 105 | (1.0 ± 0.4) × 107 | (2.2 ± 1.5) × 105 | (1.2 ± 0.3) × 107 | (6.8 ± 1.2) × 106 |
| Cefoxitin | ||||||||
| Km (μM) | 0.04 ± 0.01a | 0.022 ± 0.006a | 0.10 ± 0.03a | 0.20 ± 0.10a | 0.027 ± 0.003a | 0.20 ± 0.10a | 0.20 ± 0.06a | 0.022 ± 0.008a |
| kcat (s−1) | 0.050 ± 0.001 | 0.0025 ± 0.0005 | 0.050 ± 0.010 | 0.13 ± 0.01 | 0.026 ± 0.001 | (2.5 ± 0.1) × 10−3 | 1.8 ± 0.1 | 0.064 ± 0.004 |
| kcat/Km (M−1 s−1) | (1.2 ± 0.3) × 106 | (1.1 ± 0.5) × 105 | (5.0 ± 2.5) × 105 | (6.5 ± 3.7) × 105 | (9.6 ± 1.4) × 105 | (1.3 ± 0.7) × 104 | (9.0 ± 3.2) × 106 | (2.9 ± 1.2) × 106 |
| Cefotaxime | ||||||||
| Km (μM) | 0.050 ± 0.010a | 0.11 ± 0.02a | 0.14 ± 0.01a | 1.2 ± 0.7a | 0.0030 ± 0.0010a | 0.40 ± 0.20a | 0.019 ± 0.006a | 0.014 ± 0.007a |
| kcat (s−1) | 0.018 ± 0.002 | 0.050 ± 0.010 | 0.070 ± 0.020 | 0.20 ± 0.01 | 0.051 ± 0.002 | 0.029 ± 0.001 | 0.024 ± 0.002 | 0.016 ± 0.001 |
| kcat/Km (M−1 s−1) | (3.6 ± 1.1) × 105 | (4.5 ± 1.7) × 105 | (5.0 ± 1.8) × 105 | (1.7 ± 1.1) × 105 | (1.7 ± 0.6) × 107 | (7.3 ± 3.9) × 104 | (1.3 ± 0.5) × 106 | (1.1 ± 0.6) × 106 |
| Ceftazidime | ||||||||
| Km (μM) | 4.8 ± 1.8a | 3.6 ± 0.7a | 7.3 ± 2.2a | 19 ± 9a | 32 ± 10 | 5.4 ± 2.8a | 2.2 ± 0.9a | 0.35 ± 0.25a |
| kcat (s−1) | 0.011 ± 0.001 | 0.048 ± 0.005 | 0.010 ± 0.001 | 0.06 ± 0.01 | 0.80 ± 0.10 | (3.7 ± 0.3) × 10−3 | 0.35 ± 0.05 | 0.0029 ± 0.0004 |
| kcat/Km (M−1 s−1) | (2.3 ± 1.1) × 103 | (1.3 ± 0.4) × 104 | (1.3 ± 0.5) × 103 | (3.2 ± 2.0) × 103 | (2.6 ± 1.1) × 104 | (6.9 ± 4.1) × 102 | (1.6 ± 0.9) × 105 | (8.3 ± 7.1) × 103 |
| Aztreonamb | ||||||||
| Km (μM) | NA | NA | NA | NA | NA | NA | NA | NA |
| kcat (s−1) | NA | NA | NA | NA | NA | NA | NA | NA |
| kcat/Km (M−1 s−1) | <4.2 × 10−3 s−1 | <4.2 × 10−3 s−1 | <4.2 × 10−3 s−1 | <4.2 × 10−3 s−1 | <4.2 × 10−3 s−1 | <4.2 × 10−3 s−1 | <4.2 × 10−3 s−1 | <4.2 × 10−3 s−1 |
| Cefepime | ||||||||
| Km (μM) | >290 | >175 | >250 | >300 | >260 | >220 | >250 | >170 |
| kcat (s−1) | >0.5 | >0.7 | >0.15 | >0.17 | >0.07 | >0.14 | >1.1 | >2.1 |
| kcat/Km (M−1 s−1) | (1.7 ± 0.1) × 103 | (4.0 ± 0.2) × 103 | (6.0 ± 0.1) × 102 | (5.5 ± 0.1) × 102 | (3.0 ± 0.2) × 102 | (5.0 ± 0.5) × 102 | (4.8 ± 0.3) × 103 | (1.2 ± 0.1) × 104 |
For these drugs, the Km and kcat kinetic parameters were determined in independent experiments based on (i) hydrolysis of the tested drugs at saturating substrate concentrations for kcat and (ii) hydrolysis of nitrocefin in the presence of various concentrations of the tested drugs for Km (16).
