

Abstract
Studies have demonstrated that long non-coding RNAs (lncRNAs) play important roles in cancer development and progression. However, associations between the expression patterns and prognostic roles of lncRNAs in hepatocellular carcinoma (HCC) have not been comprehensively described. In this study, we established a prognostic model of lncRNA expression using public datasets of HCC from The Cancer Genome Atlas (TCGA) and adopted the International Cancer Genome Consortium (ICGC) as an independent cohort to validate the stability of our model. Cox regression analysis was used to explore the independent prognostic factor in both training and validation cohorts. Additionally, we explored the functional roles of lncRNAs using bioinformatic analyses. According to lncRNA consensus clusters, we resolved the distribution of molecular and clinical data and observed that individual lncRNA could function as prognostic biomarkers in HCC. Furthermore, the novel lncRNA molecular subtypes were statistically significant for predicting HCC status, which was validated by nested cross-validation. We found that lncRNA subtypes were partially related to gender, histological grade, and mutations within TP53. The lncRNA subtypes were also consistent with mRNA-based subtypes, and pathway enrichment analysis identified the involvement of multiple signaling pathways. In addition, we observed that upregulated DANCR was significantly associated with poor prognosis in HCC patients. In conclusion, our model based on lncRNA expression is statistically significant as a diagnostic and prognostic indicator for patients with HCC.
Keywords: lncRNAs, hepatocellular carcinoma, overall survival
Introduction
Hepatocellular carcinoma (HCC) has an extremely poor prognosis and is one of the most common malignancies worldwide [1,2]. Despite recent progress made with surgery and targeted therapy, recurrence, distant metastasis, and drug resistance remain clinical challenges that cause poor overall survival (OS) in HCC patients [3,4]. Thus, it is critical to explore the molecular mechanisms underlying HCC tumorigenesis and progression, and to identify new predictive biomarkers and therapeutic targets for improving HCC patient outcomes.
Increasing evidence has revealed that non-coding RNAs (ncRNAs), including microRNAs, long non-coding RNAs (lncRNAs), and circular RNA, play essential roles in tumorigenesis [5-7]. Previous studies have shown that ncRNAs can modulate target gene expression in cancer [5]. Competing endogenous RNA (ceRNA) networks have been identified as critical regulators in a variety of cancers, including HCC [8,9]. For example, Wang et al. [10] found that lncRNA MCM3AP-AS1 could play an oncogenic role in promoting HCC progression through the miR-194-5p/FOXA1 axis, and could be a novel prognostic biomarker and therapeutic target for HCC. However, associations between the expression and functional roles of lncRNAs in HCC are still under investigation.
LncRNAs, defined as transcripts of over 200 nucleotides in length, are broadly transcribed in the human genome [11]. Growing evidence suggests that lncRNAs are a category of non-coding RNAs that play essential roles in numerous biological processes, including gene transcription and mRNA splicing, cancer carcinogenesis and progression, signal transduction, and RNA activation and stability [12,13]. Deregulated lncRNAs have been implicated in a variety of malignant tumors including breast, bladder, and colorectal cancers, osteosarcoma, glioblastoma, and HCC, illustrating the broad involvement of lncRNAs in tumorigenesis and cancer progression [7,14,15]. However, a greater understanding of the significance of lncRNA expression patterns as diagnostic and prognostic biomarkers remains to be established.
In the present study, we explored the expression of lncRNAs and assessed their association with the outcomes of HCC patients. Additionally, we defined novel lncRNA-based HCC subtypes and ascertained their prognostic value. An independent cohort was adopted to validate our model. Moreover, Cox regression analysis was used to explore the independent prognostic factor in both training and validation cohorts. Collectively, our findings reveal a novel approach for prognostic prediction in HCC based on lncRNA expression.
Materials and methods
Patient cohorts
We collected the Liver Hepatocellular Carcinoma (LIHC) dataset from the data portal of The Cancer Genome Atlas (TCGA) (https://gdc.cancer.gov), as described previously [16,17]. After excluding samples with a survival time of less than 30 days, the TCGA-LIHC cohort included 278 cases with clinical information and expression of lncRNAs available for further analysis. Additionally, we obtained 212 cases from the International Cancer Genome Consortium (ICGC) with similar data available [18] (https://dcc.icgc.org/projects/LIRI-JP). After excluding samples with a survival time of less than 30 days, the ICGC-LIHC cohort included 212 cases available as an independent validation cohort. Furthermore, GEPIA (http://gepia.cancer-pku.cn/) database was adopted to detect the expression and function of DANCR.
Subtype discovery and further validation
We adopted an unsupervised learning approach for subtype discovery, and then utilized K-medoids clustering algorithm. Our optimal number of cluster (k = 2 to 10) was adopted, and we observed that k = 2 was selected utilizing a weighted silhouette index. The results of the clustering algorithm are described in Table S1 and Figure S1, with protocols described previously [19].