NA, not applicable, as hydrolysis of aztreonam (500 μM) could not be detected at the highest β-lactamase concentration tested (10 μM). Under our experimental conditions, the lower limit of detection corresponds to a turnover of 4.2 × 10−3 s−1.
Impact of N346Y and I346Y on the efficacy of AmpC cephalosporinases for hydrolysis of ceftazidime.
The N346Y substitution had a moderate impact on the kinetic parameters for hydrolysis of ceftazidime by AmpCcloacae and PDC-5 (Table 2). This observation is in agreement with the phenotypic analysis, as the substitution did not affect (≤2-fold) the MIC of ceftazidime conveyed by these enzymes in E. coli Top10 (see above, Table 1). In contrast, N346Y was associated with a large (38-fold) decrease in the catalytic efficacy of DHA-1 due to a decrease in kcat (220-fold) that was only partially compensated by a decrease in Km (6-fold). The decrease in the catalytic efficacy of DHA-1 was associated with a 4-fold decrease in the MIC of ceftazidime (from 256 μg/ml to 64 μg/ml). Likewise, introduction of I346Y in FOX-3 was associated with decreases in the MIC of ceftazidime (16-fold) and in the kcat/Km ratio (20-fold) for hydrolysis of this drug. The latter difference resulted from a large reduction in kcat (120-fold), which was only partially compensated by a 6-fold reduction in Km. Together, these results indicate that impaired efficacy of in vitro ceftazidime hydrolysis due to the introduction of N346Y and I346Y in DHA-1 and FOX-3, respectively, was associated with impaired expression of ceftazidime resistance.
Impact of the N346Y and I346Y substitutions on the hydrolysis efficacy of other β-lactams.
For AmpCcloacae, N346Y was associated with reductions in kcat/Km (10-fold) and kcat (20-fold) for hydrolysis of cefoxitin, whereas the kinetic parameters for hydrolysis of other β-lactams were only marginally affected (Table 2). For PDC-5, N346Y was associated with decreases in the catalytic efficacy of amoxicillin (16-fold) and cefamandole (14-fold) hydrolysis, mainly due to increases in Km (15- and 95-fold, respectively). The efficacy of hydrolysis of all tested β-lactams except cefepime was affected by introduction of N346Y in DHA-1 (10- to 230-fold decreases in kcat/Km) due to increases in Km and decreases in kcat alone or in combination. For FOX-3, I346Y did not impair the catalytic activity. On the contrary, hydrolysis of amoxicillin was improved with a 17-fold increase in the kcat/Km ratio resulting from a 21-fold increase in kcat. Thus, I346Y in FOX-3 specifically impaired ceftazidime hydrolysis.
Overall, decreases in the catalytic efficacy of the cephalosporinases were correlated with decreases in the MICs of β-lactams—with two major discrepancies. First, introduction of I346Y in FOX-3 was associated with large decreases in the MICs of most β-lactams, although kinetic parameters were marginally affected. This observation suggests that the I346Y substitution may affect the stability or export of FOX-3. Second, hydrolysis of aztreonam was not detected for any of the AmpC cephalosporinases, although production of these enzymes conferred significant levels of resistance (MICs ranging from 8 to 32 μg/ml) corresponding to 32- to 128-fold increases in comparison with the host strain devoid of any β-lactamase (MIC = 0.25 μg/ml). Data from the literature suggest a possible explanation for this discrepancy, as it has been reported that β-lactamases act by trapping aztreonam and thereby reducing the periplasmic drug concentration and preventing inactivation of penicillin-binding protein (PBP) targets (14).