Probably Approximately Correct (PAC) method was a non-deterministic architecture for information retrieval, applied to determine and correct the optimal cluster number. M is used to calculate the consensus moment. This PAC-learning method extracts the data of the lower triangle of the consensus matrix, and then we use this correction method to generate a curve fit to calculate the area between 0.1 and 0.9. The minimum area corresponding to k is the optimal k, which was k = 2 (Table S1).
Nested cross-validation and lncRNA subtype
We adopted a 10-fold cross-validation on our data, and the training and test datasets were divided randomly for each round as described previously [20,21]. We first adopted an unsupervised learning method on the training set, and utilized labels to train the random forest model. Similar methods were performed on the test dataset to validate our model.
Clustering of mRNA expression data
A clustering scheme similar to the lncRNA expression-based subtype identification was used for mRNA expression data clustering. We removed samples with read counts equal to zero and used a log2 read count value to normalize the data. Consistent clustering of genomic information identified that k was a robust cluster varying between 2 and 10 clusters. For each value of k, we performed Monte Carlo subsampling 1,000 times on the samples and features. We then aggregated the clustering labels of different subsampling rounds to establish a consistent matrix, and clustered samples based on this matrix. Similar to the lncRNA subtype identification, PAC method was used to find the optimal cluster number in the mRNA expression data set, in which k = 2.
Statistical analysis
All data are shown as the mean ± standard deviation (SD). Chi-squared tests were performed for association analyses. Statistical significance was calculated with R software version 3.6. Kaplan-Meier analysis was performed to analyze the correlation between lncRNA expression and survival time. Univariable and multivariable Cox regression models were adopted to analyze independent prognostic factors. A two-sided P < 0.05 was identified as statistically significant.
Results
Characteristics of lncRNA in TCGA-LIHC cohort
To characterize lncRNA expression in the TCGA-LIHC cohort, we utilized RNA sequencing and the MiTranscriptome database to annotate lncRNAs. Two subtypes of lncRNA were discovered based on expression in 278 cases of HCC using the consensus cluster method. This was validated in another independent cohort (ICGC-LIHC). A description of the notated and evaluated characteristics within the TCGA-LIHC cohort is shown in Table 1. The distribution of molecular and clinical data through an analysis of the lncRNA consensus clusters is also included (Figure 1).
Table 1.
Description of TCGA-LIHC cohort
| Number of patients | 278 |
|---|---|
| Sex | |
| Male | 193 (69.42%) |
| Female | 85 (30.58%) |
| Age | |
| Median (range) | 59 (18-82) |
| aged < 60 | 141 (50.72%) |
| AJCC stage | |
| I | 139 (50.00%) |
| II | 67 (24.10%) |
| III | 69 (24.82%) |
| IV | 3 (1.08%) |
| Invasion | |
| Invasion | 77 (27.70%) |
| None | 201 (72.30%) |
| Mutations | |
| APOB | 34 (12.23%) |
| CSMD3 | 33 (11.87%) |
| MUC16 | 65 (23.38%) |
| MUC4 | 43 (15.47%) |
| PCLO | 38 (13.67%) |
| RYR2 | 37 (13.31%) |
| TP53 | 89 (32.01%) |
Figure 1.
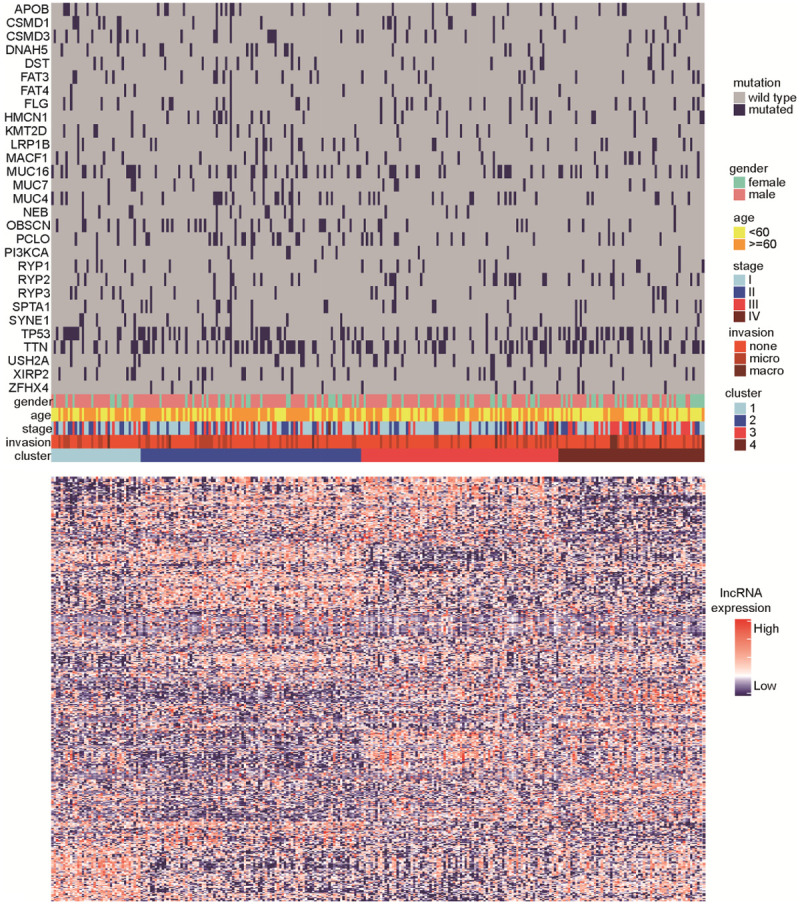
Distribution map of molecular and clinical data based on lncRNA consensus clustering. Four subtypes were identified based on lncRNA expression patterns, clinicopathological factors, and somatic mutations.
Individual lncRNA as a prognostic biomarker in HCC
We next explored the extent to which individual lncRNA were related to the outcomes of HCC patients in the TCGA-LIHC cohort. Univariate Cox regression models were adopted for individual lncRNA using time on-study as the time scale, adjusting for age, sex, tumor grade, tumor metastasis, and mutation status of APOB, CSMD3, MUC16, MUC4, PCLO, RYR2, and TP53 as covariates in the models. We found that 45 lncRNAs could serve as prognostic biomarkers of overall survival (OS) in HCC with statistical significance (Figure 2). After analyzing the data in the ICGC-LIHC cohort for validation, six lncRNAs (LINC00324, LINC01554, MIR99AHG, DNMBP-AS1, SNHG22, and DANCR) were found to be statistically significant (Figure S2), and DANCR was the only independent risk factor in two cohorts. Taken together, our results indicated that these lncRNAs could serve as novel biomarkers for prognostic prediction.
Figure 2.
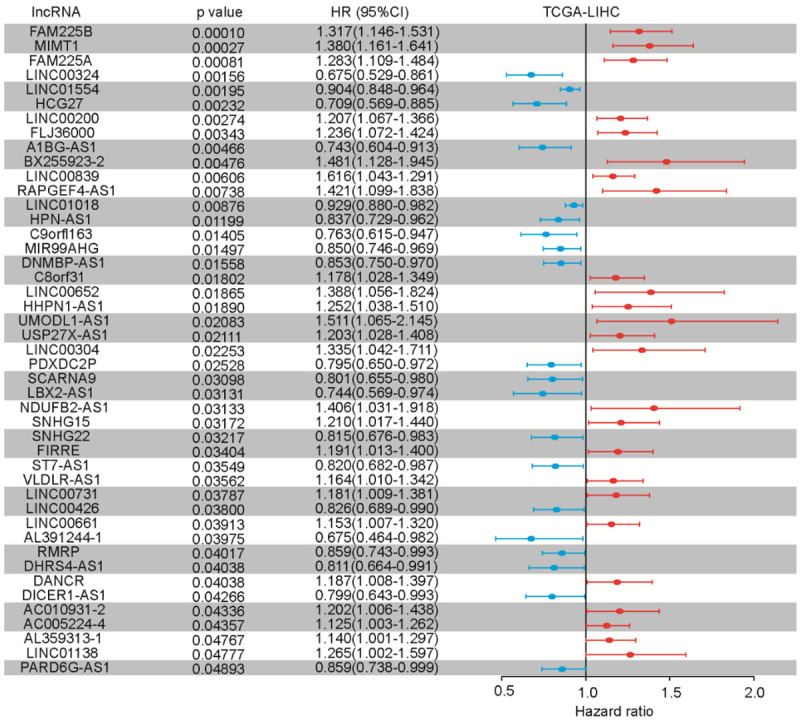
Multivariate time-to-event analysis (overall survival) of individual lncRNAs (adjusting for established risk factors) in the TCGA-LIHC cohort, and validation of 45 prognoses of lncRNAs which correlated with overall survival.
Novel molecular subtypes and prognostication based on lncRNA expression in HCC
To further resolve the role of lncRNAs in HCC, we investigated whether subgroups of HCC cases in the TCGA-LIHC cohort shared common multivariate lncRNA expression levels. We discovered two distinct lncRNA subtypes using an unsupervised learning approach based on lncRNA expression profiles. Results of consensus clustering revealed a high degree of co-clustering of subjects within these two groups (Figure S1). This demonstrated that patients from the TCGA-LIHC cohort could be divided into two distinct subtypes according to lncRNA expression. We next explored the prognostic value of the lncRNA subtypes with respect to HCC patient OS. Kaplan-Meier analysis concluded that there was a significant difference in the prognostic value for the two lncRNA-based subtypes, with Group 1 (n = 131) having a worse survival outcome than Group 2 (n = 147) (Figure 3A). To validate the prognostic value of these lncRNA subtypes, we analyzed the independent ICGC-LIHC cohort and again found the lncRNA subtypes to be statistically significant, with patients in Group 2 (n = 99) having a better OS (Figure 3B). Moreover, multivariable Cox proportional hazards models were established for both the TCGA-LIHC (Figure 3C) and ICGC-LIHC (Figure 3D) cohorts using the identified lncRNA prognostic factors, with both models providing statistically significance prognostic values.