Efficacy of AmpC cephalosporinase inhibition by avibactam.
The four AmpC enzymes were inhibited by avibactam albeit with different efficacies (DHA-1 > PDC-5 > AmpCcloacae > FOX-3 with k2/Ki ratios of 170,000, 63,000, 18,000, and 4,200 M−1 s−1, respectively) (Table 3). The N346Y substitution impaired inhibition of AmpCcloacae, DHA-1, and PDC-5 with fold decreases in the k2/Ki efficacy parameter of 270, 12,000, and 79, respectively. In contrast, the I346Y substitution had only a modest impact on the inhibition efficacy of FOX-3 by avibactam (k2/Ki of 4,200 versus 2,000 M−1 s−1 for the wild-type and I346Y variant, respectively). The k-2 rate constants were low for all enzymes, indicating that modification of the decarbamoylation efficacy is not a key element in defining the phenotypes under study (Table 3).
TABLE 3.
Kinetic parameters for β-lactamase inhibition by avibactam
| β-lactamase | Inhibition parameters |
|
|---|---|---|
| k2/Ki (M−1 s−1) | k−2 (s−1) | |
| AmpCcloacae | (1.8 ± 0.1) × 104 | (1.8 ± 0.2) × 10−3 |
| AmpCcloacae N346Y | 66 ± 8 | (2.2 ± 0.5) × 10−3 |
| PDC-5 | (6.3 ± 0.5) × 104 | (2.4 ± 0.3) × 10−3 |
| PDC-5 N346Y | (8.0 ± 2.4) ×102 | (3.1 ± 1.3) × 10−3 |
| DHA-1 | (1.7 ± 0.1) × 105 | (2.3 ± 0.3) × 10−3 |
| DHA-1 N346Y | 14 ± 1 | (1.2 ± 0.1) × 10−3 |
| FOX-3 | (4.2 ± 0.3) × 103 | (1.5 ± 0.5) × 10−3 |
| FOX-3 I346Y | (2.0 ± 0.1) × 103 | (2.7 ± 0.4) × 10−3 |
Selection of mutants resistant to the ceftazidime-avibactam combination.
We investigated the acquisition of ceftazidime-avibactam resistance by E. coli TOP10 derivatives expressing the four ampC genes. Selection was performed on agar plates containing a fixed concentration of avibactam (4 μg/ml) and 2-fold increasing concentrations of ceftazidime (0.5 to 256 μg/ml). For the AmpCcloacae-producing E. coli TOP10 strain, a single mutant was obtained on agar plates containing 4 μg/ml of ceftazidime and 4 μg/ml of avibactam (frequency of ca. 5 × 10−10). Attempts to obtain additional mutants in two additional experiments were unsuccessful. Sequencing of the ampCcloacae gene of the mutant revealed an A to T transversion at position 1096 leading to an asparagine-to-tyrosine substitution at Ambler position 346. Thus, the N346Y substitution characterized in this study is a mechanism of acquisition of ceftazidime-avibactam resistance under the selective pressure of the drug, although its frequency is very low.
For PDC-5, the N346Y substitution was not obtained in four independent experiments. This negative result may be accounted for by the modest (8-fold) increase in the MIC of ceftazidime in the presence of avibactam associated with the introduction of N346Y in PDC-5 (from 0.5 to 4 μg/ml) (Table 1). In comparison, a 64-fold increase was observed for introduction of N346Y in AmpCcloacae (from 0.25 to 16 μg/ml). The kinetic parameters that correlate with this difference between PDC-5 and AmpCcloacae are the higher efficacy of PDC-5 N346Y inhibition by avibactam in comparison to AmpCcloacae N346Y (k2/Ki ratios of 800 ± 240 versus 66 ± 8 M−1 s−1; Table 3) and the higher Km of ceftazidime (19 ± 9 versus 1.9 ± 0.7 μM; Table 2). Since avibactam and ceftazidime competitively bind to the β-lactamase active sites, differences in the k2/Ki and Km kinetic constants are both expected to contribute to the higher efficacy of ceftazidime hydrolysis by PDC-5 N346Y in the periplasm.