Figure 3.

Multivariate survival analysis (Cox proportional hazards model) of lncRNA subtypes, including clinicopathological factors and mutations, for the TCGA-LIHC and ICGC-LIHC cohorts. LncRNA expression level and the roles for lncRNA-associated survival in HCC are exhibited in (A and B). LncRNA expression-based subtypes and overall survival (OS) in the TCGA-LIHC and ICGC-LIHC cohorts are illustrated in (C and D).
Assessing the reproducibility of novel lncRNA HCC subtypes
To verify the consistency of the two lncRNA subtypes for HCC, we adopted a training set for a nested cross-validation experiment and an independent test set for model evaluation with respect to the prognostic value as described [22]. The overall correct classification rate based on test set cases was 0.9, as showed in Figure 4A. We concluded that the lncRNA subtypes were reliable as prognostic factors.
Figure 4.
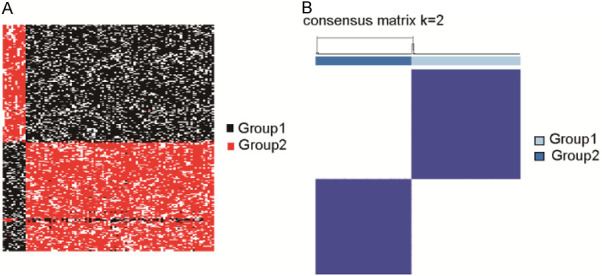
HCC model validation through the ICGC-LIHC cohort. A. Nested cross-validation cluster analysis was utilized to validate the cohort. B. Consensus clustering and unsupervised classification for profiling matrix for two groups in ICGC-LIHC (model selection results for k = 2).
To further evaluate the reproducibility of these novel lncRNA HCC subtypes, we established a random forest model for subtype classification using an independent trained ICGC-LIHC cohort. Based on lncRNA expression in 212 cases by the consensus cluster method, we discovered two subtypes (Figure 4B). In the ICGC-LIHC cohort, the prognostic value of the two lncRNA subtypes was similar to TCGA-LIHC, and the OS of patients in Group 2 was significantly longer (Figure 3C). Moreover, we established a model based on multivariable Cox proportional hazards, including age, sex, and the prognostic factors above from TCGA clinical and mutation data (Figure 3D). The value of this established model was confirmed to be remarkably significant (P < 0.05).
LncRNA subtypes were partially related to HCC pathological factors
We next performed association studies to determine whether the lncRNA subtypes were related to clinical characteristics and known genes mutation within HCC. Using the ICGC-LIHC cohort, we found a significant difference between the two lncRNA subtypes (Table 2). We observed that number of stage I patients in Group 1 was significant more than Group 2 (P = 0.0082), while number of stage III patients in Group 2 were significant more than Group 1 (P = 0.0124). These results were consistent with the findings that Group 1 had poorer OS. Additionally, we observed that TP53 mutations were strongly linked with the lncRNA subtypes, in that Group 1 contained a higher percentage of TP53 mutations (41.98%) and poorer prognosis compared with Group 2. In summary, lncRNA subtypes could provide additional levels of stratification for HCC patients.
Table 2.
Description of ICGC-LIHC cohort
| Cluster | Group 1 (%) | Group 2 (%) | p-value (Chi-squared test) | Significance |
|---|---|---|---|---|
| Patients | 131 (47.12%) | 147 (52.88%) | ||
| Sex | ||||
| Male | 66 (50.38%) | 127 (86.39%) | 1.8273e-10 | *** |
| Female | 65 (49.62%) | 20 (13.61%) | ||
| Age | ||||
| Median (range) | 57 (18-81) | 61 (41.50%) | 0.8276 | |
| aged > 60 | 55 (41.98%) | 86 (58.50%) | ||
| AJCC stage | ||||
| I | 54 (41.22%) | 85 (57.82%) | 0.0082 | ** |
| II | 34 (25.95%) | 33 (22.45%) | 0.5881 | |
| III | 42 (32.06%) | 27 (18.37%) | 0.0124 | * |
| IV | 1 (0.76%) | 2 (1.36%) | 0.9200 | |
| Invasion | ||||
| Invasion | 41 (31.30%) | 36 (24.49%) | 0.2055 | |
| None | 90 (68.70%) | 111 (75.51%) | ||
| Mutations | ||||
| APOB | 12 (9.16%) | 22 (14.97%) | 0.1965 | |
| CSMD3 | 17 (12.98%) | 16 (10.88%) | 0.7243 | |
| MUC16 | 35 (26.72%) | 30 (20.41%) | 0.2719 | |
| MUC4 | 21 (16.03%) | 22 (14.97%) | 0.9371 | |
| PCLO | 13 (9.92%) | 25 (17.01%) | 0.1233 | |
| RYR2 | 19 (14.50%) | 18 (12.24%) | 0.7065 | |
| TP53 | 55 (41.98%) | 34 (23.13%) | 0.0012 | ** |
Abbreviations: *P < 0.05, **P < 0.01, ***P < 0.001.