Although the N346Y substitution was not obtained for PDC-5, two other modifications of this β-lactamase were selected by ceftazidime combined to avibactam. First, deletion of 21 nucleotides (711ACGGGTCGGTCCCGGCCCGCT731), leading to a deletion of seven amino acids in PDC-5 (238RVGPGPL244), was obtained in three out of four independent selection experiments (mean frequency of 3 × 10−10). Second, a guanine to adenine transition at position 739, resulting in the E247K substitution, occurred in a single selection experiment (frequency of 5 × 10−10). Both modifications occurred in the Ω loop of the PDC-5 P. aeruginosa cAmpC cephalosporinase (Fig. S1).
For DHA-1 and FOX-3, no mutant was recovered from three independent selection experiments with combinations of ceftazidime and avibactam (a frequency of <10−10). The absence of mutants with the I346Y substitution in FOX-3 was expected since the plasmids encoding FOX-3 I346Y and wild-type FOX-3 conferred similar levels of resistance to ceftazidime in the presence of avibactam (MIC = 2 μg/ml versus 1 μg/ml, respectively; Table 1). In contrast, the mutation leading to the N346Y substitution in DHA-1 produced a large increase in the MIC of ceftazidime in the presence of avibactam (from 0.25 μg/ml to 16 μg/ml; Table 1). However, selection of the corresponding mutation was not obtained.
Role of Asn346 in avibactam binding.
Crystal structures have revealed that the carboxamide of Asn346 is in hydrogen interaction with the avibactam sulfonate (13). According to these structural data, loss of this interaction, possibly reinforced by steric hindrance due to the bulky Tyr side chain (13), is likely to account for the large decreases in carbamoylation efficacy caused by the N346Y substitution in AmpCcloacae, PDC-5, and DHA-1. Interestingly, FOX-3 harbors an isoleucine at position 346, a residue that cannot form any significant hydrogen interaction with the avibactam sulfonate but is similar to Asn with respect to size. For FOX-3, the carbamoylation efficacy was lower than for the N346-containing AmpCcloacae, PDC-5, and DHA-1 cephalosporinases (Table 3), possibly reflecting the absence of the carboxamide-Asn346 hydrogen interaction. Introduction of I346Y in FOX-3 was associated with a moderate (2-fold) decrease in the carbamoylation efficacy, indicating that introduction of a Tyr residue at position 346 of FOX-3 was not associated with any adverse effect involving steric hindrance.
Conclusion. Here, we show that introduction of the N346Y substitution in the AmpCcloacae, PDC-5, and DHA-1 cephalosporinases leads to drastic reductions in the efficacy of β-lactamase inactivation by avibactam but largely preserves the hydrolysis efficacy of ceftazidime (Fig. 1A). These two features are essential for the acquisition of resistance to the ceftazidime-avibactam combination, as shown by a graphical analysis of the consequences of changes in kinetics parameters on the expression of resistance (Fig. 1B). Since AmpCcloacae, PDC-5, and DHA-1 display substantial sequence diversity, our results suggest that loss of hydrogen interaction between Asn346 and the avibactam sulfonate could be a common mechanism of acquisition of resistance to the ceftazidime-avibactam combination by mutational alteration of the corresponding codon. However, the low frequency of this mutational event may limit its emergence under treatment. The relevance of Asn346 for acquisition of resistance to the combination arises from its critical role for efficacious cephalosporinase inactivation by avibactam but not for hydrolysis of ceftazidime and, to a certain extent, of other β-lactams. This appears to be a remarkable property of Asn346 in AmpC cephalosporinases, as analyses of class A β-lactamases have revealed that amino acid substitutions reducing carbamoylation efficacy often lead to large decreases in hydrolysis efficacy and, in certain instances, to hyper-susceptibility to certain β-lactams (15, 16).
FIG 1.