Pathway analysis of genes associated with lncRNA subtypes
To demonstrate which genes were related to the lncRNA subtypes, we integrated mRNA into our bioinformatic analysis. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis identified several significant pathways involved (Figure 5A), including “HPV infection” and “Neuroactive ligand-receptor interaction”. Gene ontology (GO) analysis into molecular function in cellular processes showed involvement in “Transmembrane transporter activity”, “Catabolic process”, and “Synapse and channel complex” (Figure 5B-D).
Figure 5.

Pathway enrichment and GO analyses of lncRNAs in HCC samples. A. KEGG analysis. B. GO analysis for molecular function assessment. C. GO analysis for biological process assessment. D. GO analysis for cellular component assessment.
Consistency between lncRNA and mRNA subtyping
To assess the degree that lncRNA subtypes were consistent with mRNA subtypes, we performed an identical unsupervised learning method to evaluate mRNA expression clusters similar to the lncRNA analysis, and divided mRNA expression into two groups. Despite a few cases that appeared to cross groups, most belonged to the same group (Figure 6A). These results showed that there was a significant relationship between the two models, demonstrating consistency between the mRNA and lncRNA expression profiles.
Figure 6.
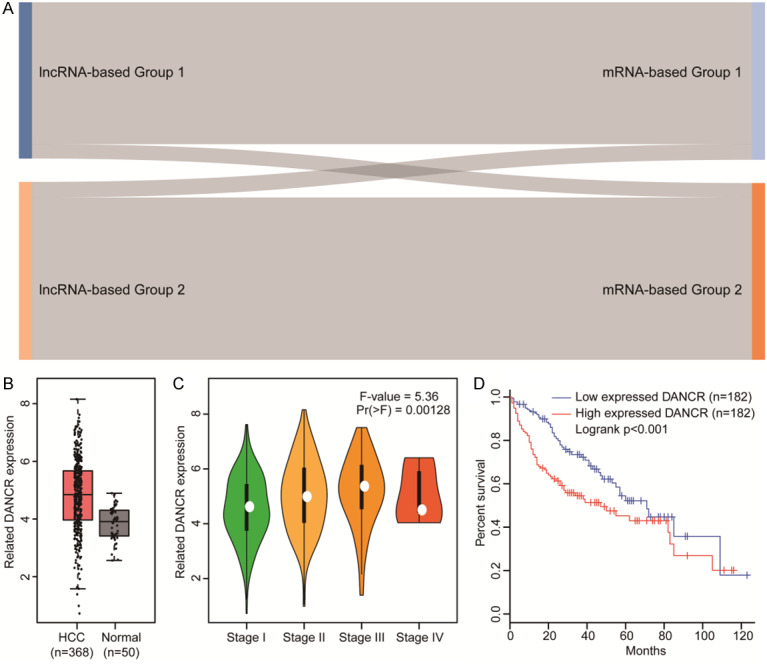
LncRNA and mRNA model validation and DANCR analyses. A. Sankey diagram of the relationship between mRNA-defined (right) and lncRNA-defined (left) subtype classifications. B. DANCR expression was significantly elevated in HCC tissues in a TCGA data analysis. C. LACTB expression was much higher in advanced TNM stages. D. Kaplan-Meier analysis showing that HCC patient OS is significantly worse with high DANCR expression than low expression.
High DANCR expression correlates with HCC patient prognosis
Considering that DANCR was the only independent risk factor in the TCGA and ICGC cohorts, we investigated the expression and function of DANCR in HCC. As shown in Figure 6B, we observed that DANCR expression was significantly elevated in HCC tissues compared to normal tissues. Moreover, we found that the expression of DANCR was much higher in advanced TNM staging (Figure 6C). Kaplan-Meier analysis showed that OS of HCC patients was significantly worse with high dancer DANCR expression than low expression (Figure 6D). Collectively, our results demonstrated that DANCR expression was elevated and contributed to poor prognosis in HCC patients.