Impact of critical amino acid substitutions on the kinetic parameters for ceftazidime hydrolysis and β-lactamase inhibition by avibactam (A) and consequences of these modifications on the MICs of ceftazidime alone or in combination with avibactam (B). For AmpCcloacae (AmpC), a modest increase in the efficacy of ceftazidime hydrolysis results in a modest increase in the MIC of ceftazidime. In combination with a large decrease in the efficacy of avibactam, this leads to resistance to the ceftazidime-avibactam combination (MIC = 16 μg/ml). The N346I substitution in DHA-1 is associated with the same level of resistance to the combination, as a reduction in the efficacy of ceftazidime hydrolysis is compensated by a very large decrease in the efficacy of avibactam. For PDC-5, the reduction in the efficacy of avibactam is relatively modest. This leads to an increase in the level of resistance to ceftazidime-avibactam but resistance to the combination is not achieved, despite a relatively low efficacy of ceftazidime hydrolysis. For Fox-3, the I346Y substitution impairs the efficacy of ceftazidime hydrolysis, leading to a large decrease in the ceftazidime MIC. However, strains producing FOX-3 and FOX-3 I346Y remain susceptible to the ceftazidime-avibactam combination since the substitution has only a moderate impact on the efficacy of avibactam. S, I, and R, susceptible, intermediary, and resistant according to EUCAST breakpoint values for P. aeruginosa.
MATERIALS AND METHODS
Construction of recombinant plasmids.
The four highly divergent class C β-lactamases evaluated in this study included cAmpC from E. cloacae P99 (designated AmpCcloacae), cAmpC PDC-5 from P. aeruginosa ATCC 27853, and the pAmpCs DHA-1 and FOX-3, which derive from chromosomally encoded enzymes from Morganella morganii and Aeromonas spp. For phenotypic analyses, the genes were amplified using primers depicted in Table S1 and cloned under the control of the isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible promoter of the plasmid vector pTRC-99k (16). Recombinant plasmids were introduced by electroporation into E. coli TOP10 with selection for resistance to kanamycin (50 μg/ml) conveyed by the vector pTRC-99k. For protein purification, gene fragments encoding soluble forms of the enzymes were amplified (see Table S1 for the sequence of the primers) and cloned under the control of the IPTG-inducible promoter of the plasmid vector pET-TEV (16). Site-directed mutagenesis was performed using the mutagenic primers depicted in Table S1, except for AmpCcloacae that was obtained by selection of a spontaneous mutant of pTRC-99kΩampCcloacae on agar containing ceftazidime (4 μg/ml) and avibactam (4 μg/ml). The sequences of the cloned genes were verified by double-strand Sanger sequencing (Eurofins Genomics).
MIC determinations.
MICs of amoxicillin, amoxicillin in the presence of clavulanate (2 μg/ml), piperacillin, cephalothin, cefamandole, cefoxitin, cefotaxime, ceftazidime, ceftazidime in the presence of avibactam (4 μg/ml; Sigma-Aldrich), cefepime, ertapenem, meropenem, and aztreonam were determined by the microdilution method in Mueller-Hinton broth (MHB) (Difco), according to the Clinical and Laboratory Standards Institute (CLSI) recommendations (17). The inoculum was prepared by growing bacteria in MHB containing IPTG (0.5 mM) for induction of the β-lactamase genes and kanamycin (50 μg/ml) for plasmid maintenance. IPTG but not kanamycin was added to MHB in the 96-well plates used for MIC determination. The experiments were performed in triplicate and the data are the medians of three experiments. Breakpoints for the susceptible, intermediary, and resistant phenotypes were those recommended by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) (version 9.0) (http://www.eucast.org/clinical_breakpoints/).
Purification of β-lactamases.