Discussion
A growing number of studies show that poor outcomes in HCC can be attributed to late diagnosis, tumor recurrence, and unsatisfactory treatment [23]. Therefore, it is urgent to develop powerful diagnostic and novel therapeutic strategies to improve the diagnosis and treatment of HCC. Studies have shown that expression levels of lncRNAs could regulate mRNA expression and act in critical roles to regulate HCC pathogenesis [9]. HOXD-AS1, MCM3AP-AS1, CASC2, and RMRP were found to promote HCC progression [10,24-26], providing novel insight into prognostic biomarker exploration. Emerging evidence shows that cancer-related lncRNAs could be diagnostic or predictive biomarkers to predict OS in HCC patients [27,28], but the functional roles of cancer-related lncRNAs has not been fully characterized for HCC.
In the present study, we characterized lncRNA expression by accessing the TCGA database to analyze a TCGA-LIHC cohort of 278 HCC patients in order to ascertain if particular lncRNAs correlated with HCC status, and if prognostic subtypes of HCC-based lncRNAs could be delineated. Our findings were then validated in a separate ICGC-LIHC cohort. DANCR was found to be the only independent risk factor in two cohorts [29-31]. Similar to our findings, studies have shown DANCR as a potential cancer-related lncRNA in different malignancies, including hepatocellular carcinoma. Yuan et al. [32] found that DANCR was overexpressed in stem-like HCC cells, and could function as a prognostic biomarker in HCC.
In the TCGA-LIHC cohort, we observed that 45 individual lncRNA could serve as independent prognosticators of OS in HCC. In previous studies, it has been determined that gene expression characteristics and prognostic models based on molecular features that drive pathogenesis were an effective means for predicting clinical outcomes [33-35]. There is increasing evidence that lncRNAs possess the potential to offer critical forecasting in the survival of HCC patients, with more lncRNAs thought to affect the evolution of HCC through their own molecular mechanisms [36,37]. Through our exploration of RNA sequencing data from the TCGA database, 23 lncRNAs were found to be of good predictive value for HCC prognosis. Further validation in the ICGC-LIHC cohort confirmed the value of lncRNAs as a new prognostic biomarker.
Our study also identified two robust lncRNA subtypes as prognostic indicators of HCC OS. In both the TCGA-LIHC and ICGC-LIHC cohorts, the survival time in Group 2 was longer than Group 1, illustrating the consistency of this subtype discovery and predictive modeling. We also trained a random forest model based on the TCGA data for subtype classification to assess the reproducibility of newly discovered HCC subtypes. Our analysis showed that lncRNA subtypes were consistent with mRNA subtypes, indicating that there may be a correlation between lncRNA and mRNA subtypes to be leveraged for diagnostics and treatment. Furthermore, the related pathways were identified through KEGG and GO analysis, indicating that lncRNAs may play special roles through specific signaling pathways in HCC. Further investigation into these signaling pathways could provide support toward the discovery of novel HCC targets.
In summary, by taking advantage of public databases to probe lncRNAs from TCGA-LIHC and ICGC-LIHC cohorts, we created a valid predictive model and identified lncRNA-based subtypes of HCC which could become novel prognostic biomarkers. Further investigation beyond this study is required to prove the clinical potential of its findings. In addition, the molecular mechanisms and biological functions of these newly identified lncRNAs must be explored further through in vitro and in vivo experiments.
Conclusions
In this study, we detected the expression of lncRNAs and assessed their association with the OS of HCC patients. We defined novel HCC subtypes based on lncRNA and ascertained their prognostic value. An independent cohort was also utilized to validate our model. Additionally, Cox regression analysis was used to explore the independent prognostic factors in both training and validation cohorts. Collectively, our findings reveal a novel approach of predicting HCC status based on lncRNA expression.
Acknowledgements
We thank Dr. Weiwei Zhu (The First affiliated Hospital, School of Medicine, Zhejiang University) for help in literatures collection, and the patients and investigators who participated in TCGA, ICGC and GEPIA for providing data. This study was supported by the National Natural Science Foundation of China (81902832 and 81672422); Open Project in State Key Laboratory for Diagnosis and Treatment of Infectious Disease (2015KF03); Zhejiang Province Health Department Program (2017KY322), and National S&T Major Project of China (2018ZX10301201).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–1314. doi: 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- 3.Bruix J, da Fonseca LG, Reig M. Insights into the success and failure of systemic therapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2019;16:617–630. doi: 10.1038/s41575-019-0179-x. [DOI] [PubMed] [Google Scholar]
- 4.Zhou ZQ, Tong DN, Guan J, Tan HW, Zhao LD, Zhu Y, Yao J, Yang J, Zhang ZY. Follicular helper T cell exhaustion induced by PD-L1 expression in hepatocellular carcinoma results in impaired cytokine expression and B cell help, and is associated with advanced tumor stages. Am J Transl Res. 2016;8:2926–2936. [PMC free article] [PubMed] [Google Scholar]
- 5.Beermann J, Piccoli MT, Viereck J, Thum T. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev. 2016;96:1297–1325. doi: 10.1152/physrev.00041.2015. [DOI] [PubMed] [Google Scholar]
- 6.Anfossi S, Babayan A, Pantel K, Calin GA. Clinical utility of circulating non-coding RNAs - an update. Nat Rev Clin Oncol. 2018;15:541–563. doi: 10.1038/s41571-018-0035-x. [DOI] [PubMed] [Google Scholar]
- 7.Yan X, Hu Z, Feng Y, Hu X, Yuan J, Zhao SD, Zhang Y, Yang L, Shan W, He Q, Fan L, Kandalaft LE, Tanyi JL, Li C, Yuan CX, Zhang D, Yuan H, Hua K, Lu Y, Katsaros D, Huang Q, Montone K, Fan Y, Coukos G, Boyd J, Sood AK, Rebbeck T, Mills GB, Dang CV, Zhang L. Comprehensive genomic characterization of long non-coding RNAs across human cancers. Cancer Cell. 2015;28:529–540. doi: 10.1016/j.ccell.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bai Y, Long J, Liu Z, Lin J, Huang H, Wang D, Yang X, Miao F, Mao Y, Sang X, Zhao H. Comprehensive analysis of a ceRNA network reveals potential prognostic cytoplasmic lncRNAs involved in HCC progression. J Cell Physiol. 2019;234:18837–18848. doi: 10.1002/jcp.28522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin P, Wen DY, Li Q, He Y, Yang H, Chen G. Genome-wide analysis of prognostic lncRNAs, miRNAs, and mRNAs forming a competing endogenous RNA network in hepatocellular carcinoma. Cell Physiol Biochem. 2018;48:1953–1967. doi: 10.1159/000492519. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Yang L, Chen T, Liu X, Guo Y, Zhu Q, Tong X, Yang W, Xu Q, Huang D, Tu K. A novel lncRNA MCM3AP-AS1 promotes the growth of hepatocellular carcinoma by targeting miR-194-5p/FOXA1 axis. Mol Cancer. 2019;18:28. doi: 10.1186/s12943-019-0957-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engreitz JM, Ollikainen N, Guttman M. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol. 2016;17:756–770. doi: 10.1038/nrm.2016.126. [DOI] [PubMed] [Google Scholar]
- 13.Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- 14.Wong CM, Tsang FH, Ng IO. Non-coding RNAs in hepatocellular carcinoma: molecular functions and pathological implications. Nat Rev Gastroenterol Hepatol. 2018;15:137–151. doi: 10.1038/nrgastro.2017.169. [DOI] [PubMed] [Google Scholar]
- 15.Zheng S, Lin F, Zhang M, Mu N, Ge X, Fu J. Long non-coding RNA AK001058 regulates tumor growth and angiogenesis in colorectal cancer via methylation of ADAMTS12. Am J Transl Res. 2019;11:6117–6123. [PMC free article] [PubMed] [Google Scholar]
- 16.Xue C, He Y, Zhu W, Chen X, Yu Y, Hu Q, Chen J, Liu L, Ren F, Ren Z, Cui G, Sun R. Low expression of LACTB promotes tumor progression and predicts poor prognosis in hepatocellular carcinoma. Am J Transl Res. 2018;10:4152–4162. [PMC free article] [PubMed] [Google Scholar]
- 17.Lee H, Palm J, Grimes SM, Ji HP. The cancer genome atlas clinical explorer: a web and mobile interface for identifying clinical-genomic driver associations. Genome Med. 2015;7:112. doi: 10.1186/s13073-015-0226-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Bajari R, Andric D, Gerthoffert F, Lepsa A, Nahal-Bose H, Stein LD, Ferretti V. The international cancer genome consortium data portal. Nat Biotechnol. 2019;37:367–369. doi: 10.1038/s41587-019-0055-9. [DOI] [PubMed] [Google Scholar]