Production and purification of AmpC β-lactamases was performed as previously described (16). Briefly, fragments of β-lactamase genes, including soluble fragments of AmpCcloacae (residues 21 to 381), PDC-5 (residues 27 to 397), DHA-1 (residues 25 to 37), and FOX-3 (residues 24 to 382) were translationally fused to the sequence of the pET-TEV vector coding for an N-terminal 6×His tag followed by a cleavage site for the TEV protease (GSSHHHHHHSSGENLYFQG). The β-lactamases were produced in E. coli BL21(DE3) and purified from clarified lysates by affinity chromatography (Ni-nitrilotriacetic acid agarose; Sigma-Aldrich) and size-exclusion chromatography in 25 mM Tris-HCl (pH 7.5) containing 300 mM NaCl (Superdex 200 HL26/60; Amersham Pharmacia Biotech). The β-lactamases were stored at –20°C in the same buffer. Protein concentration was determined by the Bio-Rad protein assay using bovine serum albumin as a standard.
Kinetic analyses.
Hydrolysis kinetics were performed at 20°C in 2-(N-morpholino)ethanesulfonic acid (MES; 100 mM, pH 6.4). Steady-state kinetic parameters for the hydrolysis of nitrocefin, amoxicillin, cephalothin, cefamandole, cefoxitin, cefotaxime, ceftazidime, cefepime, and aztreonam (kcat, Km, and kcat/Km) were determined by measuring initial enzymatic reaction rates in a Cary 300 spectrophotometer (Agilent) (15, 18). Table S2 reports the wavelength and molar extinction coefficient that were used for each antibiotic. Inhibition parameters (k2/Ki and k-2) were determined by measuring hydrolysis of nitrocefin (100 μM) in the presence of various concentrations of avibactam, as previously reported (8, 15). Inactivation of AmpC β-lactamases by avibactam was considered to proceed according to the two-step reversible reaction mechanism depicted in Fig. 2, in which E and I represent the β-lactamase and avibactam, respectively, and EI and EI* the noncovalent and covalent β-lactamase-avibactam adducts, respectively.
FIG 2.
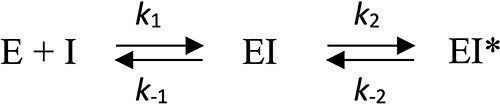
Two-step reversible reaction mechanism. E and I represent the β-lactamase and avibactam, respectively, and EI and EI* the noncovalent and covalent β-lactamase-avibactam adducts, respectively.
Equation 1 was fitted to data to determine the value of the rate constant (kobs) for various concentrations of avibactam. In equation 1, [P] represents the concentration of hydrolyzed nitrocefin, vi the initial velocity, vs the steady-state velocity, and t the time.
| (1) |
The carbamoylation efficacy (k2/Ki) was determined by plotting the values of kobs as a function of the concentration of avibactam and fitting equation 2 to the data, in which [S] represents the initial concentration of nitrocefin, Km the Michaelis constant for hydrolysis of nitrocefin by the β-lactamase, and [I] the concentration of avibactam.
| (2) |
Supplementary Material
ACKNOWLEDGMENTS
We thank Zainab Edoo for English editing.
A.D. was supported by the “Année Recherche” from the Assistance Publique-Hôpitaux de Paris.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Bush K. 2018. Past and present perspectives on β-lactamases. Antimicrob Agents Chemother 62:e01076-18. doi: 10.1128/AAC.01076-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mammeri H, Nordmann P, Berkani A, Eb F. 2008. Contribution of extended-spectrum AmpC (ESAC) beta-lactamases to carbapenem resistance in Escherichia coli. FEMS Microbiol Lett 282:238–240. doi: 10.1111/j.1574-6968.2008.01126.x. [DOI] [PubMed] [Google Scholar]
- 3.Rodríguez-Martínez J-M, Poirel L, Nordmann P. 2009. Extended-spectrum cephalosporinases in Pseudomonas aeruginosa. Antimicrob Agents Chemother 53:1766–1771. doi: 10.1128/AAC.01410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dahyot S, Broutin I, de Champs C, Guillon H, Mammeri H. 2013. Contribution of asparagine 346 residue to the carbapenemase activity of CMY-2 β-lactamase. FEMS Microbiol Lett 345:147–153. doi: 10.1111/1574-6968.12199. [DOI] [PubMed] [Google Scholar]