- 19.Damgacioglu H, Celik E, Celik N. Intra-cluster distance minimization in DNA methylation analysis using an advanced Tabu-based iterative k-medoids clustering algorithm (T-CLUST) IEEE/ACM Trans Comput Biol Bioinform. 2018 doi: 10.1109/TCBB.2018.2886006. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 20.Baumann D, Baumann K. Reliable estimation of prediction errors for QSAR models under model uncertainty using double cross-validation. J Cheminform. 2014;6:47. doi: 10.1186/s13321-014-0047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo Y, McShan D, Ray D, Matuszak M, Jolly S, Lawrence T, Ming Kong F, Ten Haken R, El Naqa I. Development of a fully cross-validated bayesian network approach for local control prediction in lung cancer. IEEE Trans Radiat Plasma Med Sci. 2019;3:232–241. doi: 10.1109/TRPMS.2018.2832609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Wang H, Mao X, Guo Q, Li W, Wang X, Li G, Lin N. A novel gene-expression-signature-based model for prediction of response to Tripterysium glycosides tablet for rheumatoid arthritis patients. J Transl Med. 2018;16:187. doi: 10.1186/s12967-018-1549-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16:589–604. doi: 10.1038/s41575-019-0186-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu S, Zhou J, Sun Y, Li N, Miao M, Jiao B, Chen H. The noncoding RNA HOXD-AS1 is a critical regulator of the metastasis and apoptosis phenotype in human hepatocellular carcinoma. Mol Cancer. 2017;16:125. doi: 10.1186/s12943-017-0676-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan JC, Zeng F, Le YG, Xin L. LncRNA CASC2 inhibited the viability and induced the apoptosis of hepatocellular carcinoma cells through regulating miR-24-3p. J Cell Biochem. 2018;119:6391–6397. doi: 10.1002/jcb.26479. [DOI] [PubMed] [Google Scholar]
- 26.Hongfeng Z, Andong J, Liwen S, Mingping B, Xiaowei Y, Mingyong L, Aimin Y. lncRNA RMRP knockdown suppress hepatocellular carcinoma biological activities via regulation miRNA-206/TACR1. J Cell Biochem. 2020;121:1690–1702. doi: 10.1002/jcb.29404. [DOI] [PubMed] [Google Scholar]
- 27.Liu H, Gu X, Wang G, Huang Y, Ju S, Huang J, Wang X. Copy number variations primed lncRNAs deregulation contribute to poor prognosis in colorectal cancer. Aging (Albany NY) 2019;11:6089–6108. doi: 10.18632/aging.102168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song L, Zhang S, Duan C, Ma S, Hussain S, Wei L, Chu M. Genome-wide identification of lncRNAs as novel prognosis biomarkers of glioma. J Cell Biochem. 2019;120:19518–19528. doi: 10.1002/jcb.29259. [DOI] [PubMed] [Google Scholar]
- 29.Lu Y, Hu Z, Mangala LS, Stine ZE, Hu X, Jiang D, Xiang Y, Zhang Y, Pradeep S, Rodriguez-Aguayo C, Lopez-Berestein G, DeMarzo AM, Sood AK, Zhang L, Dang CV. MYC targeted long noncoding RNA DANCR promotes cancer in part by reducing p21 levels. Cancer Res. 2018;78:64–74. doi: 10.1158/0008-5472.CAN-17-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thin KZ, Liu X, Feng X, Raveendran S, Tu JC. LncRNA-DANCR: a valuable cancer related long non-coding RNA for human cancers. Pathol Res Pract. 2018;214:801–805. doi: 10.1016/j.prp.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Ma X, Wang X, Yang C, Wang Z, Han B, Wu L, Zhuang L. DANCR acts as a diagnostic biomarker and promotes tumor growth and metastasis in hepatocellular carcinoma. Anticancer Res. 2016;36:6389–6398. doi: 10.21873/anticanres.11236. [DOI] [PubMed] [Google Scholar]
- 32.Yuan SX, Wang J, Yang F, Tao QF, Zhang J, Wang LL, Yang Y, Liu H, Wang ZG, Xu QG, Fan J, Liu L, Sun SH, Zhou WP. Long noncoding RNA DANCR increases stemness features of hepatocellular carcinoma by derepression of CTNNB1. Hepatology. 2016;63:499–511. doi: 10.1002/hep.27893. [DOI] [PubMed] [Google Scholar]
- 33.Franco HL, Nagari A, Malladi VS, Li W, Xi Y, Richardson D, Allton KL, Tanaka K, Li J, Murakami S, Keyomarsi K, Bedford MT, Shi X, Li W, Barton MC, Dent SYR, Kraus WL. Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res. 2018;28:159–170. doi: 10.1101/gr.226019.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karamali M, Shafabakhsh R, Ghanbari Z, Eftekhar T, Asemi Z. Molecular pathogenesis of interstitial cystitis/bladder pain syndrome based on gene expression. J Cell Physiol. 2019;234:12301–12308. doi: 10.1002/jcp.28009. [DOI] [PubMed] [Google Scholar]
- 35.Zhou N, He Z, Tang H, Jiang B, Cheng W. LncRNA RMRP/miR-613 axis is associated with poor prognosis and enhances the tumorigenesis of hepatocellular carcinoma by impacting oncogenic phenotypes. Am J Transl Res. 2019;11:2801–2815. [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Liao Z, Liu F, Su C, Zhu H, Li Y, Tao R, Liang H, Zhang B, Zhang X. Long noncoding RNA HULC promotes hepatocellular carcinoma progression. Aging (Albany NY) 2019;11:9111–9127. doi: 10.18632/aging.102378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu X, Gu J, Ding X, Ge G, Zang X, Ji R, Shao M, Mao Z, Zhang Y, Zhang J, Mao F, Qian H, Xu W, Cai H, Wang F, Zhang X. LINC00978 promotes the progression of hepatocellular carcinoma by regulating EZH2-mediated silencing of p21 and E-cadherin expression. Cell Death Dis. 2019;10:752. doi: 10.1038/s41419-019-1990-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.