- 5.Philippon A, Arlet G, Jacoby GA. 2002. Plasmid-determined AmpC-type beta-lactamases. Antimicrob Agents Chemother 46:1–11. doi: 10.1128/aac.46.1.1-11.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reisbig MD, Hanson ND. 2004. Promoter sequences necessary for high-level expression of the plasmid-associated ampC beta-lactamase gene blaMIR-1. Antimicrob Agents Chemother 48:4177–4182. doi: 10.1128/AAC.48.11.4177-4182.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacoby GA. 2009. AmpC β-lactamases. Clin Microbiol Rev 22:161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehmann DE, Jahić H, Ross PL, Gu R-F, Hu J, Kern G, Walkup GK, Fisher SL. 2012. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci U S A 109:11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodríguez-Baño J, Gutiérrez-Gutiérrez B, Machuca I, Pascual A. 2018. Treatment of infections caused by extended-spectrum-beta-lactamase-, AmpC-, and carbapenemase-producing Enterobacteriaceae. Clin Microbiol Rev 31:e00079-17. doi: 10.1128/CMR.00079-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lahiri SD, Walkup GK, Whiteaker JD, Palmer T, McCormack K, Tanudra MA, Nash TJ, Thresher J, Johnstone MR, Hajec L, Livchak S, McLaughlin RE, Alm RA. 2015. Selection and molecular characterization of ceftazidime/avibactam-resistant mutants in Pseudomonas aeruginosa strains containing derepressed AmpC. J Antimicrob Chemother 70:1650–1658. doi: 10.1093/jac/dkv004. [DOI] [PubMed] [Google Scholar]
- 11.Livermore DM, Mushtaq S, Doumith M, Jamrozy D, Nichols WW, Woodford N. 2018. Selection of mutants with resistance or diminished susceptibility to ceftazidime/avibactam from ESBL- and AmpC-producing Enterobacteriaceae. J Antimicrob Chemother 73:3336–3345. doi: 10.1093/jac/dky363. [DOI] [PubMed] [Google Scholar]
- 12.Lahiri SD, Mangani S, Durand-Reville T, Benvenuti M, De Luca F, Sanyal G, Docquier J-D. 2013. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-lactamases. Antimicrob Agents Chemother 57:2496–2505. doi: 10.1128/AAC.02247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lahiri SD, Johnstone MR, Ross PL, McLaughlin RE, Olivier NB, Alm RA. 2014. Avibactam and class C β-Lactamases: mechanism of inhibition, conservation of the binding pocket, and implications for resistance. Antimicrob Agents Chemother 58:5704–5713. doi: 10.1128/AAC.03057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papp-Wallace KM, Mallo S, Bethel CR, Taracila MA, Hujer AM, Fernández A, Gatta JA, Smith KM, Xu Y, Page MGP, Desarbre E, Bou G, Bonomo RA. 2014. A kinetic analysis of the inhibition of FOX-4 β-lactamase, a plasmid-mediated AmpC cephalosporinase, by monocyclic β-lactams and carbapenems. J Antimicrob Chemother 69:682–690. doi: 10.1093/jac/dkt434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Compain F, Arthur M. 2017. Impaired inhibition by avibactam and resistance to the ceftazidime-avibactam combination due to the D(179)Y substitution in the KPC-2 β-lactamase. Antimicrob Agents Chemother 61:e00451-17. doi: 10.1128/AAC.00451-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Compain F, Dorchène D, Arthur M. 2018. Combination of amino acid substitutions leading to CTX-M-15-mediated resistance to the ceftazidime-avibactam combination. Antimicrob Agents Chemother 62:e00357-18. doi: 10.1128/AAC.00357-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clinical and Laboratory Standards Institute. 2015. Methods for dilution susceptibility tests for bacteria that grow aerobically; approved standard. Clin Lab Stand Inst M7-A8, 10th ed Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 18.Ourghanlian C, Soroka D, Arthur M. 2017. Inhibition by avibactam and clavulanate of the β-lactamases KPC-2 and CTX-M-15 harboring the substitution N132G in the conserved motif SDN. Antimicrob Agents Chemother 61:e02510-16. doi: 10.1128/AAC.02510-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.